行政院國家科學委員會專題研究計畫 期中進度報告
利用生物活性基質進行骨軟骨及 帶之組織工程(1/3)
計畫類別: 整合型計畫 計畫編號: NSC91-2321-B-002-005- 執行期間: 91 年 08 月 01 日至 92 年 12 月 31 日 執行單位: 國立臺灣大學醫學院骨科 計畫主持人: 劉華昌 共同主持人: 楊台鴻,陳瑞明,侯勝茂,侯連團,林峰輝,黃義侑,洪士杰 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 92 年 11 月 3 日
行政院國家科學委員會專題研究計畫 期中進度報告 利用生物活性基質進行骨軟骨及 帶之組織工程(1/3) 計畫類別: 整合型計畫 計畫編號: NSC 91-2321-B-002-005 執行期間: 91 年 08 月 01 日 至 92 年 12 月 31 日 執行單位: 國立臺灣大學醫學院骨科 計畫主持人: 劉華昌 共同主持人: 楊台鴻,黃義侑,陳瑞明,洪士杰,侯勝茂,侯連團,林峰輝 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢 中 華 民 國 92 年 10 月 31 日
摘要 膝關節的軟骨缺損或退化性關節炎常常會造成膝關節之疼痛。雖然在外科 手術上有許多方法發展出來如鑽動術、骨膜皮瓣及軟骨細胞移植、馬賽克成形術 等,試圖解決這方面的問題,但效果都不盡理想。關節置換手術、自體、異體或 異種植體也用在臨床上,但關節置換手術所使用材料為非生物性,長期使用常有 鬆脫的現象;自體移植須有額外的手術,供應位置的復原及受應力情形也會受到 影響;異體或異種植體則有免疫及疾病傳染的危險。本研究擬利用組織工程技術 培養出 autogenic osteochondral grafts 及骨-韌帶-骨的植體,研究中將骨髓幹細胞 載入所發展的雙相支架中,上層為 gelatin/hyaluronan/chondroitin-6-sulfate 海綿共 聚體,下層為多孔質氫氧基陶瓷體;生長激素將以表面接枝或共聚方式導入支 架、微粒方式導入支架達到緩釋效果、或加入培養基中促進細胞的分化及增生。 組織培養將在自行設計的生物反應器下進行。骨-韌帶-骨的植體也是利用相同觀 念,將細胞值入載有生長激素的 PDLLA 纖維束中培養。 本計劃分三年進行。第一年為各種支架材料之製備與測試、幹細胞之分離 培養及分化、微粒之開發、反應器之設計組裝及測試。 關鍵詞: 組織工程,骨軟骨植體
ABSTRACT
In case of extensive arthritis, avascular necrosis, or traumatic or tumor excision-caused loss of osteochondral joint tissues, joint replacement or osteochondral defect reconstruction is often require joint function. Although there are many techniques being developed to solve the problem such as drilling chondroplasty, mosaicplasty, or periosteal flap combined with autogenous chondrocyte transplantation, all of these treatments had their limitations. Prosthetic replacement, allogenic and autogenic osteochondral grafts have been performed for this purpose. However, prosthetic joint replacement may be compromised by the limited durability of non-biological materials used due to wearing and loosening of the implant. The availability of tissue and morbidity of the donor site have limited the use of autografts. The major problems for allograft or xenograft are the potential danger of infection with HIV or hepatitis, and the necessity for immunosuppression with possible serious complications. Therefore, suitable osteochondral grafts have long been one of the goals of orthopaedic research. Tissue engineering is a recently developed science with promising results reported for reconstruction of bone, cartilage, and small joints. The project try to use the tissue engineering technique to fabricate osteochondral grafts with bone marrow-derived mesenchymal stem cells seeded on the bi-phasic scaffold. The scaffold is a bi-layer structure with gelatin/Hyaluronan/chondroitin-6-sulfate copolymer sponge on the top and porous hydroxhapaptite beneath. The growth factors will be incorporated into the system by surface graft, microsphere delivery, and dripping into the cultural medium. All the cultured system will be performed in a new designed double-chamber bioreactor. The bone-ligament-bone graft will be prepared under the same philosophy as that of osteochondral graft.
The project is a three-year study. In the first year, all kinds of scaffold used into the study will be developed to mimic the extracellular matrix of real tissue as a viable matrix. The new designed bioreactor will be assembled and tested to fit the demands of future applications. Growth factors, TGF-β1, bFGF, and PDGF, will be encapsulated with biodegradable polylactide and liposome by lyophilization and intramolecular interaction. The MSCs will harvest from the bone marrow and then preliminarily seed on the viable matrix. A PLGA fiber seeded with ligament stem cell will prepare to cooperate into the system later. The tidemark at the junction of hyaline cartilage and subchondral bone will be developed by ameoblast-seeded bovine bone
雙腔室生物反應器之製造與雙相式支架之製作,
及軟骨於該支架上之生長
Progress Report:
Development of Double-Chamber Bioreactor for
Osteochondral Tissue Engineering and the
Making of Biphasic Scaffold with Growing of
the Cartilage on the Scaffold
Design of, and rationale for, the double chamber bioreactor
The double-chamber bioreactor consists of two glass chambers, each of which has three branch tubes for medium inflow, medium outflow, and oxygen ventilation. The bioreactor is made of glass, which is completely transparent. The outer diameter of the bioreactor is 48 mm, and the inner diameter is 45 mm. The chambers are separated by a silicon septum with multiple holes to hold the biphasic scaffold. The glass
chamber can be sterilized by ethylene oxide and the silicon septum can be sterilized with 75% alcohol. Magnetic bar stirring provides the mechanical stimulation for the engineered cartilage. The whole setting can be put into a standard humidified incubator at 37°C and 5% CO2. The bioreactor has two independent medium
circulation systems (Fig. 1(a) and (b)) and thus can be used to supply different types of culture media to support different kinds of cells in co-cultures or different induction media to induce mesenchymal stem cells to differentiate into chondrocytes and
osteoblasts in the same biphasic scaffold (e.g. medium containing
beta-glycerophosphate and dexamethasone for induction of bone formation and medium containing TGF-beta 1 for induction of cartilage formation). We
to the limited diffusibility in the biphasic scaffold. However, limited diffusion of the cytokines is supposed to occur inside the scaffold, which could facilitate
micro-interactions between different cells at the interface, but little exchange will occur between the two chambers. In addition, as the cells secrete increasing amounts of matrix, diffusion would progressively decrease.
Fig 1 (a).
The design of Double-Chamber bioreactor: The double-chamber bioreactor consisted with two glass chambers. Each chamber contains three branch tubes for medium inflow, medium outflow and oxygen ventilation. The two chambers were separated with a silicon septum with multiple holes to hold the biphasic scaffold. Magnetic bar stirring provided the mechanical stimulation for the scaffold. The bioreactor contains two independent medium circulation systems.
Fig 1 (b). The Double-chamber bioreactor.
Fabrication of biphasic scaffolds and SEM and confocal microscope examination:
Cancellous bone, obtained from calf femoral condyles, was cut into small cubes of approximately 1 cm3. To avoid cracking and soot formation in the material during heat-treatment, the cubes was boiled in distilled water for 12 h. After boiling, the bone blocks were dehydrated in an alcohol series and dried at 70°C for 3 days. To remove organic matrix, the dried blocks were calcined in a conventional Ni-Cr coiled furnace by increasing the temperature to 800°C at a rate of 10°C/min and maintaining it at this temperature for 6 hours. The calcined blocks were placed on a platinum sheet and heated in a SiC heated furnace to 1350°C at a rate of 2.5°C/min and maintained at this temperature for 1 h to sinter the bone. The material, the so-called “sinbone”, was then cooled to room temperature by slow furnace cooling, then cut into a cylindrical shape with 20 mm in length and 10 mm in diameter.
Gelatin powder, extracted and purified from porcine skin, with an average molecular weight of about 60,000-100,000 daltons on SDS-PAGE (Sigma Chemical Co., USA) was dissolved as a 1% solution in deionized water in a 65°C water bath
with continuous stirring. A final concentration of 0.05 % (w/w) hyaluronan (HA) (Seikagaku Co., Tokyo, Japan) solution was then added, giving a gelatin/HA molar ratio of 9:1.
The sinbone block with 20 mm length and 10 mm diameter was soaked for 30 minutes in 5 ml of the gelatin/HA solution at a negative pressure of 10-1 torr, then the sinbone/gelatin was cross-linked overnight at room temperature with 0.01%
glutaraldehyde and lyophilized for three days. The cross-linking and lyophilization process was repeated another two times. The sinbone/gelatin was then used as a bilayer scaffold for osteochondral graft tissue engineering. The thickness of the gelatin layer is about 2 mm (Fig. 3).
The scaffolds were dehydrated by treatment with a series of graded ethanol solutions (50% for 12 h, 75% for 2 h, 85% for 2 h, and 95% for 2 h), then left overnight in a vacuum oven at 50℃ before coating with gold for scanning electron microscope (SEM) examination. One scaffold was examined using a Leica TCS-SP2 confocal microscope with an Ar/Kr laser with a wavelength of 568 nm.
