國立高雄大學應用化學系研究所
碩士口試論文
利用氮摻雜石墨烯氣凝膠負載酞菁鐵應用於
鋰氧電池正極材料
Iron Phthalocyanine supported by N-doped graphene
aerogel as a cathode material for
lithium-oxygen batteries
目錄
目錄 ... I 圖目錄 ...IV 表目錄 ... VIII 摘要 ...IX ABSTRACT ...XI 第一章 前言 ... 1 1.1 緒論 ... 1 1.2 研究動機及目的 ... 3 第二章 文獻回顧 ... 7 2.1 鋰氧電池概述 ... 7 2.2 鋰氧電池工作原理 ... 7 2.3 鋰氧電池內部組成 ... 10 2.3.1 正極 ... 11 2.3.2 催化劑 ... 14 2.3.4 M-N-C 催化劑 ... 17 2.3.3 電解液 ... 22 2.4 石墨烯於鋰氧電池的應用 ... 29 2.4.1 石墨烯本身為催化劑作為電極 ... 29第三章 實驗方法 ... 38 3.1 實驗藥品 ... 38 3.2 實驗儀器及設備 ... 40 3.3 實驗方法 ... 41 3.3.1 氧化石墨烯合成(GO) ... 42 3.3.2 氮摻雜三維石墨烯氣凝膠合成(N-GA) ... 43 3.3.3 FePc/N-GA 的合成 ... 44 3.3.4 比較材料 FePc/N-GA (π-π)材料合成 ... 45 3.4 材料鑑定及特性分析 ... 45 3.4.1 掃描式電子顯微鏡( SEM )... 45 3.4.2 X-ray 粉末分析儀( XRD ) ... 45 3.4.3 顯微拉曼光譜分析儀( Micro-Raman ) ... 46 3.4.4 高解析電子能譜圖分析儀( XPS ) ... 46 3.4.5 傅立葉轉換紅外線光譜儀分析法( FT-IR ) ... 47 3.4.6 熱重分析儀( TGA ) ... 47 3.4.7 比表面積分析儀( BET ) ... 47 3.4.8 元素分析儀(EA)... 48 3.5 鋰氧電池系統 ... 48 3.5.1 正極材料極片製備 ... 49 3.5.2 電解液配製 ... 51 3.5.3 鋰氧電池充放電測試 ... 52 3.5.4 循環伏安法(Cyclic Voltammetry, CV) ... 52
3.6 旋轉圓盤電極(Rotating disk electrode, RDE) ... 54 3.6.1 工作電極塗覆(圓盤電極) ... 54 3.6.2 旋轉圓盤電極系統 ... 55 第四章 結果與討論 ... 58 4.1 外觀及掃描式電子顯微鏡( SEM )分析 ... 58 4.2 X-ray 粉末分析儀( XRD )分析 ... 61 4.3 高解析電子能譜圖分析儀( XPS ) ... 63 4.4 顯微拉曼光譜分析儀( Micro-Raman )分析 ... 67 4.5 傅立葉轉換紅外線光譜儀分析法( FT-IR ) ... 69 4.6 熱重分析儀( TGA ) ... 71 4.7 比表面積分析儀( BET ) ... 72 4.8 元素分析儀(EA) ... 75 4.9 鋰氧電池電性測試 ... 76 4.9.1 充放電電容及庫倫效率 ... 76 4.9.2 循環壽命 ... 78 4.9.3 速率電容 ... 79 4.9.4 循環伏安法(Cyclic Voltammetry, CV) ... 81 4.9.5 交流阻抗測試(AC Impedance) ... 83
4.10 旋轉圓盤電極電性分析(Rotating disk electrode, RDE) ... 84
圖目錄
圖.1- 1 不同電池系統與汽油能量密度比較1 ... 2 圖.1- 2 不同電池系統運用於電動車可行駛距離推測圖2 ... 2 圖.1- 3 不同金屬氧化物催化劑於充電反應時過電壓表現 12... 4 圖.2- 1 鋰氧電池充放電示意圖 ... 8 圖.2- 2 以電解液分類鋰氧電池(a)非質子(b)水溶液(c)固態(d)混合13 ... 8 圖.2- 3 鋰氧電池正極三相反應示意圖 1 ... 11 圖.2- 4 不同孔洞大小與放電產物堆積示意圖29 ... 12 圖.2- 5 不同表面結構 α-MnO2 放電產物堆積示意圖 39 ... 16 圖.2- 6 左圖為 Fe/N/C 複合材料示意圖,右圖為與 α-MnO2/C 複合材料 比較充放電電位40 ... 17 圖.2- 7 液相中 FePc 反應機制15 ... 18 圖.2- 8 FePc 於 OER 催化反應機制 ... 19圖.2- 9 固相中 FePc 反應機制 (Reduction pathway i, iii) ... 20
圖.2- 10 固相中 FePc 反應機制 (Reduction pathway ii) ... 21
圖.2- 11 各溶劑於 29 ᵒC 乾燥箱內的消耗百分比比較圖 50 ... 24 圖.2- 12 碳酸酯類電解液分解機制51 ... 24 圖.2- 13 醚類電解液分解機制 ... 25 圖.2- 14 醯胺類電解液分解機制 54 ... 26 圖.2- 15 碸類電解液分解機制 58 ... 27 圖.2- 16 常見離子液體正負離子 ... 27
圖.2- 17 石墨烯與氧氣反應催化示意圖 30 ... 29
圖.2- 18 石墨烯表面 Li2O2堆積方式及鍵結能量表示59 ... 30
圖.2- 19 不同氮摻雜於石墨烯表面形態示意圖61 ... 31
圖.2- 20 石墨烯表面氮摻雜組合形式62 ... 32
圖.2- 21 Graphitic N site 進行 Path A 反應機制示意圖 ... 33
圖.2- 22 Pyridinic N site 進行 Path B 反應機制示意圖 ... 33
圖.2- 23 不同氮摻雜種類之製程方法65 ... 35 圖.2- 24 石墨烯做為催化劑 MnO2載體合成機制示意圖 ... 36 圖.2- 25 左圖為 FePc 分子結構,中圖為以 π-π 作用力複合氮摻雜石墨烯 氣凝膠,右圖為經高溫處理後之結果69 ... 36 圖.2- 26 三維石墨烯巨觀及微觀下型態 70 ... 37 圖.3- 1 實驗合成示意圖(a)石墨烯表面化學鍵結改變(b)巨觀結構改變 (c)熱處理前後三維石墨烯化學鍵結變化 ... 41 圖.3- 2 水熱法氮摻雜及三維結構形成機制72 ... 43 圖.3- 3 回流裝置示意圖 ... 44 圖.3- 4 鋰氧電池模組組裝示意圖 ... 49 圖.3- 5 減壓蒸餾裝置示意圖 ... 51
圖.3- 6 耐奎斯特圖:Re ( electrolyte resistance )、Rct ( charge-transfer resistance )、Cd (double-layer capacitance )73 ... 53
圖.4- 1 石墨烯水凝膠與凍乾後氣凝膠及熱處理後外觀 ... 58
圖.4- 2 SEM 圖譜(a) GO, (b) N-GA, (c) FePc/N-GA (放大倍率 2 K) ... 59
圖.4- 3 SEM 圖譜(a) GO, (b) N-GA, (c) FePc/N-GA (放大倍率 10 K) ... 60
圖.4- 4 XRD 測試(GO, N-GA) ... 61 圖.4- 5 XRD 測試(FePc 修飾) ... 62 圖.4- 6 XPS 全譜圖 ... 64 圖.4- 7 XPS 測試 C 1s ... 65 圖.4- 8 XPS 測試 N 1s ... 66 圖.4- 9 XPS 測試 Fe 2p ... 67 圖.4- 10 Raman 測試 ... 68
圖.4- 11 FT-IR 測試(GO, N-GA) ... 69
圖.4- 12 FT-IR 測試(FePc 修飾) ... 70 圖.4- 13 熱重損失分析(GO, N-GA) ... 71 圖.4- 14 熱重損失分析(FePc 修飾) ... 72 圖.4- 15 等溫吸脫附曲線 ... 73 圖.4- 16 孔徑大小分析 ... 73 圖.4- 17 充放電曲線 ... 77 圖.4- 18 充放電庫倫效率比較 ... 77 圖.4- 19 放電電容循環壽命分析 ... 78 圖.4- 20 不同充放電流之充放電分析圖 ... 79 圖.4- 21 循環伏安法測試測試 ... 81
圖.4- 23 於 900 rpm 下分別於氬氣及氧氣中循環伏安法測試 ... 84 圖.4- 24 不同轉速下 FePc/N-GA 於 ORR 部分曲線圖 ... 85 圖.4- 25 不同轉速下 FePc/N-GA 於 OER 部分曲線圖 ... 85 圖.4- 26 不同材料於 900 rpm 下 ORR/OER 循環伏安法測試比較圖譜 .. 86 圖.4- 27 不同材料於 900 rpm 下 ORR 部分曲線圖... 88 圖.4- 28 不同材料於 900 rpm 下 OER 部分曲線圖 ... 88
表目錄
表.1- 1 不同催化劑及複合材料於 OER/ORR 電壓表現13 ... 4 表.2- 1 鋰氧電池反應機制 ... 9 表.2- 2 各種碳材物理特性比較31 ... 13 表.2- 3 常見於鋰氧電池電解液之有機溶劑 ... 28 表.2- 4 常見氮摻雜合成方式及氮源化合物64 ... 34 表.3- 1 實驗藥品資料. ... 38 表.3- 2 實驗使用儀器設備之資料 ... 40 表.4- 1 不同材料之 ID/IG值 ... 68 表.4- 2 BET 測試結果整理: 表面積、平均孔體積及平均孔徑 ... 74 表.4- 3 元素分析測試結果 ... 75 表.4- 4 不同電流大小下放電電容量 ... 80利用氮摻雜石墨烯氣凝膠負載酞菁鐵應用於
鋰氧電池正極材料
指導教授:陳振興博士 國立高雄大學應用化學系 學生:謝子敬 國立高雄學大學應用化學系 摘要 鋰氧電池(又可作鋰空氣電池),特別在非水相的系統,由於其高能量密度對於能使 用於電動車中的能量儲存及轉換裝置有很大的期望,但在實際的應用上還是受到許多問 題的阻礙,如:充放電過程中緩慢的氧還原及氧釋出動力學反應、非導體的過氧化鋰放 電產物會生成並覆蓋電極表面、循環過程電解液的不穩定性。在這裡我們提出使用酞菁 鐵並利用配位鍵錨定於氮摻雜石墨烯氣凝膠內表面之氮原子的複合物材料,通過簡單的 水熱法以及回流反應進行合成。結合具有優良氧還原/氧釋出反應催化性能的酞菁鐵及 氮摻雜石墨烯,我們成功的合成此鋰氧電池之正極材料。 通過應用於電池系統中的測試,該 FePc/N-GA 材料在充放電電流為 100 mA/g 時可 達到 2350 mAh/g 的電容值,且充放電間的過電壓可降低至 1.57 V。再以相同電流值所電位測試也分別可得結果為 3.13 V 及 3.14 V,這樣的現象表示在充放電往返的過程中 該 FePc/N-GA 材料有助於其反應可逆性增加,有助於解決鋰氧電池中緩慢的反應動力 學及放電產物鈍化電極表面等問題。
Iron Phthalocyanine supported by N-doped graphene
aerogel as a cathode material for lithium-oxygen batteries
Advisor:Dr. Jenn-Shing Chen Department of Applied Chemistry National University of Kaohsiung
Student:Tzu-Ching Hsieh Department of Applied Chemistry National University of Kaohsiung
ABSTRACT
Li–O2 (Li–air) batteries, especially the non-aqueous type, are considered the most hopeful energy storage and conversion device candidates for the electric vehicle applications due to their high energy density. But their practical use is still hindered by several issues including the slow kinetics of both oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) during the charge/discharge process, the non-conductive Li2O2 discharge products formed and covered on the cathode surface, and the poor stability of electrolyte during the cycling. Herein, we present the composite material employed the iron phthalocyanine (FePc) with a coordinate bond anchored on the inner surface of N-doped graphene aerogel, via a simple hydrothermal method and reflux with FePc. Combining the outstanding ORR/OER catalytic activity of the FePc molecular and N-doped graphene, we successfully synthesized the bifunctional electrocatalysis material as the cathode of Li–O2 batteries. The electrochemical characteristics were investigated by battery test system, rotating disk electrode (RDE) system to determine the excellent performance.
