行政院國家科學委員會專題研究計畫 期中進度報告
中心顆粒體之蛋白質交互作用網路--(子計畫一)以比較基
因體學探討中心顆粒體之蛋白質交互作用網絡(1/3)
期中進度報告(精簡版)
計 畫 類 別 : 整合型 計 畫 編 號 : NSC 95-2627-B-002-011- 執 行 期 間 : 95 年 08 月 01 日至 96 年 07 月 31 日 執 行 單 位 : 國立臺灣大學資訊工程學系暨研究所 計 畫 主 持 人 : 高成炎 處 理 方 式 : 期中報告不提供公開查詢中 華 民 國 96 年 05 月 28 日
行政院國家科學委員會補助專題研究計畫
□ 成 果 報 告
■期中進度報告
中心顆粒體之蛋白質交互作用網路--(子計畫一)以比較基因
體學探討中心顆粒體之蛋白質交互作用網絡(1/3)
計畫類別:□ 個別型計畫 ■ 整合型計畫
計畫編號:NSC 95 - 2627 - B - 002 - 011 -
執行期間: 95 年 08 月 01 日至 96 年 07 月 31 日
計畫主持人:高成炎教授
共同主持人:
計畫參與人員: 詹鎮熊博士
成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:國立台灣大學資訊工程學系暨研究所
中 華 民 國 96 年 5 月 31 日
英文摘要
This integrated proposal is based on an interestingly observation, namely ~50% midbody proteins, which are available through proteomic screening and by literature review, are intersected with three different lung cancer microarray signature molecules. Resolving how midbody is formed, stabilized and finally resolved to produce two distinct cells may at least, in part, shed light on the way toward cancer biology field. Since a PPI requires proper spatial and/or temporal configurations, we use the midbody proteome inventories as an example to elucidate the potential PPI network occurring during cytokinesis at the midbody. Of the 190 midbody proteins examined, 98 of them can interact with other midbody proteins by using our recently up-dated protein-protein interaction database, POINT (http://point.bioinformatics.tw/). This analysis suggests that midbody proteins do not act independently at cytokinesis but form a network that modulates the cytokinesis process. This prompts us to hypothesize that could it be such organized networks are disrupted and subsequently lead to human disease. If this network is involved in lung carcinogenesis, the next question is where should we attack this network or which molecules might participate in the process of lung carcinogenesis. We have attempted to use various methods to analyze the topology of midbody network in an attempt to identify more critical nodes (genes) to the formation and functioning of midbody. One such category is the recognition of hub proteins, which are responsible for connecting numerous midbody proteins, immediately places these hub proteins as the prioritized targets. The results have been validated by other sub-projects, and provide possible link from midbody proteins to lung cancer.
Keywords: Protein-protein interaction network, Protein-protein interaction website POINT, midbody, lung cancer, systems biology
中文摘要
這個整合型計劃是建立在有關中心顆粒體近期的研究上,那就是約有 50%的中心顆粒體蛋 白質在三個不同的肺癌基因微陣列資料庫有顯著的差異。了解中心顆粒體如何形成、穩定 及最後如何分解以讓複製的細胞分離,將可能對癌症研究領域有新的啟發。蛋白質交互作 用需要適當的時間及空間上的結構,近來,有一研究團隊以蛋白質體學方式找到了 158 個 中心顆粒體的組成蛋白質,並以確認這些蛋白質的保留功能。我們利用同樣的蛋白質體學 方法產生了一個中心顆粒體在細胞質分裂時可能形成的蛋白質交互作用網路。這個網路包 含 190 個中心顆粒體蛋白,而其中有 98 個可以在POINT (http://point.bioinformatics.tw/)這個 我們近期才更新的蛋白質交互作用模擬網站中發現有和其他中心顆粒體蛋白交互作用,這 樣的結果顯示這些中心顆粒體蛋白是藉由和彼此形成交互作用網路的方式來調控細胞質分 裂的過程。因此我們假設這個由中心顆粒體蛋白所形成的網路若一旦被破壞可能導致人疾 病的發生。如果這個網路的形成在肺癌的發生的扮演著角色,那麼我們要解決的下一個問 題是我們該從這個網路中的何處著手又或者這其中有哪些分子可能參與肺癌發生的過程。 為了找出更多在中心顆粒體的形成及功能上扮演關鍵位置的基因,我們嘗試使用各種方法 去分析這個蛋白質網路的架構型態。其中一個方法是找出所謂的中心(hub)蛋白質,意即此 蛋白質可與多個網絡蛋白質有交互作用,並以其為優先研究的標的。這些結果已經由其他 子計畫驗證過,也說明了可能存在於中心顆粒體蛋白和肺癌之間的關聯性。 關鍵詞: 蛋白質交互作用網路,蛋白質交互作用網站(POINT),中心顆粒體,肺癌,系統生 物學報告內容
(1) 前言、研究目的與文獻探討
