國
立
交
通
大
學
應用化學系分子科學碩士班
碩
士
論
文
利用步進式掃描傅式轉換紅外光譜法
研究氣態 CH
3OO 的紅外吸收光譜
Infrared absorption spectra of gaseous CH
3OO detected with
step-scan Fourier-transform infrared spectroscopy
研 究 生:徐國翔 (Kuo-Hsiang Hsu)
指導教授:李遠鵬 博士 (Dr. Yuan-Pern Lee)
d 利用步進式掃描傅式轉換紅外光譜法
研究氣態 CH
3OO 的紅外吸收光譜
Infrared absorption spectra of gaseous CH
3OO detected with
step-scan Fourier-transform infrared spectroscopy
研 究 生:徐國翔 Student:Kuo-Hsiang Hsu 指導教授:李遠鵬 Advisor:Yuan-Pern Lee
國 立 交 通 大 學
應 用 化 學 系 分 子 科 學 碩 士 班 碩 士 論 文
A Thesis Submitted to M. S. Program in Molecular Science, Department of Applied Chemistry
College of Science National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in
Applied Chemistry July 2013
Hsinchu, Taiwan, Republic of China
摘要 本實驗利用波長 193 nm 的雷射光解流動的 CH3C(O)CH3/O2混和氣體及 波長 248 nm 的雷射光解流動的 CH3I/O2混和氣體,並利用步進式掃描時域 解析傅式轉換紅外光譜法搭配多重反射之 White cell 偵測共同之瞬態產物 CH3OO 之紅外吸收光譜。吾人觀測到於 3023.3 cm− 1 、2954.3 cm−1、1456.7 cm −1、1182.4 cm−1、1118.0 cm−1、910.7 cm−1、3021.4 cm−1和 1440.9 cm−1之吸收 峰依序指派為1、2、3、5、6、7、9和10之振動模吸收,與黃登瑞 等人所觀測於氣態下之 CH3OO 紅外光譜所得之最大差異不超過 0.25 %。 此結果亦和 Nandi 等人於 Ar 間質環境下得到之結果平均差異小於 1 %和 Morrison 等人於 He 奈米液滴環境下之觀測1、2、9高解析光譜之差異最 大不超過 0.1 %。與 B3LYP/aug-cc-pVTZ 非簡諧計算值平均差異於 3 %以內。 吾人以近似陀螺對稱分子之模式分析 CH3OO 的轉動譜線結構而得振動基 態之轉動常數,與 Endo 實驗組以微波測量之結果差異為 5 %。此外,吾人 根據 Just 等人計算 CH3OO 之 C–O 單鍵內轉能障,並以 PGOPHER 軟體模
擬2振動模與12振動模之熱譜帶躍遷。2振動模模擬光譜之轉動譜線與實
驗值吻合,但對於2振動模 Q 分枝結果並不一致。透過內轉動振動模之熱
譜帶躍遷模擬後,吾人認為內轉動對於 CH3OO 振動模紅外吸收光譜之貢獻
Abstract
Methylperoxy (CH3OO), the simplest alkylperoxy radical, is an important intermediate in the oxidation of methane both in the atmosphere1,2 and under combustion conditions3. In this work, CH3OO radical were produced by
irradiation of a flowing mixture of CH3I and O2 with KrF excimer laser at 248 nm. A step-scan time-resolved Fourier-transform spectrometer coupled with a multipath White cell was employed to record temporally resolved IR absorption spectra of reaction intermediate. Previously4, transient absorption bands with origins at 3032.3, 2954.3, 1456.7, 1182.6, 1118.1, 3021.4, and 1440.9 cm-1 are assigned ν1-ν3, ν5-ν6, ν9 and ν10 modes of CH3OO, respectively. Recently, ν7 band is observed with origin at 910.7 cm-1. Besides, higher resolution spectra are obtained by irradiation of a flowing mixture of CH3C(O)CH3 and O2 with ArF excimer laser at 193 nm, so that rotational constants are available by using near prolate approximation model. The rotational contours of IR spectra of CH3OO, simulated based on ratios of predicted rotational parameters for the upper and lower states and on experimental rotational parameters of the ground state, agree satisfactorily with experimental results; the mixing ratios of a-, b-, and c-types of rotational structures were evaluated based on the direction of dipole
derivatives predicted quantum chemically. Since the contribution of torsional splitting is non-negligible, we apply hot band transiton to simulate ν2 band. Though the result does not perfectly match, Q branch of ν2 band improves quite a lot.
1 G. Salisbury, A. R. Rickard, P. S. Monks, B. J. Allan, S. Bauguitte, S. A. Penkett, N. Carslaw, A. C. Lewis, D. J.
Creasey, D. E. Heard, P. J. Jacobs, and J. D. Lee, J. Geophys. Res., [Atmos.] 106, 12669(2001), and references therein.
2 G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux, G. K. Moortgat, M. J. Pilling, A. R. Ravishankara, and T. J.
Wallington, J. Geophys. Res., [Atmos.] 106, 12157 (2001), and references therein
3
G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux, G. K. Moortgat, M. J. Pilling, A. R. Ravishankara, and T. J. Wallington, J. Geophys. Res., [Atmos.] 106, 12157 (2001), and references therein
目錄 摘要 ... i 第一章 緒論 ... 1 參考資料 ... 12 第二章 實驗原理和技術 ... 14 2.1 傅式轉換紅外光譜法 ... 14 2.2 麥克生干涉儀之基本原理與傅式轉換之關係 ... 15 2.2.1 麥克生干涉儀原理 ... 15 2.2.2 傅式轉換 ... 16 2.2.3 削足函數 ... 17 2.2.4 相位校正 ... 20 2.2.5 取樣方式 ... 22 2.3 FTIR 的優點 ... 23 2.3.1 多重波長優點 ... 23 2.3.2 高光通量優點 ... 24 2.3.3 波數準確優點 ... 24 2.3.4 高解析度優點 ... 24 2.3.5 抑制散射光 ... 25 2.4 步進式掃描時間解析傅式轉換紅外光譜法 ... 25 2.4.1 工作原理 ... 25 2.4.2 跳點取樣 ... 26 2.4.3 數據擷取原理 ... 28 參考資料 ... 40 第三章 實驗裝置、步驟與參數設定 ... 41 3.1 實驗裝置 ... 41 3.1.1 光解雷射系統 ... 41 3.1.2 步進式掃描傅式轉換紅外譜儀 ... 41
3.1.3 反應系統 ... 42 3.1.4 數據擷取與儀器時序控制系統 ... 43 3.2 實驗條件 ... 46 3.2.1 光解效率評估 ... 46 3.3 實驗前準備工作 ... 51 3.3.1 反應槽中 White cell 對正 ... 51 3.3.2 更換偵測器 ... 52 3.3.3 移動鏡穩定時間量測 ... 54 3.3.4 光解雷射出光延遲時間量測 ... 56 3.4 參數設定 ... 57 3.4.1 連續式掃描模式參數設定 ... 57 3.4.2 步進式掃描模式參數設定 ... 58 3.4.3 手動操作傅式轉換 ... 60 參考資料 ... 71 第四章 結果與討論 ... 72 4.1 理論計算 ... 72 4.2 反應途徑討論 ... 73
4.3 對稱陀螺剛體轉子(symmetric top rigid rotor)模型 ... 76
4.4 實驗光譜指派和比較 ... 78 4.4.1 9振動模的分析 ... 83 4.4.2 2振動模的分析 ... 84 4.4.3 內轉動振動模(torsional mode)分析 ... 86 4.4.4 1振動模的討論 ... 87 4.5 結論 ... 89 參考資料 ... 120