As the material applied in Li-O2 batteries, the FePc/N-GA electrode exhibits 2350 mAh/g discharge capacity and the 1.57 V of overpotential at the current density of 100 mA/g. The
problem of the slow kinetic reaction and the positive electrode passivation in Li-O2 batteries.
第一章 前言
1.1 緒論
石油,又稱原油,主要由不同的碳氫化合物組成,是蘊含在地殼中 的珍貴資源且與我們現今的生活息息相關。目前所開採的石油約有 88 %作為燃料來源,其餘的 12 %則作為化工原料,在我們日常生活中所使 用交通工具的汽油、燃油皆來自於石油的提煉,而由其產生的許多副產 品如: 瀝青、石蠟、煤氣、塑膠等,更在我們生活的環境中隨處可利用 到,故其為今時今日世界上極重要的一次能源之一。不過自工業發展與 石化能源的利用以來的一項隱憂也吸引了大家的注意,隨著環保意識的 抬頭石化能源對於環境的影響也開始受到重視,不僅是開採石油對於地 表的破壞,零星的原油泄漏事件對於海洋生態更是難以復收的災難,而 干涉我們生活更深的還有溫室效應、酸雨、空氣汙染等問題,甚至有危 害到我們生存的可能。所以為了改善這些問題,替代能源的尋找及使用 逐漸變成近年科學研究發展的一項重要課題。 其中在日常生活我們所使用的交通工具由於亦是利用石化能源作為 動能故對於這方面替代能源的研究更為熱門,目前雖已有許多電動腳踏 車、電動摩托車等可應付日常生活移動的小型交通工具問世,但若需要 進行較長距離移動時,還是會考慮使用汽車等較大型的交通工具來到達 目的地,故目前關於電動車及油電混合車也是因應這樣的市場需求進行池的研究中不僅在預期可展現的理論能量密度上可更相近於汽油,其所 使用的組成材料也更加符合環保的概念,這樣藉由新型態電池作為動力 的電動車其單位里程數更有大幅度的提升,如圖 1-2 2,有成為未來電動 車動力來源的潛力。 圖.1- 1 不同電池系統與汽油能量密度比較1 圖.1- 2 不同電池系統運用於電動車可行駛距離推測圖 2
1.2 研究動機及目的
鋰空氣電池,因是利用空氣中的氧作為活性物質故又稱作鋰氧電 池,其雖然具有高理論能量密度(11680 W h Kg-1)1, 3,但要實踐這樣的功 能於電動車上還是有很多的問題需要克服。目前在鋰氧電池的研究上所 可展現能量密度還是遠低於理論值,且在於高倍率放電、循環壽命、庫 倫效率等性能上還面臨著許多挑戰2, 3。而影響鋰氧電池性能的主要部分 為空氣電極組成及電解液的穩定性,其中空氣電極的設計被視作影響內 部反應進行極重要的關鍵4。 理想的空氣電極應需要具備: (1)合適的孔洞結構以利於氧氣的擴散5, 6 (2)良好的電解液潤濕性7, 8 (3)有助於增進氧化還原動力學之催化劑9。 近年來的研究歸納出影響鋰氧電池性能的主要因素來自充放電過程反應 路徑不同而造成較高的電壓差,在放電時進行稱作氧還原反應(Oxygen Reduction Reaction, ORR)的步驟,使氧氣還原並與鋰離子結合;而充電 時則進行氧釋出反應(Oxygen Evolution Reaction, OER),此時產物分解 產生氧氣。由於在放電過程產生低導電性的放電產物 Li2O2,使得充電 過程需要更高的能量來進行放電產物的分解,而造成充電電壓的提高提 高了充放電間的電位差,而這樣的現象會導致額外的能量損失,降低了 電池循環效率且在高電壓的狀態下電解液也有分解的可能,對於電池的 壽命亦有不良的影響。目前這樣的問題可以透過添加催化放電產物 Li2O2分解的催化劑來進行改善 10, 11,降低充電過電壓來避免低循環效率圖.1- 3 不同金屬氧化物催化劑於充電反應時過電壓表現 12
其中在近期電催化劑的研究,先前常被使用於燃料電池中的催化劑 酞菁鐵(Iron Phthalocyanine, FePc)14,被發現對於鋰氧電池中氧化還原反
應有優良的催化效果15-19,故將其應用於本實驗中。酞菁鐵是一個以過 渡金屬為中心的的平面分子,常被用以π-π 作用力20 來與碳材料來進行 修飾,在研究中表示於中心金屬原子與氧氣結合的催化反應過程中,金 屬原子會被拉出分子平面而造成分子與碳材間作用力減弱,多次循環後 有脫離材料的可能21,如圖 1-4。 圖.1- 4 以理論計算展示 FePc 中心鐵原子於 ORR 反應位置變化 21 為了修正這樣的缺陷在 Cao 等人的研究21提出利用修飾垂直於碳表 面之吡啶(Pyridine)官能基的方法來進行與 FePc 分子的鍵結,如圖 1-5, 此方法利用 Pyridine 上氮原子與 FePc 的配位鍵結減少了在與氧分子鍵
圖.1-5 FePc/Py/CNT 示意圖21 而在本實驗中採用了水熱法一步合成氮摻雜石墨烯氣凝膠(graphene aerogel)的方式,在合成過程中同時加入乙二胺作為氮源來達到異質原子 摻雜的目的22,增加碳基材表面催化氧化還原的能力,再經由後續的熱 處理來調控石墨烯表面,形成有利於鋰氧電池的氮摻雜形式23,而最後 再利用加熱回流的方式使其摻雜的氮原子與 FePc 產生配位鍵結,預期 仿效上述相似於 Pyridine 官能基功能。而在水熱過程中石墨烯的自組裝 所產生的三維結構,也減少了二維平面可能造成的堆疊現象提升了碳表 面利用率,提高了材料表面積24。 總結以上,本實驗利用先行合成的氮摻雜三維石墨烯與 FePc 分子產 生配位反應,達到錨定分子的效果,最後會利用物理及化學的分析來證 明該材料的特性及組裝鋰氧電池來進行性能的檢測。
第二章 文獻回顧
2.1 鋰氧電池概述
鋰氧電池,原由鋰空氣電池而來,是藉由陽極鋰在陰極與氧氣進行 可逆化學能及電能轉換的電化學反應,在 1976 年由 Littauer 和 Tsai 提 出鋰空氣化學於水相系統中的概念,直至 1996 年 Abraham 首個提出非 水相鋰空氣電池於有機高分子電解液的研究,目前侷限於其技術而無法 達到預期的高能量密度及循環壽命,但其根據不同材料組合可達到 1000~2000 W h kg-1能量密度,理想狀態下其能量密度可超越目前鋰離 子電池甚至趨近於汽油能量密度,故鋰空氣被認為可作為未來電動車能 量來源之重要技術。13。2.2 鋰氧電池工作原理
圖 2-1 為鋰氧電池的的示意圖,在放電時於負極產生鋰離子及電 子,其中鋰離子經由電解液輸送至正極多孔材料,與通入電極的氧氣及 透過外電路傳導的電子進行反應,生成非導體的放電產物過氧化鋰 (Li2O2)堆積於正極材料表面。放電時則藉由外部電源提供能量進行放電 產物的分解反應,此時 Li2O2分解為鋰離子、電子及氧氣,鋰離子及電 子循原路徑返回負極還原為鋰金屬,而氧氣釋放回環境中。圖.2- 1 鋰氧電池充放電示意圖 根據電池所使用的電解液型態可分為四種系統13: (a)非質子 (b)水溶 液 (c)固態 (d)混合型電解液 如圖 2-2。其中以非質子電解液系統具有 潛在更高的能量密度及可充電性,且非質子電解液可保護負極的鋰金屬 避免與環境中水氣或二氧化碳接觸而產生 LiCO3或 LiOH 等副產物2。 圖.2- 2 以電解液分類鋰氧電池(a)非質子(b)水溶液(c)固態(d)混合13
