行政院國家科學委員會專題研究計畫 期中進度報告
癌症轉移抑制基因 RECK 之研究:基因調控與藥物篩選(2/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2314-B-110-001- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立中山大學生物醫學科學研究所 計畫主持人: 洪文俊 計畫參與人員: 張慧秋,潘美仁,許銘娟 報告類型: 精簡報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢 中 華 民 國 94 年 6 月 10 日行政院國家科學委員會補助專題研究計畫 期中報告 九十三年度[癌症轉移抑制基因 RECK 之研究:基因調控與藥物篩選定(2/3)] 計畫類別:█ 個別型計畫 □ 整合型計畫 計畫編號:NSC 93-2314-B-110-001 執行期間: 93 年 8 月 1 日至 94 年 7 月 31 日 計畫主持人:洪 文 俊 成果報告類型(依經費核定清單規定繳交):█精簡報告 □完整報告 本成果報告包括以下應繳交之附件: □赴國外出差或研習心得報告一份 □赴大陸地區出差或研習心得報告一份 □出席國際學術會議心得報告及發表之論文各一份 □國際合作研究計畫國外研究報告書一份 執行單位:中山大學生物醫學研究所 中 華 民 國 94 年 5 月 30 日
中 文 摘 要
RECK 癌症轉移抑制基因的產物是一細胞膜上的醣蛋白,其具有抑制間質
性金屬蛋白酶酵素活性及腫瘤轉移的能力。有研究指出致癌基因 ras 透過
RECK 啟動子上的 Sp1 結合位而抑制 RECK 的表現。所以在本計畫中,我
們將著重探討 ras 抑制 RECK 表現的分子機制。我們利用 co-transfection 的
方法發現 Sp1 及 Sp3 對於 RECK 啟動子的作用是活化者(transactivators),而 非抑制者。我們進一步探討 ras 是否促使 histone deacetylases (HDACs)結合 Sp1 進而抑制 RECK 表現,結果發現 ras 的活化確實增加 Sp1-associated HDAC1,且會促進 HDAC1 結合上 RECK 啟動子上的 Sp1 結合位,而藉由
加入 HDAC inhibitor TSA 則可有效的反轉 Ras 對 RECK 基因的抑制作用,
我們的結果更明顯在 2-12 細胞(ras-inducible)中發現致癌基因 ras 透過活化
ERK 而非 JNK 或 p38HOG kinase,且利用 MEK inhibitor PD98059 或大量表 現ERK2 dominant-negative mutant 確實可逆轉 ras 對 RECK 基因啟動子的抑
制作用,綜合我們的結果發現致癌基因 ras 是經由影響 histone deacetylation
的機制而抑制 RECK 基因表現。
關鍵詞:癌症轉移抑制基因 RECK、致癌基因 ras、histone deacetylases、Sp1、 ERK2、histone deacetylation inhibitor (TSA)
英 文 摘 要
The RECK gene encodes a membrane glycoprotein that may negatively regulate matrix metalloproteinases (MMPs) activity and potently inhibit tumor invasion and metastasis. Previous study demonstrated that oncogenic ras inhibited RECK expression via an Sp1 binding site in the RECK promoter. In this study, we investigated the molecular mechanism by which ras inhibited RECK expression. Co-transfection assay showed that Sp1 and Sp3 are transactivators, rather than repressors, for RECK gene. So, we tested whether ras activation induced the binding of histone deacetylases (HDACs) to Sp1 to repress RECK expression. Our data showed Sp1-associated HDAC1 in cells was increased after ras induction. By using DNA affinity precipitation assay, we found that induction of oncogenic ras enhanced the binding of HDAC1 to the DNA probe corresponding to the Sp1 site in the RECK promoter. Additionally, a HDAC inhibitor trichostatin A (TSA) potently antagonized the inhibitory action of ras on RECK. The signaling pathway by which ras suppresses RECK was also addressed. Induction of oncogenic ras activated extracellular signal-regulated kinase (ERK), but not c-Jun N-terminal kinase (JNK) and p38HOG kinase in 2–12 cells. Addition of PD98059 or overexpression of dominant-negative mutant of ERK2 indeed reversed ras-mediated inhibition of RECK promoter activity. Taken together, our results suggest that oncogenic ras represses RECK expression via a histone deacetylation mechanism.
Keywords:RECK, oncogenic ras, histone acetylases, Sp1, ERK2, histone acetylation inhibitor (TSA)
計畫緣由與目的
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that are involved in diverse cellular processes. The MMP gene family consists of at least 20 enzymes and may be subgrouped into different types based on substrate specificity and sequence characteristic. MMPs are synthesized as inactive precursors and can be activated by proteolytic cleavage. Therefore, MMP activity can be regulated by modulation of gene expression, control of proenzyme processing, and direct inhibition of enzymatic activity.