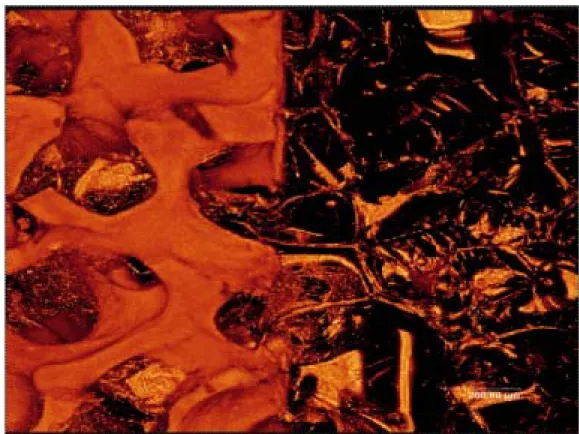
Under the SEM, the dehydrated gelatin scaffold had a uniform pore size of 180 μm and an adequate porosity of 75 % (Fig. 4(a)). This highly porous structure of the scaffold would be expected to allow cell growth and proliferation, and the size of the pore should increase even further on soaking in culture medium, allowing cell penetration and adhesion. In the calcined bone part of the scaffold, the natural trabecular pattern was preserved (Fig. 4(b)). On confocal microscope examination (Fig. 5), the porous gelatin structure could be seen, and had infiltrated into the pores of the calcium phosphate. The pores in the gelatin and calcined bone are
interconnected, which would facilitate micro-diffusion in the interface.
Fig 3.
Fig. 4 (a) Scanning electron microscopic examination of the gelatin scaffold showing a porous structure with a uniform pore size of about 180μm and porosity of 75 %. (b) Scanning electron microscopic axamination of the calcined bone showing the
preservation of the trabecular pattern.
Fig. 5 Under confocal microscope examination, porous gelatin structure can be seen, and the gelatin structure had infiltrated into the pores of calcium phosphate. The integration of cartilage part and bony part of scaffold depends on this penetration.
Isolation of Condrocytes:
Full thickness articular cartilage was harvested aseptically from adult porcine knee joints within 12 h after slaughter and chondrocytes isolated by incubating the cartilage specimens for 12 – 16 h at 37°C in DMEM medium (Hyclone Co., Logan, Utah, USA) containing 0.2 % collagenase (Sigma Co., St. Louis, USA). The isolated chondrocytes were resuspended in phosphate-buffered saline (pH 7.4), washed, and counted using a hemocytometer. Chondrocyte viability was determined using Trypan blue dye
exclusion. The monolayer culture is subcultured after a week. The chondrocytes were culture expanded for 2 weeks before seeding to the biphasic scaffold.
Seeding of Chondrocytes and Cultured in Double-Chamber Bioreactor
part) were sterilized with 75% ethanol. Chondrocytes were expanded in DMEM containing 10% fetal bovine serum (Biological Industries Ltd, Kibbutz Beit Haemek, Israel), 1 % penicillin/gentamicin (Sigma Co., St. Louis, USA), and 50 μg/ml of L-ascorbic acid (Sigma Co., St. Louis, USA). At confluence, the chondrocytes were trypsinized and resuspended at a concentration of 5.3 x 107 cells/ml DMEM, and then about 2 ml of cell suspension was injected into each scaffold (108 cells per scaffold). The cell-containing scaffolds were then cultured in double-chamber bioreactor for 2 and 4 weeks. The medium in the spinner flask was changed every week. The magnetic bars were stirred at 70 rpm for 10 hours to distribute the cells more evenly, then at 50 rpm during the rest of the culture period. Quadruplicate samples were removed at each time-point and fixed with formaldehyde, decalcified with 5% formic acid, and dehydrated by exposure to an ethanol gradient. The specimens were embedded in paraffin and cut into 5 μm slices then stained with H & E stain for histological examination. Alcian blue was used to stain glycosaminoglycan and antibodies against human S-100 protein (Novocastra Laboratories Ltd, Newcastle upon Tyne, UK) or type II collagen (Santa Cruz Biotech. Inc., CA. USA) to check the chondrocyte phenotype.
Culture results in Double-Chamber Bioreactor
After 2 weeks’ cultivation of the chondrocytes loaded biphasic scaffolds in double-chamber bioreactor, hyaline like matrix production and lacuna formation around chondrocytes were observed. Some chondrocytes can be seen distributed in the scaffold pores without matrix production, indicating the early stage of engineered cartilage formation (Fig. 6(a)). Under Alcian Blue staining, the matrix in engineered cartilage showed different depth of blue colors, showing the different stages of sulfating of the matrix (Fig. 6(b)). After 4 weeks’ cultivation in double-chamber bioreactor, the matrix stains bluer than the 2 weeks’ matrix, indicating more glycosaminoglycan production by chondrocytes (Fig. 6(c)). On the section of the decalcified specimen, the interface between cartilage and calcium phosphate can be seen. Engineered cartilage with lacuna formation on the top of the calcium phosphate was observed (Fig. 6(d)). The cartilage is well integrated to the calcium phosphate scaffold through infiltration.
Immunohistochemical staining for S-100 protein and type II collagen showed that chondrocytes cultured in double-chamber bioreactor for 4 weeks retained their phenotype in the cartilage part of biphasic scaffold. As shown in Fig. 7(a), the cells retained their round shape and stained positive for S-100 protein. The round shape of chondrocytes is an indicator of phenotype retention and is essential for matrix
formation. S-100 protein is only found in cells of ectodermal origin and chondrocytes are the only cell of mesenchymal origin to express S-100 protein; S-100 expression is lost when chondrocytes lose their phenotype. As shown in Fig. 7(b), the chondrocytes were also stained with anti-type II collagen antibody. Type II collagen, which is essential for articular cartilage, is produced by chondrocytes; these results therefore show that the chondrocytes in the scaffold were functionally active.
Fig. 6 (a)
(b)
(d)
Fig. 7 (a)
BIPHASIC EFFECTS OF NITRIC OXIDE ON CHONDROCYTE
ACTIVITIES
ABSTRACT
Chondrocytes play critical roles in cartilage formation.
Promising new therapies based on tissue engineering have
been recently developed for cartilage repair. The
development of cartilaginous tissue is dependent on the
environment that surrounds it. Nitric oxide contributes to
the regulation of chondrocyte activities. This study is aimed
to evaluate the roles of nitric oxide on human chondrocytes
using sodium nitroprusside as a nitric oxide donor.
Administration of human chondrocytes with low
concentrations of sodium nitroprusside (<0.1 mM)
significantly caused a 20-30 % increases in the levels of
nitric oxide. In parallel to the increase of nitric oxide, the cell
proliferation of chondrocytes was significantly enhanced.
Immunoblotting analyses of collagen type I and II revealed
that low concentrations of sodium nitroprusside increased
the protein levels of these tow extracellular matrixes.
Exposure of chondrocytes to 1 mM sodium nitroprusside
significantly augmented 2-fold nitric oxide and lead to cell
injury and death. Apoptotic analysis showed that a high
concentration of sodium nitroprusside significantly
increased the percentage of chondrocytes undergoing
apoptosis. This study has shown that the slight increases in
nitric oxide (20-30 %) could promote chondrocyte
proliferation and synthesis of extracellular matrix collagen
type I and II. However, when the increases of nitric oxide are
more than 2 folds, chondrocytes would be damaged and the
death mechanism is via an apoptotic pathway.
INTRODUCTION
Chondrocytes play important roles in cartilage formation. The autologous in vitro expansion of chondrocytes is a new method for the treatment of localized cartilage defect zones in humans. The association of biomaterials with autologous chondrocytes expanded in vitro can represent a useful tool to regenerate this tissue. The development of cartilaginous tissue is dependent on the environment that surrounds it. Tissue engineers have taken a cue from normal cartilage physiology and incorporated the use of mechanical stimulation into their attempts to engineer functional cartilage. Promising new therapies based on tissue engineering have been
recently developed for cartilage repair (Shieh and Athanasiou, 2003). Recent studies demonstrate that chondrocytes in native articular cartilage and in tissue-engineered constructs respond to mechanical stimuli through multiple regulatory pathways. Responses of the cells are manifested by intra- and intercellular signalling, alterations in transcription level, protein translation, post-translational modifications, and synthesis of intracellular and extracellular macromolecules (Darling and Athanasiou, 2003).
Nitric oxide (NO), produced from L-arginine by nitric oxide synthase (NOS), is involved in various physiological and toxicological processes in many tissues (Moncada et al., 1991). Our previous study had shown that NO, released from sodium nitroprusside, caused osteoblast insults through the apoptotic mechanism according to several lines of evidence including morphological shrinkage, the increase of apoptotic cells and DNA fragmentation (nuclei TUNEL and DNA ladder assays) (Chen et al., 2002). However, low levels of NO could protect osteoblast from oxidative stress-induced cell damage (Chen et al., 2001). Chondrocytes can produce NO and this oxidant contributes to the cell activity (Terauchi et al., 2003; Yoon et al., 2003). This study is aimed to sutdy the roles of NO in chondrocyte activities, including cell proliferation, production of collagen types I and II, cell injuries.