而在非質子電解液(非水相電解液)系統中反應機構如下表 2-1 1, 13. 表.2- 1 鋰氧電池反應機制 放電反應 (氧還原反應, ORR) 總反應: O2 + 2Li→ Li2O2 充電反應 (氧釋出反應, OER) 總反應: Li2O2 → O2 + 2Li 正 極 ( 半 反 應) O 2 + e - → O2-………...….(I) O 2 + Li+ → LiO 2………….…...(II) 2LiO 2 → Li2O2 + O2.….……....(III) LiO 2 + Li + + e- → Li 2O2…….…(IV) Li 2O2 → O2 + 2Li + + 2e -負 極 ( 半 反 應) Li → Li++e- Li++e- → Li 如表 2-1 反應機構所示,在放電開始時,正極的氧分子會與經由外 電路而來的電子結合形成超氧陰離子(O2 -),如式(I),該分子為以單鍵鍵 結之兩氧原子,一端為自由基而另一端為親核性陰離子,故反應性極 大,會很快的與藉內電路擴散而來的鋰離子反應形成超氧化鋰(LiO2), 如式(II),但超氧化鋰並非穩定狀態會經由化學或電化學的方式進一步 反應,化學反應為兩個超氧化鋰分子結合直接反應為一個過氧化鋰分子
的氧還原反應(ORR)中會產生過氧化鋰及部分的氧氣,氧氣會回歸於電 解液中而不溶的放電產物則堆積於正極材料中;相較於充電時正極半反 應,過氧化鋰直接分解為氧氣、鋰離子及電子,可以發現到兩者反應路 徑不同,且於放電時還出現了超氧化鋰的中間產物,這樣的情形才導致 了鋰氧電池中充放電過電壓不同的原因。
2.3 鋰氧電池內部組成
鋰氧電池主要由負極(鋰金屬)、電解液及正極(氧電極)所組成,其 中電解液是電池內部反應離子傳導不可或缺的角色,所以在電解液的選 擇上除了黏度及介電常數等影響離子傳導的特性以外,與鋰氧電池反應 中超氧陰離子的穩定性及在較高操作電壓下分解的可能性也是需要被考 慮的課題。在正極材料部份,於充放電過程所進行的三相反應是影響電 池性能的重要因素,如圖 2-3,如何能夠增加氧氣和電解液中鋰離子、 電子碰撞發生反應的機會以及減緩固態非導體放電產物過氧化鋰所造成 的電極鈍化現象更是提高電池性能的關鍵。此外,在正極材料中催化劑 的添加也能有效改進 ORR/OER 反應動力學,進而達到降低過電壓,提 高電池循環壽命等效果,而以下將分作正極、催化劑以及電解液作討 論。圖.2- 3 鋰氧電池正極三相反應示意圖1
2.3.1 正極
在非水相鋰氧電池中,正極材料作為一個提供氧分子(氣相)、鋰離 子(液相)及電子(固相)的三相反應發生位置,該材料的結構設計及底材 尤為重要,而一個優良的正極材料應該具備: (1) 高表面積及適合的孔洞性,用來容納放電產物的堆積及有利於氧氣 的通透及擴散。 (2) 高電解液潤濕性,維持電極中電解液含量保持在反應過程中離子的 傳輸不受阻。 (3) 加速氧還原反應(ORR)及氧釋出反應(OER)反應動力學的能力。 (4) 結構的穩定,能承受在充放電反應間由於放電產物的堆積及分解所 造成的體積變化而不崩解。在近期的研究中指出碳材料的孔洞大小對於鋰氧電池的放電電容有 所影響5, 25, 26。孔洞大小的定義可分為微孔(Micropore)、介孔 (Mesopore)、大孔(Macropore),其孔徑分別在小於 2 nm、2~50 nm、大 於 50 nm 之間,在 Ding 等學者5的研究發現到在限制孔徑<100 nm 的 實驗中放電產物過氧化鋰會以 7.8 nm 作單層的堆積,尚有其他研究也提 到過氧化鋰鈍化薄層厚度約在 5~10 nm27。由此可推知微孔材料較不適 合作為理想的正極,僅單層放電產物就可能堵塞孔洞減少孔洞內部表面 積的利用,如圖 2-4,故適當的孔洞大小應以尺寸大於 5 nm 的介孔或大 孔材料做為正極材料,但實際上放電產物堆積的機制及特性尚與電極材 料種類、催化劑及電流大小有密切的關係28。 圖.2- 4 不同孔洞大小與放電產物堆積示意圖29 除了電極材料的結構的設計外,構成電極本體的底材亦是影響鋰氧 電池性能的重要因素,目前多使用碳材作為製備正極材料的基底,再搭 配後續進行的修飾或改質構成目前常用的鋰氧電池氧電極,而使用碳材
作為電極材料有以下的優點: (1) 碳材料價格低廉,且合成技術及方式多樣,容易被使用 (2) 材料性質穩定,在電池系統中也不會被電解液腐蝕,在較高電壓的 操作條件下也相對穩定 (3) 結構操作性高,可藉由設計實驗方法來調整構型,達到預期的表面 積或孔洞大小 而近年來由於石墨烯的開發及合成技術趨於成熟,目前在鋰氧電池 系統的研究上也常使用石墨烯為碳材。石墨烯是一種碳原子以 sp2混成 軌域所組成六角型晶格並僅具有一碳原子厚度的二維材料,其特性包括 高表面積、高導電性、高結構強度等30,相較於其他常使用於電極材料 的碳材具有相對的優勢,如表 2-2。 表.2- 2 各種碳材物理特性比較 31
石墨烯的高導電性及高表面積的特性可以容納放電產物 Li2O2的堆 積,可提高電容量表現,而高機械強度亦可維持材料在充放電過程型態 穩定性,而其本身也具有催化劑功能32亦可有效的增加 Li 2O2 合成/分 解效率,也可預期提高電池循環效率及穩定性,故也應用於本實驗正極 材料的研究,而將於章節 2.4 更詳細描述目前石墨烯材料應用於鋰氧電 池系統中的方法及所扮演的角色。
2.3.2 催化劑
鋰氧電池正極材料的構成除了可穩定存在於系統中的碳基材外還有 另一部分是可加速ORR/OER反應動力學的催化劑。而在正極材料中添 加催化劑原是為了降低充電時OER反應所造成較高的過電壓,避免電解 液的分解產生不可逆的副產物,但在許多研究中亦有發現到催化劑對於 放電時ORR反應電位也有提升的功能,此類催化劑被稱作雙功能催化劑 (Bi-functional Electrocatalysts)9,是目前常應用在正極製備的材料。相較 於貴金屬催化劑如Pt、IrO2、RuO2等金屬或其氧化物的催化劑33-35,除了 價格高昂、取得不易等缺點外,雖然具有優良的催化功能但大部分僅提 供ORR或OER單方面的催化,故目前多以過渡金屬或碳材為主體的雙功 能催化劑有其發展的重要性,此類催化劑大致分做幾類9:(1) 非貴金屬材料(Non-precious metal-based materials): I. 單金屬氧化物(MnO2, Mn2O3, Mn3O4)
MnxCo3-xO4)、AxByC3-x-yO4型(Mn0.6Cu0.4Co2O4)
(2) 鈣鈦礦型金屬氧化物(Perovskite-type metal oxides): I. 二元鈣鈦礦氧化物(LaCoO3, h-BaTiO3)
II. 三元鈣鈦礦氧化物(La1.7Sr0.3NiO4)
III. 四元鈣鈦礦氧化物(Ba0.5Sr0.5Co0.8Fe0.2O3) (3) 其他過渡金屬化合物: 過渡金屬合金、氫氧化物、硫化物、氮化物 (4) 碳基材料: I. 無金屬碳材料(石墨烯、奈米碳管) II. M-N-C材料(FePc、CoPc) 在雙功能催化劑中大部分是以過渡金屬為主體及其延伸的化合物, 除了上述的分類法外尚有可以透過不同合成方法來調整原子結構進而達 到不同的催化活性,以金屬氧化物為例,藉由特定結構導向劑或改變反 應條件使得晶體中金屬原子與氧原子的排列晶格發生改變,進一步提高 金屬氧化物催化活性36-38。而此類過渡金屬的化合物於ORR/OER催化的 原理大多是利用表面不同原子電荷密度的差異來吸引反應過程中鋰離子 或氧分子的吸附及反應,而這樣的方式也影響了放電產物Li2O2的堆積作 用,可堆積於材料表面的均勻度增加,進而增加反應面積使分解所需能 量降低,如圖2-5所示。
圖.2- 5 不同表面結構α-MnO2 放電產物堆積示意圖 39 在雙功能催化劑中還有另一類稱為碳基材料,在無金屬的碳基材料 中有石墨烯、奈米碳管等,這類的碳材料主要能夠透過其單層碳原子表 面或其邊緣的缺陷來進行鋰離子或氧分子的鍵結進而達到在鋰氧電池中 催化的效果,此部分將於2.4.1詳細說明其反應原理。而碳基材料中的另 一部分為M-N-C複合物,是藉由金屬、氮、碳三種原子鍵結而成的催化 劑結構,而目前最常見的是以過渡金屬為中心的大環複合物分子(表示 為M-N4),在應用於鋰氧電池系統的ORR/OER催化活性研究中,在Shui 等人40經由實驗與常用於鋰氧電池中的催化劑α-MnO 2比較,證明在 OER及ORR反應的催化效能夠優於α-MnO2,如圖2-6,且該過渡金屬氧 化物在先前研究中為可降低充電過電壓至最低的材料12。
圖.2- 6 左圖為 Fe/N/C 複合材料示意圖,右圖為與α-MnO2/C 複合材料 比較充放電電位 40 .
