國 立 交 通 大 學
材料科學與工程研究所
碩士論文
披覆含有共軛樹枝狀寡噻吩配位子之釕金屬複
合物的氧化鋅奈米粒子之合成與應用
Synthesis and Applications of ZnO Nanoparticles Capped with
Ruthenium Complexes Containing Conjugated Dendritic
Oligothiophene Ligands.
研 究 生 :黃煜證
指 導 教 授:林宏洲 博士
合物的氧化鋅奈米粒子之合成與應用
Synthesis and Applications of ZnO Nanoparticles Capped with
Ruthenium Complexes Containing Conjugated Dendritic
Oligothiophene Ligands.
研 究 生: 黃煜證 Student: Yucheng-Hwang
指導教授: 林宏洲 博士 Advisor: Dr. Hong-Cheu Lin
國 立 交 通 大 學
材料科學與工程研究所
碩 士 論 文
A Thesis Submitted to Department of
Materials Science and Engineering College of Engineering National Chiao Tung University
In partial Fulfillment of the Requirements For the Degree of Master of Science In Materials Science and Engineering
June 2010 Hsinchu, Taiwan
摘要
本論文中,首先我們利用 Stille coupling reaction 合成了三個擁有軟鏈分 枝的樹枝狀寡噻吩分子(branched oligothiophene dendrons),並將三個樣品命 名為一代(3T)、二代(7T)及三代(15T);將此系列分子與 4’-(5-Bromothiophene) -2,2’,6’,2’’- terpyridine 同樣藉由 Stille coupling reaction 作結合(分別命名為 G1、G2、G3),以便可由釕金屬(Ru)形成有機-無機金屬配位鍵,最後連接 在已經有另一個具有矽氧烷基之 Terpyridine 分子(Terpy)修飾在氧化鋅奈米 粒子表面(~5 nm ZnOnanoparticle); 此系列奈米複合材( G1,2,3–Ru-Terpy- ZnO)的吸附鑑定、吸附程度及光物理性質探索,可由 1H-NMR、FT-IR、 TGA、UV-vis、PL 及 TEM 進行研究及探討。此類奈米複合材料擁有良好 的光導電荷傳輸及可溶一般溶劑(如 THF…)的良好溶解特性,但其不易成膜 的特性,易造成光電轉換效率偏低,因此成為仍須克服的問題。將此類型 奈米複合材料應用在太陽能電池上,因不具結晶態,故利用旋轉塗佈的方 式製成薄膜,在AM 1.5 的標準太陽光照射下,元件效率可達到 η=0.047 %, Jsc= 0.629 mA/cm2、Voc= 0.30 V、FF= 0.25。
Abstract
In this reaserch, we synthesized three compound of branched oligothiophene dendrons by Stille coupling reaction,and denominated 3T,7T and 15T respectively.In the same way,we utilized dendrons to link with 4’-(5-Bromothiophene) -2,2’,6’,2’’- terpyridine by Stille coupling reaction.We named G1、G2 and G3 respectively.Finally,we utilized Ru to make G1、G2 and G3 combine with ~5nm ZnO nanoparticle which modified with Terpy. Futhermore, the identification and amount of absorbent on ZnO nanoparticle as well as the investigation of photophysical properties were reaseached by
1H-NMR、FT-IR、TGA、UV-vis、PL and TEM. This kinds of nanocomposites
are very soluble in some general solvents,for instanc -e,THF…and exhibit photoinduced charge-transfer interactions between the two species.But the thin-film performance of these nanocomposites were not very good.It caused that the performances of the photovoltaics is not very well.
These three types of the nanocomposites were non-crystalline, so we investigated the potential use of the nanocomposites in bulk heterojunction solar cells by spin coating. Under the standard AM 1.5G, the power conversion efficiency has been up to η= 0.047 %(Jsc= 0.629 mA/cm2、Voc= 0.30 V、FF=
致謝
一眼瞬間,就是要與母校別離的時刻了,還記得初到交大有如大一新 鮮人般的青澀模樣,往事的種種,至今仍歷歷在目! 在碩士班兩年的求學及研究期間,首先要感謝的是我的指導教授 林宏 洲老師,很感謝老師引領我進入“有機合成及高分子"的領域,並在老師 紮實的訓練下,對於有機太陽能電池有完全不同視野的認識,開拓了我在 合成與應用學習上不間斷地追求更臻極致的領域;無論是有機合成實驗上 或無機製程上都給予相當的支持與鼓勵,使我獲益良多,終能順利的完成 學業及研究。此外,感謝交大材料系的 韋光華老師、 林建村老師及 朱治 偉老師能在百忙之中抽空審核我的論文,惠予敝人寶貴的意見,讓論文更 趨完善,對於實驗的精準度給予莫大的指導。最後要感謝吳彥興學長,其 對我的教導與許多夜晚的疑難解惑,讓我能夠順利進行實驗及量測。 還有實驗室可愛的大家,博仁學長、之之學長、威宏學長、曉萍學姊、 怡婷、阿沛妹、老魏、Sata 及學弟……因為有你們,實驗室裡的夥伴們就 像大家庭一樣溫暖,讓我感覺隨時都能有朝氣與活力面對每一次的挑戰。 還要感謝爸媽,他們用心栽培我,讓我衣食無缺的在新竹求學;最後希望 我所認識的大家在未來都能夠平安順利的渡過每一天。目錄 中文摘要 ………I 英文摘要 ………II 致謝………III 目錄………IV 圖目錄 ………V 表目錄………VI 第一章 諸論………1 1.1 前言………2 1.2 太陽能電池………4 1.2.1 簡介 ………4 1.2.2 受體-給體系統(Donor-Acceptor System) ………6 1.2.3 奈米氧化鋅之應用 ………7 1.3 太陽能電池種類………8
1. 單結晶矽太陽能電池………9 2. 多結晶矽太陽能電池………9 3. 非結晶矽太陽能電池………9 1.3.2 薄膜太陽能電池………10 1.3.3 有機太陽能電池………11 1.4 太陽光譜介紹………17 1.5 參數說明 ………19 第二章 文獻回顧及研究動機 ………22 2.1 有機太陽能電池的探討 ………23 2.2 文獻回顧 ………26 2.3 研究動機………33 第三章 實驗部分 ………35 3.1 實驗儀器 ………36 3.2 實驗藥品及溶劑 ………38 3.3 有機合成流程及 ZnO 奈米粒子製作與表面修飾 ………40
3.3.1 4’-(5-Bromothiophene)-2,2’,6’,2’’-terpyridine 的合成 ………40 3.3.2 3T,7T 及 15T.的合成 ………41 3.3.3 G1,G2 及 G3 的合成………46 3.3.4 ZnO 奈米粒子製作與表面的修飾………50 第四章 結果與討論 ………56 4.1 合成機制探討 ………57 4.2 光物理性質之探討 ………58 4.2.1 1H-NMR 鑑定 ………58 4.2.2 FT-IR 之鑑定 ………59 4.2.3 TGA 熱重分析儀之鑑定………62 4.2.4 UV-vis 吸收光譜及 PL 放光光譜之探討………66 4.2.5 TEM 穿透式電子顯微鏡 ………74 4.3 電化學性質之探討………..77 4.3.1(cyclic voltammetry,CV)循環伏安法………77 4.3.2 太陽能電池之性質……….82 4.4 結論及未來展望 ………84 參考文獻………86 附圖………91
圖1-1 光照強度圖………3 圖1-2 太陽能電池構造 ………5 圖1-3 施體(D)與受體(A)的分子軌域交互作用………7 圖1-4 有機高分子光電轉換以及元件結構………14 圖1-5 染料敏化型太陽能電池結構………15 圖1-6 有機/無機奈米複合材料太陽能電池結構………16 圖1-7 太陽能電池之種類分類及效率 ………17 圖1-8 太陽光能量光譜圖………19 圖1-9 太陽能電池於照光下的電流-電壓(I-V)特性曲線………21 圖2-1 各式奈米粒子/高分子光伏電池性質列表………27 圖2-2 奈米粒子與有機高分子電荷分離能階示意圖………27
圖2-3 Conjugated Oligothiophene-Dendron-Capped CdSe Nanoparticle 示意 圖………28 圖2-4 All-in-one 分子電荷轉移示意圖 ………29 圖2-5 All-in-one 分子結構圖 ………29 圖2-6 有機染料批覆 ZnO 奈米粒子………30 圖2-7 ZnO-CAMIZ 光激螢光、Life-Time 光譜圖………30 圖2-8 CdSe-RB/P3HT光伏裝置………31
圖 2-9 I-V 曲 線 分 布 (a) ITO/PEDOT:PSS/P3HT:(CdSe:RB)/Al, (b)ITO/ PEDOT:PSS /P3HT:CdSe/Al………32 圖2-10 Z2907及元件內部能階圖 ………32 圖2-11 分別結合有機染料釕(Ru)的金屬錯合物【G1、G2、G3】-Ru-Terpy-ZnO 奈米粒子 ………34 圖4-1 Stille coupling 可能之反應機構 ………57 圖4-2 (a)Terpy (b)Terpy-ZnO 之1H-NMR 光譜鑑定………58 圖4-3 ZnO、ZnO-Terpy、ZnO-Terpy-RuCl3之FT-IR 光譜圖………..59
