國
立
交
通
大
學
應用化學系分子科學研究所
碩
士
論
文
利用步進式掃描時間解析傅式轉換紅外光譜法研究
CH
3C(O)OO 之紅外光吸收光譜
研 究 生:陳筍陽 (Sun-Yang Chen)
指導教授:李遠鵬 博士 (Dr. Yuan-Pern Lee)
中 華 民 國 九 十 八 年 八 月
i
摘要
利用步進式掃描時間解析傅式轉換紅外光譜儀並搭配多重反射的 White cell 偵測 CH3CO 與 O2反應生成的瞬態產物 CH3C(O)OO 與
α-lactone 的紅外光吸收光譜。吾人指派 t,c-CH3C(O)OO 在氣態的ν3 (1851 cm-1) 、ν5 (1372 cm-1) 、ν6 (1169 cm-1) 、ν7 (1102 cm-1) 與 t,t-CH3C(O)OO 在氣態的ν3 (1862 cm-1)、ν6 (1142 cm-1)、ν7 (1078 cm-1) 還 有 α-lactone 在 氣 態 的 ν2 (1960 cm-1) 。 基 於 理 論 計 算 B3LYP/aug-cc-pVDZ 預測的振動基態與激發態轉動常數與偶極矩導 數,可利用光譜模擬程式SpecView 模擬出各個振動模的振轉動譜帶, 其模擬光譜與實驗光譜相當吻合。這些振動模的氣態吸收波數除了 t,t-CH3C(O)OO 的ν7振動模之外,其餘振動模的振動波數皆與 Ne 間 質隔離光譜相差 9 cm-1以內,亦與 B3LYP/aug-cc-pVDZ 的計算值相 差2 %以內。依據譜帶的吸收面積與理論計算的紅外光吸收強度,吾 人指出 t,c-CH3C(O)OO 較 t,t-CH3C(O)OO 穩定 3.0 ± 0.6 kJ mol-1,以及
CH3CO + O2 Æ α-lactone + OH 的分支比例(branch ratio)為 0.04 ± 0.01。
ii 謝誌 還記得李老師第一次為我介紹matrix 系統的情景,老師以好壞學 生為喻,指出這套系統就像是把不乖的學生關在好學生之間,讓他沒 辦法作怪。不久之後,我就被關在一群認真的好學生之間。(呵呵~) 李老師是一個對每件事情都很認真的人,也試圖讓我了解,當我在每 個小細節都更認真一點點,可以使整件事情好上數倍的重要性。(Ex: 1.110 = 2 .6) 在這兩年半內,老師給我不少教誨也點出我許多的問題。一開始 從標準低、沒雄心壯志到我不是活在一個人的世界裡,最後到擠檸檬 比喻、抗拒學習與態度不佳等。十分感謝李老師對我這不才的學生耗 費了不少心思與無限多的容忍,希望我能盡快改正這些問題,突破自 我的枷鎖,以得到更大的成長空間。 還有我得感謝已經陪伴我六年的筠寶貝,已經相識九年之久的我 們有著十分特殊的緣分,很感謝您總是幫我打理好所有的一切,讓我 可以盡情地耍賴,也希望paper 能成功在 JACS 上發表,以補償寶貝 這兩年半的辛苦。 其它我也得感謝我的堂哥白馬,沒想到清明節我們竟然在陳懷公 祠堂內上演掃墓認親的戲碼,這世界真是太小了。還有很照顧我的隆 哥、很痞的中哥與王老師實驗的籃球隊,還有大實驗室的Ella、雪兒、 惠芬、小韓、棋文大師、怪叔叔小花、山大王黃登登、活潑的郁琁、
iii
靜靜的瓊緯、自嗨的小歪、鴻菊大姊的支持與陪伴。最後我祝福勁達、 max、雅苓、崇愷、令鈞、俞範、上臻等學弟妹實驗順利。
iv 目錄 第一章 緒論 ... 1 参考文獻 ... 7 第二章 實驗原理與技術 2.1 傅式轉換紅外光譜儀 ... 11 2.2 麥克生干涉儀之基本原理與傅立葉轉換之關係 ... 12 2.3 FTIR 的優點 ... 16 2.4 步進式掃描時間解析傅氏轉換紅外光譜儀 ... 18 附圖 ... 22 附表 ... 28 参考文獻 ... 29 第三章 實驗裝置、步驟與参數設定 3.1 實驗裝置 ... 30 3.2 實驗條件 ... 34 3.3 實驗步驟 ... 35 附圖 ... 40 第四章 結果與討論 4.1 理論計算 ... 45 4.2 (CH3)2CO/O2與CH3CHO/O2/Cl2雷射光解實驗之結果 ... 47
v
4.3 t,c-CH3C(O)OO 及 t,t-CH3C(O)OO 譜帶之指派 ... 49
4.4 cyc-H2CC(=O)O (α-lactone)的指派 ... 58
4.5 反應機制與反應動力學 ... 60 4.6 結論 ... 61 附圖 ... 63 附表 ... 78 參考文獻 ... 85
1 第一章 緒論 丙酮是大氣中含量最多的含氧碳氫化合物,也是大氣中的重要分 子;其主要來自於碳水化合物的燃燒,與生質燃燒(biomass burning) 或生物界釋放(biogenic emissions)[1]。丙酮在平流層(troposphere)中, 主要的損耗方式為光解[2]。當丙酮被光解後可能會產生 CH3CO (acetyl radical) , (CH3)2CO + hν Æ CH3CO + CH3 (1-1) CH3CO 不但是大氣污染的重要中間物[3],也是燃燒化學中的重要中 間物[4]。在大氣中,CH3CO 的主要來源除了酮類的光解之外,還有 乙醛和OH 經由氫原子擷取反應所形成, CH3CHO + OH Æ CH3CO + H2O (1-2)
在大氣中,CH3CO 會與氧氣迅速反應形成 CH3C(O)OO (acetyl peroxy
radical)。
CH3CO + O2 Æ CH3C(O)OO (1-3)
倘若大氣中含有少量NO2的話,CH3C(O)OO 還會再與 NO2反應,形
成空氣汙染之重要分子PAN (peroxyacetyl nitrate,CH3C(O)OONO2),
其反應式如下所示:
CH3C(O)OO + NO2 Æ CH3C(O)OONO2 (1-4)
PAN 為光化學煙霧(photochemical smog)的成分之一,也是都市空氣汙 染的重要成分,此外PAN 還扮演著 NOX的暫時儲存與運送的角色,
2 會隨著空氣對流將 NOX傳送到遠方[5]。到目前為止,尚無人觀測到 CH3CO 與 CH3C(O)OO 在氣態下的紅外光譜。若有這些分子在氣態下 的紅外光譜資訊,不但可以對它們的鍵能與結構資訊,有更進一步的 了解,更可以直接監測其在大氣中的濃度,甚至直接研究其化學動力 學,以下分為CH3CO 與 CH3C(O)OO 兩部分來討論。 目前已經有許多研究組研究過 CH3CO 的基態分子特性,像是電 荷密度、解離能障與 CH3內轉動能障。Bennett 等人[6]利用間質隔離
(matrix isolation)技術,觀測 CH3CO 的電子自旋共振光譜 (electron
spin resonance),得到 CH3CO 的孤對電子之電荷密度分佈。他們指出
CH3CO 為σ-type 的自由基,其孤對電子主要定域化(localized)於
C=O 官能基上碳的 sp 混成軌域,其 CCO∠ 為 1300。North 等人[7] 利用光碎裂物動能譜技術(photofragment translational spectroscopy), 得知CH3CO 解離成 CH3與CO 的位能障礙為 17.8±3 kcal mol-1。Hirota
等 人 [8] 利 用 傅 式 轉 換 微 波 光 譜 (Fourier transform microwave spectroscopy)技術,得知 CH3CO 的 CH3內轉動能障為 139.958±0.018
cm-1。
關於 CH3CO 基態之振動波數(wavenumber),已經有許多研究組
利用間質隔離法,觀測 CH3CO 在間質中的紅外吸收光譜,但它們的
3 於Ar 間質中,並藉由 Li 原子擷取 CH3COCl 上的 Cl 來產生 CH3CO。 他們指派1842 cm-1的吸收峰為CH3CO 的 C=O 伸展振動模,與 1329 cm-1的吸收峰為 CH3變形振動模。1973 年 Bennett 等人[10]將 K 原子 與 CH3COCl 沉積於 CO2間質中,並藉由 K 原子擷取 CH3COCl 上的 Cl 來產生 CH3CO。他們指派出 1796 cm-1為CH3CO 的 C=O 伸展振動 模與987 cm-1為C-C 伸展振動模。Bennett 認為因為鄰近之金屬原子 及間質效應(matrix effect)的關係,使他們得觀測到的 C=O 伸展振動 波數與Shirk 不同。1982 年 Jacox [11]利用對 SF6或NF3放電來產生F
原子,並將其與CH3CHO 沉積於 Ar 間質中,藉由 F 原子擷取 CH3CHO
上的 H 來產生 CH3CO。他們藉由同位素 (13CH3CHO、CH3CDO、
CD3CHO 與 CD3CDO) 實驗的輔助,指派出 CH3CO 的 C=O 伸展振
動模為1875 cm-1與CH3變形振動模為 1420 cm-1,其結果與Shirk 並
不一致。Jacox 認為這可能是因為 LiCl 或 HF 在間質中造成干擾。另 外也有研究組觀測到CH3CO 在沸石環境下的紅外吸收。Vasenkov 等
人[12]將 1-naphthyl acetate 或 pinacolone 吸附於沸石中,以 290 nm 雷 射將其光解產生 CH3CO,並利用時間解析霍式紅外光譜儀來偵測。
他們指派出 CH3CO 的 C=O 伸展振動模為 2125 cm-1,其結果與間值
隔離法相差甚遠。Vasenkov 認為這可能是因為沸石中的 Na+拉走 CH3CO 上 O 的電子對,使得 C 上的孤對電子對往 O 上分佈而提高了
4 C=O 鍵的鍵級數(bond order)。
關於 CH3CO 的電子躍遷光譜研究尚不完整,到目前為止僅有在
可見光區535 nm[13]與紫外光區 217 nm[14、15、16、17]觀測到 CH3CO
的吸收光譜,其電子躍遷也沒有被完整的指派。Mao 等人[18]利用 MR(SD)CI/DZP 的方法計算,計算基態(X2A') CH3CO 激發到 A2A''、
B2A'、C2A'與 D2A''的電子激發態的躍遷原點波長,分別為 476 nm、 253 nm、185 nm 與 177 nm。並指出 A2A''能態為束縛態(bound state) 與B2A'能態為預解離態(predissociation state),與 HCO 相似。Rajakumar 等人[13]利用共振腔振盪衰減(cavity ring down)的技術,在 535 nm 量 測到CH3CO 的電子躍遷(A2A''←X2A'),其吸收截面積為 σ =(1.1±0.5)
×10-19 cm2 molecule-1。他們用了六種不同的反應前驅物,卻都只觀測 到一寬廣並缺乏結構的可見光吸收譜帶,其結果與 Mao 等人的束縛 態預測不一致。Rajakumar 認為這可能是因為 CH3CO 容易解離或是 耦合的關係,造成激發態生命期太短。 當 CH3CO 與 O2反應時,可能有下列 3 種反應途徑: CH3CO + O2 + M → CH3C(O)OO + M (1-6) CH3CO + O2 → OH + 其它產物 (1-7) → CH2CO + HO2 (1-8) 一般認為反應(1-6)與反應(1-7)為主要的反應途徑[19]。反應(1-6)為一