2.3.4 M-N-C 催化劑
自 1964 年 Jasinski 41發表此類化合物於燃料電池 ORR 催化劑的應 用後,該材料被廣泛的使用及研究,而當中以 Fe 或 Co 為中心原子搭配 大環分子酞菁(Phthalocyanines, Pc)作為配基的 FePc 及 CoPc 分子最常被 在文獻中討論,其中以 FePc 在 ORR 催化的表現更優良42-46,故也開始應用於鋰氧電池催化劑的研究19, 20, 47。
在Sun等人15的研究中,利用了溶解FePc於電解液中進行實驗,發現
主要的氧化還原反應是發生在FePc上而非正極的碳基材,並推導出在液 相中可能的機制,如圖2-7:
圖.2- 7 液相中 FePc 反應機制15 而在 Yu 等人19研究中,利用了 Fe-N-C 材料修飾於石墨烯海綿應用 於鋰氧電池中,電容量可達 6762 mAh/g 且充電電壓可降低至約 3.8 V, 在該研究中也推論出該催化劑於 OER 反應發生時反應機制,藉由中心 原子 Fe 所包含的兩個空軌域與放電產物 Li2O2中的氧原子鍵結,使在氧 化過程其中的 O-O 鍵結不被打斷的進行 OER 反應,如圖 2-8。
圖.2- 8 FePc 於 OER 催化反應機制
但隨後於在Gunasekara等人48的研究中參考了Sun等人的文獻,認為
在液相FePc的情形下對於陽極鋰金屬及電池系統的可充電性有不良的影 響,故藉由結合FePc與碳材(carbon black),製備出固相的FePc複合材料 (N4Fe)來進行測試,並由實驗也推導出了可能的氧化還原反應路徑機制:
Reduction pathway (i)
N4Fe + O2 + e- + Li+ N4Fe – O2Li
2N4Fe – O2Li N4Fe – O2Li2 + O2
N4Fe – O2Li2 + 2Li+ + 2e- 2Li2O + N4Fe
Reduction pathway (ii)
N4Fe + O2 + 2e- + 2Li+ N4Fe – O2Li2
Reduction pathway (iii)
Oxidation 2N4Fe – Li2O N4Fe – Li2O2 – FeN4 + 2Li+ + 2e- N4Fe – Li2O2 – FeN4 N4Fe + O2 + 2Li+ + 2e -N4Fe – LiO2 N4Fe + O2 + Li+ + e- 上述不同的反應路線可藉由不同分子之中心Fe原子間的距離來區分 在實際情況下發生的反應途徑,而圖2-9、2-10分別為不同反應途徑的示 意圖:
圖.2- 10 固相中 FePc 反應機制 (Reduction pathway ii) 而該研究中也探討到在電解液溶劑性質(DN, Donor number)的高低不 同亦會影響FePc分子中心原子Fe本身氧化還原電位的改變,導致後續鋰 氧電池中與氧氣結合的ORR反應產生不同的反應機制。故目前對於FePc 分子在鋰氧電池系統中尚未有明確反應途徑的分析及研究之原因可能是 來自於中心原子氧化數易受存在環境變化,而影響後續ORR及OER的反 應。
2.3.3 電解液
電解液在電池系統中扮演著連通電池內部,做為離子傳輸的橋梁, 做為對於電池的電容量、循環性能、安全性都有很重要的影響,電解液 可分為液態電解液及固態電解液兩種,其中液態電解質被認為在使用上 有漏液的風險性,可能會導致電池短路進而發生危險,故固態電解液有 其發展的重要性,目前研究中的材料有:聚氧化乙烯(polyethylene oxide, PEO)、聚偏氟乙烯(poly vinylidene fluoride, PVDF)、聚氯乙烯(poly vinyl chloride, PVC)、聚丙烯腈(poly acrylonitrile, PAN)等,但由於固態電解液 尚在研究階段,故目前研究上仍多以液態電解液為主。 在液態電解液部分,一般來說若做為鋰離子電解液應具備良好的離 子傳導、較大的操作電位窗、電池組件的相容性、高熱穩定性及低毒性 等優點,但由於鋰氧電池內部環境較為特殊,涉及到氧氣的反應及空間 的開放性,故還尚需考慮到以下幾點: (1) 吸濕性低,電解液做為連接正負極內電路的角色,若含有水分便有 可能腐蝕負極鋰金屬 (2) 低揮發速率(低飽和蒸汽壓),在鋰氧電池正極部分是採取開放式的 空間以提供氧氣流通而非傳統的密閉式,故電解液的揮發速率也為 重要的考慮因素 (3) 氧氣的溶解度,由 ORR 反應於正極的半反應中可以得知,反應物為 溶於電解液中的氧氣,故溶氧量越高可使反應更容易進行(4) 化學穩定性佳,在 ORR 反應的第一步即生成超氧陰離子,電解液是 否能穩定與該反應物共存而不會有分解的副反應發生,在鋰氧電池 的研究上是關鍵的一環,以下電解液溶劑的討論亦是以此為重點
而目前常用於鋰氧電池液態電解液中電解質部分的鋰鹽有:高氯酸鋰 (LiClO4)、四氟硼酸鋰(LiBF4)、六氟磷酸鋰(LiPF6)、六氟砷酸鋰
(LiAsF6)、三氟甲基磺酸鋰(LiCF3SO3)、雙三氟甲烷磺酰亞胺鋰
(Bis(trifluoromethane)sulfonimide lithium salt, LITFSI)等,而電解液中溶 劑部分以下將分為五類來進行穩定性的討論,分別是碳酸酯類(Aklyl carbonates)、醚類(Ethers)、醯胺類(Amides)、碸類(Sulfonees)、離子液體 (Ionic liquids)。 碳酸酯類(Aklyl carbonates) 雖然在常用的溶劑中碳酸酯類並非具有最低的飽和蒸汽壓,如圖 2-11,但還是常被應用於鋰離子電池系統中,碳酸丙烯酯(Propylene carbonate))、碳酸二甲酯(Dimethyl carbonate)等都是常被利用的種 類,在將其延用至鋰氧電池中時發現會被反應過程中超氧陰離子反應分 解的現象,並產生副產物 Li2CO3而使電容衰退49,而推測出分解反應的
機制,如圖 2-12,由此可知碳酸酯類溶劑並不適合使用於鋰氧電池中, 不可逆的副產物會沉積於電極表面,阻斷反應介面,最終使反應無法進
圖.2- 11 各溶劑於 29 ᵒC 乾燥箱內的消耗百分比比較圖 50
醚類(Ethers) 醚類具有相對較低的揮發速率,故也是良好電解液溶劑的選擇,在 Bryantsev 等人52的研究中透過理論計算推算醚類在對於超氧陰離子以 更好的穩定性,且其操作電位窗上限可達 4.5V,是作為電解液溶劑的合 適選項之一。但實際經過測試後發現在長時間下分解的現象還是會發 生,在 Freunberger 等人53的研究指出醚類在含氧量高的環境下會緩慢 的自動氧化(autoxidation),導致對於超氧陰離子的穩定程度將會下降而 分解,文獻中亦嘗試推導出反應機構如圖 2-13: 圖.2- 13 醚類電解液分解機制
醯胺類(Amides) 相對於碳酸酯類較為穩定,但還是有碳酸鋰的副產物形成及堆積, 對於電容量有不良的影響,在 Chen 等人54的分析及推導後得到分解機 制如圖 2-14: 圖.2- 14 醯胺類電解液分解機制54 碸類(Sulfonees) 在 Laoire 等人55的研究中證明了碸類電解液確實能夠有較長的壽命 及穩定性,但在其他的文獻中還是有發現到分解的現象發生56, 57,
Schroeder 等人58使用密度泛函理論(density functional theory, DFT)來計
算 Li2O2/DMSO 界面化學穩定性,通過活化能的計算提出分解機制如圖
圖.2- 15 碸類電解液分解機制 58 離子液體(Ionic liquids) 離子液體及低的蒸氣壓、低可燃性、高離子傳導率、優良的疏水性 及廣的電位窗,相較於其他有機電解液更有發展的潛力,其組成的分子 基團存在與氮原子相連的烷基,成為較差的離去基,故也不易與超氧陰 離子反應,可以穩定地存在於充放電過程中。但由於其黏度高的特性使 得離子的傳輸不易,且價格較為昂貴,故尚無法普遍的應用於鋰氧電池 中,常見的離子液體正負離子如圖 2-16。
由表.2-3 常用於鋰氧電池之有機溶劑之性質比較中可以觀察
