以無機分子改質雙性機丁聚糖之自組裝混成複合材料及其
生醫特性之研究
研究成果報告(精簡版)
計 畫 類 別 : 個別型 計 畫 編 號 : NSC 98-2113-M-009-004- 執 行 期 間 : 98 年 08 月 01 日至 99 年 07 月 31 日 執 行 單 位 : 國立交通大學材料科學與工程學系(所) 計 畫 主 持 人 : 劉典謨 計畫參與人員: 碩士班研究生-兼任助理人員:董簪華 碩士班研究生-兼任助理人員:劉佩鈴 處 理 方 式 : 本計畫可公開查詢中 華 民 國 99 年 09 月 23 日
期末報告
計畫編號:NSC 98-2113-M-009-004
計畫名稱:
以無機分子改質兩性幾丁聚醣之自組裝混成複合材料及其
生醫特性之研究
執行機構: 國立交通大學材料科學與工程學系
主持人: 劉典謨 副教授
09-17-2010
摘要
藉由兩性幾丁聚醣(縮酸基改質親水端,長碳鏈改質疏水端)與矽烷基偶
聯劑
APTES,(3-Aminopropyl) triethoxysilane,以化學鍵結方式結合,成功
地合成出一無機-有機混成分子,並同時具有在水溶液中自組裝的功能。我
們經由 FTIR 與
13C-, 29Si固態核磁共振儀鑑定此無機-有機混成分子之化學結
構 ; 而 在 水 溶 液 中 自 組 裝 行 為 則 由 臨 界 微 胞 濃 度 (Critical aggregation
concentration)來觀察,發現其自組裝的行為與分子在水溶液中的濃度有
關。其自組裝形成的奈米載體之外觀形貌由掃描式電子顯微鏡(
scanningelectron microscopy, SEM
)與穿透式電子顯微鏡(
transmission electron microscopy,TEM
)來觀察,其粒徑大小則由 DLS 偵測。此有機-無機混成分子在水溶液環
境中自組裝所形成的奈米微粒,由穿透式電子顯微鏡的影像,可觀察到層
狀二氧化矽的結晶相,推測在此有機-無機混成分子所形成之奈米微粒,無
機二氧化矽是以 shell 或 layer-by-layer 的形式存在
,此二氧化矽 layer
是以連續且高度整齊排列成 4~6 奈米之間的原子層,推測此連續且高度整
齊排列二氧化矽的產生,與此混成分子在水溶液中自組裝的行為所導致有
關,疏水的作用力誘引此混成分子中之原子自我組織成一整齊的排列方
式。二氧化矽層在此混成奈米微粒中,扮演著物理屏障的角色,可以減少
包覆之內容物隨著高分子在水溶液中膨潤現象的產生而擴散溢出。此二氧
化矽層的存在,顯示了一潛在的新可能性。
1. Introduction
1-1 Introduction
In recent years, development of advanced organic-inorganic hybrids has become one of
the most innovative research fields. The synthesized materials, which combine organic
macromolecules with inorganic components, allow the properties tunable from atomic,
mesoscopic, to macroscopic scale and also successfully integrated the advantages of both
organic and inorganic materials to meet various industrial demands. The ability to manipulate
outstanding chemical, physical, and biological properties from individual, i.e., organic and
inorganic, component provides opportunities to expand wider applicability in various fields,
from catalysis, optics application, to biomedical devices, etc. [1-3,18] Many approaches for the
preparation of hybrid compounds were recently reported. Among them, the synthesis of
composite through the combination of polysaccharides and silicon-based materials has been
widely reported in various fields, such as enzyme immobilization, porous materials and
electrochemical sensors [1-7]. Furthermore, chitosan-based materials have been proposed for a
range of biomedical applications [1-4, 26]. Among those relevant reports, chitosan-based hybrid
molecules capable of self assembling into well-organized architecture have been rarely found.
It is currently an important research objective by integrating diverse functions into a given
nanoobject for advanced applications. This is particularly interesting in the area of biomedical
Taking the advantage of outstanding biocompatibility of natural polymers, such as chitosan
or its modified analogs, associated with unique functions of inorganic component, it is highly
potential to design a new hybrid molecule that imparts technically desirable behavior for
biomedical demands. The use of pristine chitosan or its composite analogs has been widely
reported for drug delivery practice. However, for most polymeric drug delivery nanoparticles
reported in literature, burst-like elution of drug has frequently observed in the early-phase
delivery in-vitro and in-vivo, which is mainly due to swelling effect of the polymer. This
effect turns to be more pronounced for those water-soluble polymers or most of natural
polymers. Adsorption of drug near the surface region of a nanocarrier upon drug
encapsulation may cause uncontrollable loss or leakage upon delivery, making a final
therapeutic dose less efficacy. Moreover, many water-soluble natural polymers exert less
preference to encapsulate water-insoluble drugs, albeit some emulsification technologies or
analogous skills have been developed to compromise the deficiency. Therefore, to minimize
those undesirable effects and in the meantime, impart improved medical or pharmaceutical
potential to a new nanocarrier, it is more desirable to employ and modify a natural
macromolecule that is able to undergo self assembly to effectively entrap water-insoluble drug
for a subsequent control drug elution.
Here we report a novel hybrid molecule through the employing of an amphiphilic chitosan
previous work on the synthesis of this amphiphilic chitosan[8], where the amino groups, -NH2,
of the carboxymethyl chitosan chain were replaced partially by hydrophobic hexanoyl group,
a further chemical modification along the chain was performed using a coupling agent,
(3-aminopropyl)triethoxysilane (APTES). In this study, we successfully synthesized a new
hybrid macromolecule which was prepared through the chemical bonding between the
amphiphilic chitosan and APTES precursor. The nanostructure, physical and biological
properties, and drug encapsulation/release behavior of the novel hybrid molecule were
systematically investigated.
1-2 Literature Review
1-2-1 Introduction of Particulate Carriers Based on Chitosan
chitosan (CS), β-(1,4)-link glucosamine unit, is produced by deacetylation of chitin, which
is extracted from the shells of crabs, shrimp, and krill. It exhibits several characteristics such
as biocompatibility, nontoxicity, and bioadhesivity which make it as an ideal material for
biomedical uses, such as drug delivery [30].However, its poor solubility in water and common
organic solvents has so far limited its wide-spread utilization. The reactive amino groups in
the backbone of chitosan make it possible to chemically conjugate with various biological
molecules [31]. As a result, there have been many works about the methods to enhance the
carboxyl groups to chitosan derivatives led to the anionic or amphotheric properties, are
water-soluble, and are attracting much attention as metal collecting materials and biomaterials
[32]. Hence, hydrophilically, hydrophobically, and amphiphilically modified chitosan
derivatives are being studied to form the monodisperse self-aggregated nanoparticles by
sonication in aqueous media due to noncovalent association arising from intra- or/and
intermolecular interactions among hydrophobic segments in aqueous media [ 20, 34-35].
During the past decade there has been a growing interest in the investigation of polymeric
micelles as a potential carrier for drug delivery. It is well known that polymeric micelles have
unique core–shell architecture composing hydrophobic segments as internal core and
hydrophilic segments as surrounding corona in aqueous medium. The hydrophobic core
provides a loading space for poorly water-soluble drugs. The hydrophilic shell allows
polymeric micelle gain the stability in aqueous environment, and the active targeting to tumor
cells by further ligand modification [36-38]. Comparing with traditional micelles of
low-molecular weight surfactant, polymeric micelles are generally more stable with a
relatively lower CMC, and show slower dissociation in aqueous environment [39]. Besides,
polymeric micelles own many characteristics as the drug carrier [40]: (1) nano-order size: the
micelles can be sterilized by simple filtration, and avoided
mechanical clearance by filtration or in the spleen. (2) Long circulation: it is hard to be
and willowy shell. (3) Solubilization: solubilization of hydrophobic drug is a difficult problem
for the clinical application. Fortunately, micellar core can store the insolvable drugs to
increase its solubility.