5 個無能障的三體反應,其反應速率會隨著反應發生時的總壓力上升而 變快;當 CH3CO 與 O2 結合後會生成振動激發態的反應中間物 CH3C(O)OO*,而振動激發態的 CH3C(O)OO*可以藉由緩衝氣體碰撞 帶走能量而達到熱平衡。反應熱為-33.6 kcal/mol[19]。而在低壓環境 下,則會進行解離的反應途徑反應(1-7)。Tyndall 等人[20]研究 CH3CO 與O2的反應速率,並指出在總氣壓為 6 Torr N2時CH3C(O)OO 的產 率為50 %,在 60 Torr N2時之產率為90 %,在 100 Torr N2時之產率 則會>95 %。若總壓低於 1 mbar 時,則 OH 的產率約為 1[21]。 到目前為止,已經有許多研究組觀測到經由反應(1-7)而產生的 OH,但其共同產物(co-product)到現在仍十分不明確。Butkovskaya 等 人[22]藉由質子轉移游離法(proton-transfer ionization)來研究 CH3CO 與 O2反應的穩定產物,他們認為甲醛與 CO 為 OH 共同產物,因為 其產率(10%~15%)與 OH 相當。 但是 Hou 等人[19]用 G3MP2//CCSD /cc-pVDZ 計算 CH3CO + O2的位能曲面,並指出反應(1-7)的共同產物
很可能為 cyc-CH2CO2(cyclic α-lactone),且此反應的放熱為 30.3
kcal/mol。他們認為由於反應(1-7)大量放熱,cyc-CH2CO2分子會處於
激發態而容易解離生成甲醛與 CO,但是目前並沒有研究組在實驗上 證明cyc-CH2CO2的產生。
6
熱 6.2 kcal/mol[19]。Tyndall 等人[20]在 298K 觀測到 CH2CO 的產率
在1 torr 為 1 %,當壓力上升到 20 torr 則產率下降至 0.3 %。Lee 等人 [23]用 Density Function Theory 與 ab initio 的方法計算反應物、中間 物、過度狀態與產物的熱力學性質,並指出當溫度大於1000 K 時, CH2CO 才會成為主要的反應產物。
關於 CH3C(O)OO 基態之振動波數,目前已經有幾個研究組成功
地利用間質隔離法觀測到 CH3C(O)OO 在間質中的紅外吸收光譜。
Bruckmann 等人[24]將 PAN 熱裂解以產生 CH3C(O)OO,並將其沉積
於 Ar 間質中,首次觀測到 CH3C(O)OO 的紅外光吸收,但也留下許 多未知的吸收峰。Ahsen 等人[25]同樣以 PAN 熱裂解的方式來產生 CH3C(O)OO,並將 CH3C(O)OO 沉積於 Ar 與 Ne 間質中。他們藉由理 論計算的輔助,成功地指派出t,c-CH3C(O)OO 在 Ne 間質中的 11 個與 Ar 間質中的 12 個振動模、t,t-CH3C(O)OO 在 Ne 間質中的 7 個振動模、 與 Ar 間質中的 8 個振動模。並將 Bruckmann 觀測到的 CH3C(O)OO 紅外光吸收指派給t,c-CH3C(O)OO。 已經有許多研究組在近紅外光區(5582.5 cm-1)[26、27、28]與紫 外光區(200-260 nm)[29、30、31、32]觀測到 CH3C(O)OO 的吸收光譜。
Zahyubovsky 等人[26]利用共振腔振盪衰減(cavity ring down)技術,觀 測到CH3C(O)OO 於 5582.5 cm-1附近有吸收譜帶,並將其指派為A2A'
7 ←X2A'' 之 躍 遷 起 始 點 , 其 吸 收 截 面 積 為 σ = (1±0.5)×10-19 cm2 molecule-1。他們用同位素實驗證明觀測到的光譜並非泛頻(overtone) 或組合譜帶(combination band),並以理論計算(G2),與光譜模擬來說 明 所 觀 測 到 的 吸 收 譜 帶 為 t,c-CH3C(O)OO 之 貢 獻 。 他 們 提 出 t,c-CH3C(O)OO 在能量上較 t,t-CH3C(O)OO 結構穩定 289 cm-1,其後 一些研究組利用各種不同理論計算方法得知 t,c-CH3C(O)OO 的結構 是較穩定的構形,其異構物之間的能量差在2.5-3.5 kJ mol-1範圍[19、 25、26],而 t,c-CH3C(O)OO 轉換成 t,t-CH3C(O)OO 的能量障礙為(24-29 kJ mol-1)[25、26]。 有許多研究組研究過CH3CO + O2的反應速率常數[20、33、34、 35、36、37],一般認為其反應速率常數隨壓力增加而增加但隨溫度 增加而變小。Tyndall 等人[20]指出反應(1-6)的低壓極限之速率常數與 高壓極限之速率常數分別為6.2 × 10-30 cm6 molecule-2 s-1與3.2×10-12 cm3 molecule-1 s-1。由CH3CO 與 O2的反應速率常數有著正向壓力與負 向溫度效應可知,其反應為一個無活化能障礙的三體反應。 CH3CO 與 CH3C(O)OO 在間質中的紅外吸收光譜已被觀測到, 但是分子處於間質中的間質效應(matrix effect)的影響,使其測得的分 子振動波數與氣態不同。因此吾人利用雷射照射(CH3)2CO/O2 或 CH3CHO/Cl2/O2 的流動混合氣體來產生 CH3C(O)OO,並利用時間解
8
析步進式霍式紅外光譜儀首次偵觀到氣態下CH3C(O)OO 的紅外吸收
9 参考文獻
1. H. B. Singh, D. Herlth, D. O`Hara, W. Sachse, D. R. Blake, J. D. Bradshaw, M. Kanakidou, P. J. Crutzen, J. Geophys. Res. 99, 1805 (1994).
2. T. Gierczak, J. B. Burkholder, S. Bauerle, A. R. Ravishankara, Chem. Phys. 231, 229 (1998).
3. E. Grosjean, D. Grosjean, M. P. Fraser, G. R. Cass, Environ. Sci. Technol. 30, 2704 (1996).
4. W. C. Gardiner, Jr., Combus. Chem. (1984). 5. J. M. Roberts, Atmos. Environ. 24A, 243 (1990).
6. J. E. Bennett, B. Mile, Trans. Faraday Soc. 67, 1587 (1971).
7. S. W. North, D. A. Blank, J. D. Gezelter, C. A. Longfellow, Y. T. Lee, J. Chem. Phys. 102, 4447 (1995).
8. E. Hirota, A. Mizoguchi, Y. Ohshima, K. Katoh, Y. Sumiyoshi, Y. Endo, Mol. Phys. 105, 455 (2007).
9. J. S. Shirk, G. C. Pimentel, J. Am. Chem. Soc. 90, 3349 (1968).
10. J. E. Bennett, S. C. Graham, B. Mile, Spectrochim. Acta 29A, 375 (1973). 11. M. E. Jacox, Chem. Phys. 69, 407 (1982).
12. S. Vasenkov, H. Frei, J. Phys. Chem. A 104, 4327 (2000).
13. B. Rajakumar, J. E. Flad, T. Gierczak, A. R. Ravishankara, J. B. Burkholder, J. Phys. Chem. A 111, 8950 (2007).
14. M. M. Maricq, J. J. Szente, Chem. Phys. Lett. 253, 333 (1996).
15. M. Cameron, V. Sivakumaran, T. J. Dillon, J. N. Crowley, Phys. Chem. Chem. Phys. 4, 3628 (2002).
16. H. Adachi, N. Basco, D. G. L. James, Int. J. Chem. Kinet. 13, 1251 (1981). 17. N. Basco, S. S. Parmar, Int. J. Chem. Kinet. 17, 891 (1985).
18. W. Mao, Q. Li, F. Kong, M. Huang, Chem. Phys. Lett. 283, 114 (1998).
19. H. Hou, A. Li, H. Hu, Y. Li, H. Li, B. Wang, J. Chem. Phys. 122, 224304 (2005). 20. G. S. Tyndall, J. J. Orlando, T. J. Wallington, M. D. Hurley, Int. J. Chem. Kinet.
29, 655 (1997).
21. G. Kovács, J. Zádor, E. Farkas, R. Nádasdi, I. Szilágyi, S. Dóbé, T. Bérces, F. Márta, G. Lendvay, Phys. Chem. Chem. Phys. 9, 4142 (2007).
22. N. I. Butkovskaya, A. KuKui, G. Le Bras, J. Phys. Chem. A 108, 1160 (2004). 23. J. Lee, C. J. Chen, J. W. Bozzelli, J. Phys. Chem. A 106, 7155 (2002).
24. P. W. Bruckmann, H. Willner, Environ. Sci. Technol. 17, 352 (1983). 25. S. V. Ahsen, H. Willner, J. S. Francisco, J. Chem. Phys. 121, 2048 (2004).
26. S. J. Zalyubovsky, B. G. Glover, T. A. Miller, J. Phys. Chem. A 107, 7704 (2003). 27. H. E. Hunziker, H. R. Wendt, J. Chem. Phys. 64, 3488 (1976).
28. Y. J. Hu, H. B. Fu, E. R. Bernstein, J. Phys. Chem. A 110, 2629 (2006). 29. C. M. Roehl, D. Bauer, G. K. Moortgat, J. Phys. Chem. 100, 4038 (1996). 30. M. C. Addison, J. P. Burrows, R. A. Cox, R. Patrick, Chem. Phys. Lett. 73, 283
(1980).
31. N. Basco, S. S. Parmar, Int. J. Chem. Kinet. 17, 891 (1985).
32. G. K. Moortgat, B. Veyret, R. Lesclaux, J. Phys. Chem. 93, 2362 (1989). 33. M. A. Blitz, D. E. Heard, M. J. Pilling, Chem. Phys. Lett. 365, 374 (2002).