圖目錄 圖 1-1 甲烷光氧化的反應機構[5] ... 9 圖 2-2 不同光源之傳統光譜(右側)及其對應之干涉圖譜(左側)。 ... 31 圖 2-3 (a) 匣式截斷函數傅式轉換後之圖譜 f(~),其波形為 sinc 函數 ... 32 圖 2-4 干涉圖譜之取樣示意圖。實驗擷取單邊之 N 點干涉圖譜,並以零光程差點為 中心,相位校正時,左右各取 n 個點數以進行相位校正。 ... 33 圖 2-5 混疊示意圖 ... 34 圖 2-6 干涉圖譜及其對應之傳統光譜。(A)連續波長的紅外光源;(B) 氦氖雷射;(C) 連續白光光源。 ... 35 圖 2-7 氦氖雷射之干涉圖譜。圖中實心方格為零光程差點,實心圓點為零交叉點。 ... 36 圖 2-8 步進式掃描時間解析傅式轉換光譜之數據擷取示意圖 ... 37 圖 2-9 由 AC/DC 耦合擷取之信號導出時間解析吸收度差異譜(ΔAt( ))之步驟。 . 38 圖 3-1 實驗儀器架設圖 ... 62 圖 3-2 反應槽簡圖 ... 63 圖 3-3 White cell 工作示意圖 ... 64 圖 3-4 Vertex 80v 進行時間解析紅外吸收光譜儀器時序控制圖(DC) ... 65 圖 3-5 Vertex 80v 進行時間解析紅外吸收光譜儀器時序控制圖(AC) ... 66 圖 3-6 類比/數位轉換器之辨識晶片排線比較圖 ... 67 圖 3-7 連續模式下 TA 鏡之氦氖雷射訊號 ... 68 圖 3-8 移動鏡穩定時間測量圖 ... 69 圖 3-9 觸發雷射後雷射延遲時間測量 ... 70 圖 4-1 利用 B3LYP/aug-cc-pVTZ 計算 CH3OO 的最佳幾何結構。 ... 90 圖 4-2 利用 B3LYP/aug-cc-pVTZ 預測 CH3OO 的 12 個振動模。 ... 91 圖 4-3 800 – 3800 cm−1光區所測得之 CH3OO 光譜及對前驅物 CH3I 之吸收光譜 .. 93 圖 4-4 800 – 970 cm−1光區所測得之 CH3OO 光譜及對前驅物 CH3I 之吸收光譜修正 ... 94 圖 4-5 7振動模之模擬光譜 ... 95 圖 4-6 1080 – 1230 cm−1光區所測得之 CH3OO 光譜及前驅物 CH3I 之吸收光譜 .... 96 圖 4-7 5、6振動模之模擬光譜 ... 97 圖 4-8 1360 – 1520 cm−1光區所測得之 CH3OO 光譜及對前驅物 CH3I 之吸收光譜修 正 ... 98 圖 4-9 3、4、10振動模之模擬光譜 ... 99
圖 4-10 3、4振動模之模擬光譜 ... 100 圖 4-11 10振動模之模擬光譜 ... 101 圖 4-12 以 248 nm 雷射光解 CH3I/O2之光譜並於 2900 – 3100 cm−1光區觀測 CH3OO 之光譜及對前驅物 CH3I 之吸收光譜修正 ... 102 圖 4-13 以 193 nm 雷射光解 CH3C(O)CH3/O2 (~1/60)之光譜並於 2900 – 3100 cm−1光 區觀測 CH3OO 光譜及對前驅物 CH3C(O)CH3之吸收光譜修正 ... 103 圖 4-14 9振動模長柱形陀螺近似之能階指派 ... 104 圖 4-15 9振動模 對波數變化圖... 105 圖 4-16 2振動模長柱形陀螺近似之能階指派 ... 106 圖 4-17 2振動模基態轉動量子數 對波數及譜線間隔之變化圖 ... 107 圖 4-18 9振動模之模擬光譜 ... 108 圖 4-19 2振動模之模擬光譜 ... 109 圖 4-20 12振動模之內轉動能障計算圖 ... 110 圖 4-21 2及含12振動模熱譜帶之躍遷模擬光譜 ... 111 圖 4-22 1振動模之光譜指派 ... 112 圖 4-23 1振動模之模擬光譜 ... 113
表目錄 表 1-1 CH3OO 之低溫與常溫實驗與理論計算振動模譜線結果比較表 ... 11 表 2-1 常用之削足函數(apodization function)之削足效果與主峰半高寬之比較。 .. 39 表 4-1 CH3OO 振動頻率之理論計算和實驗值之比較表 ... 114 表 4-2 利用 B3LYP/aug-cc-pVTZ 計算 CH3OO 的振動激態(vi = 1)與基態(v = 0)轉動 常數之比例及激態轉動常數之修正值 ... 115 表 4-3 9振動模垂直躍遷譜線之指派、譜線間距和理論預測比較表 ... 116 表 4-4 2之 P、R 分枝譜線位置、譜線間距和理論計算比較表... 118 表 4-5 2及12振動模之熱譜帶躍遷模擬光譜參數表 ... 119
第一章 緒論 過氧化物自由基(ROO‧,其中 R 表烷基團或醯基團)是對流層 (troposphere)中碳氫化合物氧化的前期產物[1、2、3]。碳氫化合物 RH 與 OH 等具強氧化力之自由基和碰撞,經過擷氫反應後,產生的有機自由基 (R‧)會很快地與大氣中的氧氣進行三體反應而形成過氧化物自由基[1]。 (1-1) (1-2) 過氧化物自由基和 NO 反應後產生之 NO2,其受紫外光照射後產生之氧原 子會進一步與 O2反應形成臭氧(O3): (1-3) (1-4) (1-5) 由於 NO 和碳氫化合物為燃燒後之重要產物,因此反應式(1-3)至式(1-5)將 導致工業區中煙霧(smog)的形成[5],此乃都市空氣污染的元兇之一。而本 次實驗觀測之過氧甲基自由基(CH3OO)是有機過氧自由基中結構最簡單者, 為對流層和平流層(stratosphere)中甲烷消耗之重要中間體,如圖 1-1 所示 [5]。 近年來對於過氧甲基研究非常多。就理論方面,Walch[6]利用 CASSCF/ICCI 方法搭配 ANO 基底函數研究 CH3和 O2的反應位能面。他們 發現 CH3和電子激態為 Δ 之 O2反應生成 態的 CH3OO,其對稱面上末 端氧的類 2p 軌域只有填一個電子,因此碳上的氫原子可遷移到氧上,分解
出 OH 基生成 H2CO。而 CH3和電子激態為 之 O2生成 態的 CH3OO, 有兩個電子填在末端氧的類 2p 軌域,導致氫原子難以遷移到末端氧上。雖 然如此, 態的 CH3OO 仍可經位能面交叉(curve crossing)通到 態 CH3OO 的位能曲面,進而斷 O O 鍵產生 H2CO OH。Jafri 和 Phillips[7] 利用擬二級組態作用法(pseudo-second-order configuration interaction)計算出 CH3OO 的 C O 和 O O 鍵的平衡鍵長分別為 1.454 Å 和 1.355 Å,以及 CH3OO 斷 C O 鍵生成 CH3 O2的解離能為 194 kJ mol−1。另外他們還利用擬一級 組態作用法(pseudo-first-order configuration interaction)計算出電子基態 到第一激發態 的躍遷能量為 87 kJ mol−1(7267 cm 1)。他們由位能圖推測 CH3OO 的第一電子激發態 是束縛態,也指出 CH3OO 在 240 nm 附近的 UV 吸收峰是電子基態 躍遷到第二電子激發態 所造成。CH3OO
被激發至第二電子激發態後,會沿著 態的排斥位能面(repulsive potential
energy surface)斷 O O 鍵生成 CH3O O。 甲基與氧氣作用的主要途徑有三種: (1-6) (1-7) (1-8) 其中反應式(1-6)為無能障之三體反應(termolecular reaction),形成 CH3OO 後 放熱 129.3 kJ mole−1[8],其反應速率隨總壓而變。若 CH3OO 擁有大於 57 kJ mole−1之內能,則可越過能障進行反應式(1-7)生成 CH2O和OH[6]。若反應 物具有 120 kJ mole−1以上的能量則會發生反應式(1-8),生成 CH
此外 Zhu 等人[10]利用 RRKM 理論計算出在大氣 壓力下,溫度 1500 K 內, 反應式(1-6)產生 CH3OO 為 CH3 + O2反應之主要途徑。當溫度提升後,反應 式(1-7)和反應式(1-8)逐漸變得重要,且二者相互競爭。他們預測溫度於 2000 K 以內,主要進行反應式(1-7)產生 H2CO + OH,其反應速率常數約為 cm3 molecule 1 s 1,當溫度超過 2000 K 時,反應式(1-8)之產物 CH3O + O 的生成比例逐漸增加,其反應速 率 常 數 約 為 cm3 molecule 1 s 1 (適用溫度為 1000 3000 K)。此外該研究亦指出熱力學上最穩定的產物為
CHO + H2O,反應熱為−351.0 kJ mole
−1,但因為反應物需要越過的能障高達 200 kJ mol−1,故以動力學觀點而言,該反應較難進行。 在實驗方面,對 CH3OO 之光譜亦有許多研究。在 1967 年,Thomas[11] 利用脈衝輻射分解 CH3Br 產生 CH3自由基,並且於系統中通入大量氧氣。 他在 240 nm 附近發現一寬頻(broad band)、不具結構的吸收峰,認為是 CH3OO 之吸收。隨後 CH3OO 於波長 200 – 300 nm 之 UV 吸收光譜也被陸 續觀測到[12、13、14]。 CH3OO 的 UV 吸收光譜之所以沒有結構性,主要原因是電子躍遷到的 第二電子激發態 為一排斥態(repulsive state),導致無法得到關於分子 結構的訊息[15], CH3OO 躍遷至第一電子激發態 的光譜則有明顯的結 構[16-20]。Hunziker 和 Wendt[16]利用激發態的汞原子碰撞丙酮產生 CH3,
再與 O2反應使生成 CH3OO。他們用相位靈敏偵測技術偵測 CH3OO 躍遷至 第一電子激發態 的光譜,並且指認出躍遷譜帶的起始點約在 7375 cm 1。 此外,他們亦觀測到離起始點約 120 cm 1的 ν12振動模和 890 cm 1 的 ν7振動 模。Pushkarsky 等人[17]利用波長 193 nm 的雷射光解丙酮與氧氣之混合物 或以 248 nm 的雷射光解碘甲烷與氧氣之混合物,使產生之 CH3自由基與 O2反應,再以共振腔振盪衰減法得到 CH3OO 躍遷至第一電子激發態 的 光譜,他們得到電子基態和 激發態的轉動常數,並指認譜帶起始位置 為 7382.5 0.5 cm 1,其吸收截面積為 2.7 × 10-20 cm3 molecule 1。此外,他 們還得到了 CH3OO 的自身反應速率常數:k =4.9 × 10-13 cm3 molecule 1 s 1。 Blanksby 等 人 [18] 利 用 光 致 游 離 (photodetachment) 技 術 觀 測 CH3OO 和 CD3OO 的光電子光譜,決定出 CH3OO 躍遷至第一電子激發態 的譜帶 起始點為:0.914 0.005 eV (7372 40 cm 1);而 CD3OO 躍遷至第一電子激 發態 的譜帶起始點為:0.913 0.004 eV (7364 32 cm 1)。Fu 等人[19] 利 用 波 長 193 nm 的 準 分 子 雷 射 光 解 超 音 射 速 中 的 CH3C(O)CH3 或 CD3C(O)CD3與氧氣和氦氣之混合物使生成 CH3OO 或 CD3OO,先用一道近 紅外光雷射照射噴射流使 CH3OO 或 CD3OO 由電子基態躍遷到第一電子激 發態的各個振動能態,經過約 30 ns 後,再以另一道波長 118 nm 的真空紫 外光雷射使其游離成 CH3OO 或 CD3OO 。由於選用的吸收兩個光子的 CH3OO 或 CD3OO 有足夠內能從 CH3OO 裂解為 CH3 或由 CD3OO 裂解為