The polymeric micelles as drug carriers based on chitosan were wide-spread researched due to
the many advantages of chitosan. The linoleic acid (LA)-grafted chitosan oligosaccharide was
synthesized in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC),The
results showed that these chitosan derivatives were able to self assemble and form spherical
shape polymeric micelles with the size range of 150.7–213.9nm and the zeta potential range
of 57.9–79.9 mV, depending on molecular weight of CSO and the charged amount of LA.
Using doxorubicin (DOX) as a model drug, The drug encapsulation efficiencies (EE) of
DOX-loaded CSO-LA micelles were as high as about 75%, and the drug loading (DL)
capacity could reach up to 15%.
Liu L. [19] have prepared chitosan-based copolymers with binary grafts of hydrophobic
polycaprolactone and hydrophilic poly(ethylene glycol) (CS-g-PCL&PEG) by a
homogeneous coupling reaction of phthaloyl- protected chitosan with functional PCL-COOH
and PEG-COOH, following deprotection to regenerate free amino groups back to chitosan
backbone. They were characterized by 1H NMR, Fourier transform infrared and X-ray
diffraction analysis. These CS-g-PCL&PEG copolymers could form nano-size self-aggregates
using dynamic light scattering and transmission electron microscopy. The formed
self-aggregates become smaller with weakened stability upon pH increasing. Moreover, the
aggregates of copolymer with higher content of PEG and PCL grafts could remain stable for
over 30 days in both acid and neutral condition. A possible mechanism was proposed for the
formation of self-aggregates from CS-g-PCL&PEG and their structural changes as pH. It is
warranted to find promising application of these self-aggregates based on chitosan as drug
carriers.
In our laboratory, a new type of chitosan hollow structure, i.e., carboxymethyl-hexanoyl
chitosan (CHC), which was modified first by hydrophilic carboxymethylation to increase the
flexibility of chitosan molecular chains in water[33] followed by hydrophobic modification
with hexanoyl groups to add amphiphilic character, was employed to study its
self-aggregation behavior to form nanocapsule in aqueous solution and nanostructural
evolution. The stability of nancapsules and formation mechanism of the CHC
macromolecules were explored through the use of critical aggregation concentration (cac),
zeta potential, electron microscopy, and dynamic light scatter (DLS). By taking the advantage
of self-aggregation nature, the CHC was employed to encapsulate doxorubicin (DOX), an
anticancer agent of broad spectrum with reasonable therapeutic index and intriguing
biological and physicochemical actions,[41] to further understand its loading efficiency and
1-2-2 Inorganic-Organic Hybrid System as Drug Carriers
The development of advanced materials from the combination of macromolecules with
inorganic species has become one of the most innovative research fields. Following this
approach, the synthesized hybrid materials allow the tailoring of properties from the atomic to
the mesoscopic and macroscopic length scales. This ability to control materials’ properties
over broad length scales suggests that research on hybrids can have a significant impact in
diverse fields, such as nanoelectronics, separation techniques, catalysis, smart coatings,
sensors, immobilization of enzymes, biomedical and polymer composite applications. The
organic-inorganic hybrid materials diverse into many forms, such as composite film, gel,
particle.
To obtain functional hollow polymer spheres, various methods have been reported, which are categorized as self-assembled formation, template approaches, and complex approaches, such as emulsion polymerization, where use of a soft template coexists with self-assembled layer formation. Based on these synthetic approaches, hybrid types of capsules have also been extensively developed. The major routes for fabrication are inorganic layer formation on hard and soft templates or starting with an inorganic particle template on which to form the organic layer. Yan E. [12] have fabricated an Hybrid hollow nanospheres of chitosan–silica templated
by chitosan–poly(acrylic acid) nanoparticles in a complete aqueous system, which provide a
The hybrid hollow nanospheres show high surface area and pore volume. DOX can be
efficaciously incorporated inside the hybrid nanospheres and released slowly at physiological
condition but released more rapidly in suitable intracellular environments. This material
should have a potential application in construction of site-selective, pH-sensitive drug
delivery systems.
Takahashi T. [42] has prepared a PEG–clay hybrid in aqueous media using a PEG/polyamine
block copolymer. The dispersion stability of high ionic strength was remarkably improved by
surface modification with PEG. Improvement of the stability depends on the shielding of the
negative charge of the clay nanocrystals due to the coordination between the clay surface and
amino groups in the side chain of the PAMA segment, and the nonionic and hydrophilic
features of the PEG segment. These results obtained here satisfy some of the pharmaceutical
requirements, for example, as a drug carrier for sustained release as well as for the
improvement of bioavailability. Also, the acetal group on the perifery of the nanocrystals can
easily be hydrolyzed to an aldehyde group by acid treatment. Thus, it might be utilized as a
ligand binding site. These results strongly suggest that the PEG-modified porous clay is
anticipated to be an advanced drug carrier not only for sustained release but for targeted DDS
material by modification with ligands for the target molecules or organs.
Paul G. [43] has developed organic-silica based drug carriers by sol-gel techniques for the
encapsulated by a sol-gel process in an organic-inorganic hybrid matrix by in situ
self-assembly. In this self-assembled sol-gel synthesis, the nature of guest-host interactions
and pore structures is dictated by the synthetic parameters, pH values, and composition. The
drug release increases above and below the charge matching pH regime which is centered
around the isoelectric point of silica. The alkyl/aromatic substitution in the silica gels imparts
hydrophobic character to the gel which controls the surface interactions of the drug as well as
the diffusion barriers of the matrix. Spray drying of the self-assembled sol leads to an
enhanced dissolution process. The controlled drug release achieved from the sol-gel silica
systems can be further developed and applied into the large library of newly discovered drugs.
1-2-3 Cellular Delivery of Nanoparticles
Cellular delivery involving the transfer of various drugs and bio-active molecules (peptides, proteins and DNAs, etc.) through the cell membrane into cells has attracted increasing attention because of its importance in medicine and drug delivery. This topic has been extensively reviewed. The direct delivery of drugs and biomolecules, however, is generally inefficient and suffering from problems such as enzymic degradation of DNAs. Therefore, searching for efficient and safe transport vehicles (carriers) to delivery genes or drugs into cells has been challenging yet exciting area of research [54]. In past decades, many carriers have been developed and investigated extensively which can be generally classified into four major groups: viral carriers, organic cationic compounds, recombinant protiens and inorganic
nanoparticles. Many inorganic materials, such as calcium phosphate, gold, carbon materials, silicon oxide, iron oxide and layered double hydroxide (LDH), have been studied. Inorganic nanoparticles show low toxicity and promise for controlled delivery properties, thus presenting a new alternative to viral carriers and cationic carriers. [44, 45] To aid our
understanding of the surface properties, functionalisation of nanoparticles and their
biointeractions, it is useful to conceptually develop some generic scenarios of nanoparticles
and biomolecular interactions. Figure 1.1 is a schematic representation of
surface-functionalised inorganic nanoparticles, and the processes of biomolecule uptake and
the cellular transfer pathways [46].
There are a large number of inorganic nanoparticles that can be potentially used as carriers for the cellular delivery of various drugs including genes and proteins. However, most inorganic nanoparticles have to be subjected to chemical and/or biological modification to meet the stringent requirements for cellular delivery, such as good biocompatibility, strong affinity between the carriers and biomolecules, high charge density of the hybrids, site-specificity, etc. The functionalisation or modification depends, to a large degree, on the types of nanoparticles that provide specific functional groups on the surface, as summarized in Table 1.1. For instance, silica nanoparticles are usually modified with silane species while
gold nanoparticles are modified with thiol groups. Carbon materials can have many surface functional groups, but –COOH is often used to react with NH2– to anchor the organic
modifier.