TEGDME 除了在穩定性較佳的優點外,於其他性質如溶氧量、沸點及 蒸汽壓等性質有相對的優勢,故選擇使用 TEGDME 作為本實驗電解液 之有機溶劑,除水後與鋰鹽 LiTFSI 配製成 1 M 的電解液。
表.2- 3 常見於鋰氧電池電解液之有機溶劑 Solvent Structure Dielectric
constant ε(25 oC) Viscosity η (cP) (25oC) Oxygen solubility (mM/cm3) Boiling/ melting point ( oC) Vapor pressure (kPa, 25oC) PC 15.1 2.53 3.2 241.7/ -48.8 0.160 (55 oC) EC 16.4 1.930 (40 oC) 1.71 248.2/ 36.4 3.371 (95.21 oC) DMC 17.2 0.59 7.29 91/ 4.6 2160 TEGDME 7.79 4.05 4.43 275/ -30 <1.33 DMSO 46.45 1.991 2.1 189/ 18 56
2.4 石墨烯於鋰氧電池的應用
石墨烯除了高表面積、高導電性及本身具有催化性等有利於鋰氧電 池充放電反應的優點外,更能夠藉由不同的合成過程來進行與其他材料 的複合進一步提高本身的催化活性,材料本身的結構也能夠透過簡易的 自組裝方式來達到二維至三維間的轉換,提升表面積使用率及結構強 度,以下分作四節來講述石墨烯運用於鋰氧電池中的方法。2.4.1
石墨烯本身為催化劑作為電極
石墨烯本身即能夠做為催化劑來應用於鋰氧電池,利用石墨烯結構 上缺陷及邊緣所含σ-bonds 懸鍵 (sp3碳原子)形成活性位,而能夠與氧氣 反應分解成原子態氧,達到催化的效果如圖 2-17: 圖.2- 17 石墨烯與氧氣反應催化示意圖 30到在這些缺陷及官能基位上是不利於 Li2O2簇(cluster)的生長,而是分別 形成奈米粒子的沉積方式。此作用可限制放電產物尺寸,表示在鋰氧電 池放電過程中可減緩電極鈍化且奈米尺寸放電產物也可避免在放電過程 中電阻抗持續上升達到催化反應降低過電壓之效果。 圖.2- 18 石墨烯表面 Li2O2堆積方式及鍵結能量表示 59 在 Li 等人60的實驗中石墨烯作為電極材料時電容可達 8705.9 mAhg-1,大約是市售其他碳材的 8 倍;在 Yoo 等人30的研究中證實在放 電電壓的比較上石墨烯可達到 3.00 V,接近以 Pt/carbon black (20 wt%)
之 3.05 V,有取代貴金屬催化劑的潛力,製造這類石墨烯的方法常藉由 以 Hummer 法製備之氧化石墨烯再使用化學還原或高溫還原法 (600~1000 ℃)製成。
2.4.2
石墨烯元素(N)摻雜作為催化劑
在元素摻雜方面目前普遍以氮元素為主,是利用異質原子的摻雜來 改變石墨烯表面電荷分布形成反應位置,受 N 原子拉電子影響使周圍 C 原子子帶正電而可吸引帶負電氧氣,如圖 2-19 約可分為四種類型, pyridinic、pyrrolic、graphitic (basal /edge plane),其各有不同催化效果但 以 pyridinic 與 graphitic 最具效果23但基本原理相似。而在 Jing 等人62的研究中以密度泛函理論(DFT)及吸附能的計算來 解釋不同形式氮摻雜在 Li2O2的沉積過程及路徑,氮摻雜的方式於石墨
烯表面約可歸類存在 5 種組合( graph N1、graph N2、pyri N3、pyrro N4、pyri N5),如圖 2-20,
而其中約可分為兩種控制 Li2O2沉積的路徑 :
Path A : O2 → LiO2 → Li2O2
Path B : Li → LiO2 → Li2O2
在 graph N1、graph N2 在吸附能(Eads)較傾向 O2的吸附而由 Path A 反
應,而 pyri N3、pyrro N4、pyri N5 則反之傾向於 Path B。另外於 Yun 等人63的研究中也透過密度泛函理論(DFT)來進行計算,利用 Graphitic
及 Pyridinic 兩種形式摻雜來對於上述 A、B 路徑及鍵結方式進行表示, 如圖 2-21 及 2-22 表示:
圖.2- 21 Graphitic N site 進行 Path A 反應機制示意圖
在催化性能方面,在 Wu 等人61的研究中也可得到在 ORR 反應起始
電位接近於 Pt/C 證明有提高放電電為的效能。在該類材料的製程方法通 常是通過加入富含氮的化合物進行熱處理,如表 2-4
表.2- 4 常見氮摻雜合成方式及氮源化合物64
Process N precursor
Vapor phase growth Benzylamine, Imidazole, Ethylenediamine,
Fe-Phthalocyanine, Polymer Post-synthetic annealing Ammonia
Solution processing (aqueous/non-aqueous)
[80 ℃~250 ℃]
Urea, Ammonia, Hydrazine
[400 ℃, 500 ℃~1100 ℃] Ammonia, Melamine, Acetonitrile, Pyridine, Nitrogen, Polypyrrole
而在 Linfei 等人65的研究中表明氮摻雜形式也能夠透過不同溫度的
熱處理或改變前驅物的種類來進行調控,如使用含有六元環或五元環結 構之含氮前驅物即可能可得到較多的 Pyridinic N 或 Pyrrolic N 形式;而 經由高溫處理後則可能得到更多的 Graphitic N 的摻雜形式如圖 2-23。
圖.2- 23 不同氮摻雜種類之製程方法65
2.4.3
石墨烯作為催化劑載體
以石墨烯作為催化劑載體,主要是利用其高表面積及表面官能基的 特性來達到均勻分散及固定催化劑位置的效果,由前述催化劑的討論可 知道大部分的過渡金屬化合物可做為鋰氧電池雙功能催化劑,以金屬氧 化物為例,透過反應前驅物氧化墨烯表面帶負電含氧官能基吸附金屬陽 離子,再利用不同生成金屬氧化物方法如: 水熱法、溶劑熱法、熱處 理、熱回流、微波處理、化學還原等方法66,製備金屬氧化物催化劑與 石墨烯之複合物,如圖 2-24 67。圖.2- 24 石墨烯做為催化劑 MnO2載體合成機制示意圖 在 Wang 等人68的研究中可證明在電池方面除了所使用的 MnCo 2O4 催化劑可達到與 Pt/C 相似的效能,在這樣化學合成的方法也遠比物理混 合可表現更佳的催化效果。而在 M-N-C 催化劑材料方面,以 FePc 為 例,文獻69常利用與石墨烯表面的π-π 作用力來進行簡單的合成,或再 進一步進行高溫處理的方式形成複雜結構來與石墨烯複合,如圖 2-25。 圖.2- 25 左圖為 FePc 分子結構,中圖為以 π-π 作用力複合氮摻雜石墨烯 氣凝膠,右圖為經高溫處理後之結果69
2.4.4
三維石墨烯結構(sponge、aerogel)
在化學還原石墨烯的製備及應用中已知的缺點有: 表面缺陷、分散 性差、再堆疊現象及多層結構,故藉由凡德瓦力、氫鍵或交聯劑等作用 力使之相互連結進而將二維的石墨烯片組合成具三維網絡之結構,稱作 石墨烯氣凝膠或石墨烯海綿,用以提高其高表面積利用率,避免上述分 散性差、再堆疊現象的缺點,如圖 2-26。合成的方法大致上分為下列幾 種24: (1) 氧化石墨烯冷凍乾燥,隨後高溫熱還原(Ar, 600~1000 ℃) (2) CVD 法沉積於 3D 網絡(Ni foam)(3) 混合 Polymer (PVA 、PEO、PVP 等)
(4) 高溫高壓交聯劑還原自組裝(Py、PPy、 RF、EDA 等)
而本實驗所參考的是利用水熱法進行的交聯劑還原自組裝方式,該 方式的特點在於能夠結合前述幾種石墨烯於鋰氧中的應用方法,藉加添 加不同的前驅物經由簡易的一步合成,同時達到元素摻雜、催化劑複合 及三維結構形成的特點,是目前鋰氧電池中常用的正極材料製備技術。
第三章 實驗方法
3.1 實驗藥品
表.3- 1 實驗藥品資料.
