國 立 交 通 大 學
生物資訊及系統生物研究所
碩 士 論 文
應用參考序列檢測水平基因轉移現象的方法 :
以Stigmatella aurantiaca為例
The method of detecting Horizontal Gene Transfer
events by applying reference sequences : A case
study of Stigmatella aurantiaca.
研 究 生:陳之杭
指導教授:林勇欣 博士
應用參考序列檢測水平基因轉移現象的方法 :
以 Stigmatella aurantiaca 為例
The method of detecting Horizontal Gene Transfer events by
applying reference sequences : A case study of Stigmatella
aurantiaca.
研 究 生:陳之杭 Student:Chih-Hang Chen
指導教授:林勇欣 博士 Advisor:Yeong-Shin Lin
國 立 交 通 大 學
生 物 資 訊 及 系 統 生 物 研 究 所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology National Chia Tung University in partial Fulfillment of the Requirements
For the Degree of Master in Bioinformatics and Systems Biology
August 2012
Hsinchu, Taiwan, Republic of China
I
應用參考序列檢測水平基因轉移現象的方法 : 以
Stigmatella aurantiaca 為例
學生:陳之杭 指導教授:林勇欣
生物資訊及系統生物研究所碩士班
摘要
水平基因轉移的現象在原核與真核生物中都經常發生,且與相關基因的來源 與功能都有重要的關係。研究水平基因轉移現象,在原核生物中可以了解其如何 適應新環境、得到新的能力,如何與宿主互動等;在真核生物中則可以探究物種 的起源,和基因的功能。現今由於科技的進展,大量的基因序列與基因體資料變 得更容易取得,使我們更容易研究水平基因轉移現象。在此我們嚐試利用大量的 基因體資料,開發廣範圍搜索水平基因轉移現象候選基因的方法;利用 tBLASTn 結果的 bit-score 與比較序列的概念,在兩基因體所做的散布圖中,找出代表特定 蛋白的 outlier 位置點,加以排序之後建立該點的演化樹,由其分類上的不一致對 比樣版演化樹來確認水平基因轉移現象的存在。本研究所使用的模式物種為黏球 菌 Stigmatella aurantiaca,該菌的基因體極大,或許與其內有眾多水平基因轉移 現象有關。 最後結果顯示此方法可以廣泛找出 Stigmatella aurantiaca 內的水平基因轉移 現象候選基因,但因還未有比對的其它物種,尚無法確認 Stigmatella aurantiaca 的基因體大小與水平基因轉移現象的直接關係。II
The method of detecting Horizontal Gene Transfer events by applying
reference sequences : A case study of Stigmatella aurantiaca
Student : Chih-Hang Chen Advisor : Yeong-Shin Lin
Institute of Bioinformatics and Systems Biology
National Chiao Tung University
ABSTRACT
The Horizontal Gene Transfer (HGT) events occur frequently in both prokaryotes and eukaryotes. It is important to understand the function and origination of a gene. By researching the HGT events, we may figure out how a prokaryote adaptations the environment, gains new abilities, and interactions with its’ host. Moreover, we may use HGT events to research the origination of species in eukaryotes. Nowadays, more and more full genomic sequences and annotated genes has been identified, make us easily to discover the HGT events.
In this study, we try to apply mass amount of genomic data and develop the method of wide screen HGT events in species by using the results of tBLASTn (bit-score ) and reference sequences. We find the outlier dot from the dot plot which is made by two BLAST query hit genomes and analyze the protein of that dot, build phylogenetic tree, observe the incongruence of the tree topology, thus, find out the HGT events. We use Stigmatella aurantiaca as model organism in this research because the genomic size of this Myxococcales is abnormally large, therefor we suppose that it is an result of frequently HGT events . The final result show that our method could identify the HGT events in a wide range but the relationship between HGT events and Stigmatella
aurantiaca’s genomic size is still not clear right now, duo to the lack of contrast genome HGT events data.
III
謝致
本論文的完成,象徵我求學生涯的里程碑。從懵懵懂懂的學習階段,到能思 考與設計自己的實驗,規劃自己的時間,這是自接受者到施予者的必經歷程。感 謝先人的研究與社會的栽培,自此之後,我期許能帶給社會與國家,或整個科學 界更多的知識。 在交大生資所的求學生涯中,我碰到了很多人,結交了很多朋友。感謝生資 所的同學們與生科二館的夥伴們,讓我的碩士生活不至於只有研究,希望未來也 能繼續保持聯絡。感謝林勇欣老師實驗室的夥伴,大逵與控肉在我進實驗室開始 便指導我寫程式,帶領我認識實驗室的生活。LOKE 坐在我的旁邊,也時常陪我 聊天吃飯討論功課。小 Q 與宜佩教了我很多另一個面向的想法。火馬做為我唯 一的同屆同學陪我度過眾多在實驗室的日子,我們是生死與共的研究生夥伴!俊 霖與羿喬學長對我實驗的幫助最大,我很多對電腦不熟的地方都有賴兩位幫忙, 在此表示由衷的感激。謝謝指導教授林勇欣老師不厭其煩的替我點出各樣實驗設 計上的盲點與問題,老師讓我學到作研究者所應該有的態度與思考方法,這是最 重要的收穫。實驗室生活兩年不長,但回憶卻好的能讓我記著一輩子。 最後感謝我的父母,謝謝你們對我含辛茹苦的栽培,不厭其煩的提醒與叮囑 我各項能力與品德,我因為有你們才有今天,雖然親恩似海且不求回報,我一定 會感念在心並繼續充實自己,謝謝。IV