The organic molecules used to functionalise inorganic nanoparticles usually have two
feature groups: the anchoring group and the charging group, as listed in Table 1.1.as shown in
Step 1 of Figure 1.1. The former anchors itself onto the surface of nanoparticles while the
charging group binds with the biomolecules and also imparts the positive charge to the hybrid
nanoparticles. For example, N-(2- aminoethyl)-3-aminopropyl-trimethoxysilane is a suitable
modifier for silica nanoparticles [47, 48] in which methoxysilane (Si-OCH3) acts as the
anchoring group and amino group as the charging group. When amino groups are protonated
at pH = 7.4, the modified nanoparticles are positively charged and bound with the DNA chain
via electrostatic interactions. Similarly, thiol (–SH) is a good anchor on gold nanoparticle
surface [49]. In fact, the charging group, usually amino (–NH2), imino (–NH–) and/or =N–, is
readily protonated under the physiological condition (pH = 7.4) and thus positively charges
the nanoparticles. Further modification involves the grafting of some other functional groups
2. Experimental Section
2-1 Chemicals and Reagents
1. Chitosan (Mw=215000g/mol, deacetylation degree=85-90%, Aldrich-Sigma) 2. 2-propanol (Sigma Co.)
3. Sodium hydroxide (Sigma Co.) 4. chloroacetic acid (Sigma Co.) 5. hexanoyl anhydride (Sigma Co.)
6. (3-Aminopropyl)triethoxysilane) (APTES, purity>98%, Fluka)
7. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide methiodide (EDC, Sigma-Aldrich) 8. Dialysis tubing cellulose membrane (M.W.12400,avg. flat width 33 mm, Merck) 9. Fluorescein isothiocyanate (FITC, Sigma)
10. (S)-(+)-camptothecin (95%, CPT, Sigma) 11. Dimethyl sulfoxide (DMSO,Scharlan)
12. Phosphate buffered saline (PBS, UniRegion Bio-tech) 13. Dulbecco’s modified Eagle’s medium (DMEM, Gibco) 14. Ham’s F12 medium (GIBCO)
15. L-glutamine (Sigma)
16. HEPES(GIBCO, InvitrogenTM cell culture) 17. Sodium pyruvate (100Mm, Gibco)
18. Fetal bovine serum (FBS, Sigma) 19. Trypsin EDTA (Sigma)
20. Trypan blue (Sigma)
21. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT reagent) 22. Trixon (J.T.Beker)
23. Formaldehyde (Riedel de Haen)
24. Fluorescence mounting medium (Dako)
25. 4’-6-Diamidino-2-phenylindole (DAPI, Invitrogen) 26. Rhodamine-Phalloidin (Invitrogen)
2-2 Apparatus
1. Solid-State Nuclear Magnetic Resonance(Bruker DSX400WB NMR
spectrometer, Germany)
2. Fourier Transform Infrared Spectroscopy (FT-IR, Unican Mattson
Mod 7000)
3. Scanning Electron Microscopy(SEM 6500, JEOL2100, Japan)
4. Transmission Electron Microscopy(TEM, JEOL2100, Japan)
5. Dynamic Light Scattering(BI-200SM Goniometer DLS, Brookhaven
Inc., Holtsville, NY)
7. Flow cytometry(Becton Dickinson, USA)
8. Thermogravimeic Analyzer(TGA, TA Instrument Q500)
9. Fluorescence Microscopy(eclipse, TE2000-U, Nikon)
10. Confocal Laser Scanning Microscopy(D-eclipise C1, Nikon)
11. UV-vis spectrometer(Evolution 300, Thermo)
12. ELISA(DV990BV4,GDV)
13. Incubator(NUAIRE)
14. Autoclave(TOMIN)
2-3 Synthesis of Amphiphilic Silica-CHC Hybrid Molecules
2-3-1 Tetraethylorthosilicate(TEOS) as the precursor
Once the amphiphilic CHC was successfully prepared through both hydrophilic
carboxymethyl and hydrophobic hexanoyl substitutions, the resulting amphiphilic CHC was
then further chemically modified with the incorporation of silane precursor. A small amount, 1
g, of CHC was first dissolved and dispersed in 50 mL of D.I water (0.5 wt %) under
vigorously agitation at room temperature for 24 hours until the CHC was fully dissolved.
Aliquots of 4.071g of Tetraethylorthosilicate(TEOS) silane coupling agent was added into the
CHC solution, and then add HCl to adjust the pH value to pH2~4. The mixture was gently
ensure a well reaction control of the TEOS with the CHC, and prevent undesired reaction
from being contact with moisture28. After 24-h reaction, the resulting mixture was collected
by dialysis membrane with ethanol /water solution (75%v/v) for 24 hours and following with
ethanol solution for 24 hours. The resulting powder, with white appearance, was obtained
after evaporation and drying overnight in an oven at 50 .℃
2-3-2 (3-aminopropyl)triethoxysilane silane(APTES) as the Precursor
2-3-2-1 Control the molar ratio of (NH
2group of APTES)-to-(COOH group
of CHC)
As aforementioned, once the amphiphilic CHC was successfully prepared through both
hydrophilic carboxymethyl and hydrophobic hexanoyl substitutions, the resulting amphiphilic
CHC was then further chemically modified with the incorporation of silane precursor. A small
amount, 0.25 g, of CHC was first dissolved and dispersed in 50 mL of D.I water (0.5 wt %)
under vigorously agitation at room temperature for 24 hours until the CHC was fully
dissolved. Through the generally molar ratio measurements, aliquots of 160µL of
(3-aminopropyl)triethoxysilane silane coupling agent was added into the CHC solution, taken
as the molar ratio of (NH2 group of APTES)-to-(COOH group of CHC) was 1:1, and0.012g of
EDC as catalyst was also added17. The mixture was gently stirred to avoid air interference
28(incorporated as small bubbles), after 24-h reaction, the resulting mixture was collected by
ethanol solution for 24 hours. The resulting powder, with white-yellow appearance, was
obtained after evaporation and drying overnight in an oven at 50 .℃
2-3-2-2 Control the reaction time
In order to figure out how the reaction time affects the resulting hybrid molecule, the
reaction time was controlled. After APTES and EDC were added into the CHC solution, 8hr,
24 hr and 48hr of the reaction time was applied.
2-3-2-3 Control the added amount of the catalyst, EDC
To figure out the sufficient amount of the catalyst was added or not, the added amount of
EDC was controlled, the experimental process and the method were remain the same.
2-4 Symbol Number
Tetraethylorthosilicate(TEOS) as the precursor
“CHC-TOES” was used as the symbol for hybrid which synthesized through CHC and TEOS. The results were discrepancy with our expectation, therefore, no further exploration and discussion were focus on the CHC-TEOS hybrids.
(3-aminopropyl)triethoxysilane silane(APTES) as the Precursor
Symbol Number The amount of CHC (mg/mL) The amount of APTES (μL)
Calculated (COOH of modified chitosan: NH2 of APTES) C0.5A 0.5wt%(0.25g in 50 mL D.I water) 80 1: 0.5 CA 0.5wt%(0.25g in 50 mL D.I water) 160 1:1
C2A 0.5wt%(0.25g in 50 mL D.I water) 320 1:2 C5A 0.5wt%(0.25g in 50 mL D.I water) 800 1:5 C10A 0.5wt%(0.25g in 50 mL D.I water) 1600 1:10
2-5 Preparation of self-assembly silica-CHC nanocapsules
The silica-CHC sample was suspended in distilled water under gentle shaking at 25℃ for
24h, followed by ultrasonication using a probe type sonifier(Automatic Ultrasonic Processor
UH-500A,China) at 30W for 30 seconds. The sonication was repeated three times to get an
optically clear solution. To inhibit the heat buildup during sonication, the pulse function was
used(pulse on 5.0s; pulse off 1.0s).
2-6 Characterization of the silica-CHC Molecules
2-6-1 Fourier Transform Infrared Spectroscopy (FT-IR)
The FT-IR spectra of KBr pellets were recorded (32 scans with a resolution of 4cm-1) on a
Unican Mattson Mod 7000 FTIR.