CD3 ,故當掃描紅外雷射波長並偵測 CH3 或 CD3 質譜訊號之改變,便可得 到 CH3OO 或 CD3OO 的 躍遷光譜。他們分別指派 CH3OO 和 CD3OO 的電子躍遷譜線原點為 7381 cm 1 和 7371 cm 1。此外,他們還指派 了 CH3OO 第一電子激發態 的ν6振動模和ν7振動模分別為 1002 cm 1、 898 cm 1,以及 CD3OO 第一電子激發態 的 ν3到ν7振動模。其後,本實 驗 室 的 鍾 昭 宇 學 長 等 人 [20] 利 用 共 振 腔 振 盪 衰 減 法 觀 測 到 許 多 新 的 電振躍遷譜線,分別指派 CH3OO 在激發態 的 ν8振動模和 ν7振動模為 378 cm 1及 887 cm 1;而對 CD3OO 而言,除了指派 348 cm 1和 824 cm 1為激發態 的ν8振動模和ν7振動模為外,還指派了 ν5振動模和 ν6 振 動 模 分 別 為 954 cm 1、 971 cm 1, 這 些 振 動 波 數 的 指 派 , 均 和 UB3LYP/aug-cc-pVTZ 之理論計算結果相近。上述各個實驗組對於 CH3OO 躍遷至第一電子激發態 的譜帶原點除了精確度不同外,結果是一致的。 在微波光區的研究中,Endo 等人[21]利用傅式轉換微波光譜法觀測到 CH3OO 之振動基態之轉動 常數為 , , 。 共有三個研究組在低溫條件下觀測到 CH3OO 的紅外光譜 [22、23、24]。 Ase 等人[22]熱解 CH3I 或 CH3NNCH3產生 CH3,CH3再與 O2/Ar (1/10)作用, 產生 CH3OO 自由基於低溫間質中。他們觀測到 CH3OO 之 ν3 ν10振動模的 吸收。Nandi[23]以二隻噴嘴交替注入由熱解 CH3NNCH3/Ar (1/100)產生之
CH3自由基和 O2/Ar (3/20)沉積於 20 K 低溫靶,CH3與 O2反應生成 CH3OO, 再以傅式轉換紅外光譜儀觀測其對偏極化紅外光之吸收光譜。他們觀測到 CH3OO 之十個振動模,其中 ν1 ν8為 a 模,ν9、ν10為 a 模,其餘之 ν11、ν12 振動模未被觀測到。此外 Ase 等人和 Nandi 等人亦偵測了 CH3OO 之同位素 13 CH3OO、CH3O 18 O18、CD3OO 之光譜,其中 Ase 等人亦有觀測 CH3 18 O16O、 CH3 16 O18O、13CH3 18 O18O、CD3 18 O18O、CD3 18 O16O、CD3 18 O18O 之光譜,得 到同位素之位移資訊。Morrison 等人[24]熱解 di-tert-butyl peroxide 得到 CH3,
CH3再和 O2反應後在 He 奈米液滴環境形成 CH3OO,利用可調變式紅外光 雷射(IR-OPO)掃描其 ν1(3034.7 cm −1 )、ν2(2955.5 cm −1 )及 ν9(3024.5 cm −1 )振動 模之紅外吸收光譜。由於此三實驗組觀測環境皆於低溫固態或液滴下,因 此無法得知轉動譜線。此外,間質環境造成的微擾使得振動譜線位置亦與 氣態光譜略有差異。在氣態 CH3OO 的研究中,本實驗室的黃登瑞等人[25] 利用步進式掃描傅式紅外吸收光譜法研究 CH3OO 於電子基態的振動模。他 利用 248 nm 的雷射光光解 CH3I 和 O2之混合物,產生 CH3OO 自由基,並 且觀測到ν1 ν6、ν9和ν10振動模。雖然其觀測到之 ν1、ν2、ν9與ν3、ν4、ν10 之振動模吸收相互重疊,仍可以藉由 SpecView 軟體模擬譜線形狀去適解得 到各自的振動波數,其結果與 B3LYP/aug-cc-pVTZ 之理論計算預測的振動 波數和相對強度大致吻合。其後,本實驗室之林震洋學長[26]使用 IR-OPO 產生紅外雷射光掃描和共振腔震盪衰減光譜法測得知ν1、ν2、ν9之高解析紅
外光譜,其振動波數分別為 3031.7 cm−1、2953.4 cm−1和 3020.7 cm−1,並求 得ν2振動模之 與 之平均值為 cm−1, 為 1.71 cm−1,其結果與利用 Endo 研究組得到振動基態之轉動常數帶入理論計算得到之激態與基態之轉 動常數比值,所計算之激態轉動常數比值差異於 5 %以內。本實驗室所觀測 到之氣態 CH3OO 之振動波數研究與 Ase 等人[22]、Nandi 等人[23]和 Mossison 等人[24]在低溫環境下觀測到的結果十分接近。吾人將上述各研究 組之結果列於表 1-1。 CH3OO 的自身反應(self reaction)是造成其濃度衰減的主要原因之一。 有下列三種可能的途徑: (1- 9) (1-10) (1-11) Lightfoot 等人[27]利用閃光光解法(flash photolysis)研究 CH3OO 在溫度 248
573 K 間的自身反應,測得此溫度區間下 CH3OO 的自身反應速率常數為 k = cm3 molecule 1 s 1;在溫度 298 K 時,k = cm3 molecule 1 s 1。反應式(1- 9)和反應式(1-10)間呈現高度競爭 的情形,而反應式(1-11)可忽略。此二反應分支比和溫度有關: k9 / (k10 + k11) = (適用溫度為 388 573 K)。在 2005 年的文獻回顧[28] 列出。CH3OO 在 298 K 時的自身反應速率常數為 k =3.6 × 10-13 cm3 molecule-1 s-1,與 Lightfoot 等人的結果一致,其亦列出於 298 K 下反應分支比約為 k9 :
k10 : k11 = 0.375 : 0.625: 0。 由於目前 CH3OO 之氣態全光區(1000 4000 cm−1)紅外光譜只有黃登瑞 研究過[25],但受限於當時之儀器訊雜比,其解析度僅只有 4 cm−1,並無法 清楚解析 CH3OO 各個振動模的轉動譜線;而本實驗室之林震洋利用可調頻 之紅外雷射搭配共振腔震盪衰減光譜法搭配測量 CH3OO 之高解析紅外光 譜,但受限於紅外雷射系統之掃瞄範圍,僅能觀測到 C-H 伸展振動模之光 區,且共振腔震盪衰減光譜法波數校正不若 FTIR 精確。因此在本實驗中, 吾人使用 248 nm 雷射光解 CH3I 和 193 nm 雷射光解丙酮產生 CH3自由基, 並與 O2反應生成 CH3OO 自由基,並利用第二代的時間解析傅式轉換紅外 吸收光譜儀,以測得較黃登瑞學長[25]所得更高解析度及更優訊雜比的 CH3OO 光譜,以得到更精確之振動波數,並且希望對其轉動譜線加以解析。
表1-1 CH3OO 之低溫與常溫實驗與理論計算振動模譜線結果比較表
mode mode description Ar matrixa Ar matrixa He nanodropleta FTIRa CRDSa B3LYP/aug-cc-pVTZa,b
ν1 CH3 stretch 3032 3034.7 3033 3031.7 3150 (7.5) ν2 CH3 symmetric stretch 2968a 2954 2955.5 2954 2953.4 3050 (14.4) ν3 CH3 symmetric deformation 1453 1448 1453 1483 (9.6) ν4 CH3 umbrella 1414 1410 1408 1442 (1.6) ν5 CH3 rock OO stretch 1183 1180 1183 1216 (9.7) ν6 CH3 rock OO stretch 1112 1109 1117 1150 (2.0) ν7 CH3 O2 stretch 902 902 912 (13) ν8 CH3 O O bend 492 492 490 (6.5) ν9 CH3 stretch 3024 3024.5 3020 3020.7 3137 (10.2) ν10 CH3 antisym. deformation 1440 1434 1441 1473 (10.3)
ν11 CH3 rock unobservedc unobservedc 1127 (0.8)
ν12 CH3 torsion 134 (0.1)
reference Ase et al.[22] Nandi et al.[23] Morrison et al.[24] Huang et al.[25] 林震洋[26] Huang et al. [25]
參考資料
[1] P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat, and F. Zabel, Atmos. Environ. 26A, 1805 (1992).
[2] S. Madronich, J. Greenberg, and S. Paulson, in Atmospheric Chemistry and Global Change, edited by G. P. Brasseur, J. J. Orlando, and G. S. Tyndall
(Oxford University Press, New York, 1999), pp. 325.
[3] G. L. Bras, in Chemical Processes in Atmospheric Oxidation (Springer, Berlin, 1997), Vol. 3, pp. 13.
[4] S. B. Bertman, J. M. Roberts, D. D. Parrish, M. P. Buhr, P. D. Goldan, W. C. Kuster, F. C. Fehsenfeld, S. A. Montzka, and H. J. Westberg, Geophys. Res. 100, 22805 (1995).
[5] J. R. Barker, Progress and Problems in Atmospheric Chemistry, World Scientific, Singapore (1995).
[6] S. P. Walch, Chem. Phys. Lett. 215, 81 (1993).
[7] J. A. Jafri and D. H. Phillips, J. Am. Chem. Soc. 112, 2586 (1990). [8] I. R. Slagle and D. Gutman, J. Am. Chem. Soc. 107, 5342 (1985).
[9] J. M. W. Chase, C. A. Davies, J. J. R. Downey, D. J. Fruirip, R. A. McDonald, and A. N. Syverud, J. Phys. Chem. Ref. Data 14, Suppl. 1 (1985).
[10] R. Zhu, C.-C. Hsu, and M. C. Lin, J. Chem. Phys. 115, 195 (2001). [11] J. K. Thomas, J. Phys. Chem. 71, 1919 (1967).
[12] D. A. Parkes, D. M. Paul, C. P. Quinn, and R. C. Robson, Chem. Phys. Lett. 23, 425 (1973).
[13] C. Anastasa, I. V. M. Smith, and D. Parkes, J. Chem. Soc., Faraday Trans. 1 74, 1693 (1978).
[14] H. Adachi and N. Basco, Int. J. Chem. Kinet. 14, 1125 (1982).
[15] E. N. Sharp, P. Rupper, and T. A. Miller, Phys. Chem. Chem. Phys. 10, 3955 (2008).
[16] J. E. Hunziker and H. R. Wendt, J. Chem. Phys. 64, 3488 (1976).
[17] M. B. Pushkarsky, S. J. Zalyubovsky, and T. A. Miller, J. Chem. Phys. 112, 10695 (2000).
[18] S. J. Blanksby, T. M. Ramond, G. E. Davico, M. R. Nimlos, S. Kato, V. M. Bierbaum, W. C. Lineberger, G. B. Ellison, and M. Okumura, J. Am. Chem. Soc. 123, 9585 (2001).