34. J. Sehested, L. K. Christensen, O. J. Nielsen, T. J. Wallington, Int. J. Chem. Kinet.
10
35. E. W. Kaiser, T. J. Wallington, J. Phys. Chem. 99, 8669 (1995).
36. C. E. McDade, T. M. Lenhardt, K. D. Bayes, J. Photochem. 20, 1 (1982). 37. G. S. Tyndall, J. J. Orlando, C. S. Kegley-Owen, T. J. Wallington, M. D. Hurley,
11 第二章 實驗原理與技術 2.1 傅式轉換紅外光譜儀 分子在紅外光有特性吸收,其吸收光譜有如人類指紋般的獨特性, 因此常用於未知分子的鑑定。其吸收強度經由校正後可用於濃度測 定;其吸收頻率還可以得到分子鍵結強度與鍵結長度等訊息。由於紅 外光譜可以獲得分子相當多的資訊,因此目前被普遍地運用於高分子 材料、環境科學與化學等領域中。早期採用分光式光譜儀(dispersive spectrometer)來取得紅外光譜,在 1891 年 Michelson [1]發明了干涉儀 (interferometer),利用干涉現象及理論計算推測,干涉儀所產生的干 涉圖譜可經由傅式轉換(Fourier transform, FT)轉換成一般光譜。到 1950 年代,Fellgett [2, 3]、Jacquinot [4, 5]等人提出傅式轉換光譜技 術有兩大優點(多重波長與高光通量),可以在更短時間內取得到高解 析度的光譜。1965 年,Cooley 與 Tukey [6]發展出快速傅式轉換演算 法(FFT)得以大幅降低光譜轉換的計算量,加上微電腦處理系統大幅 加速計算速度,使其在性能上遠優於分光式光譜儀,至今FT-IR 已普 遍地取代傳統紅外光譜儀。近年來,由於時間解析傅式轉換紅外光譜 法(time-resolved FT-IR spectroscopy)技術的發展,讓研究領域不再局 限於研究穩定分子的光譜,更可以用於生命期短暫的不穩定分子。利 用時間解析的FT-IR 來鑑定不穩定分子,更可以進一步研究其化學反
12
應的動力學、動態學等,功能極其強大。
2.2 麥克生干涉儀之基本原理與傅立葉轉換之關係
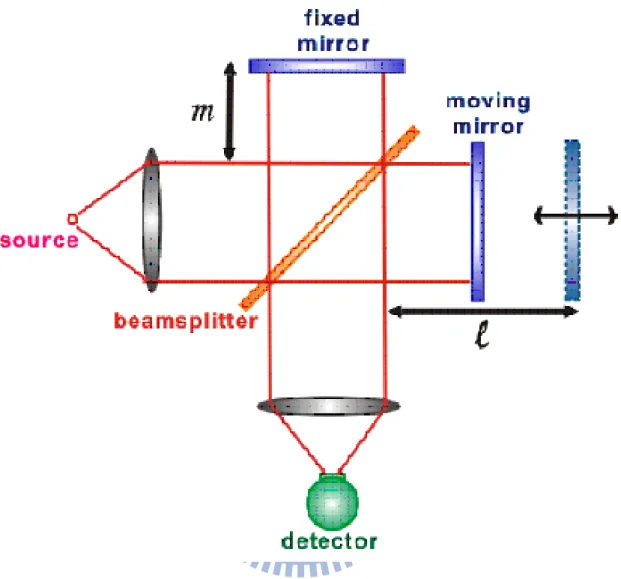
麥克生干涉儀(Michelson interferometer)是傅式轉換光譜儀內最 核心的元件,它是依照麥克森於 1891 年的理論所發展出來的。麥克 生干涉儀主要以移動鏡(moving mirror)、固定鏡(fixed mirror)及分光片 (beamsplitter)所組成,如圖 2-1。來自光源的電磁波以 45 度角入射至 分光片上,分光片將入射電磁波平均地分成兩道,一道穿過分光片射 入與光軸方向平行移動的移動鏡,另一道經由分光片反射後射至一個 固定鏡,此後兩道電磁波分別從固定鏡與移動鏡反射回分光片後重 合,一半強度的電磁波會導回光源處,另一半則可以到達偵測器中進 行偵測。重合後的電磁波會產生所謂的干涉現象。如果固定鏡和移動 鏡距離分光片分別有l 和 m 的長度,則從固定鏡與移動鏡回到分光片 重合的電磁波出現了所謂的光程差δ = 2(l-m),電磁波由於光程上的 不同會產生相位差(phase difference),進而發生干涉效應。以單色光 源(monochromatic)為例,若光程差為波長整數倍時會產生建設性 (constructive)干涉,光束強度相加;若光程差為半波長的奇數倍時會 產生破壞性(destructive)干涉,光束強度相減。因此,藉由移動鏡的移 動改變光程差,可得到光強度隨光程差之變化而改變的圖譜,即所謂 的干涉譜(interferogram)。干涉譜光譜是紀錄光強度隨光程差的變化,
13 可藉由傅式轉換與傳統光譜互相變換,如下數學式: ) v B( =
∫
∞ ∞ − δ δ e d I( ) i2πvδ (2-1) 其中I(δ)為干涉譜(光強度對光程差的函數),B(v)為傳統光譜(光強度 對頻率的函數),δ為光程差,ν為頻率。並藉由 Euler formula (eix = cos x + isin x)展開成( )
∞∫
∫
∞ − ∞ ∞ − + = I δ πvδ dδ i I δ πvδ dδ v B ( )cos(2 ) ( )sin(2 ) (2-2) )} ( { )} ( { sin cos I δ iF I δ F + = (2-3) 其中,Fcos{I(δ)}與 Fsin{I(δ)}分別為干涉譜 I(δ)經由餘弦與正弦轉換之結果。由θ(v)= )} ( { )} ( { arctan cos sin δ δ I F I F − 可得到 phase spectrum,用來進行 相位校正(phase correlation)。若只考慮實數部分並假設其為理想的左 右對稱干涉譜,則可以將(2-2)可改寫成
( )
=∫
∞ 0 ) 2 cos( ) ( 2 I δ πvδ dδ v B (2-4) 但是,現實中的移動鏡不可能移動到無限遠處,需要修正其數學式以 描述儀器所觀測得到的圖譜,因此在有限光程差內引入閘式截斷函數 (boxcar function) D (δ)。 D (δ) = 1 當 -L≦δ≦L (2-5) D (δ) = 0> 當|L∣<δ (2-6) 儀器上所觀測到的干涉譜I'(δ)為匣式截斷函數乘上理想之干涉譜14 ) (δ I , ) ( ' δ I =I(δ)×D(δ) (2-7) 故再利用傅式餘弦轉換可將上式I’(δ)轉換成傳統光譜 B(v): δ δ ν π δ δ D d I v B( )
∫
∞ ( ) ( )cos(2 ) ∞ − = (2-8) 其中I(δ)為理想干涉譜,D(δ)為匣式截斷函數。根據卷積定理 (convolution theorem),兩個函數相乘後作傅式餘弦轉換會等於兩函數 各自作傅式餘弦轉再進行卷積。其中,匣式截斷函數經過傅式餘弦轉 換後成為一sinc 函數 f(v), ) 2 ( sin 2 2 ) 2 sin( 2 ) 2 cos( ) ( ) ( L c vL L v L v L d v D v f π π π δ δ π δ = = ≡ ∞∫
∞ − (2-9) 此函數稱為儀器譜線形狀函數(instrument line shape function,ILS) , 如圖2-2(a)所示。因此儀器實際量測得到的光譜,為理想光譜與儀器 譜線形狀函數捲積的結果,如下 δ δ ν π δ δ D d I v f v B v G( ) ( )* ( )∫
∞ ( ) ( )cos(2 ) ∞ − = = (2-10) 其中G(v)為儀器實際量測到的真實光譜,*代表卷積(convolution)。以 單色光v1為例, (2-10)式可簡化成表示為 ] ) ( 2 [ sin ) ( 2 ) (v LB v1 c v1 v L G = π − (2-11) 原本無限狹窄之單色光譜因匣式截斷函數修正而使譜線變寬,解析度 變差;如圖 2-2(b)所示,主峰之半高寬(FWHM)為 0.605/L,主峰兩側 還產生一些側波(side lobe)。主峰附近的微弱吸收峰會造成混淆,為15 了減少側波造成光譜的不正確,將會引入一函數來減少側波所帶來干 擾,此函數稱為“削足”(apodization)函數。圖 2-3 為一些常用的削足函 數及削足後的光譜,而表2-1 列出數個經常使用的削足函數,其削足 效果與解析度的關係往往相反的,從圖與表中可看出這些削足函數有 一趨勢,即削足越徹底(側波強度越小),其主峰的半高寬就越大(解析 度越差)。本實驗的目的在於觀測的自由基分子的吸收光譜,而這些 自由基在結構上與前驅物相當接近,兩者的吸收頻率往往也相去不 遠,因此若使用削足能力較小的削足函數,則會造成欲觀測的自由基 吸收峰與側波互相混成,而這些側波會對極小吸收度(因為其濃度很 低) 的 自 由 基 吸 收 光 譜 受 到 嚴 重 的 干 擾 。 因 此 本 實 驗 使 用 名 為 Blackman-Harris 3-Term function 的削足函數[0.42323 + 0.49755cos(πD) +
0.07922cos(2πD)]其函數削足後主峰的半高寬為 116 %,側波最大振幅 值(Hs)與主峰高度(Hm)之百分比為 0.04 %。 一般而言,FTIR 光譜儀中至少具備三組干涉儀,分別為內部連續 式波長的紅外光源、氦氖雷射及白光光源之干涉儀,三組干涉儀共用 同一組分光片與移動鏡。其中一組干涉元件是用來定位移動鏡並決定 電腦取樣點的干涉元件,其光源是波長為 632.8 nm 的氦氖雷射。由 於氦氖雷射是單色光,其干涉圖譜會呈現餘弦函數,此餘弦波每段波 長有兩個零交叉點(zero-crossing),每兩個相鄰的零交叉點會相距
16
316.4nm。由於氦氖雷射頻率精確及穩定的特性,因此電腦可藉由這 些零交叉點精確地定位位移動鏡的位置並決定光程差;另一組干涉元 件則是用來決定零光程差(zero path difference,ZDP)的絕對位置,有 些光譜儀另外使用連續波長之白光光源(white light)來決定零光程差 的位置,由於任何波長在零光程差上均為建設性干涉,有最大光強 度;而零光程差以外的位置上,由於不同波長的光互相破壞性干涉使 光強度迅速變小,因此藉由其強而狹窄的波峰,便可以定義出零光程 差的位置並設定成光譜掃描的起點。本實驗系統在實驗前用紅外光源 (globar)對正(alignment)干涉儀,並儲存記錄波峰位置,作為零光程差 的參考點,藉此省去白光干涉儀,如圖2-4(a)所式。 2.3 FTIR 的優點 相較於傳統的分光式(dispersive)光譜儀,傅氏轉換光譜儀具有以下 的優點: (1) 多重波長優點(multiplex advantage): 分光式光譜儀是利用光柵將多色光分散開來並調整光柵角度使欲 觀測的單一波長投影在出口狹縫上,因此一次只能作單一波長的測 量。干涉儀可以同時偵測光區內所有波長,能在更短時間內擷取整個 光區的光譜。若使用同樣的光譜擷取時間、光譜解析度,掃描光譜 N 次後平均,可使訊雜比(S/N ratio)提升 N 倍,此稱之為Fellgett 優點
17 [2,3],由 Fellgett 在 1952 年提出。
