國立交通大學
照明與能源光電研究所
碩 士 論 文
含多環咪唑側團基之芴–噻吩共聚物之合成及其
在有機白光元件之應用
Synthesis of Fluorene
-
Thiophene Copolymers Containing
Polycyclic Aromatic Imidazolyl Substituents and Their
Applications in Organic White Light-Emitting Devices
研 究 生:翁國兼 Guo-Jian Wong
指導教授:楊勝雄 博士 Dr. Sheng-Hsiung Yang
含多環咪唑側團基之芴–噻吩共聚物之合成及其
在有機白光元件之應用
Synthesis of Fluorene-Thiophene Copolymers Containing
Polycyclic Aromatic Imidazolyl Substituents and Their Applications
in Organic White Light-Emitting Devices
研究生:翁國兼 Student: Guo-Jian Wong 指導教授:楊勝雄 博士 Advisor: Dr. Sheng-Hsiung Yang
國 立 交 通 大 學 照明與能源光電研究所
碩 士 論 文
A Thesis
Submitted to Institute of Lighting and Energy Photonics
College of Photonics
National Chiao Tung University
in partial Fulfillment of the Requirements
for the Degree of
Master
In Lighting and Energy Photonics
July 2013
i 含多環咪唑側團基之芴–噻吩共聚物之合成 及其在有機白光元件之應用 研究生:翁國兼 指導教授:楊勝雄 博士 國立交通大學照明與能源光電研究所碩士班 摘要 本研究之目的在於合成含多環咪唑側團基之芴–噻吩共聚物,並 探討其在熱性質、光學及電化學性質之影響,亦作為發光層材料以製 備高分子發光元件。本研究同時合成聚(9,9’-二辛基芴) (PFO)作為對 照。含剛硬咪唑側團基之聚芴衍生物分子量較 PFO 小。其熱裂解溫 度與 PFO 相差無幾,且於高溫有更多的殘留重量,證明引入咪唑側 團基可提高材料熱穩定性。其玻璃轉換溫度遠高於 PFO,有利於 PLED 元件操作之耐久性。引入咪唑側團基之聚芴衍生物吸收光譜與 PFO 相似,顯示咪唑側團基並不影響主鏈之吸收;放射光譜較 PFO 紅移,其中 P1 最大吸收峰更大幅紅移至 510 nm。電化學分析顯示 咪唑團基的引入導致材料 LUMO 下降,HOMO 亦隨之降低,P1 具 有比 PFO 更好的電子電洞注入能力,而 P2–P4 卻因 HOMO 過低導
ii 致巨大的電洞注入能障。本研究製備 ITO/PEDOT/polymer/P1-BF4/Al 之高分子發光元件,其中 P1-BF4為電子傳輸材料。材料色光分別為 P2–P4 的藍綠光及 P1 的綠光,顯示引入咪唑側團基確實可改變材料 之發光波段。本研究最後將 PFO:P1:MEH-PPV 以 300:100:1 重量比 混合以製備出白光元件,其亮度與效率分別為 509 cd/m2及 0.20 cd/A, 其 CIE’1931 座標為(0.32, 0.37)。以上結果顯示其具有固態照明之應 用潛力。
iii
Synthesis of Fluorene-Thiophene Copolymers Containing
Polycyclic Aromatic Imidazolyl Substituents and Their Applications
in Organic White Light-Emitting Devices
Student: Guo-Jian Wong Advisor: Dr. Sheng-Hsiung Yang
Institute of Lighting and Energy Photonics
National Chiao-Tung University
Abstract
The goal of this research is to synthesize fluorene-thiophene
copolymers containing polycyclic aromatic imidazolyl substituents,
and to study the influence of those substituents on thermal, optical,
and electrochemical properties of polymers. Those polymers are
used as emitting layer for fabrication of polymer light-emitting devices. Poly(9,9’-dioctylfluorene) (PFO) are also synthesized for comparison in this research. The molecular weights of polyfluorene
derivatives containing rigid imidazolyl substitutes are lower than that
of PFO. The decomposition temperatures of all polymers are almost
the same; meanwhile, those derivatives possess more weight
residues than PFO after heating, indicative of enhanced thermal
stabilities of materials by the introduction of imidazolyl substitutes
into polymer main chains. The glass transition temperatures of
iv
on operation stability of PLED. The absorption spectra of those
polyfluorene derivatives containing imidazolyl substitutes are similar
to that of PFO, referring that those substitutes have no influence on
the absorption of polymer main chains. The emission spectra of
those derivatives show red-shifts compared with PFO; among them
P1 generates a heavily red-shifted emission maximum to 510 nm.
Electrochemical analysis shows that introducing imidazolyl groups
into polymer main chains results in decreasing LUMO levels of
derivatives compared with PFO, and their HOMO levels are
decreased as well. P1 behaves better hole and electron injection
abilities than those of PFO, while P2, P3, and P4 possess huge hole
injection barrier owing to low-lying HOMO levels. Polymer
light-emitting devices with configuration of
ITO/PEDOT/polymer/P1-BF4/Al were fabricated in this research, using P1-BF4 as electron
transport material. The emissive light of those materials are
bluish-green for P2, P3, and P4, and bluish-green for P1, proving that the
introduction of imidazolyl substitutes into polymer main chains
indeed brings change on the emission bands of materials. Finally, a
polymer blend was prepared by using PFO:P1:MEH-PPV =
300:100:1 in weight ratio for fabrication of white-light-emitting
devices. The brightness and current efficiency of the device
achieved 509 cd/m2 and 0.20 cd/A, respectively, with CIE’1931
coordinating at (0.32, 0.37). The above results reveal its potential
v
誌謝 謝謝各位,下台一鞠躬。
vi 目錄 頁次 摘要 ... i Abstract ... iii 誌謝 ... v 目錄 ... vi Scheme 目錄 ... ix 圖目錄 ... x 表目錄 ... xiii 第一章 緒論 ... 1 1-1 前言 ... 1 1-2 有機發光二極體簡介 ... 1 1-3 OLED 元件結構 ... 3 1-4 高分子材料發光理論 ... 5 1-4-1 能階理論 ... 5 1-4-2 光激發光 ... 7 1-4-3 電激發光 ... 7 1-5 有機共軛高分子 ... 8 1-5-1 共軛高分子 ... 8
vii 1-5-2 聚芴高分子 ... 9 1-5-3 主鏈型聚芴衍生物 ... 10 1-5-4 含不同側取代基之聚芴衍生物 ... 13 1-5-5 鈴木耦合法 ... 21 1-6 研究動機 ... 22 第二章 實驗方法與步驟 ... 26 2-1 試藥 ... 26 2-2 鑑定儀器 ... 26 2-2-1 核磁共振光譜儀 ... 26 2-2-2 示差掃描卡計 ... 26 2-2-3 熱重分析儀 ... 27 2-2-4 凝膠滲透層析儀 ... 27 2-2-5 紫外–可見光吸收光譜儀與螢光光譜儀 ... 27 2-2-6 循環伏安計量法 ... 28 2-2-7 氣相層析質譜儀 ... 28 2-2-8 傅立葉紅外線光譜儀... 28 2-2-9 原子力顯微鏡 ... 29 2-2-10 元件量測系統 ... 29 2-3 元件製作 ... 30
