Organometallics 1994, 13, 1699-1710 1699
Synthesis, Structure, Fluxional Behavior, and Addition
Reaction of the Metal-Metal-Bonded Heterobimetallic
Phosphido-Bridged Complex C
pW(
CO)
2(p-PPh2)
M o
(CO)
5Shin-Guang Shyu,**t
Jiun-Yi Hsu,? Pei-Jung Lin,? Wen-Jin Wu,? Shie-Ming Peng,*J
Gene-Hsiang Lee,$ and Yuh-Shang Went
Institute of Chemistry, Academia Sinica, Taipei, Taiwan 11529, Republic of China, and Department of Chemistry, National Taiwan University, Taipei, Taiwan, Republic of China
Received September 20, 1993@
T h e heterobimetallic phosphido-bridged complex CpW(C0)2(p-PPh2)Mo(C0)5 (1) was prepared by the reaction of CpW(C0)3PPh2 with M o ( C ~ ) ~ ( C ~ H ~ ) . Treatment of 1 with CO produced CpW(C0)3(p-PPhz)Mo(C0)5
(2).
T h e structures of 1 and 2 were determined by single- crystal X-ray diffraction. Crystal data for 1: C ~ ~ H I ~ M O O ~ P W , space group P21/n, a = 14.464(7)A,
b = 11.6743(12)A,
c = 14.7639(14)A,
p
= 97.916(12)',V
= 2469.3(6)A3,
2 = 4. T h e structure was refined t oR
= 0.054 a n dR,
= 0.055. Crystal data for 2: C ~ ~ H I ~ M O O ~ P W , space group P21/n,a = 9.6322(16)
A,
b = 12.4673(21)A,
c = 20.813(3)A,
/3 = 94.476(12)',V
= 2472.7(7) A3, 2 = 4. T h e structure was refined t oR
= 0.035 andR,
= 0.044. T h e W-Mo distance was 3.2056(16)A
in 1, indicative of a W-Mo bond. T h e long distance between W and Mo (4.5192(14)A)
in2
indicates t h a t no metal-metal bond exists in the complex. Fluxional behavior involving the exchange of four Mo CO ligands cis t o the phosphido bridge in 1 was studied by variable- temperature 13C N M R spectroscopy. Addition reactions between 1 a n d Lewis bases L(L =
PPh2H, PMe3, P(OMe)3) produced CpW(CO)&.-PPh2)Mo(C0)4(L) (3) withL
regiospecifically and stereospecifically coordinating t o t h e Mo cis t o the phosphido bridge. T h e structure ofCpW(C0)3(p-PPh2)Mo(C0)4(PMe3)
(3a) was determined by single-crystal X-ray diffraction. Crystal data for 3a: C27H24M007PzW, space group P21/n, a = 11.756(4)A,
b = 16.423(6)A,
c= 14.968(3)
A,
/3 = 98.397(20)',V
= 2858.8(14)A3,
2 = 4. T h e structure was refined t oR
= 0.026and
R,
= 0.031. Reaction of 1 with PPh3 producedC~W(CO)~(~-PP~~)MO(CO)~(PP~~)
with a metal-metal bond a n d with PPh3 occupying the position trans t o the phosphido bridge.Complexes with similar structures were synthesized by removal of one CO from 3 t o regenerate the metal-metal bond t o produce
CpW(CO)z(p.-PPh2)Mo(CO)qL
(4;L
= PPhzH, PMe3, P(OMe)3), in whichL
regiospecifically and stereospecifically coordinates t o the Mo and is trans t o t h e phosphido bridge.-
Introduction
One special feature of binuclear metal-metal-bonded phosphido-bridged complexes is the influence of the metal- metal bond on the reacti0ns.l The metal-metal bond functions as a switch to control the reaction according to the properties of the ligand on the complex.l*,b This behavior provides not only an empty site for further ligand coordination to the binuclear complex when the metal- metal bond o p e d b but also a driving force for further reaction (e.g. migration of the ligand) when the metal- metal bond re-forms.'* This property can also be con- sidered as a cooperative effect of the two adjacent metals in the binuclear complex.
For heterobimetallic phosphido-bridged complexes, the metal-metal bond can be considered as a donor-acceptor bond.ldI2 When the metal-metal bond opens, an empty
t Academia Sinica.
f National Taiwan University.
0 Abstract published in Advance ACS Abstracts, April 1, 1994. (1) (a) Shyu, S.-G.; Wojcicki, A. Organometallics 1985, 4, 1457. (b) Mercer, W. C.; Whittle, R. R.; Burkhardt, E. W.; Geoffroy, G. L.
Organometallics 1985,4,68. (c) Powell, J.; Sawyer, J. F.; Stainer, M. V. R. Inorg. C h e n . 1989,28, 4461. (d) Powell, J.; Coutoure, C.; Gregg, M.
R. J . Chem. SOC., Chem. Commun. 1988, 1208.
site is contributed to the metal where the dative metal- metal bond was originally coordinated. Thus, the dative metal-metal bond acts as a directional switch. Such directional opening of the dative metal-metal bond was demonstrated by the addition of a Lewis base to hetero- bimetallic p h ~s ph id o-b rid ged ' ~ *~ ~ .~ and arsino-bridged complexes.4
The reactivity site of binuclear complexes can be controlled by careful selection of the metals and their ligands to construct binuclear complexes with the desired direction of the metal-metal dative bond. We have thus set out to prepare and study a series of such compounds.5 (2) (a) Breen, M. J.; Duttera, M. R.; Geoffroy, G. L.; Novotnak, G. C.; Roberta, D. A.; Shulman, P. M.; Steinmetz, G. R. Organometallics 1982,
I , 1008. (b) Jenkins, H. A;Loeb,S. J.;Stephan,D. W.1norg. Chem. 1989,
28,1998.
(3) (a) Roberta, D. A.; Steinmetz, G. R.; Breen, M. 3.; Shulman, P. M.; Morrison, E. D.; Duttera,M. R.;DeBrosse, C. W.; Whittle,R. R.; Geoffroy, G. L. Organometallics 1983,2, 846. (b) Baker, R. T.; Calabrese, J. C.; Krusic, P. J.;Therien, M. J.; Trogler, W. C. J. Am. Chem. SOC. 1988,110, 8392.
(4) (a) Langenbach,H.-J.; Vahrenkamp,H. Chem. Ber. 1979,112,3390. (b) Langenbach, H.-J.; Vahrenkamp, H. Chem. Ber. 1979,112, 3773.
(5) (a) Shyu, S.-G.; Lin, P.-J.; Wen, Y.-S. J . Organomet. Chem. 1993, 443, 115. (b) Shyu, S. G.; Hsu, J.-Y.; Wen, Y.-S. J. Organomet. Chem.
1993, 453, 97.
0276-733319412313-1699$04.50/0 0 1994 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1700 Organometallics, Vol. 13, No. 5, 1994 Scheme 1 Ph2 Ph2
co
-
hv / p \ CP(CO)3W M o t C 0 ) ~ / p \ \ Cp(CO)?UL--- M o ( C 0 ) j 1 1 k ( 0 M e ) 3 PMe3 PPhzH Shyu et al.and PPh3 were obtained from Strem; P(OMe)3 was purchased from Merck, and 13C0 (99 atom % 13C) was obtained from Isotec. Other reagents and solvents were obtained from various com- mercialsources and usedas received. Mo(CO)d(C7&),7 Na[CpW- (CO)&2DME,B and WCp(C0)~PPhzowereprepared by literature procedures. Phz 0 P c -0 cO
" \
4 3 4a, L = PMe3 4b, L = P(OMe)3 4c, L = PPhzH 4d, L = PPh3 3a, L = PMe3 3b, L = P(OMe)3 3c, L = PPhzHThe bimetallic complex CpW(C0)2(pPPhz)Mo(C0)5 (1)
with a dative Mo-W bond was synthesized, and the addition reaction of the complex toward different Lewis bases was studied. We found that the addition of the Lewis base opened the metal-metal bond. Nevertheless, the addition did not occur a t the W atom, as expected, but proceeded stereospecifically and regiospecifically at the Mo atom with the base occupying the position cis to the phosphido bridge. Reported herein are the synthesis, structure, fluxional behavior, and reactivity studies of CpW(CO)z(pPPh2)Mo(CO)5. Scheme 1 shows reactions that comprise the main focus of our work. The products of the addition reaction have been characterized spec- troscopically, and the structure of CpW(CO)s(r-PPhz)- Mo(CO)d(PMe3) (3a) was also determined by a complete single-crystal X-ray diffraction study.