(2) 高光通量優點(high throughput advantage):
分光式光譜儀波長解析度取決於光柵的色散(dispersion)與狹縫的 寬度,為了維持波長解析度,入射光必須先經過一狹縫,如此一來 便造成光強度變弱。而 FTIR 是使用圓形光圈,其入射的光量遠大於 單光儀,所以偵測器側得的訊號強度亦遠大於一般單光儀,靈敏度 因而增加,此稱為Jacquinot 優點[4,5],由 Jacquinot 在 1954 年提出。 (3) 高波數精確性(spectral accuracy advantage):
傳統光譜儀藉由光柵掃描來取得光譜,為了確定光柵轉動角度所對 應的波長與實際值是否一致,則要以外部標準波長進行校正,而光柵 旋轉的穩定及狹縫的控制,亦會影響其波數的精確性。而干涉儀以氦 氖雷射波長做基準,利用其頻率精確及穩定的特性精準地決定光程 差,使光譜的準確度可達0.001 cm–1,此稱為Connes 優點[7],由 Connes 在1958 年提出。 (4) 高解析度(high resolution): 分光式光譜儀的光譜解析度受限於狹縫寬度及光柵的線性色散率 倒數(reciprocal linear dispersion of the grating)。一般的分光式光譜儀 最高解析度為0.1 cm–1,而干涉儀的解析度則由最大光程差d 來決定: L d R 2 1 1 = = (2-12)
18 其中L 表示干涉光譜儀中移動鏡所能移動的最大距離。但光譜儀中氦 氖雷射頻率的穩定性及光束發散程度、所有光學鍵的對正程度、光圈 大小及移動鏡的穩定性亦會影響光譜解析度。目前市售FTIR 的最高 解析度可達0.001 cm–1。 (5) 抑制散射光(stray light): 分光式光譜儀往往會遇到散射光的干擾,如果用chopper,則只能 以固定頻率來調制,而連續式干涉儀的移動鏡以一個固定速率υ 移 動,因此將訊號作調頻(modulation),一波數為ν 的單色光經由干涉儀 調頻後,偵測器測到的訊號頻率為2υν ,其調制頻率隨波數而變,故 以適當的電子濾波器,可進一步過濾掉散射所造成的訊號。 2.4 步進式掃描時間解析傅氏轉換紅外光譜儀 一般干涉光譜儀的運作模式為連續式掃描,當干涉光譜儀在連續式 掃描的模式時,移動鏡以特定速率等速移動,並由氦氖雷射所造成的 零交叉點來決定移動鏡所走過的光程差,其光程差可以表示成時間的 函數δ=2vt,其中 v 為移動鏡的速率。由於連續式掃描運作時移動鏡 需要移動特定的光程差距離,才能得到完整的干涉光譜,所以每次掃 描時需要數十毫秒到數秒不等,因此無法偵測生命期短暫的物種,或 進行時間解析。 當干涉光譜儀在步進式掃描的模式下,移動鏡不是連續移動,而是
19 由電腦控制線性馬達使之停在氦氖雷射干涉譜的零交叉點上,待偵測 完畢後再移到下一個零交叉點位置。本實驗系統所使用的步進式干涉 儀(NEXUS 870, ThermoNicolet),其移動鏡位置的準確度可達±0.2 nm [8]。當移動鏡從 xi-1移動至 xi位置時,有一可設定的定位時間(settling time)用來提供移動鏡到下一個定位點 xi 的時間並待其穩定,之後再 以一脈衝式雷射觸發前驅物使反應發生,產物紅外光之放光或吸收的 時間開展(time evolution)以一適當的時間間隔 Δt 記錄下來,並多次重 複地觸發反應,用來平均訊號以提升訊雜比(S/N ratio),當 xi位置之 訊號擷取完畢後,鏡子移動至下一位置並穩定後,再次觸發反應並擷 取訊號,重複循環步驟直到掃描完畢為止。當鏡子停於 x1 位置,在 t1,t2,…… , tm 時 間 點 取 訊 號 作 ADC 轉 換 (analog-to-digital
conversion),可得到一組陣列 I(x1,t1),I(x1,t2),……I(x1,tm),用來記錄觸
發反應後光強度隨的時間變化,即時域譜(temporal profile)。當鏡子移 動至xi位置,同樣地產生(xi,t1),I(xi,t2),……I(xi,tm)的時域譜,重複循環
步驟直到掃描完畢為止。軟體會將所有時域譜組合成資料矩陣的形 式 , 並 藉 由 重 組 資 料 矩 陣 , 可 得 到 個 反 應 時 間 的 干 涉 譜 , 如 I(x1,tk),I(x2,tk),……I(xm,tk)代表時間點 k 的干涉譜。最後軟體會將各反
應時間的干涉譜進行傅式轉換,即可得到任何反應時間tk下的傳統光
20 目前,步進式掃描時間解析紅外光譜儀已經發展到相當成熟的階 段,但主要被運用於放光光譜上,其原因主要是因為放光光譜在一個 零背景(background free)的環境下擷取訊號,相較於吸收光譜是在很 大的背景信號下測量極微小變化量容易許多。藉由步進式掃描時間解 析紅外吸收光譜法可以獲得反應中間產物的吸收譜帶、反應動力學, 與放光光譜法無法提供的基態資訊與反應中間物的鑑定,其具有相當 大的價值。 本實驗系統的偵測器將訊號分成 ac 耦合與 dc 耦合[9],ac 耦合信 號用來反應觸發反應後之微小變化,而 dc 耦合信號則用來提供系統 的相位與背景資訊。其訊號處理過程如圖 2-6 所示。由偵測器的 dc 耦合端取得樣品在受到雷射激發前的靜態(static)干涉譜 I0(g),即為一 般連續式掃描所得的干涉譜,此干涉譜經傅式轉換後可得到樣品的背 景光譜 S0(ν)與其相對的相位 φ(ν),後者可用來提供 ac 耦合干涉譜作 相校正(phase correction)之用。當 dc 耦合訊號擷取完畢,光譜儀主導 觸發雷射系統以起始(initiate)反應槽內樣品的光化學反應發生,此時 偵測器的 ac 耦合訊號開始反映雷射激發後反應槽內分子對紅外光的 吸收度變化。在每一特定光程差 g 可測得一組時間解析之 ac 訊號 ΔIg(t),當掃描完所需的光程差後可得到一組完整的 ac 訊號資料矩 陣,經重排後則可得到每一特定時間 t 之干涉譜變化訊號 ΔIt(x),ΔIt(x)
21 經相位修正及傅氏轉換後,得到每一特定時間 t 之光譜強度變化量 ΔSt(v),並依照下式計算其差異吸收光譜(difference spectrum)ΔAt(v) ΔAt(v) =−log[1+ΔSt(v)/S0(v)] (2-13) 也就是說,當有新生成的產物吸收紅外光,使ΔSt(v)為負值,而 ΔAt(v) 會呈現正值;反之當反應物消失時,使ΔSt(v)為正值,故 ΔAt(v)會呈 現負值。
22
23 ν 0.605/L Hs Hm 1/L 2L f
(
ν)
(a) S=Hs/Hm x 100% 圖2-2 在有限光程差(L)下量測單色光波數之頻寬變化:(a)光程差由+L 到-L 的 boxcar 函數經傅式轉換後之圖譜波形為一 sinc x (sin x/x)函數。
(b)單色光波數為v1之正弦干涉譜經傅式轉換後的傳統光譜,此干涉譜 的最大光程差為L cm。
24
圖2-3 削足函數及其對應之儀器譜線形狀函數:(1) boxcar;(2) Bartlet;(3) Hamming;(4) 3-term Blackman-Harris;(5) N-B weak;(6) N-B medium;(6) N-B strong 函數
25
圖 2-4 FTIR 內部 IR 偵測器測到之多色光干涉譜與光二極體偵測到 之He-Ne 雷射干涉譜
26 圖2-6 步進式掃描時間解析傅式轉換光譜之數據擷取示意圖 (a)各曲線為光程差為 xi時所得之時間解析信號; (b)數據重組後,各曲線表示 ti時間下的干涉譜; (c)經 FT 後所得之時間解析光譜。 -0.02 0.00 0.02 0.04 1240 1260 1280 1300 0 10 20 30 40 50 60 Δ A b s o rb a n c e Waven um ber / c m -1 Time
(c)
27
圖2-6 由 ac/dc 耦合擷取之信號,其產生時間解析 吸收度差異譜(ΔAt(ν))之步驟。
28
表2-1 常用之削足函數(apodization function)之削足效果與主峰半高寬之比較。
Function Formula FWHM (%)c SLAM (%)d Ref.
Boxcar 1 60 2
Triangular (Bartlett) 1 - De 89 4.5 [10]
N-Ba weak 0.548 - 0.0833(1-D 2) + 0.5353(1-D2)2 72 5.8 [11]
N-B medium 0.261 – 0.154838(1-D2) + 0.894838(1-D2)2 84 1.4 [11]
N-B strong 0.09 + 0.5875(1-D2)2 + 0.3225(1-D2)3 97 0.3 [11]
Hamming (Happ-Genzel) 0.54 + 0.46cos(πD) 91 0.71 [10]
B-Hb 3-term 0.42323 + 0.49755cos(πD) + 0.07922cos(2πD) 116 0.04 [10]
B-H 4-term 0.35875 + 0.48829cos(πD) + 0.14128cos(2πD) + 0.01168cos(3πD) [10]
a表示Norton-Beer, b表示Blackman-Harris, c表示吸收峰的半高寬(單色光主峰之半高寬與所要求削足解析度之比值)
29 參考文獻
[1] A. A. Michelson, Phil. Mag. 31, 256 (1891).
[2] P. B. Fellgett, Symposium Ohio State Univ. (1952). [3] P. B. Fellgett, J. Phys. Radium 19, 187 (1958).
[4] P. Jacquinot, XVIIème Congrès du G.A.M.S., Paris (1954). [5] P. Jacquinot, J. Phys. Radium 19, 223 (1958).
[6] J. W. Cooley and J. W. Tukey, Math. Comput. 19, 297 (1965). [7] J. Connes, J. Phys. Radium 19, 197 (1958).
[8] E. Y. Jiang, Spectroscopy (Eugene, Or.) 17, 22 (2002).