圖4-4 ZnO-Terpy-RuCl3、Terpy-G1、ZnO-Terpy-Ru-G1 之 FT-IR 光譜圖…59 圖4-5 ZnO-Terpy-RuCl3、Terpy-G2、ZnO-Terpy-Ru-G2 之 FT-IR 光譜圖…60 圖4-6 ZnO-Terpy-RuCl3、Terpy-G3、ZnO-Terpy-Ru-G3 之 FT-IR 光譜圖…60 圖4-7 (1)ZnO(2)ZnO-terpy(3) ZnO-terpy-RuCl3之熱重分析圖………62
圖4-8 (ZnO-terpy-Ru-G1(Cl-)、ZnO-terpy- Ru-G2(Cl-) 、ZnO-terpy-Ru- G3(Cl-)之熱重分析圖 ………63
圖4-9 ZnO-terpy-Ru-G1(PF6-)、ZnO-terpy- Ru-G2(PF6-) 、ZnO-terpy-Ru- G3(PF6-)之熱重分析圖 ………63
圖4-10 ZnO、ZnO-Terpy、ZnO-Terpy-RuCl3在 THF 中(0.01wt%)的 UV-vis 吸收光譜圖 ………67
圖 4-12 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、 ZnO-terpy-Ru-G3 在 THF 中(0.1wt%)的(a)UV-vis 吸收光譜圖及(b)部分放大圖 ………69 圖4-13 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、 ZnO-terpy-Ru-G3薄膜狀態 吸收光譜圖 ………71 圖4-14 G1-T、G2-T、G3-T在THF 中(10-5M)的 PL 放光光譜圖………71 圖 4-15 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、 ZnO-terpy-Ru-G3 在 THF 中(0.1wt%)的 PL 放光光譜圖 ………72 圖 4-16 ZnO-terpy-Ru-G2 及 ZnO-terpy-Ru 混合 G2 在 THF 中(0.1wt%)的 (a)UV-vis 吸收光譜圖及(b)PL 放光光譜圖………74 圖4-17 ZnO-terpy-RuCl3之穿透式電子顯微鏡圖(100nm) ………74 圖4-18(a)ZnO、(b)ZnO-terpy、(c)ZnO-terpy-RuCl3、(d)ZnO-terpy-Ru-G1、(e) ZnO-terpy-Ru-G2 、 (f)ZnO-terpy-Ru-G3 之穿 透 式 電 子 顯 微 鏡 圖 (20nm) ………75 圖4-19 ZnO-terpy-RuCl3/G2 之穿透式電子顯微鏡圖(100nm)。………75 圖 4-20 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、ZnO-terpy-Ru-G3 的 HOMO 和LUMO 能階示意圖。………78 圖4-21 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、ZnO-terpy-Ru-G3在氧化電 位下之循環伏安圖。 ………79
圖4-22 ZnO-terpy-Ru-G1、ZnO-terpy-Ru-G2、ZnO-terpy-Ru-G3 在還原電位
下之循環伏安圖。 ………80
圖4-23 (a) ZnO (b)ZnO-terpy (c) ZnO-terpy-RuCl3/G2 在氧化還原電位下之
循環伏安圖。………81
圖4-24 太陽能電池元件結構圖 ………82
圖4-25 不同比混摻的 ZnO-terpy-Ru-G3:PCBM 之 J-V 曲線圖………83
表目錄
表一 ZnO 與 ZnO-Terpy-RuCl3之熱重分析………65 表二 ZnO-Terpy-Ru-G1(Cl-) 2、ZnO-Terpy-Ru-G2(Cl-)2與 ZnO-Terpy-Ru-G3 (Cl-) 2之熱重分析 ………66表三 ZnO-Terpy-Ru-G1(PF6-)2、ZnO-Terpy-Ru-G2(PF6-)2與 ZnO-Terpy-Ru-
G3(PF6-)2之熱重分析………66
表四 ZnO、ZnO-terpy、ZnO-terpy-RuCl3/G2、ZnO-terpy-Ru-G 之電化學性
質數據.………79 表五 不同混摻比例(ZnO-terpy-Ru-G3:PCBM)後之元件量測數據………83
1.1 前言
隨著科技迅速的發展,相對的能源也不斷的大量消耗,就像是石 油、天然氣、煤礦,約佔地球百分之八十五的天然能源迅速消耗;不 僅如此,在燃燒這些石化能源之後,產生的二氧化碳及一些廢棄物, 進而引發的空氣及環境的污染,漸漸的對我們的大自然產生嚴重的破 壞,相對著「全球暖化」、「能源危機」及「生物絕種」…等問題浮上 檯面,因此根據經濟部能源委員會九十一年五月「台灣能源統計年報 (九十年)」資料顯示,以目前的損耗速度估計,目前地球石油蘊藏量 保守估計只能維持四十年,天然氣可開採六十二年,煤炭可開採兩百 二十七年。使的人類開始重視「綠色環保」、「節能省碳」及「替代能 源」的問題,並在各國環保意識的抬頭下,都如火如荼地倡導、制訂 規範及開發新能源。面對著即將在二十一世紀就枯竭的石油及天然 氣,對於替代能源的開發,也需加快腳步,像是利用太陽能、生質能、 地熱能、海洋能及風力…等。其中太陽能是一種非常純淨的能源,不 會污染環境及引起全球的溫室效應,更重要的是太陽能是我們週遭” 取之不盡、用之不竭”的能源。 太陽能來自太陽內部不斷的核融合反應所產生的能量(1)。太陽能 傳送到地球大氣層以後,其中部分被大氣層吸收,部分反射回太空中,部分穿透至地表並被吸收,其照射能量為平均每平方公尺地面可 達約180 瓦特(2)。以2005 年為例,全球人類所需的電力為 45.2TWh, 而被地球吸收的太陽能(入射的總太陽能扣掉被大氣層反射的部分) 高達751296000 TWh(參見圖 1-1 光照強度圖)。由此很明確顯示,由 太陽所游離出來的能量相當巨大,如果能轉換其中部分能量,或許就 足夠人類的使用,因此如何將太陽能有效的轉換,將是各國所有研究 人員努力的目標,此外對其使用的廣泛性、製造成本及使用年限…等 各種因素,都需做最嚴謹的評估。 圖 1-1 光照強度圖
1.2 太陽能電池
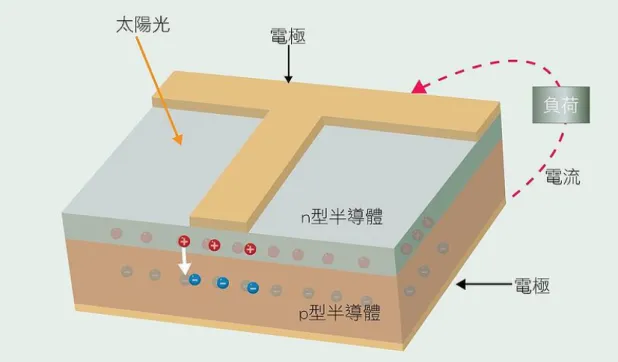
1.2.1 簡介 為了因應能源的匱乏,太陽能電池已經吸引世界各地的研究單位 注意,自1839 年 Becquerel 首次報導在電解質中發現光生伏打效應以 後,隨著科技的發展及人類的需求,目前已經進入量產的階段,太陽 能電池(solar cell)是一種將光能轉換成電能的元件(結構見圖 1-2 太陽 能電池構造),其工作原理是運用光伏效應(photovoltaic effect) (3),光 伏效應(photovoltaic effect)為在 p-n 接面附近,因電子及電洞的結合形 成的一個載子空乏區(depletion area),而 P 型及 N 型半導體分別帶有 正電及負電荷,因此形成一個內建電場。而太陽能電池發電原理是經 由太陽光照射元件,使內部的半導體活化層(active layer),瞬間將光 能轉化成電能,使N 型半導體的電子由價帶提升至傳導帶,並再與 P 型半導體接觸時,受到內建電場的影響,湧入 P 型半導體的電洞,相 對著電洞湧入 N 型半導體的方向,造成電子及電洞的分離,並由電 極形成一個迴路,產生電流及電壓,不過此種簡單接合的電池結構, 由於 p-n 接面的面積不大,效率通常不高。圖1-2 太陽能電池構造
因此,光能轉化成電能的過程,主要可以分為三個步驟:(1)光生 載流子 (2)光生載流子被半導體中的靜電場分離 (3)光生載流子被電 池兩極收集,產生電流;或是在有機/高分子太陽能電池中:(1)exciton generation,(2)charge separation,(3)carrier transport。因此若想提升光 電轉換的效率,就可以從改善成有利於這三個步驟的環境或材料著 手。近來本體異質接合 (bulk heterojunction, BHJ) 之電池結構研究引 起相當廣泛的注意,此型電池大大提高了主動層中 pn 接合介面的面 積(4a)。
1.2.2 受體-給體系統(Donor-Acceptor System)
能隙(band gap energy:Eg)是一種介於最高填滿的分子軌域
(Highest Occupied Molecular Orbital:HOMO)和最低未填滿的分子軌 域(Lowest Unoccupied Molecular Orbital:LUMO)之間的能量間隔,其 決定共軛分子的導電性。若降低分子的能隙將會增進傳導帶的熱量總 數(Thermal Population)並且增加本質的載子數目,形成真正的"有機型 金屬"(Orangic metals)。另一方面,窄能隙有較低的氧化電位時,將會 導致參雜狀態穩定,因此藉由化學修飾高分子來控制 HOMO-LUMO 之間的間隙,對於太陽能電池來說是極其重要的。 共軛高分子的電性和光學性質是來自位於 HOMO 和 LUMO 周圍 的能階所影響的。根據能帶理論來說,最高填滿帶來自於每個單體的 HOMO,也就是所謂的價帶;而最低未填滿帶則是來自於每個單體的 的 LUMO,作為傳導帶。在吸收光譜裡若要得到共軛高分子的 Eg, 則是將吸收峰進行外插法而得,因此要得到窄能隙的共軛高分子,其 吸收邊緣(Absorption edge)應在近紅外光區(Near infrared region),也就 是吸收長波長。
近年來的分子軌域計算呈現 Donor 和 Acceptor 之間的能階經過
的 HOMO 和 Acceptor 部分的 LUMO 靠的很近的話,就會使的電子 易被激發至LUMO。(如圖 1-3) 圖1-3 施體(D)與受體(A)的分子軌域交互作用 1.2.3 奈米氧化鋅之應用性 奈米氧化鋅高激子結合能之特性使氧化鋅於室溫具有高激子放 射效率,因此目前氧化鋅被認為是非常具潛力的紫外-藍光光電材 料。另外,氧化鋅可吸收較本身能隙波長短之紫外線,若在氧化鋅中 摻入鋁或鎵等 IIIA 族元素,可降低電阻率以利製作透明導電膜,用 作透明電極的應用,另可用在場發射 (field-emission) 顯示器上。 氧化鋅通電後電壓與電流間有相當高的非線性關係,可用來製作可變 電阻。於工業上可作為橡膠加速劑、顏料及陶瓷添加劑,其他還有化 妝品、紫外線吸收劑、奈米光觸媒、鋰電池、太陽能電池、和氣體感