viii 2-3-1 ITO 基板清洗步驟 ... 30 2-3-2 PLED 元件製作流程 ... 30 2-4 單體之合成 ... 32 2-5 高分子聚合 ... 40 第三章 結果與討論 ... 44 3-1 分子量測定 ... 44 3-2 NMR 光譜分析 ... 46 3-3 FTIR 分析 ... 47 3-4 熱性質分析 ... 49 3-5 光學性質分析 ... 51 3-5-1 吸收光譜分析 ... 51 3-5-2 螢光放射光譜分析 ... 53 3-5-3 螢光量子效率 ... 59 3-6 電化學性質分析 ... 61 3-7 高分子溶解度測試 ... 63 3-8 PLED 元件表現 ... 64 第四章 結論 ... 70 參考文獻 ... 72 附錄 ... 77
ix
Scheme 目錄
頁次 Scheme 1. 單體 M1–M6 之合成途徑 ... 42 Scheme 2. 高分子 PFO 及 P1–P4 之合成途徑 ... 43
x
圖目錄
頁次
Figure 1–1. Anthracene 化學結構 ... 1
Figure 1–2. Alq3與 Diamine 化學結構及其元件結構示意圖 ... 2
Figure 1–3. 多層有機發光二極體元件結構示意圖 ... 4 Figure 1–4. 基態、單重及三重激發態之電子自旋方向示意圖 ... 5 Figure 1–5. 電子躍遷示意圖 ... 7 Figure 1–6. (a)光激發光及(b)電激發光機制示意圖 ... 8 Figure 1–7. 芴酮結構形成機制示意圖 ... 9 Figure 1–8. 主鏈型聚芴衍生物化學結構 ... 12 Figure 1–9. 含分枝狀苯環結構之聚芴衍生物化學結構 ... 13 Figure 1–10. 含樹枝狀苯環結構之聚芴衍生物化學結構 ... 14 Figure 1–11. 含咔唑及樹枝狀噁二唑團基之聚芴衍生物化學結構 17 Figure 1–12. 含雙取代樹枝狀噁二唑團基之聚芴衍生物化學結構 18 Figure 1–13. 含三苯胺、噁二唑之聚芴衍生物化學結構 ... 19 Figure 1–14. (a)具側取代液晶團基之聚芴衍生物化學結構,(b)含銥 錯合物之聚芴衍生物化學結構 ... 20 Figure 1–15. 含銪錯合物之聚芴衍生物化學結構 ... 21 Figure 1–16. 鈴木耦合法反應式 ... 21
xi Figure 1–17. 鈴木耦合法反應機制 ... 22 Figure 1–18. 推測之O4 及 O5 化學結構 ... 23 Figure 1–19. O4 及 O5 螢光放射光譜 ... 23 Figure 1–20. 含咪唑團基衍生物之化學結構及其溶液態於長波 UV 照射下之放光情形... 24 Figure 1–21. 含咪唑團基之單體 M3–M6 化學結構 ... 25 Figure 3–1. 高分子 PFO 及 P1–P4 化學結構 ... 44
Figure 3–2. 高分子(a) P1、PFO、M3、(b) P2、PFO、M4、(c) P3、 PFO、M5 及(d) P4、PFO、M6 之 NMR 氫譜比較圖 ... 47
Figure 3–3. 高分子(a) PFO 及(b) P1 之 FTIR 光譜 ... 48
Figure 3–4. 高分子(a) P1、PFO、M3、(b) P2、PFO、M4、(c) P3、
PFO、M5 及(d) P4、PFO、M6 之 FTIR 光譜比較圖 ... 49
Figure 3–5. 高分子 PFO 及 P1–P4 之 TGA 曲線圖 ... 50 Figure 3–6. 高分子 PFO 及 P1–P4 之 DSC 曲線圖 ... 51 Figure 3–7. 高分子 PFO 與 P1–P4 於(a)薄膜態、(b) toluene、(c) DCM 及(d) THF 溶液之吸收光譜 ... 53
Figure 3–8. 高分子 PFO 及 P1–P4 於(a)薄膜態、(b) toluene、(c) DCM 及(d) THF 溶液之螢光放射光譜... 55
xii
Figure 3–9. 高分子 PFO 及 P1–P4 於(a)薄膜態及(b) toluene 溶液中
經長波 UV 照射之放光照片 ... 56
Figure 3–10. MEH-PPV 化學結構及其吸收與放射光譜 ... 58
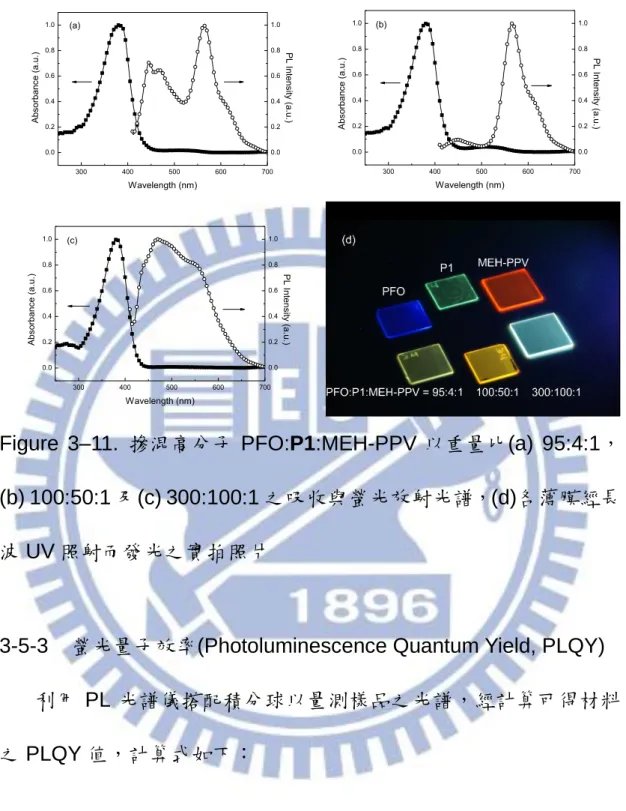
Figure 3–11. 摻混高分子 PFO:P1:MEH-PPV 以重量比(a) 95:4:1, (b) 100:50:1 及(c) 300:100:1 之吸收與螢光放射光譜,(d)各薄膜經長 波 UV 照射而發光之實拍照片 ... 59
Figure 3–12. 高分子 PFO 及 P2 於(a)薄膜態及(b) THF 溶液之光譜 ... 60
Figure 3–13. 高分子 PFO 及 P1–P4 之氧化曲線圖 ... 62
Figure 3–14. 元件使用材料之能階示意圖 ... 63
Figure 3-15. P1-BF4化學結構 ... 64
Figure 3–16. 三層元件 ITO/PEDOT/Polymer/ P1-BF4/Al 之(a) EL 光 譜、(b)電流密度–電壓、(c)亮度–電壓及(d)電流效率–電流密度曲線圖 ... 66
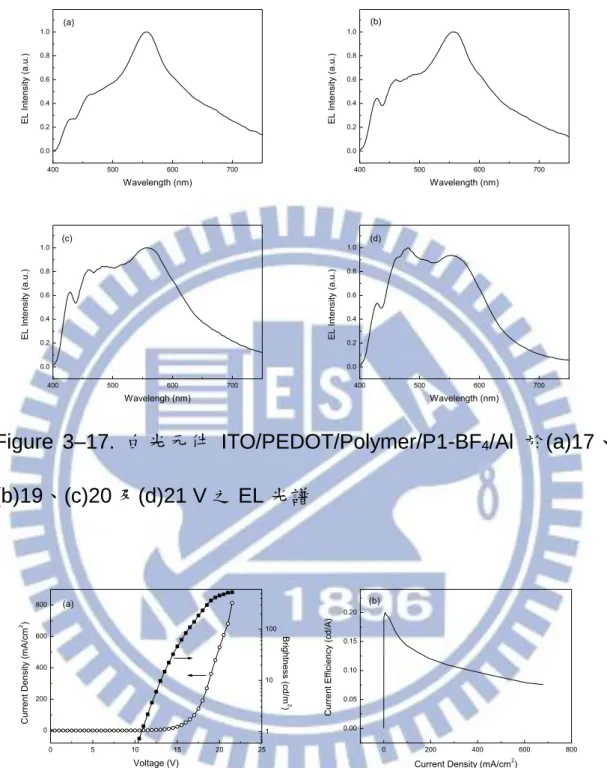
Figure 3–17. 白光元件 ITO/PEDOT/Polymer/P1-BF4/Al 於(a)17、 (b)19、(c)20 及(d)21 V 之 EL 光譜 ... 68
Figure 3–18. 白光元件 ITO/PEDOT/Polymer/P1-BF4/Al 之(a)電流密 度–亮度–電壓、(b)電流效率–電流密度曲線圖 ... 68
xiii
表目錄
頁次
Table 3–1. 高分子 PFO 及 P1–P4 之聚合結果 ... 46
Table 3–2. 高分子 PFO 及 P1–P4 之 FTIR 訊號 ... 48
Table 3–3. 高分子 PFO 及 P1–P4 之熱性質數據 ... 51
Table 3–4. 高分子 PFO 及 P1–P4 之最大吸收與螢光放射峰值 .. 56
Table 3–5. 高分子 PFO 及 P1–P4 於薄膜態及不同溶液態之螢光量 子效率 ... 61
Table 3–6. 各高分子之氧化電位、HOMO、能隙及 LUMO ... 63
Table 3–7. 高分子於不同溶劑中之溶解度比較表 ... 64
1
第一章 緒論 1-1 前言
生 活 中 隨 處 可 見 各 式 各 樣 的 顯 示 器 , 其 中 有 機 發 光 二 極 體 (Organic Light-Emitting Diode, OLED)因自發光、高對比、重量輕、
厚度薄、低耗電量、反應時間快、廣視角及可撓曲性等優點,逐漸取 代液晶顯示器(Liquid Crystal Display, LCD)成為市場新寵兒。
高分子發光二極體(Polymer Light-Emitting Diode, PLED)雖因壽 命問題不如 OLED 而較少見於市面產品,然其具有大面積製程且製 程簡單之優點,亦為可期待的未來之星。 1-2 有機發光二極體簡介 有機發光二極體的原理是藉由導入偏壓使有機發光層產生電激 發光。此現象於 1963 年由 Pope、Kallmann 等人所發現,當時以 10 至 20 微米之單晶蒽(Anthracene)作為發光層,通以 400 伏特電壓發 出微弱藍光[1]。但因為製程條件控制不易,且操作電壓過高等因素, 並沒有受到太大的重視。單晶蒽之化學結構如 Figure 1–1 所示。 Figure 1–1. Anthracene 化學結構
2
直到 1987 年柯達公司的 C. W. Tang 與 S. A. VanSlyke 等人製作
以 ITO 為陽極,Tris(8-hydroxyquinolinato)aluminum (Alq3)為發光層
與電子傳輸層,Diamine 為電洞傳輸層,鎂銀合金作為電極的雙層元
件[2],此結構能提高電子電洞對在發光層中複合的機率,於 10 V 操
作電壓下,亮度達到 1000 cd/cm2,外部量子效率(External Quantum
Efficiency, EQE)達到 1 %。為柯達公司生產 OLED 顯示器奠定了基
礎,也開啟 OLED 的蓬勃發展。元件結構與材料如 Figure 1–2 所示。 N O Al O O N N Alq3 N N H3C CH3 CH3 CH3 Diamine
Figure 1–2. Alq3與 Diamine 化學結構及其元件結構示意圖
發光高分子的研究最早於 1982 年,Patridge 利用旋轉塗佈方式 製作以 Poly(N-vinylcarbazole) (PVK)為發光層的第一個高分子電激
發光元件[3]。而後,1990 年劍橋大學的 J. H. Burroughes 等人使用
Poly(1,4-phenylenevinylene) (PPV)作為發光層[4],製作 ITO/PPV/Al
3 至此,有機發光元件開始區分為兩類:一類使用有機染料小分子 作為發光層,以熱蒸鍍方式製作元件,稱之 OLED;另一類以共軛高 分子為主,採旋轉塗佈製程,稱為 PLED。兩類材料各執長短,小分 子合成較易,螢光效率亦較高,但蒸鍍設備成本較高,材料利用率偏 低;高分子合成複雜且分子量分佈不均、色純度較低,然而成膜性佳 並可大面積製程,成本較低,且高分子中電荷載子密度較高,使元件 操作所需電壓較小分子元件低。 1-3 OLED 元件結構 現今常見 OLED 元件大部分為多層結構,如 Figure 1–3 所示。 1. 陽極(Anode): 陽極材料需具有良好的熱穩定性、透明導電度高且功函數與電洞 注入/傳輸材料的最高填滿分子軌域(Highest Occupied Molecular Orbital, HOMO)相匹配。Indium Tin Oxide (ITO)為常見的陽極材料。
2. 電洞注入層(Hole Injection Layer, HIL)
幫助電洞由陽極注入發光層,故材料選擇上 HOMO 要與陽極功 函數匹配。
3. 電洞傳輸層(Hole Transporting Layer, HTL)
4
使 其 侷 限 於 發 光 層 中 , 材 料 之 最 低 未 填 滿 分 子 軌 域 (Lowest Unoccupied Molecular Orbital, LUMO)需高於發光層之 LUMO。
4. 發光層(Emission Layer, EML)
良好發光材料除了螢光強度高外,還須具備極佳的熱穩定性,以 免因操作元件時內部溫度上升而影響發光,更需容易成膜,方便製程。 5. 電子傳輸層(Electron Transporting Layer, ETL)
須具有高電子遷移率,其 HOMO 低於發光層之 HOMO。 6. 電子注入層(Electron Injection Layer, EIL)
其 LUMO 與陰極功函數匹配,以利電子注入。 7. 陰極(Cathode)
為了降低電子注入能障,通常選用低功函數的金屬如鈣、鎂或合 金;但因為活性考量,亦會使用鋁等較高功函數但活性低之金屬。
5
1-4 高分子材料發光理論
1-4-1 能階理論
化學分子受到如光能、電能等外加能量的激發,當能量大於分子
能隙(Energy Gap, Eg),電子由基態躍遷至激發態。激發態時電子自
旋方向若相反於基態,稱之為單重激發態(Singlet Excited State, Sn);
反之,若電子自旋方向與基態時相同,則稱為三重激發態(Triplet Excited State, Tn),如 Figure 1–4 所示。