MATERIALS AND METHODS Cell culture and drug prepatration
Human chondrocytes were purchased from Cell Applications, Inc. (San Diego, CA, USA). These cells were seeded in chondrocyte growth medium (San Diego, CA, USA). All cultures were maintained at 37 oC in a humidified atmosphere of 5 % CO2-95 % air. When chondrocytes grew to a confluent monolayer, they were
sub-cultured for various experiments.
Sodium nitroprusside (SNP; Sigma, St. Louis, MO, USA) was dissolved in a PBS buffer (0.14 M NaCl, 2.6 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4) to 200
mM as a stock solution, protected from light, and stored at –20 oC for use in related experiments. Chondrocytes were treated with various concentrations of SNP for different time intervals. Cell morphologies were observed and photographed by a reverse-phase microscope.
Assay of nitrite in the culture medium
The amounts of NO in cultured medium were evaluated using enzymatic reduction of nitrate by nitrate reeducates and following the spectrophotometric
analysis of total nitrite with Greiss reagent according to the standard protocol of Bioxytech NO assay kit (OXIS International, Inc, Portland, OR, USA). In the kit, nitrate reductase was used to reduce nitrate to nitrite, and the amounts of total nitrite were determined to represent the amount of NO in the culture medium.
Determination of cell viability
Mitochondrial function of chondrocytes was assayed by the ability of viable cells to convert soluble 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into an insoluble dark blue formazan according to the method of Carmichael et al. (1987). In the bulk cell photometric MTT assay, the bulk conversion of MTT in the well of a 96-well plate was measured photometrically. MTT was dissolved in PBS at a concentration of 5 mg/ml and sterilized by passage through a 0.22-µm filter. This stock solution was added (one part to 10 parts medium) to each well of a 96-well tissue culture plate, and the plate was incubated at 37 oC for 4 hr. DMSO is added to all wells and mixed thoroughly to dissolve the dark blue crystals. After a few minutes at room temperature, to ensure that all the crystals were dissolved, the plates were read on a microplate reader at a wavelength of 570 nm. A standard curve was set up using 200-50,000 cells/well, and the absorbency was directly proportional to the number of cells over this range.
Analysis of apoptotic cells
Using a flow cytometer, apoptotic chondrocytes were determined to detect DNA fragments in nuclei stained by propidium iodide according to the method of Nicoletti et al. (1991). After treatment, chondrocytes were harvested and fixed in cold 80% ethanol. Following a process of centrifugation and washing, the fixed cells were stained with propidium iodide and analyzed using a FACScan flow-cytometer (FACS Calibur, Becton Dickinson, San Joes, CA, USA) on the basis of a 560 nm dichroic mirror and a 600 nm band pass filter.
Immunoblotting analyses of Collagen type I and II proteins
Cells were washed with PBS and lysed in ice cold RIPA buffer (Tris-HCl pH 7.2, 25 mM; SDS 0.1%; Triton X-100 1%; sodium deoxycholate 1%; NaCl 0.15 M; EDTA 1 mM) containing 1 mM of phenyl methyl sulfonyl fluoride (PMSF), 10 µg/ml of aprotinin, 1 mM of sodium orthovanadate and 5 µg/ml of leupeptin. Protein concentrations were determined with the BCA method (pierce, Rockford, U.S.A.). Protein (50 µg) was resolved on 12.5% polyacrylamide gels and blotted onto nitrocellulose sheets using the semidry blot system (TE 70; Hoefer Scientific Instruments, San Francisco, CA) at 2 mA/cm2 for 60 min in 25 mM Tris-HCl, pH 8.3;
192 mM glycine; and 20% methanol. The membrane was blocked overnight at room temperature with a blocking reagent (20 mM Tris, pH 7.4; 125 mM NaCl; 0.2% Tween 20; 4% nonfat dry milk; and 0.1% sodium azide). Then it was incubated for 1 h with the mouse anti-human collagen type I and II antibodies, washed three times, and then incubated with alkaline phosphatase-conjugated rabbit anti-mouse in PBS and 0.5% Tween 20 for another 45 min with gentle shaking. After three final washes, the proteins were made visible by the Bio-Rad NBT-BCIP color development system.
Statistical Analysis
The statistical significance of the difference between control and treated groups was evaluated by Student’s t-test. A P value < 0.05 was considered as statistically significant.
RESULTS
NO was detectable in untreated chondrocytes (Table 1). Exposure of chondrocytes to 0.1, 0.5 and 1 mM SNP significantly increased 30%, 3- and 5-fold nitrite. Administration of chondrocytes with 1 mM SNP for 6, 12 and 24 hours significantly augmented 2-, 5- and 7-fold nitrite, respectively.
Exposure of human chondrocytes to 0.1 mM SNP for 24 hours increased 40% viability of human chondrocytes (Table 2). SNP at 0.5 and 1.0 mM significantly decreased 30 and 50% cell viability, respectively. When human chondrocytes were exposed to 1 mM SNP for 6, 12 and 24 hours, the cell viability was decreased by 20, 50 and 60%, respectively.
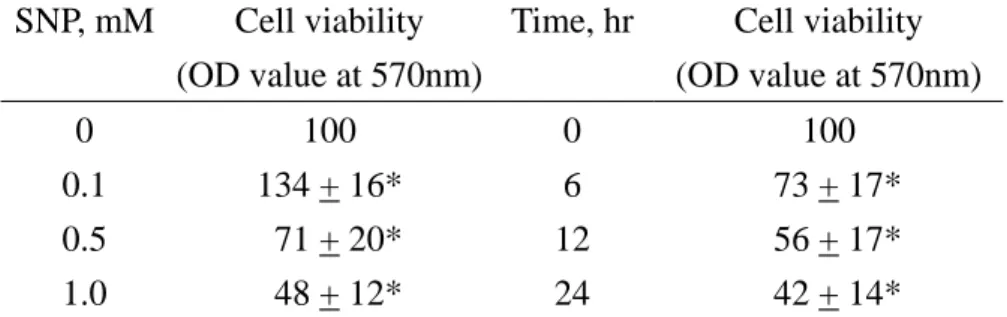
Cell proliferation was assayed using the trypan blue exclusion method to analyse the permeality of the cell membrane (Table 3). Administration of human chondrocytes with 0.1 mM SNP for 24 hours significantly increase 34% cell proliferation. However, SNP at 0.5 and 1 mM decreased 29 and 52% cell proliferation. After exposing to 1 mM SNP for 6, 12 and 24 hours, the cell proliferation of human chondrocytes was suppressed by 27, 42 and 58%, respectively.
SNP at 0.1 mM did not cause chondrocyte apoptosis (Table 4). Exposure of human chondrocyte to 0.5 and 1.5 mM SNP for 24 hours significantly increased 31 and 57% chondrocytes undergoing apoptosis. The percentages of chondrocytes undergoing apoptosis reached to 12, 49 and 61% after exposing to 1 mM SNP for 6, 12 and 24 hours.
Immunoblotting analysis revealed that collagen type I and II are detectable in human chondrocytes (Fig. 1, lane 1). Administration of human chondrocytes with 0.1 mM SNP significantly increased the levels of collagen type I and II proteins (Fig. 1,
lane 2). Quantitative analyses using β-actin as the internal standard showed that SNP at 0.1 mM significantly increase 5- and 3-fold collagen type I and II protein levels, respectively.
CONCLUSSION
This study has shown that nitric oxide released from sodium nitroprusside has biphasic effects on human chondrocyte activities: low concentrations of nitric oxide production could promote chondrocyte proliferation and synthesis of collagen type I and II, however, high concentrations of nitric oxide would lead to cell insults and death through an apoptotic pathway.
REFERENCES
Armour KE, Van'T Hof RJ, Grabowski PS, Reid DM, Ralston SH: Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Miner Res 14:2137-2142, 1999 Bates JN, Baker MT, Guerra RJr, Harrison DG: Nitric oxide generation from nitroprusside by vascular
tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem Pharmacol 42: s157-s165, 1991
Brüne B, von Knethen A, Sandau KB: Nitric oxide (NO): an effector of apoptosis. Cell Death and Diff 6: 969-975, 1999
Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB: Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 47: 936-42, 1987
Collin-Osdoby P, Nickols GA, Osdoby P: Bone cell function, regulation, and communication: a role for nitric oxide. J Cell Biochem 57(3):399-408, 1995
Damoulis PD, Hauschka PV: Cytokines induce nitric oxide production in mouse osteoblasts. Biochem Biophys Res Commun 201(2):924-931, 1994
Darling EM and Athanasiou KA. Articular cartilage bioreactors and bioprocesses. Tiss Engin. 9(1):9-26, 2003 Feb.