測器等光電之運用等多方面用途。奈米氧化鋅具有半導體催化劑的電 子結構,在光照射下,當具有一定能量的光子或者具有超過這個能隙 的光子射入半導體時,一個電子從價帶 (valence band) 激發到傳 導帶 (conduction band),而留下了一個空穴。激發態的傳導帶電子 和價帶空穴能夠重新結合消除輸入的能量和熱,電子在材料的表面態 被捕捉,價態電子躍遷到傳導帶,價帶的孔穴把周圍環境中的羥基電 子搶奪過來使羥基變成自由基,作為強氧化劑而完全對有機物 (或含 氯) 的降解,殺死病菌和病毒。2006 年 B. Q. Sun 在薄膜電晶體 (thin film transistor, TFT)上,利用氧化鋅奈米棒可製作出高 ON/OFF ratio、高移動率 (mobility)、低臨界電壓 (threshold voltage) 之高
效率薄膜電晶體元件(4b)。在有機太陽能電池 (organic solar cell) 方
面,氧化鋅可用於取代 C61-butyric acid methyl ester (PCBM) 做為
電子受體物質 (electron accepting material),具有非毒性、價格較低
廉等好處(25)。 1.3 太陽能電池種類 1.3.1 單晶矽、多晶矽及非晶矽太陽能電池 目前市場應用上大多為單結晶矽及非結晶矽,較多且廣泛,其原 因有:1.單結晶矽效率最高;2.非結晶矽價格最便宜,且無需封裝, 生產也最快;3.多結晶矽的切割及下游再加工較不易,而前述兩種都
較易於再切割及加工。 1.單結晶矽太陽能電池(5) 單晶矽電池最普遍,大多用於發電廠、充電系統、道路照明系統 及交通號誌……等,所供給之電力與電壓範圍廣、轉換效率高、使用 年限長,故導致世界主要大廠如:德國西門子、英國石油公司及日本 夏普公司均以生產此類單晶矽太陽能電池為主要產品,以現今市場評 估佔有率約五成,單晶矽電池效率從 11% ~24%,太空級(蒸鍍式)晶 片從16%~24%,當然效率愈高其價格也就愈貴。 2.多結晶矽太陽能電池 多晶矽太陽能電池,因為材料本身的多晶特性,故在切割和再加 工特性上遠比單晶和非晶矽更困難,效率方面也比單晶矽太陽能電池 的低。不過,簡單的製程和低廉的成本是它的最重要特色,且較單晶 矽電池便宜20%,所以在部分低功率的電力應用系統上,便採用這類 型的太陽能電池。 3.非結晶矽太陽能電池(6)
非晶矽電池的發展始於 1975 年,當時電池為 1 µm 厚度的矽 膜搭配p-i-n 的結構配置使其效率達 2.4%,到 1980 年已經有商品問 世,而我們現今使用的太陽能電池計算機便是此類型產品,由於非晶 矽具有較佳之光吸收能力,所以吸收層厚度較薄,一般約為 500nm 上下,對光的反應較佳,其能階約在 1.7eV,因此能量較小之紅光與 紅外光皆無法吸收,因此限制其轉換效率的發展,但微弱之陽光(非 直射、陰天)或室內的燈源已足夠使之轉換成電能,且具有較低之溫 度係數,此外因為薄膜結構具有較多之缺陷,易產生所謂光致衰敗現 象,光照使用後短時間內效率性能大幅衰退約達 15 ~ 35%之後才會 達到一個穩定的狀態。 1.3.2 薄膜太陽能電池 薄膜太陽能電池為太陽能電池產業中重要的一環,雖然薄膜電池 的效率較低,但相較於矽晶片製作而成的太陽能電池具有低成本之優 勢。因此,其具有一定比例上的佔有率約10%,就其發展來說大致上 可分為兩大類:(1)矽薄膜太陽能電池。(2)碲化鎘(CdTe)薄膜太陽能電 池。 (1)矽薄膜太陽能電池 最早在開發此型光電池時有很大的突破是在於其矽結晶層的厚
度僅5~50 毫米,可以次級矽材料、玻璃、陶瓷或石墨為基材,除矽 材料使用量可大幅降低外,此類型光電池由於電子與電洞傳導距離 短,因此矽材料的純度要求較低,材料成本可大幅降低。且由於矽材 料不如其他發展中光電池半導體材料,具有高的吸光效率,且此型光 電池矽層膜,不如矽晶圓型太陽能電池矽層厚度約達 300 微米,因 此為提高光吸收率,設計上需導入光線流滯的概念,此點是與其他薄 膜型光電池不同之處。 (2)碲化鎘(CdTe)薄膜太陽能電池(7) 此類材料最早出現是在 1982 年時由 Kodak 公司所做出之轉換 效率 10%的太陽能電池,目前以各研究實驗室最高的效率是 17%, 由美國國家再生能源實驗室實驗室所創立,而大面積之模組目前最高 約 11%,其關鍵點在吸收層主要是由 P 型 CdTe 與 N 型 CdS 形 成且為直接能隙之材料,因此僅需數個微米之厚度,即可將太陽光完 全吸收。 1.3.3 有機太陽能電池 有機太陽能電池因其具有成本低廉、製成簡易、易於大面積化及具 有可繞曲性,因此吸引許多研究員投入,尤其具有可繞曲性的特點, 在未來可將此有機太陽能電池材料以直接塗抹的方式,塗佈於可繞式
基板表面,因此大大減低其成本及製程,然而有機太陽能電池至今尚 未取代一般半導體材料的太陽能電池,主要因為其轉換效率太低及熱 穩定性的問題仍需克服。因此提高轉換效率及熱穩定性,並開發新型 的太陽能電池,能是目前研究的目標。 有機太陽能電池大致上可分為三大類: (1)有機高分子太陽能電池、(2)染料敏化型太陽能電池、(3)有機/無機 奈米複合材料。 (1)有機高分子太陽能電池(5)
University of California, Santa Barbara Alan Heeger 研究團隊所發 表 第 一 個 具 備 較 高 效 率 的 高 分 子 有 機 太 陽 能 電 池 , 其 使 用 MEHPPV/C60 Bilayer Hetrojucton 的概念,所製作出第一個高分子碳
材 太 陽 能 電 池 , 於 1995 年 在 Science 期 刊 發 表 並 以 其 Bulk Hetero-junction (BHJ)概念為主要製程的 MEH-PPV/C60 太陽能電池 後,此類型的太陽能電池才真正受到重視並發展 高分子/ 碳材太陽能電池原型(Prototype),迄今高分子有機太陽能電 池仍沿襲著此 BHJ 觀念,搭配導電高分子/碳材上的變化來提昇效 率。 高分子太陽能電池發電原理為:光主要由Donor 材料(共軛高分子;
Conjugated Polymer)吸收,由於共軛高分子材料具高的吸收係數, 因此其元件的厚度為 100 nm (Polycrystalline CuInSe,CdTe: 1µm、 Crystalline Silcon:100 µm),為最輕薄的太陽能電池,光電轉換詳細作 用機制已經在1.2 敘述,利用 Donor-type 材料與 Acceptor-type 材料 進行混摻,藉由太陽光的照射,以產生電子與電洞對(Electron/Hole Pair),最後電子與電洞分離,並分別經由電子與電洞傳導材料,傳輸 至 陰 陽 電 極 而 形 成 電 壓 降 產 生 電 能 , 由 於 有 機 半 導 體 材 料 激 態 (Exciton)下,有較高的束縛能(Binding Energy:約在 0.2~1.0 eV)與
無機材料(矽的Binding Energy;約 0.015 eV)相比,其束縛能約大
上一兩個等級,故室溫條件下,有機材料無法形成自由的電子或電洞 (Free Carriers),必須藉由 N 型與 P 型材料界面的勢能差,才能達到 電子與電洞分離的效果,目前最常見之有機混成太陽光電系統,主要
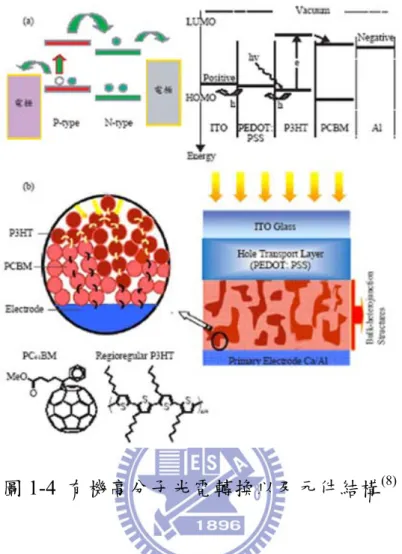
採Heeger A. J. 與 Wudl F.所設計的 BHJ 結構,其元件結構如圖 1-2
圖1-4 有機高分子光電轉換以及元件結構(8)
以高分子/碳材混摻系統(Poly(3-hexylthiophene) (P3HT)為 N-type 而[6,6]-phenyl-C61-butyric acid methyl ester (PCBM)為 P-type 材料)所
組成的主動層材料,配合ITO 基材與 Poly(3,4-ethylenedioxythiophe ne):
poly(styrenesulfonate) (PEDOT: PSS)組成的陽極及以陰極鋁(Al 所構 成。
(2) 染料敏化型太陽能電池(9)
的特色,整體的成本約為傳統矽基太陽能電池的1/5~1/10,因而引起
能源界積極地投入這方面的技術研究開發。電池結構如圖1-5
1991 年以瑞士洛桑聯邦理工學院 Grätzel 教授為首的研究小
組,以奈米多孔TiO2 膜為半導體電極,以過渡金屬Ru 及 Os 等有機
化合物作敏化劑,並選用適當的氧化−還原電位,發展出一種新型的 太陽能電池,稱之染料敏化太陽能電池(Dye-Sensitized Solar Cells; DSSC),其在 AM1.5 的模擬日光照射下轉化率可達 7.1%。 1993 年 Grätzel 等再次報導了光電能量轉換率達 10%的 DSSC 電池。 1997 年其轉換效率達到 10~11%。 1998 年他們又研製出全固態 DSSC 電池,更進一步朝實用 化邁進。 圖1-5 染料敏化型太陽能電池結構(10)
(3) 有機/無機奈米複合材料太陽能電池
目前有機/無機奈米複合材料太陽能電池的研究,幾乎都是合成
有機高分子,如P3HT、MEH-PPV 、MDMO-PPV…等,並將有機高
分子充當 Donor 混合金屬奈米粒子,或是塗抹、填充於金屬奈米棒
上,其選擇的金屬如ZnO(11)、CdSe(12)、PbS(13)、PbSe(14)、TiO2(15)…等
金屬則作為 Acceptor,且探討其物理性質、電化學性質及元件性質。
其有機/無機奈米複合材料太陽能電池結構(如圖 1-6)。
圖 1-6 有機/無機奈米複合材料太陽能電池結構(16)
及 型 態(morphology) 的 程 度 並 不 佳 , 易 造 成 電 子 及 電 洞 再 結 合 (recombination)。因此,有機/無機奈米複合材料太陽能電池,其轉換 效率僅有大約2%。有鑑於有機/無機奈米複合材料太陽能電池的效率 或許仍然有發展的空間,因此如何提高有機/無機奈米複合材料太陽 能電池的轉換效率及開發新型的太陽能電池已成一個課題。 圖1-7 太陽能電池之種類分類及效率(17)
1.4 太陽光譜介紹
太陽光譜能讓我們了解如何有效的捕捉到光子,並且對太陽能電 池的效率有相當大的影響。太陽表面溫度接近6000 K,因此其放射光譜幾乎等同於該溫度下的黑體輻射,並且光譜照射並無方向性,地球 與太陽相距約1億5千萬公里遠,能到達地球表面的光子,幾乎只有正 向入射至地球表面的光譜所貢獻,到達地球大氣圈表面的光譜輻射能 量定義為太陽常數(solar constant),其數值大約1.353 kW/m2,因此大 氣圈外的太陽光譜定義為AM 0,其中大氣質量(air mass, AM)用來估 量因為大氣層吸收後,所導致影響太陽光譜表現與總體能量值,而這 些能量值亦是地球表面應用的太陽電池元件所能運用的,而大氣質量 的計算方法是使用air mass = 1/cosθ來計算,其中θ = 0所代表的是太陽 光線從頭頂上方直射下來,而由上述的計算式中可知,地球表面用以 衡量太陽光譜的大氣質量值是大於等於1,目前被慣以使用的太陽光 譜AM 1.5,即是太陽光入射角偏離頭頂46.8度,當太陽光照射到地球 表面時,由於大氣層與地表景物的散射與折射的因素,會多增加20% 的太陽光入射量,抵達地表上所使用的太陽電池表面,其中這些能量 稱之為擴散部份(diffusion component),因此針對地表上的太陽光譜能 量有AM 1.5G (Global)與AM 1.5D (Direct)之分,其中AM 1.5G 即是有 包含擴散部分的太陽光能量,而AM 1.5D則沒有。圖1-7表示的即是 大氣圈外(AM 0)與地表上(AM 1.5)太陽光能量光譜。太空用的太陽電 池元件電性量測所使用的標準光譜是採AM 0,而地面上應用的太陽 電池元件電性量測所使用的標準光譜,依其應用性之不同,可採用