Figure 1–4. 基態、單重及三重激發態之電子自旋方向示意圖 電子由激發態降回基態過程中會以非輻射形式及輻射形式釋放 能量,如 Figure 1–5 所示: 非輻射形式 1. 振動鬆弛(Vibrational Relaxation) 激發態的電子受分子間碰撞影響,由較高的振動能階落至同一激 發態的較低能階,並以熱能形式放出能量。
6 2. 內轉換(Internal Conversion) 若激發態的能階相當接近,導致振動能階彼此重疊,電子可能由 較高激發態躍遷至較低激發態,同樣以熱能形式釋放能量。 3. 外轉換(External Conversion) 激發態的分子與其他分子(如:溶劑分子)相互作用致使能量散失, 影響發光強度降低。 4. 系統間跨越(Intersystem Crossing) 激發態電子自旋方向改變,由單重激發態轉換為三重激發態。常 見於含重原子(如:碘、溴)的材料。 輻射形式 輻射形式的能量釋放可分為螢光放射與磷光放射兩種,螢光與磷 光產生的比例約為 1:3,故螢光能達到的最高內部量子效率(Internal Quantum Efficiency)為 25 %。 1. 螢光(Fluorescence) 電子由單重激發態降回基態(Sn→ S0)釋放的光能稱為螢光,生命 週期約 10-7–10-9秒。 2. 磷光(Phosphorescence) 單重激發態的電子經由系統間跨越落至較低能階的三重激發態,
7 再降回基態(Sn→ Tn → S0)輻射的光能為磷光。生命週期在 10-4至數 秒間,由於輻射時間過長,容易導致非輻射方式損耗能量。 Figure 1–5. 電子躍遷示意圖 1-4-2 光激發光(Photoluminescence, PL) 有機共軛高分子的發光可依能量來源分為光激發光與電激發光。 光激發光是以能量高於分子能隙的光源照射,使電子由基態躍遷至激 發態,隨即以輻射形式釋放光能並返回基態。如 Figure 1–6 (a)所示。 1-4-3 電激發光(Electroluminescence, EL) 電激發光是以外加偏壓作為激發源施加於 OLED 元件,驅使電子 與電洞分別由陰極與陽極注入,於發光層複合(Recombination)形成
8 電子電洞對,再由激發態落回基態,並以光能形式放出能量。釋放的 光波長取決於發光層的能隙,利用不同材料可得到由紫外光到紅外光 各波段的光,如 Figure 1–6 (b)所示。 Figure 1–6. (a)光激發光及(b)電激發光機制示意圖 1-5 有機共軛高分子 1-5-1 共軛高分子(Conjugated Polymer) 共軛高分子是指化學結構上單–雙鍵相互交替,使 p 軌域上的 π 電子可沿著分子軌域形成非定域化電子雲,藉此傳導電子。高分子共 軛長度越長,所輻射的光波長越短。薄膜態時,高分子鏈與鏈間會互 相堆疊,共軛π 電子雲亦相互重疊,使激發態能階降低,導致放出光 波長產生紅位移。 此外,高分子螢光材料一大特點就是可藉由連接不同側取代基改 變能隙,進而發出不同波長的光。
9 1-5-2 聚芴高分子(Polyfluorene, PF) 高能隙的藍光材料一向是很受矚目的高分子材料,因其能用於全 彩顯示上,且能夠做為主體材料提供能量轉移至客體材料以增加發光 效率[5]。其中 PF 是現今最常見的藍光材料[6–8],其發光波長落在 400– 460 nm[9–11],同時具有極佳的溶解度,良好的化學、熱穩定性以及螢 光量子效率,並可經由改變 9 號碳位置上的團基而調變材料之光學性 質[12]。
1991 年,Y. Ohmori 等人首先製作以 PF 為發光層,Mg/In 合金
為陰極的單層元件[13]。PF 元件加入偏壓後,常於 540 nm 產生新的
放射肩峰,色光也由藍光紅移為藍綠光[14]。有文獻認為此現象發生原
因是分子鏈之間產生堆疊(Aggregation)或激化雙體(Excimer)所導致
[15–18]。2002 年,E. J. W. List 與 U. Scherf 等人提出另一種解釋[19],
主張 PF 在 9 號碳位置受外加偏壓影響而氧化,產生酮基的缺陷,形 成芴酮(Fluorenone)團基,稱為 Keto Effect,如 Figure 1–7 所示。
R H + e -- [H] R R -OO O2 O - RO -Figure 1–7. 芴酮結構形成機制示意圖
10
1-5-3 主鏈型聚芴衍生物(Main-Chain Type PF Derivatives)
聚芴高分子發展出許多衍生物,大部分為改變主鏈結構之共聚高 分子,此類文獻資料數量相當繁多,在此僅約略介紹與本研究相關的 色光調變型之聚芴衍生物。
C. Ego 等人將紅光材料之駢苯二醯亞胺(Perylene Bisimide, PBI)
與芴共聚[20],其中 PBI 於高分子中僅佔 1%比例,高分子結構如 Figure 1–8 (a)所示,經由能量轉移,此高分子最大放射波長為 600 nm,為 一紅光高分子。Q. Hou 等人將(噻吩–苯并噻二唑–噻吩) (4,7-di-2-Thienyl-2,1,3-Benzothiadiazole)與芴合成共聚高分子[21],化學結構如 Figure1–8 (b)所示,噻吩與苯并噻二唑分別作為施、受體延長了主鏈 之 共 軛 , 使 高 分 子 放 光 紅 移 至 628–674 nm , 以 結 構 ITO/PEDT/polymer/ Ba/Al 製備元件,其最大亮度為 259 cd/m2。X.
H. Zhou 等人受 Keto Effect 特性啟發,直接合成芴酮與芴之共聚高
分子[22],化學結構如 Figure 1–8 (c)所示,隨芴酮所佔比例由 0.1%逐 步提昇至 25%,螢光最大放射波長也由 540 nm 紅移至 579 nm,以 結構為 ITO/PEDOT/ polymer/Ba/Al 製作之高分子元件最大亮度為 1156 cd/m2。P. Herguth 等人引入苯并噻二唑與芴共聚以提高電子傳 導能力[23],高分子結構如 Figure 1–8 (d)所示,其螢光最大放射波長 為 548 nm,以結構 ITO/ BTPD-PFCB/polymer/Ca/Al 製備元件,其
11
最大亮度達 59400 cd/m2,效率為 18.5 cd/A。W. Yang 等人引入二
苯并噻吩(Dibenzothiophene)與芴共聚[24],分子結構如 Figure 1–8 (e)
所示,使 PF 堆疊情形減少,當二苯并噻吩與芴以 1:1 比例共聚,該 高分子於長波波段(約 550 nm)的發光完全消失,成為純藍光之材料。 T. Miteva 等人於 PF 高分子鏈兩端引入具良好電洞傳輸特性的三芳 胺(Triarylamine)[25],化學結構如 Figure 1–8 (f)所示,為藍光高分子 材料,以結構 ITO/PEDOT/ polymer/Ca 製備元件,其最大亮度為 1600 cd/m2,效率為 1.1 cd/A。
12 N N O O O O O O O O R R R R C8H17 C8H17 n m R = N S N S S C8H17 C8H17 m n n = 1%, 5%, 10%, 15%, 25%, or 35% C8H17 C8H17 O m n n= 0.1%, 0.5%, 1%, 2%, 5%, 10%, or 25% N S N C8H17 C8H17 m n C8H17O OC8H17 p m:n:p = 75:12.5:12.5 S C8H17 C8H17 m n n= 5%, 10%, 20%, 30%, or 50% (a) (b) (c) (d) (e) (f) C8H17 C8H17 n N N Figure 1–8. 主鏈型聚芴衍生物化學結構[20–25]
13
1-5-4 含不同側取代基之聚芴衍生物 (PF Derivatives Containing
Different Side Substituents)
除了改變主鏈結構與芴共聚而成之高分子外,另有一類為引入側 取代基的聚芴衍生物,此類研究相對而言僅佔少數。 為防止 PF 發生堆疊,影響色光純度及發光效率,引入多枝的團 基為一可行方法;S. Setayesh 等人引入分枝狀的苯基於 PF 的 9 號 碳位置[26],分子結構如 Figure 1–9 所示,以結構 ITO/PEDOT/ polymer/Ca/Al 製作元件,並未發現大於 500 nm 之放射峰,元件效 率為 0.2 cd/A。D. Marsitzky 等人同樣於 PF 的 9 號碳位置引入苯環 的樹枝狀結構以防止堆疊[27],化學結構如 Figure 1–10 所示,其中第 二代樹枝狀結構能提升 PL 發光效率。上述兩篇文獻的取代基皆不影 響主鏈之共軛,僅改變分子結構以減少堆疊,故材料光色並無改變; 此類研究隨後發展出含不同官能基之側取代基或引入磷光材料,改變 聚芴衍生物之電、光學等特性。 Ph Ph Ph Ph Ph Ph Ph Ph n Figure 1–9. 含分枝狀苯環結構之聚芴衍生物化學結構[26]
14 Gx Gx n G1 = O O O O O O O O O O O O O O O O O O O O O O G2 = G3 = Figure 1–10. 含樹枝狀苯環結構之聚芴衍生物化學結構[27]
15 H. C. Lin 等人將咔唑(Carbazole)及樹枝狀噁二唑團基引入芴分 子 9 號碳位置[28],高分子結構如 Figure 1–11,以結構 ITO/PEDOT/ polymer/LiF/Al 製備元件,其最大亮度為 2446 cd/m2,效率為 0.24 cd/A;同一團隊亦合成含噁二唑樹枝狀團基的聚芴衍生物[29],分子結 構如 Figure 1–12,使 PL 發光效率提高,且放射峰半高寬(Full-Width at Half Maximum, FWHM) 變 窄 。 R. Tang 等 人 合 成 含 三 苯 胺
(Triphenylamine)側取代基之聚芴衍生物[30],化學結構如 Figure 1–13 (a)所示,三苯胺可提昇材料的電洞傳輸能力,且以側取代基方式可使 高分子鏈不易堆積而提高發光效率,光激發光最大放射峰紅移至 485 nm,為藍綠光材料,以結構 ITO/PEDOT/polymer/Alq3/Al 製備元件, 其最大亮度為 3460 cd/m2,效率達 2.08 cd/A。F. I. Wu 等人引入具 良好電子傳輸能力之噁二唑(Oxadiazole)於芴分子 9 號碳位置[31],高 分子結構如 Figure 1–13 (b)所示,取代基並未改變材料之光學特性, 最大螢光放射峰位於 440 nm,為主鏈之放光;玻璃轉換溫度由 PFO 的 67 oC 大幅提昇至 213 oC,有助於元件的耐久性[32],以結構 ITO/PEDOT/polymer/Ca/Ag 製備元件,其最大亮度為 2770 cd/m2, 效率為 0.25 cd/A。C. F. Shu 等人引入三苯胺及惡二唑於芴分子 9 號 碳位置並共聚之[33],分子結構如 Figure 1–13 (c),惡二唑及三苯胺團 基平衡了電子電洞的注入,元件結構同前一篇文獻,其效率達 0.63
16
cd/A,最大亮度為 4080 cd/m2。Y. H. Yao 等人合成具側面液晶團基
之聚芴衍生物[34],化學結構如 Figure 1–14 (a)所示,此高分子具高度
偏振特性,且混合不同光色之共聚物可開發出白光材料,以結構 ITO/
PEDOT/polymer/Ca/Al 製備元件,其最大亮度為 2454 cd/m2。除了
上述官能基外,PF 亦曾引入不同磷光發光團基合成共聚高分子。Y.