2-6-2
13C- and
29Si- Solid State Nuclear Magnetic Resonance
magnetic resonance (NMR). (13C and 29Si solid-state NMR spectra were recorded at 79.49 and
100.62 MHz, on a (9.4T) Bruker DSX400 WB NMR spectrometer).
2-6-3 Thermogravimetric analysis (TGA)
The dynamic weight loss test of hybrids macromolecules were conducted on a DuPont
2050 thermogravimetric analyzer. All tests were conducted in a N2 purge gas using sample
weights of 5~7mg over a temperature range 30℃ to 600℃ at a scan rate of 10℃/min.
2-6-4 Self-assembly Behavior (CAC concentration)
Pyrene has been frequently chosen as a fluorescence probe to monitor the self-aggregation
behavior of surfactants and polymers because of its high chemical affinity to those
hydrophobic microdomains. Pyrene molecule emits only small fluorescence intensity in the
polar environments (for example: water) due to its poor solubility, but strongly emits when the
hydrophobic microdomains are evolved in an aqueous solution. In measurement, the intensity
ratio of the first peak (372 nm) and the third peak (385 nm), i.e., I372/I385, in its fluorescent
spectrum is used since this ratio has been verified to be relatively sensitive to the environment
surrounding the pyrene molecules and has been frequently used as an indicator for the stable
change of its surrounding environments. Pyrene-containingsolution (1.0×10-4 M) in methanol
was added into the test tube and evaporated under nitrogen gas to remove the solvents. Then,
solutions of hybrid nanoparticles in D.I water were added into the above test tube, bringing
pyrene in water at 22℃. The mixture were sonicated for 30 min in an ultrasonic bath and
shaken in a shaking air bath for 1 hour at ambient temperature. Pyrene emission spectra were
obtained using a fluorescence spectrophotometer (Hitachi FL-4500, Japan). The probe was
excited at 343 nm, and the emission spectra were recorded in the range of 350-500nm at an
integration time of 1.0 s. The excitation and emission slit opening were 10 and 10 nm,
respectively.
2-6-5 Morphology (Scanning Electron Microscopy, Transmission Electron
Microscopy)
The morphology of the self-assembly hybrid nanoparticles were examined by field-emission
scanning electron microscopy(FE-SEM, JAM-6500F). The microstructure observations were
performed using transmission electron microscope(TEM, JEOL2100, Japan) operated at
200keV.
2-6-6 Dynamic Light Scattering(DLS) Analysis
The mean size and size distribution of the self-assembly hybrid nanoparticles were
measured by dynamic light scattering(BI-200SM Goniometer DLS, Brookhaven Inc.,
Holtsville, NY).
2-6-7 Swelling Ratio
The swelling ratio of the self-assembly hybrid nanoparticles were primary determined by
dynamic light scatter (DLS).
2-6-8 Zeta potential
The zeta potential of the self-assembly CHC and the hybrids nanoparticles were measured
3 Characterization, Analysis and Discussion
3-1 The effect of different silane precursor on the hybrid
molecules
In order to synthesis the chitosan-silica hybrid molecule, two types of silane precursors
have been test, and the 13C-, 29Si- solid state nuclear magnetic resonance (solid-state NMR)
was used to estimate if the siloxane groups were grafted with chitosan main chain or not.
3-1-1 Tetraethylorthosilicate(TEOS) as the precursor
Herein, tetraethylorthosilicate(TEOS) was also used for the preparation of the silica-CHC
hybrid molecule, as mentioned in the experimental section, the pH value and the reaction time
were also adjust to the most suitable environment. The initial idea was that the siloxane
groups of TEOS hydrolysis under the acidic environment and then combine with the COOH
groups of the modified chitosan. However, through the analysis of 13C- and 29Si- solid state
NMR, it indicated that the siloxane groups was preferrentially hydrolyzed and condensed with
each other rather than connected with the modified chitosan, and, hence, formed bulky silica
network. From the 29Si-NMR spectra, Figure3.1, there are three characteristic peaks shown at
Q2,Q3,Q4…. ppm,which was represent Si(OSi)2(OH)2, Si(OSi)3(OH), Si(OSi)4, indicated that
the most of the siloxane groups were condensated and formed the silica network, and none of
that it’s failed to connect the inorganic silica part with the modified chitosan via the use of
tetraethylorthosilicate(TEOS) as the precursor.
3-1-2 (3-aminopropyl)triethoxysilane (APTES) as the Precursor
Qualitative Analysis
Figure 3.2a shows the 13C solid state NMR spectra of neat CHC and the CA hybrid. For the
hybrids, the linkage of the siloxane to the CHC polymer is confirmed by the appearance of
new signals at ca. 11, 23, 44 and 160-170 ppm. The peaks located at near 11, 23, and 43 ppm
are characteristic peaks of the (CH2)3 aliphatic chains and the ester group carbons bonded to
silicon atoms (-Si-CH2CH3). The peak at around 164 ppm due to C=O amide functions is
clearly evidenced in all the spectra [9,13,14]. This observation supports the incorporation of
inorganic lattice with the organic CHC macromolecule where the carboxylic acid of the CHC
is chemically coupled with the –NH2 group from the the silane. The chemical reaction of the
modified chitosan with the APTES was schematically shown in Scheme 3.1.
Figure 3.2b shows the 29Si solid state NMR spectra of the hybrid. The hybrids exhibit
characteristic signals of T1 (-48 to -50 ppm), T2 (-58 to -59 ppm), T3 ( -66 to -68 ppm) [9,15,16].
These sites are labeled using the conventional Tn notation, where n (n=1, 2, 3) is the number
of Si-bridging oxygen atoms, which represents the –Si in different environments. The spectra
respectively) indicating the presence of two main types of local structures: (SiO)2Si(CH2)3OH
and (SiO)3Si(CH2)3, respectively. The small peak at ca.-48 ppm is ascribed to T1 site, which is
translated as (SiO)Si(CH2)3(OH)2 structure. Therefore, it is clear that the silica lattice
chemically attached with the CHC chain is virtually a mixture of small amount of Si-OH and
large population of Si-O-Si groups. From the T2 and T3 structures of the 29Si-NMR spectra, it
is reasonably to conclude that the siloxane groups (Si-OH) were subsequently condensated to
form Si-O-Si network. The degree of condensation will be discussed in the next section.