Dypbukt JM, Ankarcrona M, Burkitt M, Sjöholm Ä, Ström K, Orrenius S, Nicotera P: Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells: the role of intracellular polyamines. J Biol Chem 269: 30553-60, 1994
Evans DM, Ralston SH: Nitric oxide and bone. J Bone Miner Res 11:300-305, 1996
Heflich MH, Evans DE, Grabowski PS, Pollock JS, Ohshima H, Ralston SH: Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J Bone Miner Res 12: 1108-1115, 1997
Hikiji H, Shin WS, Oida S, Takato T, Koizumi T, Toyo-oka T: Direct action of nitric oxide on osteoblastic differentiation. FEBS Lett 410:238-42, 1997
Hortelano S, Bosca L: 6-Mercaptopurine decreases the Bcl-2/Bax ratio and induces apoptosis in activated splenic B lymphocytes. Mol Pharmacol 51: 414-421, 1997
Johnson DL, McAllister TN, Frangos JA: Fluid flow stimulates rapid and continuous release of nitric oxide in osteoblasts. Am J Physiol 271:E205-8, 1996
Löwik CW, Nibbering PH, van de Ruit M, Papapoulos SE: Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J Clin Invest 93:1465-72, 1994
Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Mijake S, Tohyama M: Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem 73: 2037-2046, 1999
Messmer UK, Brüen B: Nitric oxide – induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem J 319: 299-305, 1996
Mogi M, Kinpara K, Kondo A, Togari A: Involvement of nitric oxide and biopterin in proinflammatory cytokine-induced apoptotic cell death in mouse osteoblastic cell line MC3T3-E1. Biochem Pharmacol 58(4):649-654, 1999
Moncada S, Higgs A: The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002-2012, 1993 Okuno S, Shimizu S, Ito T, Nomura M, Hamada E, Tsujimoto Y, Matsuda H: Bcl-2 prevents
caspase-independent cell death. J Biol Chem 273: 34272-34277, 1998
Otsuka E, Hirano K, Matsushita S, Inoue A, Hirose S, Yamaguchi A, Hagiwara H : Effects of nitric oxide from exogenous nitric oxide donors on osteoblastic metabolism. Eur J Pharmacol 349:345-350, 1998
Parfitt AM: Bone remodeling and bone loss: understanding the pathophysiology of osteoporosis. Clin Obstet Gynecol 30: 789-811, 1987
Partridge NC, Alcorn D, Michelangeli VP, Kemp BE, Ryan GB, Martin TJ: Functional properties of hormonally responsive cultured normal and malignant rat osteoblastic cells. Endocrinology 108: 213-219, 1981
Ralston SH, Todd D, Helfrich M, Benjamin N, Grabowski PS: Human osteoblast-like cells produce nitric oxide and express inducible nitric oxide synthase. Endocrinology. 135:330-336, 1994 Riancho JA, Zarrabeitia MT, Fernandez-Luna JL, Gonzalez-Macias J: Mechanisms controlling nitric
oxide synthesis in osteoblasts. Mol Cell Endocrinol 107(1):87-92, 1995
Shieh AC, Athanasiou KA. Principles of cell mechanics for cartilage tissue engineering. Ann Biomedl Engin 31(1):1-11, 2003 Jan.
Terauchi R. Takahashi KA. Arai Y. Ikeda T. Ohashi S. Imanishi J. Mazda O. Kubo T. Hsp70 prevents nitric oxide-induced apoptosis in articular chondrocytes. Arthritis & Rheumatism. 48(6):1562-8, 2003 Jun.
cytoprotective mechanisms of nitric oxide. Free Radic Biol Med 25: 434-456, 1998
Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, Nathan C: Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 1256: 225-8, 1992
Yoon JB, Kim SJ, Hwang SG, Chang S, Kang SS, Chun JS. Non-steroidal anti-inflammatory drugs inhibit nitric oxide-induced apoptosis and dedifferentiation of articular chondrocytes independent of cyclooxygenase activity. Journal of Biological Chemistry. 278(17):15319-25, 2003 Apr 25.
Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, Platts LA, Hukkanen M, Polak JM, Lanyon LE: Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res 14:1123-1131, 1999
TABLES AND FIGURES
Table 1 Concentration- and time-dependent effects of SNP on
nitrite
production in human chondrocytes
SNP, mM Nitrite, µM Time, hr Nitrite, µM
0 2.9 + 0.2 0 2.9 + 0.3
0.1 4.0 + 0.3 6 7.9 + 0.9*
0.5 8.1 + 0.5* 12 16.5 + 2.1*
1.0 16.2 + 0.7* 24 19.4 + 3.5*
Human chondrocytes were treated with 0, 0.1, 0.5 and 1.0 mM sodium nitroprusside (SNP) for 16 hours or with 1 mM SNP for 6, 12 and 24 hours. The levels of nitrite in culture medium were assayed by the Griess reaction. Each value was represented Mean + SE for n=6. *Value significantly different from the respective control, P < 0.05.
effects of SNP on viability of human
chondrocytes analyzed by MTT assay
SNP, mM Cell viability (OD value at 570nm)
Time, hr Cell viability (OD value at 570nm)
0 0.86 + 0.08 0 1.03 + 0.09
0.1 1.20 + 0.09* 6 0.84 + 0.03*
0.5 0.61 + 0.07* 12 0.52 + 0.03*
1.0 0.46 + 0.03* 24 0.42 + 0.05*
Human chondrocytes were treated with 0, 0.1, 0.5 and 1.0 mM sodium nitroprusside (SNP) for 24 hours or with 1 mM SNP for 6, 12 and 24 hours. Cell viability was assayed by the colorimetric MTT test as described in Materials and methods. Each value was represented Mean + SE for n=12. *Value significantly different from the respective control, P < 0.05.
Table 3 Concentration- and time-dependent
effects of SNP on viability of human
chondrocytes analyzed by trypan blue exclusion
assay
SNP, mM Cell viability (OD value at 570nm)
Time, hr Cell viability (OD value at 570nm) 0 100 0 100
0.1 134 + 16* 6 73 + 17*
0.5 71 + 20* 12 56 + 17*
1.0 48 + 12* 24 42 + 14*
Human chondrocytes were treated with 0, 0.1, 0.5 and 1.0 mM sodium nitroprusside (SNP) for 24 hours or with 1 mM SNP for 6, 12 and 24 hours. Cell viability was assayed by the trypan blue exclusion method as described in Materials and methods. Each value was represented Mean + SE for n=5. *Value significantly different from the respective control, P < 0.05.
Table 4 Concentration- and
time-dependent effects of SNP on
human chondrocyte apoptosis
Apoptotic cells, % mM hour 0 2.1 + 0.4 0 1.3 + 0.4 0.1 1.9 + 0.3 8 12.1 + 2.7* 0.5 31.9 + 12.1* 12 48.7 + 10.3* 1.0 57.1 + 13.4* 24 60.6 + 12.5* Human chondrocytes were treated with 0, 0.1, 0.5 and 1.0 mM sodium nitroprusside (SNP) for 24 hours or with 1 mM SNP for 6, 12 and 24 hours. The percentages of apoptotic cells in human chondrocytes were determined by a flow cytometric method as described in Materials and Methods. Each value was represented Mean + SE for n=6. *Value significantly different from the respective control, P < 0.05.
Collagen I
Fig. 1 Effects of SNP on collagen type I and II protein. Human chondrocytes were treated with 0.1 mM SNP for 24 hours and cytosolic proteins were isolated. Immunoblotting analyses of collagen type I and II were carried out using monoclonal antibodies against human collagen type I and II protein.
利用生物活性基質進行軟骨級韌帶之組織工程
Using hydrogel as drug carrier and scaffold for cartilage regeneration
一、中文摘要(關鍵詞:聚乳酸、微小球、幾丁聚醣、藥物釋放、軟骨再生) 組織工程中生長因子的傳遞與制放在組織再生及組織培養中扮演著相當重要的地 位。近年使用的基材偏向使用方便的水膠,因此本實驗模擬組織在體內培養的狀態,利 用聚乳酸微小球包覆藥物樣本 lidocaine,並將之分散於去乙醯幾丁聚醣凝膠中以研究藥 物釋放的情形及其機制,並探討幾丁聚醣凝膠作為細胞培養基材的潛力。實驗結果顯示 經由幾丁聚醣基材之藥物樣本在凝膠中可減緩其釋放速率,達到長效性藥物釋放之目 的,而細胞在幾丁聚醣凝膠中的生長情形良好並可建構為立體的結構。因此幾丁聚醣凝 膠是一個良好的控制釋藥與組織重建的材料。
Growth factor delivery in tissue engineering affects the success of tissue repair. Recent studies turned to choose hydrogel as matrix for its convenience in use. Our experiments mimic the conditions of tissue growing in human body; therefore, the mechanisms of drug delivery system by using PLLA microspheres encapsulating lidocaine were studied; each of which was mixed in chitosan gel, and the potential of chitosan as tissue matrix was also investigated. The results demonstrate that sample drugs in chitosan gel make the release rate diminish and achieve long-term release. In addition, chondrocytes performed well in chitosan gel and constructed 3D structure with matrix. In conclusion, chitosan gel is an appropriate material for both controlled release and tissue regeneration.