Cao 等人將含銥(Iridium)之官能基與芴共聚[35],分子結構如 Figure 1–
14 (b),以結構 ITO/PEDOT/PVK/ polymer/Ba/Al 製備元件,其最大 亮度為 737 cd/m2,效率為 0.70 cd/A;同一團隊亦引入銪(Europium) 於聚芴衍生物中[36],高分子結構如 Figure 1–15 (a)所示,元件結構同 上一篇文獻,其亮度為 25 cd/m2。Q. D. Ling 等人亦曾發表含銪之聚 芴衍生物[37],分子結構如 Figure 1–15 (b)所示,該高分子薄膜態放射 光譜為紅光,且有極小的 FWHM (約 4 nm)。
17 C6H13 C6H13 C6H13 C6H13 Gx Gx N N m n G1 = G2 = G3 = N N O OC8H17 N N O O OC8H17 O N N O O OC8H17 O O O N O N OC8H17 O O N O N N N O OC8H17 OC8H17 N N O OC8H17 Figure 1–11. 含咔唑及樹枝狀噁二唑團基之聚芴衍生物化學結構[28]
18 G1 = G2 = G3 = N N O OC8H17 N N O O OC8H17 O N N O O OC8H17 O O O N O N OC8H17 O O N O N N N O OC8H17 OC8H17 N N O OC8H17 Gx Gx n C6H13 C6H13 Figure 1–12. 含雙取代樹枝狀噁二唑團基之聚芴衍生物化學結構[29]
19 N C6H13 C6H13 n N C6H13 C6H13 n OC8H17 C8H17O O N N N N O C8H17 C8H17 n C8H17 C8H17 n C8H17 C8H17 O N N N N O N N C4H9 C4H9 C4H9 C4H9 m (a) (b) (c) Figure 1–13. 含三苯胺、噁二唑之聚芴衍生物化學結構[30, 31, 33]
20 R m n C8H17 C8H17 random polymer R = C4H9 C4H9 OCH3 OC8H16O H3CO C8H17 C8H17 N S N N S N S S n C8H17 C8H17 Ir O O N m m:n=95:5 2 (a) (b) Figure 1–14. (a)具側取代液晶團基之聚芴衍生物化學結構[34],(b)含 銥錯合物之聚芴衍生物化學結構[35]
21 C6H13 C6H13 O O N N O O Eu n 2 (b) C6H13 C6H13 O C6H13 C6H13 O O (CH2)9 O (CH2)9 N N N N Eu O O m n 3 (a) Figure 1–15. 含銪錯合物之聚芴衍生物化學結構[36, 37] 1-5-5 鈴木耦合法(Suzuki Coupling)
1981 年 Suzuki N.與 Miyaura A.提出此種聚合方法[38],反應式如
Figure 1–16 所示。 B(OH)2 Z + X Z' Pd(PPh3)4 Na2CO3 H2O-benxene Z Z' X = Br, I Z = functional groups Figure 1–16. 鈴木耦合法反應式 反應於鹼性環境下進行,以鈀金屬作為觸媒催化,使芳香鹵化物 (Aryl Halides)與有機硼酯(Boronic Ester)化合物進行耦合反應。此聚
22 鈴木耦合法反應機制主要有四個步驟[39–41],如 Figure 1–17 所示: 1. 鹵化物受零價鈀金屬活化,形成含兩價鈀金屬中間體。 2. 反應環境中的鹼帶走活化中間體的鹵素離子,由氫氧根取代。 3. 硼酯與鹼反應後再和中間體反應,形成的硼酯根移走,兩價鈀 金屬與 Ar 及 Ar1形成中間體。 4. 兩價鈀金屬脫離回到零價,重複第一步驟。 Figure 1–17. 鈴木耦合法反應機制 1-6 研究動機 本 實 驗 室 李 國 志 學 長 進 行 含咪 唑(Imidazole)側 團 基 之 噻 吩 (Thiophene)聚合時,發現兩種含咪唑側團基的噻吩共聚效果不佳, 形成分子量只有一千左右的寡聚物 O4 與 O5,但卻意外發現此兩種
23 寡聚物分別具有黃綠光與紅光的發光特性,其推測化學結構見 Figure 1–18,薄膜態螢光光譜如 Figure 1–19 所示。 S S S C6H13 C6H13 N N N N S C6H13 H H S S S S S N N C8H17O H H N N C8H17O C6H13 C6H13 C6H13 O4 O5 Figure 1–18. 推測之O4 及 O5 化學結構 400 500 600 700 0.0 0.2 0.4 0.6 0.8 1.0 N orm aliz ed PL Int ens ity (a. u. ) Wavelength (nm) O4 O5 510 nm 624 nm Figure 1–19. O4 及 O5 螢光放射光譜 大部分共軛高分子之電洞傳輸能力較電子為佳,不平衡的電子電 洞傳輸能力對於發光元件的效率有很大的限制,文獻上已報導咪唑具 有降低材料 LUMO 的特性,能夠提高材料的電子傳輸能力[42],含咪 唑的 1,3,5-tris(N-phenylbenzimiazole-2-yl)benzene (TPBi)即為常見
24 的電子傳輸材料[43]。此外,咪唑團基亦曾應用於小分子 OLED 材料, 其色光可由深藍推廣至橘光[44, 45],其結構及溶液光色如 Figure 1–20 所示。 Figure 1–20. 含咪唑團基衍生物之化學結構及其溶液態於長波 UV 照射下之放光情形 本研究改採用芴與含不同咪唑團基的噻吩進行聚合,期望能獲得 新的螢光共軛高分子材料,並推廣其放射光譜範圍。本實驗使用五種 不 同 含 咪 唑 團 基 之 噻 吩 單 體 , 包 含 Triphenyl-imidazole (M3) 、 Phenanthrenyl-imidazole (M4) 、 Pyrenyl-imidazole (M5) 、
25 N N S Br Br M3 N N S Br Br M4 N N S Br Br M5 N N S N N Br Br M6 Figure 1–21. 含咪唑團基之單體 M3–M6 化學結構 上述咪唑衍生物之中,M3 帶有三個自由苯基取代;M4 及 M5 限 制側邊苯環的轉動,可提高平面性;M6 引入氮原子,除探討雜原子 的影響外,未來亦可做為配位基引入磷光材料;M7 的環張力比 M4 更大,平面性更高,預期能導致紅位移的產生。 本研究利用核磁共振儀及紅外線光譜探討化學結構、以吸收光 譜及螢光光譜量測光學性質、以循環電位儀偵測氧化還原電位與能 階、以熱重分析儀與示差掃瞄卡計決定熱性質,並製作有機發光元 件探討其光電應用。
26
第二章 實驗方法與步驟
2-1 試藥
本實驗試藥及溶劑皆自聯工、Merck、Aldrich 與 Alfa Aesar 購入, 不經純化直接使用。反應用之無水 Tetrahydrofuran (THF)是在氮氣環 境下加入 Sodium (Na)除水,並加入 Benzophenone 作為指示劑,經 除水 2 天後蒸出使用;無水 Toluene 是在氮氣環境下加入 Calcium hydride 除水,經除水 3 天後蒸出使用。
2-2 鑑定儀器
2-2-1 核 磁 共 振 光 譜 儀 (Nuclear Magnetic Resonance
Spectrometer, NMR)
係使用 Bruker Avance 600 MHz NMR 型,並使用 d-DMSO、d-Chloroform (CDCl3)與 d-Toluene 作為溶劑,Tetramethylsilane (TMS)
作為標定基準點。在光譜中 s 代表單峰(Singlet),d 代表雙重峰 (Doublet), t 代表三重峰 (Triplet) , dd 代 表 二 雙 重 峰 (Doublet of
Doublet),m 代表多重峰(Multiplet),光譜單位為 ppm。
2-2-2 示差掃描卡計(Differential Scanning Calorimeter, DSC)
27
度及其焓熱值,熔點(Melting Point, Tm)取其極值,玻璃轉移溫度
(Glass Transition Temperature, Tg)則取其反曲點。溫度以Indium及
Tin作校正。秤取樣品5–10 mg,於氮氣流量100 mL/min下加熱量測,
溫度範圍為-20 – 200 oC,升溫速率為10 oC/min。
2-2-3 熱重分析儀(Thermogravimetric Analyzer, TGA)
係使用SII TG/DTA 7200型。實驗時取樣品5–10 mg,於氮氣流量
100 mL/min下測量其熱裂解溫度(Decomposition Temperature, Td),
溫度範圍為100–600 oC,加熱速率為10 oC/min。
2-2-4 凝膠滲透層析儀(Gel Permeation Chromatography, GPC)
係使用 Viscotek VE3580 GPC 型。使用 Polystyrene (PS)標準品 製作分子量檢量線,THF 為沖提液,流速為 1 mL/min,管柱保持在
32 oC 之恆溫槽內。樣品溶液配置濃度為 4 mg/2 mL。
2-2-5 紫 外– 可 見 光 吸 收 光 譜 儀 (Ultraviolet-visible Absorption Spectrometer)與螢光光譜儀(Fluorescence Spectrophotometer)
此兩種光譜儀皆採用 Princeton Instruments Acton 2150 型。薄 膜樣品的製備:將高分子以濃度 10 mg/mL 配製溶液,再以旋轉塗佈 法成膜於已清洗之玻璃基板上。溶液樣品的製備:將高分子以 1
28 mg/100 mL 之濃度分別溶於 Toluene、Dichloromethane (DCM)及 THF 中,再將溶液滴入石英槽中量測,光譜單位為 nm。 2-2-6 循環伏安計量法(Cyclic Voltammetry, CV) 係 使 用 Autolab PGSTAT30 型 。 配 製 0.1 M n-Tetrabutylammonium tetrafluoroborate/Acetonitirle 之電解液,參考 電極為 Ag/AgCl,另準備兩片面積相同之 ITO 基板作為工作電極與對 應電極。將待測高分子溶液以滴乾成膜法(Drop Casting)塗佈於 ITO 工作電極上,測量範圍從 3 V 到–3 V。
2-2-7 氣相層析質譜儀(Gas Chromatography-Mass Spectrometer,
GC-MS)
係使用Micromass TRIO–2000 GC-MS型。係將樣品氣化後導入 質譜儀以獲得分子量及結構之資訊。
2-2-8 傅 立 葉 紅 外 線 光 譜 儀 (Fourier Transform Infrared
Spectrometer, FTIR)
係使用Thermo Scientific Nicolet iS–10光譜儀。利用干涉波照射 至樣品後,經由傅立葉轉換得到化合物之紅外線光譜。樣品製備以滴 乾成膜法將樣品溶液均勻地分佈於空白KBr碇上,於氮氣環境下自然
29
乾燥成膜,光譜單位為cm-1。
2-2-9 原子力顯微鏡(Atomic Force Microscope, AFM)
使用 Bruker Innova AFM 型。固定一振幅使用敲擊模式(Tapping
Mode),藉由雷射聚焦於探針上緣,當探針針尖與待測物表面產生交
互作用力(主要為凡徳瓦爾力)而改變探針高度,由反射之雷射訊號換 算,得到待測物之表面形貌。亦可使用手術刀將薄膜表面製作出階高 以量測薄膜厚度。樣品大小為 1.5×1.5 cm×cm。
2-2-10 元件量測系統(Device Measurement System)
元件量測系統包含光電二極體檢測器、光纖光譜儀及半導體參數 分析儀。光電二極體檢測器購於OSI Optoelectronics,為能將光訊號 轉換為電訊號的半導體元件,測量元件所發出的光子能量並計算其電 流密度、量子效率、亮度、發光效率及電流效率等數據;光纖光譜儀 係使用Ocean Optics USB2000+型,由2-MHz模數(A/D)轉換器、可 程式編輯模塊、CCD陣列探測器組成,可於每毫秒截取並儲存一張光 譜圖;半導體參數分析儀使用Agilent 4155C型,量測解析度約可達