Cytokinesis, a process by which a dividing cell splits in two, partitioning the cytoplasm into two cellular packages plays a central part in cell division. Cytokinesis is an event common to all organisms that involves the precise coordination of independent pathways. Failures in cytokinesis can cause cell death and age-related disorders or lead to a genome amplification, characteristic of many cancers. Midbody consists of a compact, dense matrix of proteins, which are indispensable for cytokinesis. Recently, a proteomic screen identifies 158 mammalian midbody proteins [1]. In addition, many midbody proteins are not in this proteome inventory. Therefore, through literature review, we have expanded the midbody proteome collections into 190. Subsequently, we have converted these handful individual proteins into protein networks by using our newly up-dated protein-protein interaction database POINT (http://point.bioinformatics.tw/). The result identifies several central proteins (or referred to as “hub” proteins), which are connecting with numerous midbody proteins. These hub proteins might govern the process of cytokinesis. Since the list of midbody proteomes is not saturate yet and their interaction networks are far from completion, we propose to use the concept of interlogs to fill in the missing gaps of midbody network followed by empirical validation and to design new software to visualize the conserved interactions as well as the hub proteins.
We, as an integrated team, have united computer science, bioinformatics, and traditional
laboratory researchers to engage this program project in multi-disciplinary team research settings. The proposed study aims to (A) decipher the connection of midbody proteomes and lung cancer by addressing could it be that such organized midbody networks are disrupted in human diseases, and (B) overcome the bottleneck of how to convert the huge protein-protein interaction datasets into a complex but well organized biological network.
(2) 研究方法、結果與討論(含結論與建議)
(A) Establishment of a protein-protein interaction dataset collector, POINT
Rationale: The management of various PPI datasets, which have diverse data formats, is a
difficult task. Creation of an infrastructure to collect and annotate the PPI datasets is an essential step to analyze the PPI network topology.
Approach and Result: New version of POINT has collected PPI datasets from the public
domain. In addition, POINT has applied the concept of interlogs to predict the potential human protein-protein interactions [2]. So far, the prediction rate of ours as well as other reports remains unsatisfactory (less than 10%) [3], raising the question to what extent can we extract protein-protein interaction data by using the concept of interlogs. This prompts us to clarify the applicability and the theoretical upper-bound of orthologs-based PPI prediction (brief description in accepted or submitted manuscript section).
(B) Illustrating midbody PPI network by combining different criteria to select the important target, “hub protein”
Rationale: The gene regulatory relationships and protein-protein interactions are the keys to
perturbation. This network construction will provide us powerful layout to target the critical molecules.
Approach and result: Based on the availability of midbody proteomes, we have developed a
computation program to convert a seemly random and an independent protein-protein interaction (PPI) datasets into biologically meaningful networks (see later in Figure 2). This tool has
apparently a wide range of applications. For example, if one wants to elucidate the relationship among hundreds or thousands of differentially expressed genes or proteins identified from
microarray or proteomics (e.g. protoarray from Invitrogen) analyses, it is almost impossible to go over the literatures to find the link among these hundreds or thousands of genes. Transformation into protein networks might be an alternative choice to resolve the long-term challenge in how to make sense of microarray data. Our datasets provide an excellent opportunity to detect direct protein-protein interactions between two given genes and to prioritize the target selections by targeting those “hub proteins”.