,
Experimental Section
Unless otherwise stated, all reactions and manipulations of air-sensitive compounds were carried out at ambient temperatures under an atmosphere of purified N2 with standard procedures. A 450-W Hanovia medium-pressure quartz mercury-vapor lamp (Ace Glass) and a Pyrex Schlenk tube as a reaction vessel were used in the photoreactions. Infrared (IR) spectra were recorded on a Perkin-Elmer 882 infrared spectrophotometer. lH,
lac,
and 31P NMR spectra were measured by using Bruker AMX-500, MSL-200, AC-200, and AC-300 spectrometers. 31P NMR shifts are referenced to 85 % H3P04. Except as noted, NMR spectra were collected at room temperature. Electron impact (EI) and fast-atom bombardment (FAB) mass spectra were recorded on a VG 70-2505 or a JEOL JMS-HX 110 mass spectrometer. Microanalyses were performed by the Microanalytic Laboratory at National Cheng Kung University, Tainan, Taiwan.Materials. THF was distilled from potassium and benzophe- none under an atmosphere of NZ immediately before use. Other solvents were purified according to established procedures.6 The metal carbonyls M(C0)e (M = Mo, W), PPhZC1, PMe3, PPh2H,
(6) Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. Purification of Laboratory Chemicals; Pergamon: Oxford, U.K., 1966.
Synthesis of CpW(CO)2(pPPh2)Mo(CO)a (1). A yellow suspension of Na[CpW(CO)&2DME (0.36 g, 0.68 mmol) in 50
mL of toluene was cooled to 0 "C. A solution of 0.12 mL (0.65 mmol) of PPh2Cl in 25 mL of toluene was then added slowly to the above solution. After 1 h, the solution turned orange-red. MO(CO)~(C~H~) (0.18 g, 0.60 mmol) was then added to the above solution. The solution turned red immediately. After the solution was stirred overnight, solvent was removed and the residue was chromatographed on silica gel. Elution with CH&lz/hexane (1:
4) afforded two fractions. The solvent was removed. A trace amount of yellow solid was obtained from the first band and was not identified. The purple solid 1 was obtained from the second band. Yield: 0.14 g (32%). Anal. Calcd for C24H1607PMoW C, 39.67; H, 2.07. Found: C, 39.83; H, 1.67. IR spectrum (THF, v(C0)): 2071 m, 1957 s, 1930 m, 1861 w cm-1. IR spectrum (hexane, v(C0)): 2073 m, 2004 vw, 1984 m, 1957 s, 1937 m, 1876 w cm-l. lH NMR spectrum (CDCl3): 6 7.77 (m, 2H), 7.42 (m, 3H), 7.20 (m, 3H), 6.98 (m, 2H), 5.17 (s, 5H). 31P(1H) NMR spectrum (THF): 6 170.46 (Jp-w = 342.6 Hz). 13C{lH) NMR spectrum (CDCl3): 6 226.73 (d, 2 J p ~ = 7.51 Hz, CO), 221.80 (s,
CO), 208.28 (d, VP_C = 12.08 Hz, CO), 206.43 (br, CO), 143.15 (d,
J p x = 11.07 Hz, ips0-C PPhz), 142.31 (d, Jpx = 12.83
Hz,
ipso- C', PPhZ), 133.94 (d, 2 J p ~ = 8.45 Hz, 0-C, PPhz), 131.68 (d, V p _ c= 11.2 Hz, 0-C', PPhZ), 129.81 ( 8 , p-C, PPhZ), 129.03 (9, p-C', PPh2), 128.31 (m, m-C, m-C', PPhz), 91.75 (s, C5H5). MS (FAB): M+ m/t 726.
Synthesis of CpW(CO)&PPh,)Mo(CO)b (2). A solution of 1 (0.27 g, 0.30 mmol) in 15 mL of THF was stirred under 1 atm of CO overnight. The solution changed from purple to yellowish brown. The solvent was then removed, and the residue was chromatographed on silica gel. Elution with CHzCldhexane (1:
4) afforded two fractions. The first band, which was purple, was unreacted 1. The second band was yellow. After the solvent was removed, 2 was obtained as a yellow solid. Yield: 0.10 g (44%). Anal. Calcd for CBH160pMoW: C, 39.82; H, 1.99. Found C, 39.56; H, 2.26. IR spectrum (THF, v(C0)): 2067 m, 2025 m, 1948
s, 1938 sh, 1914 m cm-l. 1H NMR spectrum (CDC13): 6 7.60 (br, 4H), 7.34 (m, 6H), 5.32 (br, 5H). 31P(1H) NMRspectrum (THF):
6 -41.66 (8). MS (FAB): M+ m/z 754.
Thermolysis of 2. A solution of 1.80 g of 2 in 100 mL of THF was heated at reflux temperature for 6 h under Nz. The solution changed from yellow to purple. The solvent was then removed, and the residue was chromatographed on silica gel and eluted with CHzClp to afford a purple band. After the solvent was
removed, 1 was obtained as a purple solid. Yield 1.10 g (64%). Synthesis of CpW(CO)s(p-PPh2)Mo(CO)~(PMe~) (3a).
Complex 1 (0.40 g, 0.55 mmol) was dissolved in 25 mL of THF under N2 at room temperature. To this solution was added 70
p L of PMe3 (0.69 mmol). After 1 h, the solution changed from purple to reddish brown. After 90 min, the solvent was removed and the residue was chromatographed on grade I11 A1203 and eluted with CHzCldhexane (1:4) to afford two fractions. The first band was unreacted 1. The second band was yellow. After the solvent was removed, 3a was obtained as a yellow solid. Yield: 0.25 g (57 % ). Anal. Calcd for CZ,HNO,P*MOW C, 40.43;
H, 2.99. Found: C, 39.97; H, 2.91. IR spectrum (THF, v(C0)): 2026 w, 2007 s, 1951 8,1901 s, 1888 sh, 1853 m cm-l. lH NMR spectrum (CDC13): 6 7.60 (m, 4H), 7.28 (m, 6H), 5.49 (s,5H), 0.86 (7) Bennett, M. A,; Pratt, L.; Wilkinson, G. J. Chem. Soc. 1961,2037.
(8) Bender, R.; Braunstein, P.; Jud, J.-M.; Dusausoy, Y. Inorg. Chem. 1983,22, 3394.
(9) (a) Adams, H.; Bailey, N. A.; Day, A. N.; Morris, M. J.; Harrison,
M. M. J. Organomet. Chem. 1991,407,247. (b) Malisch, W.; Maisch, R.; Colquhoun, I. J.; McFarlane, W. J . Organomet. Chem. 1981,220, C1.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
-
Structure and Reactions of C p
W ( C O ) Z ( ~ - P P ~ ~ ) M O ( C O ) ~
(d, VP-H = 6.7 Hz, 9H). 31P(1H) NMR spectrum (THF): 6 -36.20 (d), -22.27 (d, Vp-p = 26.0 Hz). MS(FAB): (M
-
CO)+ m/z 776.Synthesis of CpW(CO)3(pPPhz)Mo(CO)4(P(OMe)3) (3b).
To a purple solution of 1 (0.60 g, 0.83 mmol) in 30 mL of THF was added 153 pL of P(OMe)3 (1.30 mmol) under Nz at room temperature. After 2 h, the solution changed to brown. The solvent was then removed, and the residue was chromatographed on grade I11 A1203 and eluted with CHzClz/hexane (1:4) to afford four fractions. The first band was unreacted 1, the second band was yellow, and the third band was purple-red. Only trace amounts of products were obtained from the second and third bands, and the products were not identified. The fourth band was yellow. After the solvent was removed, 3b was obtained as a greenish yellow solid. Yield: 0.44 g (65%). Anal. Calcd for Cz,Hz4010PzMoW: C, 38.12; H, 2.82. Found: C, 37.82; H, 2.71. IR spectrum (THF, v(C0)): 2030 s, 2013 m, 1931 s, 1900 m, 1871 cm-l. lH NMR spectrum (CDCl3): 6 7.66 (m, 4H), 7.30 (m, 6H), 5.37 (br, 5H), 3.36 (d, 3Jp-H = 11.0 Hz, 9H). 31P(1H) NMR spectrum (THF): 6 157.73 (d), -38.11 (d, V p - p = 26.0 Hz, Jp-w = 82 Hz). MS(FAB): (M - CO)+ mlz 822. Synthesis of CpW(C0)3(pL-PPh~)Mo(CO)4(PPhzH) (3c).