The results of the 13C- and 29Si-NMR shown that through the carbodiimide coupling
reaction, the NH2 groups of APTES precursor was successfully connect with the COOH
groups of modified chitosan(CHC) and, hence, the modified chitosan-silica hybrid molecule
was formed. The further semi-quantitative analysis of the hybrid molecules will be discussed
: hydrophilic silanol group : hydrophobic hexanoyl group : main chain of chitosan
O HO HN C O CH2 OCH2 O O HO OH HNC O CH3 O O HO NH2 C O NH (CH2)3 Si OH O O CH2 C O NH (CH2)3 Si OH O O O O HO OH HN CH2 C O OH O O HO HN C O CH2 OCH2 C NH (CH2)3 Si OH O O O O O Si Si Si m Si Si Si H2N CH2 H2C Si O H2C CH3 O H2C CH3 O CH2 CH3 + EDC Self-assemble into nanoparticles O O NH C O CH2 CH2 CH2 CH2 CH3 O O OH OH O OH NH2 O O OH OH O OH OH NH C O CH2 CH2 CH2 CH2 CH3 O m NHCH2COOH NHCH2COOH CH2COOH CH2COOH Scheme3.1 -40 -60 -80 -100 -120 -140 -160
ppm
Q2 : ‐91ppm
Q3: ‐101ppm
Q4: ‐110ppm
200 180 160 140 120 100 80 60 40 20 0 11.3 22.9 44.6 83.5 164.6
ppm
CHC
CA
(a)
0 -20 -40 -60 -80 -100 -120 -140 T1:-48~-50 T2: -58~-59ppm
T3: -66~-68(b)
Figure3.2 (a)
13C solid state NMR spectra of CHC and silica-CHC(CA)3-2 The physical and chemical properties of the silica-CHC hybrid
molecule
3-2-1 Fourier Transform Infrared Spectroscopy (FT-IR)
As aforementioned in the NMR analysis, through the use of APTES silane, the silica-CHC
hybrid molecules can be successfully synthesized. The hybrid was prepared through the
carbodiimide coupling reaction, where the carboxylic groups from the carboxymethyl ligands
of the CHC is expected to chemically react with the amide group (-NH2) of the APTES
precursor. On the other hand, with the used of tetraethylorthosilicate(TEOS), the siloxane
groups were hydrolyzed and condensedn with each other rather than connected with the
modified chitosan, hence, failed to synthesize the silica-CHC hybrid molecule. The chemical
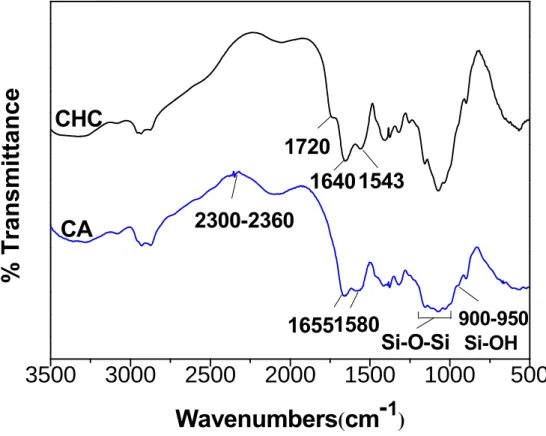
structure of the hybrid molecules was characterized by FTIR, as illustrated in Figure 3.3, The
presence of C=O stretching bands near 1720 cm-1 and a peak near 1200 cm-1 assigned to the
CO single bond. The absorption band at 2700-3300 cm-1 is referred to the stretching mode of
OH group in the CHC which became broader than that in silica-CHC (hereinafter termed as
CA) hybrid. This may be ascribed to the OH stretching of the unmodified COOH groups
along the CHC chain. After modification with the silane molecule, the absence of the peak at
extent by the silane. The displacements of the NH (1590-1560 cm-1) and CO (1652-1633cm-1)
were shifted in the CA hybrid, which may translate as a result of the presence of urethane
groups in the hybrids. Fu et al [9] reported that the regions of the urea groups vibrations, the
so-called “amide Ι” and “amide ΙΙ“, were highly complex vibrations: the “amide Ι” mode
(1800-1600 cm-1) involved the contribution of the C=O stretching, the CN stretching, and the
C-C-N deformation. The “amide ΙΙ” mode (1600-1500 cm-1) is a mixed contribution of the
in-plane N-H bending, C-N stretching, and C-C stretching vibrations. The shifts of the NH
and CO peaks in CA hybrid also indicated that the “amide Ι” mode is predominant, revealed a
chemical link between N-H and C=O groups. The presence of the peak at 2300-2360 cm-1 of
the spectrum for the CA hybrid represents the C=N stretch vibration. The peak showed up at
900-950 cm-1 in the hybrids, indicated a Si-OH stretch mode displacement, revealed that the
uncompleted polycondensation of the silanol groups. In other words, the hybrids synthesized
are not just a polymeric Si-O-Si network structure but with some unbounded Si-OH groups
exposed along the hybrid chain. In comparison, the 1000-1200 cm-1 band in the hybrids
became broader than that at the CHC spectrum which may due to the peak of Si-O-Si (stretch
3500
3000
2500
2000
1500
1000
500
%
T
ran
sm
ittan
ce
Wavenumbers
(
cm-
1)
1720
900-950 Si-OH2300-2360
1543
1640
16551580
Si-O-Si
CHC
CA
Figure 3.3. FTIR spectra of CHC and silica-CHC hybrid(CA).
3-2-2
13C- and
29Si- Solid State Nuclear Magnetic Resonance
As we have mentioned in the previous section, through the qualitative NMR analysis of the
hybrid molecules, we can specifically identify the chemical structure of the hybrid silica-CHC
molecule. In order to understand the effect of the added amount of silane precursor on the
3-2-2-1 Semi-quantitative Analysis
The semi-quantitative analysis of the hybrid macromolecule was also performed by 13C-,
29Si-nuclear magnetic resonance, where the weight of each sample was fixed 0.29 g and the
frequency was controlled at 6230 Hz. However, since the variant, such as degree of
substitution, in molecular structure of the hybrids between each sample should present,
together with the low natural abundance of 13C, 29Si nuclei, smaller gyromagnetic ratio and
long T1 relaxation time make the acquisition of quantitative spectra with adequate
signal-to-noise ratio impractical. It is thus inherently difficult to precisely perform a
quantitative analysis via current technical protocol in this work, instead, semi-quantitative
analysis was applied.
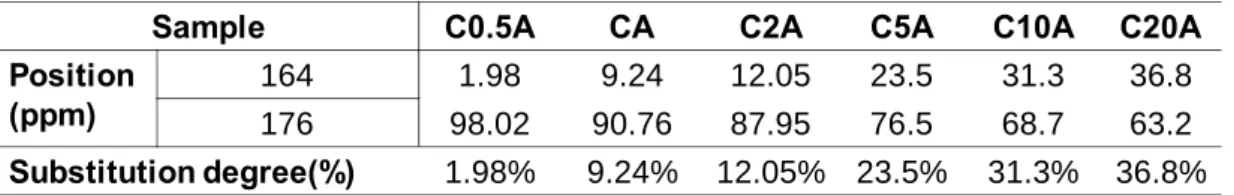
The substitution of the siloxane group to the COOH group of chitosan main chain was
calculated through the 13C-NMR spectra. Since we have mentioned that the the 13C-NMR
spectra exhibited several resonances at 160-180 ppm, associated with carbonyl groups in
different environments, the peak at 176 ppm was attributed to the COOH group of the
chitosan, which was present in the CHC 13C NMR spectra. After the siloxan groups grafted to
the COOH groups of the chitosan and formed the CONH bond, the peak at 164 ppm appeared
in the hybrid spectra. With the mole ratio of APTES: CHC increased, the growth of the peak
at 164ppm and the decline of the peak at 176ppm become more and more pronounced.
assignment and the relative population of each peak, as shown in Figure3.4.It is also assumed
that the decrease in the integral area of the COOH peak was considered to be a result of the
reaction toward CONH bond formation. Therefore, through estimation of the integral area of
the two peaks (164 ppm and 176ppm), the increment in the area at 164 ppm was estimated
and considered to be the substitution degree.
Substitution degree (%) = (the integral area of peak at 164ppm/ the integral area of peaks at
164 and 176ppm) * 100%
The Gaussian peak fitting results of each hybrid sample were illlustrated in Figure 3.4and
the estimated substitution degree was given inTable 3.1.From the 13C spectra,the increase of
silane precursor (APTES) molar ratio gives rise to a higher substitution.