Keywords: PLLA, microsphere, chitosan, drug delivery, cartilage regeneration
二、計畫的緣由與目的 利用組織工程的技術,重建修補身體的組織缺陷是目前研究發展的目標。組織重建 所需要的元素包括細胞、胞外間質及生長因子[1]。生物可分解性高分子可以在生物體內 自行分解並代謝,一些特定的材料如聚乳酸[1]亦有良好的生物適應性。 近年的組織工程研究趨向於使用在低溫為液狀、升溫可成膠狀的材料,優點是可填 覆各種形狀的傷口、避免複雜手術的操作,亦可輕易結合藥物治療。本計畫是以水膠 PluronicF-127 及幾丁聚醣凝膠作為熱可逆性支架探討其熱成膠現象,並加入聚乳酸微粒 當作藥物載體,包覆水溶性與油溶性作為控制釋藥,此外,將軟骨細胞培養於凝膠,以 評估水膠作為細胞培養及控制釋藥基材的能力。 三、研究方法 實驗所選用的水膠是一種熱成膠性凝膠,因此須先決定材料最適當的濃度及酸鹼 值。首先我們測定 Pluronic F-127 以及幾丁聚醣成膠溫度。幾丁聚醣凝膠是將 36%的
Glycerophosphate 溶液滴入 2%的 chitosan/HCl 溶液而形成 pH=7.2 的熱敏感性凝膠。另 外配置不同濃度的 Pluronic F-127 水溶液,利用黏度計測定不同溫度下兩種溶液黏度的 變化值。
在藥物釋放實驗方面,以 PLLA 包覆油溶性藥物 lidocaine,混合在凝膠中進行釋放, 另外則以 free drug 從基材釋放、PLLA 微小球包覆藥物從透析膜釋放作為對照,每種條 件皆加入固定量 PBS 溶液。在固定時間測定藥物( lidocaine at UV 215nm)的濃度。 我們使用豬軟骨細胞在較低溫下與幾丁聚醣凝膠混合均勻,置入細胞培養箱中使之 形 成 失 去 流 動 性 的 人 工 組 織 , 以 評 估 水 膠 作 為 組 織 工 程 支 架 的 可 能 性 。 以 DMEM+10%FBS 為培養液,培養 3 週後觀察細胞的型態。 四、結果與討論 (一)熱成膠現象探討
Pluronic-F127 是經 FDA 核准且在藥劑中常被用到的一種高分子材料,具有 sol-gel 的特性,在低溫時溶液呈液狀;而在接近體溫時會變成水膠狀,不同濃度 Pluronic-F127 溫度對黏度的關係如圖一,顯示濃度越高,成膠溫度越低,如表一所示。形成凝膠的過 程一般認為是親水性的 PEO 鏈在外部,疏水性的 PPO 鏈在核心形成微小球。當微小球 不斷累積糾結即形成大塊的膠狀物。在體溫 37o C 下成膠最適當濃度為 15%至 20%,因 為過高濃度造成操作不易,在液態下與細胞混和時會因控制的溫度過低而造成細胞的死 亡。然而,因為 Pluronic F-127 優良的親水性,使其於水溶液中極易溶解,無法維持固 定的形狀。因此根據評估的結果,Pluronic F-127 水膠不適宜作為體外藥物釋放或體內 組織重建的基材。 幾丁聚醣是一種半結晶狀的高分子,在 pH>6.2 時會和陰離子交聯而沉澱,因而限 制了其用途。低 pH 值的幾丁聚醣溶液培養細胞或直接注入體內皆因酸度過高會造成細 胞的死亡。Chenite [2]製作出在 pH=7 時仍為液體,且當溫度升高時,溶膠轉變為凝膠的 幾丁聚醣溶液,僅須將調整 chitosan 溶液酸鹼度的溶液改為弱鹼性的 Glycerophosphate 即可。造成幾丁聚醣凝膠成膠的原因推測為隨著溫度的上昇,幾丁聚醣分子疏水性的作 用力大於分子內氫鍵力,使幾丁聚醣分子彼此及與 Glycerophosphate 分子之間聚集形成 凝塊[2]。實驗所選用的去乙醯化程度為 85%,以黏度計測量所得的成膠溫度為 26.8℃, 如圖二,且在 PBS 溶液及培養液中均能維持其形狀,因此可以應用在人體進行藥物釋放 或組織工程基材。 (二)體外藥物釋放實驗 實驗中選用的代表藥物為油溶性的 lidocaine。Lidocaine 是一種具有抗發炎效果的油 溶液藥物,可利用 lidocaine 在 UV 215nm 處的吸光值測量固定時間下藥物的釋放情形。 首先利用 PLLA 包覆 lidocaine 後,於幾丁聚醣凝膠中進行藥物釋放實驗。控制釋藥的能 力是 drug in PLLA from matrix >free drug from matrix>drug in PLLA from dialysis membrane,如圖三。Burst release 的現象在 48 小時內發生,但從基材釋放出來的藥較少, 因凝膠本身就可當成控制釋藥的基材。可減緩其釋放速率、增加藥物滯留時間。
藥物初始釋放量皆很明顯上升,主要是因為幾丁聚醣在成膠過程中,發生體積驟縮 現象,使得原本懸浮在 PLLA 微小球內的藥物或懸浮藥物因體積的變化而游離出來,因 此造成最初的 24 小時內就達到高的釋放量。 (三)幾丁聚醣凝膠的細胞培養評估 軟骨的組織中最主要胞外間質為 GAGs,而幾丁聚醣的和 GAGs 的分子結構相當類 似,因此將此材料用於模擬體內軟骨細胞的生長環境是一個合理的方法。 在培養 2、7、14 天時測試細胞增殖 MTS test 以評估材料的生物適應性,初始豬軟 骨細胞數為1.2 ×106 cells/ml。在 2 天內,培養於細胞培養盤的細胞較多,但之後 因細胞凋亡使數量劇減,而培養在凝膠內的細胞在過了適應期後,細胞數明顯增 加,顯示幾丁聚醣凝膠對於軟骨細胞培養有正面的影響,如圖四。 此外,如圖五所示,培養於凝膠中的軟骨細胞保持圓形,因此仍具有軟骨細 胞的表型與功能,但在培養盤上的細胞則呈細長紡錘型而可能喪失了軟骨細胞的 特性。而細胞在凝膠中形成了立體的結構,圖六即為同一個視野不同的焦距的連 續圖,在不同位置的焦距可觀察到不同的細胞,顯示這樣的組織是一個良好的 3D 結構,可應用在真實的軟骨傷害中。 五、結論 綜合上述,幾丁聚醣凝膠是一極優的長效性藥物釋放系統,而細胞在幾丁聚醣凝膠 中的生長情形良好並建構為立體的結構。因此幾丁聚醣凝膠是一個良好的控制釋藥與組 織重建的材料。 六、參考文獻
1. Burg K J L, et al., Biomaterials 21 (2000) 2347-2359. 2. Chenite A, et al., Biomaterials 21(2000) 2155-2161.
3. Cao Y, et al., J Biomaterials Sci. Polymer Edition, vol. 9(5) (1998) 475-87. 4. Malmsten, M, et al., Micromolecules 25 (1992) 5440-5445.
