國立臺灣大學生命科學院基因體與系統生物學學位學程 博士論文
Genome and Systems Biology Degree Program College of Life Science
National Taiwan University Doctoral Dissertation
全面性分析在自閉症表現量異常的環狀核糖核酸及環狀 核糖核酸–微小核糖核酸–信使核糖核酸間相互調控網路
Integrative analysis of circular RNA dysregulation and circular RNA-microRNA-mRNA regulatory axes in autism
陳彥如 Yen-Ju Chen
指導教授: 莊樹諄 博士
Advisor: Trees-Juen Chuang, Ph.D.
中華民國 109 年 5 月 May 2020
口試委員審定書
致謝
在寫下誌謝的同時,代表博士班的時光即將畫下句點,這麼多年的時間就 在不知不覺中飛逝。回首這幾年的研究時光多是平靜且踏實的,並不像許多人在 博班過程中經歷後悔掙扎,因為我總認為當下定決心後就沒有什麼好後悔猶豫的,
當目標堅定就能一步步走過這條漫長的道路。在完成論文後也體會到,學習過程 中有太多無形的收穫比實質的學位更可貴。而一個讓自己變得更好的過程中,需 要感謝的是身邊支持著你,讓你有足夠能量堅持到底的每個人。
最要感謝的是莊樹諄老師,在博士班期間給予我充分的指導與支援。在科 學的領域中當我還在看著小河流的時候,老師帶著我經過一次次的討論,逐步調 整研究方向並擬定新的研究策略,最後帶我看見大海。他嚴謹細緻的科學態度和 按部就班的工作風格,都將成為我學習的榜樣。此外,也感謝研究過程中給予我 協助和建議的每一位口試委員及教授。
這段期間, 讓我覺得最幸運的是實驗室的同仁們都如同小天使般幫助著我。
在這個大數據分析實驗室,要學習的領域有很多,如果單靠我一個人的力量是沒 有辦法完成這篇論文的。其中尤其感謝嘉瑩、德倫和泰緯,他們在統計和程式撰 寫上給予我很多協助和指導,幫助我一步步克服研究上的難關,在此深表感謝。
最後想要感謝我的家人,一直以來對我所做的決定都給予支持與鼓勵。因 為你們我才能完成自己的理想和目標,因為你們我才能成為今天的我。小時候想 當一個科學家,覺得科學家很厲害,能發現很多驚奇的事物。長大後發現,原來 想當個真正的科學家並不是這麼容易啊。但如今完成了這個博士學位, 也算是圓 了小時候的一個心願吧。
中文摘要
自閉症譜系障礙是一種腦部發展障礙所導致的複雜疾病,患者特徵有社交 溝通與互動障礙,侷限且重複的行為或興趣,有些伴隨不同程度語言發展障礙。
在已開發國家中約有 1-2% 孩童被診斷罹患自閉症。普遍認為自閉症與遺傳因素
有相當大的關係,然而患者間在基因變異上有很大的差異,因此目前對自閉症的 致病機轉仍不甚了解。許多研究發現自閉症與特定的基因變異有關,其所影響的 功能多和神經元活性及可塑性、突觸連結以及免疫和發炎反應等相關。而在核糖
核酸 1 層次上,後轉錄調控機制是否參與在自閉症致病機轉仍不甚了解,尚待進
一步探討。
藉由人腦組織的轉錄體與表觀基因體分析發現,許多自閉症患者上表現異
常的生物標記,如信使 RNA (mRNA)、 微小 RNA (miRNA) 、長非編碼
RNA (lncRNA)、多樣性切割以及各種表觀遺傳因子等。近年來陸續有研究指
出,環狀 RNA(circRNA)與許多神經疾病的發生與神經發育有關,因此具有重
要研究價值。環狀RNA 是一種非線性 RNA,經由先導 mRNA 反式剪接而成,具
有共價閉合的單鏈環狀結構。circRNA 能扮演一種 miRNA 海綿效應,結合互補
的 miRNA,使其無法抑制下游基因轉錄,而這樣的機轉也被報導在許多神經疾
病中,但circRNA 是否參與在自閉症調控機轉中目前尚未被探討。
本篇研究中,我們整合上百筆人腦組織的轉錄體定序數據,揭開自閉症患
者和非自閉症大腦中 circRNA 的表現圖譜,發現自閉症患者大腦皮質中存在六十
個表現量異常的環狀 RNA 以及三群共同表達的 circRNA。經由整合 mRNA、
miRNA 和 circRNA 表現量資料,以及預測 miRNA 結合為結合位,建立出自閉症
相關的 circRNA–miRNA–基因調控網路。最後我們證實一個在自閉症患者腦部表
現量明顯上升的環狀 RNA(circARID1A),它能吸附 miR-204-3p 進而影響多個
自閉症相關基因的表達。這顯示自閉症除了受到風險基因突變影響外,也可能藉 circRNA 調控 miRNA,進而影響下游基因表達。而 circRNA–miRNA–基因調控網 路的預測方法,未來也可應用在其他複雜神經疾病中,為複雜疾病診斷、追蹤及 治療提供新的思考方向。
關鍵字:自閉症、環狀核糖核酸、微小核糖核酸、基因調控網路
Abstract
Autism spectrum disorders (ASDs) are a heterogeneous group of complex neurodevelopmental disorders characterized by impairment in social, communication, and restricted or repetitive behaviors. Despite remarkable genetic heterogeneity, ASD- associated genes have been suggested to target a few convergent biological processes, including synaptic transmission and plasticity, neural activity, and metabolism-related.
However, the role of post-transcriptional mechanisms in ASD is largely uncharacterized.
In recent years, through analysis of transcriptome and epitranscriptome of human brain tissues, many biomarkers such as messenger RNA (mRNA), microRNA (miRNA), long non-coding RNA, alternatively spliced transcript and various epigenetic factors have been found in ASD patients. Several studies have suggested that circRNAs are involved in the occurrence and development of neurological diseases. Currently, much less is known about the contribution of circRNA in regulatory mechanisms of ASD. Circular RNA (circRNA) is a type of endogenous non-co-linear RNA, which are covalently closed single-stranded RNA molecules derived from the backsplicing of pre-mRNAs.
CircRNAs play a regulatory role as miRNA sponges to suppress the downstream targets of complementary miRNAs.
In this study, we performed genome-wide circRNAs expression profiling in post- mortem brains from ASD and non-ASD samples. Our analysis revealed 60 differential expressed circRNAs and three perturbed co-regulated modules in ASD. We explored ASD-associated circRNA–miRNA–mRNA interactions, in which target genes were
particularly enriched for ASD risk genes and genes encoding inhibitory postsynaptic density proteins. Furthermore, we confirmed that some ASD risk genes were indeed regulated by circARID1A via sponging miR-204-3p in human neuronal cells. Our genome-wide analysis provides a deeper insight into the role of dysregulated circRNAs, as well as the corresponding circRNA–miRNA–mRNA axes in ASD pathophysiology.