藥品名稱 化學式 製造廠商
Graphite C Aldrich
Sodium nitrate NaNO3 Acros
Sulfuric acid H2SO4 J.T. Baker
Potassium permanganate KMnO4 J.T. Baker
Hydrogen peroxide H2O2 Aldrich
Hydrogen chloride HCl J.T.Baker Iron(II) phthalocyanine
(FePc)
C32H16FeN8 Aldrich
Super P C TIMCAL
Poly vinylidene difluoride (PVDF)
N-Methy-2-pyrrolidone (NMP)
C5H9NO
Riedel-de Haën® Lithium foil Li Aldrich Bis(trifluoromethane)sulfonimide lithium salt (LiTFSI) CF3SO2NLiSO2CF3 Aldrich Tetraethylene glycol dimethylether (TEGDME) CH3O(CH2CH2O)4CH Aldrich
3.2 實驗儀器及設備
表.3- 2 實驗使用儀器設備之資料 儀器設備名稱 儀器廠商及型號 高溫管狀煅燒爐 Lindberg/Blue HTF 555322A X-ray 粉末分析儀 RigakuMultiflex 2kw 高解析電子能譜圖分析儀 ULVAC-PHI XPS 掃描式電子顯微鏡 HITACHI S-3400NJEOL-6330 Field-Emission SEM
鍍金機 Hitachi E1010
顯微拉曼光譜分析儀 PROTRUSTECH CO.,Ltd. (PTT)/BWП RAMaker 比表面積及孔洞分析儀 Micromeritics ASAP 2020 熱重/差式掃描量熱分析儀 SETARAM Labsys
八通道充放電測試儀 Acu tech Systems BAT-750B 雙恆電位儀 CHI Model 704A 手套箱及純化系統 E-LIEN GB-125
烘箱附真空幫浦 泛群科技 VO-30
3.3 實驗方法
實驗流程先由 Hummer 法71進行石墨的氧化製成氧化石墨烯分散液 (GO),再經由添加乙二胺的水熱法及後續的凍乾和高溫熱處理製得碳材 氮摻雜石墨烯氣凝膠(N-GA),最後進行回流法複合 FePc 分子,,即可 得到最終產物 FePc/N-GA,反應的機制如圖 3-1: 圖.3- 1 實驗合成示意圖(a)石墨烯表面化學鍵結改變(b)巨觀結構改變 (c)熱處理前後三維石墨烯化學鍵結變化3.3.1 氧化石墨烯合成(GO)
利用改良 Hummers 法71由石墨粉為前驅物製備氧化石墨烯(Graphene Oxide),實驗步驟如下: 1. 反應容器中加入 23 ml 硫酸,於冰浴 (0 ℃)下與 0.5 g 石墨粉及 0.5 g 硝酸鈉磁力攪拌混合約 30 分鐘的預氧化過程。 2. 持續於冰浴(0 ℃)下攪拌,緩慢加入 3 g 過錳酸鉀氧化反應,加入完 畢後維持攪拌 30 分鐘。 3. 將反應容器轉移至 35 ℃恆溫水槽中持續磁力攪拌約 1 小時,隨後 緩慢加入 40 ml 去離子水。 4. 將水浴溫度提升至 95 ℃,持續攪拌至溶液由棕色轉為黃色約 需 30 分鐘。 5. 反應結束後加入 30 %過氧化氫水溶液 3 ml 以還原多餘的過錳酸 根,反應約 10 分鐘後再加入 100 ml 去離子水再持續攪拌約 10 分 鐘。 6. 抽氣過濾後收集濾餅,以去離子水利用離心洗至中性,最後以離心 2000 rpm 分離出固體氧化石墨烯產物。 7. 將產物以冷凍乾燥-80 ℃, 72 h 乾燥後收集備用。3.3.2 氮摻雜三維石墨烯氣凝膠合成(N-GA)
水熱法 N-GA 的合成機制如圖 3-2 及實驗步驟如下22: 1. GO 分散液 (3 mg/ml, 30 ml)加入 180 μl 之 EDA,超聲波震盪 30 min 使其分散均勻。 2. 將溶液轉移至高壓釜進行 160 ℃水熱法 12 h,得水凝膠 GH。 3. 將水凝膠冷凍乾燥-80 ℃, 72 h 得後氣凝膠 GA。(約可得產物 70~80 mg) 4. 將氣凝膠置入水平管狀爐中,在氬氣環境下熱處裡 1000 ℃, 8 h 得 N-GA 產物。(GA 前驅物 100 mg 約可得產物 20~30 mg) 圖.3- 2 水熱法氮摻雜及三維結構形成機制723.3.3 FePc/N-GA 的合成
FePc/N-GA 的合成採用回流法21,步驟如下: 1. 架設回流裝置,如圖 3-3, 先於反應容器中放入 FePc 粉末、碳材及 攪拌子(FePc、N-GA 各 100 mg,FePc 濃度約 1 mg/ml) 2. 以針筒注入無水 THF 於反應容器中, 以攪拌子攪拌至粉末溶解 3. 同時持續通入 Ar 約 30 min, 以保持反應環境於 Ar 中 4. 加熱回流 3 h (THF 沸點 66 ℃) 5. 反應結束後取出產物以 THF 抽氣過濾,於 110 ℃烘箱乾燥,得 FePc/N-GA 產物。(約可得產物 170~180 mg) 圖.3- 3 回流裝置示意圖3.3.4 比較材料 FePc/N-GA (π-π)材料合成
FePc/N-GA 材料的合成主要是利用物理的混合,利用 π-π 作用力固定 FePc 分子於 N-GA 表面,是用於比較與 N-GA 表面氮原子配位對於材料 性能造成影響,合成方法如下20:
1. 混合 N-GA、FePc 粉末於無水乙醇中(FePc、N-GA 各 100 mg,FePc 濃度約 1 mg/ml) 2. 超音波震盪約一小時,並隨後以磁石攪拌混合液約一小時 3. 反應結束後取出產物以無水乙醇及 THF 抽氣過濾,於 110 ℃烘箱 乾燥,得 FePc/N-GA (π-π)產物。(約可得產物 170~180 mg)
3.4 材料鑑定及特性分析
3.4.1 掃描式電子顯微鏡( SEM )
取微量待測樣品粉末放入適量的無水乙醇中並利用超音波震盪機進 行樣品的分散,待樣品顆粒均勻分散後利用滴管取出分散液滴至矽基板 上(事先以雙面銅膠固定於金屬板上),以烘箱 110 ℃烘乾隔夜後即可送 入 SEM 系統中抽真空,在 SEM 中利用不同的放大倍率來進行對樣品表 面的觀察。3.4.2 X-ray 粉末分析儀( XRD )
kV/30 mA,D.S. ( Divergence slit ) = 0.5 mm、S.S. ( Scanttering slit ) = 1 mm、R.S. ( Receiving slit ) = 0.15 mm;掃描速率為 1 o/min,偵測間隔為 0.1 o/point,掃瞄範圍(2θ)為 5 o ~ 80 o。將分析後的繞射圖譜與 JCPDS ( Joint Committee on Power Different Standards )資料庫做比對,可確認單個產物 晶型結構及複合物中所含的不同材料之晶型。
3.4.3 顯微拉曼光譜分析儀( Micro-Raman )
取適量研磨後之樣品粉末利用打片模具壓製成薄片並放於玻璃試片 上,再將玻璃試片置入顯微拉曼光譜儀( PTT BWII RAMaker )進行分 析,使用 532 nm( He-Ne Laser )光源其能量為 2 mW,以 10 %光通量垂 直照射於樣品表面,設定掃描波長範圍 500~1800 cm-1,掃描時間單次 15 秒共 15 次,分析待測樣品中原子與原子間鍵結模式與強度。3.4.4 高解析電子能譜圖分析儀( XPS )
待測樣品粉末經過打片模具壓製成薄片,取面積小於 7.5 cm×7.5 cm,厚度小於 2.0 cm 之薄片,送入儀器中抽真空後方可進行測試,X-ray 光源為掃描式單光器( Scanning Monochromated ) Al anode,能量分析 儀為 180 o Spherical Capacitor Analyzer+32Channel Detector,真空度為<5×10-10 torr。最後使用軟體( XPSPEAK41 )進行曲線的擬合分析材料中鍵 結的種類。
3.4.5 傅立葉轉換紅外線光譜儀分析法( FT-IR )
使用傅立葉轉換紅外線光譜儀( PerkinElmer Spectrum RX1 )進行分 析。秤取重量比約 100 : 1 之溴化鉀及待測樣品,使用研缽均勻研磨混合 後利用油壓打片模具壓製成薄片,薄片呈現透明狀即可進行傅立葉轉換 紅外線光譜儀的分析。利用得到的 FT-IR 圖譜,可得知待測樣品中原子 和原子之間的振動模式,來分析樣品中的鍵結形式。3.4.6 熱重分析儀( TGA )
測量樣品在特定氣氛下的升溫過程中,材料隨著溫度變化的重量損 失趨勢,用以判斷材料的熱分解特性。秤取適量待測樣品置於坩鍋內, 並將放入熱重分析儀當中,高溫煅燒以氮氣為反應氣體,升溫設定 5 ℃ /min,由室溫 30 ℃煅燒至 800 ℃進行分析。3.4.7 比表面積分析儀( BET )
秤取樣品粉末(>0.2 g)置於試樣管中,於真空下以升溫速率 10 ℃/min 加熱至 180 ℃進行除氣,並持溫 12 小時,將樣品中的水分與吸附雜質去 除,除氣後秤量實際樣品重以降低分析誤差。接著將試樣管移至分析端 進行分析,分析時試樣管浸於液態氮中,並於填入定量氮氣,進行不同相 對壓力下的氮氣吸附量分析。最後分析結果經由 Brunauer-Emmentt-Teller ( BET )方法計算出樣品的比表面積、孔徑分布趨勢、孔體積相關物理性3.4.8 元素分析儀(EA)