[19] H. B. Fu, Y. J. Hu, and E. R. Bernstein, J. Chem. Phys. 125, 7 (2006). [20] C.-Y. Chung, C.-W. Cheng, Y.-P. Lee, H. Y. Liao, E. N. Sharp, P. Rupper, and T. A. Miller, J. Chem. Phys. 127, 14 (2007).
[21] Private communication with Prof. Y. Endo.
[22] P. Ase, W. Bock, and A.Snelson, J. Phys. Chem. 90, 2099 (1986).
[23] S. Nandi, S. J. Blanksby, X. Zhang, M. R. Nimlos, D. C. Dayton, and G. B. Ellison, J. Phys. Chem. A 106, 7547 (2001).
[24] A. M. Morrison, J. Agarwal, H. F. Schaefer, and G. E. Douberly, J. Phys. Chem. A 116, 5299 (2012).
[25] D. -R. Huang, L. -K. Chu, and Y. -P. Lee, J. Chem. Phys. 127, 234318 (2007).
[26] 林震洋,國立交通大學碩士論文,民國一百零一年。
[27] P. D. Lightfoot, R. Lesclaux, and B. Veyret, J. Phys. Chem. 94, 700 (1990). [28] D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker, and J. Warnatz, J. Phys. Chem. Ref. Data 34, 757 (2005).
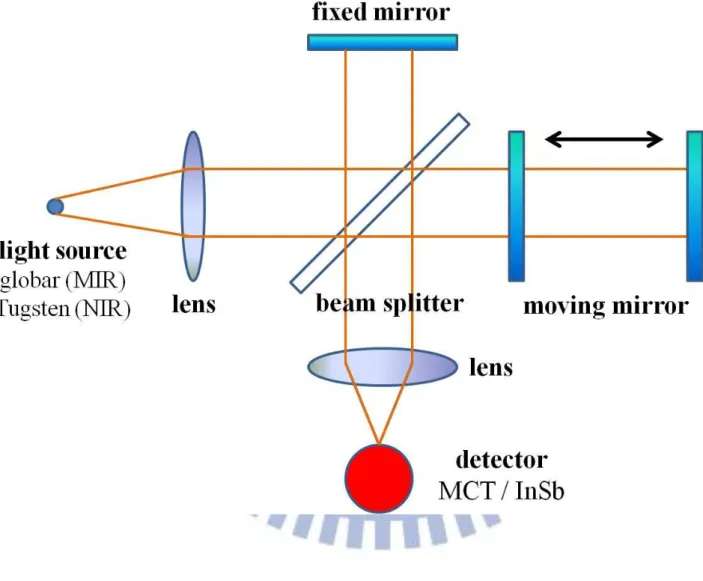
第二章 實驗原理和技術 多數分子在紅外光譜中有特徵吸收,其獨特性就如人類的指紋般,可 用於分子的鑑定;若吸收強度經校正後,則可量測其濃度。分子吸收的頻 率則可推測所具有之官能基(functional group);若用適當的解析度觀測分子, 得到其轉動能階間距,便可計算分子的轉動常數得到其結構的訊息。由於 紅外光譜法易得知分子諸多特性,因此普遍用於環境科學、大氣化學、工 業生產等領域中。 2.1 傅式轉換紅外光譜法 在早期,一般是使用分光式光譜儀(dispersive spectrometer)來量測光譜。 而在 1891 年麥克生(A. A. Michelson,1852-1931) 發明干涉儀[1],開啟了光 譜量測的新方法。干涉儀主要由分光片(beam splitter)、移動鏡(moving mirror) 及固定鏡(fixed mirror)所組成,如圖 2-2 所示。光源經拋物面鏡後,形成平 行光並導向分光片。理想情況下,分光片將入射平行光平均分成強度相同 的兩道光束,其中一道光束穿透分光鏡,經由固定鏡反射後再經由分光片 反射導向偵測器,另外一道光束則由分光片反射至移動鏡,之後再經由移 動鏡反射後穿透分光片並導向偵測器。當移動鏡沿著光軸移動時,匯集於 偵測器之兩道光束所經過的光程便會不同,造成相位差(phase difference)的 改變,因而產生干涉現象。
對於單色光而言,當光程差為半波長的偶數倍(δ = n × λ/2;n = 0,±2, ±4,…)時,兩道光束抵達偵測器時為同相位(in phase),形成建設性 (constructive)干涉,此時匯集之光強度最強;若光程差為半波長之奇數倍(δ = n ×λ/2;n = ±1,±3,…),則兩道光束抵達偵測器時為反相位(out of phase), 形成破壞性(destructive)干涉,光束強度最弱。當移動鏡以定速(v)來回移動 時,由於光程差的改變,匯集光束重複地經過建設性及破壞性干涉,而光 程差之變化為時間之函數(δ = 2vt),因此可由偵測器測得一隨時間變化之干 涉圖譜。此干涉圖譜可經由傅式轉換轉成傳統光譜。 2.2 麥克生干涉儀之基本原理與傅式轉換之關係 2.2.1 麥克生干涉儀原理 光是電磁波的一種,因此可利用電磁波的電場變化函數來表示:
0 2 ~ φ0 0 φ 0 ,t Eekr t Eekr c t r E (2-1) 其中 k 為波向量(wave vector)、r 為位置向量(position vector)、ω 為角頻率 (angular frequency)、t 為時間點(time)、0為初始時間下的相位(phase)、c 為光速、~為波數(wavenumber),而光束的強度 0
, 2 2 1 t r E c I 成正比。以一 固定波數~的單色光源為例,當其經由分光片平分成兩道光束時,各個光束 的電場變化函數就改變為
2 ~ φ0 0 2 1 ,t E ekr c t r E ,又光程為dct,故上式可 改寫為
2 ~ φ0 0 2 1 ,d E ekr d r E 。因此, 經由分光片分開最後匯集於偵測器 之光束強度為:
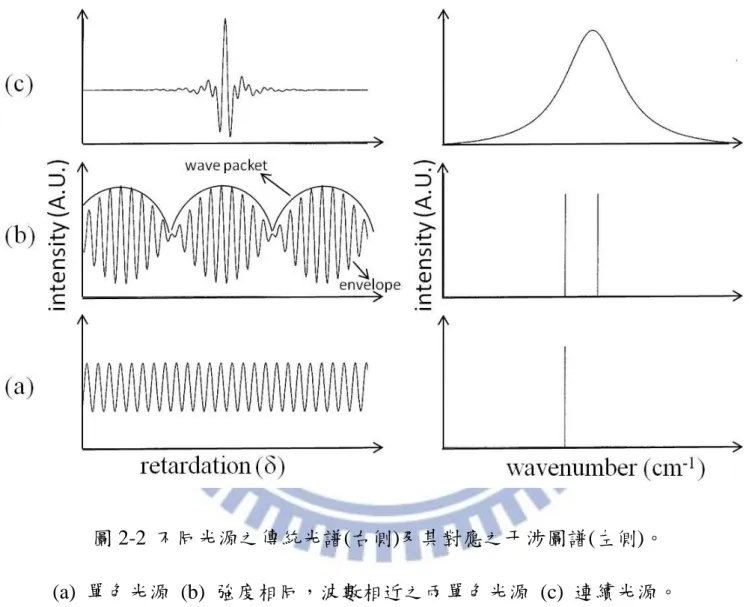
2 cos 0 2 1 0 2 1 2 cos 1 ) 2 1 ( 2 1 2 1 2 1 2 1 , , 2 1 , 2 1 2 0 0 2 φ ~ 2 0 φ ~ 2 0 0 2 2 1 0 2 0 0 2 0 1 I I E c e E e E c d r E d r E c d r E c I d r k d r k (2-2) 其中光程差δ 。即單色光的干涉圖譜為一個向上平移的餘弦函數, 其光程差變化與頻率的關係為 f c~。圖 2-3 為不同光源及其對應的干涉 圖譜,圖 2-3 (a)為單色光源的干涉圖譜,為一餘弦波;圖 2-3 (b)為兩道頻 率相近之單色光源的干涉圖譜,可視為兩個餘弦函數疊加,因其具相近的 頻率、相位發生干涉現象可觀察到波包(wave packet)與波列(envelope),而 波包與波包之間相隔不遠;圖 2-3 (c)為一連續光源之干涉圖譜,因其為多 個強度、相位、頻率不同之餘弦函數疊加導致複雜的干涉現象。在δ= 0 時, 所有頻率的光均為同相,因此可觀測到一極強的訊號,此點為零光程差點 (zero path difference);而δ漸漸變大時,各個頻率的光相互抵消,使光強度 減弱。 2.2.2 傅式轉換 利用傅式轉換方式即可將干涉圖譜轉換成傳統光譜,其數學式如下:
~ Im ~ Re ~ 2 sin ~ 2 cos ~ 2 ~ i d I i d I d e I B i
(2-3) 式(2-3)之實數部分可描述干涉圖譜經傅式餘弦轉換所得到之傳統光譜,如下表示:
I
d B
cos 2 ~ ~ (2-4) 理想之干涉圖譜應為一左右對稱圖形,因此可將式(2-4)改寫成兩倍的 0 到
積分表示:
I
d B
0 ~ 2 cos 2 ~ (2-5) 但實際上干涉譜的取樣點並非連續的,因此式(2-3)中的積分式應改成總和 式:
j x j i j x e I B ~ 2~ (2-6)其中取樣區間為x,此方法稱為離散傅式轉換(discrete Fourier transform,
DFT),此取樣方法得到的光譜會以 x 1 的週期出現,如下式:
~ ~ 2 (~ ) 2 ~ 2 2 ~ x j x j i j j j p i x j i j j x j x p i j x B e I e e I e I x p B
(2-7) 此現象稱為混疊(folding)或失真(aliasing)。 2.2.3 削足函數 上述式子均含無限大的序列或積分範圍,然而移動鏡移動的距離有限, 光程差無法達到無限大,實驗上只能得到 2 L 到 2 L 之間的干涉圖譜,其中 L 為移動鏡所能移動之最大距離。該干涉圖譜以I
表示,就如同理想之干涉圖譜I
乘上一匣式截斷函數(boxcar truncation function) ( )D ,如圖
2 0 2 1 L D L D 當 當 (2-8) 因此,偵測器所測得的光束強度隨光程差的變化函數可改寫成下式: I