Keywords: Autism spectrum disorder, ASD, circular RNA, circRNA-microRNA- mRNA, circARID1A, has-miR-204-3p
Table of contents
口試委員審定書 ... II 致謝 ... III 中文摘要 ... IV Abstract ... VI Table of contents ... VIII List of Figures ... X List of Tables ... XII
CHAPTER 1. Introduction ... 1
1.1 Autism spectrum disorder (ASD) ... 1
1.1.1 Overview of ASD ... 1
1.1.2 Neurobiology and co-occurring conditions of ASD ... 2
1.1.3 The heritability and genetic basis of Autism ... 3
1.2 Circular RNA (circRNA) ... 5
1.2.1 Characteristics of circRNAs ... 5
1.2.2 Biogenesis of circRNAs ... 6
1.2.3 Expression of circRNAs ... 8
1.2.4 Regulation of circRNAs ... 9
1.2.5 CircRNAs in neurological diseases ... 11
1.3 Detection and validation of circRNAs ... 12
1.3.1 Detection of circRNAs by bioinformatics and statistical methods ... 12
1.3.2 Validation of circRNAs by experimental methods ... 13
1.4 Purpose of this study ... 14
CHAPTER 2. Materials and methods ... 15
2.1 Identification and quantification of circRNAs in human brain ... 15
2.2 Identification of differentially expressed circRNAs (DE-circRNAs) ... 16
2.3 Weighted gene co-expression network analysis (WGCNA) analysis ... 17
2.4 MicroRNA binding sites prediction ... 17
2.5 Construction of ASD-associated circRNA–miRNA–mRNA networks ... 20
2.6 Gene set enrichment analysis ... 21
2.7 Cell culture ... 21
2.8 Total RNA isolation, RNase R treatment and subcellular localization ... 22
2.9 cDNA synthesis and RT-qPCR ... 23
2.10 Construction of vector ... 24
2.11 Cell transfection ... 25
2.12 Luciferase reporter assays ... 26
2.13 Microarray analysis ... 26
2.14 Neuronal differentiation and Immunostaining ... 27
2.15 Statistical analysis ... 28
2.16 Code availability ... 28
CHAPTER 3. Results ... 29
3.1 Identification of circRNAs in autism and healthy cortex ... 29
3.2 Differential expression of circRNAs in ASD cortex ... 31
3.3 Construction of co-expression networks of circRNA dysregulation in ASD ... 36
3.4 Profiling of ASD-associated circRNA–miRNA–mRNA regulatory axes ... 39
3.5 Experimental validation of circARID1A interacting with miR-204-3p ... 45
3.6 Regulation of ASD risk genes via the identified circRNA–miRNA interaction ... 54
CHAPTER 4. Discussion ... 61
CHAPTER 5. Future works ... 65
CHAPTER 6. Reference ... 68
List of Figures
Figure 1. (A) DSM-5 classification system for ASD. (B) Classification of ASD based on severity and
intellectual development. ... 2
Figure 2. Schematic representation of the biogenesis of circRNAs. ... 6
Figure 3. Identification processes of the potential ASD-associated circRNA-miRNA-mRNA axes. ... 19
Figure 4. Comparison of normalized numbers of circRNAs in ASD and non-ASD control samples from different brain regions. ... 29
Figure 5. The workflow of the identification of DE-circRNAs and DE-modules. ... 30
Figure 6. Comparisons of the 1,060 circRNAs and human/mouse circRNAs collected in the well-known databases. ... 30
Figure 7. Principal component plots of circRNA expression profiles of the 1,060 circRNAs in samples from FC, TC, and CV. ... 31
Figure 8. DE-circRNAs between ASD and CTL cortex. ... 32
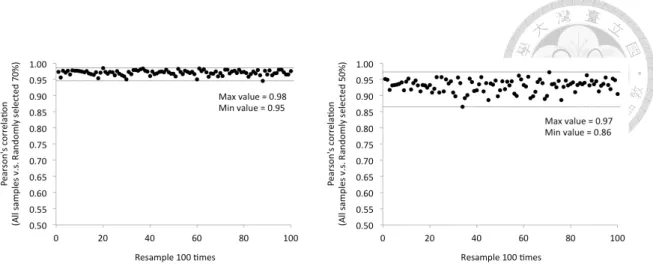
Figure 9. Resampling analysis with 100 rounds of random sampling of 70% and 50% of the samples examined. ... 33
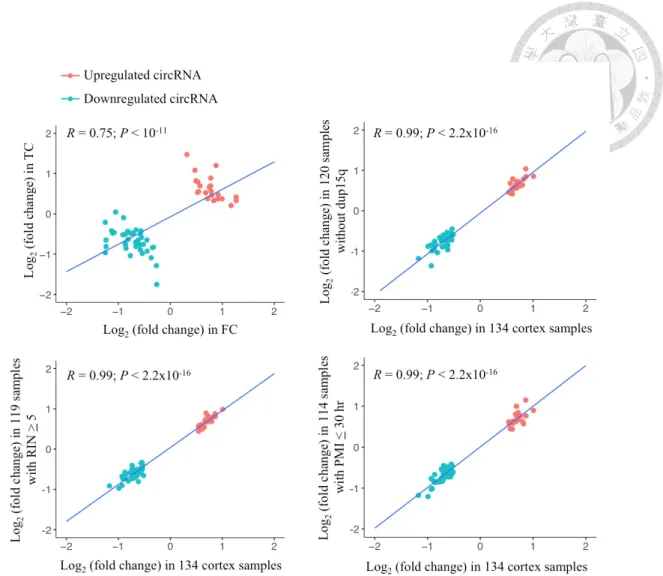
Figure 10. Comparison of DE-circRNA expression fold changes in the FC and TC samples and the corresponding small number of samples and all 134 samples combined. ... 34
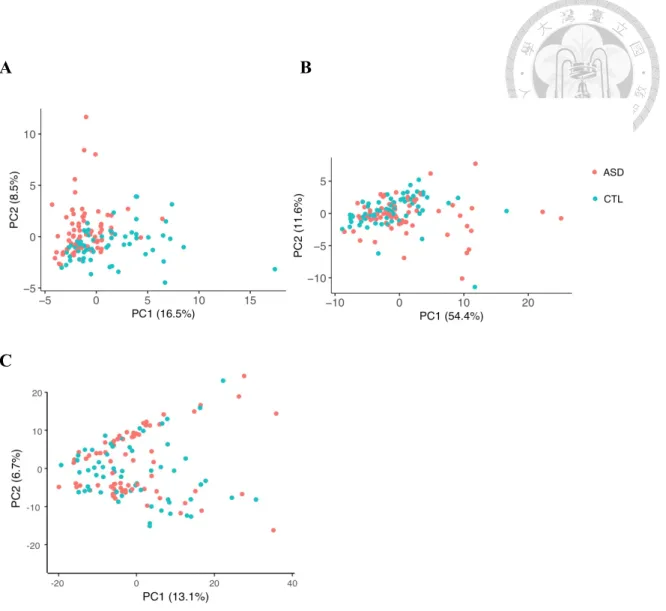
Figure 11. PCA based on the 60 DE-circRNAs and their host genes in ASD and non-ASD samples. ... 35
Figure 12. Clustered heatmap of 60 DE-circRNAs in ASD and non-ASD. ... 36
Figure 13. Hierarchical cluster tree showing 14 modules of co-expressed circRNAs. ... 37
Figure 14. Dysregulation of circRNA coexpression networks in ASD cortex. ... 38
Figure 15. Module preservation Z-summary statistics of 14 modules. ... 39
Figure 16. Schematic diagram representing the criteria for the identified ASD-associated circRNA– miRNA–mRNA axes. ... 41
Figure 17. The 8170 ASD-associated circRNA–miRNA–mRNA interactions. ... 41
Figure 18. The four categories of circRNA-involved ASD-associated circRNA–miRNA–mRNA interactions. ... 43
Figure 19. Enrichment analyses of phenotype ontology (A) and 14 group of gene list (B) among the target genes of the identified ASD-associated circRNA–miRNA–mRNA interactions. ... 44
Figure 20. CircARID1A serves as a sponge for miR-204-3p. ... 46
Figure 21. Validation of the back-spliced junction of circARID1A through RT-PCR and Sanger sequencing. ... 47
Figure 22. Resistance of circARID1A, ARID1A and GAPDH after the RNase R treatment. ... 47
Figure 23. Experimental examination of the evolutionary conservation of circARID1A across vertebrate brains. ... 48
Figure 24. The relative expression of circARID1A and its corresponding co-linear mRNA counterpart in 10 normal human tissues. ... 49 Figure 25. RT-PCR analysis of circARID1A expression in 24 human brain regions. ... 49 Figure 26. Subcellular localization of circARID1A and ARID1A. ... 50 Figure 27. qRT-PCR analyses of the expression of circARID1A and ARID1A after circARID1A
knockdown or overexpression in various neuronal cell lines. ... 50 Figure 28. qRT-PCR analyses of the correlations between the expression of circARID1A and miR-204-3p