To a purple solution of 1 (0.30 g, 0.41 mmol) in 30 mL of THF was added 87 pL of PPhzH (0.50 mmol) under NZ at room temperature. After 90 min, the solution changed to reddish brown. The solvent was then removed, and the residue was chromatographed on grade I11 A1203 and eluted with CHzClzI hexane (1:4) to afford three fractions. The first band was unreacted 1, and the second band was yellow. Only a trace amount of product was obtained from the second band, and it was not identified. The third band was greenish yellow. After the solvent was removed, 3c was obtained as a greenish yellow solid. Yield: 0.24 g (63%). Anal. Calcd for C~~HZ&P~MOW: C, 47.37; H, 2.85. Found: C, 47.26; H, 2.89. IRspectrum (THF, v(C0)): 2028 m,2012m, 1932s,1908s, 1896sh,1863mcm-l. lHNMRspectrum (CDC13): 6 7.63 (br), 7.30 (br), 5.45 (br, 5H). 4.74 (dd,Jp-H = 326 Hz, 3Jp..H = 6.9 Hz, 1H). 31P{lH) NMR spectrum (CDCl3): 6 20.98 (s, br), -36.29 (d, 2Jp-p = 21.4 Hz). MS(FAB): (M - CO)+ m/z 885. Synthesis of CpW(Co)z(~-PPhz)Mo(C0)4(PPh3) (4d). To a purple solution of 1 (0.30 g, 0.41 mmol) in 20 mL of THF was added PPh3 (0.11 g, 0.42 mmol) under NZ at room temperature. The mixture was stirred overnight. A cloudy brownish purple mixture was obtained. The solvent was then removed, and the residue was chromatographed on grade I11 A1203 and eluted with CH&lz/hexane (1:4) to afford three fractions. The first band was unreacted I, and the second band was yellow. After the solvent was removed, a gray solid was obtained. The compound was identified as Mo(C0)5PPh31° (yield 0.11 g, 54%) according to its spectroscopic data. The third band was red. After the solvent was removed, 4d was obtained as a deep red solid. The yield was 0.12 g (30%). Anal. Calcd for C~~H~&.PZMOW: C, 51.29; H, 3.12. Found: C, 50.97; H, 3.20. IR spectrum (THF, v(C0)): 2029 w, 1968 vw, 1928 s, 1916 s, 1844 m cm-l. lH NMR spectrum (CDC13): 6 7.41 (m, 25H), 5.06 (s, 5H). 31P{1H) NMR spectrum (THF): 6 43.27 (d), 168.26 (d, Vp-p = 27.1 Hz, J p - w = 325.5 Hz). l3C(lH) NMR spectrum (CDCl3): 6 230.95 (d, Vp-c
= 15.87 Hz, CO), 223.54 (s, CO), 211.50 (vbr, CO), 91.24 (s, C5H5). MS (FAB): M+ mlz 960.
-
Synthesis of CpW(CO)z(p-PPhz)Mo(CO)~(PMe~) (4a). A solution of 3a (0.10 g) in THF (35 mL) was irradiated with UV for 15 min at 10 "C. The solution changed from yellow to red. The solvent was then removed, and the residue was chromato- graphed on silica gel. Elution with CHzCldhexane (1:4) afforded three fractions. After the solvent was removed, trace amounts of products were obtained from the first and the third bands, which were purple and yellow, respectively. They were not identified. The second band was red. After the solvent was removed, 4a was obtained as a red solid. Yield 35 mg (36%).
(10) Grim, S. 0.; Wheatland, D. A.; McFarlane, W. J . Am. Chem. SOC.
1967,89, 5573.
Organometallics, Vol. 13, No. 5, 1994 1701 Anal. Calcd for Cz~Hz&PzMoW: C, 40.34; H, 3.13. Found: C, 40.41; H, 2.74. IR spectrum (THF, u(C0)): 2025 m, 1959 w, 1923 s, 1905 8,1840 m cm-l. lH NMR spectrum (CDC13): 8 7.80 (m, 2H), 7.35 (m, 3H), 7.22 (m, 3H), 7.00 (m, 2H), 5.02 (s,5H), 1.70 (d, 'JP-H = 8.3 Hz). 31P(1H} NMR spectrum (THF): 6 -7.21 (d), 171.79 (d, V p - p = 24.4 Hz, Jp-w = 322.0 Hz). MS (FAB): M+ m/z 776. Synthesis of C~W(CO)Z(N-PP~Z)MO(CO)~(P(OM~)~) (4b).
A solution of 3b (0.10 g) in THF (35 mL) was irradiated with UV for 15 min at 10 "C. The solution changed from yellow to red. The solvent was then removed, and the residue was chromato- graphed on silica gel with CHzClz/hexane (1:4) as the eluent to afford two fractions. The solvent was removed from the second major band. A red solid was obtained. Yield 51 mg (54%). Anal. Calcd for CaH2409PzMoW: C, 37.98; H, 2.95. Found: C, 38.10; H, 2.99. IR spectrum (THF, v(C0)): 2036 m, 1969 vw, 1925 vs, 1846 m cm-l. 1H NMR spectrum (CDC13): 6 7.85 (m, 2H), 7.39 (m, 3H), 7.21 (m, 2H), 7.02 (m, 3H), 5.07 (d, 3 J p - ~ = 11.7 Hz, 5H), 3.72 (dd, 3 J p - ~ = 11.7 Hz, 5 J p 4 = 1.2 Hz). 3lP(lH) NMR spectrum (THF): 6 169.70 (d), 162.36 (d, 2Jp-p = 49.5 Hz, J p - w = 329.6 Hz). MS (FAB): M+ m/z 822. Synthesis of C~W(CO)Z(~-PP~Z)MO(CO)~(PP~~H) (4c).
A solution of 3c (0.10 g) in THF (35 mL) was irradiated with UV for 15 min at 10 "C. The solution changed from yellow to red. The solvent was then removed, and the residue was chromato- graphed on silica gel with CHzCldhexane (1:4) as eluent to afford two fractions. The solvent was removed from the second band. A red solid was obtained. Yield: 51 mg (54%). Anal. Calcd for C ~ ~ H Z & ~ P ~ M O W :
c,
47.54; H, 2.94. Found: C, 47.12; H, 2.72.IR spectrum (THF, v(C0)): 2034 m, 1930 s, 1920 sh, 1846 m cm-l. 3lP{lHj NMR spectrum (THF): 6 16.18 (d), 166.94 (d, V p - p = 26.2 Hz, Jp-w = 326.7 Hz). MS (FAB): M+ m/z 886.
Preparation of WO-Enriched 2. A solution of 1 (3.0 g, 4.1 mmol) in THF (100 mL) in a 250-mL Schlenk flask was stirred under 1 atm of 13C0 for 4 days. The solution changed from purple to yellowish brown. After chromatography on silica gel, 2.80 g (93% yield) of 13CO-enriched 2 was obtained. IR spectrum (THF, v(C0)): 2059 m, 2024 m, 1935 s, 1912 sh cm-l. 31P(1H) NMR spectrum (THF): 6 -141.17. MS (FAB): M+ m/z 758.
Over 11 atom % enrichment was obtained, according to the parent peak pattern of the mass spectrum of the enriched 2.
Preparation of WO-Enriched 1. A solution of 115 mg of WO-enriched 2 in 10 mL of THF was irradiated with UV for 20 min between freeze-thaw cycles. The solution changed from yellow to purple. 13CO-enriched 1 was separated after chroma- tography on silica gel. Yield: 18 mg (16 % ). IR spectrum (THF,
v(C0)): 2070m, 1951 s, vbr, 1860mcm-l. 31P{1H) NMRspectrum (THF): 6 170.50 (Jp-w = 347.2 Hz). MS (FAB): M+ m/z 730. About 9 atom % enrichment was obtained according to the parent peak pattern of the mass spectrum of the enriched 1.
Reaction of 2 with PR3 (R = Ph, Me, OMe) and PPhaH.
To a yellow solution containing 200 mg (0.27 mmol) of 2 in 20 mL of THF was added 34 pL (36 mg, 0.15 mmol) of P(OMe)3. The solution was stirred in the dark at room temperature overnight. No color change was observed. Results of a NMR study of the reaction mixture indicated the existence of the unreacted 2 and P(OMe)3 and small amounts of unidentified impurities.
Similar reaction conditions were applied to the reaction between 1 and PPh3, PPhzH, and PMe3. No complex 3 was observed in the reaction product, according to 3lP NMR spectra of the reaction mixtures.
Reaction between 4 and CO. A solution of 4b (60 mg) in THF was stirred under 1 atm of CO overnight at room temperature. A 31P NMR study of the solution indicated that no reaction took place between 4b and CO. Similar conditions were applied to complexes 4a,c,d. No reaction was observed, as indicated by a 31P NMR study of the reaction mixture.
Structure Determination of 1, 2, and 3a. Crystals of complexes 1,2, and 3a were grown by slow diffusion of hexanes into the saturated solutions of the relevant complexes (1 in CH2-
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1702 Organometallics, Vol. 13, No. 5, 1994
Table 1. Crystal and Intensity Collection Data for 1, 2, and 3a
Shyu et al. 1 2 3a mol formula mol wt ;PFG group b (A) c (A) B (deg)
v
(A31 p(calcd) (Mg m3) Z cryst dimens (mm) abs coeff p(Mo Ka) (mm-I) temp radiation 28 range (deg) scan type no. of reflns no. of obsd rflns variables R RW S A F (e/A3) ( A / g ) m a x C ~ ~ H ~ S M O W O ~ P 726.02 14.464(7) 11.6743(12) 14.7639( 14) 97.916( 12) 2469.3(6) 1.913 4 0.10 X 0.20 X 0.38 5.35 room temp Mo K a (A = 0.709 30 A) 45 P21/n 28- 3220 2546 (>2.5u (I)) 3 20 0.054 0.055 3.39 <1.370 0.043Clz; 2 and 3a in THF) at -15 "C. Cell dimensions and space group data were obtained by standard methods on an Enraf- Nonius CAD4 diffractometer. Details of data collection and refinement are given in Table 1.