I D (2-9) 即傳統光譜B
~ 為 B
I
D
d 0 ~ 2 cos 2 ~ (2-10)根據傅式分析卷積定理(convolution theorem of Fourier analysis),當 f
x 和
x g 均為可積分函數,則兩個函數之乘積的傅式轉換為此兩個函數個別傅式 轉換後之卷積(convolution),如下式所示: F
f
x g x
F
f
x F
g
x (2-11) 其中*表示卷積。卷積數學式定義則如下式所示: f
x g x
t f
x g x
d 0 (2-12) 而匣式截斷函數 ( )D 作傅式餘弦轉換後為一 sinc 函數 f
~ ,此函數稱為儀器譜線形狀函數(instrumental line shape function,ILS),其數學表示式如下: f
L
L c
L
s i n ~ ~ ~ s i n ~ (2-13) I(δ)作傅式餘弦轉換後成為B
~ ,因此,實驗所得到的真實光譜B'
~ 為理 想傳統光譜和儀器譜線形狀函數卷積的結果: B'
~ F
I
D
B~ f ~ (2-14) 對於單色光~1而言,上式可簡化為:
L c LB f B B ~ ~ sin ~ ~ ~ ~ ' 1 1 (2-15) 因此,如圖 2-3 所示,原本應為單一波數~1且無限窄頻寬的圖譜,由於移 動鏡無法移動至無限遠處而經匣式截斷函數修正,使得譜線變寬,主峰之 半高寬(full width at half maximum,FWHM)為0.605L ,此半高寬常被用來表
示傅式紅外光譜的理論解析度(theoretical resolution)。此外,干涉譜經匣式 截斷函數修正及傅式轉換後所得之傳統光譜亦會在主峰兩側產生額外的側 波;側波最大振幅值(Hs,side lobe amplitude maximum,SLAM)與主峰高度
Hm的比值為 s 21.7% m H H ,當主峰附近有其他微弱訊號則易與此側波混淆。 為了除去匣式截斷函數造成的側波干擾,可用其他函數取代匣式截斷 函數,其作用彷彿削去主峰旁的足部一樣,故稱此類函數為削足函數 (apodization function)[2]。表 2-1 列出幾種簡單的削足函數,從中可發現削 足函數雖然可以降低側波之干擾,但卻也導致主峰的頻寬增加。因此,如 果頻寬不是重要的考量,則可選擇 s m H H 值較小的削足函數;反之,若頻寬 是主要的考量因素,則可選用 FWHM 較小的削足函數。除了這些考慮因素 外,還要考量解析度與譜線密度,再選擇適合該實驗條件的削足函數。本 實驗使用的削足函數為 Blackmann-Harris 3-term 其 FWHM 為 L 16 . 1 ,而 % 04 . 0 m s H H
其儀器解析度,其主峰之半高寬為 L 9 . 0 ,而 4.5% m s H H 。此光譜儀之移動鏡 所能造成之最大光程差(optical retardation)L 為 12 cm,在未使用削足函數之 干涉譜,其實際解析度(nominal resolution) R 定義為最大光程差 L 之倒數: 0.083 12 1 1 L R cm−1 (2-16) 若使用 triangular 削足函數則可得解析度為 0.075 cm−1;除了削足函數對解 析度有影響外,光圈大小也會影響到解析度,此稱為光圈增寬 (aperture broadening)[3]。當光圈愈大則立體角愈大,此時通過光圈的離軸光(off-axis ray)會和正軸光(on-axis ray)干涉,當移動鏡移動距離愈長,光圈邊緣的光會 逐漸和光圈中心的光形成破壞性干涉,即使再增加移動鏡距離也無法增加 干涉譜的資訊,因此光圈大小和解析度之關係必須符合下式: d max max ~ 200 ~ 2 R R f (mm) (2-17) 其中 d 表示為光圈直徑、f 為光源所使用之拋物面鏡焦距(f = 10 cm)、R 為理 論解析度(未受削足函數影響)、~max為觀測光區之最大波數。此稱為自削足 現象(self-apodization)[4]。假設欲觀測之光區最大波數為 4000 cm−1,觀測解 析度 R = 1 cm−1,則光圈大小至多為 3.1 mm;若改變解析度 R = 0.15 cm−1, 則光圈大小至多為 1.2 mm。 2.2.4 相位校正 由於電子濾波器、光學元件及不當取樣等因素會造成相位差 (phase
error),影響干涉圖譜之對稱性。以電子濾波器為例,其對於不同頻率的光 會產生不同的相位延遲(
~ ,phase lag)效應,因此,必須利用相位修正 (phase correction)來修正此誤差。亦即式(2-4)必須加上 ~ 以進行相位修正, 才能描述真實之干涉圖譜:
~ ~ sin ~ 2 sin ~ cos ~ 2 cos ~ ~ ~ ~ 2 cos ~ d B d B I
(2-18) 上式中之 ~ 效應相當於原餘弦函數中引入一正弦函數成分,使原本以 0 對稱之干涉譜變得稍不對稱。如果只是以餘弦傅式轉換將會導致光譜 上的誤差,因此式(2-3)中,將干涉圖譜進行傅式餘弦及正弦轉換後即可得 相位角:
~ Re ~ Im arctan ~ (2-19) 以此資訊代入式(2-18),可進一步得到修正後之傳統光譜。 此外,不當取樣也會影響所量測之干涉圖譜。當理想的干涉圖譜對稱於 0 ,但如果第一個取樣點並非於 0,而是在 時,實際之干涉圖 譜應修正表示為[5]:
0 ~ ~ 2 cos ~ B d I (2-20) 此因素所造成的誤差也和有相位差類似。因此,無論是不當取樣或濾波等 因素所產生干涉譜的相位誤差均可以相位修正之數學步驟加以修正,以避 免光譜轉換後所得之傳統光譜發生嚴重的誤差。本實驗所使用的相位修正 方法為 Mertz method[6、7]。通常實驗為了節省時間及縮短傅式轉換運算量和時間僅擷取單邊之干 涉圖譜(single-sided interferogram),故相位校正程序相當重要。實驗時於干 涉圖譜 0左側多取 n 個數據點,得到一個含 2n 個數據點的雙邊干涉圖 譜,如圖 2-4 所示,再將對稱區域進行 FFT 轉換,以取得相位誤差資訊, 作為相位校正。如同 N 點數多寡決定光譜解析度,n 點數多寡決定隨波數 改變之相位解析度 Rθ
~ (phase resolution)。所幸相位角
~ 隨波數變化緩慢, 因此 Rθ
~ 不需和 R
~ 相若,一般而言,Rθ
~ 約為 R
~ 的八倍。 2.2.5 取樣方式 一般的傅式轉換紅外光譜儀有三組不同光源之干涉儀,三組干涉儀共 用分光鏡和移動鏡,三組光源包括具連續波長的紅外光源、氦氖雷射以及 連續白光光源,分別做為偵測樣品光譜、測量取樣之相對光程差以及定義 零光程差之用途;圖 2-6 為其干涉圖譜及傳統光譜。氦氖雷射可提供頻率 極為穩定之單色光源(波長 λvacuum= 632.9 nm),故其干涉圖譜為一餘弦函數, 如圖 2-7 所示。由於此餘弦波之頻率與移動鏡速率成正比,若移動鏡速率 稍有變動時,則餘弦波之頻率亦隨之改變。因此,電腦隨時調整取樣時間, 才能確保每一個取樣點之光程差的準確性。餘弦波每段波長有兩個零交叉 點(zero-crossing),其固定間隔為 316.5 nm,傅式轉換紅外光譜儀以氦氖雷 射干涉圖譜的零交叉點做為定位點,而不以固定時間間隔來取樣,可建立 一個固定光程差的量度法,並以之作為取樣的間隔。由於氦氖雷射只能定位移動鏡位移每段距離的相對位置,故利用白光光源之干涉圖譜的最高點 作為零光程差位置的訂定。圖 2-6 (c)所示,連續波長的白光,其干涉圖譜 在 0時,為完全建設性干涉,強度最大,而 0時其強度迅速減弱,故 其干涉圖譜為一個強而窄的訊號,以此定位取樣的起始點。然而本實驗所 使用的 Bruker FTIR 是利用步進式馬達來驅動移動鏡,可精準地定位移動鏡 的位置,因此僅使用兩組干涉儀,不需使用連續白光干涉儀。在實驗正式 擷取數據前,先利用光譜儀的紅外光源對正(align)干涉儀,並儲存干涉圖譜 的波峰之光程差位置,作為零光程差的參考基準點,以確保每次干涉圖譜 擷取訊號的起始點一致,才可在多次掃瞄光譜時可以精確地疊加。 2.3 FTIR 的優點 相較於一般分光式光譜儀,傅式轉換光譜儀具有以下的優點: 2.3.1 多重波長優點 分光式光譜儀利用光柵把多色光分散開,透過狹縫選擇單一波長的光 出射,此單一波長光強度較容易不穩,因此訊雜比較差,且一次只能量測 單一波長,掃描一張光譜十分費時;而干涉儀得以同時量測欲偵測光區中 所有波長,光強度遠大於分光式光譜儀,且雜訊經過傅式轉換後會平均於 整張光譜中,因此光譜訊雜比較佳。而除上述外,透過此優點亦能在更短 時間內擷取完整光區之光譜。因此當使用同樣光譜擷取時間及光譜解析度, 干涉儀得以較分光式光譜儀多掃描 N 次並加以平均。如果雜訊以隨機形式