The RECK gene was isolated as a transformation suppressor gene by using an expression cloning strategy designed to identify human cDNA inducing flat reversion in a v-Ki-ras-transformed NIH/3T3 cell line. This gene encodes a membrane glycoprotein that may negatively expressed in most of human tissues and untransformed cells, it is undetectable in many cancer cell lines or in cells artificially expressed active oncogenes. Recent study showed that ras oncogene suppressed RECK expression via inhibition of transcription. Additionally, the authors indicated that an Sp1 binding site in the RECK promoter serves as a negative target for the ras signal. However, the detailed mechanism by which ras induces down-regulation of RECK is not clear at present. In this study, we tried to characterize the molecular pathway that mediates the inhibitory effect of ras on RECK.
結果與討論
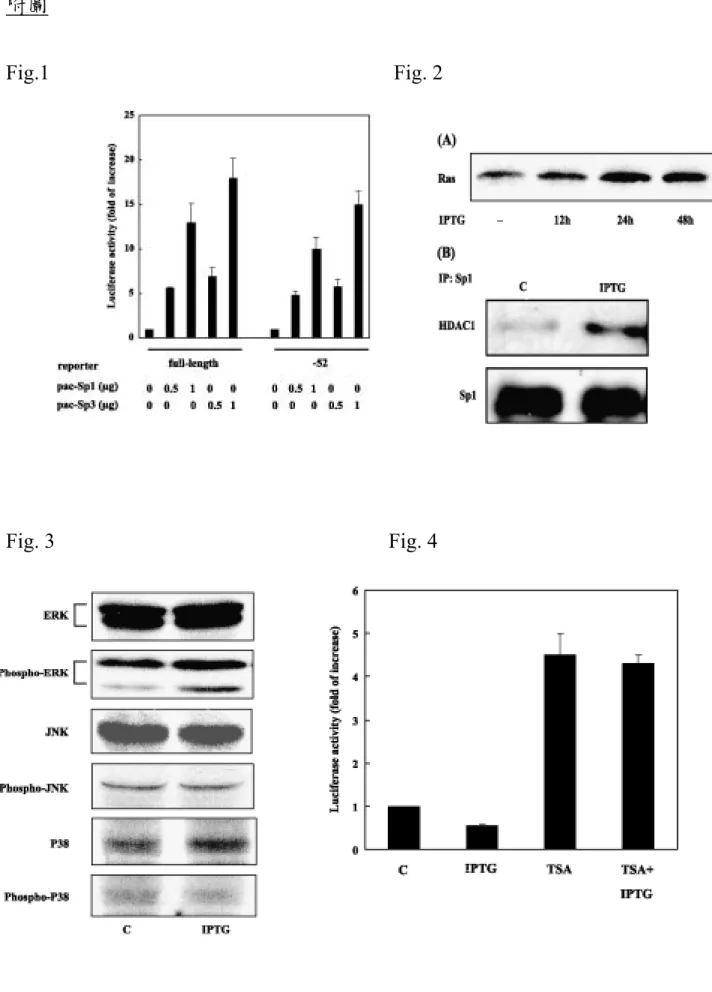
Previous study has indicated that ras suppressed RECK via an Sp1 binding site located within the proximal 52 bp from transcription start site of the RECK gene promoter. Therefore, we tested whether Sp1 or Sp3 might directly suppress
RECK promoter activity in SL2 cells. As shown in Fig. 1, Sp1 and Sp3 activated the full-length RECK promoter as well as its deletion mutant containing the proximal 52 bp region in a dose-dependent manner. These data indicate that Sp1 and Sp3 are transactivators, rather than repressors, for RECK gene.
Recent studies demonstrate that HDACs may bind to Sp1 to repress gene expression. Therefore, we test the possibility that ras may inhibit RECK expression via recruitment of HDACs to Sp1. Addition of IPTG increased the expression of Ras protein in 2–12 cells in a time-dependent manner (Fig. 2A). We next investigated the interaction between Sp1 and HDAC1 in cells by using immunoprecipitation/western blotting assay. Fig. 2B demonstrated that Sp1-associated HDAC1 was increased after IPTG treatment. This increase is not due to change of HDAC1 or Sp1 protein level because the amount of these two proteins in control or IPTG-treated 2–12 cells is similar.