The coordinates of heavy atoms were obtained from Patterson syntheses. The positions of the remaining non-hydrogen atoms were obtained from Fourier syntheses. For complex 1, inspection of a difference Fourier revealed severe disorders (Figure 1). There were dominant and minor fractions; the ratio was determined to be 0.85:0.15. Except for W (anisotropic) and P (isotropic), the minor portion was fixed in the final refinement. The final model of the major fraction was obtained with all non-hydrogen atoms refined anisotropically and the H atom at idealized positions with R = 5.4% and R , = 5.0%. The final positional parameters are listed in Tables 2 ( l ) , 3 (2), and 4 (3a). Selected interatomic distances and bond angles are given in Tables 5 (1 and 2) and
6 (3a). The thermal parametersfor these complexes are provided in the supplementary material.
R e s u l t s and Discussion
Syntheses, Spectroscopic Characterization, a n d Molecular S t r u c t u r e s of 1 a n d 2. T h e new W-Mo complex 1 was synthesized by the reaction of CpW- (C0)3PPh2 with M o ( C O ) ~ ( C ~ H ~ ) . The metallophosphine CpW(C0)3PPh2 acted as a ligand to replace the C& t o form the complex:
CpW(C0)3PPh2 + Mo(CO)&H*
-
PPh2/ \
Cp(CO)2W
-
Mo(CO)5+
C7H81
When 1 was stirred under 1 atm of CO, complex 2, which lacks a metal-metal bond, was obtained. The addition of CO to complex 1 was reversible. When 2 was heated in T H F a t reflux temperature under N2 or irradiated with UV light, CO was removed from 2 t o regenerate 1. Both complexes were stable a t room temperature in air in the solid state.
The 31P(1HJ NMR spectrum of 1 in THF a t room temperature shows a resonance a t 170.4 ppm with Jp-w
= 342 Hz. This relatively downfield resonance reveals the existence of a metal-metal bond in the complex." In
CzsHlsMoOsPW 754.15 9.6322(16) 12.4673(21) 20.814(3) 94.476( 12) 2472.7(7) 2.026 4 0.25 X 0.16 X 0.315 5.36 room temp Mo K a (A = 0.709 30 A) 45 28-w 3220 2337 (>2.0a (I)) 325 0.035 0.044 1.93 C0.810 0.039 P21ln C ~ ~ H Z ~ M O O ~ P ~ W 802.22 11.756(4) 16.423(6) 14.968(3) 98.397(20) 2858.8(14) 1.864 4 0.39 X 0.41 X 0.35 4.69 room temp Mo K a (A = 0.709 30 A) 45 28- 3723 3022 (>2.5u (I)) 343 0.026 0.031 1.49 <0.830 0.006 P21/n
contrast, the relatively upfield resonance a t -41.7 ppm in the 31P{1H} NMR of 2 indicates the opening of the W-P- Mo triangle in the complex." Nevertheless, the Jp-w value cannot be observed because of the broad signal.
The W-Mo complexes 1 and 2 were further characterized by single-crystal X-ray diffraction methods. Structures of them are shown in Figures 2 and 3.
In 2, a W-Mo distance of 4.5192(14)
A
indicates that there is no metal-metal bond. One can consider the metallophosphine CpW(C0)3PPh2 to be a ligand similar to PR3. Thus, five CO's and CpW(C0)3PPh2 coordinate to the Moo atom t o form a distorted octahedron. Similar examples of the metallophosphine ligand CpFe(C0)zPPha coordinated to M(C0)5 (M = Cr, Mo, W) to form CpFe- (CO)2(pL-PPh2)M(C0)5 with structures similar t o that of 2 has been reported.5aIn complex 1, the Mo-C(4)-0(4) angle 169.3(13)' indicates a semibridging carbonyl. The observed IR a t 1876 cm-l a t room temperature and 13Cj1H} NMR a t 218.69 ppm (see below) at 210 K indicate a real interaction between the W atom and the CO(4) ligand. The W-Mo distance (3.2054(16)
A)
in 1 falls between the W-Mo distances reported for the complexes CpMo(CO)2(p-SMe)-w(co)5
(3.131(1)A)12a
and [HB(pz)31 (CO)zW(p-CS)Mo- (CO)s(Ind) (3.3102(4)A;
Ind = 9-CgH7, indenyl; HB(pz)3= hydrotris(l-pyrazolyl)borate).12b It is, however, sig- nificantly longer than the W-Mo distance in MoW2(p-
CCsH4Me-4)2(p-C0)2(CO)~(9-C5H5)2 (2.938(1) A)12c and
is similar to that in
(~-C~H~)(CO)~MOW(CO)~(C-~-(CH~)~-
CH2}(9-CgHg)} (3.239(4)A),12d
which has no bridging ligand.I
1 I
(11) (a) Carty, A. J.; Maclaughlin,S. A.; Nucciarone, D. Inphosphorus- 31 NMR Spectroscopy in Stereochemical Analysis: Organic Compounds and Metal Complexes; Verkade, J. G., Quin, L. P., Eds.; VCH: New York, 1987; Chapter 16, and references cited therein. (b) Carty, A. J.
Adu. Chem. Ser. 1982, No. 196,163. (c) Garrou, P. E. Chem. Reu. 1981, 81, 229.
(12) (a) Guerchais, J. E.; LeQuere, J. L.; Petillon, F. Y.; Manojlovic- Muir, L.; Muir, K. W.; Sharp, D. W. A. J. Chem. SOC., Dalton Trans. 1982, 283. (b) Doyle, R. A,; Daniels, L. M.; Angelici, R. J. J. Am. Chem. SOC. 1989, 111, 4995. (c) Garriedo, G. A.; Howard, J. A. K.; Marsden, K.;
Stone, F. G. A.; Woodward, P. J . Chem. Soc., Dalton Trans. 1984,1589. (d) Adams, H.; Bailey, N. A.; Winter, M. J. J. Chem. Soc., Dalton Trans. 1984, 273.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
Structure and Reactions of C p W ( C O ) , ( p - P p h , ) ~ o ( C O ) 5
0 2
Figure 1.
ORTEP
drawing of the disordered molecular structure of 1.Table 2. Atomic Coordinates and Isotropic Thermal Parameters (A2) for 1