Contents
Contents ... IV List of Figures ...V List of Tables ...V Chapter 1 Introduction ... 1 1.1 研究動機 ... 1 1.2 水平基因轉移... 2 1.3 水平基因轉移之特徵 ... 4 1.4 檢測水平基因轉移現象的方法 ... 6Chapter 2 Material and Methods ... 7
2.1 主要實驗構想... 7 2.2 tBLASTn ... 8 2.3 all-against-all 比對 ... 9 2.4 比較序列 ... 10 2.5 Outlier 檢測 ... 10 2.6 排序與 Alignment ... 11 2.7 結果驗證 ... 12 Chapter 3 Result ... 14 3.1 tBLASTn 的適用性 ... 14 3.2 Mahalanobis 距離輸出與排序... 14 3.3 STAUR_2131 散布圖分析 ... 15 3.4 STAUR_2131 演化樹分析 ... 16 3.5 其餘的水平基因轉移現象候選基因 ... 17 Chapter 4 Conclusion ... 19 Chapter 5 Disscussion ... 20
V
List of Figures
Figure 1 :利用演化距離不一致確認水平基因轉移現象 ... 22
Figure 2 :利用三條列間的距離建立比較組散布圖 ... 23
Figure 3 :方法流程圖 ... 24
Figure 4 :all-against-all 比對與比較組 ... 25
Figure 5 : 比較序列與 outlier ... 26
Figure 6:STAUR_2131 之演化樹... 27
Figure 7:NC_014844.1 對 NC_014148.1 比較組散布圖 ... 29
Figure 8:STAUR_2131 比較組之散布圖 ... 31
Figure 9:YP_003951762.1(STAUR_2131)演化樹 ... 32
Figure 10:YP_003951762.1(STAUR_2131)樣版演化樹 ... 32
Figure 11:YP_003950678 與 YP_003954354 之演化樹... 33
Figure 12:YP_003955618 與 YP_003953284 演化樹 ... 34
List of Tables
Table 1 :tBLASTn 效力驗證 ... 28
1
Chapter 1 Introduction
1.1 研究動機
水平基因轉移的現象在原核與真核生物中都經常發生,且與相關基因的來源 與功能都有重要的關係。本研究的主體 Stigmatella aurantiaca 屬於黏球菌目 (Myxococcales),是一種「兼性補食菌」(facultative predatory bacteria),這類細菌 棲息在土壤、水中、與人體內,以共同(或單獨)溶解並捕食其它的細菌維生,對 象極廣且不論死活[1]。此類細菌之基因體大小較大,相較於一般細菌 Genome 大小介於 1Mb~9Mb 間[2],Stigmatella aurantiaca (NC_014623) 的 Genome 有 10.26Mb 之多[3]。這樣基因體大小的差距是為了對抗獵物的防禦機制,自行變異 並保留的?或起因於其補食的特性,使其較易發生水平基因轉移現象,從而得到 補食對象的廣泛選擇能力,便是值得討論的課題。
本實驗室之前已確認一組由 Aspergillus clavatus 與 Stigmatella aurantiaca 構 成水平基因轉移的例子─基因「STAUR_2131」,但基因的來源與 Stigmatella
aurantiaca 是否還有其它的基因受到水平基因轉移現象的影響則屬未知,因此本
實驗希望能對 Stigmatella aurantiaca 全基因進行掃描,找出可能發生水平基因轉 移現象的其它基因,判斷其來源以試著解答基因體大小異常的原因,並且發展出 一套新方法以利後續對其它的物種進行水平基因轉移現象的研究。
2
1.2
水平基因轉移
水平基因轉移(Horizontal Gene Transfer, HGT)又被稱做 Laterial Gene
Transfer(LGT),是指遺傳物質在物種間水平轉移的特定現象。在達爾文理論基礎 的遺傳學中,基因物質轉移應該是一代一代在演化樹的枝幹(clade)上發生,即垂 直基因轉移(Vertical Gene Transfer),但在一些情況下,遺傳物質的轉移是枝幹對 枝幹的(clade to clade),尤其是在原核生物中更為明顯[4],這樣的水平基因轉移 現象可以大大加速演化的速率、讓原核生物得到新的特性(比如抗生素的抵抗)。 事實上,水平基因轉移的速率往往高於我們能觀察到的[5],且是原核生物基因 體變異性的一個很重要影響因子─大約有 81 ± 15%的基因都曾發生過水平基因 轉移現象[6],顯示出雖然某些特定的水平基因轉移有其限制(如與宿主相關的毒 性基因),但大部分的基因都是可以被轉移的[7]。水平基因轉移在原核生物中可 以經由轉形(transformation)、結合(conjugation)、轉導(transduction) [8],和細菌 間的 Nanotube[9]來達成,另外一些海生的 Rhodobacter 可以自行吐出類似噬菌體 的結構來隨機轉移自己的某部分基因序列給其它 Rhodobacter,即 GTA(Gene transfer agent)[10, 11],這樣的基因轉移是完全隨機挑選與隨機插入的,因此驅 動變異的速度非常的快[12]。而真核生物則可藉由吞噬作用(phagocytosis)或內共 生(endosymbiont)的交互作用造成。 在近期關於海洋細菌的研究中,一個特殊的水平基因轉移現象「GTA」,解 釋了海洋細菌如何快速得到新的特性以有彈性的與環境進行互動,如代謝環境中
3 的化學物質、溫室氣體,製造特殊的養分和扮演海洋生物鏈中的特殊功能,其基 因交換的頻率比起以往所預測的高出了驚人的 47%之多[12]。真核生物方面最近 也有研究指出,真菌類雖然因為有細胞壁且失去了胞吞作用的能力,但仍然能經 由水平基因轉移取得新的基因[13]。 之前的研究對於真核生物的起源與真細菌(Eubacteria)和古生菌(Archaea)之 間的關係有不同看法,比如將真核生物視為古生菌的 sister group、將真核生物分 在古生菌內、或認為古生菌與真細菌都是由一種類似真核生物的祖先 (Eukaryote-like ancestor)來的[14]。而最近關於水平基因轉移的研究協助我們了解 真核生物的基因組成是一種合成的形式(Chimeric),包含了細菌與古生菌特徵的 基因。進一步由演化樹分析,解決了多序列 alignment 的問題之後,可以建立出 網路形式的演化樹[15],得到真核生物中由古生菌而來的多半是訊息類 (informational)的基因,而自真細菌來的多半是功能性(operational)基因的結論。 訊息類的基因雖然數量比較少,可是在 protein-protein interaction 中扮演著重要 的角色,且這樣的蛋白質更容易和相同源的蛋白質交互作用[16]。所以自水平基 因轉移現象為起始的研究不只可以探究生物的新特性從何而來,甚至可以探究起 源與基因功能性的領域。
4
1.3 水平基因轉移之特徵
