野生種白色念珠菌與cph1/cph1 efg1/efg1雙基因突變種於可誘發野生種形成菌絲環境下之基因表現比較
54
0
0
全文
(2) 野生種白色念珠菌與 cph1/cph1 efg1/efg1 雙基因突變種於可誘發野生種 形成菌絲環境下之基因表現比較. 學生:徐嘉瞳. 指導教授:楊昀良 教授. 國立交通大學生物科技學系 (研究所) 碩士班. 摘要 白色念珠菌 (Candida albicans) 是一種真菌類伺機性病源菌,在免疫系統不健全的人 體中引起病害,嚴重時足以致命。在型態上,野生種白色念珠菌會形成菌絲,並且對寄 主有致病力,一般也認為形成菌絲與否跟有無致病能力有相當程度的關係;而在過去的 研究中發現一株 CPH1 EFG1 雙基因突變種沒有形成菌絲的能力,亦無法致病。 本研究藉由抑制相減雜交技術 (Suppression Subtractive hybridization, SSH) 得到 在能誘發野生種產生菌絲的條件下,野生種和突變種中表現不同之基因。其中包含影響 形態或/及致病之基因,我們期望從中篩選出確實賦予白色念珠菌致病能力之基因,提供 藥物設計者更精準的藥物標的 (drug target)。針對白色念珠菌特有之基因設計藥物,可 以減少對寄主本身的危害,並且增加治療的效率,避免抗藥性的產生。 在本實驗之 SHH 結果中, 逢機挑選 32 個基因片段進行分析, 分離出種 12 種不同基因。 其中包括與形成菌絲有關的 EFG1 基因,證實了此技術的可行性。在已分析的 32 個基 因片段中,EFG1 共出現 18 次,可能表示該基因在野生種細胞內所被轉譯之套數。其他 十二種基因,有些已有相關文獻或是可能牽涉形態改變或致病,例如:serine/threonine kinase 已知與真菌形成菌絲有關;白色念珠菌之 squalene epoxidase (CAERG1) 因為 與細胞膜生合成有關,所以可能涉及型態上的轉換;以及白色念珠菌之 hxt6 gene for galactose/glucose transporter 與能量的代謝可能影響致病機制….等等。在未來的研究 中,本實驗室將設計更多試驗以尋出與白色念珠菌致病直接相關之基因。. i.
(3) Comparison of Gene Expression between Wild-Type Candida albicans and cph1/cph1 efg1/efg1 Double Mutant under the Condition of Inducing Filament Formation of Wild Type Student:Chia-Tung Hsu. Advisor:Dr. Yung-Liang Yang. Department (Institute) of Biological Science and Technology National Chiao Tung University. ABSTRACT Candida albicans (C. albicans) is an opportunistic pathogenic fungus and one of the. most. commonly. isolated. human. pathogens.. Especially. immunocompromised patients, it can lead to lethal candidiasis. that filamentous growth is involved in virulence.. in. It is believed. Researchers have developed. the avirulent cph1/cph1 efg1/efg1 C. albicans, which is unable to form filaments. In recent years, the abuse of antibiotics and the emerging of resistance to existing antifungal drugs have made the treatment of fungal infection more and more difficult.. To search for a new drug with higher efficiency and less toxicity, I. have screened and isolated the virulence genes of C. albicans with the technique of suppression subtractive hybridization in the hope of unveiling suitable new targets for drug development. Two. sets. of. suppression. substrate. hybridization. were. performed.. One. subtracted mutant RNA from wild type RNA (WT-Mut). The other subtracted wild-type RNA from mutant RNA (Mut-WT).. Genes in the WT-Mut group are. expressed only in the wild type C. albicans or expressed at a higher level in wild-type than in mutant strain.. Virulence genes could be in this group.. islated cDNA fragments were subjected for sequence analysis. 18 were EFG1 DNA fragments.. 32. Among them,. Since EFG1 gene was included in the WT-Mut. group as expected, this performance of this technique is reliable.. It. demonstrated that this suppression subtractive hybridization can indicate not only whether certain genes are expressed or not but also the difference in the expression level. In the remaining 14 clones, grouped into 12 different genes, some have been reported to be associated in morphology or virulence: Serine/threonine kinase is involved in filamentous growth; squalene epoxidase (CAERG1) is in the pathway of synthesis of cell membrane; hxt6 gene for galactose/glucose transporter is responsible for the metabolism of energy. ii.