AM 1.5G 或是AM 1.5D,其中AM 1.5G光譜的總照度為963.75W/m2, 而AM 1.5D光譜的總照度為768.31W/m2,在量測計算應用上方便,常 會將此二值做normalize至1000 W/m2(18)。 太陽輻射的光譜主要是以可見光為中心,波長從 0.3 μm 的紫外 光到數微米的紅外光是主要的分布範圍。對於太陽光的模擬,本研究 採用汞燈(Hg lamp)做為光源作為模擬太陽光之光源。 圖1-8 太陽光能量光譜圖
1.5 參數說明
藉由可調變電源負載提供,來量測元件在照光清況下的電流值與 電壓值,圖 1-9 所示則為常見的太陽能電池元件的電性量測結果。從所測得I-V 曲線圖裡,可分別萃取出元件的開路電壓、短路電流、填 充因子、轉換效率等物理參數。其個別定義如下所述(19): 1.開路電壓(Voc):當太陽能電池元件電流等於零時,所得之電壓值。 2.短路電流(Jsc):當太陽能電池元件電壓等於零時,所得之電流值。 3.轉換效率(Efficiency):η%=(元件的最大功率輸出值/(入射光譜 能量* 元件面積))*100 Efficiency η = VOC * ISC * FF / Pinput 其中Pinput 為入射光強度。
4.填充因子(Fill Factor):FF =(元件的最大功率輸出值/(Voc*Jsc)) *100
FF = (I * V)max/( Jsc* VOC)
5.入射光子-電流轉換效率(IPCE):其值是代表在單色光照射下入射光 子轉換成電子的效能,其公式定義為:
IPCE=1.24*103*光電流密度(μA/cm2) / λ(nm)*I(W/m2) 式中λ 為單色光的激發波長,I 為入射光子通量。
2.1 有機太陽能電池的探討
目前光敏化劑即有機染料釕(Ru)的金屬錯合物其最高光電轉換
效率已達到11%(19),而純有機染料(Metal-free)其最高光電轉換效率可
達 9%(20)。此外,典型有機高分子太陽能電池(Poly(3-hexylthiophene)
(P3HT) 為 N-type 混 合 [6,6]-phenyl-C61-butyric acid methyl ester (PCBM)為 P-type 材料)所組成的主動層材料,其光電轉換效率可達 約5%(21)。然而有機/無機奈米複合材料太陽能電池,其轉換效率僅有 大約2%(22)。有鑑於有機/無機奈米複合材料太陽能電池的效率或許仍 然有發展的空間,因此如何提高有機/無機奈米複合材料太陽能電池 的轉換效率及開發新型的太陽能電池為主要實驗目標。 以高分子太陽能電池而言,主要是利用高分子主鏈中單雙鍵組成 的重複單元,π 或 π*鍵通過形成電荷遷移,因而具有導電性。為了提 高導電性,除了分子設計使分子軌道能強烈離域和相互重疊,最好的 方法就是使材料具有電子給體(donor)和受體(accepter)的系統。一個給 體材料可以充當另一個給體性更強的材料的受體,因此可以在高分子 主鏈或側鏈上設計具有受體性質的官能基團,可以產生推拉電子的作 用 , 若 是 延 長 了 高 分 子 的 共 軛 體 系 , 也 可 以 降 低 高 分 子 的 能 隙 (bandgap),增加可見光吸收的範圍。除此之外,就是將共軛高分子作 為電子給體,有機小分子或無機半導體作為電子受體,形成的複合材
料,給體與受體間的界面面積大,接觸處形成異質結,整體可視為一 個大異質結,這種類型的太陽能電池,被稱為本體異質結太陽能電池 (bulk heterojunction solar cell)(BHJSC)。(23)
BHJSC,近來最多的研究當然就是添加 C60或C70的衍生物PCBM
作為電子受體材料(24),這種添加半導體奈米粒子的型式,也已屬於奈
米粒子/高分子光伏電池(nanoparticle/polymer photovoltaic cell)的範
疇,但因為PCBM 價格昂貴,以其他半導體奈米粒子取而代之的類似 研究,也逐漸受到重視。(25)這種有機/無機奈米複合材料,除了承繼了 高分子材料原本的柔韌性、溶解度及成膜性,基本的機械性質和熱性 質由於奈米粒子與高分子間的界面作用力也將有效提昇(26);在光伏裝 置方面,無機奈米粒子能增加可見光區的吸收,並由於無機奈米粒子 本身擁有的高載流子遷移效率,其對電子的吸引力會大於有機的共軛 高分子,再加上共軛高分子傳導電洞的能力極高於傳導電子的能力, 因此在有機高分子與無機奈米粒子的界面,容易發生電荷的轉移,進 而提升整體材料的載流子遷移率及光電轉化效率。 理論上無機半導體奈米粒子的添加的確是能提昇光電轉化效 率,條件是增加給體與受體的接觸界面面積,使電荷轉移確實發生。 其 方 法 可 以 是 改 質 奈 米 粒 子 的 表 面 , 使 具 備 有 機 的 官 能 基 (surfactant),增加奈米粒子與有機共軛高分子的相容性,但這些表面
官能基通常為絕緣,為了減少這些絕緣 ligand 的影響,儘可能控制 ligand 分子鏈的長度,並想辦法增加奈米粒子與共軛高分子間的作用 力,是主要研究的課題。同時並有研究指出,微相分離的複合材料通 常具有較好的光電轉化效率,原因是奈米粒子之間聚集形成連續通 道,比較於奈米粒子能夠在高分子中達到均勻分散,增加給體與受體 的接觸界面的原理互相矛盾,在兩者優缺點相互競爭下,通常必須嚐 試有機ligand 的種類及濃度,或是控制奈米粒子與共軛高分子掺混的 比例,才能得到適合材料本身理想的條件。(27) 此外當無機半導體奈米粒子的粒徑大小,小於內部電子的費米波 長(Fermi wavelength)時,即可稱作量子點(Quantum dot),量子點奈米 顆粒會表現出新的光、電、聲、磁等體積效應,隨著粒徑減小,比表 面積變大、能隙變寬、能帶彎曲減小、光譜藍位移,以及光生載流子 從體內擴散到表面所需的時間縮短,光生電荷分離的效果就越高,電 子電洞的復合率(Recombinations)越小。這些效應對太陽能電池的影響 其實有好有壞,所以在奈米粒子/高分子光伏電池的研究中,量子尺寸 效應也是必須考量的要素。(28)
2.2 文獻回顧
2008 年 , Saunders 等 學 者 在 所 發 表 的 文 獻 回
顧 ”Nanoparticle-polymer photovoltaic cell”中(25),將以往各式無機奈米
粒子與各式有機共軛高分子掺混的太陽能電池效果列表比較,(如圖 2-1),並對奈米粒子/高分子光伏電池大致整理出一些有效的規則,如 下:
I. The nanoparticles and polymers should have high electron and hole mobilities, respectively. The interfacial area of the bulk heterojunction should be high.
II. The polymer and nanoparticles should have high extinction coefficients and absorb light from as much of the solar spectrum as possible in a complementary manner.
III. The energy levels for the nanoparticles and polymer should be chosen so that they promote charge separation and transfer (圖 2-2). Optimum values could be determined using the work of Scharber et al. in the case of PCBM nanoparticles.
IV. The nanoparticles should be ligand-free (naked) within the photoactive layer and form interconnected bicontinuous solid dispersions or a vertically aligned structure.
圖2-1 各式奈米粒子/高分子光伏電池性質列表。(25)
圖2-2 奈米粒子與有機高分子電荷分離能階示意圖。(25)
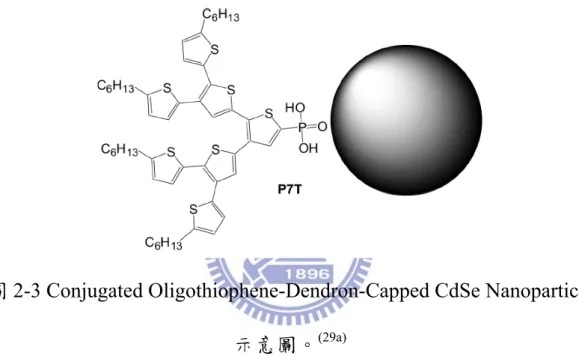
2004 年,Locklin 等學者成功利用 ligand exchange 的方法將 oligothiophene 的 dendrimer 披覆在 CdSe 奈米粒子表面上(如圖 2-3)。
此light-harvesting dendrimer 可以增加可見光區的吸收,並使披覆的奈 米粒子擁有的良好的溶解度,良好的光生電荷分離效果,被應用在 BHJ 太陽能電池中,得到 0.29%的光電轉化效率,並預期能夠藉由增
加 dendrimer 的代數或與 polythiophene 結合,能夠得到更好的光電轉
化效率。(29a)
圖 2-3 Conjugated Oligothiophene-Dendron-Capped CdSe Nanoparticle
示意圖。(29a)
2007年,Zhiqun Lin 等學者利用Vinyl-Terminated P3HT修飾在 [(4-bromophenyl)methyl] dioctylphosphine Oxide(DOPO-Br)-Functiona -lized CdSe QD,設計理念囊括了增加吸收光範圍及良好的分子間電 荷傳輸。由圖2-5可知,直接鍵結半導体粒子的發光幾乎完全被抑制 (Quench),並且螢光的 life-time明顯下降,證明了直接鍵結半導体粒
圖2-4 利用Vinyl-Terminated P3HT修飾在[(4-Bromophenyl)methyl]
dioctylphosphine Oxide (DOPO-Br)-Functionalized CdSe QD之合成。(30)
圖2-5 P3HT/CdSe composite與P3HT-CdSe nanocomposite之PL放光光
譜及Life-Time光譜圖。(30) 2009年,Marczak等學者,以sol-gel的方法製備約5 nm大小的ZnO 奈米粒子,並利用catechol官能基對金屬氧化物的吸附能力,將有機染 料porphyrinatozinc接枝到ZnO奈米粒子的表面(如圖2-6)。離心分離相 分離的奈米粒子後,發現有機染料可大幅提升ZnO可見光區的吸收, 且若是增加有機染料的濃度,可以使ZnO的發光幾乎完全被抑制