10-14 A及10-7 V,利用探針分別連接元件之陰極與陽極,量測元件的
30 2-3 元件製作 2-3-1 ITO 基板清洗步驟 1. 將 ITO 基板切割為 1.7×1.7 cm×cm; 2. 配置 Detergent 與去離子水(體積比 1:5)的溶液; 3. 以牙刷沾上述溶液清潔 ITO 玻璃基板; 4. 將 ITO 板置入 Detergent 溶液中施以超音波震洗 30 分鐘; 5. 取出 ITO 基板置入去離子水以超音波震洗 30 分鐘;
6. 依此類推,將溶液依序換為 Acetone 與 Isopropyl alcohol (IPA)
震洗 30 分鐘; 7. 取出 ITO 基板以氮氣槍吹乾; 8. 置入真空烘箱以 120 oC 烘烤 30 分鐘; 9. 待其冷卻後施以 UV-ozone 照射 30 分鐘。 2-3-2 PLED 元件製作流程 1. 將電洞傳輸材料 PEDOT 旋轉塗佈於 ITO 基板上。配置溶液:
PEDOT Aldrich 408395:IPA:Detergent=1.95:0.5:0.1 wt%;
塗佈參數設定如下:以 5800 rpm/30 sec 塗佈於 ITO 基板上,
再放入真空烘箱中以 120 oC 烘烤 30 分鐘。
31 rpm/30 sec 的參數設定塗佈於 ITO/PEDOT 上,再放入真空 烘箱中以 90 oC 烘烤 30 分鐘。 3. 將電子傳輸層溶液(濃度為 0.05 mg/mL 溶於 Acetonitrile,材 料由本實驗室蔡佳昇學長提供)以 2000 rpm/30 sec 的參數設 定塗佈於發光層之上,接著放入真空烘箱中以 60 oC 烘烤 30 分鐘。 4. 以熱蒸鍍方式蒸鍍鋁電極(腔體壓力為 8×10-6 torr)。
32 2-4 單體之合成 各中間物及單體 M1–M6 之合成流程如 Schemes 1 所示。 2,5-Dibromo-3-thiophenecarboxaldehyde (1) 取 3-Thiophenecarboxaldehyde (2.0 g, 17.83 mmol) 與 N-Bromosuccinimide (NBS) (7.0 g, 39.33 mmol) 溶 於 N,N’-Dimethylformamide (DMF) (30 mL),於室溫下攪拌 24 小時。接著以
大 量 Ethyl acetate (EA) 與 飽 和 食 鹽 水 萃 取 , 有 機 層 以 無 水
Magnesium sulfate (MgSO4)除水並濃縮,再以管柱層析法純化(沖提
液 EA:Hexane = 1:10 體積比),得黃色固體 3.0 g,產率 62%。 1H–NMR (CDCl 3, ppm): 9.78 (s, 1H, aromatic proton), 7.32 (s, 1H, aromatic proton). 13C–NMR (CDCl 3, ppm): 183.10, 139.27, 128.62, 124.17, 113.33. Mass (EI): m/z 269. Tm = 48 oC. 2,7-Dibromofluorene (2)
取Fluorene (20.0 g, 120.48 mmol)及NBS (42.0 g, 235.96 mmol) 置 入 雙 頸 瓶 中 , 依 序 加 入 Acetic acid (AcOH) (240 mL) 及 48 % Hydrobromic acid (4 mL),於室溫下攪拌16小時。反應完成後加入大
量純水使固體析出,減壓抽濾收集米白色粗產物,再使用Ethanol (EtOH)進行再結晶純化,得白色固體26.5 g,產率68%。
1H–NMR (CDCl
33 (d, J = 8.4 Hz, 2H, aromatic protons), 7.54–7.53 (d, J = 8.4 Hz, 2H, aromatic protons), 3.79 (s, 2H, –CH2). 13C–NMR (CDCl3, ppm): 144.72, 139.60, 130.06, 128.20, 121.09, 120.89, 36.49. Mass (EI): m/z 324. Tm = 164 oC. 2,7-Dibromo-9,9-dioctylfluorene (M1) 取化合物(2) (2.0 g, 6.17 mmol)、n-Bromooctane (9.0 g, 46.6 mmol)與 Sodium hydroxide (NaOH) (1 g, 25.0 mmol)溶於 Dimethyl
sulfoxide (DMSO) (40 mL),於 80 oC 下反應 12 小時。反應完成後
加入 EA 及飽和食鹽水萃取,有機層以無水 MgSO4除水並濃縮,粗
產物利用 EtOH 進行再結晶純化,得白色固體 1.4 g,產率 41%。
1H–NMR (CDCl
3, ppm): 7.53–7.51 (d, J = 8.6 Hz, 2H, aromatic
protons), 7.46–7.45 (d, J = 6.7 Hz, 4H, aromatic protons), 1.94–1.91 (m, 4H, Fluorene–CH2), 1.28–1.20 (m, 4H, –CH2CH2(CH2)5CH3), 1.18–1.06 (m, 16H, –CH2CH2(CH2)4CH2CH3), 0.85–0.83 (t, J = 7.1 Hz, 3H, –(CH2)7CH3), 0.62–0.61 (t, J = 7.2 Hz, 4H, –(CH2)6CH2CH3). 13C–NMR (CDCl 3, ppm): 152.54, 139.05, 130.13, 126.17, 121.47, 121.08, 55.67, 40.13, 31.74, 29.84, 29.70, 23.61, 22.57, 14.46. Mass (EI): m/z 548. Tm = 53 oC. 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene (M2) 取化合物M1 (3.3 g, 6 mmol)置入乾燥雙頸瓶中,以針筒抽取無水
34 THF (50 mL)打入雙頸瓶中,待其溶解後置於–78 oC下持續攪拌10分 鐘,之後以針筒抽取1.6 M n-Butyllithium (9.5 mL, 15.2 mmol)緩慢滴 入雙頸瓶中,於–78 o C下持續攪拌1小時;再以針筒抽取2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.25 mL, 40.4 mmol)滴入 上述溶液,之後緩慢回升至室溫並攪拌反應12小時。反應完成後濃縮, 再加入EA與純水萃取,有機層以無水MgSO4除水,濃縮後之粗產物 使用Hexane進行再結晶純化,得白色固體2.3 g,產率60%。 1H–NMR (CDCl 3, ppm): 7.81–7.80 (d, J = 7.7 Hz, 2H, aromatic
protons), 7.75 (s, 2H, aromatic protons), 7.72–7.71 (d, J = 7.5 Hz, 2H, aromatic protons), 2.01–1.98 (m, 4H, –CH2(CH2)6CH3), 1.39 (s, 24H, –CH3), 1.20–1.16 (m, 4H, –CH2CH2(CH2)5CH3), 1.13–1.00 (m, 16H, –CH2CH2(CH2)4CH2CH3), 0.82–0.80 (t, 6H, J = 7.2 Hz, – (CH2)7CH3), 0.56–0.55 (d, J = 6.6 Hz, 4H, –(CH2)6CH2CH3). 13C– NMR (CDCl3, ppm): 150.47, 143.91, 133.64, 128.91, 119.36, 83.70, 55.17, 40.08, 31.77, 29.92, 29.18, 24.93, 23.59, 22.58, 14.05. Mass (EI): m/z 642. Tm = 128 oC. 2-(2,5-Dibromothiophen-3-yl)-1,4,5-triphenyl-1H-imidazole (M3) 取化合物(1) (2.0 g, 7.43 mmol)、Aniline (0.692 g, 7.43 mmol)、 Benzil (1.56 g, 7.43 mmol)、Ammonium acetate (3.67 g, 47.66 mmol)
與 AcOH (20 mL),於 120 oC 進行迴流 6 小時。待反應完成後加入
35
1:1 體積比),得黃色固體 3.6 g,產率 91%。
1H–NMR (CDCl
3, ppm): 7.58–7.57 (d, J = 7.5 Hz, 2H, aromatic
protons), 7.28–7.23 (m, 8H, aromatic protons), 7.20–7.18 (t, J = 7.26 Hz, 1H, aromatic proton), 7.15–7.14 (d, J = 6.48 Hz, 1H, aromatic proton), 6.99–6.97 (d, J = 7.38, 1H, aromatic proton), 6.77 (s, 1H, aromatic proton). 13C–NMR (CDCl3, ppm): 140.23, 138.42, 136.03, 134.09, 132.45, 131.92, 130.97, 130.24, 130.18, 128.87, 128.45, 128.27, 128.14, 127.76, 127.33, 126.75, 113.52, 111.12. Mass (EI): m/z 535. Tm = 230 oC. 2-(2,5-Dibromothiophen-3-yl)-1-phenyl-1H-phenanthro[9,10-d]imidazole (M4) 參考 M3 之合成步驟,取化合物(1) (2.0 g, 7.43 mmol)、Aniline (0.692 g, 7.43 mmol)、Phenanthrenequinone (1.54 g, 7.43 mmol)、
Ammonium acetate (3.67 g, 47.66 mmol)與 AcOH (20 mL)進行反應,
得白色固體 3.0 g,產率 76%。
1H–NMR (CDCl
3, ppm): 8.83–8.82 (dd, J1 = 7.92 Hz, J2 = 0.9 Hz, 1H,
aromatic proton), 8.77–8.76 (d, J = 8.4 Hz, 1H, aromatic proton), 8.71–8.69 (d, J = 8.28, 1H, aromatic proton), 7.75–7.72 (t, J = 7.56 Hz, 1H, aromatic proton), 7.67–7.64 (t, J = 8.1 Hz, 1H, aromatic proton), 7.59–7.55 (m, 3H, aromatic protons), 7.54–7.51 (t, J = 6.84 Hz, 1H, aromatic proton), 7.47–7.46 (dd, J1 = 7.02 Hz, J2 = 1.62 Hz,
36
proton), 7.24–7.23 (dd, J1 = 8.34 Hz, J2 = 0.78 Hz, 1H, aromatic
proton), 6.76 (s, 1H, aromatic proton). 13C–NMR (CDCl
3, ppm):
144.29, 137.41, 132.32, 131.52, 129.89, 129.40, 128.36, 128.30,
127.49, 127.41, 127.08, 126.34, 125.78, 125.27, 124.10, 123.09,
122.73, 120.97, 114.81, 111.31. Mass (EI): m/z 533. Tm = 182 oC.