待測樣品除去水份及溶劑後取約 2~3 mg 盛於錫金屬容器內,瞬間升 溫至 1800 ℃使樣品完全燃燒,再經過銅還原處理後,生成之 N2、CO2、 H2O 混合物經過特殊之分離管分離後, 可利用熱傳導偵檢器(TCD) 分別 測定其含量,再經資料處理機運算,即可自動列計碳、氫、氮及硫之重量 百分比。3.5 鋰氧電池系統
鋰氧電池模組為 MTI Corporation 的 Lithium-Air battery ( EQ-STC-LI-AIR ),組裝,如圖 3-4,組裝過程皆在氬氣環境之手套箱( H2O < 1 ppm, O2 < 1 ppm )中進行,電解液為 1 M LiTFSI/TEGDME,隔離膜為 Celgard 2300 的多孔隔離膜。組裝過程依序如下: 1. 將於底座上放入鋰金屬並輾平,確認其完全貼合電池底座。 2. 將圓形隔離膜(直徑約 3.5 cm)浸漬於電解液中吸附至呈現半透明狀後 放置,並完整覆蓋鋰金屬(避免氣泡)。 3. 放置正極 Ni foam,將材料塗覆面貼合隔離膜,並添加 1~2 滴電解液 潤濕極片。 4. 放置密封組件,最後再將上蓋鎖緊。 5. 送出手套箱後通入高純氧(99.999 %)至壓力讀表達 0.15 MPa (約 2.5 atm)。
圖.3- 4 鋰氧電池模組組裝示意圖
3.5.1 正極材料極片製備
Ni foam 前處理 1. Ni foam 裁切為 1x1 cm。 2. 以丙酮及乙醇超音波震盪 20 min 清洗。 3. 以 0.5 M HCl 溶液超音波震盪 10 min 去除氧化層。 4. 再利用乙醇及去離子水超音波震盪清洗。 5. 以真空烘箱 110 ℃隔夜烘乾。 漿料製備
本實驗用於鋰氧電池系統極片漿料配置活性物質、助導劑(Super P, SP)及黏著劑(polyvinylidene difluoride, PVDF)各比例為:
Super P : PVDF = 9 : 1
Carbon material (N-GA : SP) : PVDF = 9 (7 : 3) : 1
Carbon material (FePc/N-GA or FePc/N-GA (π-π) : SP) : PVDF = 9 (7 : 3) : 1
配置步驟如下: 1. 碳材料預先於 110 ℃烘箱中烘乾隔夜。 2. 預先利用均質機混合 PVDF 及 NMP (2500 rpm)製成澄清(淡黃) 膠液。 3. 再加入碳材料同樣以均質機攪拌(2500 rpm)至漿料呈現均勻狀(無 顆粒、表面平滑反光)。 極片塗覆 1. 將先前漿料取出適量,以刮刀均勻塗佈於 Ni foam 上。 2. 將極片風乾隔夜後再於 110 ℃烘箱中再次烘乾隔夜。 3. 組裝電池前先放入真空瓶中以真空烘箱 90 ℃烘乾約 5 h,立即 取出後送入手套箱備用。
3.5.2 電解液配製
鋰鹽 Bis(trifluoromethane)sulfonimide lithium salt ( LiTFSI )在使用前 先於真空烘箱 90 ℃ 8 小時烘乾除去水份,隨後保存於氬氣環境手套箱 中。有機溶劑 Tetraethylene glycol dimethyl ether ( TEGDME )在使用前先 進行除水,裝置如圖 3-5,先在氬氣環境下與 CaH2進行除水約 12 小時,
再進行減壓蒸餾法收集,最後加入 4 Å 分子篩靜置 2 天進行除水。取除 水 後 LiTFSI 以 50 mL 定 量 瓶 與 除 水 後 TEGDME 配 置 成 1 M LiTFSI/TEGDME 電解液,配製好的電解液需以 4 Å 的分子篩混合靜置除 水 24 小時再使用。
3.5.3 鋰氧電池充放電測試
本實驗藉由鋰氧電池進行充放電測試,利用電腦紀錄電流-電壓變 化,了解材料對於充放電的電容量與過電壓的關係。
充放電參數設定以 100 mA/g、200 mA/g、300 mA/g 設置充放電電 流(單位克數以活性物質重量為計),採用先放電再充電為一循環,並設 置截止電壓分別為放電電壓小於 2000 mV、充電電壓大於 4500 mV 及額 外的充電截止條件為充電容量大於放電電容 105 %,每次循環結束後中 斷 10 分鐘使電解液內離子均勻分散,再回到放電階段進行下一個循 環,進行數個循環的測試。 而循換壽命則以電流 100 mA/g 進行測試,並限制充放電電容為 500 mAh/g,藉以觀察電容衰退及循環壽命。
3.5.4 循環伏安法(Cyclic Voltammetry, CV)
將組裝好的鋰氧電池通入氧氣後靜置 8 小時,再利用雙恆電位儀進 行循環伏安法分析,分別以鋰金屬(負極)當作輔助電極和相對電極,正 極材料極片作為工作電極。而儀器參數設定為掃描電壓範圍 2.0~ 4.5 V,掃描速度 1 mV/sec,進行 5 個循環測試。3.5.5 交流阻抗測試(AC Impedance)
交流阻抗測試又稱為電化學阻抗頻譜法(Electrochemical impedance spectroscopy,EIS)。由電位儀與可變頻正弦波供應器結合,利用小振福的 交流電壓或電流對電極進行擾動從而獲得交流阻抗數據,而其電化學阻抗可藉由等效電路圖模擬得到,其結果可轉換為複數平面耐奎斯特圖, 如圖 3-6 所示。對於等效電路提出不同的掃描頻率範圍可用以解釋不同 的阻抗行為: (1) 高頻區( > 1 kHz )屬於鈍化層的範圍 (2) 中頻區( 10 Hz~1 kHz )屬於電荷轉移範圍 (3) 低頻區( < 1 Hz )與電子傳遞相關 耐奎斯特圖中高頻區的半圓對應電荷轉移阻抗及電極和電解液之間 的界面阻抗,半圓直徑越小,其電荷轉移阻抗越小,低頻區的斜直線對應 鋰離子在固態電極中的擴散阻抗,斜率越大,則鋰離子的擴散阻抗越小。
圖.3- 6 耐奎斯特圖:Re ( electrolyte resistance )、Rct ( charge-transfer resistance )、Cd (double-layer capacitance )73
操作時將通入氧氣的鋰氧電池靜置 5 小時再進行交流阻抗分析,使 電解液完全潤濕電極及氧氣的溶入,開路電壓約為 3.2 V,頻率範圍設
3.6 旋轉圓盤電極(Rotating disk electrode, RDE)
旋轉圓盤電極系統是結合流體動力學方程式(hydrodynamic equation) 和對流擴散方程式(convective-diffusion equation)在穩定狀態(steady state) 下解釋及量化的流體動力學電化學系統。藉由旋轉帶動內部物質的移 動,降低擴散層影響使能更容易觀察動力學控制的反應,並使盤電極表 面電流分布均勻提高實驗數據準確性。以下介紹 RDE 系統的前處理及 操作。
3.6.1 工作電極塗覆(圓盤電極)
圓盤電極(Disc)塗覆方法如下圖 3-7 而工作電極漿料配置重量比為: Super P : PVDF = 6 : 4Carbon material (N-GA : SP) : PVDF = 6 (9 : 1) : 4
Carbon material (FePc/N-GA or FePc/N-GA (π-π) : SP) : PVDF = 6 (9 : 1) : 4
3.6.2 旋轉圓盤電極系統
實驗前置 (a) 手套箱環境: 本實驗於 Ar 環境下的手套箱中進行,利用燈泡中 燈絲測試水氧環境(恆亮>20 s),若未達標準則利用 Ar 流通換氣 至達到標準。 (b) 工作電極: 使用玻璃碳(Glassy Csrbon, GC)作為電極,材料塗覆 後先以烘箱 110 ℃隔夜烘乾,在開始實驗前再利用真空烘箱 90 ℃烘乾約 3 h 再送入手套箱。 (c) 參考電極: 以銀線浸置於所配置溶有 0.1 M Tetrabutylammoniumhexafluorophosphate (TBAPF6)和 0.01 M Silver nitrate (AgNO3)的
ACN 溶劑做為參考電極。參考電極於本實驗 1 M LiTFSI/TEGDME 電解液系統下與鋰金屬量測為 0 VLi=-3.45 ± 0.01 VAg/Ag+。實驗數據所出現電壓直接以置換對鋰為標準 (V=VLi/Li+)。保存於 Ar 環境之手套箱,使用前存放於密封血清 瓶中置於傳遞箱中真空保存。 (d) 相對電極: 以鋰金屬包覆部分銅絲並以隔離膜袋密封保護鋰金 屬,如圖 3-8,於手套箱內完成製作並封存於密封的血清瓶放置 於傳遞箱中真空保存。 (e) 電解液: 1 M LITFSI/TEGDME,使用前先配置完成並保存於溶劑 儲存瓶中同參考電極置放
圖.3- 8 相對電極製作示意圖 實驗步驟 (a) 組裝儀器: 依序組裝 RDE 頭、相對電極、電解液、參考電極、F 形玻璃管(O2通入用) ,並注意電極及組件間相對位置(高度、距 離相近,且測試部分完全浸漬於電解液中),裝置完成圖如圖 3-9。 (b) 氬氣實驗: 完成組裝約 30 min(待相對電極浸潤)即開始測試,轉 速轉換間平衡約 10 min 可開始進行電化學分析。 (c) 氧氣實驗: 實驗開始前必須先通氧 90 min 才可開始進行測試, 實驗進行過程中注意氣泡不可於通氧時停留於工作電極表面, 實驗操作方法如氬氣實驗。 (d) 實驗結束: 將各部分歸回原位,RDE 組件金屬部分及 Disc 利用 丙酮震盪清洗,塑料部分則利用擦拭紙及乙醇(95 %)清潔。
第四章 結果與討論