水平基因轉移現像可以藉由演化分析上的不一致與相關基因組成的不一致 觀察到。所謂演化分析上的不一致指的是有水平基因轉移現象的基因所呈現出的 演化樹分類(pattern),和拿來做為對照的標準演化樹分類會有明顯的不同。舉例 來說,當我們把一群有類似功能(或同源)但不同物種的基因 align 在一起之後建立 演化樹,再將此樹拿來和其他基因或者物種的演化樹(細菌中通常是利用小核醣 體 RNA─16S rRNA[17]來建)做比較,如果可以發現不一致的分類─通常是距離比 較遠的物種在水平基因轉移現像發生的演化樹中被插進原本近似的物種群中,就 可以直接證明水平基因轉移現像[18]。另一方面,基因組成的不一致也可以做為 水平基因轉移的現象的證據(Composition-based methods),比如 GC 含量(G+C content)、二聯核苷酸相對豐富度(Dinucleotide Relative Abundance, DRA)、和密碼 子偏性(Codon usage bias)等。二聯核苷酸相對豐富度是 1995 年由 Karlin S.和 Ladunga I.提出的,表示兩兩 核苷酸共同出現的相對機率,假設序列是完全隨機的,該值應為一常數,所以一 條序列的值相對於該常數的偏差;同 GC content 一樣,可以做為該條序列的特徵 [19]。因此檢測 HGT 候選基因的這些數值,與相鄰基因、HGT 可能來源基因做比 較,較接近後者的結果便預示了水平基因轉移的可能性。密碼子偏性則是基於 tRNA pool,由於大部分的胺基酸都是被一個以上的密碼子定義的,不同的物種 對於同一個胺基酸所喜好使用的密碼子比例甚至密碼子的組成可能不相同,距離
5 越遠的物種其差距會越大,且基因內 coding sequences 的密碼子組成會反映此一 偏性,才能使基因內容有效率且無誤的轉譯出來,因此研究密碼子的組成與使用 頻率便可以相當程度代表該 coding sequences 是來自哪個物種[19],從而得知與 水平基因轉移有關的訊息。 但是關於水平基因轉移之基因組成分析,都會受到轉移現象發生之後慢慢與接受 者的基因體同化現象的影響,而造成判斷上的困難。比如久遠以前的轉移或是接 受者的演化速率較快,都會造成比對原始來源與現今觀察者基因組成時,可能與 原始來源已較不相像的情況發生。因此,本實驗所利用的方法之一便是將每個基 因之同源基因都與另一條同源基因進行比對,用相對比較並找出歧異點的方法來 解決直接比較組成會碰到的問題。
6
1.4 檢測水平基因轉移現象的方法
由於 BLAST 本身即是兩序列同源性的應用,因此傳統針對單一基因之水平基 因轉移檢測可以經由全基因組或蛋白資料庫的 BLAST 結果,過濾掉相同或相近的 同源基因,找出序列近似度高但物種關係很遠的基因來當做水平基因轉移現象的 候選者,並進行接下來的演化與基因組成分析。 但是這樣的分析速度較慢且必須耗費人力去確認”關係很遠的物種”,因此有些檢 查 HGT 現象的方法便被開發了。 有別於傳統達爾文演化觀念中樹狀的表示法,在原核與真核生物中廣泛發生 的水平基因轉移現象會使演化樹變成網路的形式─即 Phylogenetic network。 Phylogenetic network 的建立讓我們能一目瞭然的知道基因的來源,而其建立法主 要分為:利用 SPR distance 與 Maximum Parsimony[20]。SPR 是一種融合重建演化 樹的方法,將兩棵或以上的樹以最少的移動次數(即 SPR distance)接在一起並建立 internal node 形成 network 的形式,而 Maximum Parsimony 則是在序列改變次數 最少的前提下嘗試加入 inter node 以使一個圖可以符合眾多分類法。但這樣改變 樹的型態的方法的前提都必須是原圖的不一致性是起因於演化上如基因轉移現 象,建樹的基因選擇困難[20],且無法廣泛的針對 Genome 進行檢測。因此我們 嚐試開發一套方法流程完成廣泛的水平基因轉移現象檢測。7
Chapter 2 Material and Methods
2.1 主要實驗構想
我們的主要實驗構想是利用三條序列─分別為 Stigmatella aurantiaca (或者 其他輸入的 query 序列)上的基因序列 (假設為物種 A),和我們的 Genome database 中與該序列為同源關係的另外兩條序列 (假設為物種 B、C),來進行比 較。此三條序列可以做出類似[Figure.1]上圖的演化樹形式,假設此樹為此三物種 的 Species tree 或一般認知的基因建立的 common tree (如 16SrRNA)。在正常的演 化情況下,此三物種內的任何同源基因樹應該都會維持這樣的排列法,也就是 A 至 B 的演化距離與 A 至 C 的演化距離應該會呈現一固定的值[Figure1.up]。但有水 平基因轉移現象發生在 A 與 B、或 C 之間的時候,該 A 的基因和 B、C 內的同源 基因所建立的演化樹將與原本的發生不一致的現象,因此其距離的比值便會和原 本的不同[Figure1.below]。 另外,利用前述的 A、B、C 三條序列兩個距離的關係,我們可以建立出一 個散布圖上的點[Figure2.up]。假設要研究 A 物種的水平基因轉移現象,那就可以 利用 B 為 Y 軸,A 至 B 的距離為 Y 座標,以及 C 為 X 軸,A 至 C 的距離為 X 座標, 則此圖上代表 A 的基因的點,其座標便是 (A-C,A-B)。綜合了許多 A、B、C 物種 內同源基因的點之後,便可以建立一個以 B、C 為 X-Y 軸,A 之基因為點的多點 散布圖[Figure2.below]。此圖內的點如之前所假設,大部分 X/Y 的比值應為定值,
8
也就是呈線性關係。但假設有些點(基因)發生了水平基因轉移現象的話,由於其 比值與原本不同,在圖上便會產生 outlier 的現象。根據這樣的假設,只要能夠 建立所有 Genome 對 Genome 的比較組(即 all-against-all 比較),抓出其同源基因 中的 outlier 點,我們便能大量檢測水平基因轉移現象。
本方法利用 PhP 程式語言為基礎,應用了 BLAST 的資料,R 語言計算 Mahalanobis 距離,CLUSTALW 和 MEGA 進行 alignment 與演化樹的建立,詳細的流程圖為 [Figure.3]。
2.2 tBLASTn
大部分尋找水平基因轉移方法應用的是 BLASTp。但 BLASTp 精準卻很容易受 到蛋白資料缺失的影響,且水平基因轉移現象亦不一定是以基因為單位跳躍,因 此在動機為廣泛大量搜尋的情況下,我們便選擇以 Genome 為 nucleotide database 的 BLASTn,如此便可利用完整的基因體做搜尋。在 Query 的部分,由於演化時 間較長時核酸序列的改變容易飽和,便無法觀察到相應的改變,因此我們依然使 用蛋白序列為 Query,應用的 BLAST 便是 tBLASTn。
本實驗 tBLASTn database 所取用之基因體序列皆來自 NCBI FTP,「Genomes」資料 夾下的「Fungi」及「Bacteria」 (實際上包含了 Eubacteria 及 Archaea)下的.fna(FASTA Nucleic Acid file)檔案,BLAST 之 query 為「Bacteria」資料夾下 Stigmatella aurantiaca 的.faa(FASTA Amino Acid file)檔案,其內含所有 Stigmatella aurantiaca 已確認之蛋
9
白序列。利用 BLAST+做為 BLAST 工具[21],其原始碼可以在 NCBI FTP 下載[22]; e-value cutoff 為 10-5、輸出型式為 .csv。
根據 The NCBI Handbook 的介紹,BLAST 之 score 可以代表 alignment 程度的好壞, 換句話說可以代表兩序列間的同源性,而經過標準化後的 bit-score 更可以用來比 較不同 alignments 間的關係,即使它們應用不同的 scoring matrix 亦可[23]。因此, 我們將 tBLASTn 結果之 bit-score 輸出之後用來當做後續研究之參數。
2.3 all-against-all 比對