after circARID1A knockdown or overexpression in various human neuronal cell lines. ... 51 Figure 29. Luciferase reporter assay for the interaction between circARID1A and miR-204-3p. ... 52 Figure 30. Site-directed mutagenesis of the potential binding sites of miR-204-3p in GLuc-circARID1A
reporter construct. ... 53 Figure 31. The expression level of circARID1A after overexpression of miR-204-3p in ReN and NHA
cells. ... 53 Figure 32. Examination of predicted target gene expression after perturbed circARID1A and miR-204-3p
using microarray analyses. ... 54 Figure 33. The correlations between log2 (fold change) of target genes expression after knockdown
circARID1A, overexpress circARID1A, and overexpress miR-204-3p. ... 55 Figure 34. Distribution of the target mRNA log2 (fold change) in response to knockdown of circARID1A,
overexpression of circARID1A, and overexpression of miR-204-3p. ... 56 Figure 35. Heat map of the 12 ASD risk mRNA expression in response to knockdown of circARID1A,
overexpression of circARID1A, and overexpression of miR-204-3p, respectively. ... 56 Figure 36. qRT-PCR analyses of ASD risk gene expression in ReNc (top) and NHA (bottom) cells after
circARID1A knockdown, miR-204-3p overexpression, and miR-204-3p overexpression with circARID1A overexpression, respectively. ... 57 Figure 37. Fluorescent imaging of ReNcell differentiated for 14 days. ... 58 Figure 38. Relative expression of circARID1A and two ASD risk genes (NLGN1 and STAG1) during
ReNcell differentiation. ... 59 Figure 39. Enrichment of high-confidence ASD risk genes for the targets of the identified circRNA–
miRNA–mRNA interactions. ... 62
List of Tables
Table 1. Common concurrent clinical disorders in ASD. ... 3
Table 2. Potential functions of circRNAs. ... 11
Table 3. Representative circRNAs and related regulatory interactions in neurological diseases. ... 12
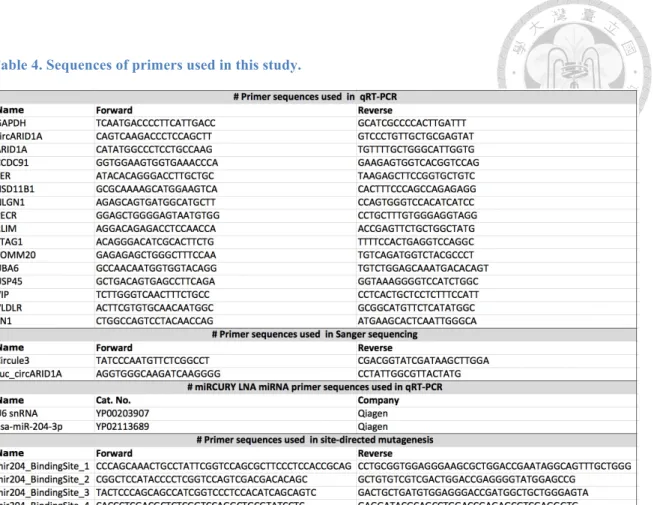
Table 4. Sequences of primers used in this study. ... 24
Table 5. Sequences of oligonucleotides used in this study. ... 25
Table 6. The antibodies used for immunofluorescence. ... 27
CHAPTER 1. Introduction
1.1 Autism spectrum disorder (ASD)
1.1.1 Overview of ASD
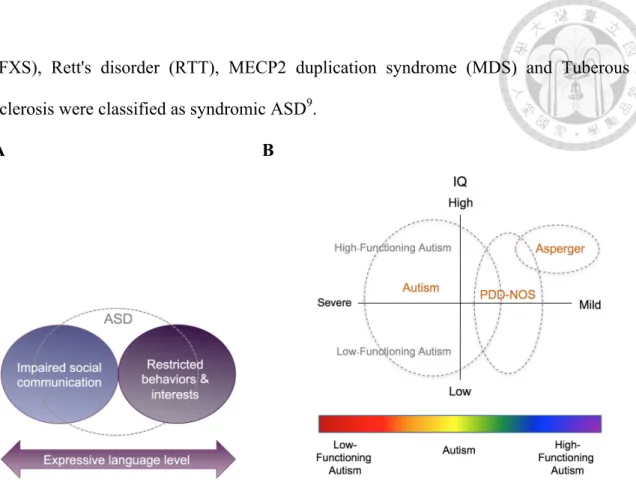
ASD is the most common neurodevelopmental disorders observed in childhood, which is a group of complex neurodevelopment disorders. ASD is defined by a deficit in social communication and interaction, restricted interests and repetitive behaviors2-4 (Fig. 1A).
In the new systems, language ability is not a core diagnostic criterion of ASD, because level of language skill is highly variable in ASD. The worldwide prevalence of autism is about 1-2%, which has continued to rise in the past decades5-7.
The two new diagnostic systems for autism characteristics are the Diagnostic and Statistical Manual of Mental Disorders (DSM-5), fifth edition, which was compiled by the American Psychiatric Association (APA), and the 11th revision of the International Classification of Diseases (ICD-11), which was published by the World Health Organization (WHO). Autism is known as a “spectrum” diagnosis because there is wide variation in the severities and abilities (IQ, language, etc), which can be subgrouped into autistic disorder, Asperger syndrome, pervasive developmental disorder not otherwise specified (PDD-NOS), and childhood disintegrative disorder (CDD)8 (Fig. 1B).
Autism was categorized as syndromic or non-syndromic. Most cases of ASD are non- syndromic which influenced by the small effects of many genes. A small part of ASD
(FXS), Rett's disorder (RTT), MECP2 duplication syndrome (MDS) and Tuberous sclerosis were classified as syndromic ASD9.
A B
Figure 1. (A) DSM-5 classification system for ASD. (B) Classification of ASD based on severity and intellectual development.
(A) The definition of ASD by DSM-5. The figure is adapted from10. (B) The relationship between degree of atypicality severity and IQ of ASD.
1.1.2 Neurobiology and co-occurring conditions of ASD
Functional magnetic resonance imaging (fMRI) studies have revealed that children with ASD have increased volume of brain and amygdala and reduced volume of corpus callosum11. The post-mortem studies observed that the neuron number in the amygdala, fusiform gyrus and cerebellum were reduced12,13. However, the neuron number in the prefrontal cortex was increased14. In addition, frontal lobes, temporal lobes and cerebellar vermis have been reported to be the main regions implicated in dysfunction in autism15,16. The medial prefrontal cortex, superior temporal sulcus, temporoparietal junction, amygdala, and fusiform gyrus are hypo-active in autism17-19. Several lines of
evidence implied that autism is characterised by abnormal brain connectivity20. The brain of autism often reduced global connectivity and increased local connectivity21, decreased frontal-posterior cortical connectivity, and enhanced parietal-occipital connectivity22,23. Local information processing involves sensory and perceptual inputs;
global information processing integrates higher-level cortical control, which is important in social interaction and communication24.
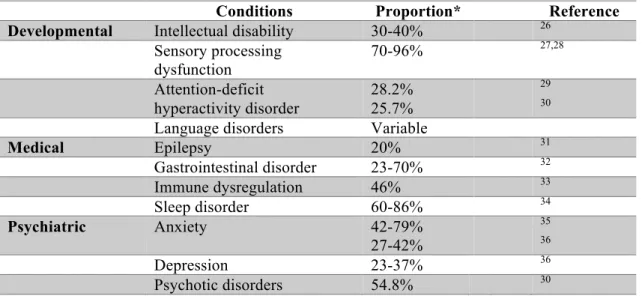
Additionally, more than 70% of people with ASD tend to co-occur with ASD-associated concurrent developmental, medical and psychiatric conditions24,25. The detailed information was listed in Table 1.