(Quench),且螢光的 life-time明顯下降(如圖2-7),顯示電子有效轉移, 證明有機染料的披覆,不僅可增加奈米粒子在有機相的相容性、提升 可見光區吸收,也使電子能有效轉移。(31) 圖2-6 有機染料批覆ZnO奈米粒子。(31) Increasing of μM 圖2-7 ZnO-CAMIZ光激螢光、Life-Time光譜圖。(31) 2009年,Guchhait等學者合成表面具有amine官能基的 CdSe奈米 粒子,大小約為2.5 nm,使與有機染料Rose Bengal (RB)之間形成化學 鍵結,形成無機Core/有機Shell的CdSe-RB奈米複合粒子(如圖2-8)。有
機染料的披覆,再一次證明可以增加奈米粒子在有機相的相容性、提 升可見光區吸收,也使電子能有效轉移;此外,進一步以CdSe-RB奈 米複合粒子做為電子受體,P3HT為電洞傳輸層,進行光伏裝置的應 用。在最佳摻混條件下,有機染料披覆的CdSe,相對於未披覆的CdSe, 皆大幅提升了光伏裝置的短路電流、光電轉化效率和external quantum efficiency(如圖2-9),證明有機染料披覆造成光生電荷轉移的效率的提 升,對光伏裝置有正面的影響。(32) 圖2-8 CdSe-RB/P3HT光伏裝置。(32)
圖2-9 I-V 曲線分布(a) ITO/PEDOT:PSS/P3HT:(CdSe:RB)/Al,
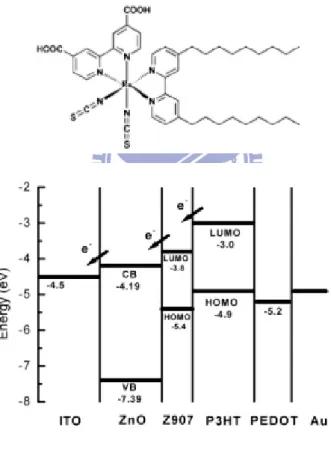
在 2006 年 Punniamoorthy Ravirajan 和 Jenny Nelson 在 ZnO 及 P3HT 間置入有機染料釕(Ru)的金屬錯合物 Z2907 (如圖 2-10),以致 於降低電子及電洞再結合(Recombination),此外更延長其可見光區的 吸收範圍,以增加外部量子的轉換效率,進而增加其光電子數,並且
探討ZnO nanoparticle 及 ZnO nanorod 個別得情形,效率分別達 0.05%
及0.2%。
2.3 研究動機 去 基於以上文獻回顧,所得到的訊息,我們製備BHJSC的活性層 時,最好加入半導體粒子作為電子受體,有效的改質半導體粒子(有 機染料的批覆或過渡金屬配位鍵的生成),可增加在有機相的相容 性、提升可見光區吸收,也使電子能有效轉移,可以嚐試直接接枝共 軛高分子,或是摻混理想的共軛高分子,作為電洞傳輸層,以茲光伏 裝置的研究,為了增加研究的新穎性,和加強奈米粒子與共軛高分子 間的界面作用力,超分子化學鍵,是值得運用的不二法門。如前文, 我們有鑑於在染料敏化型太陽能電池的有機染料釕(Ru)的金屬錯合 物與ZnO具有可觀的傳導電子的能力,因此我們將設計有機染料釕 (Ru)的金屬錯合物G1、G2、G3 修飾ZnO奈米粒子(nanoparticle)的 表面[如圖 2-11],以減小其有機及無機奈米粒子間的距離(domain size),因此利用三個碳的碳鏈結合有機染料釕(Ru)的金屬錯合物與 ZnO,由於三個碳的長碳鏈對其吸收係數(extinction coefficient)影響效 果較為良好,以便期待能改善電子傳遞能力,並且控制其奈米粒子大 小於2~5 nm(34) 。利用有機染料釕(Ru)的金屬錯合物為Donor,除了前 文所提及的電子與電洞分離效應外,能考慮到有機染料釕(Ru)的金屬 錯合物具有良好的電子傳導能力,以往都製成染料敏化型太陽能電池
且有良好的熱穩定性(35),於是將利用樹枝狀的oligothiophene(36)去提升 光電量子數及可見光吸收範圍,並給予適當的長碳鏈數以致於修飾其 溶解性質;如此一來,期待能改善其表面相分離(Phase separation)及 型 態 (Morphology) 的 程 度 , 進 而 降 低 電 子 及 電 洞 再 結 合 (Recombination)。 圖 2-11:分別結合有機染料釕(Ru)的金屬錯合物【G1、G2、G3】 -Ru-Terpy-ZnO 奈米粒子
3.1 實驗儀器
1、真空系統 (Vacuum Line & Schlenk Line)
2、核磁共振光譜儀 (Nuclear Megnetic Resonance):
型號:Varian 300 型。將 sample 溶於 d-solvent 中,利用所測得 1H
與 13C 光譜判斷化合物之結構與純度。化學位移單位為 ppm,偶 合常數單位為 Hz,並以 d-solvent 值為內標 (d-CHCl31H: δ = 7.24 ppm, 13C: δ = 77 ppm)。s 代表 singlet, d 代表 doublet, t 代表 triplet, m 代表 multiplet。 3.紫外光-可見光-近紅外光光譜儀(Ultraviolet-Visible-Near Infrared Spectrophotometer): 型號:Lambda 950型。紫外光-可見光吸收光譜可以用來偵測軌域 中之電子被激發而產生躍遷的情況,所以放射光的波長和物質內 的電子結構有關。操作方法為先將溶劑放入方形的石英管且放入 儀器的樣品槽中,並設定好儀器參數作基線掃描,再將溶劑倒掉, 放入樣品溶液進行掃描。 4. 螢光光譜儀 (Fluorescence Spectormeter): 型號:Hitachi F-4500 型。螢光光譜可以用來偵測發光團分子之電 子被激發後以發光的型式回到基態的情況,所以發射光的波長和物 質內的電子結構有關。操作方法為配置適當濃度並設定其激發波
長,再放入螢光光譜儀掃描。 5.循環伏安電化學儀 (Cyclic Voltammeter,CV): 型 號 : AutoLab 型 。 先 配 置 電 解 質 溶 液 為 0.1M tetra-n-butylammonium hexafluorophosphate的無水乙睛溶液,再將 各個樣品配置成10 mg/1 ml的氯苯溶液。本次實驗所用到的CV是 屬於三電極式,工作電極是Pt薄膜,參考電極是Ag/AgCl參考電 極,輔助電極是Pt線,而量測時先將高分子溶液滴加在Pt薄膜上, 等自然風乾成薄膜後,再放入電解質溶液裡,並用100 mV/s的掃 描速率去進行電化學的量測。我們可以由循環伏安法來得知高分 子的氧化還原電位、最高填滿分子軌域(HOMO)、最低未填滿分子 軌域(LUMO),並利用HOMO和LUMO的電位相減進而得知電化學 能隙。
6.紅外線光譜儀 (Infrared Spectrometer, IR): 型號:Perkin-Elmer Spectrum 100 型。 7 質譜儀 (Mass Spectrometer):
型號:Micromass Trio-2000 型。用FAB (Fast Atom Bombardment)
或 EI (Electron Ionization)來將樣品離子化。此外,由於分子量較大
的分子非常難以被離子化,在其離子化的過程中很容易就分散掉
Desorption/Ionization-Time Of Flight)來取得圖譜。由交通大學貴重 儀器中心代測樣品。
8.元素分析儀 (Elemental Analyzer):
型號:Perkin-Elmer 240C 型。由交通大學貴重儀器中心代測樣品。
9.熱重分析儀(Thermogravimetry Analysis, TGA)
型號:Du Pont TGA 2920 型 測量方法:取 2~5mg 樣品,在氮氣環
境下以 10°C/min 從室溫加熱至 900°C,從曲線尖端外切兩條斜線
相交而得其熱裂解溫度 Td。
3.2 實驗藥品及溶劑
藥品名稱 廠牌
Ammomium acetate SHOWA 2-Acetyl pyridine ACROS
Bromine MERCK Butyllithium 2.5M Chemetall 1-Bromohexane ACROS 3-Bromothiophene ACROS 4-Bromobenzaldehyde ACROS 5-Bromo-2-thiophene carboxaldehy de ACROS 1,3-Bis(diphenylphosphinopropane)
Nickel(II) chloride ACROS Dibutyltin dilaurate TCI Hydrochloric acid 37% Fisher Scientific
4-Hydroxy-benzaldehyde ACROS
Magnesium Scharlau Magnesium sulfate anhydrous SHOWA
N-Bromosuccinimide Fluka
Sodium hydroxide SHOWA Tri-n-butyltin chloride ACROS
Thiophene Aldrich 3-(Triethoxysilyl)propyl isocyanate TCI
Tetrakis(triphenylphosphine)palladium(0) Ultra Fine Chemical Technology Corp. 3-(Trimethoxysilyl)propylmethacrylate Aldrich
Zinc Acetate SHOWA
溶劑名稱 廠牌
Acetone ECHO n-Butyl Alcohol Showa
Dichloromethane Seechem
Ethyl Acetate ECHO
n-Hexane ECHO Tetrahydrofuran Mallinckrodt
Ethanol Showa
Diethyl Ether TEDIA
Toluene TEDIA
反應用的溶劑,如:dichloromethane、tetrahydrofuran(THF)所有溶 劑均經由標準程序蒸餾除水、除氧後氮氣下保存。
3.3 有機合成流程及 ZnO 奈米粒子製作與表面修飾
3.3.1 4’-(5-Bromothiophene)-2,2’,6’,2’’-terpyridine 的合成 將5.158 g 的 5-bromo-2-thiophene carboxaldehyde 溶於 150 ml 乙醇 中,再注入9.9 g 的 2-Acetyl pyridine,並於攪拌 20 分鐘後,注入已 溶在50 ml 水中的 7.57 g NaOH 攪拌 24 小時後,再用減壓濃縮裝置 移除乙醇,並用DCM:H2O=1:1 萃取且收集有機層,經無水硫酸鎂 除水後過濾,用減壓濃縮裝置移除 DCM 得中間產物 1。將 70 g 的 NH4OAc 及中間產物 1 溶入擁有 100 ml 醋酸及 20 ml 乙醇的混合溶液 中,在130 ℃下加熱迴流 24 小時後,冷卻至室溫並加入 3 公升水並 過濾出固體產物,最後利用甲醇再結晶且真空抽乾得咖啡色產物 3.7 g,產率約 35%。 1H NMR (CDCl 3,300 MHz): 8.72 (d, 2H, J = 4.8 Hz), 8.62 (d, 2H, J = 7.8 Hz), 8.58 (s, 2H), 7.87 (td,2H, J = 1.8 Hz, 7.5 Hz, 7.5 Hz), 7.51 (d, 1H, 3.9Hz), 7.35 (td, 2H, J = 1 Hz, 4.8 Hz, 7.8 Hz), 7.11 (d, 1H, 3.9 Hz).