(4) 本論文は天国にいる父親に捧げる この些細な成績と努力の結果を認めてくれるよう心から願っておる. iii.
(5) Table of Contents Page 中文摘要. i. Abstract. ii. Dedication. iii. Table of contents. iv. List of Tables. v. List of Figures. vi. I.. Introduction. II.. Materials and Methods. 10. III.. Results and Discussion. 22. IV.. Reference. 26. 1. iv.
(6) List of Tables Page Table 1.. OD600 of Cell Culture. Table 2.. Groups of Restriction Pattern in WT-Mut Group. 33. Digested by Hae III Table 3.. 34-35. DNA Sequencing Results of Randomly Chosen Clones from WT-Mut Group. Table 4.. pCRII-TOPO Vector Fragments Digested by HaeIII and Sau3AI. Table 5.. 36. 37. Groups of Restriction Mapping Pattern in Mut-WT Group Digested by HaeIII. v. 38-39.
(7) List of Figures Page Figure 1.. Wild Type and cph1/cph1 efg1/efg1 Candida albicans grown in the media. Figure 2.. 40. 1% Agarose Gel of RNA Isolates from Wild Type And Double Mutant Strains. Figure 3.. Different DNA Fragments of Candida albicans EFG1 Gene. Figure 4.. Figure 5.. 41. 42-45. Sequences of the PCR-Select cDNA Synthesis Primer, Adaptors, and PCR Primers. 46. Map of pCR®II-TOPO®. 47. vi.
(8) Introduction A. Candida albicans Candida albicans (C. albicans) is an opportunistic fungal pathogen and one of the most commonly isolated human pathogens (Tsarfaty et al, 2000).. Especially in immunocompromised patients, it can lead to lethal. candidiasis, which started in the digestive system by penetrating the mucous lining of gastrointestinal walls.. In the previous study, a mouse. can be killed by injection of adequate quantity of wild-type C. albicans (Lo et al, 1997). The cells of C. albicans can have different morphology.. For instance, it. can switch from a unicellular budding yeast to a filamentous form. fiamentous form may consist of hyphal or pseduohyphal cells.. The. The ability. of changing morphology is suspected to be involved in the virulence (Kobayashi and Cutler, 1998).. B. Filament Formation Filamentous C. albicans have been observed to adhere to and invade host cells more readily than the yeast ones, suggesting that filamentous growth may contribute to the virulence of C. albicans (Leberer et al, 1997). The genetic regulation of cell morphology in C. albicans has been discovered.. The gene products of CPH1, HST7, and CST 20 produced in C. albicans are respectively homologous to those of STE12, STE7, and STE20 in S. cerevisiae. mutated. have. C. albicans strains with either one of these genes demonstrated. retarded. filamentous. growth. but. no. impairment of serum-induced germ tube and hyphae formation (Liu et al, 1994; Kron et al, 1995).. The study of efg1/efg1, cph1/cph1 efg1/efg1,. and tup1/tup1 mutants, whose capability to form hyphae were impaired. 1.
(9) by the mutations, indicated that capabilities to invade and injure endothelial cells are significantly reduced (Phan et al, 2000).. C. Adherence One critical step in the pathogenic process is their adherence to host tissues.. When C. albicans cells disseminate in the bloodstream of hosts,. the organisms are likely to adhere to and then penetrate the endothelial cell lining of blood vessels to invade the tissues (Filler et al, 1996). Adherence of C. albicans to epithelial tissues is mediated primarily by specific adhesin-receptor interactions.. Two major isolates among Candida. species, C. albicans and C. tropicalis, (Asakura et al, 1991) adhere to host cells in vitro to a greater extent than the relatively nonpathogenic species. The correlation between adhesion and virulence has been confirmed in a comparative study of the adherence ability in different Candida species, including C. albicans serotype A, serotype B, C. stellatoidea, C. tropicalis, C. krusei and C. glabrata.. The results showed that both serotypes of. Candida albicans adhere to buccal epithelial cells to a significantly greater degree than the other species tested and there were no differences between C. albicans serotypes A and B (Ghezzi et al, 1986). A study of strains of C. albicans with reduced ability to adhere in vitro has shown that they also have impaired abilities to cause infection in animal models (Calderone et al, 1991). Strains which had an increased ability to adhere to buccal epithelial cells and produced increased amounts of extracellular proteinase activity were shown to have increased lethality in a mouse model.. The C. albicans isolates that adhered most strongly to. buccal epithelial cells had the highest relative proteinase activities and were most pathogenic (Ghannoum et al, 1986). Int1p is a C. albicans surface protein with limited similarity to vertebrate integrins. In S. cerevisiae, the expression of INT1 was sufficient to direct the adhesion of this normally nonadherent yeast to human epithelial cells. 2.