利用 PhP 程式語言,將 BLAST 後得到的所有結果依照不同的 Genome 進行分 類,並建立兩兩 all-against-all 之索引。想法是每個 Genome 皆和另一個 Genome 進行一次比對,找出共同 Stigmatella aurantiaca 蛋白質之 hit 當做一個資料點, 並佐以該點在各自 Genome 上的 bit-score 當做該點的坐標,如此便可得到以兩 Genomes 為 X-Y 軸、對應之 Stigmatella aurantiaca 蛋白質為資料點的散布圖 [Figure.4]。
但用此方法偵測水平基因轉移時會受到 Paralogous 的影響,增加分析的難度 與時間。基於水平基因轉移之序列相似度應該會大於 Paralogous,因此同一個蛋 白在特定 Genome 上的 hit 只取最高 bit-score 分數的結果,進行過濾後接著下面 的分析。
10
2.4 比較序列
得到兩 Genome 比對之散布圖之後 (後稱此兩 Genome 為一「比較組」),此 圖便有比較序列之意義,在上面的蛋白資料點在兩 Genome 上都有同源的基因。 假設完全沒有水平基因轉移現象,影響此兩 Genome 上資料點之 bit-score 值的因 素只有 Paralogous(最高 bit-score 分數的)和 Orthologuous。雖然各物種間演化速 率不同,但這些同源基因在物種內的演化速率應是一致的,因此資料點應該會呈 現一線性的圖形[Figure.5 ]。 以演化上的角度來說,水平基因轉移是非垂直的現象,有水平基因轉移的基 因與其基因來源的序列距離會小於無水平基因轉移的同源基因,假設完全沒有水 平基因轉移現象發生,兩同源序列到該基因的距離應是一致的,可參考 [Figure.11.up]。 因此有水平基引轉移現象發生的資料點,與其有水平基因轉移關係的 Genome 和另一正常同源關係的 Genome 所形成之資料點便會產生群組中偏移; 有水平基因轉移關係的 Genome 上的 bit-score 值應會比理論上來的大,在圖上便 會呈現 Outlier 的形式。如此,只要能偵測每個比較圖中的 Outlier 點,便能找出 可能是水平基因轉移現象的候選基因[Figure.5]。
2.5 Outlier 檢測
11 較多研究利用的多變量方法(multivariate methods)。多變量分析方法中依據取得 資訊的不同又可分為統計法(Statistical),基於距離(Distance)與分群(Clustering)的 數值採礦(Data-Mining)法[24]。先前的研究指出統計法在處理大量而複雜的數值 資料上優於數值採礦法[25],雖然會受到覆蓋(Masking)與圍困(Swamping)效應的 影響,但我們並不執行任何 outlier 的去除,因此受到的影響不大。[26] 統計法中最常被利用的距離標準是 Mahalanobis 法[27]。假設有n組數據,影響
的維度為p,整組的 mean vector 為𝑥̅n,其 covariance matrix 為:
則每個點與其 mean vector 之經過標準化後的 Mahalanobis 距離為:
2.6 排序與 Alignment
得到 Mahalanobis 距離後我們自動進行排序,挑出距離比較大的點做為水平 基因轉移候選並進行分析。我們可以指定要瀏覽的組數,程式會自動將此範圍內 相同的水平基因轉移候選(來自不同比較組)視為一個群組 (此後稱為「候選基因 群組」),並且取得該該水平基因轉移候選蛋白質的 nucleotide coding sequence、 及兩 Genome 上 BLAST hit 的 nucleotide 序列,也就是說,當此候選基因群組內只
12
有一組比較組時,裡面的序列便是該 Stigmatella aurantiaca 水平基因轉移候選與 比較組兩 Genome 序列各一,當群組內有兩組比較組時,裡面的序列便是一條
Stigmatella aurantiaca 與四條Genome 序列,但程式會自動刪除重複的序列資料
(比較組的其中一個 Genome 可能和另一個比較組的 Genome 重複,基於同一個 蛋白在特定 Genome 上的 hit 只取一組,這兩個由同一蛋白對到同一 Genome 的 序列會是完全相同的)。
之後,自動利用 CLUSTALW[28],將 nucleotide 序列轉成胺基酸後進行 Multiple Alignment,並輸出 nucleotide 形式的 Alignment 結果。
2.7 結果驗證
我們應用 MEGA 做為分析工具[29],此軟體協助我們針對每個水平基因轉移 候選建立演化樹,演化樹的種類為 Neighbor-Join、Amino acid distance、Scoring Matrix 為 PAM。假設有水平基因轉移現像的話,此樹已經可以看出分類上的不一 致性,但仍須對照樣版演化樹才能真正確定。 用來對照的樣版演化樹,可以利用 Housekeeping gene 建成,微生物中最常被利 用的是小核醣體 RNA(16S rRNA),由於它廣泛存在於所有細菌中,且功能沒有 隨著時間轉變,1500bp 的大小也足以涵蓋必要的資訊[17]。但有研究指出在物種 太相近或是分化的時間點不長的話,小核醣體 RNA 沒有辦法有效的分類出這些 物種[30]。因此,我們利用近似於 MLST(Multilocus sequence typing)微生物分類
13
法的概念[31],將好幾條序列串接在一起來建立樣版演化樹。方法是隨機在比較 組內挑選十條正常非 Outlier 的 Stigmatella aurantiaca 基因,且這些基因必須對候 選基因群組內的每個 Genome 都有 hit,因此可以抓出代表每條 Genome 的序列。 將這些 Stigmatella aurantiaca 基因各自與其候選基因群組內的 Genome 序列進行 Multiple Alignment 後再串接起來(為了避免不同基因間互相干擾),便完成樣版演 化樹。但假如沒有十條共同的基因可以選擇的話,仍然要使用小核醣體 RNA 來 建立演化樹。
最後比較這兩者之間演化樹分類上的不同,觀察是否有類似水平基因轉移的插入 現象。
14
Chapter 3 Result
3.1 tBLASTn 的適用性
本實驗承接何宜佩的發現[32],Stigmatella aurantiaca 基因 STAUR_2131 (YP 003951762.1)與 Fungi 類有水平基因轉移現像[Figure.6]。此實驗與前述的其他方
法大部分都是利用 BLASTp 做為研究發現的基礎,因此要利用 Stigmatella
aurantiaca 來開發使用 tBLASTn 為基礎的新方法勢必要確認 tBLASTn 也能找到
BLASTp 可以找到的結果。
由於本方法目標是掃描全 Genome,而 Figure.4 中只有 A.oryzae、S. aurantiaca、
A.fumigatus、A.nidulans、N.crassa (紅色與藍色邊框)有完整的基因組。利用 Figure.4
中的蛋白為 query 進行 tBLASTn 對應本實驗的 database 後,可以發現 Figure.4 該有的 hit 都可以被找到,但同樣以核酸為 database 的 BLASTn 就只能掃到 STAUR_2131 了 [Table.1],這表示蛋白序列所含有的資訊較核酸多,因此本方法
選用 tBLASTn 做為研究基礎。
3.2 Mahalanobis 距離輸出與排序
由於同樣值點的 Mahalanobis 距離在不同的比較組中,其大小會不相同,因 此單純按照距離大小排序並不足以判斷水平基因轉移現像的顯著性。事實上,序