With midbody PPI network (3,084 PPIs, Figure 1A) at hand, we want to know that which nodes are more important in the PPI network. These 3,084 PPIs from POINT are not all available in mitosis and in the midbody. To obtain a relatively confident PPI network for midbody, the following criteria are used: PPIs among 190 midbody proteins (query-query PPIs), PPIs between proteins with similar/identical GO annotations, and PPIs conserved in multiple organisms (with interologs available). Query-query PPIs may be confident in terms of temporal and spatial constraints (at the midbody stage and within the midbody proteome). Because this PPI network will be the basis for further analysis, we performed PubMed curation of 184 query-query PPIs. As a result, we have validated 125 PPIs (Figure 1B). 198 PPIs sharing similar GO annotations
provide more reliable PPIs in the spindle and membrane (spatial configuration). The interolog PPIs (289 PPIs) gives us insights to the evolutionary conserved network of midbody among different species. Figure 1 depicts the overlaps and coverage of midbody PPI network with various constraints.
(A)
(A)
190 midbody proteins183 midbody proteins with P P Is
Q uery‐query P P Is (184 P P Is ) G O P P Is (197 P P Is ) Interologs P P Is (288 P P Is ) 3,084 P P Is 544 P P Is 700 H ubs Q uery‐als o H ubs (49 Hubs ) G O H ubs (75 Hubs ) Interologs H ubs (87 H ubs ) 161 H ubs P ubmed curation 125 P P Is
(A)
190 midbody proteins183 midbody proteins with P P Is
Q uery‐query P P Is (184 P P Is ) G O P P Is (197 P P Is ) Interologs P P Is (288 P P Is ) 3,084 P P Is 544 P P Is 700 H ubs Q uery‐als o H ubs (49 Hubs ) G O H ubs (75 Hubs ) Interologs H ubs (87 H ubs ) 161 H ubs 190 midbody proteins
183 midbody proteins with P P Is
Q uery‐query P P Is (184 P P Is ) G O P P Is (197 P P Is ) Interologs P P Is (288 P P Is ) 3,084 P P Is 544 P P Is 700 H ubs Q uery‐als o H ubs (49 Hubs ) G O H ubs (75 Hubs ) Interologs H ubs (87 H ubs ) 161 H ubs P ubmed curation 125 P P Is
(B) hrough n l 1, y Figure 1. An architectural map of the midbody through a protein-protein interaction network (A)
Midbody PPI networks.
Among 190 midbody proteins, 183 of them have at least one experimental PPI record. To sieve authentic PPIs out of 3,084 experimental PPIs of the midbody protein network, three criteria are therefore set, resulting in identification of 544 highly confident PPIs, including 184 query-query PPIs (PPIs between 121 known midbody nodes), 197 GO PPIs (PPIs between nodes that correspond to the similar cellular components in GO), and 288 interlogs PPIs (PPIs that can also be observed in organisms other than human). To classify hubs that may play a role in the midbody network from the total of 700, three criteria similar to what we set to analyze PPIs are used here: namely, query-also hubs, GO hubs and interlogs hubs. Accordingly, a total of 161 hubs emerged, including 49 query-also hubs, 75 GO hubs and 87 interlogs hubs. (B) To validate the PPI datasets, we have preformed PubMed curation. Of 211 papers analyzed, 125 PPIs,
involved in 98 midbody query proteins, are verified from 155 papers. Of particular interest is that there are 70 phosphorylated proteins (nodes in blue) within 125 PPIs.