3.3.2 3T,7T and 15T.的合成
(1) 2-Hexylthiophene
在雙頸瓶中,將15 g 的 thiophene 溶入 150 ml THF(dry solvent)
中,在-78 ℃氮氣環境下注入 60 ml 的 2.5 M n-butyllithium 並攪拌 45 分鐘。再將27 g 1-bromohexane 注入以上的溶液中並移開冰浴且攪拌 5 小時,加入水並用 Ether:H2O=1:1 萃取且收集有機層,經無水硫 酸鎂除水後過濾,用減壓濃縮裝置移除 Ether,產物再以矽膠管柱層 析純化,以 hexane 為沖堤液,真空抽乾得液體產物 22 g,產率 72.22%。 1H NMR (CDCl3,300 MHz): 7.09 (dd, 1H, J=5.1Hz,1.2Hz), 6.90 (dd, 1H, J=5.1Hz,3.4Hz), 6.77 (dd, 1H, J=3.4Hz, 1.1Hz), 2.82 (t, 2H, J=7.7Hz), 1.67 (p, 2H, J=7.7Hz), 1.34 (m, 6H), 0.89 (t, 3H, J=6.6Hz) (2)2-Bromo-5-hexylthiophenre 在陰暗的環境下,將25.6 g NBS 溶在 50 ml DMF 中,在 0 ℃下 逐漸滴入已溶於150 ml DMF 的 2-hexylthiophene 中,並攪拌至室溫
達 10 小時以上,置入水並用 DCM:H2O=1:1 萃取且收集有機層, 經無水硫酸鎂除水後過濾,用減壓濃縮裝置移除 DCM,產物再以矽 膠管柱層析純化,以hexane 為沖堤液,真空抽乾得液體產物 27.3 g, 產率78.1 %。 1H NMR (CDCl3,300 MHz): 6.85 (d, 1H, J=3.6Hz), 6.54 (dt, 1H, J=3.6Hz, 0.9Hz), 2.74 (td, J=7.6Hz, 0.9Hz), 1.63 (p, 2H, J=8.1Hz), 1.31(m, 6H), 0.90 (t, 3H, J=6.7Hz). (3)5,5’’-Dihexyl-[2,2’;3’,2’’]terthiophene (3T) 在0℃氮氣環境下,將 3.6 g Mg 及 100 ml Ether 混合於雙頸瓶中, 並將12.4 g 2-Bromo-5-hexylthiophenre 緩慢注入,攪拌五個小時。在 另一個雙頸瓶裡氮氣環境下,將4.2 g 2,3-dibromothiophene 及 150 mg NidpppCl2 混合溶於 100 ml Ether,五小時後再將上述的格林那試劑打 入此混合液中,攪拌 24 小時。之後將 5 %HCl 溶液緩慢加入,用 Ether: H2O=1:1 萃取且收集有機層,經無水硫酸鎂除水後過濾,用減壓濃 縮裝置移除Ether,產物再以矽膠管柱層析純化,以 hexane 為沖堤液, 真空抽乾得液體產物4.7 g,產率 65.1 %。 1H NMR (CDCl3,300 MHz): 7.20 (d, 1H, J=5.3Hz), 7.11 (d, 2H, J=5.3Hz), 6.93 (d, 1H, J=3.5Hz), 6.86 (d, 1H, J=3.5Hz), 6.67 (d, 1H, J=3.5Hz), 6.64 (d, 1H, J=3.5Hz), 2.77 (t, 4H), 1.65 (m, 4H), 1.34 (m, 12H), 0.88 (t, 6H, 6.5Hz).
PS:2,3-Dibromothiophene 在反應瓶中,將 10 g (5.74 ml) 3-bromothiophene 溶於 250 ml 醋 酸中,並將3 ml Bromine 稀釋成 50 ml,並在室溫下由加料漏斗緩慢 滴入以上溶液中,反應 24 小時後,用 Ether:H2O=1:1 萃取且收集 有機層,經無水硫酸鎂除水後過濾,用減壓濃縮裝置移除 Ether,產 物再以矽膠管柱層析純化,以 hexane 為沖堤液,真空抽乾得液體產 物11.9 g,產率約 80 %。 1H NMR (CDCl3,300 MHz): 7.24 (d,1H, J=3.6Hz), 6.88 (d,1H, J=3.6Hz). (4)Tributyl-(5,5’’-dihexyl-[2,2’;3’,2’’]terthiophen-5’-yl)stannane
用 150 mlTHF(dry solvent)溶解 6 g 的 5,5’’-dihexyl-[2,2’;3’,2’’] terthiophene 於雙頸瓶裡,在-78℃且氮氣環境下注入 9 ml 的 2.5 M n-butyllithium 攪拌兩個小時後,將 9.45 ml (10 g) tributyltin chloride
萃取且收集有機層,經無水硫酸鎂除水後過濾,用減壓濃縮裝置移除 Ether,真空抽乾並直接進行下一步(5)合成。 (5)2,3-Di(5,5’’-dihexyl-[2,2’;3’,2’’]terthiophen-5’-yl)thiophene (7T) 將上述的產物(4)、150 mg Pd(PPh3)4及 30 ml DMF 置入雙頸瓶 中,並抽真空灌氮氣數次後,注入1.71 g 的 2,3-dibromothiophene, 並維持100 ℃下加熱迴流 24 小時。24 小時之後回到室溫,置入水並 用DCM:H2O=1:1 萃取且收集有機層,再加入 KF 飽和水溶液進行 萃取且收集有機層,經無水硫酸鎂除水後過濾,用減壓濃縮裝置移除 DCM,產物再以矽膠管柱層析純化,以 hexane 為沖堤液,真空抽乾 得液體產物5.17 g,產率 39.28%。 1H NMR (CDCl3,300 MHz): 7.28 (d,1H, J=5.3Hz), 7.21 (s, 1H), 7.18 (s, 1H), 7.17 (d, 1H, J=5.3Hz), 6.94 (d, 1H, J=1.7Hz), 6.93 (d, 1H, J=1.7Hz), 6.88 (d, 1H, J=3.5Hz), 6.86 (d, 1H, J=3.5Hz), 6.51 (m, 4H), 2.76 (t, 8H), 1.66 (m, 8H), 1.34 (m, 24H), 0.88 (t, 6H, J=6.7Hz).
(6)Tributyl-{2,3-di(5,5’’-dihexyl-[2,2’;3’,2’’]terthiophen-5’-yl)thiophe n-5-yl}stannane
用 150 ml THF(dry solvent) 溶 解 4 g 2,3-di(5,5’’-dihexyl-
[2,2’;3’,2’’]terthiophen -5’-yl)thiophene(7T)於雙頸瓶裡,在-78 ℃且 氮氣環境下注入2.1 ml 的 2.5 M n-butyllithium 攪拌兩個小時後,將 2.4 ml(2.88 g) Tributyltin chloride 注入並放置於室溫下,攪拌 3 小時後 加入水,並用 Ether:H2O=1:1 萃取且收集有機層,經無水硫酸鎂除 水後過濾,用減壓濃縮裝置移除 Ether,真空抽乾並直接進行下一步 (7)合成。 (7)15T 將上述的產物(6)、150 mg Pd(PPh3)4及 30 ml DMF 置入雙頸瓶 中,並抽真空灌氮氣數次後,注入0.53 g 的 2,3-dibromothiophene, 並維持100 ℃下加熱迴流 24 小時。24 小時之後回到室溫,置入水並 用DCM:H2O=1:1 萃取且收集有機層,再加入 KF 飽和水溶液進行 萃取且收集有機層,經無水硫酸鎂除水後過濾,用減壓濃縮裝置移除
DCM,產物再以矽膠管柱層析純化,以 hexane:EA=10:1 為沖堤液, 真空抽乾得液體產物2.35 g,產率 28.21 %。 1H NMR (CDCl3,300 MHz): 7.33 (d,1H,5.3Hz),7.28 (s,1H),7.24 (s,1H), 7.237 (s,1H), 7.23 (s,1H), 7.21 (s,1H), 7.20 (d,1H,J=5.3Hz), 7.19(s,1H), 6.93 (m,4H), 6.84 (m,4H),6.62 (m,8H), 2.74 (t,16H,J=7.4Hz), 1.61 (m,16H), 1.29 (m,48H), 0.88 (t,24H, 5.4Hz). 3.3.3 G1,G2 及 G3 的合成
(8)3T-SnBu3 同上述步驟(4)。 (9)G1 將上述的中間產物、150 mg Pd(PPh3)4及30 ml DMF 置入雙頸瓶 中,並抽真空灌氮氣數次後,注入 5.67 g 的 4’-(5-Bromothiophene) -2,2’,6’,2’’-terpyridine,並維持 100℃下加熱迴流 24 小時。24 小時之 後回到室溫,置入水並用DCM:H2O=1:1 萃取且收集有機層,再加 入 KF 飽和水溶液進行萃取且收集有機層,經無水硫酸鎂除水後過
濾,用減壓濃縮裝置移除 DCM,產物再以鋁膠管柱層析純化,以 hexane:EA=20:1 為沖堤液,真空抽乾得液體黏稠產物 6.04 g,產 率57.06 %。 1H NMR (CDCl3,300 MHz): 8.74 (d, 2H, J=0.9Hz), 8.64 (m, 2H), 8.62 (m, 2H), 7.88 (td, 2H, J=7.8Hz), 7.72 (d, 2H, J=3.9Hz), 7.34 (m, 2H), 7.32 (m, 1H), 7.23 (m, 1H), 6.96 (d, 1H, J=3.6Hz), 6.90 (d, 1H, J=3.3Hz), 6.67 (d, 2H, J=3.3Hz), 2.78 (m, 4H), 1.68 (m, 4H), 1.30 (m, 12H), 0.88 (m, 6H).
Elemental analysis for C43H43N3S4: Calculated : (%)C, 70.74; H, 5.94; N, 5.76. Found : (%) C,70.29; H, 5.93 ; N,5.90。 MS (FAB): m/z 730 (calcd [M]+). (10)7T-SnBu3 同上述步驟(6)。 (11)G2 將上述的中間產物、150 mg Pd(PPh3)4及30 ml DMF 置入雙頸瓶 中,並抽真空灌氮氣數次後,注入 1.76 g 的 4’-(5-Bromothiophene) -2,2’,6’,2’’-terpyridine,並維持 100 ℃下加熱迴流 24 小時。24 小時之 後回到室溫,置入水並用DCM:H2O=1:1 萃取且收集有機層,再加 入 KF 飽和水溶液進行萃取且收集有機層,經無水硫酸鎂除水後過 濾,用減壓濃縮裝置移除 DCM,產物再以鋁膠管柱層析純化,以 hexane:EA=15:1 為沖堤液,真空抽乾得液體黏稠產物 2.67 g,產
率49.64 %。 1H NMR (CDCl3,300 MHz): 8.75 (d, 2H, J=3.0Hz), 8.68 (m, 2H), 8.65 (m, 2H), 7.88 (td, 2H, J=7.5Hz), 7.73 (d, 1H, J=3.0Hz), 7.36 (m, 2H), 7.29(m, 1H), 7.23 (m, 1H), 7.22 (m, 2H), 6.99 (m, 2H), 6.89 (d, 2H, J=6.0Hz), 6.67 (m, 4H), 2.77 (m, 8H), 1.65 (m, 8H) , 1.32 (m, 24H), 0.88 (m, 12H).
Elemental analysis for C71H75N3S8 : Calculated : (%) C, 69.51;H, 6.16;N, 3.42. Found : (%) C, 69.32; H, 6.63 ; N,3.41。 MS (FAB): m/z 1227 (calcd [M]+). (12) 15T-SnBu3 用150 mlTHF(dry solvent)溶解 3 g 的 15T 於雙頸瓶裡,在-78 ℃ 且氮氣環境下注入1 ml 的 2.5 M n-butyllithium 攪拌兩個小時後,將 1 ml Tributyltin chloride 注入並放置於室溫下,攪拌 3 小時後加入水, 並用Ether:H2O=1:1 萃取且收集有機層,經無水硫酸鎂除水後過濾, 用減壓濃縮裝置移除Ether,真空抽乾並直接進行下一步(13)合成。 (13) G3 將上述的中間產物、150 mg Pd(PPh3)4及30ml DMF 置入雙頸瓶 中,並抽真空灌氮氣數次後,注入 0.62 g 的 4’-(5-Bromothiophene) -2,2’,6’,2’’-terpyridine,並維持 100℃下加熱迴流 24 小時。24 小時之 後回到室溫,置入水並用DCM:H2O=1:1 萃取且收集有機層,再加
入 KF 飽和水溶液進行萃取且收集有機層,經無水硫酸鎂除水後過 濾,用減壓濃縮裝置移除 DCM,產物再以鋁膠管柱層析純化,以 hexane:EA=15:1 為沖堤液,真空抽乾得液體黏稠產物 1.4 g,產率 40.24 %。 1H NMR (CDCl3,300 MHz): 8.74 (d, 2H, J=4.2Hz), 8.69 (m, 2H), 8.64 (m, 2H), 7.87 (td, 2H, J=7.5Hz), 7.73 (d, 1H, J=3.9Hz), 7.36 (m, 2H), 7.31(m, 1H), 7.28 (m, 1H), 7.25 (m, 4H), 6.92 (m, 4H), 6.67 (m, 4H), 6.61 (d, 8H), 2.74 (m, 16H), 1.61 (m, 16H), 1.30 (m, 48H), 0.87(m, 24H).