[9] W. Uhmann, A. Becker, C. Taran, and F. Siebert, Appl. Spectrosc. 45, 390 (1991).
[10] E. H. W. Meijering, W. J. Niessen, and M. A. Viergever, Medical Image Analysis 5, 111 (2001).
30 第三章 實驗裝置、步驟與參數設定 3.1 實驗裝置
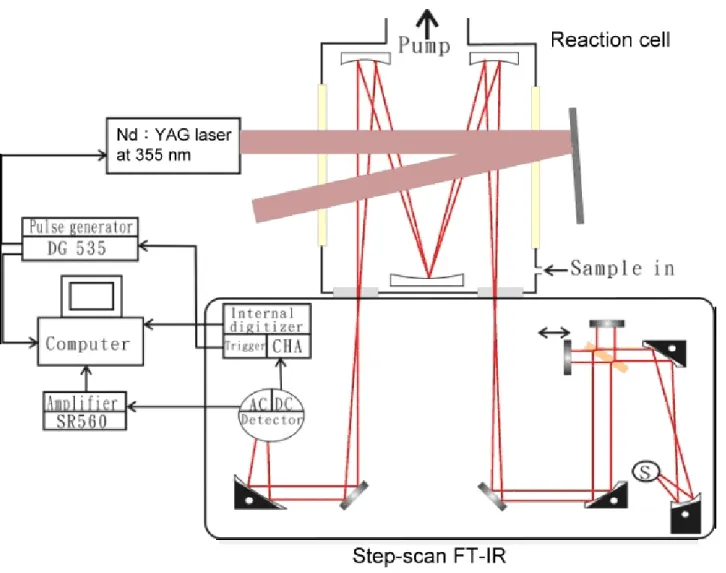
圖3-1 為本實系統之儀器裝置示意圖,其主要有:(1)光解雷射系 統(Nd:YAG laser)、(2)步進式掃描傅式轉換紅外譜儀(step-scan Fourier -transform spectrometer)、(3) 反應系統、(4)數據擷取與儀器時序控制 系統,茲分述如下:
(1)光解雷射系統
本實驗使用Nd:YAG 雷射(LOTIS TII,LS-2137/20)三倍頻波長 355 nm 的雷射光來光解前驅物以產生待測分子。此 Nd:YAG 雷射以 10 Hz 的重複頻率來照射反應槽內的反應前驅物。每發雷射光束到達 反應槽的窗口前能量約為69 mJ,其雷射光束形狀為一圓形光圈(雷射 光圈面積約為0.5 cm2)。吾人使用兩平面雷射反射鏡(3 cm × 12.5 cm) 分別置於反應槽後方與前方的光解光窗口,用來增加雷射通過反應槽 的次數,藉此增加雷射光的光解效率以提高訊號強度。 (2) 步進式掃描傅式轉換紅外譜儀 吾人使用NEXUS 870 (ThermoNicolet)的步進式傅式轉換紅外光 譜儀,其光源可選用 tungsten-halogen 或 globar、分光片可選用 CaF2
或 KBr、偵測器可選用 MCT 或 InSb。本實驗系統用 globar 來產生 mid-IR (9600−20 cm-1),此氣冷式 globar 無須外接冷卻水;分光片則
31
選用KBr (7400−350 cm-1)。由於光譜儀內部無法抽真空,吾人接氮氣 管線到光譜儀的 purge port,藉由氮氣排出光譜儀內的水氣及二氧化 碳,以降低其所帶來的干擾。偵測器則使用光電壓型(photovoltaic)的 MCT (Mercury Cadmium Telluride,1×1 mm2的偵測面積,20 MHz 的
頻寬,響應時間約為50 ns),使用前必須充填液態氮降低溫度到 77 K。 (3)反應系統 不鏽鋼反應槽置於光譜儀的樣品室中,其體積約為1600 cm3,且 反應槽底座有兩片圓形 BaF2光窗,將反應槽與光譜儀區隔開來。反 應槽之設計如圖3-2 所示,其夾層設計目的在於可通入高溫或低溫流 體來改變反應槽內氣體的溫度。反應槽內裝一熱偶溫度計用來測量反 應槽內氣體的實際溫度。氣體反應物經由反應槽內一個上有數個平均 刺穿小洞的teflon 圓環(sample injection ring)進入反應槽內,如此可使 樣品較均勻散佈於反應槽內,減少反應槽內的擾流(turbulence)發生, 以減少光譜的雜訊。反應槽內石英光窗下方放置兩teflon 擋板,當通 入的purge 氣體經過擋板會形成噴射氣流沖刷光窗,用來避免光解產 生的碳化物附著在石英光窗上,使雷射光穿透不易。
為了增加反應所產生的瞬間產物之吸收度,反應槽內放置 White cell (Infrared Analysis,model M-3-8V)鏡片來增加吸收路徑。White cell 包含了三片表面鍍金且相同曲率半徑的球面鏡,其中兩片由一個半徑
32 為20 cm 的球面鏡切為兩半圓形(M2 及 M3),另一片由另一個球面鏡 切為 T 形(M1),如圖 3-3 所示。半圓形鏡與 T 形鏡間的距離(20 cm) 即為球面鏡的兩倍焦距。入射光源由 T 型鏡的缺口處導入,在 T 型 鏡與兩半圓鏡間進行32 次反射,可使 IR 光在反應槽內行進 6.4 m 的 路徑,再由另一缺口射出。若要改變 IR 光束之反射次數,可調整兩 個半圓鏡之夾角。通常 White cell 中所使用反射 IR 的球面鏡需要有很 高的反射率,因此用高反射率的金做為鍍膜(coating)材質。本實驗系 統所用的金鏡,其反射率在 2.5-20 um 約大於 98%,IR 光經過反射 32 次後仍有 52%以上的強度。
反應槽上接兩個電容式壓力計分別為10 Torr (MKS,model 622A) 及1000 Torr (MKS,model 122AA)以量測系統壓力。反應樣品的流量 則使用針閥控制,並以質量流量器(mass flow transducer,MKS,model 0258C,最大流量為 1000 sccm)進行測量。反應槽上方連接抽氣速率 約800 l min−1的乾式幫浦(TAIKO KIKAI,model SLT-333P)保持氣體 流動。
氧氣由高壓鋼瓶流出後,分成兩路:一路為大流量同時作為反應 物與焠息體,直接經流量計及針閥組,從兩片石英光窗下方進入反應 槽,經由入口處放置的兩 teflon 擋板形成小噴射氣流,以避免光解後 的碳化物沉積在光窗上造成光解過程中雷射能量逐漸下降的問題;另
33
一路則為小流量,通往樣品瓶,用以帶出反應前驅物。稀釋的反應前 驅物經由上述之圓環注入到反應槽中。
(4)數據擷取系統與儀器時序控制
偵測器之信號輸出端分成ac 與 dc 耦合。ac 耦合信號可利用前置 電壓放大器(Stanford Research System,SR560) 以特定電子濾波頻寬 (通常為 100−1 M Hz)放大 20 倍,放大後的信號經一 14 位元之類比/ 數 位 轉 換 器(Gage Applied Technology , CompuScope 14100 , 108 samples s−1,最快時間解析為10 ns)擷取後由電腦處理。而 dc 耦合信 號則不經過放大器,直接由 FTIR 內部的 16 位元類比/數位轉換器 (2×105 samples s−1,最快時間解析為5 μs)進行訊號擷取。
本實驗使用的NEXUS 870 (ThermoNicolet)步進式傅式轉換紅外 光譜儀被設定成主動模式(master mode),也就是由 FTIR 主導所有動 作時間順序的開始。如圖 3-4 所示,當移動鏡由 1 號位置移往已預先 決定的2 號位置時(時間由 aÆb),會造成 He-Ne 雷射訊號的調變。移 動鏡抵達2 號位置後可設定一定位時間 A (settling time,A=c-b)待移 動鏡穩定,並在時間點c 之後設定一段時間 B (static average time, B=d-c)擷取反應槽內的靜態背景光譜;此時取得的訊號由偵測器的 dc 耦合端輸出。而後FTIR 送出一觸發來啟動光解雷射,由於雷射接受 觸發到雷射出光會有延遲,此段延遲時間F (post-trigger delay,F=e1-d)
34
可依所使用的雷射類型設定,使其在時間點e1時雷射剛好到達反應槽
並激發反應,FTIR 並於 e1開始擷取信號(take data,TKDA),開始紀
錄反應槽內光強度量隨時間的變化,此時取得的訊號由偵測器的 ac 耦合端輸出到外部的類比/數位轉換器(analog to digital convertor)。為 了增加訊雜比,在同一位置可進行多次雷射觸發並平均,雷射擊發的 重覆頻率為 1/E。圖 3-4 中顯示在同一鏡片位置擷取三次相同的化學 反應事件用以平均,之後再移往下一個鏡片位置進行上述相同的步驟 直到得到完整的干涉譜。時域譜Ix(t)中,C 代表每一積分閘門大小, D 代表時域切割(time slice)的個數,故 C×D 即為所擷取的總時間。吾 人進行步進式掃描時間解析實驗的詳細參數列於3.3 節末。 3.2 實驗條件 1. 光解效率的評估 本實驗利用波長355 nm 雷射光解 Cl2以產生Cl 原子,雷射光到達 反應槽的窗口前其能量為~69 mJ plus-1、光束截面積為~0.5 cm2,其在 反應槽中來回反射,達到108 cm(18×6)的總路徑長。由 Cl2在 355 nm 波長的吸收截面積σ(Cl2)355nm = 1.6×10-19 cm2 molecule-1與雷射光通量 (fluence) 2.5×1017photons cm-2,則可知Cl 2的光解效率為~4 %。本實 驗使用分壓為1 Torr (= 3.24×1016 molecules cm-3 在 298 K) 的 Cl2,假 設量子產率Φ=2 (Cl2吸收一 355 nm 的光子產生兩個 Cl 自由基),則
35 可知雷射光解後產生了分壓為~0.8 Torr 的 Cl 原子。 2. 樣品及反應條件 本實驗以Cl 擷取 CH3CHO 上的 H 來產生 CH3CO,而反應生成的 CH3CO 會迅速與 O2反應形成CH3C(O)OO,如下所示: CH3CHO + Cl ⎯⎯→ CH3CO + HCl (3-1) CH3CO + O2 + M ⎯⎯→ CH3C(O)OO + M (3-2) 各樣品在標準狀態(S.T.P.表示 273.15 K 及 1 atm)下的流速分別為: Fbubble ~ 0.08、FCl2 ~ 2.0、FO2 ~ 19.5 STP cm3 s-1。所使用的試劑CH3CHO
(Riedel-de Haën,99.5 %) 、Cl2 (99.99 %,AGA Specialty Gases)及 O2
(99.995 %,AGA Specialty Gases)皆未加以純化,其蒸氣壓分別為 CH3CHO 約 0.4 Torr、Cl2約1 Torr、O2約 100 Torr。
3.3 實驗步驟 (1)反應槽中 White cell 鏡片之對正 由於人眼看不見在White cell 內反射的紅外光束,因此在對光時 必須先將光源切換成可見光,並更換成 CaF2之分光片,調整適當 的光圈大小,以方便作初步對正: 1. 調整 White cell 鏡片在鏡架上的位置及入射光窗下方反射鏡的角 度,使可見光束聚焦於反射鏡 M1 的延伸平面上,如圖 3-3 中標 記為0 的位子。