Figure 2. Curated 125
query-query PPIs of midbody network. Among 190 midbody
proteins, 98 of them can interact with each other to form 125 PPIs, which are further verified t
PubMed curation. There are 70 phosphorylated proteins (nodes i blue) and 11 kinases (red edges) within 125 PPIs. Several nove molecules, including SEPT FLJ10540, KIAA1377, KIAA0133, and PIN1, which might be involved in the midbod network, are also indicated in red.
(4) Evaluating ranks of important nodes in the midbody PPI network
ore important in th
lity distribution
irtual Knockout Entropy
in silico knockout, and evaluate the effects of
gene
With midbody PPI network at hand, we want to know that which nodes are m
e PPI network. We use various methods to analyze the midbody PPI network, in an attempt to identify more critical nodes to the formation and functioning of midbody. These methods are listed and described below.
• Virtual knockout entropy • Sub-network degree centra • Betweenness Centrality (BC) • Closeness Centrality (CC) • Degree Centrality (DC)
V
With PPI networks, it is possible to perform
s on the network. The effects of the specified gene on midbody network and global network (human interactome) can be assessed and compared. For example, if we knockout CDC2 in midbody network, the 15 interactions associated with CDC2 in midbody network will be removed as well. Using the following formulation, the effect of the knockout can be evaluated. Given that N is the number of interactions in the midbody network, and M is that in the global network, En and Em are the entropy of changes in interactions after the knockout, and Vk is the
resulting score to rank the nodes.
,
Vk e phasis the effect of gene on the midbody network (En), and minimize the effect of the
glob
ub-network Degree Centrality Distribution
associated with a gene) used to be a critical meas
m
al network (Em). Genes ranked high by Vk are supposed to be more critical to the midbody
network than to the global network.
S
Degree centrality (number of interactions
ures on the importance of genes. Comparisons between sub-network degree centrality (interactions associated with a gene in a given sub-network, e.g. midbody network) and global network degree centrality. The bootstrap method has been applied to random sample sub-networks with the same size as the midbody network. The sampling process was repeated 10,000 times for each node, resulted in a distribution of 10,000 data points. The SDC value can be obtained with the following equation:
, where d is the degree of gene in the midbody network, μ and σ are the mean and standard deviation of the degree distribution, respectively.
The SDC score also attempts to maximize the effect of a gene in the midbody sub-network relative to the global network.
PPI Network Centrality
The centrality values of each node in the midbody PPI network can be evaluated using different definitions. Generally, the following centralities are used in network analysis.
• Degree centrality (DC): the number of degrees associated with a gene normalized by highest degree
• Closeness centrality (CC): the normalized distance of a gene to all other nodes in the network
• Betweenness centrality (BC): the degree of a node lies between two or more sub-networks Note that BC and CC must be evaluated in the connected graph. Therefore, BC and CC of a gene are evaluated in the sub-network containing this gene. The definitions of these centralities are given below.
Degree Centrality (DC)
Closeness Centrality (CC)
In a network, a node is central, if it is close (on average) to all other nodes. In following equation, is each node of closeness centrality value, and is the normalize results, where is each node of the network.
Betweenness Centrality (CC)
In a network, a node is central, if it is between many pairs of other nodes. In following
equation, is each node of betweenness centrality value, and is the normalize
results, where is each node of the network.
The 190 midbody genes are ranked using the five methods, respectively. The rankings given by the five methods differ dramatically. However, Vk and SDC rankings are highly correlated, and
BC and CC are correlated. Our results suggest that DC alone may not be the best score to rank genes in sub-networks, contrary to other network analysis approaches. However, most network analysis focus on global networks, and may not be appropriate for sub-network analysis such as midbody PPI network.
參考文獻
1. Skop, A. R., Liu, H., Yates III, J. R., Meyer, B. J., and Heald, R. (2004). Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanism. Science 305, 61-66.
2. Huang T., Tien A., Huang W., Lee Y., Peng C., Tseng H., Kao C., and Huang C. * (2004). POINT: a database for the prediction of protein-protein interactions based on the orthologous interactome. Bioinformatics 20, 3273-3276.