Table 1. Common concurrent clinical disorders in ASD.
Conditions Proportion* Reference
Developmental Intellectual disability 30-40% 26
Sensory processing
dysfunction 70-96% 27,28
Attention-deficit hyperactivity disorder
28.2%
25.7%
29
30
Language disorders Variable
Medical Epilepsy 20% 31
Gastrointestinal disorder 23-70% 32
Immune dysregulation 46% 33
Sleep disorder 60-86% 34
Psychiatric Anxiety 42-79%
27-42%
35
36
Depression 23-37% 36
Psychotic disorders 54.8% 30
* Proportion of individuals with ASD present co-occurring conditions.
1.1.3 The heritability and genetic basis of Autism
Many causes are reported to be implicated in ASD, including genetic (e.g., rare, common or de novo mutated37,38 and copy number variations39,40) and non-genetic (e.g.,
environment3,41, parental reproductive age42,43, and gestational factors44,45) factors.
Although the exact mechanism of ASD still remains unknown, the weight of evidence suggests that genetic factors may be the major cause for autism spectrum disorders.
Twin studies have suggested that autism has approximately 80% heritability46,47. The concordance rates of ASD for monozygotic twins (50–90%) are much higher than dizygotic twins (0–36%)47-50, again indicating strong genetic influences on ASD. On the other hand, the recurrence rate of ASD in families who already have one affected sibling, with recurrence estimates ranging from 5.8% to 18.7%51-53.
Over the past decade, several studies showed that thousands of genes and thousands of rare variants were associated with ASD susceptibility48,54,55. Several strongly ASD- associated genes have been identified, including postsynaptic scaffolding genes (SHANK3)56, synaptic plasticity (SYNGAP1)57, and neurexin family genes (CNTNAP2)58. Besides, some of individuals with ASD were found to be linked to chromosomal rearrangements or single gene disorder, such as fragile X syndrome59 (CGG trinucleotide repeat expansion in the FMR1 promoter) and Rett syndrome60 (mutations that inactivate MECP2).Recently, many genome-wide studies have reported ASD-associated dysregulation of gene expression15,16,61 and epigenetic factors such as alternatively spliced transcripts15,16,62, long non-coding RNAs16,63, miRNAs64,65, DNA methylation66, histone trimethylation67, acetylation68 and so on. The contribution of these genetic factors to this complex disease is highly heterogeneous. Despite remarkable genetic heterogeneity, ASD-associated genes have been suggested to target
convergent biological pathways69, such as synaptic function15,70,71, transcriptional regulation71, neural cell adhesion72,73, immune/inflammatory responses74,75 and excitatory/inhibitory (E/I) neuronal balance76,77.
Several studies in small population have explored gene expression profiles in ASD brain regions78-81. Most of the genes surveyed were exposed to be consistently up- or down- regulated in different studies82. This strongly implies that these genes are not coincidentally up- or down- regulated, but might actually have roles in the underlying pathogenesis of ASD. Recently, Daniel H. Geschwind’s group performed large-scale of genome-wide mRNA16 and miRNA64 expression profiling of post-mortem brains in ASD. Now, integration and dissection of the role of co-/post-transcriptional regulatory mechanisms in the etiology of ASD await further investigation.
1.2 Circular RNA (circRNA)
1.2.1 Characteristics of circRNAs
CircRNAs are a large class of non-coding RNAs produced by thousands of protein- coding genes, which are circularized by non-canonical “backsplicing” of pre- mRNAs83,84. CircRNAs are single-strand circular molecules without 5’ cap or 3’ poly(A) tail (Fig. 2). Owing to this structure, circRNAs are resistant to degradation by exonucleases RNase R85-87 and relatively stable than their corresponding co-linear mRNA isoforms86-89. In 1970s, circRNAs were firstly discovered in RNA viruses by electron microscopy90 and later in eukaryotic cells in 197991. Until 1991, circRNAs
92
product of pre-RNA splicing. These recent years, increasing evidence shows the roles of circRNAs in the development and diseases.
Figure 2. Schematic representation of the biogenesis of circRNAs.
1.2.2 Biogenesis of circRNAs
According to biogenesis from different genomic regions, circRNAs can be classified into three subtypes: exonic circular RNAs (ecircRNAs), exon-intron circular RNAs (EIciRNAs), and circular intronic RNAs (ciRNAs) (Fig. 2). More than 80% of circRNAs are derived from exons of protein-coding genes without introns, constituting ecircRNAs86. Approximately 20% of circRNAs are derived from both exonic and intronic regions, constituting exon-intron circular RNAs (EIciRNAs)93. A small fraction of circRNAs contain only introns regions, termed circular intronic RNAs (ciRNAs), which stem from spliced out lariats94.
CircRNAs formation can be modulated by cis-elements, trans-factors and other factors.
1) Cis-elements (i.e., DNA sequences):
CircRNA biogenesis can be promoted by RNA pairing between the reversely complementary sequences (RCSs) across flanking introns87,95 (Fig. 2). Further studies indicated that the flanking introns of circRNA junctions were longer than background introns and harbored more repetitive elements87,96. Our previous study has demonstrated that RCSs (Alu repeats) across flanking introns can affect the formation of both circRNA and trans-spliced RNA isoforms89. Nevertheless, no specific motifs for circRNA formation have been identified in exonic and flanking intron sequences.
2) Trans-factors (i.e., RNA binding proteins, RBPs):
In addition to cis-element, circRNAs can also be facilitated by trans-factors to bridge flanking introns pairing97,98. Some splicing factors (e.g. Muscleblind (MBL)83, Quaking (QKI)97) and RNA-binding proteins (e.g. FUS99, ADAR100) can affect circRNA formation. MBL was shown to bind to its own pre-mRNA and bridging between the two flanking introns to induce backsplicing, which stimulates circMbl production and decrease the expression level of MBL mRNA83. Another regulator of circRNA biogenesis is QKI which binding on intronic QKI binding motifs, then significantly increased circRNA formation97. FUS promote backsplicing by binding the flanking introns of circularized exons99. Conversely, ADAR1 protein suppresses circRNA formation by disrupting the stem structure. ADARs catalyze A-to-I RNA editing within double stranded RNA pairing structures, resulting in reduced RNA pairing100.
3) Spliceosome activity and Pol II elongation rate:
A recent study has found that depletion of spliceosome activity increased long and repeat-rich flanking intron101 and non-complementary sequences102 to pair, facilitating
circRNA formation. Besides, the process of circRNA formation was also influenced by the transcription rate of the corresponding gene. The average Pol II transcription elongation rate of circRNA host genes is higher than that of non-circRNA genes84.
1.2.3 Expression of circRNAs
CircRNAs exhibit evolutionary conservation across multiple species, and exists widely in eukaryotes87. Several studies reported that circRNAs are enriched in neuronal tissues compared with other tissues103-105, especially synaptosomes105. That prompting many researchers to explore the role of circRNAs in neurological diseases. Most of circRNAs are enriched in the cytoplasm85,106, suggesting that circRNAs regulate gene expression through interfering with miRNAs. In general, circRNAs are expressed in a tissue- or age-dependent manner107,108, and also show dynamic expression during neuronal differentiation, depolarization and development104,109,110.
Multiple studies have demonstrated that the expression of circular isoform was not correlated with the expression of its cognate linear mRNA103. Although most circRNAs are expressed at a much lower level compared with their host gene81,111,112. However, in some cases, circRNAs even more abundant than their co-linear counterparts88,89. For example, Morten, et al. found that the expression of three circRNA (circCSPP1, circHDAC2, and circRIMS2) were much higher than those of the linear transcript in porcine brain, and their host gene were associated with synaptic plasticity or brain development104.