4,5-Phenanthrenedicarboxylic Acid (3)
取 Pyrene (20.0 g, 98.88 mmol)與 Tungstic acid (1.02 g, 4.08 mmol)溶於 Chlorobenzene (CB) (90 mL)中,再依序加入 Aliquat 336
(2 mL, 4.37 mmol)、Phosphoric acid (1 mL, 19.2 mmol),接著緩慢
倒入 35 % Hydrogen peroxide (80 mL),維持在 80 oC 反應 24 小時。 待冷卻至常溫後,加入大量蒸餾水並攪拌 2 小時,收集之固體粗產物 以大量 EA 攪拌清洗後,得棕黑色固體 21.0 g,產率 80%。 1H–NMR (d 6–DMSO, ppm): 8.09–8.08 (dd, J1 = 7.86 Hz, J2 = 0.78 Hz, 2H, aromatic protons), 7.96–7.95 (dd, J1 = 7.2 Hz, J2 = 1.14 Hz,
2H, aromatic protons), 7.87 (s, 2H, aromatic protons), 7.65–7.63 (t,
J = 7.56 Hz, 2H, aromatic protons), 3.36 (s, 2H, Ar–COOH). 13C–
NMR (d6–DMSO, ppm): 169.33, 134.13, 133.81, 131.27, 127.99,
127.34, 126.38. Mass (EI): m/z 266. Tm = 246 oC.
Dimethyl 4,5-phenanthrenedicarboxylate (4)
取化合物(3) (10.0 g, 37.59 mmol)溶解於 DMSO (50 mL)與 Methanol (MeOH) (60 mL)混合溶液中,緩慢滴入 98% Sulfuric acid
37 (H2SO4) (15 mL),維持 80 oC 反應 24 小時。待反應結束後,於冰浴 下加入大量 EA 及蒸餾水,抽氣過濾後收集濾液,再以飽和食鹽水萃 取,有機層以無水 MgSO4乾燥並濃縮,粗產物再以 Hexane 攪拌清 洗,得棕色固體 3.84 g,產率 35%。 1H–NMR (CDCl 3, ppm): 8.03–8.02 (d, J = 7.2 Hz, 2H, aromatic
protons), 7.99–7.97 (d, J = 7.86 Hz, 2H, aromatic protons), 7.74 (s, 2H, aromatic protons), 7.62–7.60 (t, J = 7.5 Hz, 2H, aromatic protons), 3.80 (6H, s, COOCH3). 13C–NMR (CDCl3, ppm): 169.29, 134.00, 132.51, 131.68, 128.70, 127.44, 127.36, 126.24, 52.25. Mass (EI): m/z 294. Tm = 248 oC. Pyrene-4,5-dione (5) 取化合物(4) (8.0 g, 34.48 mmol)溶於無水 THF (60 mL),再加入 Na (3.15 g, 136.95 mmol)後,於氮氣及室溫環境下攪拌 24 小時。待 反應結束後緩慢加入大量水以除去多餘的 Na,接著以大量 EA 與飽 和食鹽水萃取,有機層以無水 MgSO4乾燥並濃縮,再以 Hexane 攪 拌清洗,得橘色固體 3.2 g,產率 49%。 1H–NMR (CDCl 3, ppm): 8.40–8.38 (d, J = 7.38 Hz, 2H, aromatic
protons), 8.10–8.08 (d, J = 7.86 Hz, 2H, aromatic protons), 7.76 (s, 2H, aromatic protons), 7.69–7.67 (t, J = 7.62 Hz, 2H, aromatic protons). 13C–NMR (CDCl
3, ppm): 180.28, 135.65, 131.94, 130.06,
38
2-(2,5-Dibromothiophen-3-yl)-1-phenyl-1H-pyreno[9,10-d]imidazole (M5)
參考化合物 M3 之合成步驟,取化合物(1) (3.5 g, 12.96 mmol)、 Aniline (1.21 g, 12.99 mmol)、化合物(5) (3.0 g, 12.93 mmol)、
Ammonium acetate (4.0 g, 51.94 mmol)與 AcOH (30 mL)進行反應,
得黃色固體 5.0 g,產率 69%。
1H–NMR (CDCl
3, ppm): 9.07–9.06 (d, J = 7.5 Hz, 1H, aromatic
proton), 8.21–8.19 (d, J = 7.5 Hz, 1H, aromatic proton), 8.15–8.12 (t, J = 7.68 Hz, 1H, aromatic proton), 8.11–8.10 (d, J = 8.88 Hz, 1H, aromatic proton), 8.07–8.05 (d, J = 7.62 Hz, 1H, aromatic proton), 8.04–8.03 (d, J = 8.88, 1H, aromatic proton), 7.68–7.65 (t, J = 7.86 Hz, 1H, aromatic proton), 7.64–7.63 (dd, J1 = 7.08 Hz, J2 = 0.96 Hz,
1H, aromatic proton), 7.62–7.60 (t, J = 7.62 Hz, 2H, aromatic protons), 7.55–7.54 (d, J = 6.9 Hz, 2H, aromatic protons), 7.47–7.45 (d, J = 7.92 Hz, 1H, aromatic proton), 6.83 (s, 1H, aromatic proton).
13C–NMR (CDCl
3, ppm): 144.56, 137.90, 137.46, 131.59, 129.94,
128.50, 128.00, 127.52, 126.50, 125.34, 124.73, 124.63, 119.85,
118.09, 114.91, 111.40. Mass (EI): m/z 558. Tm = 195 oC.
1,10-Phenanthroline-5,6-dione (6)
取化合物 1,10-Phenanthroline (5.0 g, 27.74 mmol)與 Potassium
bromide (KBr) (5.0 g, 42.02 mmol)於冰浴下緩慢加入 H2SO4(50 mL)
39 待反應結束後於冰浴下將溶液緩慢滴入蒸餾水(300 mL)中,再緩慢滴 入 50% NaOH 水溶液直至 pH 呈中性,之後以 Chloroform 萃取,有 機層以無水 MgSO4乾燥並濃縮,粗產物再以 EA 攪拌清洗,得黃色 固體 3.56 g,產率 61%。 1H–NMR (CDCl 3, ppm): 9.11–9.10 (dd, J1 = 4.68 Hz, J2 = 1.5 Hz, 2H, aromatic protons), 8.50–8.49 (dd, J1 = 7.86 Hz, J2 = 1.44 Hz, 2H, aromatic protons), 7.59–7.57 (dd, J1 = 7.8 Hz, J2 = 4.62 Hz, 2H, aromatic protons). 13C–NMR (CDCl 3, ppm): 156.39, 152.90, 137.29, 128.07, 125.58. Mass (EI): m/z 210. Tm = 261 oC. 2-(2,5-Dibromothiophen-3-yl)-1-phenyl-1H-phenanthro[9,10-d]imidazole (M6) 參考化合物 M3 之合成步驟,取化合物(1) (2.0 g, 7.43 mmol)、 Aniline (0.692 g, 7.43 mmol)、化合物(6) (1.56 g, 7.43 mmol)、
Ammonium acetate (3.67 g, 47.66 mmol)與 AcOH (20 mL)進行反應,
得灰白色固體 1.86 g,產率 47%。 1H–NMR (CDCl 3, ppm): 9.17–9.16 (dd, J1 = 4.5 Hz, J2 = 1.8 Hz, 1H, aromatic proton), 9.10–9.08 (dd, J1 = 7.98 Hz, J2 = 1.44 Hz, 1H, aromatic proton), 9.04–9.03 (dd, J1 = 4.2 Hz, J2 = 1.32 Hz, 1H, aromatic proton), 7.75–7.73 (dd, J1 = 7.98 Hz, J2 = 4.2 Hz, 1H,
aromatic proton), 7.64–7.59 (m, 3H, aromatic protons), 7.52–7.50 (dd, J1 = 8.46 Hz, J2 = 1.56 Hz, 1H, aromatic proton), 7.48–7.47 (dd,
40
J1 = 7.14 Hz, J2 = 1.38 Hz, 2H, aromatic protons), 7.31–7.29 (dd, J1
= 8.46 Hz, J2 = 4.44 Hz, 1H, aromatic proton), 6.76 (s, 1H, aromatic
proton). 13C–NMR (CDCl3, ppm): 149.17, 148.32, 145.65, 145.07, 144.43, 136.60, 136.09, 131.53, 131.25, 130.54, 130.43, 130.30, 128.17, 128.09, 126.27, 123.91, 123.67, 122.21, 119.52, 115.06, 111.78. Mass (EI): m/z 535. Tm = 335 oC. 2-5 高分子聚合 高分子 PFO、P1–P4 之合成途徑如 Scheme 2 所示。所有高分 子皆採用 Suzuki 耦合法進行聚合,P1–P4 聚合比例為:單體 M3– M6、M1 與 M2 之當量比為 1 比 4 比 5,詳細合成步驟以 PFO 為 例,說明如下: 取化合物 M1 (0.548 g, 1 mmol)、M2 (0.642 g, 1 mmol) 及 Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (0.04 g, 0.034
mmol),於氮氣環境下打入無水 Toluene (10 mL)與 2 M Potassium
carbonate(aq) (10 mL),加熱至 90 oC 迴流反應 120 小時。反應完成
後將溶液滴入 MeOH 中,收集之固體溶解於 THF,再滴入 THF(100 mL)與 MeOH (50 mL)混合溶液進行再沉澱,重複上述步驟一至二次,
41
P1:取 M1 (0.438 g, 0.8 mmol)、M3 (0.107 g, 0.2 mmol)、M2 (0.642