15 列太相似的比較組(常是同一種菌的不同品系),只要序列稍微不同就會造成相當 大的 Mahalanobis 距離。 序列間微小的差異不是此方法研究的重點,且這樣的輸出結果會大大增加研 究者篩選可能的轉移現象的時間。為了避免這樣的誤判我們對比較組的基因體相 似程度做了限制,在基因體相似度<70%時,前幾名的 Mahalanobis 距離輸出結果 之序列差異與比較組物種的差異才比較接近可能為水平基因轉移現象的結果。 另外,為了節省運算時間,我們也想找到 Mahalanobis 距離能成為 outlier 的最低 點。在所有輸出結果中有距離為 10 的基因點的比較組是 NC_014844.1 對 NC_014148.1 蛋白點名稱為「YP_003951646」,如[Figure.7]可以看出圖上已有更 明顯的 outlier「YP_003956356」,而該點的 Mahalanobis 距離為 39.83429,其餘 圖上明顯的 outlier 皆介於此值之間,因此我們先設定輸出的 outlier 閾值必須大 於 10。 如此,最後輸出的 Stigmatella aurantiaca 水平基因轉移現象候選基因一共有 3733 個。
3.3 STAUR_2131 散布圖分析
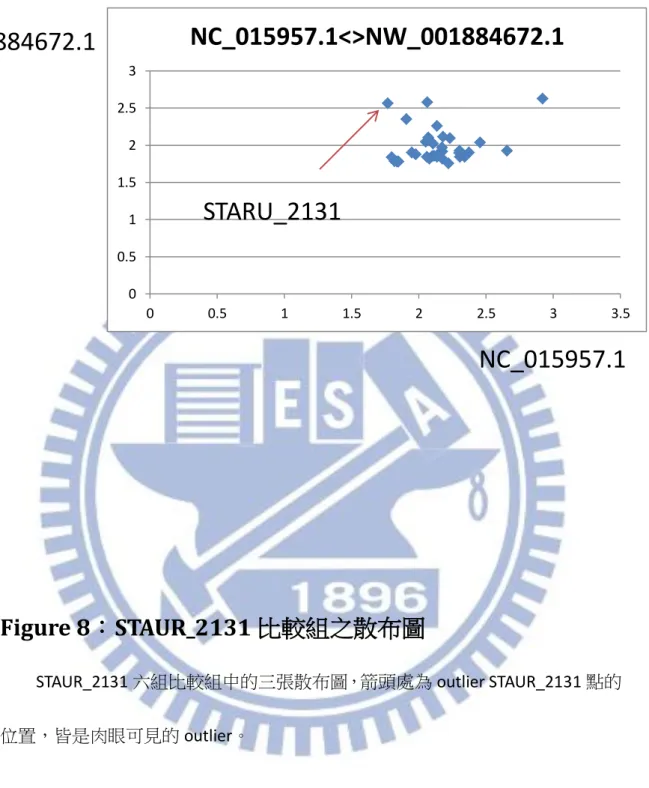
最終結果中與 STAUR_2131 有關係的比較組一共有六組,如[Table.2]所示, 其中三點的散布圖[Figure.8],皆可以看出 STAUR_2131 在這樣的比較組中是明顯 的 outlier(圖上箭頭位置),且圖形大致上呈線性分布,因此此方法確實能有效的16
將有水平基因轉移現象的點規類為 outlier。但當比較組中的物種較遠時,比如 NC_015957(Bacteria)和 NW_001884672(Fungi),由於本身序列差異就比較大,因 此有共同對 Stigmatella aurantiaca 基因的 Hit 自然就很少,點數少的情況下看起 來線性的狀態就不佳,但 Mahalanobis 距離大的點仍然可以看出是 outlier(STAUR_2131)。
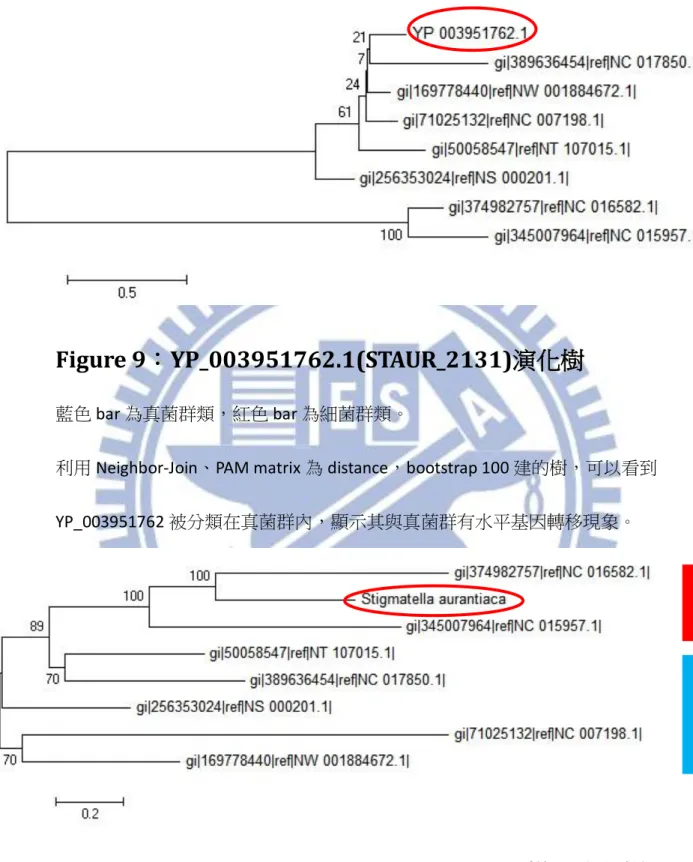
3.4 STAUR_2131 演化樹分析
將表二的六組比較組融合成 STAUR_2131 的候選基因群組,便可以建立一組 含有八組物種的群組,其演化樹為[Figure.9]。YP_003951762.1 (STAUR_2131)被分 在 Fungi 群中,最接近的是 NC_017850.1(M.oryzae)。在外面則是前述演化樹中被 認為最接近 STAUR_2131 的 NC_007198 (A.fumigatus)。此數整體的分類和前述演 化樹一樣是將 STAUR_2131 分累入了真菌群(藍色條)內,是明顯的水平基因轉移 現象,此外,圖上的另外兩個細菌類(紅色條)則 group 在一起後成為 outgroup, 間接說明此分類的正確性。 我們仍應檢視樣版演化樹,但此候選基因群組其物種間差異太大,因此 STAUR_2131 的候選基因群組中只有一個共同基因「YP_003951700」,這樣建出來 的樣版演化樹[Figure.10]或許沒有很強的效力,但仍然可以看到預想的結果─細 菌群與真菌群被完整的分離開來,且 Stigmatella aurantiaca 的位置是在細菌群內 的。而一般狀況下無法連接共同基因來建立樣版演化樹時應該要利用小核醣體17
RNA,但此處的問題是 Fungi 類並沒有此一基因,因此無法利用小核醣體 RNA
建立樣版演化樹。
3.5 其餘的水平基因轉移現象候選基因
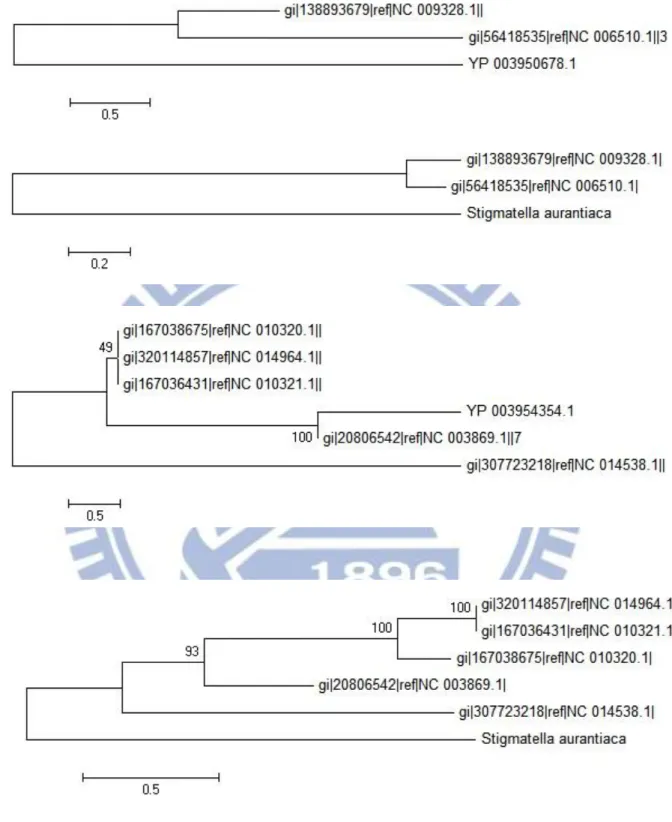
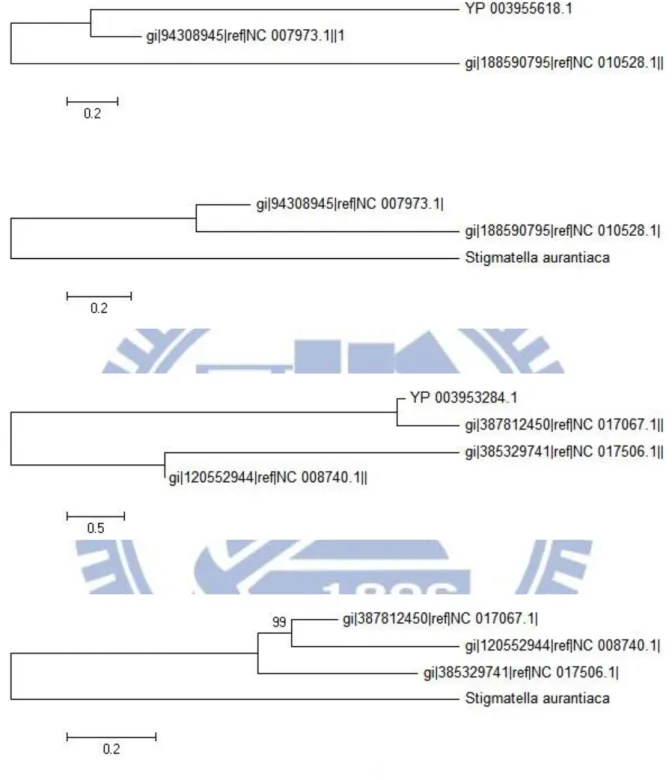
此次實驗篩選出了 3733 個水平基因轉移現象的候選基因,前一千組資料, Mahalanobis 距離最大前三名分別為 YP_003950678.1、YP_003955618.1、 YP_003954354.1、YP_003953284.1,分數分別為 405、308、303、295 分。其演 化樹及樣版演化樹都顯示出分類上不一致的地方。 首先 YP_003950678.1 [Figure11.up] 的演化樹呈現了其與 NC_009328.1 (Geobacillus thermodenitrificans)、NC_006510.1(Geobacillus kaustophilus)演化距離 的差距,但在樣版演化樹上此差距變得相當小,顯示 YP_003950678.1 可能與 NC_009328.1 有水平基因轉移現象。YP_003955618.1 [Figure12.up] 則呈現了更極端的現象,顯示 YP_003955618.1 與 NC_007973.1(Cupriavidus metallidurans)分類在一起,而樣版演化樹則是正常的
Cupriavidus 屬:NC_007973.1 與 NC_010528.1 (Cupriavidus taiwanensis)分類在一
起,足見 YP_003955618.1 與 NC_007973.1 有水平基因轉移現象。 YP_003954354.1 [Figure11.below]與 NC_003869.1(Caldanaerobacter
subterraneus)分類在一起,但樣版演化樹顯示 Caldanaerobacter subterraneus 的分
18
上,連在原演化樹中看似 outgroup 的 NC_014538.1 都是屬於
Thermoanaerobacteraceae 科的,和 Stigmatella aurantiaca 在分類上「門」的位階
就不同,因此樣版演化樹的結果應是可信,如此一來原演化樹極有可能是由
Thermoanaerobacteraceae 科的細菌水平基因轉移入 Stigmatella aurantiaca 的
YP_003954354.1 結果。
YP_003953284.1 [Figure12.below] 則是另一個水平基因轉移的例子,
YP_003953284.1 與 NC_017067.1(Marinobacter hydrocarbonoclasticus)群組在一起, 而未與其餘的 Marinobacter NC_008740.1、NC_017506.1 合在一起,似乎是水平 基因轉入的現象。
綜合以上的觀察我們可以說此方法確實在一定程度內能自動且大範圍的檢測出 水平基因轉移現象候選基因。
19
Chapter 4 Conclusion
本研究的目的在開發一套方法,能夠自動大量檢測特定物種的全基因組與所 有有完整 genome 序列的物種之間是否有水平基因轉移現象產生,降低之前的方 法所需的人力與時間成本,跳脫之前的方法會受到未確認基因 missing data 的限 制,得到更多轉移候選基因來進行接下來的實驗與分析工作。 由本研究的結果顯示此方法確實能夠大量的挑出有水平基因轉移現象的候 選基因,並建立出可供後續研究的演化樹。但是本方法在針對跨物種距離較遠或 組數太多、太少的候選基因群組時會因為找不到共有基因而使樣版演化樹的效果 不盡理想,但仍然可以嘗試利用 16srRNA 或尋找特定的研究以建立樣版演化樹。 本研究完成後確認 Stigmatella aurantiaca 有 3733 個基因曾經與其他物種發生水 平基因轉移現象,且此數目可能還會因為除去本實驗中的限制而增加。20
Chapter 5 Disscussion
本方法雖然可以廣泛尋找水平基因轉移現象,但在開發階段 all-against-all 比較的分析時間,tBLASTn 的時間和最後處理跑 CLUSTALW 與 MEGA 的時間過久, 勢必需要找出能夠更快分析的方法。另外,最後的實驗輸出仍然需要人工去確認 是否有水平基因轉移現象。
本方法候選基因從哪裡來,甚至是之後到哪裡去的問題。建立了眾多可能有 水平基因轉移現象的演化樹之後便可以採取前述 SPR 方法以建立 phlogenetic network,如此或許可以完整的呈現一個基因的旅程。因此,本方法可以視作是 建立 phlogenetic network 得前置工作,可以大量得到有效的 data 以供後續研究。 另外,雖然此法可以篩選出水平基因轉移候選,但水平基因轉移現象的確認仍需 結合其他方法,比如 GC content 與 Codon usage 等方法。