5. Huang YY, et al., The Int. J. Pharmaceu. 156 (1997) 9-15.
Pluronic F-127 Gelation Temperature Temp (o C) 5 10 15 20 25 30 35 40 V u scosit y (cp) 1 10 100 1000 10000 10% Pluronic 15% Pluronic 20% Pluronic 22% Pluronic 25% Pluronic 圖一、不同濃度 Pluronic-F127 溫度對黏度的關係 表一、Pluronic-F127 之成膠溫度 Pluronic-F127 溶液濃度 10% 15% 20% 22% 25% 成膠溫度(o C) no 26.7 20.1 18.0 15.5
Chitosan/GP Gelation Temperature Determination Temperature (o C) 5 10 15 20 25 30 Viscosity (cps) 0 200 400 600 800 1000 1200 1400 1600 1800 2000 2200 圖二、pH 7.2 幾丁聚醣凝膠黏度與溫度變化圖 Time (hr) 0 100 200 300 400 500 600 A c c u m ilation Release(%) 0 20 40 60 80 100 120
Drug in PLLA release from dyalisis membrane Free drug release from chitosan matrix Drug in PLLA release from chitosan matrix
Days
2 days 7 days 14 days
OD 490nm 0 1 2 3 4 polystyrene chitosan/GP 圖四、細胞增殖 MTS Test (a) (b) 圖五、細胞型態培養於 (a)幾丁聚醣凝膠 (b)細胞 培養盤 圖六、不同焦距相同視野的軟骨細胞,顯示整體構成了立體的結構
韌帶組織工程之研究
I.簡介(Introduction)與背景說明 根據 Langer 的統計,美國每年至少有十二萬的病人需要進行肌腱或韌帶置換的手 術,在台灣也因為機車為普遍之交通工具,導致車禍前十字韌帶斷裂之情形較國外更為 常見。目前臨床上處理韌帶或肌腱的斷裂大多是採用自體或異體移植的方式,或是以人 工合成之材料作為取代物。然而目前為止,以合成材料作為取代物之手術雖然在臨床上 相當普及,但材料經過長時間之後仍會有老化等問題發生,而異體移植目前仍處於臨床 試驗之階段,主要的問題為仍無法有效抑制免疫及發炎反應的發生。因此,找尋合適的 方法幫助撕裂肌腱及韌帶之修復仍是臨床上亟欲解決之課題。 一般生醫材料多為身體組織損壞時之替代品,因此需具有良好之穩定與持久性、適 當的物理化學性質,及良好的生物相容性。並於生醫材料植入人體後能維持其生理機 能。近幾年來由於生醫材料大部分需植入人體,因此多往生物裂解方面研究,許多脂肪 族聚酯類(aliphatic polyesters)聚合物諸如聚乳酸(polylatic acid,PLA)、聚甘醇酸(polyglycolic acid,PGA)、聚己內酯等均具有生物可降解性及生物可吸收性,而此類聚 合物已發展在止血材料、可降解之手術縫線、固定用之骨釘、骨板等牙科及骨科材料上, 可作為修補及促進軟骨再生材料,待組織修復後材料易被分解吸收。 近年來國外專家學者對細胞培養的研究結果顯示,生醫材料的應用中,生物體往往 只接觸到生醫材料的表面,故材料表面構造的重要性遠大於整體(bulk)構造,(整體構 造與機械性質或其他性質較有關聯),而根據過去本實驗室的研究也發現,材料表面結 構的改變確實會對於細胞的貼附及生長有很大的影響,因此實驗的初步將先探討不同表 面結構之聚乳酸薄膜對於韌帶及肌腱細胞貼附及生長的影響。
組織中的活細胞是藉由一層細胞外基質(extracellular matrices, ECMs)進行貼附和生 長行為,ECM 大部分是由蛋白質(protein) 和聚葡萄胺(glycosaminoglycans)所構成。而 ECM 已經被確認能調節細胞的多種行為,例如細胞的貼附、生長、移動和分化等等。 而構成這些 ECM 的生物分子成份包含有如 fibronectin, collagen, vitronectin, polylysine 和 laminine 諸如此類具有黏著性的蛋白質。而有鑑於過去學者認為聚乳酸為疏水性之生 醫材料,其生物適合性並不佳,因此本計畫在聚乳酸的表面作改質,塗佈較適合韌帶及 肌腱細胞生長之細胞外間質(Extracellular matrix 簡稱 ECM)。根據文獻,肌腱為連接骨 骼與肌肉的組織,而韌帶則為連接骨骼與骨骼之間的組織,在肌腱及韌帶的周圍都被大 量的第一型膠原蛋白(type-I collagen)及少量的聚葡萄胺(glycosaminoglycans)所環繞。因 此,本研究探討在聚乳酸薄膜表面塗佈不同的 ECM 如 type I collagen、fibronectin 等, 找尋最適於內側副韌帶細胞及前十字韌帶細胞之 ECM。
II 關鍵材料及方法(Subject and Methods)
※薄膜製備 本研究使用相轉變法(phase inversion)來製備薄膜,聚乳酸高分子於二氯甲烷,於 室溫下充分攪拌約一天,使其充分溶解均勻。 將混合均勻的高分子溶液,取一玻璃,將此溶液倒至玻璃板,以刮刀將溶液均勻塗 在玻璃板上,再將此玻璃板置於bath槽中或室溫下一天後脫膜。 ※膜性質測試 掃描式電子顯微鏡(SEM)觀察 將薄膜表面以雙面膠或銀膠黏於載台上,將薄膜浸泡在液氮中淬斷,也以雙面膠黏 於載台上,以掃描式電子顯微鏡觀察試片表面和斷切面的形態。
※ 生物適應性 細胞培養
細胞培養主要分為細胞貼附(cell adhesion)和細胞成長(cell growth)兩種。而細 胞培養乃是評估材料生物適合性主要的方法之一。
若材料表面的生物適合性不佳,則細胞不易與材料的表面產生交互作用,也就是細 胞不易貼附及成長。反之,若材料表面的生物適合性較佳,則材料表面會貼附細胞並進 而增生。
實驗使用的細胞為取自純種紐西蘭白兔的前十字韌帶細胞及內側副韌帶細胞。細胞 培養基乃為市售的 MEM 培養液,混合 10﹪胎牛血清 FBS(Fetal Bovine Serum)並加入 1%的 sodium pyruvate 以及 1﹪的三合一抗生素(antibiotics)所調配而成。
初代培養步驟:此初代培養步驟之主要目的是取得前十字韌帶(Anterior Cruciate Ligament)ACL 之韌帶細胞,並將其種在不同表面結構之薄膜上。此韌帶細胞是取自膝 關節處,其過程如下: 韌帶及肌腱細胞之取得: 韌帶或肌腱的纖維細胞培養,採用 Nagineni CN etal 的方法加以改進而來。膝 前十字韌帶組織取自人工膝關節置換的病人所切除的前十字韌帶或前十字韌帶斷 裂病人,肌腱組織取自接受前十字韌帶重建的病人的髖股韌帶或大腿後方內側的半 腱狀肌,係手術中修剪剩下的肌腱、清除相連的肌肉和結締組織,在培養盤上切成 小塊的 explant cultures。 純化細胞: 將取得之組織以 PBS 潤洗 2 次,切碎至 1mm2,置放於培養盤上。置放於 37 ℃,5%CO2之培養箱中。待細胞長出後,移除小塊的組織,讓細胞繼續增生,直 到滿盤。每三天更換新鮮之培養基,直到有足夠之韌帶細胞,再種至準備好之材料 上。 將細胞"撒"入有試片的細胞培養皿上,細胞數量控制為 8 ×104/ml,置於細胞培養箱 中培養。其每個 well 培養基為 1 ml/well。每組薄膜試片均重覆 6 次實驗。 評估肌腱及韌帶細胞生長情形: 光學顯微鏡觀察 因為光學顯微鏡只能觀察光線可通過的樣本,所以只能用光學顯微鏡觀察培養在對照組 和乾式膜上神經細胞生長的情形。 MTT 試驗 MTT test 是利用活細胞中的粒線體可以將黃色的 MTT 代謝為藍色的 formazan 晶體,可以用來測量細胞的存活率,利用 Mosmann et al 所使用之方法,經改善後 如下: 1. 將培養盤中的 medium 抽出並以 PBS 溶液沖洗兩次,將 0.1g 的 MTT 粉末溶於 50ml 的 PBS 溶液中,過濾滅菌,然後在每一個 well 中加入 0.1ml 的 MTT 溶液,置於 incubator 中作用 3 小時,整個過程需避光。
2. 在每個 well 中加入 0.2ml DMSO 溶液,在搖盪器(shaker)上搖 15 分鐘,使其均勻。 將細胞內的 MTT 產物 formazan 溶出。
3. 取 0.1ml 溶液置於 96well 的培養盤中,以 ELISA Reader 讀取波長 570 nm 的吸光值。 電子顯微鏡觀察:
掃瞄式電子顯微鏡觀察前(SEM)必須先作細胞固定。培養箱裡,培養皿中薄膜試片 上的細胞,從細胞培養箱中拿出,去除培養基,先用 PBS 清洗約兩次,以戊二醛 (Glutaraldehyde)固定,然後,進行臨界點乾燥(Critical Point Dryer,CPD)處理,
以防細胞變形。將其表面用鍍金處理,鍍金後以掃描式電子顯微鏡觀察細胞形態。 III.重要之結果 (Results) 圖一三種不同結構之PLLA薄膜之上表面SEM圖(a)顆粒膜(b)多孔膜(c)緻密膜 內側副韌帶細胞(100X) 內側副韌帶細胞(100X) 前十字韌帶細胞(100X) 前十字韌帶細胞(100X) 圖二、內側副韌帶細胞(MCL cell)與前十字韌帶細胞(ACL cell)之光學顯微鏡照片 由光學顯微鏡的照片可看出細胞會自組織的周圍遷徙出來,組織的周圍有較多的細 胞,距離組織愈遠則細胞呈現愈稀疏的狀態。照片顯示內側副韌帶的細胞較為細而小, 而前十字韌帶的細胞則是較為細而長,若觀察其自組織遷徙出來的速度是MCL較ACL 快速,此情形均與paper所描述雷同(9),因此可以確定我們所取得之組織確實是內側副 韌帶及前十字韌帶 。
MCL cells isolated from New Zealand rabbit culture on PLLA membranes
4h day1 day7 OD val ue 0.0 0.1 0.2 0.3 0.4 particulate porous dense
MCL cells isolated from New Zealand rabbit culture on PLLA membranes with coating fibronectin
4h day1 day7 OD value 0.0 0.1 0.2 0.3 0.4 0.5 0.6 particulate+F porous+F dense+F (a)未塗佈ECM (b)塗佈fibronectin
MCL cells isolated from New Zealand rabbit culture on PLLA membranes with coating collagen
4h day1 day7 ODvalue 0.0 0.1 0.2 0.3 0.4 0.5 particulate+C porous+C dense+C (c)塗佈collagen 圖三、內側副韌帶細胞(MCL)於不同表面結構之聚乳酸薄膜上培養4小時、一天、及七 天的MTT結果(n=4) (a)未塗佈之顆粒膜 (b)未塗佈之多孔膜 (c)未塗佈之緻密膜