Elemental analysis for C127H139N3S16 : Calculated : (%) C, 68.69;H, 6.31;N,1.89 Found : (%) C, 67.86; H, 6.75 ; N,1.73。 MS (MALDI-TOF): m/z 2220.7 (calcd [M]+). 3.3.4 ZnO 奈米粒子製作與表面的修飾 將 3.29 g (15 mmol) 之 Zn(OAc)2.2H2O 溶於 150 mL 之 ethanol 中,在氮氣下加熱至 80 ℃ 迴流 3 小時。在迴流 2 小時 後,開始藉由 Dean-Stark 裝置移除 90 mL 之 ethanol,接著再加入 同體積 (90 mL) 的新鮮 ethanol,並冷卻至 0 ℃,形成前驅物溶液。 分別將 0.87 g (20.7 mmol) 之 LiOH.H2O 溶於 90 mL 之 ethanol 中,在 0 ℃ 兩小時內緩慢加入前驅物溶液中並均勻攪拌, 回至室溫後,再加熱至40 ℃反應兩小時,待溶液變為澄清,停止加
熱。最後使用 0.1 μm 之 glass fiber filter 過濾,以便移除不可溶的不
純物。為了穩定 ZnO 奈米粒子的表面將 6.0 g 3-(trimethoxysilyl)
propylmethacrylate 溶於 10 mL 的 ethanol,在1 小時內 0 ℃ 下緩慢
加入上述溶液中,加完後保持在室溫反應 12 小時。反應結束後使用 0.1 μm 之 glass fiber filter 過濾出不純物,並以 3:1 的體積比加入 heptane 將濾液中的氧化鋅奈米粒子沈澱析出,使用 6000 rpm 的速 度,將奈米粒子分離。最後將之溶回 ethanol 保存。 (14)4-[2,2';6',2'']Terpyridin-4'-yl-phenol 將3.29 g 的 4-Hydroxy-benzaldehyde 溶於 150 ml 乙醇中,再注入 9.82 g 的 2-Acetyl pyridine,並於攪拌 20 分鐘後,注入已溶在 50 ml 水中的7.566 g NaOH 攪拌 24 小時後,加入 1 wt% HCl 溶液,並用 DCM:H2O=1:1 萃取且收集有機層,經無水硫酸鎂除水後過濾,用
減壓濃縮裝置移除DCM 得中間產物 2。將 70 g 的 NH4OAc 及中間產 物1 溶入擁有 100 ml 醋酸及 20 ml 乙醇的混合溶液中,在 130℃下加 熱迴流24 小時後,冷卻至室溫並加入 3L 水並過濾出固體產物,最後 利用甲醇再結晶且真空抽乾得咖啡色產物約5 g,產率約 75 %。 1H NMR (DMSO,300 MHz): 10.07 (d, 1H, J=7.2Hz), 9.15 (m, 2H), 8.69 (m, 4H), 8.02 (t, 2H, J=7.5Hz), 7.79 (d, 2H, J=8.7Hz), 7.51 (m, 2H), 7.21 (m, 2H). (15)Terpy- 在 迴 流 裝 置 雙 頸 瓶 裡 置 入 1.872g (5.76mmol) 4-[2,2';6',2''] Terpyridin-4'-yl- phenol 並抽真空灌氮氣數次後,注入 15ml THF 均勻 攪 拌 後 , 再 注 入 另 外 溶 在 15mlTHF 的 1.99 g (8.064mmol)
dibutyltin dilanurate 且在氮氣環境下維持 60℃加熱迴流 24 小時;反應 完放冷並用DCM:H2O=1:1 萃取且收集有機層,經無水硫酸鎂除水 後過濾,用減壓濃縮裝置移除 DCM,產物再以鋁膠管柱層析純化, 以 DCM:EA=5:1 為沖堤液,真空抽乾得固體產物 2.18g,產率約 66.1%。 NMR (CDCl3,300 MHz): 8.75 (d, 2H, J=3.2Hz), 8.66 (m, 2H), 8.64 (m,2H), 8.13 (s, 1H), 8.00 (t, 2H, J=6.3Hz), 7.79 (d, 2H, J=8.7Hz) , 7.52(m, 2H), 6.97(d, 2H, J=8.4Hz), 3.87 (m, 6H), 2.48 (m, 6H), 1.22 (m, 9H) . (16)Terpy-ZnO(38) 在250 ml 雙頸瓶中,將 0.3 g ZnO 溶於 20ml toluene 中,緩慢加入溶 有1 g Terpy 的 10 ml THF 溶液,並維持 100℃加熱迴流 24 小時;反 應結束後冷卻,再用離心出固體且以 toluene、THF 個別數次清洗至 澄清溶液後,離心並在真空下烘乾得 0.37 g 固體,再置入乙醇中保 存。(4.2.1 1H-NMR 鑑定) (17)ZnO-Terpy-RuCl3 在250 ml 雙頸瓶中,將 0.37 g Terpy-ZnO 分散於 25ml 乙醇中, 同時0.9 g RuCl3過量溶於30 ml 乙醇中並緩慢滴入,最後在氮氣環境 下,攪拌 24 小時;反應結束後離心出固體,並以乙醇數次清洗過多
的RuCl3,最後在真空下烘乾得約 0.45 g 固體。(由 4.2 之 FT-IR、TGA、 TEM 鑑定) (18)ZnO-Terpy-Ru-G1(PF6)2 在250 ml 雙頸瓶中,將 0.1 g ZnO-Terpy-RuCl3溶於10 ml 的丁 醇中,同時0.3 g G1 溶在 10 ml 己醇中並緩慢滴入,最後維持 130℃ 加熱迴流三天;反應完成後冷卻,加入過量的[NH4]PF6 並攪拌 3 小 時,離心出固體並用hexane、乙醇個別清洗數次至澄清溶液並離心出 固體,最後在真空下烘乾得0.06 g 固體。(38) (由 4.2 之 FT-IR、TGA、 TEM 鑑定) (19)ZnO-Terpy-Ru-G2(PF6)2 在250 ml 雙頸瓶中,將 0.1 g ZnO-Terpy-RuCl3溶於10 ml 的丁 醇中,同時0.3 g G2 溶在 10 ml 丁醇中並緩慢滴入,最後維持 130℃ 加熱迴流三天;反應完成後冷卻,加入過量的[NH4]PF6 並攪拌 3 小 時,離心出固體並用hexane、乙醇個別清洗數次至澄清溶液並離心出 固體,最後在真空下烘乾得0.09 g 固體。(38) (由 4.2 之 FT-IR、TGA、 TEM 鑑定)
(20)ZnO-Terpy-Ru-G3(PF6)2 在250 ml 雙頸瓶中,將 0.1 g ZnO-Terpy-RuCl3溶於10 ml 的丁 醇中,同時0.3 g G3 溶在 10 ml 丁醇中並緩慢滴入,最後維持 130℃ 加熱迴流三天;反應完成後冷卻,加入過量的[NH4]PF6 並攪拌 3 小 時,離心出固體並用hexane、乙醇個別清洗數次至澄清溶液並離心出 固體,最後在真空下烘乾得0.10 g 固體。(38)(由 4.2 之 FT-IR、TGA、 TEM 鑑定)
4.1 合成機制探討
Stille coupling reaction
Stille 反應是一個利用鈀金屬催化有機錫化合物與有機鹵化物,
而形成C-C 鍵化學偶合的反應,反應式如下所示:
R1-Sn(Bu)3 + R2-X R1-R2 + X-Sn(Bu)3
本 文 中 , 使 用 的 鹵 化 物 是 以 溴 為 主 , 故 必 須 先 使 用 NBS
(N-bromosuccinimide)或 bromine 將化合物進行溴化反應(bromination)
後,和有機錫化合物再進一步行偶合反應,可能反應機構如圖4-1。
但由於金屬錫毒性甚大,除非必要應避免使用,可以其他有機反應可
取代之,如Suzuki reaction、Negishi reaction 等。
PdⅡ Pd0Ln XSnBu3 R2SnBu3 R1-PdⅡLm-R 2 R1-PdⅡLm-X R1-R2 1 2 3 4 5 6 7 8 R1-X 圖 4-1 Stille coupling 可能之反應機構
4.2 光物理性質之探討
4.2.1 1H-NMR 鑑定 (a) (b) 圖4-2 (a)Terpy (b)Terpy-ZnO 之1H-NMR 光譜 實驗過程中,利用 Terpy 上的矽氧基去修飾在 ZnO 奈米粒子上 以後,則Terpy 上 a 位置的乙基軟段將會斷裂;如圖 4-2(a)的 3.872 ppm 即為 a 位置上的六個氫,在修飾之後如圖 4-2(b),a 位置上六個氫消 失了,因此鑑定(37)利用 Terpy 去修飾在 ZnO 奈米粒子。4.2.2 FT-IR 之鑑定