3. Gandhi, T.K., Zhong, J., Mathivanan, S., Karthick, L., Chandrika, K.N., Mohan, S.S., Sharma, S., Pinkert, S., Nagaraju, S., Periaswamy, B., Mishra, G., Nandakumar, K., Shen, B.,
Deshpande, N., Nayak, R., Sarker, M., Boeke, J.D., Parmigiani, G., Schultz, J., Bader, J.S. and Pandey, A. (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38, 285-293.
計畫成果自評
In the first year of this integrated proposal, sub-project 1 have contributed two submitted manuscripts. The titles, abstracts, and key findings are listed below. PIs, who participate in this integrated project, are highlighted with bold.
Accepted or Submitted Manuscripts
1. Sheng-An Lee, Cheng-hsiung Chan, Chia-Ying Yang, Cheng-Yan Kao, Kun-Mao Chao,
Jin-Mei Lai, Feng-Sheng Wang, and Chi-Ying F. Huang. The Applicability and Inference
Power of Orthologs-Based Protein-Protein Interaction Prediction (submitted)
Background: Although the human genome has been fully sequenced, only less than 10,000 gene
products have protein-protein interaction (PPI) information available. Predictions of PPI thus provide insights to the underlying mechanisms of biological pathways. Among various PPI prediction schemes, orthologs-based approaches have been widely applied, where human PPIs are inferred from those in model organisms through orthologsous relationships. However, to what extend could orthologs-based PPI predictions be applied has not been analyzed. In this work, we analyze the applicability of orthologs-based PPI prediction and provide the theoretical upper-bound of this approach.
Results: Using orthologs information for 18 eukaryotic species (including human), we expand all
pairing relationships between any two orthologs groups. With these relationships, we can predict all possible ‘interologs’, which are PPIs derived from orthologs information. The predicting power of orthologs-based PPI prediction highly relies on phylogenetic distances. Though obvious, this dependency has not been clearly described and quantified. We have found that simpler model organisms can only infer smaller fraction of human PPIs. Moreover, nearly 95% of human PPIs can, in theory, be inferred from 17 eukaryotic species, given that the PPIs of these species are completely unveiled. However, in current experimental human PPIs, only less than 10% have ‘interologs’ available. Based on current data, we have identified the applicability and the theoretical upper-bound of orthologs-based PPI prediction. Our results also provide insights to the evolution of eukaryotic protein-protein interaction networks.
Availability: Protein-protein interaction data and supplementary data can be accessed in
http://point.bioinformatics.tw/
interologs inferred from various species. One would expect to observe most of these interologs from species with high theoretical coverage (grey box). However, the PPIs of these species are incomplete. Besides these species, D. melanogaster (fruit fly) and S. cerevisiae (yeast) have rather unusually high coverage. This may be contributed by their near complete interactome data.
As shown in Figure 3, although mouse, rat, P. troglodytes (chimpanzee), C. familiaris (dog), and G. gallus (chicken) have high theoretical interologs coverage, the experimental coverage for chimpanzee, dog, and chicken are completely missing. On the contrary, the theoretical interologs for D. melanogaster (fruit fly) and yeast are low, but the experimental interologs coverage is exceptionally high. This can be easily explained by costs and techniques of PPI experiments. These results suggest that the prediction powers of orthologs-based PPI predictions are highly relied on the selection of model organisms. For human, the prediction power of a less abundant mouse model is larger than that of a yeast model with more complete interactome.
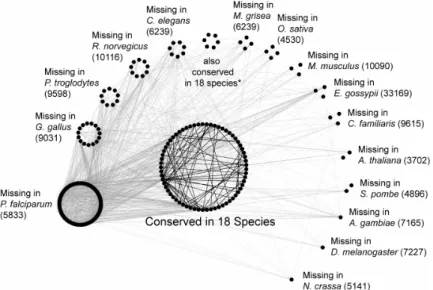
Figure 4. Interaction networks among 397 orthologs groups conserved in 17 and 18 species. The
missing genes for each species are labeled. Black lines indicate the interactions among orthologs groups shared by 18 species. There are 243 orthologs groups missing P. falciparum, suggesting that many cellular processes and functions are absent and may be compensated by parasite-host interactions. All 397 orthologs groups are available for S. cerevisiae and K. lactis. *These orthologs groups are conserved in 18 species, but currently there are no interologs among these orthologs groups.