(d)塗佈collagen之顆粒膜 (e)塗佈collagen之多孔膜 (f)塗佈collagen之緻密膜
圖四、內側副韌帶細胞(MCL)於不同表面結構之聚乳酸薄膜上培養4小時之SEM
之差異,甚至有多孔膜較差的趨勢。由(b)圖可以看出添加fibronectin(纖維網蛋白)對於初 期的貼附並沒有明顯的幫助,但由第七天的結果可以看出其MTT讀值都超過0.4,而(a) 圖的第七天讀值均不到0.4,因此可以推測添加纖維網蛋白有助於內側副韌帶細胞後期之 生長。若比較三種薄膜在二(b)圖的差異,可以看出第七天的顆粒型薄膜讀值最高,推測 可能是因為顆粒型薄膜的表面積最高,因此可塗佈之面積最大的關係。由圖二(c)可以看 出塗佈collagen(膠原蛋白)對於內側副韌帶細胞的四小時貼附有較為明顯的幫助,但對於 後期的生長似乎並無助益。而顆粒型薄膜之所以有較高的讀值,我們推測其原因也是因 為表面積較大的關係。 由圖四的SEM可以看出內側副韌帶細胞在未塗佈任何ECM的三種薄膜上,其貼附的 型態並無明顯的差異,都是呈細長型的型態,但在塗佈collagen之後,可以看出細胞呈 現更為攤平的貼附型態,與MTT結果相吻合。由此可以再次證明在四小時的時間點上塗 佈collagen是有幫助的。 VI.討論 (Discussion)與成果之貢獻 由以上的實驗結果可以發現將內側副韌帶細胞培養在三種不同型態的聚乳酸薄膜 上並無明顯的差異,推測多孔膜略差的原因可能是因為此細胞一顆約25μm,但孔洞卻 只有8μm,因此細胞在多孔膜上的貼附情形反而不如緻密膜。而較少被人討論到的顆粒 型薄膜雖然在未塗佈ECM時並沒有較佳的貼附及生長效果,但因具有較大表面積的優 勢,因此對於塗佈collagen時有助於細胞的初期貼附及塗佈fibronectin時有助於細胞的後 期生長都有優於其他兩種薄膜的結果。至於為何collagen有助於此細胞之貼附,而 fibronectin有助於後期的生長呢?我們推測原因是type I collagen本來就是內側副韌帶細胞 周遭最重要的一種細胞外間質,因此初期細胞尚未自我分泌collagen時,塗佈collagen有 模擬體內韌帶環境的效果。而後期由於韌帶細胞本身即具有分泌collagen的功能,因此 塗佈collagen並沒有太大的幫助。而韌帶細胞有可以和纖維網蛋白結合的domain,因此 塗佈纖維網蛋白有幫助韌帶細胞後期生長之效果。 此研究發展出一種較為新式的顆粒型薄膜,可以提供更大的表面積以吸附更多的細 胞外間質,並發現collagen有助於細胞之貼附,fibronectin而有助於細胞之後期生長,此 機制可以進一步應用在斷裂韌帶之輔助修復上。 參考文獻 (References)
1. C.C. Schmidt, H.I. Georgescu, C.K. Kwoh, G.L. Blomstrom, C.P. Engle, L.A. Larkin, C.H. Evans, S.Y. Woo, “Effect of growth factors on the proliferation of fibroblasts from the medical collateral and anterior cruciate ligaments”, Journal of Orthop Res
13(1995)184-190
2. D.S. Torres, T.M. Freyman, I.V. Ioannis, and Myron Spector, “Tendon cell contraction of collagen-GAG matrices in vitro:effect of cross-linking”, Biomaterials
21(2000)1607-1619
3. D.L. Butler, and H.A. Awad, “Perspectives on cell and collagen composites for tendon repair”, Clinical Orthopaedics and Related Research 367S(1999)S324-S332
4. J.D. Goldstein, A.J. Tria, J.P. Zawadshy, Y.P. Kato, D. Christriansen, and F.H. Silver, ” Development of a reconstituted collagen tendon prosthesis: a preliminary study”, Journal of Bone Joint Surg 71A(1989)1183-1191
5. W.J. Grzesik, B. Ivanov, F.A. Robey, J. Southerland, and M. Yamauchi, “ Synthetic integrin-binding peptides promote adhesion and proliferation of human periodontal ligament cells in vitro” , Journal of dental research 77-8(1998)1606-1612
6. K. Kobayassy, R.M. Heley, R.L. Sah, J.J. Clark, P.Tu, R.S. Goomer, W.H. Akeson, H. Moryia, D. Amiel, “Novel Method for the quantitative assessment of cell migration:A study on the motility of rabbit anterior cruciate(ACL) and Medial collateral
ligament(MCL) cells”, Tissue Engineering 6(2000)29-38
7. R. Langer, and J.P. Vacanti, “ Tissue Engineering”, Science 260 (1993)920-926
8. L.D. Bellincampi, R.F. Closkey, R. Prasad, J.P. Zawadsky, and M.G. Dunn, ”Viability of fibroblast-seeded ligament analogs after autogenous implantation”, Journal of
Orthopaedic Research 16(1998)414-420
9. C.N. Nagineni, D. Amiel, M. H. Green, M. Berchuck, and W.H.Akeson,
“Characterization of the Intrinsic Properties of the Anterior Cruciate and Medial
Collateral ligament cells: an in vitro cell culture study”, Journal of orthopaedic research
AdV Transduction & CAR Expression in hMSC
Summary
Previous reports debated on the effects of differentiation on adenoviral vectors (AdV)
transduction efficiency and Coxsackie-adenovirus receptor (CAR) expression. This prompted
us to investigate the efficiency of AdV transduction and CAR expression in human
mesenchymal stem cells (hMSC) and their differentiated progeny. Current results revealed
high efficiency (> 90%) of AdV transduction and a consistent level of CAR expression in
hMSC by the use of AdV carrying the green fluorescent protein (GFP) reporter gene.
Competition of CAR with blocking monoclonal antibody RmcB resulted in a reduction in
transduction efficiency; indicating the CAR involvement in transduction of hMSC.The cells
were then induced to differentiate into bone, fat or neural cells, and results demonstrated that
the differentiation was accompanied with a consistent decline in AdV transduction and a
decrement in CAR expression. According to the present investigation, undifferentiated hMSC
can serve as a gene-delivering system and gene transfer into hMSC before differentiation can
resolve the difficulties in transduction of their differentiated progeny.
Key Words
Introduction
Adenoviral vectors (AdV) are well known in its stability, ease handling and highly
uptake by many cell types including non-cycling cells. In addition, AdV can be concentrated
to extremely high titers as well.1-3 Recent advances in AdV modifications offer a less
immunogenic strain (defective recombinants) and an increase in the recombinant genome
insert size.4,5 Therefore, the application of AdV is more relevant for human gene therapy than
other means.