1.2.4 Regulation of circRNAs
Although the function of circRNAs is not entirely clear, the recent studies have shown that circRNAs may have the ability to regulate gene expression through multiple mechanisms106 (Table 2). The most understood function of circRNAs is the regulatory role of miRNA sponges, suggesting a previously underappreciated regulatory pathway of circRNA-miRNA-mRNA axes. For example, CDR1as is one of the most widely studied circRNA, which has more than 70 binding sites for miR-7 and function as an miRNA sponge to compete with mRNA for miRNA binding113. In addition, certain circRNAs can regulate transcription. For example, circMbl negatively regulate MBL pre-mRNA splicing by competing the splicing factors83. EIciRNAs are dominantly located in the nucleus and interact with a spliceosomal component U1 snRNPs, which can recruit RNA polymerase II on the promoter of their host genes and thus promote the transcription of the host gene, such as EIciEIF3J, EIciPAIP2 and Ci-ankrd5293,94. Another study demonstrated that circERBB2 can promotes ribosomal DNA transcription114. In contrast, circSamd4 can represses transcription of the myosin heavy chain protein family by associated with PURA and PURB, two repressors of myogenesis115.
On the other hand, many circRNAs interact with proteins through specific binding sites116. In their function as protein decoys, circPABPN1 serve as a decoy for HuR and suppresses PABPN1 translation117. Besides, circ-Amotl1 bind to c-Myc, promoting their nuclear translocation, and upregulated its targets118. Additionally, circANRIL has similar secondary structure to pre-rRNA, which decoys PES1 to suppress rRNA
processing and maturation119. CircRNAs can also function as scaffolds to facilitate subcellular co-localization of their substrates. Circ-Foxo3 acts as scaffold to interact with CDK2 and p21, and then leading to the inhibition of the CDK function120. Circ- Foxo3 also promotes the interaction between MDM2 and p53 to induces apoptosis121. Specifically, circ-Foxo3 affected ID1, E2F1, HIF1α and FAK subcellular translocation122; circ-Amotl1 interacts with PDK1 and AKT1 to facilitate their nuclear translocation123. Moreover, some protein-coding circRNAs contain internal ribosome entry site (IRES) and open reading frame, such as circ-ZNF609124, circ-FBXW7125, circ- AKT3126 and circPPP1R12A127 can be translated to produce peptides (Table 2).
One of the most well-known functions of circRNAs is the role of miRNA sponge to regulate target gene expression113. Several data indicated that the interactions between circRNAs and miRNAs were important for normal brain function. For example, CDR1as knockdown mice displays impaired sensorimotor gating and abnormal synaptic transmission128. Therefore, it is believed that circRNAs can mediate miRNAs and thus regulate the downstream genes at the post-transcriptional level. While some cases of ASD-associated miRNA–mRNA regulatory interactions have been reported64,129, circRNA–miRNA–mRNA regulatory system may play an important mechanism of epigenetic control over gene expression in ASD and healthy samples.
Table 2. Potential functions of circRNAs.
Function circRNAs References
miRNA sponge CDR1as / Sry / circHIPK3/ circMTO1 /
circITCH / circCCDC66 / circTP63
86,88,113,130-133
Regulation of transcription circMbl / EIciEIF3J / EIciPAIP2 / Ci- ankrd52 / circERBB2 / circSamd4
83,93,94,114,115
Protein decoys circPABPN1 / circ-Amotl1 / circANRIL 117-119
Protein scaffolds circ-Foxo3 / circ-Amotl1 120-123
Translation peptides circ-ZNF609 / circ-FBXW7 / circ-AKT3 / circPPP1R12A
124-127
1.2.5 CircRNAs in neurological diseases
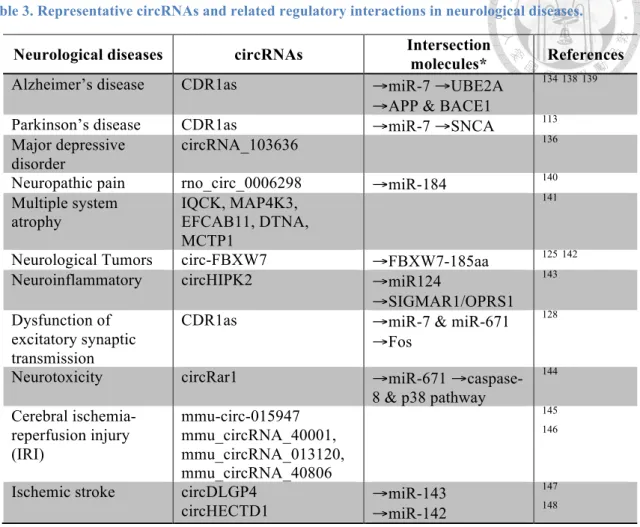
CircRNAs expressed in fly heads or mouse brains are enriched in genes that code for neuronal proteins and synaptic factors, suggesting a potential role for circRNA in the central nervous system105,107. Moreover, several cases of circRNAs show distinct localization in different parts of neurons103, their host genes are related to several synaptic functions, including neurogenesis, neural differentiation, WNT signaling, and synaptic plasticity during neurogenesis103-105. Therefore, circRNAs have the potential to serve as novel therapeutic targets and diagnosis biomarkers to treat neurological diseases, such as Alzheimer's disease134, Parkinson’s disease135, major depressive disorder136, and many nervous system disorders137 (Table 3).
These studies highlight a potential function of circRNAs in the nervous systems and suggest their relevance to pathogenesis of neurodegenerative. However, the biological functions of circRNA regulatory mechanisms in ASD are largely unknown.
Table 3. Representative circRNAs and related regulatory interactions in neurological diseases.
Neurological diseases circRNAs Intersection
molecules* References
Alzheimer’s disease CDR1as →miR-7 →UBE2A
→APP & BACE1
134138139
Parkinson’s disease CDR1as →miR-7 →SNCA 113
Major depressive disorder
circRNA_103636 136
Neuropathic pain rno_circ_0006298 →miR-184 140
Multiple system
atrophy IQCK, MAP4K3,
EFCAB11, DTNA, MCTP1
141
Neurological Tumors circ-FBXW7 →FBXW7-185aa 125142
Neuroinflammatory circHIPK2 →miR124
→SIGMAR1/OPRS1
143
Dysfunction of excitatory synaptic transmission
CDR1as →miR-7 & miR-671
→Fos
128
Neurotoxicity circRar1 →miR-671 →caspase-
8 & p38 pathway
144
Cerebral ischemia- reperfusion injury (IRI)
mmu-circ-015947 mmu_circRNA_40001, mmu_circRNA_013120, mmu_circRNA_40806
145 146
Ischemic stroke circDLGP4
circHECTD1 →miR-143
→miR-142
147 148
* Intersection molecules represent the downstream pathway of circRNAs.
1.3 Detection and validation of circRNAs
1.3.1 Detection of circRNAs by bioinformatics and statistical methods
CircRNAs can be detected from RNA sequencing (RNA-seq) and microarray using computational approaches to identify the back-spliced junction (BSJ). Total RNA-seq with ribosomal RNA (rRNA) depletion or RNase R treatment are most commonly used for circRNA profiling149. Usage of paired-end reads for identifying circRNAs could help us to filter out false positive circRNAs, if the paired-end of a read out of the predicted circles150.
Currently, numerous circRNA detection tools (circRNA_finder, CIRCexplorer, find_circ, etc.) have been developed. However, there are great inconsistencies in the results among different tools151. A recent article compared the performance of several published algorithms, finding dramatic differences between sensitivity and specificity152. Notable, NCLScan153 was a conservative method with the highest precision compared with currently circRNA detectors154. It constructs the putative non-co-linear (NCL) references from the unmapped paired-end reads and BLAT-aligning the concatenated sequences to the reference genome. Then, removing concatenated sequences with an alternative co-linear explanation153. The pipeline carries out several alignments and filtering and integrates with BWA, Novoalign and BLAT to reduce false positives.