出現,則可將訊雜比 S/N 提升 N1/2倍。此優點首次由 Fellgett 提出,又稱為 Fellgett 優點[8]。 2.3.2 高光通量優點 分光式光譜儀波長解析度取決於光柵的色散(dispersion)與狹縫的寬度, 為了維持波長解析度,入射光必須先經過一 0.1–1 mm 寬的狹縫,因此便造 成可利用之光強度減弱。相較之下 FTIR 是使用 1–10 mm 之圓形光圈,其 入射的光量遠大於分光式光譜儀,所以偵測器測得的訊號強度亦遠大於一 般單光儀,靈敏度因而增加,此優點於 1954 年由 Jacquinot 提出,稱為 Jacquinot 優點[9、10]。 2.3.3 波數準確優點 分光式光譜儀的波數準確性是由外部標準波長校正、光柵角度控制的 穩定性和光學對正來決定。而干涉儀是透過內部的氦氖雷射波長為基準, 其頻率精確及穩定的特性精準地決定光程差,使光譜的準確度可達 0.001 cm–1,此由 Connes 所提出,稱為 Connes 優點[11]。 2.3.4 高解析度優點 分光式光譜儀的光譜解析度受限於狹縫寬度及光柵的線性色散率倒數 (reciprocal linear dispersion of the grating),其解析度一般為 0.1 cm-1,而干 涉儀的解析度則主要取決於最大光程差 L,目前市售 FTIR 所能達到最高解 析度<0.001 cm-1。
2.3.5 抑制散射光 分光式光譜儀往往會受散射光的干擾,如果用 chopper,則只能以固定 頻率來調制,無法分出不同波長之散射光。而連續式干涉儀的移動鏡以一 個固定速率 v 移動,因此將訊號作調頻(modulation),一波數為~的單色光 經由干涉儀調頻後,偵測器測到的訊號頻率 f 為2v~,其調制頻率隨波數 而變,故以適當的電子濾波器,可進一步過濾在待測光區外之散射光訊號。 2.4 步進式掃描時間解析傅式轉換紅外光譜法 2.4.1 工作原理 一般而言,當光譜儀在連續掃描模式中,干涉儀的移動鏡是以同一速 度 v 進行,光程差 δ 亦隨時間而變,可表示成 δ = 2vt。而連續掃描模式中, 一張光譜需花數十毫秒到數秒不等,對短暫生命的物種無法偵測,也無法 做時間解析之偵測。 當干涉儀在步進式掃描模式下,移動鏡並非連續移動,而是在氦氖雷 射的零交叉點停下,並取時間解析之訊號,且可重複擷取以進行訊號平均。 接著移動鏡移動,在下一個零交叉點停止並如前一步驟記錄隨時間變化的 訊號。當移動鏡從第 xn-1個零交叉點移動至第 xn個零交叉點並於適當的穩 定時間(stabilization time)後,FTIR 會觸發內建的類比/數位轉換器 (analog-to-digital convertor,ADC),使其開始擷取於第 xn個零交叉點時間變
化的時間記錄(temporal profile),如圖 2-8(a)所示;當完成第 xn個零交叉點 的訊號擷取後,移動鏡會移動到第 xn+1個零交叉點處,進行相同步驟,直 到干涉譜的最後一點。移動鏡在氦氖雷射的第 x 個零交叉點所得到的時間 記錄,以陣列 Ix(t)表示,透過電腦內部重組成陣列 It(x),即時間 t 下之干涉 譜,如圖 2-8 (b),而再將每個時間 t 下所擷取的干涉譜經過傅式轉換得到 一組時間解析的傳統光譜 At(~),如圖 2-8 (c)。 目前,步進式掃描時間解析紅外光譜儀已經發展到相當成熟的階段。 在氣態研究中,其主要應用於放光光譜,探測因光分解或自由基反應中激 發態分子的放光,以研究反應動態學[12、13]。放光光譜法是測量零背景的 環境中之微小訊號,因此具有較高靈敏度。相較之下,吸收光譜法必須在 極大的背景訊號中擷取極微小的變化量,故實驗上較為困難。但藉由步進 式掃描時間解析紅外吸收光譜法可以獲得反應中間產物的吸收譜帶並進而 研究其反應動力學[14、15、16];如果可以得到轉動解析之光譜,尚可得知 分子的轉動常數。相較於放光光譜法無法提供的基態資訊與反應中間物的 鑑定,吸收光譜法具有相當大的應用價值。 2.4.2 跳點取樣 除前述僅擷取單邊干涉譜外,另一個節省時間的方法為跳點取樣。氦 氖雷射波長於真空中為 632.99 nm,每個零交叉點間距為 316.50 nm,假設 移動鏡在每個零交叉點取樣,則根據 Nyquist criterion[17],任何週期性訊號
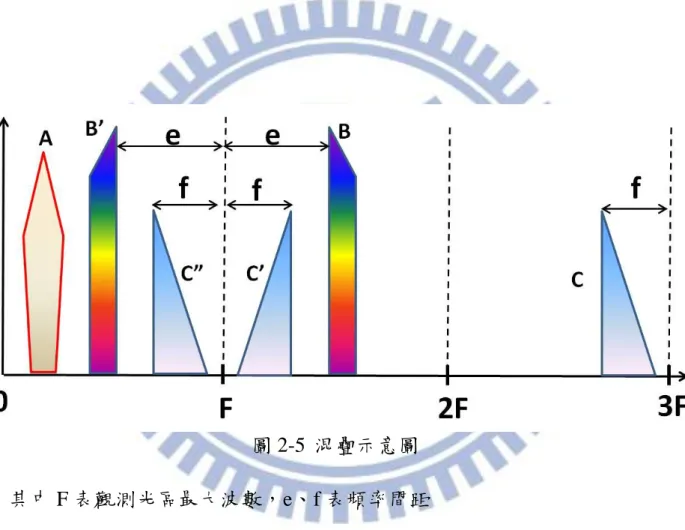
的 數 位 化 取樣 ,其 取 樣 頻 率必 須大 於 等 於 偵測 訊號 2 倍 以 上 之 頻寬 (bandwidth)才可得到正確資訊,因此如於每個零交叉點取樣,可觀測光區範 圍為 0 – 15798.01 cm-1;若每隔 2 個零交叉點取樣,則可觀測光區範圍為 0 – 7899.00 cm-1及 7899.00 – 15798.01 cm-1。跳點數和可測光區的關係可表示 為下式: ) ~ ~ ( ~ min max , He Nelaser under s N (2-21) 而離散干涉譜的點數由光區和解析度決定。假設欲觀測的光區範圍介於~max 和~min之間,且解析度為~,至少需要之總取樣點數 s N 可表為下式: ~ ) ~ ~ ( 2 max min s N (2-22) 假設解析度為 0.15 cm-1,取的光區為 0 – 1560 cm-1,跳點數為 under s N , 13 . 10 0 1560 01 . 15798 ,跳點數為 10 點,即每隔 5λHeNe取樣,表可能的觀測範圍 為 0 – 1579.8 cm−1、1579.8 – 3159.6 cm−1、3159.6 – 4739.4 cm−1。可估算不 包含相位校正(phase correction)所需之取樣點數為 21064 個點。觀測 0 – 1579.8 cm-1的光區中,如式(2-7)所示,訊號每次間隔 x 1 訊號重複。在圖 2-5 中,F 為觀測區間之最大波數為 1579.8 cm-1,若其中於 0–F、F–2F、2F–3F 區間有 A、B、C 之訊號,若不使用濾光片則於 F – 2F 區間之訊號 B 會混疊 至 0 – F 形成 、在 2F – 3F 區間之訊號 C 則會混疊至 F – 2F 如 ,而 再 混疊至 0 – F 形成 ,因此若觀測光區為 0 – F 則會觀測到實際之訊號 A、 混疊之訊號 和 、 ,故為了避免發生混疊的現象必須加濾光片以限制所
能通過的光區使與所欲偵測之光區一致。 2.4.3 數據擷取原理 本實驗系統的偵測器將訊號分成 AC 耦合與 DC 耦合[18],如圖 2-9 所 示,因其輸出之訊號通常很小,通常會先經過前置放大器(pre-amplifier)放 大後才輸入至類比/數位轉換器轉換為數位訊號,然而內建類比/數位轉換器 無法於一次量測中同時取得 AC 和 DC 耦合訊號,只得做單一頻道(single channel)訊號記錄,故必須先在第一次量測中取無雷射激發之 DC 干涉譜, 再於第二次量測中使用脈衝產生器觸發雷射以擷取 AC 干涉譜,而透過脈衝 產生器之儀器時序將於第三章 3.1.4 中詳述。DC 耦合端取得樣品在未受到 雷射激發之干涉譜 I0(δ),此干涉譜經傅式轉換後可得到樣品的背景光譜 Bt(~)與其相對的相位 θt(~)。而 DC 譜之訊號未受雷射干擾因此訊號,前者 可經由平均後降低雜訊,以 B0(~)表示,後者可用來提供 AC 耦合干涉譜作 相位校正(phase correction)之用;由 AC 耦合端得到的訊號則會反映雷射激 發後反應槽內分子對紅外光的吸收度變化,在每一特定光程差 δ 可測得一 組時間解析之 AC 訊號 ΔIδ(t),當掃描完所需的光程差後可得到一組完整的 AC 訊號資料陣列,經重組後則可得到每一特定時間 t 之干涉譜變化訊號 ΔIt(δ),ΔIt(δ)經相位修正及傅式轉換後,得到每一特定時間 t 之光譜強度變 化 量 ΔBt(~ ) ,並 依 照 下 式 計 算 AC 和 DC 差 異 吸 收 光 譜 (difference spectrum)ΔAt(~)