atom X Y Z BCC W Mo P c 1 c 2 c 3 c 4 c 5 C6 c 7 C8 c 9 c 1 0 c 1 1 c 1 2 C13 C14 C15 C16 C17 C18 C19 c 2 0 c 2 1 c 2 2 C23 C24 0 1 0 2 0 3 0 4 0 5 0 6 0 7 0.30641(5) 0.191 1 l(9) 0.1976(3) 0.3640(12) 0.4144(12) 0.1 539( 12) 0.2843(11) 0.2917( 12) 0.0977( 11) 0.0881(12) 0.2336( 12) 0.3 139( 13) 0.3454(15) 0.2805(15) 0.1930( 15) 0.1669( 14) 0.0894( 10) 0.0014(11) -0.0727(11) -0.0639( 11) 0.0219( 12) 0.0997(11) 0.3592( 16) 0.3400( 14) 0.2484( 13) 0.2064(18) 0.2766( 16) 0.4005(9) 0.4763(8) 0.1341( 11) 0.3285(9) 0.3437(8) 0.0483(9) 0.0333(9) 0.13387(7) 0.24447(12) 0.2872(4) 0.2393(15) 0.1963( 16) 0.2398(17) 0.1 146( 13) 0.3699( 14) 0.3750( 12) 0.1248( 15) 0.43 18( 14) 0.4666(15) 0.5728(21) 0.6493( 17) 0.6145(17) 0.5050( 15) 0.2614( 13) 0.2789(18) 0.2686( 16) 0.2449(18) 0.2306( 19) 0.2377( 18) -0.0354(18) -0.0548(20) -0.0493( 14) -0.01 57( 17) -0.0010( 16) 0.2799( 13) 0.2342( 12) 0.2354( 14) 0.0486(11) 0.4318(11) 0.4476(11) 0.0592( 11) 0.03996(5) -0.13982(9) 0.0298(3) 0.1401 (1 2) -0.01 42( 13) -0).2786( 11) -0.1636(10) 4 . 1 543( 10) -0).1465(10) -0.1286( 10) 0.06 14( 10) 0.0470( 12) 0.0679( 13) 0.1057( 12) 0.1214( 12) 0.0982(12) 0.0798( 10) 0.0333(10) 0.0804( 10) 0.1708( 11) 0.2174(10) 0.1749( 10) 0.1120(15) 0.0208(15) -0.0045( 12) 0.07 17( 16) 0.1461 (1 3) 0.2003( 8) -0.0454(9) -0.3538(7) -0.1897(8) 4 . 1 67 3( 8) -0.1570( 10) -0.1254( 10) 3.06(3) 3.12(6) 2.70(20) 6.0(10) 4.3(10) 5.3(10) 4.1(8) 4.3(8) 4.1(8) 4.8(9) 2.9(8) 3.8(9) 6.9(14) 5.5(11) 5.3(11) 4.5 ( 10) 5.7(11) 5.5 ( 10) 6.1(11) 5.8 ( 10) 6.5(12) 6.8(13) 3.8(9) 7.3(15) 5.5 ( 12) 7.9(8) 7.4(8) 9.7( 10) 6.9(7) 6.6(7) 8.1(8) 8.5(9) 3.5(7) 4.7(9)
The metal-metal bond in 1 can be considered as a covalent bond between Mol and W1 (structure a) or a donor-acceptor bond from Moo to W" (structure b). We
Ph2 Ph2
JP\
/ p \CP(CO)~% Mo(CO)S C p ( C O ) 2 W Mo(CO)s
a b
prefer the latter assignment, which is similar to descrip-
Organometallics, Vol. 13, No. 5, 1994 Table 3. Atomic Coordinates and Isotropic Thermal
1703
Parameters (A2) for 2
atom X Y Z B , W Mo P 0 1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 c 1 c 2 c 3 c 4 c 5 C6 c 7 C8 c 9 c 1 0 c 1 1 c 1 2 C13 c 2 1 c 2 2 C23 C24 C25 C26 C3 1 C32 c 3 3 c 3 4 c 3 5 C36 0.50793(6) 0.791 80( 12) 0.74705(34) 0.2663( 10) 0.4328(11) 0.6604(11) 0.8461(13) 0.8717(12) 1.1 186(11) 0.7245( 14) 0.4759(11) 0.3546(15) 0.4608( 14) 0.6067(14) 0.8282(15) 0.8393(15) 1.0027(16) 0.7475( 15) 0.5887(16) 0.3492( 15) 0.4323( 19) 0.5758(17) 0.5745( 16) 0.4347( 17) 0.777 1 (13) 0.6767( 14) 0.7045(16) 0.8327(18) 0.9363( 17) 0.9082(15) 0.8879( 12) 0.9459( 14) 1.0489( 15) 1.0974(15) 1.0393(15) 0.9340( 14) 0.75999(4) 0.78949(9) 0.7 1501 (25) 0.6059(8) 0.6367(8) 0.6235(8) 1.0346(8) 0.8551(10) 0.7494( 10) 0.5554(9) 0.8513(9) 0.661 1( 11) 0.6801(11) 0.6705(10) 0.9448( 12) 0.8308(12) 0.7594(12) 0.6333( 13) 0.8268(11) 0.8999( 11) 0.9254( 11) 0.9402(10) 0.9196(11) 0.8976( 11) 0.5672(9) 0.49 13(9) 0.3830(10) 0.3491(11) 0.4231( 12) 0.5307(11) 0.7578( 10) 0.6898(11) 0.7240(13) 0.8291(14) 0.8978(11) 0.8645(10) 0.15894(3) 0.00525(6) 0.1 1909( 16) 0.1857(6) 0.0271(5) 0.2743(4) 0.0398(6) -0.1 306(6) 0.0492(6) -0.0534(6) -0.0453(6) 0.1756(7) 0.0753(6) 0.2302(8) 0.03 12(7) -0.08 19(8) 0.0348(6) 4.0293(7) 4).0253(7) 0.1 594( 10) 0.1136(8) 0.1461( 10) 0.21 16(9) 0.2207(8) 0.1204(6) 0.1287(6) 0.1 257 (6) 0.1137(8) 0.1058(7) 0.1092(7) 0.1 85 5 (6) 0.2343(7) 0.2818(7) 0.2843(7) 0.2352(7) 0.1872(7) 2.40(2) 2.64(5) 2.33(14) 5.3(6) 4.5(5) 3.9(5) 6.3(7) 6.0(7) 6.0(6) 7.0(7) 5.8(6) 3.2(7) 2.9(6) 3.6(7) 3.9(8) 3.8(7) 3.7(8) 4.0(7) 4.8(9) 4.9(10) 4.1(8) 4.1(8) 2.3(6) 2.8(6) 3.4(7) 4.7(9) 4.3(8) 3.5(7) 2.7(6) 3.4(6) 4.3(8) 3.8(8) 3.8(7) 3.5(7) 3.4(7) 4.3(9)
tions given earlier for (PPh3)(C0)3Fe(p-PPhz)~r(CO)~- (PPh3)3* and
(CO)bW(p-PPhz)Re(C0)4.1b
The Mo-W bond in 1 can be considered as the donation of an electron pair from one of the filled tag orbitals of Mo to the adjacent W such that the Mo-W dative bond acts as the fifth ligand, donating two electrons to W, in addition to the two CO's, the p-PPhz, and the Cp ligands coordinated to W. Consistent with this view is the observation that the Mo-W vector bisects an edge of the distorted molybdenum octahedron and the Mo atom lies on the least-squares plane consisting of P, C(3), C(4), and C(6). The Mo-W vector was only 1.34' off the plane.13Variable-Temperature 13C NMR a n d Fluxional Behavior of
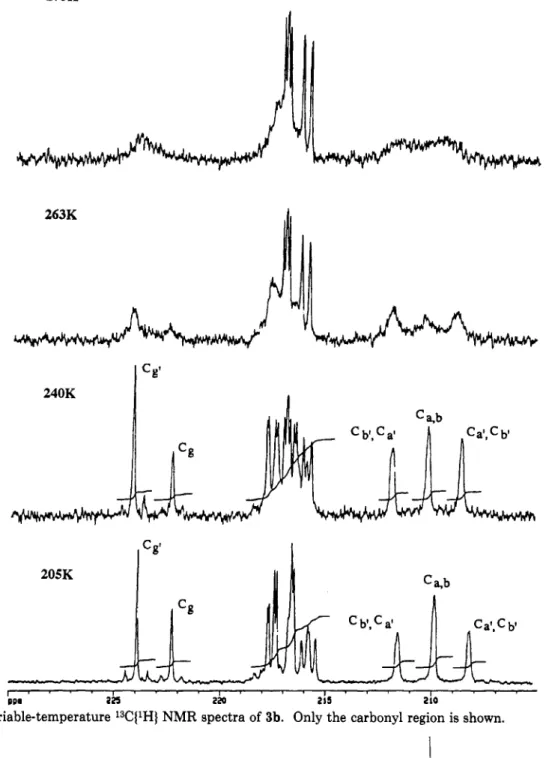
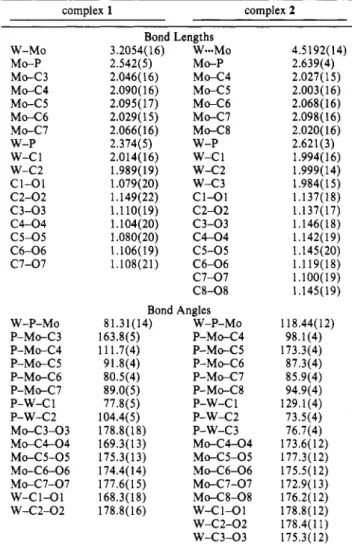
C~W(CO)~(~-PP~~)MO(CO)S.
The broad hump at 206.43 ppm observed in the 13C{lHJ NMR of complex 1 a t room temperature indicates a possible exchange of metal carbonyl ligands. In order to understand this fluxional behavior, variable-temperature 13C NMR of enriched 1 was undertaken (Figure 4).A t 210 K, the two CO signals, C1 (6 228.11,2Jc-p = 15.0 Hz, Jc-w = 120.9
Hz)
and C2 (6 223.11,Jc-w
= 133.9 Hz), are assigned to CO ligands coordinated to the W on the basis of observations of their tungsten satellites. The signal C4 (218.69 ppm) belongs to the semibridging carbonyl because bridging carbonyl has a relatively downfield (13) Equation of the plane: [10.65(6)1%+
[7.86(5)1y-
[2.46(5)1z = 4.292(5). Distances (A) to the plane from the atoms in the plane: P, -0.0039 C(3), -0.081(24); C(4), 0.039(22); C(6), 0.057(21). x* for this plane IS 22.399. Distances (A) to the plane from the atoms out of the plane: W, -0.075(14); Mo, O.OlO(8).(14) Smith, J. G.; Thompson, T. D. J . Chem. SOC. A 1967, 1694.