g, 1 mmol)及 Pd(PPh3)4 (0.04 g, 0.034 mmol),反應步驟參照 PFO,
得黃色固體 144 mg,產率 33%。
P2:取 M1 (0.438 g, 0.8 mmol)、M4 (0.106 g, 0.2 mmol)、M2 (0.642
g, 1 mmol)及 Pd(PPh3)4 (0.04 g, 0.034 mmol),反應步驟參照 PFO,
得綠色固體 128 mg,產率 30%。
P3:取 M1 (0.438 g, 0.8 mmol)、M5 (0.111 g, 0.2 mmol)、M2 (0.642
g, 1 mmol)及 Pd(PPh3)4 (0.04 g, 0.034 mmol),反應步驟參照 PFO,
得綠色固體 130 mg,產率 30%。
P4:取 M1 (0.438 g, 0.8 mmol)、M6 (0.111 g, 0.2 mmol)、M2(0.642
g, 1 mmol)及 Pd(PPh3)4 (0.04 g, 0.034 mmol),反應步驟參照 PFO,
42 S CHO S CHO Br Br NBS DMF 1 NaOH/DMSO Br C8H17 C8H17 n-BuLi/THF C8H17 C8H17 B B M1 M2 2 Br Br NBS Br Br O O O O O O B AcOH NH2 O O N N S Br Br NH4OAc M3 (1) AcOH NH2 O O N N S Br Br M4 NH4OAc (1) AcOH 1. H2WO4, Chlorobenzene 2. Aliquat 336, H3PO4, H2O2, 80 oC COOH COOH COOMe COOMe MeOH H2SO4 Na THF O O NH2 N N S Br Br AcOH 3 4 5 M5 (1) (5) DMSO NH4OAc NH2 N N S N N Br Br AcOH O O N N N N 1. HNO3, KBr, H2SO4, O oC 2. 70oC M6 6 (1) (6) NH4OAc Scheme 1. 單體 M1–M6 之合成途徑
43 N N N N N N N N N N P1 P2 P3 P4 R = Pd(PPh3)4, K2CO3 S R C8H17 C8H17 n M3 M4 M5 M6 M1 + M2 Pd(PPh3)4, K2CO3 toluene C8H17 C8H17 n PF toluene + M1 + M2 m m:n=1:9 Scheme 2. 高分子 PFO 及 P1–P4 之合成途徑
44 第三章 實驗結果與討論 3-1 分子量測定 本研究旨在合成四種含不同咪唑側取代基之噻吩–芴共聚物,並 探討其對於聚芴高分子在熱性質、光學性質及元件表現上有何差異。 不同於小分子具有明確的分子量,每條高分子鏈長度都參差不齊,因 此高分子分子量是由數量平均分子量(Number-Average Molecular
Weight, Mn)、重量平均分子量(Weight-Average Molecular Weight,
Mw)及分子量分佈(Polydispersity Index, PDI)來表示。其中分子量分
佈定義為重量平均分子量與數量平均分子量之比值,亦即 PDI = Mw Mn ⁄ 本研究藉由 GPC 測定高分子之分子量大小及分佈, PFO 與 P1– P4 高分子結構如 Figure 3–1 所示: C8H17 C8H17 n PFO P1 S C8H17 C8H17 n N N m P2 S C8H17 C8H17 n N N m P3 S C8H17 C8H17 n N N m m:n=1:9 m:n=1:9 m:n=1:9 P4 S C8H17 C8H17 n N N m N N m:n=1:9 Figure 3–1. 高分子 PFO 及 P1–P4 化學結構
45
高分子 PFO 及 P1–P4 之聚合結果整理於Table 3–1。PFO 具有
最高的Mn與Mw,分別為 4.21×104與 1.53 ×105 g/mol,PDI 為 3.63。 而 P1–P4 (m:n=1:9)的Mn分別為 1.41×104、3.01×104、3.20×104及 0.43×104 g/mol; Mw為 3.65×104、5.92×104、7.07×104及 0.85×104 g/mol;PDI 為 2.59、1.97、2.21 及 1.99,P1–P4 的分子量較 PFO 為低,歸因於引入剛硬的咪唑側取代基,且其缺乏長碳鏈,導致高分 子產物溶解度降低,分子量表現亦較小;尤其 P4 之單體 M6 於 THF 溶劑中溶解度較低,且 Phenanthro[9,10-d]imidazole 上之兩個氮原 子可能於聚合過程中螯合住 Pd 催化劑,使其失去效用而導致聚合程 度不佳,分子量尚不滿一萬。研究過程中亦曾將含咪唑側取代基之噻 吩單體(M3–M6)與芴單體(M2)以同等比例進行聚合(m:n=1:1),惟只 有 M4 可與 M2 共聚成高分子,且分子量較 PFO 為高,而 M3、M5 及 M6 與 M2 共聚皆無法成功得到高分子產物,為求實驗一致性,故 本研究接下來之探討皆採用 m:n=1:9 之比例。
46
Table 3–1. 高分子 PFO 及 P1–P4 之聚合結果
Polymers Mn×104 (g/mol) Mw×104 (g/mol) PDI
PFO P1 (m:n=1:9) P2 (m:n=1:9) P2 (m:n=1:1) P3 (m:n=1:9) P4 (m:n=1:9) 4.21 1.41 3.01 8.31 3.20 0.43 15.29 3.65 5.92 17.20 7.07 0.85 3.63 2.59 1.97 2.07 2.21 1.99 3-2 NMR 光譜分析 本研究利用 1H–NMR 光譜分析材料結構,並比較高分子 P1–P4、 PFO 與各對應單體之差異,對照圖如 Figure 3–2 所示。以 P1 對照 圖為例,P1 光譜分別於δ 8 ppm 及 2 ppm 附近觀察到芴主鏈之氫訊 號及芴分子 9 號碳上的長碳鏈 1 號碳上之氫訊號,在 PFO 光譜亦可 觀察到相同訊號;M3 光譜於δ 6.78 ppm 顯現噻吩之氫訊號,P1 光 譜亦有相同訊號,然因單體 M3 與芴單體之共聚比例為 1:9,前者所 佔比例較少,故訊號強度較弱,而芴單體比例佔大多數,故 P1 與 PFO 光譜極為相似,其餘 P2–P4 光譜亦有相同現象。
47
Figure 3–2. 高分子(a) P1、PFO、M3、(b) P2、PFO、M4、(c) P3、 PFO、M5 及(d) P4、PFO、M6 之 NMR 氫譜比較圖 3-3 FTIR 分析 本研究利用 FTIR 光譜分析所合成高分子之化學鍵結,以 PFO 及 P1 為例,其 FTIR 光譜如 Figure 3–3 所示,芳香環之 C–H 吸收峰位 於3054–3063 cm-1,長碳鏈上 C–H 吸收峰落於 2954–2853 cm-1, 芳香環之 C=C 吸收峰位於 1608–1617、1458 cm-1,末端 CH 3之吸 收峰則落於 1394–1377 cm-1,指紋區之 813 cm-1 為芳香環平面外 (Out-of-Plane)之 C–H 振動吸收峰[46],各吸收波峰值列表於 Table 3– 2。與 NMR 光譜表現相同,高分子 P1–P4 與 PFO 有著高度相似的 光譜表現。高分子 P1–P4、PFO 與相對應單體之光譜對照圖見 Figure
48 3–4,各單體分別於 740–694 cm-1及 1661–1645 cm-1處顯現 C–S 及 C=N 之吸收峰[47],然而在共聚物中可能由於比例較低,上述吸收峰 未能清楚顯現於高分子之 FTIR 光譜。 4000 3000 2000 1000 20 40 60 80 100 T rans m itt anc e (%) Wavenumber (cm-1 ) Ar C-H Ar C=C Aliphatic C-H (a) 4000 3000 2000 1000 40 60 80 100 (b) Ar C=C Ar C-H T rans m itt anc e (%) Wavenumber (cm-1) Aliphatic C-H
Figure 3–3. 高分子(a) PFO 及(b) P1 之 FTIR 光譜
Table 3–2.高分子 PFO 及 P1–P4 之 FTIR 訊號
Polymers Ar C–H Aliphatic C–H Ar C=C –CH3 PFO P1 P2 P3 P4 3057 3057 3054 3054 3063 2953, 2926, 2854 2954, 2926, 2854 2953, 2926, 2853 2953, 2926, 2853 2953, 2925, 2853 1608, 1458 1610, 1458 1617, 1458 1617, 1458 1609, 1458 1377 1389 1388 1389 1394
49 4000 3000 2000 1000 Wavenumber (cm-1 ) P1 PFO M3 (a) T rans m itt anc e 4000 3000 2000 1000 Wavenumber (cm-1 ) P2 PFO M4 C-S C=N (b) T rans m itt anc e 4000 3000 2000 1000 Wavenumber (cm-1 ) P3 PFO M5 (c) T rans m itt anc e 4000 3000 2000 1000 T rans m itt anc e (%) Wavenumber (cm-1 ) P4 PFO M6 (d)
Figure 3–4. 高分子(a) P1、PFO、M3、(b) P2、PFO、M4、(c) P3、
PFO、M5 及(d) P4、PFO、M6 之 FTIR 光譜比較圖
3-4 熱性質分析
本研究利用 TGA 及 DSC 測定高分子之熱性質。Figure 3–5 為高
分子 PFO、P1–P4 之 TGA 曲線圖,分析圖譜可知 PFO、P1–P4 之
熱裂解溫度(Td,定義為重量損失 5 %時溫度)分別為 412、412、413、
409 及 399 oC,皆落於 400 oC 附近,顯示出良好的熱穩定性。一般
而言,高分子的分子量愈大,則熱穩定性愈佳,本研究 P1–P4 分子
量雖然較低,但其 Td卻與 PFO 數值相近。此外,比較各高分子升溫
50 52、54、57%,皆高於 PFO;P4 之分子量最低,故殘留比率亦較低, 但與 PFO 相差無幾。上述兩項 TGA 數據皆顯示了引入含剛硬咪唑 側取代基,確實有助於增強材料的熱穩定性。 DSC 量測方面,Figure 3–6 為高分子 PFO、P1–P4 之 DSC 曲 線圖,由圖譜可知 PFO 之玻璃轉換溫度(Tg)為 67 oC,與文獻記載相 同[11, 48, 49],P1–P4 之 T g則提高至 91–125 oC。PFO 雖有較高之分子 量,但其 Tg卻較 P1–P4 為低,證明引入剛硬的咪唑團基可提高主鏈 之剛硬性,且較高之 Tg使高分子不易受熱而擾動,有助於 PLED 操 作時之穩定性[32]。PFO 之熔點表現於 154 oC[11],而 P1 及 P3 加熱至 250 oC 仍未發現熔點,P2 及 P4 之熔點分別為 220 及 231 oC。所有 高分子之熱性質數據整理於 Table 3–3。 100 200 300 400 500 600 40 60 80 100 W eight R es idure (%) Temperature (oC) PFO P1 P2 P3 P4