) ~ ( ) ~ ( 1 log ) ~ ( 0 B B A t t (2-23) 而 AC 耦合訊號通常較 DC 耦合訊號來的小,因此 AC 耦合訊號之放大倍率 會大於 DC 耦合訊號之放大倍率;假設 AC 耦合訊號放大 j 倍、DC 耦合訊 號放大 k 倍,則計算吸收度時須將式(2-23)改為: j k B B A t t ) ~ ( ) ~ ( 1 log ) ~ ( 0 (2-24) 本實驗偵測器之前置放大器放大倍率於 3.1.4 中詳述。因此,當有新生成之 產物吸收紅外光,使光譜變化量St(~)為負值,而吸收度變化量 ΔAt(~)會 呈現正值;反之當反應物消失時,使St(~)為正值,故 ΔAt(~)會呈現負值。
圖 2-2 不同光源之傳統光譜(右側)及其對應之干涉圖譜(左側)。 (a) 單色光源 (b) 強度相同,波數相近之兩單色光源 (c) 連續光源。
(a) 0. 605/ L Hm Hs 2L 1/ L (a) f( )~ (cm-1) ~ (b) 1/L 1 ~ ~1 2L 1 (cm- 1) ~ ~1 2L 1 B( )~ (b) 圖 2-3 (a) 匣式截斷函數傅式轉換後之圖譜 f(~),其波形為 sinc 函數 (b) 在移動鏡之有限位移±L 下,單色光波數
1之干涉圖譜以匣式截斷函 數進行傅式轉換後之圖譜B(~)。其中 Hm為主波強度之絕對值;Hs為測 波強度之絕對值
L c
L
f ~ 2 sin 2~
LB
c
L
B~ 2 ~1 sin 2 ~1 ~
圖 2-4 干涉圖譜之取樣示意圖。實驗擷取單邊之 N 點干涉圖譜,並以零光程 差點為中心,相位校正時,左右各取 n 個點數以進行相位校正。
n
n
N points full precision
interferogram
2n points double-sided interferogram
ZPD
圖 2-5 混疊示意圖 其中 F 表觀測光區最大波數,e、f 表頻率間距
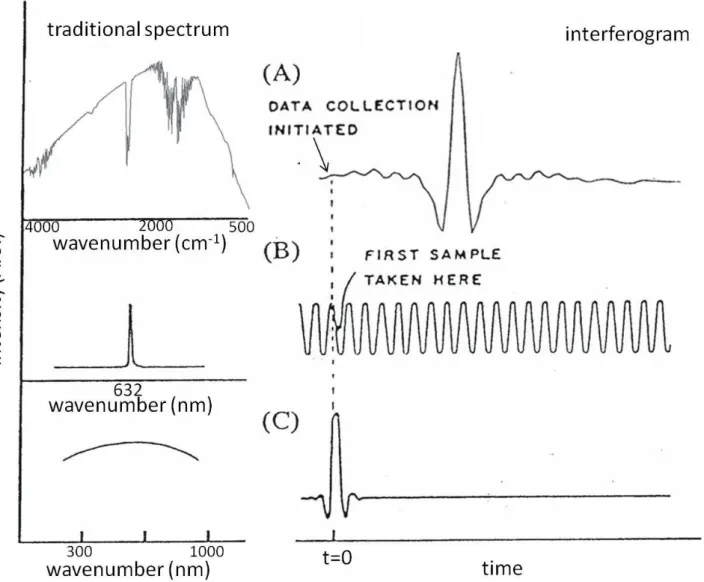
圖 2-6 干涉圖譜及其對應之傳統光譜。(A)連續波長的紅外光源;(B) 氦氖雷 射;(C) 連續白光光源。
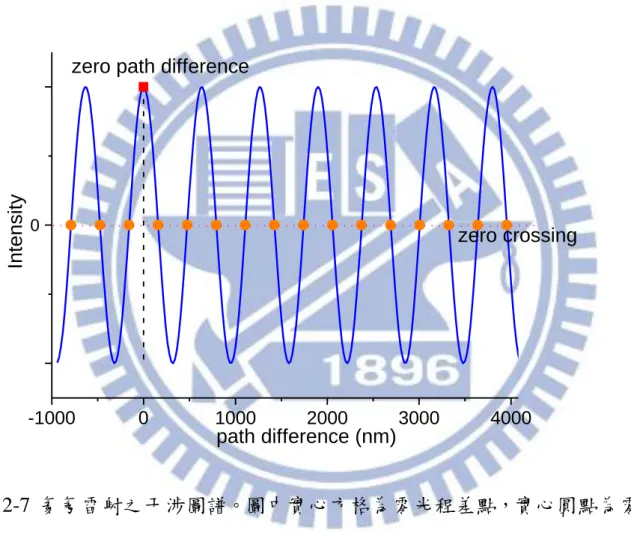
圖 2-7 氦氖雷射之干涉圖譜。圖中實心方格為零光程差點,實心圓點為零交叉 點。 -1000 0 1000 2000 3000 4000 0 path difference (nm) Int ens it y
zero path difference
圖 2-8 步進式掃描時間解析傅式轉換光譜之數據擷取示意圖 (a)各曲線為光程差為 xn時所得之時間解析信號; (b)數據重組後,各曲線表示 tm時間下的干涉譜; (c)經 FT 後所得之時間解析光譜。 -0.02 0.00 0.02 0.04 1240 1260 1280 1300 0 10 20 30 40 50 60 Ab sorbance Wave nu mber / cm -1 Time
(c)
圖 2-9 由 AC/DC 耦合擷取之信號導出時間解析吸收度差異譜(ΔAt( )) 之步驟。
表2-1 常用之削足函數(apodization function)之削足效果與主峰半高寬之比較。
Function Formula FWHM (%)c SLAM (%)d Ref.
Boxcar 1 60 2 Triangular (Bartlett) 1 - De 89 4.5 [19] N-Ba weak 0.548 - 0.0833(1-D 2) + 0.5353(1-D2)2 72 5.8 [20] N-B medium 0.261 – 0.154838(1-D2) + 0.894838(1-D2)2 84 1.4 [20] N-B strong 0.09 + 0.5875(1-D2)2 + 0.3225(1-D2)3 97 0.3 [20] Hamming (Happ-Genzel) 0.54 + 0.46cos(πD) 91 0.71 [19]
B-Hb 3-term 0.42323 + 0.49755cos(πD) + 0.07922cos(2πD) 116 0.04 [19] B-H 4-term 0.35875 + 0.48829cos(πD) + 0.14128cos(2πD) +
0.01168cos(3πD)
[19]
a表示 Norton-Beer,b表示 Blackman-Harris,c表示吸收峰的半高寬比(單色光主峰之半高寬與所要求削足解析度之比值)
參考資料
[1] A. A. Michelson, Phil. Mag., Ser. 5, 31, 256 (1891).
[2] C. A. Carere, W. S. Neil, and J. J. Sloan, Appl. Opt. 35, 2857 (1996).
[3] J. Kauppinen and J. Partanen, Fourier transforms in spectroscopy, 1st edition, Wiley-VCH (2001).
[4] E. G. Codding and G. Horlick, Appl. Spectrosc. 27, 85 (1973).
[5] P. R. Griffith and J. A. de Haseth, Fourier Transform Infrared Spectroscopy (John Wiley & Sons, Inc., New York, 1986).
[6] L. Mertz, Transformations in Optics, Wiley, New York (1965). [7] L. Mertz, Infrared Phys. 7, 17 (1967).
[8] P. B. Fellgett, J. Phys. Radium 19, 187 (1958). [9] P. Jacquinot, Rep. Progr. Phys. 23, 267 (1960).
[10] P. Jacquinot, XVII ème Congrès du G.A.M.S., Paris (1954).
[11] J. Connes, and P. Connes, J. Opt. Soc. Am. 56, 896 (1966).
[12] C.-K. Huang, Z.-F. Xu, M. Nakajima, Hue M. T. Nguyen, M. C. Lin, S. Tsuchiya, and Y.-P. Lee, J. Chem. Phys. 137, 164307 (2012).
[13] A. Bagchi, Y.-H. Huang, Z. F. Xu, P. Raghunath, Y. T. Lee, C.-K. Ni, M. C. Lin, and Y.-P. Lee Chem. Asian J. 6 2961 (2011).
[14] P. Y. Chen and R. A. Palmer, Appl. Spectrosc. 51 580 (1997).
[15] P. Y. Chen, R. A. Palmer, and T. J. Meyer, J. Phys. Chem. A 102, 3042 (1998).
[16] J. Eberhard, P. S. Yeh, and Y.-P. Lee, J. Phys. Chem. 107, 649 (1997). [17] H. Nyquist, AIEE Trans. 617 (1928).
[18] W. Uhmann, A. Becker, C. Taran, and F. Siebert, Appl. Spectrosc. 45, 390 (1991).
[19] E. H. W. Meijering, W. J. Niessen, and M. A. Viergever, Medical Image Analysis 5, 111 (2001).