From the qualitative analysis of the 29Si-NMR spectra, we have already known that the
silica lattice chemically attached with the CHC chain resulting in virtually a mixture of small
amount of Si-OH and large population of Si-O-Si groups. The degree of condensation (c)
involves the population of the distinct Si (T1, T2, T3) environments and it was calculated using
the equation,
c = 1/3(%T1+2%T2+3%T3)
Peak assignment and the relative population of the different Si sites, estimating after
in the molar ratio of silane precursor (APTES) (refer to the experimental section) caused an
increase the population of the T2 and T3 sites but reduce the population of T1 site, which is
interpretated as a result of higher degree of condensation and , hence, either a more bulky
silica lattice developed or a structurally more compacted network formed. The results
indicated that the degree of poly(siloxane) formation varies from hybrid to hybrid with
190 180 170 160 150 ppm 190 180 170 160 150 ppm 190 180 ppm170 160 150 190 180 170 160 150 ppm 190 180 170 160 150 ppm 190 180 170 160 150 ppm
C0.5A
CA
C5A
C2A
C10A
C20A
-40 -50 -60 -70 -80 ppm A Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
-40 -50 -60 -70 -80 ppm A Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
-40 -50 -60 -70 -80 ppm A Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
-40 -50 -60 -70 -80 ppm B Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
-40 -50 -60 -70 -80 ppm B Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
-40 -50 -60 -70 -80 ppm B Baseline Fit Peak 1 Fit Peak 2 Fit Peak 3 Cumulative Fit Peak
C0.5A
CA
C2A
C5A
C10A
C20A
Ι Ι Ι
Ι Ι
Ι
Ι
Ι
Ι Ι
Ι Ι
Ι Ι
Ι Ι
Ι Ι
Ι Ι Ι
Ι Ι Ι
Ι Ι Ι
Ι Ι Ι
Ι Ι Ι
Ι
Ι
Ι
Sample C0.5A CA C2A C5A C10A C20A Position (ppm) 164 1.98 9.24 12.05 23.5 31.3 36.8 176 98.02 90.76 87.95 76.5 68.7 63.2 Substitution degree(%) 1.98% 9.24% 12.05% 23.5% 31.3% 36.8%
Peak Position(ppm) Sample C0.5A CA C2A C5A C10A C20A
Ι -48 ~ -50 T1: (SiO)Si(CH2)3(OH)2 7.5% 6.6% 4.04% 3.53% 2.68% 2.2%
ΙΙ -58 ~ -59 T2: (SiO)2Si(CH2)3OH 34.1% 34.1% 31.37% 27.87% 25.77% 21.5% ΙΙΙ -66 ~ -68 T3: (SiO)3Si(CH2)3 58.4% 59.3% 64.59% 68.6% 71.55% 76.3%
Degree of condensation,c(%) 83.67 84.25 86.85 88.35 89.63 91.35
Table 3.1 The substitution degree of each hybrid sample, estimated from
13
C-NMR spectra.
Table 3.2 The condensation degree of each hybrid sample, calculated from
29
Si-NMR spectra.
3-2-2-3 Effect of the catalyst on the Hybrid synthesis
The catalyst, EDC, provided a lower chemical potential pathway to the combination of
COOH group of modified chitosan to the NH2 group of APTES precursor. However, if the
amount of the EDC was not sufficient to catalyze the reaction, the NH2 groups of APTES are
not easily to combine the COOH groups of modified chitosan, and the unreacted APTES will
hydrolyze and condensate quickly. In order to avoid such unwant reactions, different amount
amount of EDC increased 5 times and even 20 times, which was also evident that the original
amount of EDC was sufficient to catalyze the reaction.
3-2-2-2 Effect of reaction time on the hybrid synthesis
As aforementioned, the substitution degree of the siloxane group to the COOH group of
chitosan and the condensation degree of silica group were measured through the
semi-quantitative solid state NMR analysis. The substitution degree was increased with the
increased of the added amount of APTES precursor, from 1.98% to 36.8%. The condensation
degree of the silica-CHC hybrid samples all reached above 80%, indicated that a large amount
of the siloxane group were condensated and formed the bulky silica network, which might
reduce the hydrophilic force and with the bulky groups increased, might retard the
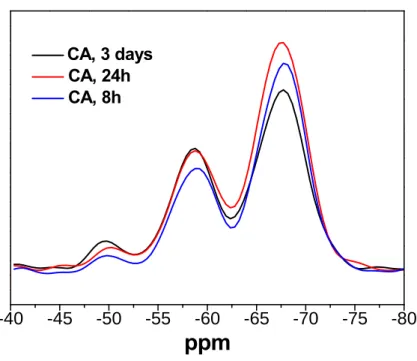
self-assembly behavior. In order to estimate if the reaction time could affect the condensation
degree of the silica-CHC hybrid or not, we chose the component of CA and control the
different reaction time, from 8h, 24h, to 48h. The result was shown in Figure 3.7, indicated
that the reaction time does not have significant effect on the condensation degree of the silica
group. With the reaction time reduced to 8 hour, the condensation degree was still keep at
around 80%, indicated that the reaction time is not the dominant key-factor on the
150 160 170 180 190 200
ppm
CA, EDC normal, 24h CA, EDC 20times, 24h
-40 -45 -50 -55 -60 -65 -70 -75 -80
ppm
CA,24h CA,EDC 5 times,24h CA,EDC20times,24h(a)
(b)
Figure3.6 (a) The solid-state 13C- NMR spectra(150~200 ppm) of CA hybrid synthesized under different amount of EDC added. (b) The solid-state 29Si- NMR spectra of CA hybrid synthesized under different amount of EDC added.
-40 -45 -50 -55 -60 -65 -70 -75 -80
ppm
CA, 3 days CA, 24h CA, 8hFigure 3.7 Solid-state 29Si-NMR spectra of the hybrids synthesized under different reaction time.
3-2-3 Thermogravimetric analysis (TGA)
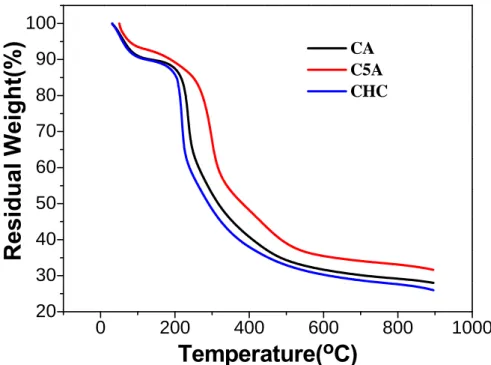
Through the thermogravimetric analysis, the ratio of the organic and inorganic can be
identified. With the modify of the siloxane group on chitosan main chain, the thermal
degradable temperature(TD) should be raised due to the introduction of the inorganic
group.The curve in Figure 3.9 was shifted to the higher temperature with the increased
silica-to-CHC ratio, indicating that the TD temperature was increased as the silica-to-CHC
ratio increased. This indicated that the inorganic components were intimately combined with
the organic groups and formed the singular hybrid macromolecules, the increased of the TD
temperature was induced by the encoring of the inorganic Si element, the needed energy to
break the
3-2-4 Self-assembly Behavior (CAC concentration)
The structural development upon dispersing the hybrid molecules into water proved the self
assembled nature of the amphiphilic hybrid [19,21], which is the same as that previously
observed in its organic counterpart [8]. To further elucidate the CAC profile of the hybrids in
water medium, pyrene was employed as a fluorescence probe, to investigate the
self-aggregation behavior of the hybrid at a molecular level. Pyrene has been frequently
chosen as a fluorescence probe to monitor the self-aggregation behavior of surfactants and
The critical aggregation concentration (CAC), which was defined as the threshold
concentration of self-aggregate formation by intra- and/or intermolecular association, can be
determined from the variation of the I372/I385 value of pyrene in the presence of polymeric
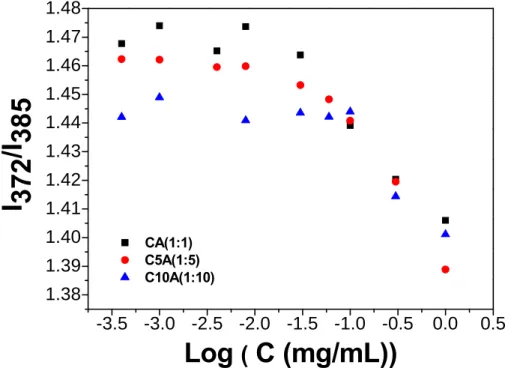
amphiphiles. Figure 3.10 illustrates the variation of I372/I385 ratio of pyrene as a function of the
concentration of the hybrids with various -COOH-to-NH2 ratios, corresponding to
CHC-to-silica ratio, ranging from 1:1 to 1:10.
The CAC value is increased with the silica/CHC ratio, and while comparing with neat
amphiphilic CHC macromolecule, higher CAC values were always detected, i.e., the lowest
CAC is 0.0186 mg/mL for 1:1 composition, compared with 0.01 mg/mL for neat CHC.