This allows the comparisons of genomes and interactomes among P. falciparum and its hosts, human and A. gambiae (African malaria mosquito), to be carried out. The integrated genome and interactome comparison should be able to provide insights to the interplays between malaria parasite and its hosts, and shed lights to the prevention of malaria outbreaks in the third world. there are only 1189 orthologs groups shared by human and yeast, and there should be many human genes missing in yeast genome. All 398 of the highly conserved orthologs groups are available in human and yeast, and represents 33.47% of the 1189 orthologs groups shared by human and yeast. Genes missing in yeast do not appear until less conserved orthologs groups emerged. This implies that the yeast interactions among these genes can be used to infer human interologs confidently. These orthologs groups are highly conserved in these eukaryotes (except P.
falciparum). Therefore, these interologs may also be conserved as well.
In this works, we have investigated the interologs coverage of various model organisms, and provided the theoretical upper bound of each species in predicting human interologs. There is currently a large gap between theoretical limits and experimental observed interologs coverage. Our results suggest that orthologs-based approaches have the potential to cover large proportion of human interactions when the interactomes of various model organisms are complete. The PPI network constructed from currently available interologs also provides insights to the evolution of eukaryotic PPI networks, notably the interactions between malaria parasite P. falciparum and its hosts, human and African malaria mosquito. With current available PPI data, orthologs-based human PPI prediction should be benefited by complementary kernel-based methods and other machine learning approaches.
2. Sheng-An Lee, Chen-Hsiung Chan, Hsiao-Hsuan Kuo, Jin-Mei Lai, Cheng-Yan Kao,
Feng-Sheng Wang, and Chi-Ying F. Huang. POINT to the midbody: elucidation of the
protein-protein interaction network during cytokinesis. (submitted)
Motivation: Since a PPI requires proper spatial and/or temporal configurations, we use the
midbody proteome inventories, which are available through proteomic screening and by literature review, as an example to elucidate the potential PPI network occurring during cytokinesis at the midbody. Conventional network analyses postulate hub degree as an indicator for essentialness in a network of global scale. However, in a sub-proteomic network such as the midbody network, ranking the hubs solely on the centrality of hubs may not reflect the significance of certain hubs in the sub-network.
Results: Of the 180 midbody proteins examined, 90 of them can interact with other midbody
proteins, suggesting midbody proteins do not act independently at cytokinesis but form a network that modulates the cytokinesis process. Moreover, many proteins involved in cytokinesis appear to share functional homology, raising the possibility that interologs can be used to confidently assign experimental PPIs and to fill in the missing gaps in the midbody network. A survey of the midbody protein interaction network reveals that there are 476 hubs, including 61 midbody proteins, in the network and each hub connects with 2-18 midbody proteins. Instead of sorting the essentialness of hubs by the number of interactions, we implemented a z-score, a standard
statistic score, to rank the hubs based on their associations within the midbody network. This approach significantly enhances the novel target selection, especially for proteins with fewer known interacting proteins. This sub-proteome network construction not only sheds light on the intimate interactions of the midbody proteomes, but also prioritizes novel midbody hubs that may govern the process of cytokinesis.
Since the hubs with high degrees of interactions may not be suitable candidates for functional characterization, we therefore implemented a z-score analysis to re-evaluate the importance of hubs with particular interest in those that are poorly annotated proteins. The z-score, a standard statistical score, can be converted to probabilities of observing specified hub degrees in random networks. Higher z-scores mean that the associations of hubs with a particular network are statistically significant and the probabilities of forming such associations by chance are smaller.