36 2. 調整可見光反射角度,使由聚焦點發散的可見光束能完整覆蓋整 個M3。 3. 藉由調整 M3 背面的三顆螺絲來改變 M3 的角度,使可見光聚焦於 M1 上標記為 2 的位置。 4. 藉由調整 M1 背面的三顆螺絲來改變 M1 的角度,使得反射後再次 發散的可見光能完全覆蓋整個M2。 5. 藉由調整 M2 背面的三顆螺絲來改變 M2 的角度,使得可見光再次 聚焦於M1 上標記為 4 的位置。 6. 微調 M2 與 M3 的反射角度,使得可見光在 M1 上呈現兩列平行排 列的聚焦點,在反射達 32 次後導到出口光窗。此時必須檢查 M1 上的聚焦點形狀與大小是否完全一致,若否,則須繼續微調螺絲 直到形狀大小均一致為止。 7. 調整出口光窗下方反射鏡的角度,直到偵測器測得的可見光強度 達到最大。 8. 微調偵測器前方的反射鏡,直到偵測器所測的可見光強度達到最 大。 9. 再將光源切換成 IR 光,更換分光片成 KBr,置入一減光片(screen B) 於反應槽與偵測器之間的IR 光徑上,並設定光圈為 32,此時到達 偵測器的 IR 光束會與偵測器的光窗同大(直徑為 0.94 mm),才可
37 進行auto align。 10.將所測得的 IR 強度作紀錄,若訊號強度遠小於 3.7 V,則重複步 驟1−9 進行檢查及微調,使訊號強度恢復至以前的大小。 (2)流量計校正 由於本實驗需要測量氣體樣品的流量,因此需要對氣體流量計作校 正以方便日後的定量實驗。首先對流量計的 readout 作校正,先用提 供 0 伏電壓給 readout 的訊號線並調整 readout 的歸零可變電阻將其 讀值歸零;再提供 5 伏電壓給訊號線並調整 readout 的放大可變電 阻 , 將 讀 值 調 至 流 量 計 的 最 大 流 量 。 吾 人 採 用 濕 式 測 量 法(wet testmeter)來校正流量計,利用排水法測定的原理,測量單位時間內排 出的氣體體積,再校正成STP 標準狀態(0°C, 1 atm)下的流量,如下 式: room water room a N
T
P
P
F
F
273
760
×
−
×
=
其中FN為標準狀態(0°C, 1 atm)下之標準流量(cm3 s-1),Fa、Proom、Pwater、Troom分別表示為readout 的流量讀值(cm3 s-1)、周遭的壓力(torr)、
水的飽和蒸汽壓力(torr)與周遭的溫度(K)。 (3)光譜儀的參數設定
1. 連續式掃描設定
38
進入連續式掃描之參數設定的視窗,此時於 Collect 欄下設定掃描次 數,解析度等;在Bench 欄下可觀測到偵測器接收電壓最大值及最小 值、零光程差位置與目前所使用的偵測器及分光片資訊等,並可設定 移動鏡掃描的速度(1.8988 cm/s)、光圈大小及光源選擇與記錄光區範 圍;在 Advanced 欄裡設定 zero filling=1 level(表示在兩個數據點間插 入一數據點,使干涉譜圖形較為平滑但不會影響解析度),削足函數 =Blackman-Harris,跳點取樣區間(sample spacing) =1(表示每一個零交 差點取一個數據點),相位校正方法=Mertz,並選擇只掃描單邊的干 涉譜,所有條件確認無誤後按 OK 回到原視窗。按下 Collect→Collect Background 即可用連續掃描模式取得光譜。 2. 步進式掃描設定 按下 SST→Step-Scan Time-Resolved 來設定時間解析實驗之參數, 如圖 3-5 所示。本實驗用紅外光濾光片(IR fillter)縮限實驗光區為 4100−800 cm-1,其點取樣區間設為 3 (每三個零交差點取一個數據 點),光譜解析度(spectral resolution)設為 2.0 cm-1,零光程差前多取的 點數(IFG points before ZPD)設為 64 點,則移動鏡需停 5560 點。時序 設定相關設定:移動鏡定位時間(settling time)設為 150 ms,擷取背景 訊號的時間(static average time)設為 100 ms 與 post-trigger delay 設為 130 μs (雷射從收到 trigger 訊號到發出雷射光至反應槽前所需要的時
39
間)。雷射重覆速率為 10 Hz (trigger interval=0.1 s),在每個移動鏡暫 停的零交叉點進行 10 次擊發(number of triggers per step)以平均信 號。訊號擷取相關設定:時間解析度設1 μs,而 average data points 設100 (表示每張間格 1 μs 的光譜是由 100 個 10 ns 的積分視窗所組 成),並設定 300 個時間紀錄點(number of time slices) (表示可以獲得 雷射光解後300 × 1 μs 時間內的光譜資訊)。當移動鏡全程掃描 5560 個點約需2 小時。設定完所有參數按 OK 回到原視窗,然後按下 SST →Collect Sample 即可使用步進式掃描模式擷取光譜。
40
41
圖 3-2 反應槽設計圖:(a)為透視圖;(b)為俯視圖,其中 White cell 的鏡面M1、M2 及 M3 分別在反應槽的下方及上方
42
圖3-3 White cell 光線行進示意圖(僅表示前 4 個反射光徑),其中各數 字表示光線抵達M1 鏡面時已通過反射槽的次數。
43
44
45 第四章 結果與討論
4.1 理論計算
吾人使用Gaussian 03 軟體[1],以密度泛函理論(density function theory)B3LYP[2,3] 方 法 搭 配 aug-cc-pVDZ 基 底 函 數 , 預 測 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 在基態平衡時的最佳幾何結構、簡 諧振動波數、紅外光吸收強度、振動基態(v = 0)與激發態(v = 1)的轉 動常數,及兩異構物之間的相對能量,與異構化位能障礙。 藉由理論計算可以得知 CH3C(O)OO 有兩種穩定的幾何結構 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO , 如 圖 4-1 所 示 。 吾 人 並 將 B3LYP/aug-cc-pVD 與 B3LYP/6-311G(d,p)[4]預測的構形結果列於圖 中以比較其差異,由圖可知其構形差異小於0.7 %。 近期的理論計算指出 t,c-CH3C(O)OO 的分子結構在能量上比 t,t-CH3C(O)OO 穩定,其可能是因為 t,c-CH3C(O)OO 的末端氧原子會 與甲基上的氫形成微弱的作用力所導致。Zalyubovsky 等人[5]利用 G2 計算出t,c-CH3C(O)OO 在能量上較 t,t-CH3C(O)OO 穩定 3.5 kJ mol-1;
Ahsen 等人[4]利用 B3LYP/6-311G(d,p) 計算得其相對能量為 2.3 kJ mol-1;Hou 等人[6]利用 G3MP2 計算得其相對能量為 2.5 kJ mol-1。吾 人 利 用 理 論 計 算 B3LYP/aug-cc-pVDZ 並 考 慮 振 動 零 點 能 量 (zero-point-energy)修正可知 t,c-CH3C(O)OO 比 t,t-CH3C(O)OO 穩定 3.4
46
kJ mol-1,相較於先前的文獻數據,其都在誤差範圍內。在室溫下, CH3C(O)OO 的兩個異構物可以藉由轉動 C-O 鍵並越過一個位能障礙
來互相轉換。吾人利用 B3LYP/aug-cc-pVDZ 計算 t,c-CH3C(O)OO 轉
換到t,t-CH3C(O)OO 的位能障礙為 24.3 kJ mol-1,與Zalyubovsky 等人
[5] 利 用 G2 計 算 所 得 之 24.2 kJ mol-1、Ahsen 等 人 [4] 利 用 B3LYP/6-311G(d ,p) 計算所得之 23.8 kJ mol-1 及 Lee 等人[7]利用 CBS//B3LYP/6-31G(d,p) 計算所得之 29.3 kJ mol-1一致。 表 4-1 與 4-2 分 別 為 t,c-CH3C(O)OO 及 t,t-CH3C(O)OO 在 B3LYP/aug-cc-pVDZ 與 6-311G(d,p)所預測的簡諧振動波數[4],以及 其在Ar 與 Ne 間質下觀測到的振動波數[4,8]。從間質實驗所得之紅 外光吸收強度來看,在吾人的實驗光區內 (1000-4000 cm-1)內, t,c-CH3C(O)OO 前四個較強的紅外光吸收振動模ν3、ν5、ν6 及ν7,分 別為1850、1372、1160 與 1106 cm-1。而 t,t-CH3C(O)OO 前三個較強 的紅外光吸收的振動模ν3、ν5及ν7,分別為 1861、1141 與 1096 cm-1。
吾人以B3LYP/aug-cc-pVDZ 預測 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的
振動波數,其與 B3LYP/6-311G(d,p)[4]預測的結果之差異分別不超過 1.8 %與 1.7 %,與 Bruckmann 等人[8]及 Ahsen[4]等人在 Ar 間質下測 得的振動波數分別相差不到3.0 %與 2.7 %,與 Ahsen[4]在 Ne 間質測 得的振動波數都相差不到2.5 %。
47
圖 4-2 為利用 B3LYP/aug-cc-pVDZ 預測 t,c-CH3C(O)OO 的ν3、
ν5、ν6及ν7與 t,t-CH3C(O)OO 的ν3、ν5、ν6及ν7振動模在振動時的位
移向量及偶極矩導數在分子 a、b、c 轉軸上的分量。表 4-3 與 4-4 分 別為利用B3LYP/aug-cc-pVDZ 計算 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO
的基態(v =0)及振動激發態(vi =1)轉動常數之列表,其中 A"、B"、C" 代表基態的轉動常數,而A'、B'、C'代表振動激發態的轉動常數,並 列出A'/ A"、B'/ B"、C'/ C"的比例。 4.2 (CH3)2CO/O2與CH3CHO/O2/Cl2雷射光解實驗之結果 本實驗利用雷射激發(CH3)2CO/O2或CH3CHO/O2/Cl2之混合氣體 來產生CH3C(O)OO,並利用步進式掃描時間解析傅式轉換紅外吸收 光譜儀偵測CH3C(O)OO 在氣態下的紅外吸收光譜。以下為實驗結果 與討論。 4.2.1 (CH3)2CO/O2於248 nm 雷射照射後的時間解析差異光譜 利用 248 nm 雷射照射(CH3)2CO/O2 (1/36)的混合氣體,其總壓力 約為150 Torr、平均溫度為 298 K。雷射激發後 0-200μs,以 50 μs 為 時間間隔之時間解析差異光譜示於圖 4-3;藉由時間解析差異光譜可 以清楚地表示譜帶隨時間的消長趨勢。在此差異光譜中向上的吸收 (正吸收)表示有產物生成,而向下的吸收(負吸收)表示有反應物消 耗。其中,在1800-1700 cm-1、1475-1325 cm-1與1240-1190 cm-1一帶