(Figure 3.11) These comparatively higher CAC values suggested that the e substitution
of –NH2 (from APTES) to the -COOH of the CHC may undergo a subsequent condensation
reaction, as evidenced in the Si-NMR spectrum, Figure 3.2b, where a bulky lattice along the
chain is expected. The OH groups, i.e., (SiO)Si(CH2)3(OH)2 may lead to more hydrated
force with water molecules, forming hydrogen bonds, which, in turn, imparts more
hydrophilic nature than the Si-O-Si network, i.e. T3 structure. However, it is clear that from
the NMR spectra aforementioned, the attached silica lattice has much less Si-OH groups than
Si-O-Si groups along the hybrid chain. This may alter the hydrophobic-hydrophilic interaction
originally evolved in the neat amphiphilic CHC macromolecule, with respect to water
CHC. Moreover, the condensed bulk groups, having a form of Si-O-Si network, may also
modify the self-assembled behavior of the hybrid macromolecules; which we assume that
more silica substitutions, the more bulky groups exist in the hybrids, resulting in higher CAC
values. This is further evidenced by the measurement of particle size using DLS data
aforementioned (Table. S1). 0 200 400 600 800 1000 20 30 40 50 60 70 80 90 100
Residual Weight(%)
Temperature(
oC)
CA C5A CHC-3.5 -3.0 -2.5 -2.0 -1.5 -1.0 -0.5 0.0 0.5 1.38 1.39 1.40 1.41 1.42 1.43 1.44 1.45 1.46 1.47 1.48 CA(1:1) C5A(1:5) C10A(1:10)
I 372
/I 385
Log
(
C (mg/mL))
Figure.3.
10
The change of I
372/I
385value of pyrene with the concentration
of various composition of silica-CHC hybrids.
0 2 4 6 8 10 12 cac*10 -2 (mg/mL )
Ratio of (COOHof CHC):(NH2 of APTES)
1:1
1:0 1:5 1:10
Figure 3.11
The critical aggregation concentration (CAC) of different
3-2-5 Morphology
Scanning Electron Microscopy (SEM)
Based on both the FTIR and NMR analyses aforementioned, the molecular structure of
resulting hybrids can be drawn schematically in Scheme 3.1. From the spectroscopic analyses,
the siloxane group was covalently bridged to the carboxylic acid of the CHC, which was
considered as a hydrophilic substitution, forming both Si-OH and Si-O-Si groups along the
CHC chain, whilst the hydrophobic hexanoyl groups along the other side of the hybrid
molecule remained intact. Accordingly, such a hydrophobic-hydrophilic nature imparts a self
assembly force when the hybrid molecule is subjecting to an aqueous or solvent medium
[19,20,21,22,27], forming a thermodynamically stable nanoarchitecture in order to minimize the
free energy of the suspension system. After dissolving in D.I water, the silica-CHC hybrid
molecules were found self assembling into nanoparticles, show a spherical appearance with an
average size around 65 nm in diameter, as illustrated in scanning electron micrograph (SEM)
image (Figure 3.12).
Transmission Electron Microscopy (TEM)
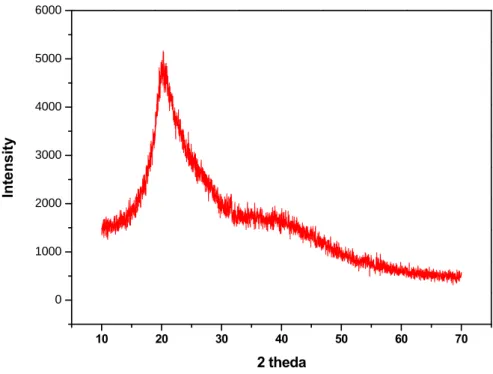
What is more than surprisingly found in this study is that the silica lattice (which has been
confirmed using X-ray diffraction analysis, where a broad silica reflection peak at around
20~30° was clearly detected (Figure 3.13), after self assembly in aqueous medium, formed a
highly-ordered nanophase of ~6 nm in width surrounding the nanoparticle (Figure 3.14a), for
A layer-like nanostructure formed the hybrid nanoparticle through self assembly, where the
highly-ordered silica lattice (Figure 3.14b), i.e., as a crystal layer, should be arisen due to self
assembly of the hybrid macromolecule, and according to the TEM examination, such an
assembly should be relatively self-organized, rather than randomly aggregated. Having
amphiphilic property, the layer-like nanoarchitecture of the hybrid, also evidenced by the
TEM examination, can be translated to be a result of the hydrophobic-hydrophilic
arrangement. Upon such an arrangement, regions of hydrophobic nature, i.e., hexanoyl groups,
are assembled and those with hydrophilic nature, i.e., silica lattice, were fused into the
layer-like configuration. This arrangement also suggests that the crystal silica nanophase is
atomically directed as a result of the hydrophobically-induced assembly of each single hybrid
macromolecule in a well-regulated manner. Such a silica nanolayer not only reinforces the
resulting nanoparticles but also renders an irreversible character of the nanoparticles from
undesirable disassembly while the concentration is below the CAC, this was evidenced from
an extreme dilution, i.e., greater than 10 times below the CAC, of the suspension prepared for
TEM observation. This finding has its clinical significance of being diluted in circulation
while being administrated for medication, which ensures retaining of the encapsulated drug
from unwanted loss as a result of significant structural swelling and rupture upon a
progressive dilution in blood vessel. Comparing with existing chitosan-based hybrid
stabilize the resulting assembled nanoobject, the hybrid nanoparticles developed in this study
do provide unique potential for biomedical application.
3-2-6 The Mean Size, Zeta Potential and Swelling Ratio
The mean size of the hybrid nanoparticles was measured by dynamic light scatter (DLS).
As aforementioned, with the increase of silica substitutions, the more bulky groups exist in
the hybrids, which may hinder the self-assemble behavior, resulting in higher CAC values and
larger nanoparticle size, shown in Table 3.3. The zeta potential of CHC and the hybrids
nanoparticles was also shown in Table3.3.
The swelling ratio was primarily estimated by comparing the size under SEM image with
the size measured by dynamic light scatter (DLS). The self-assembly silica-CHC
nanoparticles show a spherical appearance with an average size around 65 nm in diameter in
SEM images. Besides, from the dynamic light scattering (DLS) measurement (Table 3.3), the
mean size of the hybrid nanoparticles was around 133 nm in diameter, which is greater by
about 2 times in volume than that of SEM measurement. This is mainly due to swelling effect
in water medium. In comparison with neat CHC, where a swelling of nearly 8 times was
observed in an earlier study [8], indicating a considerable reduction in the swelling behavior, to
nearly 4 times, when an equivalent amount of the -NH2 groups from the APTES was
chemically modified with the –COOH groups of the CHC macromolecule. Such a silica
substitution should enhance the rigidity of the resulting hybrid entity, thus, reduce
10 20 30 40 50 60 70 0 1000 2000 3000 4000 5000 6000 Inte ns it y 2 theda
Figure 3.13 X-ray diffraction pattern of silica-CHC.
Table 3.3 The mean size and zeta potential of the different ratio of (COOH of
CHC): (NH
2of APTES) hybrids.
sample mean size Zeta potential(mV)
CHC 122 -33.47
CA 133 17.59
C2A 302 34.78
C5A 500 45.26
Figure 3.14 (a) TEM image of the silica-CHC nanoparticle (b) The
interior TEM image of silica-CHC nanoparticle, showing a layer-like
nanoarchitecture where a highly- ordered silica phase of ~6 nm in width
surrounding the nanoparticle.
Conclusions
A novel hybrid macromolecule based on a chemical modification along the –COOH groups
of amphiphilic chitosan (CHC) with (3-aminopropyl) triethoxysilane molecules was
successfully designed and synthesized. This hybrid macromolecule showed a
concentration-dependent self-assembly behavior making a final hybrid nanoparticle tunable in
size, drug encapsulation efficiency and release profile. Formation of a highly-ordered
crystalline silica layer of ~6 nm in thickness, upon self-assembly of the hybrid molecule
rendered a prolonged, sustained release of a model drug, CPT, compared with the neat CHC
molecules. Such an continuous highly-ordered silica nanoarchitecture evolved surrounding
the hybrid nanopartciles provided not only a well-stabilized nanostructure without using
crosslinker to prevent undesirable diluting disassembly, but proved to be a result of highly
self-organization rather than random arrangement of the hybrid macromolecule upon the
natural assembly operation.