-
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1704 Organometallics, Vol. 13, No. 5, 1994
Table 4. Atomic Coordinates and Isotropic Thermal Parameters
(A*)
for 3aShyu et al. Table 5. Selected Bond Lengths
(A)
and Bond Angles (deg)in Complexes 1 and 2 atom X Y Z B , W Mo P1 P2 0 1 0 2 0 3 0 4 0 5 0 6 0 7 c 1 c 2 c 3 c 4 c 5 C6 c 7 C8 c 9 c 1 0 c 1 1 c 1 2 C13 C14 C15 c 2 1 c 2 2 C23 C24 C25 C26 C3 1 C32 c 3 3 c 3 4 c 3 5 C36 0.74191(2) 0.75461(5) 0.67827( 14) 0.91 553( 15) 0.5788(5) 0.8 365 (5) 0.9344(5) 0.5901 ( 5 ) 0.5968(5) 0.9780(4) 0.8823(6) 0.6397 (6) 0.8069(6) 0.871 l(6) 0.6463(6) 0.6497(7) 0.8899(7) 0.8311(8) 0.7786(6) 0.7660(7) 0.6534(7) 0.5944(6) 0.6715(7) 1.05 17(7) 0.9664(6) 0.8976(8) 0.7086(5) 0.7811(6) 0.8031(7) 0.7516(8) 0.6802(7) 0.6594(6) 0.5187(5) 0.4593(6) 0.3408(6) 0.28 12(6) 0.337 l(6) 0.4559(5) 0.63069(2) 0.38405(3) 0.53626(9) 0.40525(11) 0.3340(4) 0.2035(3) 0.3996(4) 0.3102(3) 0.7527(3) 0.5774(4) 0.7929(4) 0.3557(4) 0.2702(4) 0.3993(4) 0.3420(4) 0.7062(4) 0.597 5 (4) 0.7347(5) 0.5495(5) 0.6307(5) 0.6555(4) 0.5895(5) 0.5257(4) 0.3667(5) 0.5009(4) 0.3474(5) 0.5844(4) 0.6515(4) 0.68 04 (4) 0.6446(5) 0.5793(5) 0.5481 (4) 0.5442(3) 0.6038(4) 0.6116(4) 0.5613(5) 0.5007(4) 0.4924(4) 0.38926(2) 0.25710(4) 0.24870( 11) 0.16171 (12) 0.0834(4) 0.2566(4) 0.4351(4) 0.3828(4) 0.2579(4) 0.3330(4) 0.4037(4) 0.1442(5) 0.2565(5) 0.3705(5) 0.3365(5) 0.3019(5) 0.3488(5) 0.3983(5) 0.5204(4) 0.5456( 5) 0.5158(5) 0.4677(5) 0.4716(5) 0.221 2(6) 0.1208(5) 0.0580(6) 0.1427(4) 0.1392(4) 0.0559(5) -0.0238( 5) -0.0 196(5) 0.0622(4) 0.2321(4) 0.1778(5) 0.1745(5) 0.2225(5) 0.2730(5) 0.2775(4) 2.79(1) 2.51(2) 2.37(7) 3.08(8) 6.6(3) 5.1(3) 6.1(3) 5.2(3) 5.8(3) 5.3(3) 8.5(4) 3.5(3) 3.4(3) 3.5(3) 3.9(4) 3.8(3) 5.5(4) 3.8(3) 4.7(4) 4.2(4) 3.9(3) 3.9(4) 5.8(5) 4.3(4) 6.2(5) 2.6(3) 3.4(3) 4.5(4) 4.6(4) 4.5(4) 3.3(3) 4.3(4) 3.7(3) 3.6(3) 2.4(3) 3.2(3) 4.2(4) 3.0(3)
position in comparison with the corresponding terminal carbonyls. The doublet C3 (208.58 ppm, 2Jc-p = 11.09 Hz) is assigned to the molybdenum CO trans to the phosphido bridge on the basis of its relatively downfield position in comparison with the resonance positions of the other terminal CO’s of Mo.15 The remaining signals (C5, C6, C7) are assigned to the other CO ligands on Mo cis to the phosphido-bridge ligand. The observed similar- ity of the 2Jp-c value (11.09 Hz) of the cis CO signal C5 to the 2Jp-c value (11.09 Hz) of the trans CO signal C3 for Mo is not common. However, a similar observation of a
2Jp-c value of cis CO larger than the 2Jp-c value of trans CO for Mo has been reported for (CO)5MoP(O-i-Pr)3.16 We interpret the l3C NMR observations as follows. A t 210 K, all carbonyls were rigid and no intramolecular exchange occurred among carbonyl ligands. A t 223 K, exchange took place among the three cis molybdenum carbonyl ligands and the semibridging CO, as indicated by the broadening of their NMR signals (C4, C5, C6, C7). A t 320 K, the signal for C1 broadened. This indicates an additional exchange process of the CO ligands. Two kinds of exchange are possible. One is the exchange between this tungsten CO and the other cis CO ligands on Mo. A t higher temperature (330 K), however, the com- pound decomposed and the coalescence point could not
(15) (a) Hawkes, G. E.; Sales, K. D.; Aime, S.; Gobetto, R.; Lian, L. Y.
Inorg. Chem. 1991,30, 1489. (b) Gansow, 0. A.; Burke, A. R.; Vernon, W. D. J . Am. Chem. SOC. 1976,98, 5817. (c) Todd, L. J.; Wilkinson, J.
R. J . Organomet. Chem. 1974, 77, 1.
(16) Braterman, P. S.; Milne, D. W.; Randall, E. W.; Rosenberg, E. J. Chem. SOC., Dalton Trans. 1973, 1027.
complex 1 complex 2 Bond Lengths W-MO 3.2054(16) W-*MO 4.5 192( 14) Mo-P 2.542(5) Mo-P 2.639(4) Mo-C3 2.046(16) Mo-C4 2.027( 15) M+C4 2.090(16) M e C 5 2.003( 16) Mo-C5 2.095(17) M e C 6 2.068( 16) M 4 6 2.029( 15) Mo-C7 2.098( 16) Mo-C7 2.066(16) Mo-C8 2.020( 16) w-P 2.374(5) w-P 2.621 (3)
w-c
1 2.014(16) W-C1 1.994(16) w - c 2 1.989(19) W-C2 1.999( 14) c1-01 1.079(20) W-C3 1.984( 15) c2-02 1.149(22) C1-01 1.137( 18) C3-03 1.110(19) C2-02 1.1 37( 17) C4-04 1.104(20) C3-03 1.146( 18) C5-05 1.080(20) C4-04 1.142( 19) C6-06 1.106(19) C5-05 1.145(20) C7-07 1.108(21) C6-06 1.1 19( 18) C7-07 1 .loo( 19) 1.145(19) C8-08 Bond Angles W-P-MO 8 1.3 1 (1 4) W-P-MO 118.44(12) P - M e C 3 163.8(5) P-Mc-C~ 98.1(4) P-MeC5 91.8(4) P-Mc-C~ 8 7.3 (4) P - M e C 6 80.5(4) P-Mo-C~ 8 5.9( 4) P-Mo-C~ 89.0(5) P-Mc-CI 94.9(4) P-w-c 1 77.8(5) P-w-c 1 129.1(4) P-w-c2 104.4(5) P-w-c2 7 3.5 (4) M+C3-03 178.8(18) P-w-c3 76.7(4) M 4 4 - 0 4 169.3(13) Mo-C4-04 173.6(12) P - M e C 4 11 1.7(4) P-Mo-CS 173.3(4) Mc-C~-05 175.3(13) M e C 5 - 0 5 177.3(12) M e C 6 - 0 6 174.4( 14) Mo-C6-06 175.5(12) M e C 7 - 0 7 177.6(15) MwC7-07 172.9( 13) W-C1-01 168.3(18) Mo-C8-08 176.2(12) W-C2-02 178.8(16) W-C1-01 178.8(12) W-C2-02 178.4(11) W-C3-03 175.3(12) Table 6. Selected Bond Lengths(A)
and Bond Angles (deg)in Complex 3a Bond Lengths W*-MO 4.5198( 17) W-C6 2.000(8) Mo-P1 2.6527(18) w - c 7 1.998(9) M e P 2 2.5549( 19) 01-c1 1.129(9) Mo-C 1 2.057(8) 02-c2 1.149(9) Mc-C2 1.969(7) 03-C3 1.13 1 (10) Mo-C3 2.035(8) 04-C4 1.15 l(9) Mo-C4 1.988(7) 05-C5 1.133 (1 0) w-PI 2.6327( 17) 06-C6 1.144( 10) w - c 5 2.002(8) 07-C7 1.127(11) Bond Angles W-P 1 -Mo 117.55(6) PI-w-c7 129.99(23) P 1 -M e P 2 96.78(6) MC-C1-01 174.8(6) P1-MeC 1 89.87(18) M e C 2 - 0 2 179.3(6) P l - M e C 2 176.83(20) M e C 3 - 0 3 173.0(6) P l - M e C 3 96.03(19) W-C4-04 1 72.8 (6) P l - M e C 4 96.56(19) W-C5-05 174.8(6) P1-w-c5 77.25 (20) W-C6-06 174.3(6) PI-W-C6 75.86(20) W-C7-07 179.3(8)
be obtained. The other possibility is the dissociation and reassociation of this tungsten CO in solution a t high temperature. The ligand exchange experiment was un- successful for this purpose because the reaction between 1 and 13C0 resulted in the formation of the non-metal- metal-bonded complex 2.
The exchange of the semibridging carbonyl ligand with the other three cis CO ligands may proceed through the rotation of the Mo-P bond (Scheme 2). The rotation of the Mo-P bond requires the cleavage and the re-formation of the metal-metal dative bond. The cleavage of the
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
-
Structure and Reactions of C p W(CO)z(p-PPh&l4o(CO),
@
O4Figure 2. ORTEP drawing of 1. Hydrogen atoms are omitted.
Figure 3. ORTEP drawing of 2. Hydrogen atoms are omitted.
metal-metal bond in phosphido-bridged complexes is usually accompanied by an upfield shift of the 3lP NMR signal. Variable-temperature 31P NMR of 1 did not show any change of resonance position of the phosphido phosphorus. This indicates that the cleavage and the re- formation of the Mo-W bond are so rapid that the exchange time scale is beyond the NMR detection limit.