Accordingly, we identified circRNAs by the NCLscan (version 1.6) to detect BSJ reads from RNA-seq data.
1.3.2 Validation of circRNAs by experimental methods
CircRNAs can be validated by some experimental methods. First, the most basic approach is to detect the BSJ reads by divergent primer PCR (Fig. 2). However, trans- splicing transcripts155 (splicing between two separate pre-mRNA) and template switching156 can lead to false positive events of circRNA. Therefore, researcher can use MMLV- and AMV- derived RTases to exclude template switching by reverse transcription (RT)157, and use exoribonuclease RNase R to degrade linear form of trans- splicing transcripts158. Sanger sequencing can validate the BSJ sequence, and the expression level of circRNAs can be quantified by quantitative real-time PCR.
Moreover, if circular and linear RNA exhibit different sizes, circRNA can be confirmed by northern blots and fluorescence in situ hybridization 37,159. The procedures of northern blots and FISH do not involve RT or PCR amplification that can avoid detecting false positives from RT-based artifacts. However, it can only detect highly expressed candidates.
For functional study, circRNAs overexpression and knockdown are two typical ways.
For mechanism study, luciferase reporter assay, RNA pull down assay, RNA immunoprecipitation, and mass spectrometry are performed to uncover circRNA interactions106.
1.4 Purpose of this study
To systematically determine regulatory role of circRNAs in ASD, we investigated the expression profile and potential regulatory role of circRNAs in ASD. Our study aimed to integrate the expression of circRNA, miRNA and mRNA to investigate circRNA dysregulation in ASD, and construct the ASD-associated circRNA–miRNA–mRNA regulatory networks according to the common target miRNAs of the circRNAs and mRNAs. That may open a new approach to take a global view of heterogeneous diseases, and unveil the potential mechanisms of circRNAs and may lead to improve ASD diagnosis and treatment in the future.
CHAPTER 2. Materials and methods
2.1 Identification and quantification of circRNAs in human brain
We collected the rRNA-depleted total RNA-seq data from Synapse (https://www.synapse.org) under accession number syn4587609. A total of 236 post- mortem samples included frontal cortex (FC, Brodmann area 9), temporal cortex (TC, Brodmann area 22, 41 and 42), and cerebellar vermis (CV) from 48 individuals with ASD and 49 non-ASD controls (CTL)16.
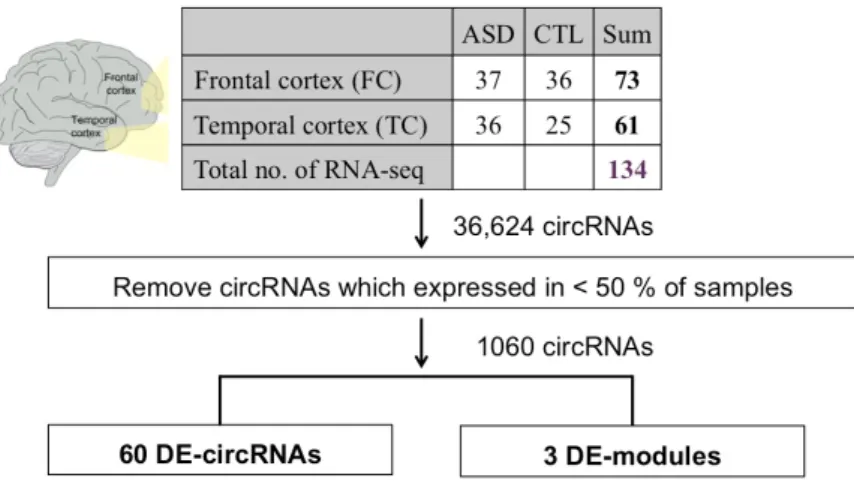
For circRNA quantification, we used some criteria to improve accuracy. Fist, the samples were not considered in the following analysis if the number of the identified circRNAs of these samples were one standard deviation below the mean. Therefore, 202 samples (73 FC, 61 TC, and 68 CV samples) were retained. Second, since several studies of ASD have illustrated that human cortex has been implicated in the pathophysiology of ASD160,161, and changes in transcriptome were more evident in the cortex than in the cerebellum16. Accordingly, our following analysis focused on frontal and temporal cortex. Third, to reduce potentially spurious events, we only considered the circRNAs that were expressed (≥ 10 reads) in more than 50% of the 134 cortex samples. Thus, a total of 1,060 circRNAs were used in the following analyses.
CircRNAs were identified by NCLscan program which was reported to exhibit the greatest precision among currently circRNA-detection tools153,154,162,163. To align the reads to the human reference genome (GRCh38, Ensembl 90) with default parameters.
The previously identified human and mouse circRNAs were obtained from circBase164 and CIRCpedia165 version 2 database. Since circBase use the hg19 assembly for human circRNA, so we converted the genomic coordinates of circRNAs from hg19 to GRCh38 assembly using the liftOver tool166. Also, the coordinates of mouse circRNAs collected in circBase (mm9) and CIRCpedia (mm10) were transformed to the corresponding GRCh38 coordinates by the liftOver166.
2.2 Identification of differentially expressed circRNAs (DE-circRNAs)
For estimated of circRNA abundance in different dataset, the total number of BSJ- spanning reads per million uniquely mapped reads (RPM) was used for measuring circRNA expression level104. Therefore, RPM = number of junction reads / (number of mapped reads/106). The read counts of the host genes were calculated by the STAR aligner167, followed by the RSEM tool. The expression level of genes were measured by log2 normalized FPKM (normalized GC content, gene length, and library size) using the cqn package in R168.
Many methods have been proposed to identify differentially expressed expression. The expression of circRNA is potentially affected by many confounding factors. Here we used the ‘nlme’ package in R169 to control potential confounding factors, including sex, age, brain region (FC or TC), RNA quality (RNA integrity number; RIN), post-mortem interval (PMI), host gene expression, sequencing batch, and brain bank batch. We performed LME model to identified DE-circRNAs between ASD and non-ASD samples with controlling for potential confounding factors:
lme (RPM~ diagnosis + sex + age + brain region + RIN + host gene expression + sequencing batch + brain bank batch, rand = ~1|individual ID)
The model can fix different factors to estimate p values for each circRNAs. P-values <
0.05 and | log2 (fold change) | > 0.5 were considered evidence of significant difference.
2.3 Weighted gene co-expression network analysis (WGCNA) analysis
Gene co-expression network was constructed by the R package WGCNA170 to identify co-expression modules in this study. It is a data mining method for studying biological networks with pairwise correlations between variables, and to identify groups of highly correlated genes that co-express across samples. We present the Dynamic Tree Cut R library with function cutreeDynamic, in which the paramenters are: method = “hybrid”, deepSplit = 3, pamStage = T, pamRespectsDendro = T, minClusterSize = 10, for detecting clusters in a dendrogram depending on their shape. The expression of each module was summarized by the eigengene (ME), which can be considered as the first principal component of a given module. To identify diagnosis status, the Pearson Coefficient between circRNAs and several confounding factors (diagnosis, age, sex, brain region, RIN) were calculated. All circRNA–miRNA–mRNA networks were visualized by Cytoscape software171 (https://cytoscape.org/).