(10) (Gale et al, 1998).. Furthermore, disruption of INT1 in C. albicans. suppressed hyphal growth, adhesion to epithelial cells, and virulence in mice (Gale et al, 1998). Whether the hydrophobicity (CSH) of cell surface of the yeast cells may also contribute to virulence has not been definitively demonstrated. Nineteen isolates of C. albicans were grown in Sabouraud dextrose broth at either 23℃ or 37℃ and tested for CSH by a polystyrene microsphere assay and for the ability to adhere to HeLa cells, a human cervical epithelioid carcinoma cell line (Hazen, 1989).. Growth temperature did. not appear to determine adherence ability, as all isolates that did not differ in CSH after growth at either temperature also did not differ in the ability to adhere.. No correlation (r = 0.44) was obtained between CSH. and adherence when the isolates grown at 23 ℃ were evaluated as a group.. However, higher correlation (r = 0.65) was obtained when the. isolates were grown at 37 ℃ .. Interestingly, a significantly positive. correlation between CSH and adherence was obtained when individual isolates were analyzed (Hazen, 1989). It has been concluded that CSH had little effect on adherence once a moderately high level of CSH was attained.. These results indicated that. CSH is involved in adherence but is not the predominant mechanism and that the effect of CSH on adherence is isolate dependent (Hazen, 1989).. D. Proteinase Activity The hydrophobicity on the cell surface is one of the physiological characteristics of C. albicans.. Studies have revealed that hydrophobic C.. albicans are more virulent than hydrophilic ones (Antley et al, 1988).. The. extracellular proteinase activity of C. albicans is thought to be associated with virulence.. Secretory acid proteinase antigen and antibody can be. demonstrated in patients infected with C. albicans (Macdonald et al, 1980). The enzyme may function in two ways affecting virulence.. 3. One is to.
(11) invade tissue by the proteolytic activity; the other is to colonize host tissues with production of enzymes (McCullough et al, 1996). Previous studies showed that C. albicans could use serum as nitrogen source, and produce proteinase (Ganesan et al, 1991). publications focus on aspartyl acid proteinase.. Most of the. Scientists found that. different strains of C. albicans can secrete proteinases with similar molecular weight, but with different pH optima, substrate specificity, and isoelectric point.. This means the secreted proteinases are strain-specific. (reviewed by Cutler, 1991). Virulence of C. albicans strains with targeted disruption of secretory aspartyl. proteinase. genes. (SAP1. to. SAP6). was. assessed. in. estrogen-dependent rat vaginitis model (De Bernardis et al, 1999).. an Null. sap1 to sap3 mutants lost most of the virulence of their parental strain SC5314. In particular, the sap2 mutant was almost avirulent in this model. Reinsertion of the SAP2 into this latter mutant led to the recovery of the vaginopathic potential. The vaginal fluids of the animals infected by the wild-type. strain. or. by. the. sap1. or. sap3. mutants. expressed. a. pepstatin-sensitive proteinase activity in vitro. No trace of this activity was found in the vaginal fluid of rats challenged by the sap2 mutant. strains were capable of developing true hyphae during infection.. All. Thus,. members of SAP family, in particular SAP2, play a clear pathogenic role in vaginitis and may constitute a novel target for chemoimmunotherapy of this infection.. At least three proteinases have been found in the. intracellular compartments of C. albicans (Portillo et al, 1986).. They have. distinctly different pH optima and other enzymatic characteristics.. One of. these proteinases has many characteristics in common with the putative virulence factor, aspartyl acid proteinase.. Like the secreted enzyme, the. intracellular one is a glycoprotein, an aspartyl acid proteinase that can act on a wide variety of proteins.. It is not inhibited significantly by. phenylmethylsulfonyl fluoride and is inhibited by pepstatin.. But the. molecular weight and pH optimum of the intracellular form is 60kd and. 4.