This type of fluxional behavior seems general for mono- (phosphid0)-bridged carbonyl c ~ m p l e x e s . ~ J ~ Note that the mechanism involving cleavage of the metal-phosphido bridge bond and the rotation of the metal-metal bond has been proposed to explain the fluxional behavior which involves the exchanges of terminal CO on one of the metal moieties in the bis(phosphid0)-bridged complex.18
Addition of Phosphine (PMe3, PPhZH, P(0Me)S) a n d CO t o 1. Reaction of 1 with phosphine L (L = PMe3, PPhzH, P(OMe)3) a t room temperature yielded CpW- (17) Shyu, S. G.; Lin, P.-J.; Dong, T.-Y.; Wen, Y.-S. J. Organomet.
(18) Finke, R. G.; Gaughan, G.; Pierpont, C.; Cass, M . E. J. Am. Chem. Chem. 1993,460, 229.
SOC. 1981, 103,1394.
Organometallics, Vol. 13, NO. 5, 1994 1705 ( C O ) ~ ( P - P P ~ ~ ) M ~ ( C O ) ~ L (3) with L regiospecific and stereospecific on the Mo site cis to the phosphido-bridge ligand (Scheme 1). The regiospecific assignment is revealed by the absence of Jp-w for the signal of L in the 31P NMR of 3. A downfield shift of the phosphido-bridge phosphorus signal in the 31P NMR indicates the cleavage of the metal-metal bond. The structure of CpW(CO)3- (~-PPhz)Mo(C0)4(PMe3) was further characterized by a single-crystal X-ray diffraction study (Figure 5 ) .
The long distance between W and Mo (4.5198(14)
A)
indicates that no metal-metal bond exists between the two metals. The PMe3 ligand is coordinated to the Mo and is cis to the phosphido bridge. The replacement of CO in 2 withPMe3 does not increase the repulsion between the W and the Mo moieties in 3a. This is shown by the observation that the distance between W and Mo and the W-P-Mo angle in 3a are almost the same as the distance between W and Mo and the W-P-Mo angle in 2.
The regiospecific and stereospecific addition of phos- phines to 1 is of interest because the addition of a Lewis base to heterobimetallic phosphido-bridged complexes with a metal-metal dative bond usually produces com- plexes with the base coordinated to the metal at the place where the metal-metal dative bond originally coordinated (if the metal-metal bond ~ l e a v e d ) . ~ ~ ~ ~ b ~ ~ * ~ In this case, the phosphine should coordinate to the W atom. However, the phosphine may initially coordinate to W to form the kinetic product and further migration of the phosphine to the adjacent Mo may occur as in the case of (C0)4Fe(p- AsMe2)Co(CO)zLz (L = PMe3, P(OMe)3).4* The regiospe- cific addition on Mo may also be due to the steric effect because of the bulky Cp and p-PPhz ligands and the incoming phosphine. In order to evaluate these factors, we used W O in the addition reaction.
The 13C(lH} NMR spectrum of 13CO-enriched 2 shows only one doublet at 206.8 ppm with 2 J p - ~ = 6.3 Hz. No other signals in the terminal carbonyl region were observed. Although the W { l H } NMR of nonenriched 2 cannot be obtained because of the low solubility and slow decom- position of the complex in solution in long-term NMR measurements, the doublet is assigned to the cis-CO of Mo. The assignment is based on the absence of Jc-w satellites and the favorable comparison with the reported resonance a t 6 206.5 (Jp-c = 7.7 Hz) of for the cis CO signal in
Cp(C0)2Fe(~-PPhz)Mo(C0)5.~
This indicates that CO addition to 1 was regiospecific and stereospecific on the Mo site and that the addition was cis to the phosphido- bridged ligands. This result excludes both intramolecular ligand migration from W to Mo and the steric influence of Cp and p-PPhz ligands, because no tungsten terminal CO signal was observed. Formation of 3 from 1 requires the loss of one CO from Mo and the addition of one CO to W. There are two possible sources for this added W CO ligand. One possibility is that carbon monoxide on Mo may first be substituted by the phosphine ligand to form the metal-metal-bonded complex 5 (Scheme 3). Free CO from the environment may react with 5 to form 2. The other possibility is the intramolecular migration of the semibridging CO on Mo to the adjacent W during the reaction. A reaction between 1 and P(OMe)3 under 13C0 was carried out to produce 3b. Both the mass spectrum and 13C NMR of the product indicate no 13C0 was introduced into the product. In addition, the complex 4(trans isomer of 5 ) prepared from the irradiation of the corresponding 3 did not react with CO to regenerate 3.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1706 Organometallics, Vol. 13, No. 5, 1994 Shyu et al, 223K
1
238Kfi
298Km\-
I
1
320K DDn 230 225 220 215 210 205 200Figure 4. Variable-temperature l3C{lH] NMR spectra of 1 in CDzClz. Only the carbonyl region is shown.
These observations exclude the intermolecular CO addi- tion to the W atom in the reaction.
Role of the Metal-Metal Dative Bond in the Ad-
dition Reaction. If we consider Cp(C0)aWPPhz in
cp(co)2y-
Mo''.
complexes 2 and 3 as a ligand, complex 3 can be considered as a disubstituted Mo(C014LL' complex with L' = Cp- (C0)3WPPh2 and L = PPhzH, P(OMe)3, PMe3. The substitution of group VI metal carbonyl complexes usually requires high temperature.lg The metallophosphine ligand Cp(C0)3WPPhz did not activate the Mo(C0)5 moiety for further substitution, since no complex 3 was observed when complex 2 was allowed to react with P(OMe)3 in T H F phosphine ligands to form 3 proceeded a t room tempera- ture within several hours.
Scheme 2 Phz CO Ph2 CO P co co
/
+e,,..I
*&.*...
cd
I
kCO
/p% ',,,I
.*.,.
8 CP(CO)~W- MoJ
I'co1
co O co 0overnight. However, addition reactions between 1 and Ph2 CO
(19) Keiter, R. L.; Keiter, E. A,; Mittelberg, K. N.; Martin, J. S.; Meyers, V. M.; Wang, J . G . Organometallics 1989, 8 , 1399.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
Structure and Reactions of C p r ; t ' ( C O ) , ( r - P P h , ) ~ o ( C O ) ~
A
Organometallics, Vol. 13, No. 5, 1994 1707
205K
I
Figure 5. ORTEP drawing of 3a. Hydrogen atoms are omitted. Scheme 3 L = P(OMe)3
***eHIcL*.uJLJ
263KL
L
,A,
I. 165 160 I I-..-J!-
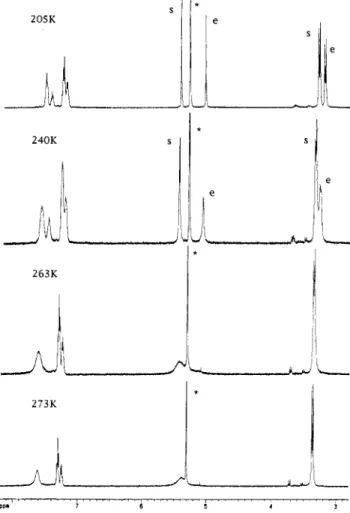
Dp -30 -a -40 273KFigure 6. Variable-temperature 31P{1H) NMR spectra of 3b
in CDzClz. t
Ph, cn
5
The Mo(C0)s moiety in 1 was probably activated by the formation of the metal-metal bond. The metal-metal bond can influence the Mo(C0)b fragment in two ways. One way is electron donation from the filled tzg orbital of the Mo atom to the W atom through the dative metal- metal bond. Powell suggested that the net result of this donation will be a decrease in d,, a* CO bonding to the equatorial CO's (C6, C4, C3).ld This may result in the weakening of the Mo-CO bond in 1. The second way is that the dative metal-metal bond brings two metals together such that the adjacent tungsten is able to activate one of the Mo carbonyl ligands through the donation of the electron from the electron-rich tungsten atom to the
P* orbital of the adjacent molybdenum CO to form a
semibridging CO ligand.20
Formation of the metal-metal dative bond in 1 thus can be considered as a switch, which triggers the substitution of the Mo carbonyl by the activation of one of the Mo carbonyls through the adjacent W atom. The labilization of the metal-CO group by the metal-metal-bonded adjacent metal in phosphido-bridged complexes has been suggested in the (C0)4Ru(~-PPhz)Co(C0)3 system.2l This phenomenon of activation of one of the metal fragments
-
(20) Cotton, F. A. Prog. Inorg. Chem. 1976, 21, 1.
(21) Regragui, R.; Dixneuf, P. H.; Taylor, N. J.; Carty, A. J. Orgu-
nometallics 1986, 5, 1.
by the adjacent metal through metal-metal dative bond formation can also be considered as a cooperativity effect of the adjacent metal in heterobimetallic complexes.