51 50 100 150 200 250 H eat F low (uW ) Temperature (oC) PFO P1 P2 P3 P4 Figure 3–6. 高分子 PFO 及 P1–P4 之 DSC 曲線圖 Table 3–3. 高分子 PFO 及 P1–P4 之熱性質數據 Polymers Td (oC) Tg (oC) Tm (oC) PFO P1 P2 P3 P4 412 412 413 409 399 67 120 119 91 125 154 N/A 220 N/A 217 3-5 光學性質分析 3-5-1 吸收光譜分析 高分子 PFO、P1–P4 薄膜態與溶液態之吸收光譜整理於 Figure 3–7 中。所有高分子薄膜態最大吸收峰落於 382–385 nm,屬於 PF
52 主鏈之 π–π*躍遷[50];Toluene 溶液中,高分子最大吸收峰位於 382– 385 nm,DCM 與 THF 溶液為 386–389 nm,最大吸收峰隨溶劑極性 提高而呈現些許紅位移[51]。PFO 薄膜態最大放射峰與溶液態相比並 無紅移現象,此情形亦見於其他文獻[11, 12, 52, 53],推測是由於芴分子 9 號碳為 sp3軌域,長碳鏈與芴分子之平面的四面體構形於空間上形成 相當大的立體阻礙,使高分子鏈間不易產生交互作用,導致 PFO 較 易表現出單一高分子鏈行為,故薄膜態與溶液態吸收峰相近,高分子 P1–P4 亦顯示相似現象。 PFO 與 P1–P4 相比,五種高分子於三種溶液態及薄膜態中皆表 現出相似的吸收特性,顯示咪唑團基並未影響主鏈的吸收,惟 P1–P4 吸收波形較 PFO 為寬,乃因於引入咪唑側取代基可延伸材料之吸收 波段導致,類似吸收變寬現象 Wei et al 亦曾報導,其於聚噻吩主鏈 引入咪唑側取代基,使得吸收範圍增大些[54]。 由薄膜態吸收光譜以切線方式得吸收截止波長(λonset),可推算出 高分子之能隙(Eg),其計算式為: Eg (eV) = 1240/λonset PFO 與 P1–P4 之λonset分別為 428、435、438、437 及 438 nm,
53 求得能隙為 2.90、2.85、2.83、2.84 及 2.83 eV,顯見 PF 主鏈之共 軛可延伸至咪唑團基上,因此降低材料之能隙。 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Abs orbanc e (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in Thin Film (a) 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Abs orbanc e (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in Toluene (b) 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Abs orbanc e (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in DCM (c) 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Abs orbanc e (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in THF (d)
Figure 3–7. 高分子 PFO 與 P1–P4 於(a)薄膜態、(b) toluene、(c) DCM 及(d) THF 溶液之吸收光譜 3-5-2 螢光放射光譜分析 Figure 3–8 為高分子 PFO、P1–P4 薄膜態與溶液態之螢光放射 光譜。溶液態中,PFO 最大放射峰表現於 419–421 nm,為主鏈 S10 →S00之放射,另有一明顯肩峰落於 450 nm 附近,為主鏈 S10→S01 之放射[55];P1–P4 最大放射峰位於 447–459 nm,為主鏈之主要螢光
54 放射峰,於 420 nm 附近有一肩峰,為多個芴單體組成區段之放射, 尤其 P4 的肩峰特別明顯,可能是由於單體溶解度較差,高分子鏈中 導入之單體比例較低,故顯現更強烈之芴鏈段放射(420 nm)。P1–P4 放射峰相對於 PFO 較為紅移,證明咪唑團基可有效延長高分子之共 軛長度[54, 56]。為確認其主放射峰是否為芴–咪唑團基間之能量轉移, 本研究亦曾將芴單體(M1)與咪唑單體(M3)混合後測量其螢光放射光 譜,並未發現其於 480 nm 產生放射峰,故推論其並非芴–咪唑團基 間之能量轉移。 所有高分子薄膜態放射光譜相較於溶液態皆有紅移情形,起因於 薄膜態高分子鏈易產生堆疊導致能階的簡併態(Degeneracy)[57]。以 THF 溶液與薄膜態放射光譜比較,PFO 由 421 nm 位移至 427 nm; P1 由 456 nm 大幅紅移至 510 nm,於長波 UV 照射下為綠光發光; P2 從 457 nm 紅位移至 471 nm;P3 由 455 nm 紅位移至 468 nm; P4 則由 447 nm 紅位移至 468 nm。其中 P1 紅位移程度遠大於 P2– P4,推測因 P2、P3 及 P4 之咪唑側取代基分別含有聯環的菲基
(Phenanthrenyl)、芘基(Pyrenyl)和鄰菲羅啉(Phenanthroline),雖具
的較高共軛性,但其共平面性高,導致團基太過剛硬而於薄膜態阻礙 堆疊,使發光未能有效紅移;P1 之咪唑側取代基含有三自由苯基 (Triphenyl),與聯環團基相比較不阻礙堆疊,且主鏈之共軛延伸至側
55 取代基上,使 P1 薄膜態放光紅移至綠光波段。所有高分子之薄膜與 溶液經長波 UV 照射而發光之實拍照片列於 Figure 3–9。 400 500 600 700 0.0 0.2 0.4 0.6 0.8 1.0 N orm aliz ed PL Int ens ity (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in Thin Film (a) 400 450 500 550 600 0.0 0.2 0.4 0.6 0.8 1.0 N orm aliz ed PL Int ens ity (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in Toluene (b) 400 450 500 550 600 0.0 0.2 0.4 0.6 0.8 1.0 N orm aliz ed PL Int ens ity (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in DCM (c) 400 450 500 550 600 0.0 0.2 0.4 0.6 0.8 1.0 N orm aliz ed PL Int ens ity (a. u. ) Wavelength (nm) PFO P1 P2 P3 P4 in THF (d)
Figure 3–8. 高分子 PFO 及 P1–P4 於(a)薄膜態、(b) toluene、(c) DCM 及(d) THF 溶液之螢光放射光譜
56
Figure 3–9. 高分子 PFO 及 P1–P4 於(a)薄膜態及(b) toluene 溶液中 經長波 UV 照射之放光照片
Table 3–4. 高分子 PFO 及 P1–P4 之最大吸收與螢光放射峰值
Polymers
Abs λmax (nm) PLλmax (nm)
Toluene DCM THF Film Toluene DCM THF Film PFO P1 P2 P3 P4 382 384 385 383 384 386 387 389 386 386 386 388 389 387 388 382 385 384 384 385 419 454 455 455 449 421 454 459 457 451 421 456 457 455 447 427 510 471 468 468
57
另外,本研究嘗試 PFO:P1:MEH-PPV 以不同比例混合製備溶液 (MEH-PPV 由實驗室陳韋齊同學提供,化學結構及吸收與放射光譜如
Figure3–10),再塗佈以得到白光薄膜,其吸收與放射光譜如 Figure 3–11 所示。Figure 3–11 (a)為 PFO:P1:MEH-PPV 以 95:4:1 wt%混 合之吸收、放射光譜,其吸收曲線與 PFO 相似,因 PFO 所佔比例最 高;螢光放射光譜涵蓋紅、藍光波段,其中 P1 之放射峰值約 510 nm 並未顯現,乃因於 MEH-PPV 的吸收峰值為 506 nm,故 P1 之放射 大部分被 MEH-PPV 所吸收導致。為了提高綠光波段的發光強度,接 著本實驗大幅增加 P1 的比例,將 PFO:P1:MEH-PPV 以 100:50:1 wt%混合,其吸收、放射光譜如 Figure 3–11 (b)所示,P1 放光的提 高亦促使 MEH-PPV 之吸收增加而使紅光強度明顯提升,藍光波段相 對減弱,此結果與本實驗所期望之白光元件大相逕庭。由此可知,除 了提高綠光的強度外,藍光波段的發光亦需增加,以均衡紅光發光的 增強。故採用 PFO:P1:MEH-PPV 以 300:100:1 wt%混合,其吸收放 射光譜如 Figure 3–11 (c)所示,成功得到一涵蓋可見光波段之白光薄 膜,其薄膜經長波 UV 照射而發光之實拍照片列於 Figure 3–11 (d), 以肉眼觀察三種來源高分子 PFO、P1 及 MEH-PPV 薄膜分別呈現深 藍光、綠光及紅光螢光,高分子摻混 PFO:P1:MEH-PPV = 95:4:1 wt% 之薄膜呈現淡黃色螢光,而 PFO:P1:MEH-PPV = 100:50:1 wt%之薄
58 膜因 P1 比例提高而轉變為明顯黃色螢光,最後 PFO:P1:MEH-PPV = 300:100:1 wt%之薄膜終呈現偏藍的冷白色螢光。將其製備成元件 之表現與 EL 光譜參見 3–7 節。 O O n MEH-PPV 400 500 600 700 0.0 0.2 0.4 0.6 0.8 1.0 Wavelength (nm) Abs orbanc e (a. u. ) 0.0 0.2 0.4 0.6 0.8 1.0 PL Int ens ity (a. u. ) Figure 3–10. MEH-PPV 化學結構及其吸收與放射光譜
![Figure 3–7. 高分子 PFO 與 P1–P4 於(a)薄膜態、(b) toluene、(c) DCM 及(d) THF 溶液之吸收光譜 3-5-2 螢光放射光譜分析 Figure 3–8 為高分子 PFO、P1–P4 薄膜態與溶液態之螢光放射 光譜。溶液態中,PFO 最大放射峰表現於 419–421 nm,為主鏈 S 10 →S 00 之放射,另有一明顯肩峰落於 450 nm 附近,為主鏈 S 10 →S 01 之放射 [55] ;P1–P4 最大放射峰位於 447–459 nm](https://thumb-ap.123doks.com/thumbv2/9libinfo/8362287.176910/68.892.131.754.275.896/Figure高分子溶液態之螢光放射光譜溶液態中PFO最大放射→有一明顯肩.webp)