48
之雜亂吸收峰是因為前驅物(CH3)2CO 在這些光區的飽和吸收所造成
的影響,因此在這些光區內無法提供可信的資訊。在 2349 cm-1附近 可以觀測到CO2的吸收譜帶,而在 1850 cm-1、1160 cm-1與1090 cm-1
附近,有三個立即生成的吸收譜帶隨著時間衰減。為了更清楚表示這 些譜帶,吾人將其分別標記成A1(1850 cm-1)、A2(1160 cm-1)、A3(1090 cm-1)。 4.2.2 CH3CHO/O2/Cl2於355 nm 雷射照射後的時間解析差異光譜 利用 355 nm 雷射照射 CH3CHO/Cl2/O2(2/5/500)的混合氣體,其總 壓力約為100 Torr、平均溫度為 298 K。雷射激發後 0-200μs,以 50 μs 為時間間隔的時間解析之差異光譜示於圖 4-4。其中,在 1746 cm-1 附近的向下吸收譜帶表示前驅物CH3CHO 的消耗,而 2349 cm-1附近 有CO2的吸收譜帶。在1850 cm-1、1370 cm-1、1160 cm-1與1090 cm-1 附近,有四個快速生成的吸收譜帶隨著時間的衰減。其中 1850 cm-1、 1160 cm-1與 1090 cm-1附近的吸收譜帶與(CH3)2CO/O2之雷射光解實 驗所觀測到的光譜相同,由於CH3CHO/O2/Cl2之雷射光解實驗沒有前 驅物飽和吸收的問題,因此可以在 1370 cm-1附近多觀測到一個吸收 譜帶。由於這四個吸收譜帶的衰減趨勢一致,因此其很有可能為同一 物種所造成的貢獻,為了更清楚表示這些吸收譜帶,吾人將其標記成 A1 (1850 cm-1)、A2 (1160 cm-1)、 A3 (1090 cm-1) 、A4 (1370 cm-1)。
49 4.3 t,c-CH3C(O)OO 及 t,t-CH3C(O)OO 譜帶之指派 4.3.1 (CH3)2CO/O2之雷射光解反應系統討論 當248 nm 雷射激發光解 (CH3)2CO 以產生 CH3CO 及 CH3自由基 後,實驗系統中可能發生的反應如下: CH3CO + O2 Æ CH3C(O)OO (4-1) CH3 + O2 Æ CH3OO (4-2) CH3 + CH3 Æ C2H6 (4-3) CH3CO + CH3CO Æ (CH3CO)2 (4-4) 由於吾人的實驗條件中,O2的濃度大過(CH3)2CO 約 36 倍,再考慮 (CH3)2CO 之光解率為 0.1 %後,可知 O2的濃度會大於 CH3CO 及 CH3 約36000 倍,吾人並考慮 k1=4.0×10-12 cm3 molecule-1 s-1 (150 Torr N2, 298K) [9,10]、k2=4.8×10-13 cm3 molecule-1 s-1 (150 Torr O2,298K) [11]、 k3=5.5×10-11 cm3 molecule-1 s-1[11]與 k4=1.75×10-11 cm3 molecule-1 s-1[12] 可預期反應(4-3)及(4-4)無法與反應(4-2)、(4-1)競爭而不重要,而且其 紅外光吸收也與 A1-A3 譜帶不符。根據反應速率 k1與氧氣濃度,吾
人可估計CH3CO 的生命期(life time)將小於 55 ns,其與 A1-A3 譜帶
不符,因此吾人所觀測到的 A1-A3 吸收譜帶很可能為 CH3OO 或
CH3C(O)OO 的貢獻。本實驗室已經利用同一套系統研究 CH3OO 在氣
態下的紅外吸收光譜[13],其 ν1-ν6、ν9與 ν10的吸收峰分別在 3033、
50 收峰會干擾A2 與 A3 譜帶,但是 CH3OO 的紅外光吸收強度微弱,而 且其光譜輪廓與吾人觀測到的A2、A3 譜帶不符,可預期 CH3OO 在 A2 與 A3 譜帶所造成的影響應該不大。因此吾人所觀測到的 A1、A2、 A3 譜帶很可能是 CH3C(O)OO 所造成的貢獻。 4.3.2 CH3CHO/O2/Cl2之雷射光解反應系統討論 利用 355 nm 雷射激發光解 Cl2以產生Cl 原子後,實驗系統中可 能發生的反應如下: CH3CHO + Cl Æ CH3CO + HCl (4-5) Æ CH2CHO + HCl (4-6) CH3CO + O2 + M Æ CH3C(O)OO + M (4-1) CH3CO + Cl Æ CH2CO + HCl (4-7) Cl + O2 + M Æ ClOO + M (4-8) CH3CO + Cl2 Æ CH3C(O)Cl + Cl (4-9) 吾人藉由 Cl 擷取 CH3CHO 上的 H 以產生 CH3CO,其會與大量的氧 氣反應生成CH3C(O)OO。由 Niki[14]的研究可知,反應(4-5)的產率大 於99 %,因此經由反應(4-6)所產生的 CH2CHO 之濃度必定極小。由 於CH3CHO 光解成 HCO 與 CH3所需的波長門檻為315 nm[15],而且 其在355 nm 的吸收截面積僅為~4×10-23 cm2,因此不會對本反應系統 造成影響。此外,本實驗條件下的O2濃度大於 Cl 一千倍以上,並考 慮k7 = 1.8×10-10 cm3 molecule-1 s-1 [11]可知反應(4-7)無法與反應(4-1) 競爭CH3CO。而 Cl 會與 O2反應生成ClOO,其為三體反應,反應速
51 率常數為k8 = 1.7×10-33 cm6 molecule-2 s-1[16],可估計其在總氣壓為 100 Torr 的實驗條件下二級反應速率約為 k′8 = 1.7×10-33 cm3 molecule-1 s-1,相較於 k5 = 7.6×10-11 cm3 molecule-1 s-1 [14]約小了一萬倍以上而無 法與其競爭。除了O2之外,Cl2也會與 CH3CO 反應並生成 CH3C(O)Cl 分子,其反應速率為k9 = 2.4×10-11 cm3 molecule-1 s-1[11]。由於 O2與 Cl2的濃度均大於CH3CO 十倍以上,藉由擬一級反應(pseudo-first order) 來推論,可估計本實驗條件中反應(4-1)的反應速率快過反應(4-9)約 15 倍,因此在本實驗條件下大部份的 CH3CO 會與 O2 作用生成 CH3C(O)OO,僅有少數的 CH3CO 會與 Cl2反應生成 CH3C(O)Cl。因 此吾人觀測到的吸收譜帶A1、A2、A3、A4 很可能是 CH3C(O)OO 所 造成的貢獻。 由圖 4-3 與圖 4-4 兩種不同反應前驅物之光解實驗光譜可知,在 這兩個實驗中都觀測到三個相同的吸收譜帶A1、A2、A3,其不論在 吸收頻率或相對吸收強度上都很一致。吾人考慮兩種實驗之可能反應 機構可知,其共同的反應中間物為CH3CO,而在大量 O2環境下,最 可能的共同產物為 CH3C(O)OO,因此吾人所觀測到的吸收譜帶極有 可能為CH3C(O)OO 的紅外光吸收。由於 CH3CHO/O2/Cl2的雷射光解 實驗中沒有前驅物飽和吸收的干擾,不但可以多觀測到一個吸收譜帶 A4,還可以減少 CH3OO 在光譜中所造成的干擾,因此吾人以
52 CH3CHO/O2/Cl2的10-50 us 之差異吸收光譜來進行 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的光譜指派,示於圖 4-5(a)。 4.3.3 t,c-CH3C(O)OO 及 t,t-CH3C(O)OO 的光譜指派 除了由實驗反應系統之討論可知,所觀測的吸收譜帶應為兩系統 之共同前驅物 CH3CO 所致,而在大量氧氣環境中,其最有可能為 CH3C(O)OO 之貢獻。除此之外,由量子化學計算亦可以提供更進一 步的佐證。利用 B3LYP/aug-cc-pVDZ 方法預測 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 在光區 1000-2000 cm-1內的振動波數(以實線表示) , 及其吸收強度(以線條之高度表示)與在 Ar 在間質隔離法所觀測到的 振動波數(以虛線表示並標示為 m),及其吸收強度(以線條高度表示) 分別示於圖4-5(d)與圖 4-5(e)。比較圖 4-5(a)、4-5(d)及 4-5(e)可發現, 吾人所觀測到的譜帶與 t,c-CH3C(O)OO 的 Ar 間質光譜,不論是振動 波 數 或 紅 外 光 吸 收 強 度 上 都 相 當 接 近 , 但 尚 未 能 完 全 排 除 t,t-CH3C(O)OO 之可能性,因為這兩個異構物在這個光區內的吸收峰 十分接近,由差異光譜上可以觀測到,譜帶A2 的紅外光吸收強度比 譜帶A3 強,與間質隔離光譜一致,但與 B3LYP/aug-cc-pVDZ 的預測 相反,其很可能是因為 ν6與 ν7的振動模嚴重混合,造成理論計算在 預測分子的紅外光吸收強度上會有較大的誤差所致。 4.3.4 t,c-CH3C(O)OO 及 t,t-CH3C(O)OO 的光譜模擬
53 就目前吾人所使用的光譜解析度並不足以解析CH3C(O)OO 的轉動-振動光譜,因此吾人使用光譜模擬程式SpecView[17]進行光譜模擬, 並與實驗結果做比較。吾人所使用的轉動常數為 B3LYP/aug-cc-pVDZ 預測之結果,如表4-3 所示。而其它所需的光譜參數為:Jmax=100、 T=298 K,都普勒半高寬= 2.0 cm-1。在進行模擬之前須將前驅物 CH3CHO 的吸收干擾消除,為了避免 1746 cm-1的吸收峰有飽和吸收 的問題,吾人以2715 cm-1之向下吸收為基準,將CH3CHO 的吸收峰, 乘上一比例作回補,使2715 cm-1之向下吸收峰完全補平,其回補後 的光譜如圖4-5(c)所示。圖 4-5(b)為 CH3CHO 的紅外吸收光譜。 在 1900-1800 cm-1 光 區 附 近 應 有 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 之 C=O 伸展振動模(ν3),而這兩個譜帶的位置相當接 近。吾人使用 B3LYP/aug-cc-pVDZ 預測的轉動常數:A"= 0.30527 cm-1、B"= 0.15827 cm-1、C"= 0.10628 cm-1及A'= 0.30461 cm-1、B'= 0.15808 cm-1、C'= 0.10612 cm-1對t,c-CH3C(O)OO 的 ν3振動模進行模 擬,其 a、b、c 的混成(hybride)躍遷的比例為 0.54:0.46:0。吾人使 用B3LYP /aug-cc-pVDZ 所預測之轉動常數:A"= 0.34385 cm-1、B"= 0.14516 cm-1、C"= 0.10406 cm-1及 A'= 0.34261 cm-1、B'= 0.14508 cm-1、 C'= 0.10392 cm-1對t,t-CH3C(O)OO 的 ν3振動模進行模擬,其a、b、c