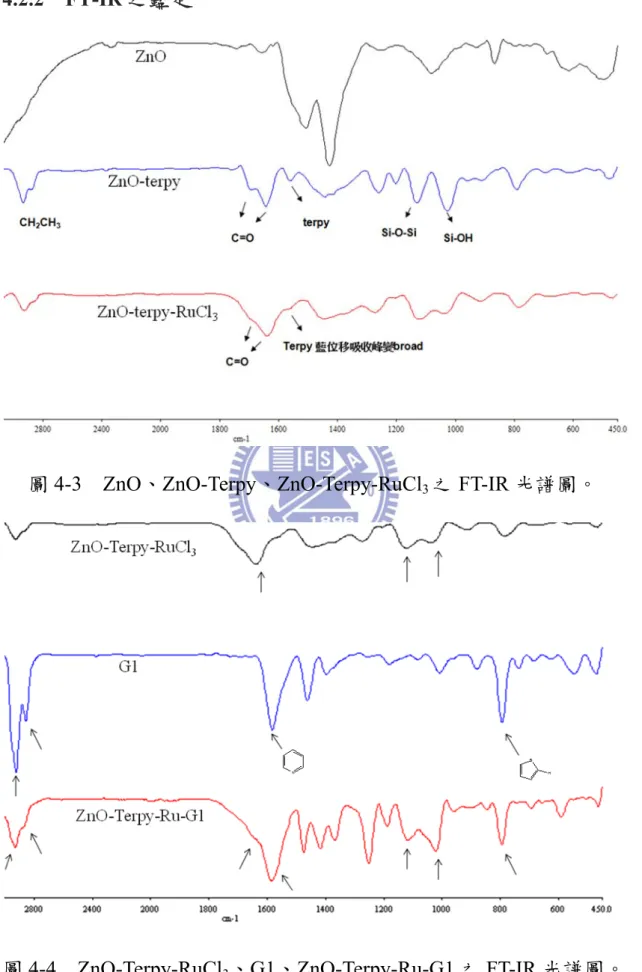
圖4-3 ZnO、ZnO-Terpy、ZnO-Terpy-RuCl3之FT-IR 光譜圖。
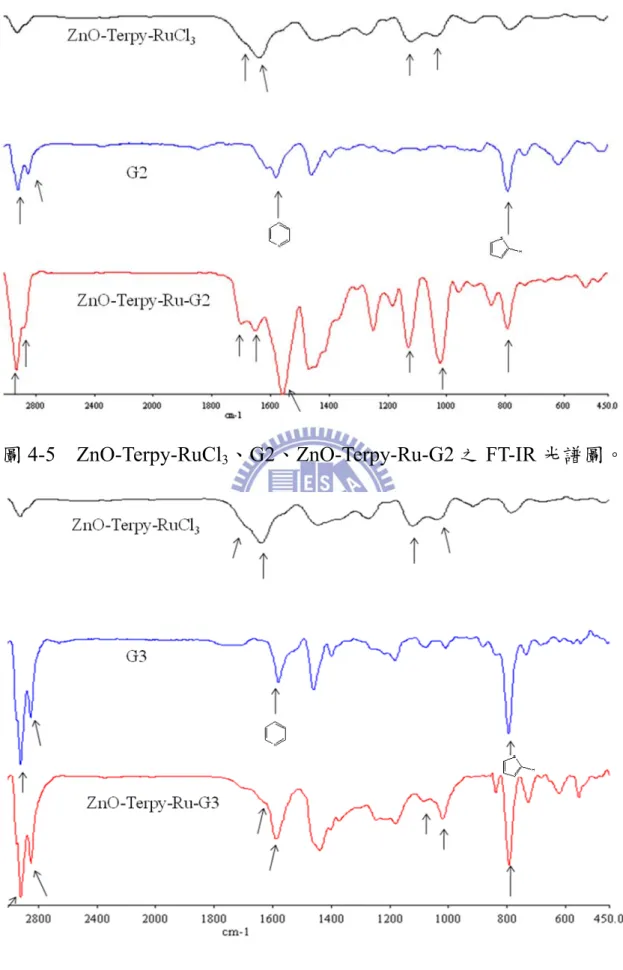
圖4-5 ZnO-Terpy-RuCl3、G2、ZnO-Terpy-Ru-G2 之 FT-IR 光譜圖。
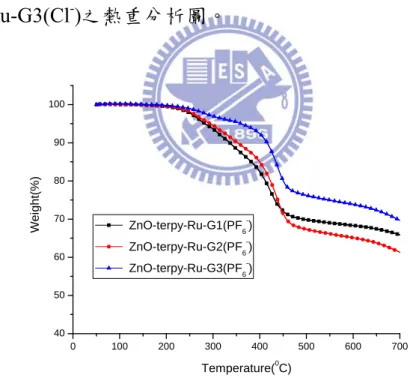
由圖 4-3 可知,當 Terpy 去修飾 ZnO nanoparticle 表面後,發現 urethane 上 C=O 的伸縮(1696.87cm-1、1643.13cm-1) 、具有芳香性之 Terpyridine 的 伸 縮 (1574.21 cm-1) 、 Si-O-Si 的 非 對 稱 伸 縮 (1125.03cm-1) 、silanol 上 Si-O 的伸縮(1023.99 cm-1);並且在與 Ru 形成有機-無機金屬配位鍵後,對於吸收峰變得較寬以外,原來在 1574.21 cm-1 Terpyridine 的吸收峰位移至 1582.44 cm-1。由圖 4-4 可 知,對於 G1 可發現-CH2-的伸縮(2935.34、2853.44 cm-1)、具有芳香 性之Terpyridine 的伸縮(1587.23 cm-1) (29b)、樹枝狀thiophene 上造成的 C-H 伸縮(791.81 cm-1) (29a);同樣地,在圖 4-5 發現 G2 可發現-CH2 -的伸縮(2931.03、2862.06 cm-1)、具有芳香性之 Terpyridine 的伸縮 (1576.49 cm-1) (29b)、樹枝狀 thiophene 上造成的 C-H 伸縮(789.66 cm-1) (29a)及在圖4-6 發現 G3 可發現-CH 2-的伸縮(2918.10、2857.75 cm-1)、 具有芳香性之Terpyridine 的伸縮(1585.09 cm-1) (29b)、樹枝狀thiophene 上造成的C-H 伸縮(789.41 cm-1) (29a);由圖4-4、圖 4-5、圖 4-6 可知其 G1、G2、G3 的特徵峰在與 ZnO-Terpy-Ru 鍵結後,從 FT-IR 圖可看 出其各官能基在鍵結後仍保有特徵峰,以 ZnO-Terpy-RuCl3、G1、 ZnO-Terpy-Ru-G1 系列為例,對於 ZnO-Terpy-Ru-G1 可發現-CH2-的 伸 縮 (2932.93 、 2834.41 cm-1) 、 urethane 上 C=O 的 伸 縮
(1659.22cm-1)、具有芳香性之 Terpyridine 的伸縮(1587.32 cm-1) (29b)、
Si-O-Si 的非對稱伸縮 (1116.43cm-1) 、silanol 上 Si-O 的伸縮(1021.84
cm-1)、樹枝狀 thiophene 上造成的 C-H 伸縮(790.92 cm-1) (29a),相對於 ZnO-Terpy-RuCl3、G1 的特徵峰,大致上都只有稍微的位移並保有其 特徵峰;如此一來,也間接證明ZnO-Terpy-Ru-G1、 ZnO-Terpy-Ru-G2 及ZnO-Terpy-Ru- G3 的形成。 註: O NH O R2 R1 urethane 4.2.3 TGA 熱重分析儀 0 100 200 300 400 500 600 700 74 76 78 80 82 84 86 88 90 92 94 96 98 100 102 Temperature(0C) W e ight(%) ZnO ZnO-terpy ZnO-terpy-RuCl3 圖 4-7 ZnO、ZnO-terpy、 ZnO-terpy-RuCl3之熱重分析圖。
0 100 200 300 400 500 600 700 50 60 70 80 90 100 Weigh t(%) Temperature(0C) ZnO-terpy-Ru-G1(Cl-) ZnO-terpy-Ru-G2(Cl-) ZnO-terpy-Ru-G3(Cl-)
圖4-8 ZnO-terpy-Ru-G1(Cl-)、ZnO-terpy- Ru-G2(Cl-) 、ZnO-terpy
-Ru-G3(Cl-)之熱重分析圖。 0 100 200 300 400 500 600 700 40 50 60 70 80 90 100 Temperature(0C) W ei ght(%) ZnO-terpy-Ru-G1(PF6-) ZnO-terpy-Ru-G2(PF6-) ZnO-terpy-Ru-G3(PF6-)
圖4-9 ZnO-terpy-Ru-G1(PF6-)、ZnO-terpy- Ru-G2(PF6-) 、ZnO-terpy
-Ru-G3(PF6-)之熱重分析圖。
-Ru-G3 大致的吸附情形,因此藉由熱重分析儀(TGA)去分析(如圖 4-7),並由圖 4-7 中的(b)得知在溫度 200℃至 400℃內,在 ZnO 奈米
粒子上的Terpy 熱重損失約 20%,相對地其 ZnO 奈米粒子約為 80%;
藉 由 以 上 的 比 例 ZnO : Terpy =80% : 20% , 在 圖 4-7 中 其
ZnO-terpy-RuCl3的 Terpy 熱重損失約 16%,故換算其 ZnO 奈米粒子
為64%及 RuCl3為 20%(其值整理如表一);除此之外,在反應完成後, 在未加入NH4+PF6-去置換Cl-之前,由於terpyridine 所配位的 Ru2+ 將 相對應兩個Cl- ,故經由分子量比RuCl3:Ru2+(Cl-)2=207.4:171.97 可換算出其熱重損失比由ZnO:RuCl3 =64%:20%變成 ZnO:Ru2+(Cl - )2 =64%:16.58%;相同地,利用 ZnO 與 Ru2+(Cl-)2的比例及ZnO 與Terpy 的比例,由圖 4-8 中在 200℃至 500℃內所得的有機化合物之 熱重損失,可換算出ZnO-Terpy-Ru-G1(Cl- )2、ZnO-Terpy-Ru-G2(Cl-)2 與ZnO-Terpy-Ru-G3(Cl- )2的各自ZnO 奈米粒子分別為 51.4%、51.6% 及 53.2% ;Terpy 含量分別為 12.9%、12.9%及 13.3%;G1、G2 及 G3 分別為 22.5%、22.1% 及 19.7%。(數據整理如表二);相對地在加 入NH4+PF6-去置換Cl-之後,同樣地 terptridine 所配位的 Ru2+ 將相對 應兩個PF6-,因此同文獻所說 PF6-能幾乎完全置換Cl-(38),故經由分 子量比 RuCl3:Ru2+(PF6-)2=207.4:492.1 可換算出其熱重損失比為 ZnO:Ru2+(PF6-)2 =64%:47.45%;相同地,利用 ZnO 與 Ru2+(PF6-)2
的比例及 ZnO 與 Terpy 的比例,由圖 4-9 中在 200℃至 500℃內所得
的 有 機 化 合 物 之 熱 重 損 失 , 可 換 算 出 ZnO-Terpy-Ru-G1(PF6-)2、
ZnO-Terpy-Ru-G2(PF6-)2與 ZnO-Terpy-Ru-G3(PF6-)2的 ZnO 奈米粒子
分別為 39.5%、37.5% 及 42.6% ;Terpy 含量分別為 9.9%、9.4%及 10.7%;G1、G2 及 G3 分別為 21.5%、25.2% 及 15%。(數據整理如 表三)。由以上換算所得到的數據,可以瞭解到在製作過程中熱重損 失的趨勢,由表一及圖4-7 可知在接上 RuCl3之後,使得有機物質在 溫度 200℃至 400℃內之熱重損失將會提升;表二及圖 4-8 可得知當 G1、G2、G3 接上之後,在各代的 Terpy 幾乎有相同熱重損失下, G1、 G2 及 G3 分別為 22.5%、22.1% 及 19.7%,將其值對照其各代之分子 量G1:G2:G3=730.1:1226.9:2220.7,最後我們將表二中 G1、G2、 G3 換算成莫耳比以後 G1:G2:G3=1:0.58:0.25 可間接得知隨著 代數上升其吸附量是相對下降,可能由於立體效應所造成;最後我們 將表三中G1、G2、G3 換算成莫耳比以後,得到 G1:G2:G3=1:0.67: 0.22,同樣的在置換陰離子之後,也有相同的趨勢,如表三及圖 4-9。 表一 ZnO 與 ZnO-Terpy-RuCl3之熱重分析
ZnO Terpy RuCl3
ZnO-Terpy 80% 20% ---