另外當比較組中的 Genome 序列太相近時,一點點序列差異便會使 Mahalanobis 距離變得相當大,本次實驗中最大的距離高達 923,但此兩組 genome 為同種不同品系、其序列差異僅只有 1 個 nucleotide,這樣的差異極有可能只是 普通演化現象造成的多樣性,並不是此方法所要討論的重點,因此我們限制了序 列間的相似度必須<70%。但,這樣的限制雖然讓前幾名的輸出能呈現物種的差 異性,仍不能排除有漏掉可能基因轉移現象的可能性。
21 本次研究觀察到 Stigmatella aurantiaca 的水平基因轉移率有 3733 個,並不 算特別突出,因此無法直接證明其基因體大小的異常現象是否和水平基因轉移現 象有關,且我們的方法內仍有需要完全確認的地方,比如 BLAST 的準確性、e-value 條件的設定是否真的可以完整的找到所有的同源基因,又或者 Mahalanobis 的計 算方法,是否能夠完整的找出所有的 outlier,又或者有些 Genome 的構造會使此 方法產生誤差,如真菌類的 Intron 等。 最後,為了真正確認 Genome 大小與水平基因轉移的關聯性,我們仍然需要 用此方法來檢視比較一般的菌種,如 E.coli,來做為對照以說明此假說是否成立。
22
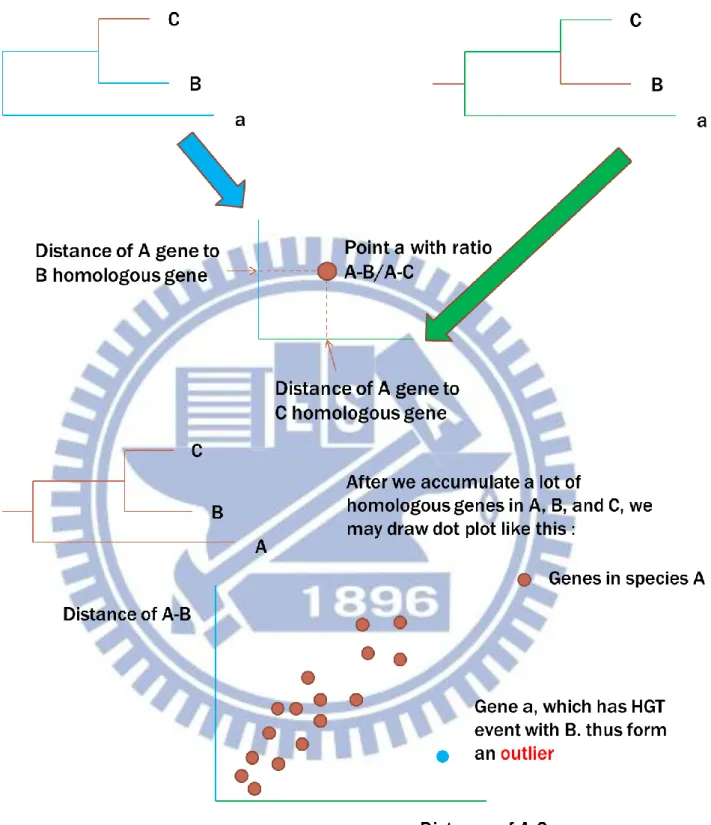
Figure 1 :利用演化距離不一致確認水平基因轉移現象
給定 Genome A、B、C,如果物種演化樹為上圖,A、B、C 的共有同源 基因之演化樹形式應該呈如此排列,因此 A-B/A-C 應為定值。 在有水平基因轉移現象發生的時候,由於該特殊的基因是經由轉移現象獲得 其距離必定與一般演化得到的基因不同,A-B/A-C 的比值因此而產生差異。 這樣的差異便可以化為座標差異應用在後面的散布圖觀念中。23
Figure 2 :利用三條列間的距離建立比較組散布圖
24
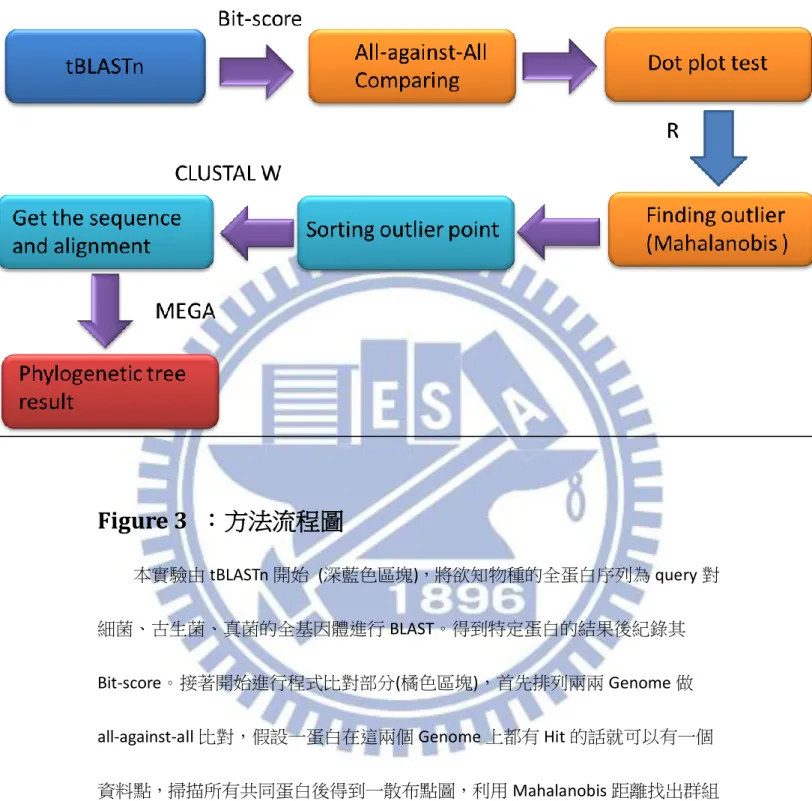
Figure 3 :方法流程圖
本實驗由 tBLASTn 開始 (深藍色區塊),將欲知物種的全蛋白序列為 query 對 細菌、古生菌、真菌的全基因體進行 BLAST。得到特定蛋白的結果後紀錄其 Bit-score。接著開始進行程式比對部分(橘色區塊),首先排列兩兩 Genome 做 all-against-all 比對,假設一蛋白在這兩個 Genome 上都有 Hit 的話就可以有一個 資料點,掃描所有共同蛋白後得到一散布點圖,利用 Mahalanobis 距離找出群組 中 outlier 的點。最後的是排序與分析部分(淺藍色區塊),將 Mahalanobis 距離大 的點挑出利用 CLUSTAL W alignment 後,以 MEGA 建立演化樹,輸出最後的結果。
25
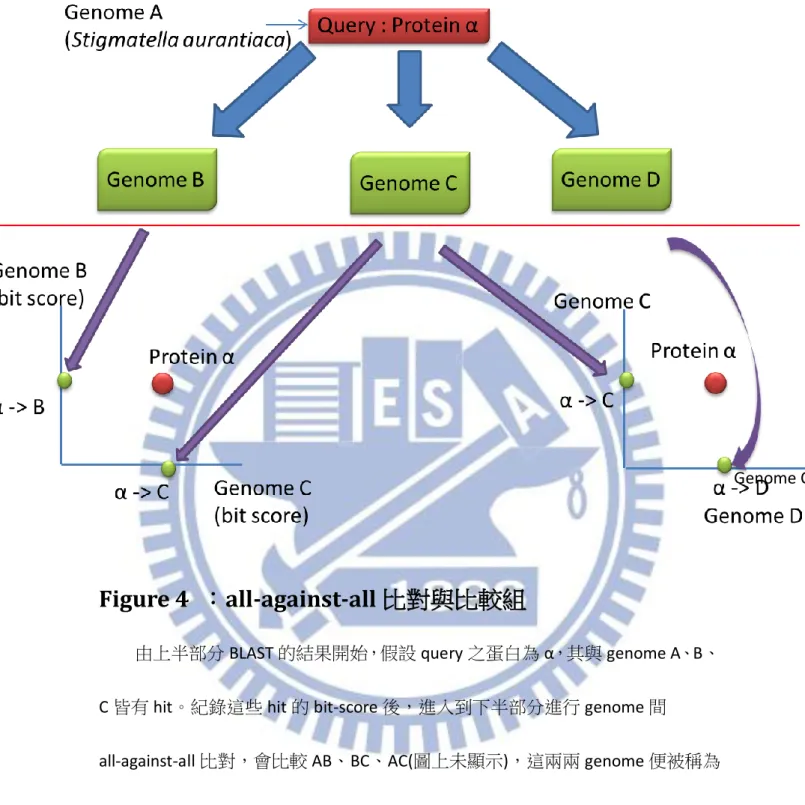
Figure 4 :all-against-all 比對與比較組
由上半部分 BLAST 的結果開始,假設 query 之蛋白為 α,其與 genome A、B、 C 皆有 hit。紀錄這些 hit 的 bit-score 後,進入到下半部分進行 genome 間
all-against-all 比對,會比較 AB、BC、AC(圖上未顯示),這兩兩 genome 便被稱為 「比較組」,並可以畫出一個以兩 genome 為軸,bit-score 為刻度之散布圖。共 同對應到的蛋白 α 便以在各自 Genome 上的 bit-score 為 X-Y 坐標,在圖上形成一 資料點。
26
Figure 5 : 比較序列與 outlier
當我們得到比較組,畫出散布圖,標出所有在其上有 hit 的蛋白點之後,便 可以得到一個有眾多點的群組,一般情況下 Genome B、C,到共同同源的蛋白點 的距離應該是成比例的線性形態(上左),只是距離較遠的線性可能會有偏向(上 右),如此群中 outlier 的點便可能是水平基因轉移現象造成的,下圖為一實際的 例子,在 NC_005957.1 與 NC_011658.1 genome 比較組中出現的 outlier 蛋白。B
C
C
D
27
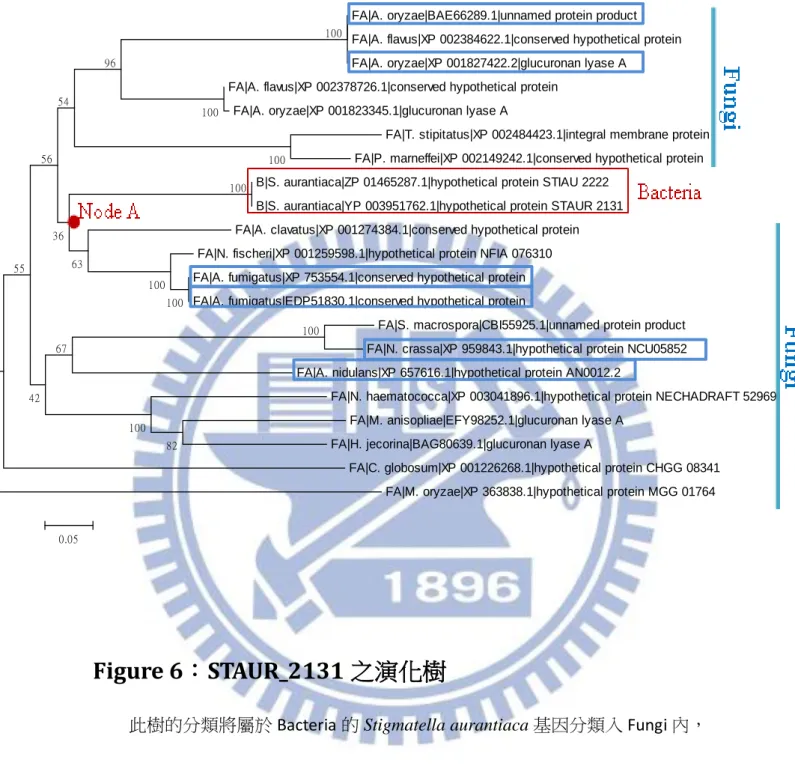
Figure 6:STAUR_2131 之演化樹
此樹的分類將屬於 Bacteria 的 Stigmatella aurantiaca 基因分類入 Fungi 內, 顯示出此基因是由 fungi 轉移過來的。
紅色與藍色邊框是本實驗有包含的 dataset,紅色為 Bacteria,藍色為 Fungi。 原圖來自何宜佩發表之碩士論文[32]。
FA|A. oryzae|BAE66289.1|unnamed protein product FA|A. flavus|XP 002384622.1|conserved hypothetical protein FA|A. oryzae|XP 001827422.2|glucuronan lyase A
FA|A. flavus|XP 002378726.1|conserved hypothetical protein FA|A. oryzae|XP 001823345.1|glucuronan lyase A
FA|T. stipitatus|XP 002484423.1|integral membrane protein FA|P. marneffei|XP 002149242.1|conserved hypothetical protein B|S. aurantiaca|ZP 01465287.1|hypothetical protein STIAU 2222
B|S. aurantiaca|YP 003951762.1|hypothetical protein STAUR 2131 FA|A. clavatus|XP 001274384.1|conserved hypothetical protein FA|N. fischeri|XP 001259598.1|hypothetical protein NFIA 076310 FA|A. fumigatus|XP 753554.1|conserved hypothetical protein FA|A. fumigatus|EDP51830.1|conserved hypothetical protein
FA|S. macrospora|CBI55925.1|unnamed protein product FA|N. crassa|XP 959843.1|hypothetical protein NCU05852 FA|A. nidulans|XP 657616.1|hypothetical protein AN0012.2
FA|N. haematococca|XP 003041896.1|hypothetical protein NECHADRAFT 52969 FA|M. anisopliae|EFY98252.1|glucuronan lyase A
FA|H. jecorina|BAG80639.1|glucuronan lyase A
FA|C. globosum|XP 001226268.1|hypothetical protein CHGG 08341 FA|M. oryzae|XP 363838.1|hypothetical protein MGG 01764 100 100 100 100 100 82 100 100 100 96 67 42 55 63 54 56 36 0.05
28
BLAST type
Query Hit target Subject
seq start Subject seq end Blastn c2627104-2626334 (STAUR-2131 coding region) NC_007198.1 (the only one hit)