2.4 MicroRNA binding sites prediction
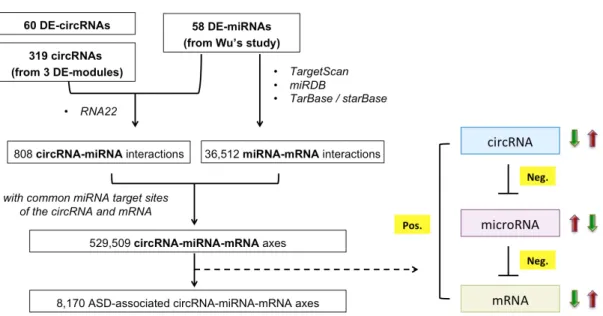
In summary, we identified 60 DE-circRNAs (22 upregulated and 38 downregulated circRNAs) and three DE-modules (one upregulated module included 21 circRNAs and
identified ASD-associated circRNA–miRNA–mRNA regulatory axes. We collected previous study which have evaluated 58 DE-miRNAs (41 upregulated and 17 downregulated miRNAs) in ASD cortex64. These ASD-affected miRNAs were identified from 95 human cortex samples (47 ASD and 48 non-ASD samples), of which 73 samples overlapped with the samples examined in this study.
To investigate the interaction between dysregulated circRNA and miRNA, we used RNA22172 (version 2.0) with default parameters to predict 58 miRNA binding sites on 60 DE-circRNAs and the circRNAs defined in the DE-modules (Fig.3). RNA22 allows identifying putative target sites of novel miRNAs on any sequence of interest (i.e.
protein-coding mRNA, long non-coding RNA or non-canonical targets). Thus, a total of 808 potential circRNA–miRNA interactions were identified.
Next, to investigate putative target mRNA of 58 dysregulated miRNA, we combine the miRNA–mRNA pairs from multiple sources. For 37 well annotated miRNAs, we identified downstream targets by the microRNA Target Filter in Ingenuity Pathway Analysis package173,174 (QIAGEN Inc.) and DIANA-TarBase v8175. The IPA provides experimentally validated interactions collected by Ingenuity Expert Findings, as well as predicted microRNA-mRNA interactions by TargetScan176. TarBase collected experimentally confirmed miRNA targets (Fig.3). To obtain reliable interactions, the miRNA–mRNA binding events were considered if one of the following three criteria is satisfied:
1) TargetScan predicted interaction with context++ score < -0.16, and also identified by Wu et al.64.
2) Experimentally confirmed events collected by Ingenuity Expert Findings, which were manually curated by the IPA experts.
3) Experimentally confirmed events collected in TarBase.
For the other 21 novel miRNAs, we use miRDB177,178 to predicted downstream targets with prediction scores > 80. The mature miRNA sequences were downloaded from the study of Wu et al64. For accuracy, we only considered the predicted miRNA–mRNA interactions that were also previously identified by Wu et al.64. Thus, a total of 36,512 miRNA–mRNA interactions were determined (Fig.3).
Figure 3. Identification processes of the potential ASD-associated circRNA-miRNA-mRNA axes.
“Neg.” represents a negative correlation; “Pos.” represents a positive correlation. The red arrow represents upregulation; the green arrow represents downregulation.
2.5 Construction of ASD-associated circRNA–miRNA–mRNA networks
The expression level of the target genes were measured by log2 normalized FPKM, which accounted for gene read counts, GC content, gene length, and library size, using the cqn program in the R package. The miRNAs expression data was obtained from Prof. Daniel H. Geschwind and Ye E. Wu64, which was measured by normalized read counts with controlling library size, GC content, batch effect, and other technical covariates (RIN, PMI, and batch bank).
We integrated the identified interactions for 808 circRNA–miRNA interactions and 36,512 miRNA–mRNA interactions; we determined 529,509 circRNA–miRNA–mRNA interactions according to the common miRNA target sites of the circRNAs and mRNAs (Fig.3). We then calculated the correlations between circRNA and miRNA expression, between miRNA and mRNA expression, and between circRNA and mRNA expression based on the same set of cortex samples (i.e., 73 samples). Only the circRNA–miRNA–
mRNA interactions were considered if they simultaneously satisfied the following rules:
1) Both the circRNA–miRNA and miRNA–mRNA pairs should exhibit a significantly negative correlation (one-tailed Spearman’s P < 0.05) of expression profile, between circRNAs and the corresponding predicted regulated miRNAs and between miRNAs and target mRNAs, respectively.
2) The circRNA expression should be positively correlated with the corresponding mRNA expression.
3) The Fisher’s combined P values179 of the above three independent Spearman’s correlation tests should be less than 0.05.
Finally, 8,170 ASD-associated circRNA–miRNA–mRNA regulatory axes were determined, which included 2,302 target genes (Fig.3).
2.6 Gene set enrichment analysis
The SFARI180 gene list was downloaded from https://gene.sfari.org/. The high- confidence ASD genetic risk genes (102 genes)181 were derived from an enhanced Bayesian analytic framework based on a large dataset of whole-exome sequencing (35,584 ASD subjects). The epilepsy-related gene list was downloaded from the EpilepsyGene database182 (all epilepsy-related genes). The schizophrenia-related gene lists was downloaded from the SZgene database183. The genes associated with human height were downloaded from the study of Lango Allen et al.184. The gene lists of AustismKB, iPSD, ePSD, and other brain disorder risk genes were downloaded from the study of Wang, P., et al.185 Gene set enrichment analysis were performed using two- tailed Fisher’s exact test with the fisher.test R function. Phenotype ontology analysis was performed by the ToppFun module of ToppGene Suite software186. P values were false discovery rate (FDR) adjusted across 14 target groups for each gene list using Bonferroni correction.
2.7 Cell culture
To study the function of circRNAs for neural cells interact, we relies on the use of human neural progenitor (ReNcell VM) cell187,188, primary normal human astrocytes cell (NHA), human glioblastoma cell (U118) and human neuroblastoma cell (SH- SY5Y). ReNcell is a valuable material for investigating neurodevelopmental
pathway189, which provided by Prof. Jean Lu. The ReNCell was grown on 20 µg/ml laminin (Merck) coated culture plates containing ReNCell NSC maintenance medium (Merck) supplemented with 20 ng/ml of bFGF and EGF (Merck). NHA cell line was purchased from Gibco (N7805100) and cultured in Human Astrocytes Growth medium (Cell Applications). U118 cell line was purchased from ATCC (HTB-15) maintained in Dulbecco’s modified Eagle’s medium. SH-SY5Y cell line was obtained from ATCC (CRL-2266) and cultured in DMEM/F12 medium (Gibco). All culture media contain 10% fetal bovine serum (FBS) and 5 mg/ml antibiotic-antimycotic (Gibco), and growth at 37% and 5% CO2. Cells were passaged when the confluence reached 80% of the culture plate every three to four days. Briefly, cells were rinsed with PBS and then incubated in Accutase (Millipore) for 3 minutes until cell detached. We used the culture medium to inhibit enzymatic reaction and centrifuged the suspension at 500×g for 5 minutes, and resuspend the cell pellet in fresh medium.
2.8 Total RNA isolation, RNase R treatment and subcellular localization
Total RNA was extracted using PureLink RNA Mini Kit (Thermo Fisher Scientific) and PureLink DNase Set (Ambion). Total RNAs of normal 10 human tissues were obtained from Ambion Inc. For RNase R treatment, total RNA was incubated with or without RNase R (Epicentre) for 45 minutes at 37 °C to deplete linear and enrich circular RNAs.
To validate the subcellular localization preference, we used the NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific) to separated nuclear and cytoplasmic fractions. Total RNA was then extracted using TRIzol reagent according to
the manufacturer’s instructions. qRT-PCR analyses were performed to examine the relative expression of cytoplasmic and nuclear localization for each gene. GAPDH and U6 snRNA were served as controls for cytoplasmic and nuclear RNAs, respectively.
2.9 cDNA synthesis and RT-qPCR
For mRNA and circRNA quantitation, RNA was reverse-transcribed into cDNA using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). QRT-PCR was performed using Luminaris Color HiGreen High ROX qPCR Master Mix (Thermo Fisher Scientific). Quantity of gene expression was normalized by GAPDH. For miRNA quantitation, cDNA synthesis was carried out by or miRCURY LNA RT Kit (Qiagen).