To further confirm that HDACs are indeed involved in ras-induced repression of RECK, we used a specific HDAC inhibitor trichostatin A (TSA) in our subsequent works. Our results strongly suggest that histone deacetylation is involved in the inhibition of RECK by ras (Fig. 3).
We next tried to clarify the signaling pathway by which ras inhibited RECK expression. Because mitogen-activated protein kinases (MAPKs) are major downstream effectors of ras, we tested whether these kinases might be involved in the down-regulation of RECK by ras. Control and IPTG treated 2–12 cells were harvested and the kinase activity of ERK, JNK, and p38 was investigated by phospho-specific antibodies. As shown in Fig. 4, IPTG treatment stimulated ERK, but not JNK and p38, activity in 2–12 cells. Addition of PD98059 (inhibitor for ERK signaling pathway) might counteract the inhibition of RECK promoter activity by ras (Fig. 5). Taken together, we provide the first evidence
that oncogenic ras may inhibit the metastasis suppressor RECK via a histone deacetylation mechanism. 計畫成果自評 於人類癌症細胞中 RECK 的表現很明顯的受到抑制,所以能清楚釐清 此抑癌基因受調控的機轉將有助於尋找能增加 RECK 表現的天然物質或藥 物,進而達到抑制腫瘤侵犯或轉移的能力,而於本年度計畫成果中,我們 提出第一個證據證明致癌基因ras 透過 histone acetylation 抑制 RECK 基因表
現並促進腫瘤侵犯和轉移的能力,所以提供了 HDAC inhibitor 在臨床上應 用於恢復 RECK 的表現進而抑制 MMPs 的活化和腫瘤細胞的侵犯及轉移的 可行性。 成果發表 Cellular Signaling (2004) 16, 675-679. 參考文獻
[1] M.D. Sternlicht, Z. Werb, Annu. Rev. Cell Dev. Biol. 17 (2001) 463– 516. [2] H. Birkedal-Hansen, Curr. Opin. Cell Biol. 7 (1995) 728– 735.
[3] H. Kitayama, Y. Sugimoto, T. Matsuzaki, Y. Ikawa, M. Noda, Cell 56 (1989) 77–84. [4] J. Oh, R. Takahashi, S. Kondo, A. Mizoguchi, E. Adachi, R.M. Sasahara, et al., Cell 107 (2001) 789– 800.
[5] C. Takahashi, Z. Sheng, T.P. Horan, H. Kitayama, M. Maki, K. Hitomi, et al., Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 13221–13226.
[6] R.M. Sasahara, C. Takahashi, M. Noda, Biochem. Biophys. Res. Commun. 264 (1999) 668– 675.
[7] H.S. Liu, H. Scrable, D.B. Villaret, M.A. Lieberman, P.J. Stambrook, Cancer Res. 52 (1992) 983– 989.
[8] M.R. Pan, W.C. Hung, J. Biol. Chem. 277 (2002) 32775– 32780.
[9] Y.C. Huang, L.Y. Chuang, W.C. Hung, Mol. Pharmacol. 62 (2002) 1515– 1521. [10] A. Doetzlhofer, H. Rotheneder, G. Lagger, M. Koranda, V. Kurtev, G. Brosch, et al., Mol. Cell. Biol. 19 (1999) 5504– 5511.
[11] J. Won, J. Yim, T.K. Kim, J. Biol. Chem. 277 (2002) 38230–38238.
[12] M. Oft, J. Peli, C. Rudaz, H. Schwarz, H. Beug, E. Reichmann, Genes Dev. 10 (1996) 2462– 2477.
[13] P.J. Ross, M. George, D. Cunningham, F. DiStefano, H.J. Andreyev, P. Workman, et al., Mol. Cancer Ther. 1 (2001) 29– 41.
[14] T.H. Vu, Z. Werb, Genes Dev. 14 (2000) 2123– 2133.
[15] J.L. Arbiser, M.A. Moses, C.A. Fernandez, N. Ghiso, Y. Cao, N. Klauber, et al., Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 861–866.
[16] T. Andreu, T. Beckers, E. Thoenes, P. Hilgard, H. von Melchner, J. Biol. Chem. 273 (1998) 13848– 13854.
[17] X. Zhou, V.M. Richon, A.H. Wang, X.J. Yang, R.A. Rifkind, P.A. Marks, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 14329– 14333.
[18] L.T. Liu, H.C. Chang, L.C. Chiang, W.C. Hung, Oncogene 21 (2002) 8347–8350. [19] L.T. Liu, H.C. Chang, L.C. Chiang, W.C. Hung, Cancer Res. 63 (2003) 3069– 3072.
附圖
Fig.1 Fig. 2