Since the efficacy of virus entry is restricted by interaction of viral fibers and cell surface
receptors, the abundance and the variety of viral associated surface molecules expressed in
cells are particularly important. The entry pathway for AdV consists of initial binding to the
cells, which is mediated by the association of the adenoviral fiber protein and a 46 kDa
membrane protein known as Coxsackie-adenovirus receptor (CAR),6 followed by
internalization, which is through an interaction of viral penton arginine-glycine-aspartate
(RGD) sequence to the αvβ3 and αvβ5 integrins.7
The CAR is a primary passage mediated the uptake of adenovirus in 293 cell line, HeLa
cell line and the cells derived from airways. However, cellular function of CAR is not clear
and CAR is only expressed in some types of cells and tissues. It has been shown that CAR
expression correlated not only with the undifferentiated state, but also with efficiency of AdV
transduction in oropharyngeal epithelial cells that the superficial layer (more developed) had
demonstrated in a small subset of CD34+ bone marrow (BM) and mobilized blood cells,9,10
while AdV transduction efficiency and CAR expression increase as the cells differentiate into
erythroid and myeloid haematopoietic cells.11 CAR is expressed in human mesenchymal stem
cells (hMSC),12 but not expressed in certain kind of mesenchymal cells, such as primary
human fibroblast.13
Since hMSC can be induced to differentiate into certain kinds of mesenchymal and
nonmesenchymal tissues, they provide a model to study the effects of differentiation on AdV
transduction and CAR expression. Therefore, we investigated AdV tranduction efficiency and
CAR expression in hMSC and their differentiated progeny. Efforts were also made to
characterize the tranduced hMSC in the differentiation potential and to know the persistence
Results
In vitro AdV-mediated gene transfer into hMSC
For these studies, gene transfer was demonstrated by AdV transduction with the
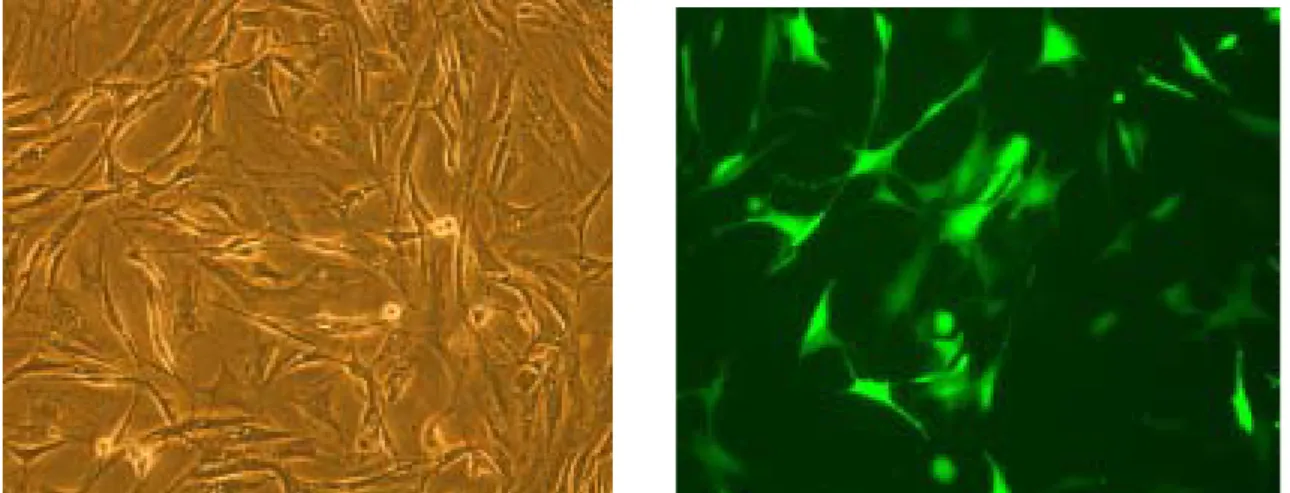
enhanced green fluorescent protein (GFP) gene. The hMSC cultures were infected with
Ad-PGK-GFP; and 2 days later, transduction was assessed by transgene expression. As seen
in Figure 1, after exposure to Ad-PGK-GFP for 2 days, a population of transduced cells was
detected under both fluorescent microscope (Figure 1a & b) and by flow cytometry analysis
(Figure 1c), displaying high fluorescent signal for GFP. At this time, transduced cells
encoding GFP fluorescence were found to have a good viability and no disturbance in
adherence. Transgene was retained and expressed by a small fraction of hMSC progeny even
up to 4 weeks after infection (data not shown). A strong green fluorescence in the infected
hMSC indicated a high efficiency of virus particles entering in cells. The efficiency of
AdV-mediated gene transfer into hMSC as analyzed by flow cytometry was 91.7 ± 6.6% (Table 1). High AdV transduction efficiency was also attained by the use of Ad-CMV-GFP, a
vector directing the expression of GFP under the control of a cytomegalovirus (CMV)
promoter (data not shown).
Expression and participation of CAR in AdV-mediated gene transfer into hMSC
Conget and Minguell12 have reported that the attachment of CAR receptor but not
we investigated by flow cytometry whether these cells express CAR. As shown in Figure 1d,
CAR expression was demonstrated in hMSC by using CAR specific monoclonal antibody
RmcB. The pattern of CAR expression was homogeneous, and near half of the cells had
moderate expression of CAR receptor. To determine the participation of CAR in AdV
infection, hMSC were infected with Ad-PGK-GFP in either presence or absence of
monoclonal antibody RmcB for competing CAR receptor. As shown in Figure 2, incubating
specific monoclonal antibody RmcB against CAR receptor immediately before AdV infection
was able to inhibit AdV infection by nearly 40%. Moreover, the effect of CAR competition by
specific monoclonal antibody to inhibit AdV infection in hMSC was a dose-dependent
manner.
Reduced AdV transduction efficiency and CAR expression in
osteogenic differentiated hMSC
The hMSC culture was induced to differentiate along the osteogenic, adipogenic or
neurogenic lineage as previously described.14,15 As shown in Figure 3 and Table 1, hMSC had
a relatively low efficiency of AdV transduction with 31%, 14% and 59% at 10 days, 17 days
and 23 days respectively after osteogenic induction. The low efficiency of AdV transduction
in the differentiated cells was correlated with a decrease in CAR expression after 10 days of
induction, but a regain of CAR was observed in 23-days induced cells.
Reduced AdV transduction efficiency and CAR expression in adipogenic differentiated hMSC
The AdV transduction efficiency was measured in hMSC on day 3, 7 and 10 after
adipogenic induction. As shown in Figure 4, the adipogenic differentiation in hMSC was
accompanied by a decrease in AdV transduction efficiency and a decrement in CAR
expression. There was an inverse correlation between the maturation of adipogenic
differentiation and the efficiency of adenoviral transduction. The observed efficiency was
44%, 30% and 19% at 3 days, 7 days and 10 days respectively after adipogenic induction
(Table 1). CAR expression was only observed in a subpopulation of adipogenic differentiated
hMSC, equal to 5% or less at 3 days after adipogenic induction, but CAR expression was
undetectable if the induction was continued more than 7 days.
Reduced AdV transduction efficiency and CAR expression in neurogenic differentiated hMSC
Unlike osteogenic and adipogenic inductions, neurogenic induction of hMSC had only
minor influence on AdV transduction efficiency. As shown in Figure 5 and Table 1, the
respectively. However, the prolonged neurogenic culture for 5 days had a lower fluorescent
intensity compared to those had neurogenic induction for 5 hours. In addition, hMSC
undergone neurogenic induction had less CAR expression in total population as compared to
Discussion
We confirm in this report that AdV efficiently infect hMSC, and that transduced
sequences are retained and expressed by a small fraction of hMSC progeny up to 4 weeks after
infection. The 90% transduction rate we have obtained, assessed by transgene expression, is
much higher than that observed by Conget et al in hMSC (19% transduction rate even at MOI
2000)12 and also higher than bone marrow,9 cord blood, and mobilized blood CD34+ cells.10
Moreover, the fluorescence intensity of the transduced cell population is high, and maintained
over several days in culture. This high expression rate associated with both Ad-PGK-GFP and
Ad-CMV-GFP indicate that efficient key factor in AdV-mediated transduction is probably not
on the vector or the promoter the vector used, but dependent on the content or purity of the
cells. Bone marrow, a mixture of many cell populations, contained haematopoietic stem cells
and nonhaematopoietic stem cells that include MSC. Previous reports debated on the
homogeneity of hMSC, but the purity of hMSC depends on the method used to isolate and
enrich them. The hMSC that we have isolated and characterized in previous report has
attained 98% of homogeneity.14 In contrast to undifferentiated hMSC, we did not achieve a
high ratio of transgene expression in hMSC that have been treated by a different induction
medium, where the cells underwent lineage-specific differentiation. Furthermore, several
attempts to improve upon this infection efficiency by manipulating serum and divalention
hMSC.
Factors determining the susceptibility of the target undifferentiated and induced cells to
the AdV infection are not entirely known. AdV utilizes fiber protein to attach to a recently
identified cell surface CAR receptor,6 and vitronectin-binding ανβ3 and ανβ5 integrins function as secondary receptors to mediate virus internalization.7 The level of CAR expression
but not integrins expression on the cell surface of hMSC determined the efficiency of
AdV-mediated gene transfer12 and the induction of CAR expression on fibroblast promoted
the binding of penton base protein, and facilitated AdV-mediated gene delivery.13 Based on
these observations, we have evaluated CAR expression on hMSC surface before and after
incubation with different induction mediums, and attempted to relate CAR expression with
transgene expression. We reasoned that the initiation of differentiation in hMSC should affect
the level of CAR expression and indirectly have effects on AdV-mediated gene transfer into
the target cells. We observed that, before any induction, hMSC expressed a high level of CAR
expression and more than 90% of hMSC were permissive for AdV infection; moreover,
treatment with any induction medium effectively decreased surface expression of CAR
receptor, and reduced expression of the transgene. Our results suggest that, as reported in
other cell types,6 CAR expression is necessarily predictive of the extent of transgene
expression in hMSC. We also observed that incubation of hMSC with CAR specific
confirming the participation of CAR on AdV entry and transgene expression in hMSCs.12
Similar with other reports, however, competition of CAR receptor with monoclonal antibody
did not totally hinder the AdV-mediated gene transfer, suggesting that the necessity of CAR to
infection may be relative rather than absolute, and possibly other, not yet defined, mechanisms
have participated in AdV-mediated gene transfer into hMSC.
The degree of commitment to lineage-specific differentiation coincided with a
reduction in CAR expression in hMSC, suggesting the role of CAR in differentiation-related
pathway. CAR expression is also reciprocally correlated with the maturity of cellular
differentiation in oropharyngeal cells that the superficial layer (more developed) had less
CAR expression than the basal layer (less developed).8 But CAR expression is upregulated
during maturation and lineage-specific differentiation in haematopoietic cells. Cellular
function of CAR is not investigated here and slight expression of CAR was observed again in
the late osteogenic differentiation of hMSC. Therefore, efforts still should be made to know
more about the involvement of CAR in cell proliferation and differentiation by the use of
overexpression or deletion of CAR in hMSC.
According to our previous data, hMSC isolated from 5-10 ml of bone marrow, could be
expanded to 1014 in an additional 15 weeks of culture, equal to the total cells of an individual
human body.14 The hMSC could be efficiently induced to undergo differentiation by different