Next we analyzed the top 29 hubs sorting by degree and compared with the top 29 hubs sorting by z-score. Figure B shows that only four of them, sorting by degree, belong to query-also-hubs (17%), whereas 13 query-also-hubs (45%) can be identified by z-score. In contrast, nine out of 29 hubs (31%), sorting by degree, do not have any information related to midbody and cytokinesis are classify into “other”. On the other hand, only two hubs (7%) sorting by z-score belong to the category “other”. Together, these analyses raise the possibility that the implementation of z-score to prioritize hubs might have a better chance to identify novel molecules participating in
cytokinesis. These findings may provide a foundation on which research can be based to study potential new cytokinesis players in the post-genomic era through a systematic bioinformatics approach.
(A) (A)
(B) Top 29 Top 44
(B) Top 29 Top 44
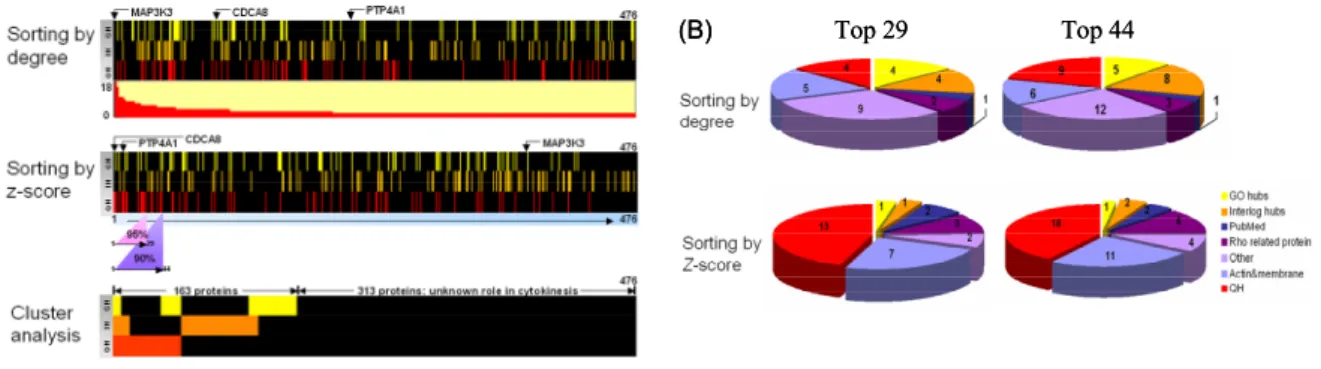
Figure 5. (A) Distributions of midbody hubs sorted by degree, z-score and cluster analysis.
(Top column) In total, 476 hubs are sorting by degree, ranging from 18 to 2. (Middle column) 476 hubs are sorting by z-score with the top 29 proteins within the 95% confidence level and 44 proteins within the 90% confidence level. (Bottom column) The 476 hubs are rearranged using cluster analysis and only 34% of them can be classified into GH for GO hubs (yellow), IH for interologs hubs (orange), and QH for query-also-hubs (red). MAP3K3, the number one ranking protein as sorted by degree, belongs to none of the three hub categories; whereas for CDCA8, the number one ranking protein as sorted by z-score is one of the query-also-hubs. Comparing the top and the middle columns reveals a great difference in protein rankings as well as distributions.
(B) Midbody hubs sorting by z-score. Midbody hubs, sorting by z-score with 1.96 and 1.645 as
the cut-off values, generate 29 (left) and 44 (right) highly confident hubs for cytokinesis. The top 29 and 44 midbody hubs, either sorting by degree or z-score, are divided into several categories. (Bottom) sorting by z-score: of these 29 hubs, there are 13 QH (45%, labeled in red). Seven of them are actin and membrane associated proteins (24%, watchet blue). The rest of them include three Rho related proteins (10%, purple), two PubMed reported (7%) (blue), one GO hub (yellow), and one interologs hub (orange). (Top) sorting by degree: of the top 29 hubs, there are four query-also-hubs (QH, 17%) (red), and nine proteins do not have any characteristics related to the midbody and/or cytokinesis.