QRT-PCR was performed by miRCURY LNA SYBR Green PCR Kit (Qiagen) with miRCURY LNA miRNA PCR primer (Qiagen) for each candidate miRNAs, and small nuclear U6 RNA served as an internal standard. QPCR was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems). The primers are provided in Table 4. qRT-PCR reactions were performed using two independent biological replicates, with each having three technical replicates.
Table 4. Sequences of primers used in this study.
2.10 Construction of vector
circARID1A overexpressed plasmid was constructed the exon sequence of circARID1A (E2-E3-E4) into Circle3 (pCIRC2) vector132 (provided by Dr. Laising Yen), which can circularized the transcript to produce circARID1A. The exon sequence of circARID1A was amplified via PCR from the human cortex, and the PCR product was cloned into the Circle3 vector between Mfe-I and Age-I sites.
ReNcell was knockdown of circARID1A by lentivirus carring shRNA targeting the BSJ of circARID1A, and overexpress circARID1A by lentivirus carring circARID1A sequence, respectively. For dual-luciferase assay, ReNcell was transfected by lentivirus
carring dual-luciferase and circARID1A sequence. The lentiviral vectors were constructed by the National RNAi Core Facility at Academia Sinica. All constructs are sequence-verified.
2.11 Cell transfection
Has-miR-204-3p is overexpressed by mimic transfection (Tools) using Lipofectamine RNAiMAX (Invitrogen) for 48 hours. The sequences of oligonucleotides are shown in Table 5. CircARID1A was knockdown by siRNA (MDBio) to target the BSJ, and overexpress by circARID1A overexpressed plasmid in NHA, SH-SY5Y and U118 cells.
Transfection of plasmid was carried out using TransIT-LT1 Reagent (Mirus) according to the transfection manufacturer’s instructions for 48 hours.
For lentiviral transduction, the lentiviruses were infected into ReNcell at an MOI of 2 with 8 µg/ml polybrene for 24 hours. Transfected cells were selected by 0.25 µg/ml puromycin for 3 days. The efficiency of knockdown or overexpression in stable expressing cell lines was verified by RT-qPCR.
Table 5. Sequences of oligonucleotides used in this study.
2.12 Luciferase reporter assays
The luciferase reporter was constructed by subcloning the circARID1A sequence into the Secrete-Pair Dual Luminescence vector (GeneCopoeia). Cells were seeded in 24- well plate 24 hours prior to transfection. By comparing overexpression of miR-204-3p, we used a scramble miRNA mimic in control group. Dual Luminescence vectors that contained circARID1A sequence were co-transfected with miR-204-3p mimic or scramble mimic, respectively, by TransIT-X2 transfection reagent (Mirus). After 48 hours of transfection, cell culture medium were collected. Gaussia luciferase (GLuc) and secreted alkaline phosphatase (SEAP) activities were measured by Secrete-Pair Dual Luminescence Assay System (GeneCopoeia) according to the manufacturer’s protocol. The luciferase activity of GLuc was normalized with SEAP.
We mutated miR-204-3p binding sites in the luciferase-circARID1A reporter vector using Quick-Change Lightning Site-Directed Mutagenesis Kit (Agilent Technologies).
The four binding sites were selected because they were also identified by MiRanda190 (version 3.3a). The mutated GLuc-circARID1A reporter was confirmed by Sanger sequencing. The primers used for mutations of these miR-204-3p binding regions were listed in Table 4.
2.13 Microarray analysis
The microarray hybridization and the data collection were performed with the help of Affymetrix GeneChip System Service center in Genomics Research Center, Academia Sinica. The assessment of purity and integrity of RNA were evaluated by 2100
Bioanalyzer (Agilent Technologies). Total RNA was hybridized to Affymetrix Human Genome Plus 2.0 Array (Affymetrix). The microarray raw data was analyzed using Transcriptome Analysis Console (TAC 4.0) software (Affymetrix). The microarray results of the target genes affected by circARID1A and miR-204-3p are provided at GitHub (https://github.com/TreesLab/circRNA_ASD/tree/master/Microarray_data).
2.14 Neuronal differentiation and Immunostaining
For differentiation of ReNCell into neurons and glial cells, ReNcell was incubated with maintenance medium without containing FGF-2 and EGF growth factors. Maintenance basal media was changed every 3 days for 2 weeks. After 2 weeks, we categorized the neuronal subtypes by immunofluorescence. Cells were plated on glass slides coated with laminin overnight. Cells were fixed with 4% formaldehyde at 37 oC for 25 min, permeabilized with 0.05% Triton X-100 for 15 min, and incubated with 3% FBS for blocking for 1 hr. Cells were then incubated with primary antibody against MKI67 (cell proliferation marker, cat. no. ab15580, Abcam) βIII-tubulin (neuronal marker; ab18207, Abcam) or GFAP (glial marker; cat. no. 13-0300, Invitrogen) at 4oC overnight. Next day, cells were incubated with fluorescently labeled secondary antibodies at 37oC for 1.5 hours. Nuclei were stained with SlowFade Diamond Antifade Mountant with DAPI (Invotrogen). Details for the antibodies are provided in Table 6.
Table 6. The antibodies used for immunofluorescence.
2.15 Statistical analysis
The data were presented as mean ± standard deviation. Statistically significant differences were calculated using Student’s t-test, LME and Pearson’s correlation, as appropriate. In all cases differences were defined statistically significant when p values
< 0.05. A single * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
NS indicates not significant (p > 0.05).
2.16 Code availability
The code for the DE-circRNA analysis, the related input data and results were publicly accessible at GitHub (https://github.com/TreesLab/circRNA_ASD).
CHAPTER 3. Results
3.1 Identification of circRNAs in autism and healthy cortex
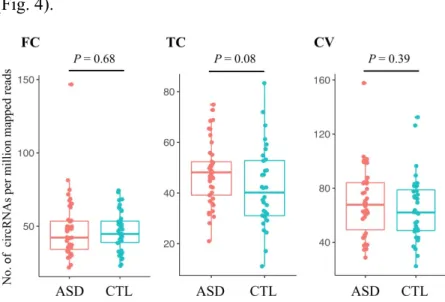
To assess the potential role of circRNAs, we retrieved 236 total RNA-seq data of temporal cortex, frontal cortex and cerebellar vermis from individuals with ASD and non-ASD (CTL)9. First, we characterized circRNA transcripts using NCLscan, which was reported to exhibit the greatest precision among currently accessible circRNA- detection tools153,154,162,163. To improve accuracy, we removed the number of detected circRNAs of this sample was less than one standard deviations away from the mean. In total, 53,427 circRNAs were identified in 202 remained samples. Comparison of the number of detected circRNAs per million mapped reads between ASD and CTL samples, there were no statistically significant differences between three brain regions (Fig. 4).
Figure 4. Comparison of normalized numbers of circRNAs in ASD and non-ASD control samples from different brain regions.
Temporal cortex; TC, Frontal cortex; FC, cerebellar vermis; CV. P values were determined by two-tailed Wilcoxon rank-sum test.
Of the 134 cortex (TC and FC) samples, 36,624 circRNA events were detected, 40%
were found in one sample only. To reduce potentially spurious events, we only remained the circRNAs detected in more than 50% of the 134 samples. After filtering, a total of 1,060 circRNAs were considered for the following analyses (Fig. 5).
Figure 5. The workflow of the identification of DE-circRNAs and DE-modules.
Of note, 61.4% of the 1,060 circRNAs were detected in mouse and 47% were observed in mouse brain (Fig. 6), indicating that circRNAs were highly conserved between human and mouse.
Figure 6. Comparisons of the 1,060 circRNAs and human/mouse circRNAs collected in the well- known databases.