第三章 實驗裝置、步驟與參數設定
3.1 實驗裝置
圖 3-1 為本實驗系統之儀器裝置示意圖,其主要有:(1)光解雷射系統、 (2)步進式掃描傅式轉換紅外譜儀(step-scan Fourier-transform spectrometer)、 (3) 反應系統、(4)數據擷取與儀器時序控制系統,茲分述如下:
3.1.1 光解雷射系統
本實驗使用準分子雷射(Coherent Compex Pro 102F)之波長 193 nm 和 248 nm 的雷射光來光解前驅物以產生待測分子。當波長為 193 nm 時,每發 雷射光束到達反應槽的窗口前能量約為 100 mJ,其雷射光束形狀為一長方 形光圈(雷射光解面積約為 3 cm 1 cm)。當波長使用 248 nm 時,每發雷射 光束到達反應槽的窗口前能量約為 200 mJ,其雷射光束形狀為一長方形光 圈(雷射光解面積約為 1.5 cm 1.1 cm)。吾人使用兩平面反射率為 95 %之雷 射反射鏡(3 cm 12.5 cm)分別置於反應槽後方與前方的光解光窗口,用以 增加雷射通過反應槽的次數,藉此增加雷射光的光解效率以提高訊號強 度。 3.1.2 步進式掃描傅式轉換紅外譜儀 吾人使用 Bruker Vertex 80v 的步進式傅式轉換紅外光譜儀,其最高解析 度可至 0.075 cm−1 (削足函數為 triangular)。光譜儀為可抽真空型,利用真空
幫浦可將 FTIR 腔體抽至~2.6 Torr 以避免水氣及二氧化碳等干擾。光源可選 用 tungsten (NIR)或 globar (MIR);本實驗因光區分為二區而使用不同的分光 片及偵測器:針對 1800 cm−1以上之光區,分光片使用 CaF2 (1250 – 14500 cm−1)而偵測器使用光電壓型的 InSb (11 mm2的偵測面積,Indium
antimonide,InfraRed Associates, Inc.,D413/6),對於 1800 cm−1以下的光區, 分光片則使用 KBr(350 – 7400 cm−1)且偵測器為 MCT(11 mm2的偵測面積, Mercury Cadmium Telluride,Kolmar Technology,KV-100-B7/190)。二者使 用前均需充填液態氮降低溫度至 77 K。 3.1.3 反應系統 反應系統使用的反應槽為不鏽鋼材質,體積約為 1370 cm3,且反應槽 底座有兩片圓形 KBr 光窗,可將反應槽內氣體與外界隔離,但可使偵測的 紅外光穿透。反應槽之設計如圖 3-2 所示,為一圓柱體側接二面長方形之氣 體進出管路板以及二面長方形光窗。圓柱體反應槽外為一長方體金屬外層, 可通入高溫或低溫流體來改變反應槽內氣體之溫度。前驅物氣體由圖中右 邊(y 軸)進入反應槽,入口處有二片金屬片所形成約 1 mm 的狹縫,使氣體 進入反應槽時較均勻散佈,可減少反應槽內的擾流(turbulence)發生,以減少 光譜的雜訊。反應槽於圖中左邊接上 1/2 英吋之抽氣管路將氣體抽走。光解 雷射自 x 軸方向進入石英光窗;光窗兩側通入 purge 氣體,用以沖刷光窗來 減緩光解產生的碳化物附著在石英光窗上,可避免光解雷射通過光窗之能
量逐步遞減。紅外光偵測方向為圖中 z 軸。為增加反應所產生之瞬態產物 之吸收度,反應槽內放置 White cell 鏡片來增加吸收路徑。如圖 3-3 所示, White cell 包含了三片表面鍍金且相同曲率半徑(15 cm)的球面鏡,其中兩片 由一面球面鏡切為兩半圓形(M2 及 M3),另一片由另一面球面鏡切為 T 形 (M1)。M1 與 M2、M3 鏡的距離為 15 cm(即焦距的二倍)。IR 光源由 T 型鏡 的缺口處導入,在 T 型鏡與兩半圓鏡間進行 24 次反射,即行進了 3.6 m, 再由另一缺口射出。若要改變 IR 光束之反射次數,可調整兩個半圓鏡之夾 角。通常 White cell 中所使用反射 IR 的球面鏡需要有很高的反射率,因此 用高反射率的金做為鍍膜(coating)材質。本實驗系統所用的金鏡,其反射率 在 2.5 – 20 μm 之光區約大於 98 %,IR 光經過反射 24 次後仍有 62 %以上的 強度。
反 應 槽 上 接 電 容 式 壓 力 計 (capacitance manometer , MKS , model 626A13TEE,1000 Torr)測量系統壓力。反應樣品的流量使用針閥控制,鋼 瓶 的 氣 體 則 透 過 質 量 流 量 控 制 器 (mass flow controller , MKS , model 1179A-27472,最大流量為 1500 STP cm3 min−1)控制。反應槽內連接抽氣速 率約 1600 l min 1的乾式幫浦(dry pump,Hanbell,model PS80-A)保持氣體 流動。
3.1.4 數據擷取與儀器時序控制系統
測器訊號擷取方式可分為 AC 耦合和 DC 耦合。AC 耦合模式用來偵測反應 槽內紅外光強度隨時間之變化量,輸出訊號利用內建的前置訊號放大器進 行放大。本實驗所使用 MCT 之前置放大器( LN-MCT Photovoltaic 1mm 8H Fast Sn:MCP0183{4102-2})AC 訊號端放大倍率為 2.2,DC 訊號端並不放大, 而 InSb 之前置放大器(LN-InSb Fast Sn:ISB0073 {K-21981-IS})AC 訊號端放 大倍率為 5.1 倍,DC 訊號端並不放大。放大後之訊號經由內建的 24 位元類 比/數位轉換器(ADC,analog-to-digital convertor,8×104 sample s−1)進行轉換。 DC 耦合模式用來偵測未受雷射激發之背景光譜,其訊號處理作用模式同 AC 耦合模式,唯其訊號並不放大。 儀器時序控制分為 DC 耦合訊號和 AC 耦合訊號二部分。DC 耦合訊號 透過 FTIR 內部觸發(internal trigger)來取干涉譜,實驗開始時,測量驅動訊 號(measurement trigger)保持為 high 直到實驗結束,如圖 3-4 所示。當測量 驅動訊號為 high 後,FTIR 便觸發移動鏡移動訊號(step trigger),當其為 high 時,移動鏡開始移動,約需時 8 ms。移動到定點後,移動鏡移動訊號則會 降為 low,並等待移動鏡穩定時間 T(stabilization time,其量測方法於 3.3.3 中詳述)後,接著 FTIR 觸發序列驅動訊號(sequence trigger)時間,經過 25 ns 延遲後開始每隔時間解析度(time resolution)12.5 μs 之觸發取樣訊號
(sampling point trigger)擷取訊號,取樣結束後,經過 25 ns 序列驅動訊號變 為 low。此序列驅動訊號即為於此取樣點之時間解析譜。間隔 4 ms 之擷取
關閉時間(acquisition closure time)後 FTIR 可再次觸發序列驅動訊號,並擷取 訊號。同一移動鏡位置處可重複觸發序列驅動訊號數次,並平均所得序列 訊號以降低雜訊;而訊號平均的次數由 OPUS 軟體控制,於 3.4.2 中詳述。 接著 FTIR 會再次觸發移動鏡移動訊號(step trigger),使移動鏡開始移動並經 過移動鏡穩定時間 T 後,繼續下一點取樣,上述步驟會重複直至干涉譜擷 取完成。除了前述透過在同一移動鏡位置取樣多次降低雜訊外,由於 DC 譜是偵測未照射雷射之背景光譜,因此不論何者時間點下 DC 譜結果均應一 致,便可將此 DC 譜平均。若 DC 譜時間解析之間隔為 12.5 μs,且取樣數 通常於軟體中設定為 800 張,平均後便可得到雜訊較低之 DC 光譜。若如 2.3.1 所述,雜訊以隨機形式存在,則訊雜比可提升約 28 倍。
AC 耦合訊號則透過脈衝產生器(pulse generator,Stanford Research Systems,DG535)於外部觸發,此觸發訊號其阻抗為 high impedance、波形 TTL 、 形 狀 normal ( 正 值 ) 。 和 DC 耦 合 訊 號 不 同 的 是 測 量 驅 動 訊 號 (measurement trigger)只有在為 high 時才會接收來自 DG535 之訊號。如圖 3-5 所示,觸發訊號 E1,雖其訊號阻抗、波形、形狀正確,但此時測量驅動訊 號訊號為 low,故此觸發訊號將被 FTIR 忽略,無法正確觸發取樣訊號。待 FTIR 之測量驅動訊號為 high 後,以 DG535 產生 E2 訊號,才可成功觸發 FTIR 之序列驅動訊號(sequence trigger),待約 40 ns 後以每次間隔 12.5 μs 取樣,記錄 AC 訊號隨時間的變化,並且於同一移動鏡位置處重複觸發序列
驅動訊號擷取隨時間改變之訊號,並平均之以降低雜訊。當 FTIR 的序列驅 動訊號觸發後,吾人控制 DG535 觸發雷射光解先驅物。但為確定 AC 訊號 穩定,會在觸發雷射前取得二十張空白 AC 光譜,即 12.5 μs × 20 = 250 μs, 此光譜訊號應小於 0.001。DG535 延遲觸發雷射(laser trigger delay)約 248.3 μs(另外的 1.7μs 為雷射觸發後出光之延遲時間,測量方法於 3.3.4 詳述)。經 序列驅動訊號因此吾人可由 FTIR 中得到 AC 受雷射激發後訊號隨時間的變 化。完成擷取此移動鏡位置之重複取樣次數後,FTIR 會觸發移動鏡移動訊 號(step trigger),使移動鏡開始移動並經過移動鏡穩定時間 T 後,繼續下一 點取樣,不斷重複至干涉譜擷取完成。 3.2 實驗條件 3.2.1 光解效率評估 本實驗透過兩種方法產生 CH3 自由基分子,第一種方法利用波長 248 nm 的雷射光解 CH3I;第二種方法使用波長 193 nm 的雷射光解 CH3C(O)CH3, 二者產生之 CH3再和 O2反應產生 CH3OO。 在使用 CH3I 和 O2的實驗中,CH3I 的分壓為 2 Torr,O2的分壓為 98 Torr, 總壓維持在約 100 Torr,反應溫度維持在 298 K,在標準狀態下總流速為 25 STP cm3 s−1。雷射光到達反應窗口前,其能量約為 200 mJ pulse−1、光束截 面積約為 1.5 × 1.1 cm2、雷射頻率為 7 Hz,並且使用一面長方形反射鏡(3 cm × 12.5 cm)使其在反應腔體中來回反射 4 次,而反應槽長度為 14 cm,因此
雷射光解氣體體積為 92 cm3,以增加產生 CH 3自由基的濃度。然而在波長 248 nm 雷射光通過石英光窗時,部分能量會被其吸收,能量穿透率為 90 %, 而雷射反射鏡反射率 95 %。但反應槽內氣體亦會吸收能量,故雷射能會隨 著雷射進入反應槽能量開始遞減,導致光解率亦會下降,若不考慮雷射發 散的情況,可預估雷射初進入反應槽能量為2000.9180 mJ pulse−1。吾人 定義在反應槽入口前,每個光子能量為 E J photon−1,雷射光通量(fluence) 為 F J cm−2,雷射光解截面積為 A cm2,光子通量為 F/E photon cm−2,前驅物 在波長λ nm 下之吸收截面積為 σ cm2 molecule−1,量子產率為Φ,反應槽長 度為 l cm。根據 Beer-Lambert law,吾人可估計在離入口 x cm 處,雷射初 入反應槽雷射之光解產率 y 可表示為: Φ 3-1) 在波長 248 nm 下,每個光子能量 E 為 8.02 × 10−19 J photon−1,雷射光通量 (fluence) F 在能量 180 mJ pulse−1時為 1.1×10−1 J cm−2,光子通量 F/E = 1.32 1017photon cm−2。CH3I 在波長 248 nm 下之吸收截面積為(absorption cross section) 8.45×10−19 cm2 molecule−1[1],假設量子產率 Φ = 1 (一個碘甲烷分子 光解後產生一個 CH3自由基分子),將式(3-1)積分後可得到第一道雷射之平 均光解產率: Φ (3-2) 此時能量氣體吸收而能量遞減,根據 Beer-Lambert law 可計算第一道雷射出