(12) 5.0; the secreted proteinase is 40-45kd and 2.2-4.5.. The aspartyl acid. proteinase can work at some particular tissues, which is acidic, because of the low pH optimum (Remold et al, 1968; Ruchel, 1981). Factors. involved. in. adherence,. proteinase. activity. and. filament. formation were always considered important contributors to virulence. However, the study on molecular level has just begun.. Recently, some. scientists have begun to discover gene whose activities are related to the virulence of Candida (reviewed by Cutler, 1991).. E. CPH1 Gene Cph1p is the homologue of Ste12p.. Ste12p in S. cerevisiae is an. important transcription factor that regulates several signal transduction pathways (Burchett et al, 2001). The genetic pathway of the yeast-hyphal dimorphism in C. albicans has not been established, but Cph1p has been shown to be involved in hyphal growth of C albicans (Liu et al, 1994).. F. EFG1 Gene EFG1p is the homologue of Phd1p.. The overexpression of phd1 in S.. cerevisiae resulted in enhanced pseudohyphae growth and the phd1 ste12 double mutants are more defective in pseudohyphal growth than the ste12 mutant (reviewed by Ernst, 2000). Homozygous efg1 mutants form only pseudohyphae on solid media and do not germinate at all in liquid media, even in the presence of serum stimulation (Lo et al, 1997).. G. Subtractive Hybridization a. Background. 5.
(13) In 1994, a PCR-based technique called representational difference analysis has been announced.. It does not require physical separation of. single-stranded (ss) and double-stranded (ds) cDNAs.. Representational. difference analysis has been applied to enrich for genomic fragments that differ in size or representation (Lisitsyn et al, 1993) and to clone differentially. expressed. cDNAs. (Hubank. et. al,. 1994).. However,. representational difference analysis does not solve the problem of the wide differences in abundance of individual mRNA species.. Consequently,. multiple rounds of subtraction are still needed (Hubank et al, 1994). The mRNA differential display (Liang et al, 1992) and RNA fingerprinting by arbitrary primed PCR (Welsh et al, 1992) are potentially faster methods for identifying differentially expressed genes.. However, both of these. methods have a high level of false positives (Bauer et al, 1994; Sompayrac et al, 1995), biased for high-copy-number mRNA (Bertioli et al, 1995) and might be inappropriate in experiments in which only a few genes are expected to vary (Sompayrac et al, 1995). Numerous cDNA subtraction methods have been reported. In general, they involve hybridization of cDNA from one population (tester) to excess of mRNA (cDNA) from other population (driver) and then separation of the unhybridized fraction (target) from hybridized common sequences.. The. latter step is usually accomplished by hydroxylapatite chromatography (Hedrick et al, 1984), avidin-biotin binding (Duguid et al, 1990; Sargent et al, 1983;Davis, 1984), or oligo(dT)30-latex beads (Hara et al, 1991). Despite the successful identification of numerous important genes such as the T-cell receptors (Hedrick et al, 1984) by these methods, they are usually inefficient for obtaining low abundance transcripts.. These. subtraction techniques often require greater then 20 μg of poly(A)+ RNA, involve multiple or repeated subtraction steps, and are labor intensive. b. Suppression Subtractive Hybridization, SSH In 1996, Diatchenko et al. presented a new PCR-based cDNA subtraction. 6.
(14) method,. termed. suppression. demonstrated its effectiveness.. subtractive. hybridization. (SSH),. and. SSH is used to selectively amplify target. cDNA fragments (differentially expressed) and simultaneously suppress nontarget DNA amplification.. The method is based on the suppression. PCR effect previously described by Diatchenko’s laboratories (Diatchenko et al, 1996). This highly effective method, suppression subtractive hybridization (SSH), has been developed for the generation of subtracted cDNA libraries. It is based primarily on a recently described technique called suppression PCR and combines normalization and subtraction in a single procedure. The suppression PCR is mediated by long inverted terminal repeats. When attached to the ends of DNA fragments, these inverted repeats form stable panhandle-like loop structure after each denaturation and annealing cycle.. In a PCR with primers derived from the sequences of the long. inverted repeats, the panhandle-like structures cannot be amplified exponentially because intramolecular annealing of the long inverted terminal repeats is highly favored and is more stable than intermolecular annealing of the shorter PCR primer to the long inverted repeats (Lukyanov et al, 1995).. By incooperating this suppression effect in a PCR. amplification scheme, undesirable DNA fragments can be eliminated from a mixture of target sequences (Chenchik, 1996; Siebert, 1995). In the process of SSH, the normalization step equalizes the abundance of cDNAs within the target population and the subtraction step excludes the common sequences between the target and driver populations.. In a. model system, the SSH technique enriched for rare sequences over 1,000-fold in one round of subtractive hybridization (Diatchenko et al, 1996). c. Application The suppression PCR effect has been applied in chromosome walking (Siebert et al, 1995) and rapid amplification of cDNA ends (Chenchik et al,. 7.