54 t,t-CH3C(O)OO 的 ν3 振動模之光譜模擬與實驗光譜比對可看出, t,c-CH3C(O)OO 的 ν3 振 動 模 在 光 譜 中 有 較 多 的 貢 獻 。 故 先 將 t,c-CH3C(O)OO 的 ν3 固 定 在 1851 cm-1 的 位 子 , 再 藉 由 調 整 t,t-CH3C(O)OO 的 ν3的吸收位置與相對強度,使其大約符合實驗光譜 的譜帶輪廓。可以得到t,c-CH3C(O)OO 的 ν3譜帶原點為1851 cm-1, 與 t,t-CH3C(O)OO 的 ν3譜帶原點為 1862 cm-1,其實驗光譜與模擬光 譜之比對示於圖4-6。圖 4-7 為 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的 ν3 振動模之個別 a、b 及 c 型躍遷的模擬光譜,最底下則是用來與實驗 光譜比較的a、b 混合躍遷之模擬光譜。吾人所觀測到 t,c-CH3C(O)OO
與 t,t-CH3C(O)OO 的振動波數與 B3LYP /aug-cc-pVDZ 預測之振動波
數分別相差1.5 %與 2%;其與 Ne 間質隔離光譜都相差 1 cm-1;而Ar 與間質隔離光譜分別相差9 cm-1與10 cm-1。藉由個別積分模擬光譜的 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的吸收峰面積,可知其相對吸收度
為3.7:1.0。由先前進行 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 之相對能
量討論(t,c-CH3C(O)OO 較 t,t-CH3C(O)OO 穩定 2.3-3.5 kJ mol-1)與相對
紅外光吸收強度之理論計算(t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的 ν3
吸收強度比例為 1.1:1)可知 t,c-CH3C(O)OO 之 ν3在光譜中會有較多
的貢獻,其與吾人觀測到的光譜一致。
55 t,t-CH3C(O)OO 之 ν5振動模,由於先前的研究組在間質隔離光譜法中 觀測到比ν5振動模更微弱的吸收峰,卻沒有觀測到t,t-CH3C(O)OO 之 ν5振動模,而且 t,t-CH3C(O)OO 的光譜輪廓與 A4 譜帶相差較遠,因 此 吾 人 暫 時 排 除 其 在 本 實 驗 光 譜 中 造 成 貢 獻 的 可 能 。 吾 人 使 用 B3LYP/aug-cc-pVDZ 預測之轉動常數:A"= 0.30527 cm-1、B"= 0.15827 cm-1、C"= 0.10628 cm-1及A'= 0.30291 cm-1、B'= 0.15828 cm-1、C'= 0.10595 cm-1,其 a、b、c 的混成躍遷比例為 0.22:0.78:0。固定以 上轉動常數,再藉由調整ν5之吸收位置與強度,使其大約符合實驗光 譜的譜帶形狀。可以得到t,c-CH3C(O)OO 之 ν5譜帶原點為1372 cm-1, 而實驗光譜與模擬光譜之比對示於圖 4-8。圖 4-9 為 t,c-CH3C(O)OO 的 ν5之 a、b 及 c 型躍遷之模擬光譜,最底下則是用來與實驗光譜比 較的 a、b 混合躍遷之模擬光譜。吾人所觀測到 t,c-CH3C(O)OO 的 ν5 振動波數與 B3LYP/aug-cc-pVDZ 預測之振動波數相差 0.2 %,而與 Ne 間質隔離光譜法相同,相較於 Ar 間質隔離光譜法則相差 5 cm-1。 在 1250-1000 cm-1 光 區 附 近 應 有 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 之 ν6、ν7振動模。利用B3LYP/aug-cc-pVDZ 所預測之 t,c-CH3C(O)OO 的振動模 ν6之轉動常數為:A"= 0.30527 cm-1、B"= 0.15827 cm-1、C"= 0.10628 cm-1及 A'= 0.30498 cm-1、B'= 0.15799 cm-1、 C'= 0.10610 cm-1,其 a、b、c 的混和躍遷的比例為 0.5:0.5:0。而
56 t,c-CH3C(O)OO 的振動模 ν7之轉動常數為:A"= 0.30527 cm-1、B"= 0.15827 cm-1、C"= 0.10628 cm-1及 A'= 0.30465 cm-1、B'= 0.15808 cm-1、 C'= 0.10600 cm-1,其 a、b、c 的混成躍遷的比例為 0.86:0.14:0。而 t,t-CH3C(O)OO 的振動模 ν6 之轉動常數為:A"= 0.34385 cm-1、B"= 0.14516 cm-1、C"= 0.10406 cm-1及 A'= 0.34391 cm-1、B'= 0.14476 cm-1、 C'= 0.10384 cm-1,其 a、b、c 的混和躍遷的比例為 0.44:0.56:0。而 t,t-CH3C(O)OO 的振動模 ν7 之轉動常數為:A"= 0.34385 cm-1、B"= 0.14516 cm-1、C"= 0.10406 cm-1及 A'= 0.34347 cm-1、B'= 0.14495 cm-1、 C'= 0.10384 cm-1,其 a、b、c 的混成躍遷的比例為 0.84:0.16:0。固 定以上轉動常數,藉由調整譜帶間的吸收位置與相對強度,可得到 t,c-CH3C(O)OO 的 ν6與 ν7之譜帶原點為 1169 cm-1與 1102 cm-1,與 t,t-CH3C(O)OO 的 ν6與ν7之譜帶原點為1142 cm-1與1078 cm-1,實驗 光譜與模擬光譜之比對示於圖 4-10。圖 4-11 與圖 4-12 分別為 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 的 ν6、ν7之個別a、b 及 c 型躍遷之
模擬光譜,最底下則是用來與實驗光譜比較的 a、b 混合躍遷之模擬 光 譜 。 吾 人 所 觀 測 到 t,c-CH3C(O)OO 的 ν6 及 ν7 振 動 波 數 與
B3LYP/aug-cc-pVDZ 所預測之振動波數都相差 2 %,相較於 Ne 間質 隔離光譜法上各相差9 cm-1與4 cm-1,而與 Ar 間質隔離光譜法各相 差 16 cm-1 與 3 cm-1。而 t,t-CH3C(O)OO 的 ν6 及 ν7 振動波數與
57 B3LYP/aug-cc-pVDZ 所預測之振動波數分別相差 2%與 4%,相較於 Ne 間質隔離光譜法上各相差 1 cm-1與18 cm-1,而與 Ar 間質隔離光譜 法各相差5 cm-1與16 cm-1。 表 4-2 列出本實驗所測得之 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 之 振動波數與吸收強度;各振動模的吸收強度均為 ν3積分面積之相對比 例(ν3吸收強度定為 100),並與理論計算 B3LYP/aug-cc-pVDZ 及 Ne 與 Ar 間質隔離光譜法做比較。本實驗所觀測到的振動波數與計算值 相差 0.2-4.2 %,符合以 B3LYP 方法計算振動頻率的誤差值內(2-4 %),與 Ne 及 Ar 間質內的振動頻率之誤差值都在 16 cm-1以內。 由圖4-5 可知,CH3C(O)OO 除了 ν3之外,ν5、ν6與 ν7都受到前 驅物 CH3CHO 的干擾,其會造成光譜模擬的誤差。因此吾人利用 t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 之 ν3振動模來估計兩者之間的相對 濃度比例。由於紅外光吸收面積會正比於分子的紅外光吸收強度(IR intensity)與分子濃度。由先前吾人於譜帶 A1 所得到的 t,c-CH3C(O)OO 相對於 t,t-CH3C(O)OO 的紅外光吸收面積比為 3.7:1.0,並考慮
t,c-CH3C(O)OO 與 t,t-CH3C(O)OO 在 B3LYP/aug-cc-pVDZ 下所預測的
紅 外 光 強 吸 收 強 度 比 為 1.1 : 1.0 , 可 推 斷 t,c-CH3C(O)OO 與
t,t-CH3C(O)OO 的濃度比約為~3.4:1.0。假設 t,c-CH3C(O)OO 與
58
可估計t,c-CH3C(O)OO 在能量上較 t,t-CH3C(O)OO 約低了~3.0±0.6 kJ
mol-1。其結果與理論計算之2.3-3.5 kJ mol-1一致[4,5,6]。 4.4 cyc-H2CC(=O)O (α-lactone)的指派
由圖4-4 可以看到於 1960 cm-1有一微弱吸收峰 B1,其吸收峰約 維持了100 μs 後才消失。由理論可知 CH3CO + O2的反應途徑主要為
CH3C(O)OO 之生成,另一個次要的反應途徑會產生其它產物 + OH,
而可能生成的其他產物有•CH2C(=O)O• (diradical)、cyc-H2CC(=O)O
(α-lactone)、H2CO + CO、HOO(C=O)CHO。Maranzana 等人[20]利用
B3LYP/6-311G(2df,p)研究 CH3CO 與 O2的反應位能曲面,作者提出
CH3CO 可能會與兩個 O2反應而產生下列分子 HOOC(=O)CH2OO、
OOC(=O)CH2OOH 或 HOOC(=O)C(H)OOH. 。 吾 人 利 用 理 論 計 算
B3LYP/aug-cc-pVDZ 計 算 上 述 分 子 與 CH3C(O)OO 的 異 構 物 HOOC(=O)CH2之振動波數並列於表4-5(除 H2CO 之外)。由表 4-5 可 知,除了α-lactone 之外其他分子的振動波數皆不可能對 1960 cm-1的 B1 吸收峰造成貢獻,因此吾人指派 B1 吸收峰為α-lactone 的ν2振動 模,其振動波數與Milligan 等人[18]在 CO2間質隔離光譜法所測得的 1967 cm-1一致。而且Ruggiero 等人[19]利用理論計算 MP2/6-31+G(d,p) 提出α-lactone 為氣態 C2H2O2最穩定的結構。 為了更加確定B1 譜帶為α-lactone 的貢獻,吾人利用光譜模擬程