References
[1] Gamys CG, Beyou E, Bourgeat-Lami E. Journal of Polymer Science: Part A: Polymer Chemistry 2010; 48: 784–93.
[2] Mu¨llner M, Schallon A, Walther A, Freitag R, Mu¨llner AHE. Biomacromolecules 2010; 11: 390–6.
[3] Qi XY, Xue C, Huang X, Huang Y, Zhou XZ, Li H, et al. Adv. Funct. Mater. 2010; 20: 43–9.
[4] Murakami K, Aoki H, Nakamura S, Nakamura SI, Takikawa M, Hanzawa M, et al.Biomaterials 2010; 31: 83-90.
[5] Wang GH, Zhang LM. J. Phys. Chem. B 2006; 110: 24864-8.
[6] Tripathi BP, Shahi VK. J. Phys. Chem. B 2008; 112 (49): 15678–90. [7] Guo XH, Zheng D, Hu N. J.Phys. Chem. B 2008; 112 (48): 15513–20. [8] Liu KH, Chen SY, Liu DM, Liu TY. Macromolecules 2008; 41: 6511-6.
[9] Fu L, Sa´ Ferreira RA, Silva NJO, Carlos LD. Chem. Mater. 2004; 16: 1507-16. [10] Bermudez VZ, Carlos LD, Alca´cer L. Chem. Mater. 1999; 11: 569-80.
[11] Mahapatro A, Johnson DM, Patel DN, Feldman MD, Ayon AA, Agrawal CM. Langmuir
2006; 22: 901-5.
[12] Yan E, Ding Y, Chen CJ, Li R, Hu Y, Jiang X. Chem. Commun. 2009; 12: 2718–20.
[13] Franville AC, Mahiou R, Zambon D, Cousseins JC. Solid State Sciences 2001; 3: 211–22.
[14] Goncüalves MC, Bermudez VZ, Sa´ Ferreira RA, Carlos LD, Ostrovskii D, Rocha J. Chem. Mater. 2004; 16: 2530-43.
Chem. 2005; 15: 3952–61.
[16] Carlos LD, Sa´ Ferreira RA, Orion I, Bermudez VZ, Rocha J. J. Lumin. 2000; 87: 702-5. [17] Lin CL, Shih CL, Chau LK. Anal. Chem. 2007; 79 : 3757-63.
[18] Yavuz MS, Cheng YY, Chen JY, Cobley CM, Zhang Q, Rycenga M, et al. Nature 2009; 8: 935-9.
[19] Liu Li, Xu Xian, Guo S, Han W. Carbohydrate Polymers 2009; 75: 401–7. [20] Zhu A, Chan-Park MB, Dai S, Li L. Colloids Surf. B 2005; 43: 143–9. [21] Wang Y, Liu L, Weng J, Zhang Q. Carbohydr. Polym. 2007; 69: 597–606. [22] Li G, Zhuang Y, Mu Q, Wang M, Fang Y. Carbohydr. Polym. 2008; 72: 60–6. [23] Wilhelm M, Zhao C, Wang Y, Xu R, Winnik MA, Mura J. Macromolecules
1991; 24: 1033–40.
[24] Amiji MM. Carbohydr. Polym. 1995; 26: 211–3. [25] Lee KY, Jo WH. Langmuir 1998; 14: 2329–32.
[26] Hu FQ, Liu LN, Du YZ, Yuan H. Biomaterials 2009; 30: 6955-63.
[27] Sun ZC, Bai F, Wu HM, Schmitt SK, Boye DM, Fan HY. J. Amer. Chem. Soc.
2009; 131: 13594–5.
[28] Wen J, Wilkes GL. Chem. Mater. 1996; 8: 1667-81.
[29] Ritger PL, Peppas NA. J. Controlled Release 1987; 5: 37-42. [30] Fini A, Orienti I. Am. J. Drug Delivery 2003; 1: 43–59.
[31] Lee M, Nah JW, Kwon Y, Koh JJ, Lo KS, Kim SW. Pharm. Res. 2001; 18: 427–31. [32] Muzzarelli RAA, Muzzarelli C. Adv. Polym. Sci. 2005; 186: 151–209.
[33] Zhao AJ, Yuan XB, Chang J. Polym. Bull. 2004; 4: 59–63.
[34] Chae SY, Son S, Lee M, Jang MK, Nah JW. J. Controlled Release 2005; 109:330–44. [35] Suia W, Songa G, Chenb G, Xuc G. Colloids Surf., A 2005; 256: 29–33.
[38] Sezgin Z, Yu ksel N, Baykara T, Eur. J. Pharm. Biopharm. 2006; 64: 261–8.
[39] Zuccari G, Carosio R, Fini A, Montaldo PG, Orienti I. J. Control. Release 2005;103: 369–80.
[40] Liu P, Wang BC, Li J, Qiao WL. Med. Hypotheses 2008; 71: 379–81. [41] Young C, Ozols RF, Myers CE. New Engl. J. Med. 1981;30: 139–153.
[42] Takahashi T, Yamada Y, Kataoka K, Nagasaki Y. Journal of Controlled Release 2005; 107: 408-16.
[43] Paul G, Heimink J, Koller H. Chem. Mater. 2008; 20: 5083-9. [44] Azzam T, Domb AJ. Current Drug Delivery 2004; 1: 165-193.
[45] Garnett MC. Critical Review in Therapeutic Drug Carrier Systems 2004; 16: 147-207. [46] Rolland A. Harwood, Amsterdam 1999.
[47] Kneuer C, Sameti M, Bakowsky U, Schiestel T, Schirra H, Schmidt H, Lehr CS. Bioconjugate chemistry 2000; 11: 926-32.
[48] Kneuer C, Sameti M, Haltner EG, Schiestel T, Schirra H, Schmidt H, Lehr CS.International Journal of Pharmaceutical 2000; 196: 257-261.
[49] Sanhu KK, McIntosh CM, Simard JM, Smith SW, Rotello VM. Bioconjugate Chemistry
2002; 13: 3-6.
[50] Kubo T, Sugita T, Shimose S, Nitta Y, Ikuta Y, Murakami T. International Journal of Oncology 2001; 18: 121-5.
[51] Berry CC, Curtis ASG. Journal of Physics D: Applied Physics 2003; 36: R198-206. [52] Zhu SG, Xiang JJ, Li XL, Shen SR, Lu HB, Zhou J, et al. Biotechnology of Applied Biochemistry 2004; 39: 179-187.
計畫名稱:以無機分子改質雙性機丁聚糖之自組裝混成複合材料及其生醫特性之研究 量化 成果項目 實際已達成 數(被接受 或已發表) 預期總達成 數(含實際已 達成數) 本計畫實 際貢獻百 分比 單位 備 註 ( 質 化 說 明:如 數 個 計 畫 共 同 成 果、成 果 列 為 該 期 刊 之 封 面 故 事 ... 等) 期刊論文 1 1 100% 研究報告/技術報告 0 0 100% 研討會論文 0 0 100% 篇 論文著作 專書 0 0 100% 申請中件數 2 2 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 2 2 100% 博士生 0 0 100% 博士後研究員 0 0 100% 國內 參與計畫人力 (本國籍) 專任助理 0 0 100% 人次 期刊論文 0 0 100% 研究報告/技術報告 0 0 100% 研討會論文 0 0 100% 篇 論文著作 專書 0 0 100% 章/本 申請中件數 0 0 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 0 0 100% 博士生 0 0 100% 博士後研究員 0 0 100% 國外 參與計畫人力 (外國籍) 專任助理 0 0 100% 人次