本章節探討在材料間物性及電性的變化,前驅碳材部分主要觀察氧 化石墨烯(GO)及氮摻雜石墨烯氣凝膠(N-GA)合成過程材料性質變化,並 針對該實驗過程中高溫熱處理前後氣凝膠(GAN-GA)內部氮摻雜形式 的調整進行驗證。其他合成部分則對於 N-GA、FePc/N-GA (π-π)、 FePc/N-GA 等材料進行物性測試及實際應用至鋰氧系統中的電性表現。4.1 外觀及掃描式電子顯微鏡( SEM )分析
在實驗過程中主要的結構形成在於將懸浮的氧化石墨烯片經由水熱 法還原並自組裝成石墨烯水凝膠,由於在高溫烘乾的狀態下水分的抽離 會帶動水凝膠維結構的破壞而失去原本預期的孔洞結構,故在此步驟利 用冷凍乾燥法進行水分的抽離,由圖 4-1 可看出冷凍乾燥前後結構上並 無太大的改變,而在熱處理後雖尚可維持該立體結構但體積縮小,可能 來自於高溫熱處理脫去了剩餘的含氧官能基及水熱法過程產生的含氮官 能基(章節 3.3.2 之圖 3-2)。 圖.4- 1 石墨烯水凝膠與凍乾後氣凝膠及熱處理後外觀其中也能夠透過在掃描式電子顯微鏡(SEM)下進行觀察由氧化石墨 烯轉變為石墨烯氣凝膠結構的變化,如圖 4-2 (a)、(b)、(c),圖中可觀察 到在 GON-GA 過程材料立體度的增加,由平鋪相疊的形式轉換為較 立體的結構並富有平面片狀結構所形成的孔洞,可視為由二維結構轉變 至三維結構的過程,減少可能因為堆疊而失去可進行反應的表面積,而 在經過了修飾反應還是能夠維持三維的形態。
材料表面的變化也可由圖 4-3 來觀察,反應前的 GO 在 SEM 下呈現 平滑且帶有皺褶的表面結構,經由水熱法及高溫處裡後表面呈現較粗糙 且破損的結構,這樣的改變可能來自於還原過程官能基的剝離,使得表 面產生缺陷。而在最終材料 FePc/N-GA 的 SEM 圖譜下可以觀察到所添 加的 FePc 顆粒可均勻的分佈在石墨烯的表面。
4.2 X-ray 粉末分析儀( XRD )分析
圖 4-4 為 XRD 測試結果,由圖中可以看出符合氧化石墨烯特徵的 訊號約在 11 0,在經由水熱法及後續的高溫處理後可發現訊號再轉移至 25.6 0。這樣的結果表示該含氧官能基可大部分的被除去,由此可證明 還原反應的成功。藉由布拉格繞射公式n λ = 2 d sinθ的比較也能夠推 導出在晶格間距 d 值可隨著水熱還原及高溫還原有明顯的減少,主要是 來自於該官能基的除去使石墨烯層結構間距離縮短。 圖.4- 4 XRD 測試(GO, N-GA) 在後續與 FePc 材料的複合反應過程,也可由圖 4-5 中觀察,可利用 純 FePc 特徵峰值及數據資料庫中對應於 α-FePc 之 JCPDS No.022-1771前述布拉格繞射公式來堆導出晶格間距的增加,表示可藉由石墨烯表面 來達到 FePc 分子的分散。而其中在 FePc/N-GA 約 13.8 0有出現訊號, 其來源為利用π-π 作用力錨定於石墨烯表面產生堆疊的 FePc 分子所造 成74 ,相較於配位鍵結的 FePc/N-GA 材料,其堆疊現象明顯減少,證明 了兩材料間 FePc 分子與石墨烯表面作用力的改變。 圖.4- 5 XRD 測試(FePc 修飾)
4.3 高解析電子能譜圖分析儀( XPS )
圖 4-6 為對實驗中各種材料所進行的 XPS 的全譜圖測試,圖中可以 觀察到在 C、N、O、Fe 等元素的訊號變化,其中在 O 1s 部分可以觀察 到隨著由 GO 為前驅物進行的水熱還原及高溫熱處理,該訊號比例隨著 GO GA N-GA 的反應途徑遞減,證明了還原過程中含氧官能基的 脫去,在 N 1s 部分也能夠觀察到在經由添加乙二胺為氮源的水熱法所 製得的 GA 產物相較於其前驅物 GO 有所增加,而經過了高溫處理後該 N 1s 訊號比例的下降也可能來自結構中部分含氮官能基的除去,這部分 可參考章節 3.3.2 之圖 3-2 的三維結構形成機制中石墨烯結構間的含氮 官能基鍵結,其在高溫處理過程中可能產生脫離石墨烯或改變其氮摻雜 結構而造成的損失。在後續的 FePc 的複合中也可明顯觀察到 N 1s 及 Fe 2p 部分訊號的增加,來自於所修飾的 FePc 分子。圖.4- 6 XPS 全譜圖 本實驗在 XPS 對於各材料的解析中主要是觀察 C、N、Fe 三種元素 在各自不同的鍵結中呈現的束縛能進行判別。在 C 1s 的部分中主要觀 察的有 C-C/C=C (284.4~285.6 eV)、C-O (285.8~286.7 eV)、C=O (287.1~287.9 eV)、O-C=O (288.8~290.2 eV)等鍵結,在 N1s 的部分觀察 的是 Pyridinic N (398.4~399 eV)、Pyrrolic N (399.5~400 eV) 、Graphitic N (401~403 eV)等峰值。製備石墨烯氣凝膠的過程中除了材料三維型態 的確定外,主要是將氧化石墨烯還原為可導電的石墨烯表面及氮原子的 摻雜,由下圖 4-7 中可以在 C 1s 的測試中可以觀察到還原過程
GOGAN-GA 中碳氧鍵結的變化,明顯在含氧官能基的鍵結部分分 別在水熱及高溫還原兩製程後都有明顯下降的趨勢,由此可證明兩步驟 還原反應的成功。 圖.4- 7 XPS 測試 C 1s 而在實驗過程中氮摻雜的形式則可經由 N 1s 的測試來進行觀察, 首先在 N-GA 碳材料的合成中如圖 4-8,先藉由水熱法合成之 GA 可得 到多屬 Pyrrolic 形式的氮摻雜,其中於 406.5 eV 出現之峰值可歸類於 N-O 間鍵結所造成,可能來自於水熱法中乙二胺與氧化石墨烯表面含氧官
Pyridinic 及 Graphitic 兩種形式,除了可證明了氮摻雜形式的可調控性 外,該兩種氮摻雜形式於催化鋰氧電池 ORR 及 OER 反應也較 Pyrrolic 形式更具優勢。 圖.4- 8 XPS 測試 N 1s 在最後的 FePc 材料的複合部分,藉由與材料 FePc/N-GA 的訊號觀 察比較利用π-π 作用力以及在配位鍵結存在下不同的的複合方式,在 XPS 測試中的差異,如圖 4-9,在圖中可觀察到相同在 Fe 2p 部分的測 試中於訊號 Fe 2p3/2的位置發生位移的現象,在配位鍵的影響下峰值由 709 eV 位置偏移至約 711 eV,與文獻描述相符21,故也可藉由此部分來 證實材料合成的成功性。
圖.4- 9 XPS 測試 Fe 2p
4.4 顯微拉曼光譜分析儀( Micro-Raman )分析
由 Raman 光譜的分析中可以藉由觀察 G band 及 D band 來分析其表 面碳原子組成的模式,其中 G band 表示 sp2混成的碳原子鍵結,用來表 示無異質原子及官能基的石墨烯表面,位置約在 1580 cm-1;而 D band 則表示 sp3的混成鍵結,來自於石墨烯表面官能基或異質原子的鍵結, 破壞原本了石墨烯表面結構,位置約在 1350 cm-1。由所得的測試圖 4-10,可以觀察到實驗過程中該訊號值的變化,並藉由計算 D band 及 G band 的訊號強度比值(ID/IG)探討反應過程對於材料的影響,如表 4-1。 在 GO 的氧化合成過程,由其 ID/IG比值為 0.92 可以看出在石墨(無 D band 訊號)氧化過程雖使 sp3的混成鍵結增加但尚保留較大部分原先之結 構。而在接續合成 N-GA 的還原及氮摻雜反應進行使得 ID/IG比值增加證 明了含氧官能基去除及氮摻雜的成功,使得石墨烯表面更多的 sp2部分
圖.4- 10 Raman 測試
4.5 傅立葉轉換紅外線光譜儀分析法( FT-IR )
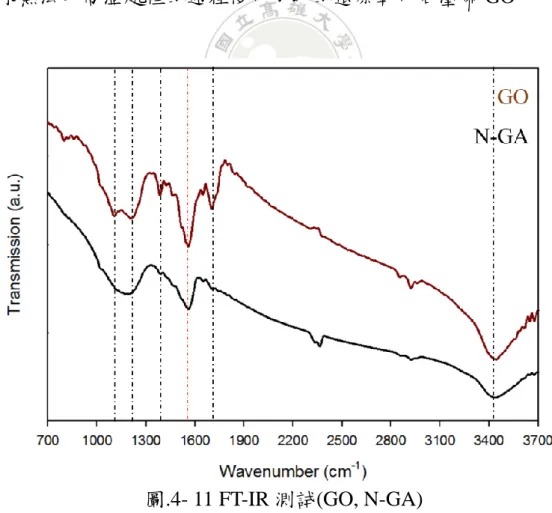
圖 4-11 為製備氮摻雜石墨烯氣凝膠實驗過程以 FT-IR 觀察反應前後 材料鍵結形式變化,在 GO 轉變至 N-GA 的過程是為一還原反應,目的 為除去原 GO 上的含氧官能基,使其表面回復為石墨烯結構,始能展現 其高導電性的特性。由圖中可看到在黑色虛線部分標記為碳氧原子間不 同鍵結之特徵訊號,分別為 C-O (1100 cm-1)、C-O-C (1210 cm-1)、C-OH
(1380 cm-1)、C=O (1705 cm-1)、O-H (3400 cm-1),紅色虛線部分對應於石 墨烯表面結構 C=C (1550 cm-1),由兩曲線間的比較可以發現到該含氧官
能基部分訊號在經還原後的 N-GA 部分有明顯的減少,由此可證明在經 由該水熱法及高溫處裡的過程後可成功的還原氧化石墨烯 GO。
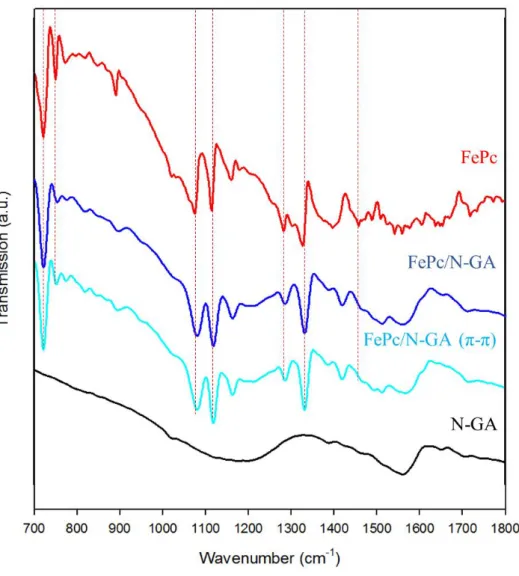
圖 4- 12 為以 N-GA 為前驅物進行後續修飾 FePc 分子催化劑之反應 過程材料鍵結結構於 FT-IR 中測試結果,其中在材料 FePc/NGA 及 FePc/N-GA (π-π)部分能夠觀察到 FePc 分子之特徵峰值(紅色虛線部分)其 位置約在 720 cm-1
(C–H out-of-plane bending vibration)、750 cm-1(benzene and isoindole in-plane deformation and Fe–N stretching) 、1084 cm-1 (C–N stretching) 、1117 cm-1 (Fe–N of pyrrole) 、
1283 cm-1 (C–H in-plane bending vibrations) 、1326、1456 cm-1(C=C or C=N stretching of pyrrole ring and isoindole stretching in the plane) 等位置,能夠在所製備材料的測試圖譜中對應
其特徵峰值,藉此證明 FePc 複合的成功。