2798658 2799151 tBLASTn YP_003951762.1 (SRAUR-2131 Protein) NC_007198.1 2798586 2799350 tBLASTn XP_753554.1 (Aspergillus fumigatus Af293) NC_007198.1 2798523 2799350 tBLASTn YP_003951762.1 (SRAUR-2131 Protein) NW_001884672.1 1345381 1344695 tBLASTn XP_001827422.2
(Aspergillus oryzae RIB40)
NW_001884672.1 1345381 1344695 tBLASTn YP_003951762.1 (SRAUR-2131 Protein) NT_107015.1 42199 42885 tBLASTn XP_657616.1 (Aspergillus nidulans FGSC A4) NT_107015.1 42130 42885 tBLASTn YP_003951762.1 (SRAUR-2131 Protein) NW_001092400.1 14676 13987 tBLASTn XP_959843.1 (Neurospora crassa OR74A) NW_001092400.1 14745 13987
Table 1 :tBLASTn 效力驗證
利用 Figure.4 之蛋白為 Query 進行 BLASTn 與 tBLASTnQuery 欄對應 Figure.4 中的蛋 白,Hit target 欄為 BLAST 後 hit 到的 Genome,標示紅色為 Figure.4 中有的蛋白,在本實 驗中也有被找到。可以看出圖中的蛋白全部都能夠利用 tBLASTn 的方法找出。
29
Figure 7:NC_014844.1 對 NC_014148.1 比較組散布圖
箭頭處為 outlier 點 M.distance 比較組 A Species B Species 16.8986814 NT_107015.1 A.nidulans NC_017850.1 M.oryzae 15.6519429 NC_016582.1 S.bingchenggensis NC_007198.1 A.fumigatus 13.9237548 NC_015957.1 S.violaceusniger NC_007198.1 A.fumigatus 11.8339654 NS_000201.1 P.chrysogenum NT_107015.1 A.nidulans 11.2209951 NS_000201.1 P.chrysogenum NC_007198.1 A.fumigatus 10.6673748 NC_015957.1 S.violaceusniger NW_001884672.1 A.oryzaeTable 2:STAUR_2131 之比較組
比較組之 A、B 各為 Genome,Species 欄為該 Genome 之種名,M.distance 為該點之 Mahalanobis 距離,橘色區塊為 Bacteria,其餘皆為 Fungi。
NC_014148.1
NC_014844.1
YP_003956356
YP_003951646
30 0 0.5 1 1.5 2 2.5 3 0 0.5 1 1.5 2 2.5 3
NT_107015.1<>NC_017850.1
NT_107015.1
NC_017850.1
STARU_2131
0 0.5 1 1.5 2 2.5 3 0 0.5 1 1.5 2 2.5 3NS_000201.1<>NC_007198.1
NS_000201.1
NC_007198.1
STARU_2131
31
Figure 8:STAUR_2131 比較組之散布圖
STAUR_2131 六組比較組中的三張散布圖,箭頭處為 outlier STAUR_2131 點的 位置,皆是肉眼可見的 outlier。 0 0.5 1 1.5 2 2.5 3 0 0.5 1 1.5 2 2.5 3 3.5
NC_015957.1<>NW_001884672.1
NC_015957.1
NW_001884672.1
STARU_2131
32
Figure 9:YP_003951762.1(STAUR_2131)演化樹
藍色 bar 為真菌群類,紅色 bar 為細菌群類。
利用 Neighbor-Join、PAM matrix 為 distance,bootstrap 100 建的樹,可以看到 YP_003951762 被分類在真菌群內,顯示其與真菌群有水平基因轉移現象。
Figure 10:YP_003951762.1(STAUR_2131)樣版演化樹
藍色 bar 為真菌群類,紅色 bar 為細菌群類。建樹條件如上。
33
Figure 11:YP_003950678 與 YP_003954354 之演化樹
其演化樹與樣版演化樹皆同,建樹方法為 Neighbor-Join、PAM matrix distance、 bootstrap 100。
34
Figure 12:YP_003955618 與 YP_003953284 演化樹
其演化樹與樣版演化樹皆同,建樹方法為 Neighbor-Join、PAM matrix distance、 bootstrap 100。
35
Reference
1. Jurkevitch, E., Predatory Behaviors in Bacteria—Diversity and Transitions. Microbe, 2007. 2(2): p. 67-73.
2. Trevors, J.T., Genome size in bacteria. Antonie van Leeuwenhoek, 1996. 69(4): p. 293-303.
3. Stuart Huntley, et al., Comparative Genomic Analysis of Fruiting Body
Formation in Myxococcales. Mol Biol Evol, 2011. 28 (2): p. 1083-1097.
4. Andam, C.P. and J.P. Gogarten, Biased gene transfer in microbial evolution. Nat Rev Micro, 2011. 9(7): p. 543-555.
5. N. B. Shoemaker, H.V., K. Hayes, and A. A. Salyers, Evidence for Extensive
Resistance Gene Transfer amongBacteroides spp. and among Bacteroides and Other Genera in the Human Colon Appl. Environ. Microbiol. , 2001. Vol. 67 no. 2 p. 561-568
6. William, D.T.A.-R.Y.M., Modular networks and cumulative impact of lateral
transfer in prokaryote genome evolution. PNAS, 2008. vol. 105 no. 29: p.
10039-10044.
7. Sorek, R., Genome-wide experimental determination of barriers to horizontal
gene transfer, 2007.
8. Brown, J.R., Ancient horizontal gene transfer. Nat Rev Genet, 2003. 4(2): p. 121-132.
9. Dubey, G.P. and S. Ben-Yehuda, Intercellular Nanotubes Mediate Bacterial
Communication. Cell, 2011. 144(4): p. 590-600.
10. Beatty, A.S.L.a.J.T., Genetic analysis of a bacterial genetic exchange element:
The gene transfer agent of Rhodobacter capsulatus. PNAS USA, 2000. 97(2): p.
859-864.
11. Zhao, Y., et al., Gene transfer agent (GTA) genes reveal diverse and dynamic
Roseobacter and Rhodobacter populations in the Chesapeake Bay. ISME J,
2008. 3(3): p. 364-373.
12. McDaniel, L.D., et al., High Frequency of Horizontal Gene Transfer in the
Oceans. Science, 2010. 330(6000): p. 50.
13. Richards, T.A., et al., Gene transfer into the fungi. Fungal Biology Reviews, 2011. 25(2): p. 98-110.
14. David Alvarez-Ponce, E.B., Phylogenomic networks provide insights into the
chimerical origin of eukaryotes, in SMBE2012; 2012: Dublin, Ireland.
15. Martin, T.D.a.W., Getting a better picture of microbial evolution en route to a
36 2187-2196
16. McInerney, D.A.-P.a.J.O., The Human Genome Retains Relics of Its Prokaryotic
Ancestry: Human Genes of Archaebacterial and Eubacterial Origin Exhibit Remarkable Differences Genome Biol Evol, 2011. 3 p. 782-790. .
17. Abbott, J.M.J.a.S.L., 16S rRNA Gene Sequencing for Bacterial Identification in
the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J Clin Microbiol., 2007. 45(9): p. 2761–2764.
18. John W. Whitaker, G.A.M.a.D.R.W., Prediction of horizontal gene transfers in
eukaryotes: approaches and challenges. Biochemical Society Transactions,
2009. 37: p. 792–795.
19. Kariin, S. and C. Burge, Dinucleotide relative abundance extremes: a genomic
signature. Trends in Genetics, 1995. 11(7): p. 283-290.
20. Nakhleh, L., Evolutionary Phylogenetic Networks: Models and Issues, in The
Problem Solving Handbook for Computational Biology and Bioinformatics, L.
Heath, Ramakrishnan, N, Editor 2010, Springer. p. 125-158.
21. Christiam Camacho, G.C., Vahram Avagyan, Ning Ma, Jason Papadopoulos, Kevin Bealer and Thomas L Madden BLAST+: architecture and applications. BMC Bioinformatics, 2009. 10:421.
22. BLAST+. Available from:
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ . .
23. Madden, T., Chapter 16 The BLAST Sequence Analysis Tool, O.J. McEntyre J, Editor 2002, National Center for Biotechnology Information (US): The NCBI Handbook [Internet].
24. Rodriguez, E.A.a.C., A Meta analysis study of outlier detection methods in
classification. 2004.
25. Williams, G., et al. A comparative study of RNN for outlier detection in data
mining. in Data Mining, 2002. ICDM 2003. Proceedings. 2002 IEEE International Conference on. 2002.
26. Rodriguez, E.A.a.C., On detection of outliers and their effect in supervised
classification. 2006.
27. Ben-Gal, I., Outlier Detection, in Data Mining and Knowledge Discovery
Handbook: A Complete Guide for Practitioners and Researchers2005, Kluwer
Academic Publishers.
28. J D Thompson, D.G.H., and T J Gibson, CLUSTAL W: improving the sensitivity of
progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res,
1994. 22(22): p. 4673-4680.
37
Analysis (MEGA) Software Version 4.0. Mol Biol Evol, 2007. 24(8): p.
1596-1599.
30. George E. Fox, J.D.W.a.P.J.J., How Close Is Close: 16S rRNA Sequence Identity
May Not Be Sufficient To Guarantee Species Identity. IJSEM, 1992. 42(1): p.
166-170
31. BG, S., Multilocus sequence typing:molecular typing of bacterial pathogens in
an era of rapid DNA sequencing and the Internet. Current Opinion in
Biotechnology, 1999. 3.
32. Ho, Y.-P., The horizontal gene transfer events and alterations of genomic GC
content in the genus Aspergillus, in Institute of molecular medicine and bioengineering2011, National Chiao Tung University.