Conformational Isomers of 3. Both variable-tem- perature 31P(1HJ NMR (Figure 6) and variable-temperature lH NMR (Figure 7) of 3 indicate that two isomers exist in equilibrium in solution. The upfield phosphido-bridge signal in the 31P{1H) NMR indicates that a metal-metal bond does not,exist in either isomer. Low-temperature l3C{lH) NMR of the W0-enriched 3b indicates that there were two conformational isomers with one having the phosphine on the Mo eclipsed with the W moiety (isomer e) and the other having all ligands on the Mo staggered with respect to the W moiety (isomer s, Figure 8) in the solution.
Figure 9 shows the 13C(lHJ NMR spectrum of 3b in the metal carbonyl region a t 205 and 240 K. Signals C, and C,! are assigned to CO, in isomer e and CO, in isomer s,
respectively, due to the observedJcw satellite. The signal
Ca,b is assigned to the two equivalent CO, and
cob
ligandsin isomer e. Signals
cat
and cb' are assigned to the CO,and
cob
ligands in isomer 8 , respectively. On the basisof the integration of signals, the ratio of isomer e to isomer s was 8:lO a t 205 K. When the temperature was raised to 240K, the ratio of isomer e to isomers in solution changed to 6: 10. This observation further supports the assignment, since the eclipsed form should have higher energy due to the steric hindrance of the ligands in the complex.
The 13C(1HJ resonance positions in 3b corresponding to the ipso-C carbons of the phenyl group (Pha and Phb) in the diphenylphosphido-bridged ligand further support that the isomers represent eclipsed and staggered con- formational isomers. In isomer e, the signals of ipSO-Ca and ipso-Cbshould be equivalent. In isomers, they should
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1708 Organometallics, Vol. 13, No. 5, 1994 Shyu et al.
ipSO-C,b signal corresponding to the equivalent ipso-c, and ipso-Cb in the e isomer and two signals corresponding to ipso-car and iPSO-Cb’ in the s isomer. Similarly, signals corresponding to 0-C, m-C, andp-C should follow the same argument. In all, there should be 1 2 sets of phenyl signals, with 8 sets corresponding to the staggered isomer and 4 sets corresponding to the eclipsed isomer. Indeed, 6 sets of signals corresponding to ipSO-Ca,b (d, 140.27 ppm, J p - c
= 9.05 Hz), ipSO-Caf (8, 145.0 ppm), ipso-cb’ (d, 141.18
ppm, J p - c = 3.40
Hz),
O-Car (d, 134.71 ppm, J p - c = 3.40Hz),
o-cbi (d, 131.76 ppm, J p - c = 7.55Hz),
and o-ca,b (d, 132.16 ppm, JP-c = 6.04Hz)
are clearly observed. For m-C and p-C, the signals cannot be clearly assigned due to overlap but are clear enough to support the above argument (Figure 10).The interconversion between isomers e and s probably occurs through the rotation of the M-P(phosphid0) bond (M = Mo, W). From the variable-temperature NMR data, the isomer s is more stable a t higher temperatures. The equilibrium also depends on the phosphine ligands. On the basis of 13C NMR, the ratios of e isomer to s isomer were 1:5 for 3a (L = PMe3, 205 K, CDzClz), 3:5 for 3b (L
= P(OMe)3,205 K, CDzClz), and 1:6 for 3c (L = PPhzH, Syntheses and Spectroscopic Characterization of 210 K, CDCl3).
205K s i l * l e
263K
__
DO. 7 6 5 4 3
Figure 7. Variable-temperature lH NMR spectra of 3b in CDZC12. Solvent signals are indicated with asterisks.
view view a view a CO,? I:
.
co, W ‘% view be
f o r ms
f o r mFigure 8. Conformation isomers of 3.
not be equivalent. In the solution mixture of isomer e and isomer s, three i p s o 4 signals should be observed with one
CpW(CO)2(p-PPhz)Mo(CO)d(L)
(4; L = PPh3, PPhZH, PMe3, P(OMe)3). Reaction of 1 with PPh3 produces 4d (Scheme 1). The downfield resonance in the 31P NMR of the phosphido phosphorus a t 168.26 ppm (2Jp-p = 27.1 Hz, J p - w = 325.5 Hz) indicates the presence of the metal- metal bond. The PPh3 is coordinated to the Mo, because no coupling between W and P is observed. Although we did not obtain a single-crystal X-ray structure determina- tion of 4d, the PPh3 is believed to occupy the position trans to the phosphido bridge. This assignment is based on 13C NMR of the complex. One doublet a t 230.95 ppmP J p - c = 15.87 Hz) and one singlet at 223.54 ppm are observed in the terminal carbonyl region. These two signals are assigned to two terminal CO’s on tungsten, since they compared favorably with 226.73 ppm (2Jp-c =
17.6 Hz) and 221.80 ppm for the tungsten terminal CO resonances of 1. No Mo terminal CO signal was observed. Because the exchange of cis CO’s on Mo in 1 results in the flattening of their CO signals in the NMR and trans CO is not involved in the fluxional behavior, the absence of observable Mo terminal CO groups indicates the absence of a trans CO and the presence of four cis CO ligands. Thus, the phosphine occupies the trans position.
One of the carbonyl ligands in 3 can be removed by photolysis togenerate 4. The downfield resonance position of the phosphido phosphorus indicates the presence of the metal-metal bond. On the basis of the similarity of the IR to that of 4d, the phosphine ligands in 4 occupy the position trans to the phosphido bridge.
The reaction between 1 and PPh3 to produce 4d instead of opening the metal-metal bond to form CpW(CO)&- PPhz)Mo(CO)d(PPhs) (as in the case of the other phos- phine ligands PMe3, P(OMe)3, and PPhZH) can be explained by the steric hindrance of the bulky PPh3 group. Therefore, PPh3 may initially react with 1 to form 3d. Because of the steric hindrance of the bulky PPh3, re- formation of the metal-metal bond following loss of one CO from 3d produces 4d as the thermodynamic product. The direct substitution of the Mo carbonyl ligand in 1 by PPh3 is unlikely, because no complex 4 is observed in the
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
-
Structure and Reactions of
C~W(CO)Z(~-PP~Z)MO(CO),
273K
Organometallics, Vol. 13, No. 5, 1994 1709
I " ' . l ' ' ~ - I ' ' ~ . l
D D I 215 22Q 215 2 IO
Figure 9. Variable-temperature l3C(lH) NMR spectra of 3b. Only the carbonyl region is shown.
205K O-' a,b
I ' " ' " " ' , . . ~ ~
Pp. tb I4 133 IN Ib
Figure 10. Phenyl region of the 13C{lH] NMR spectrum of 3b at 205 K. reaction between the other phosphine ligand and 1 when
the reaction is followed by 31P NMR spectroscopy. The strain from the repulsion between the cis phosphine ligand and ligands on the adjacent tungsten can also be
released by the formation of the trans isomer 3-trans since trans isomers were the thermodynamic products of bis- (ph0sphine)molybdenum carbonyls.22 However, pyrolysis of 3b results in the formation of 4b. These results indicate
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
1710 Organometallics, Vol. 13, No. 5, 1994
that the reaction path to CO removal to form the metal- metal bonded complex is favored a t elevated temperatures. The steric hindrance in the proposed complex 3d may also be released by the fragmentation of the product, as indicated by the isolation of a large amount of Mo- (C0)5PPh3 as the side product. Reaction between l and the bulky PEt3 at room temperature overnight resulted in fragmentation of the complex, further supporting the above statement.
Shyu et al. of the M-P bond in 1 was observed by 13C NMR spectrometry.
Addition of CO and a Lewis base to 2 was regiospecific and stereospecific on the Mo and was cis to the phosphido bridge. The structure of 3a was determined by single- crystal X-ray methods. Results from the addition reaction under 13C0 demonstrated that the transfer of CO from Mo to W was intramolecular. The adjacent metal was believed to assist the addition reaction through the formation of a metal-metal dative bond.
Conclusions
Heterobimetallic phosphido-bridged complexes with metal-metal dative bond,
CpW(CO)z(p-PPhz)Mlo(C0)5,
and without a metal-metal bond, CpW(CO)&-PPhz)Mo- (CO)5, were synthesized and their structures were deter- mined by single-crystal X-ray diffraction methods. Flux- ional behavior involving the exchange of four Mo carbonyl ligands cis to the phosphido bridge through the rotation (22) (a) Magee, T. A.; Matthews, C. N.; Wang, T. S.; Wotiz, J. H. J.
Am. Chem. SOC. 1961, 83, 3200. (b) Darensbourg, D. J. Inorg. Chem.
1979, 18, 14.
Acknowledgment. We wish to thank the National Science Council, Republic of China, and Academia Sinica for financial support of this work.
Supplementary Material Available: Listings of crystal data and refinement details, calculated atomic coordinates, anisotropic thermal parameters, and bond distances and angles and figures giving additional views of compounds 1, 2, and 3a (34 pages). Ordering information is given on any current masthead page. OM930649V
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009



![Figure 4. Variable-temperature l3C{lH] NMR spectra of 1 in CDzClz. Only the carbonyl region is shown](https://thumb-ap.123doks.com/thumbv2/9libinfo/8671135.196020/8.924.194.769.60.848/figure-variable-temperature-spectra-cdzclz-carbonyl-region-shown.webp)