(15) 1996).. The subtraction method described here overcomes the problem of. differences in mRNA abundance by incorporating a hybridization step that normalizes. (equalizes). subtraction. by. sequence. standard. abundance. hybridization. during. kinetics.. It. the. course. eliminates. of any. intermediate step(s) for physical separation of single strand and double strand cDNAs, requires only one subtractive hybridization round, and can achieve greater than 1,000-fold enrichment for differentially expressed cDNAs.. The effectiveness of the SSH method has been demonstrated by. generating a testis-specific cDNA library and characterizing selected cDNA clones.. Furthermore, the subtracted cDNA mixture can be used directly. as a hybridization probe for screening recombinant DNA libraries, such as a. human. Y. chromosome. cosmid. library,. thereby. chromosome-specific and tissue-specific expressed sequences.. identifying Results of. some studies have suggested that the SSH technique is applicable to many studies in molecular genetics and positional cloning for the identification. of. disease,. developmental,. tissue-specific,. or. other. differentially expressed genes (Diatchenko et al, 1996).. H. Pharmaceutical Treatments for C. albicans Recent years, the abuse of antibiotics and the increased number of immunocompromised cases had increased the prevalence of opportunistic fungal pathogens and made diagnosis and treatment important issues. Clinically applied antifungal drugs could bring side effects and develop resistance easily (Reviewed by Yang, 2003). Three major categories of antifungal drugs are 5-flucytosine, polyenes and ergosterol biosynthesis inhibitors (Kerridge et al, 1986).. The. synthetic pyrimidine, 5-flucytosine inhibits the synthesis of DNA and protein in yeasts. The other two types of inhibitors target the main component of fungal plasma membranes, ergosterol.. Since cholesterol of. human cell membranes is very similar to cholesterol, polyenes are toxic.. 8.
(16) The polyene macrolide, amphotericin B is one of the effectively and frequently applied antifungal drugs (reviewed by Gallis et al, 1990). Amphotericin B acts to slow down the growth and multiplication of susceptible fungi.. If the concentrations of this medication are high. enough, they can also destroy the fungi. It is approved by the FDA for the treatment of certain types of serious fungal infections, but there are many possible side effects, such as fever, chills, nausea, vomiting, headaches, electrolyte imbalance and renal failure.. I. The Aim of This Study Molecular biology and genetics have been applied to the studies of Saccharomyces cerevisiae because of its genome stability and well-studied properties.. Unlike Saccharomyces, life cycle of C. albicans is still unclear,. and this makes the application of molecular biology and genetics difficult. Researchers have compared the same Candida strain with different morphologies resulted from different growth conditions or media and revealed the association between morphology and virulence.. But these. conditions, such as temperature, media or even interactions changed not only the virulence but also other phenotypes.. To understand the. virulence of C. albicans on the molecular level, it is necessary to identify and isolate the virulent genes.. The technique of subtractive hybridization. was used to isolate virulence factors that are involved by comparing the virulent (wild-type) strain and avirulent mutant strains under the same condition. The aim of this study is to sieve out genes encoding virulence factors of C. albicans to pave the way for understanding the mechanisms of virulence and also provide information of more specified potential targets for drug design to develop higher efficiency and less toxicity antifungal drugs.. 9.
(17) Materials and Methods A. Culture of Candida albicans The wild-type C. albicans (phenotype:CPH1/CPH1 EFG1/EFG1) strain SC5314 (Gillum et al, 1984) and cph1/cph1 efg1/efg1 double mutant strain HLC54 (Lo et al, 1997) were used throughout this study. Yeast was precultured in 10 ml of YEPD medium (Clontech, pH 7) at 30°C overnight. Two milliliter of the overnight culture were transferred to 60 ml of YEPD containing 10% serum (Gibco BRL) and incubated at 37°C with shaking until the OD600 reaches 0.4-0.7. B. 1. RNA Isolation Cell was cultured overnight in YEPD containing 10% serum at 37°C. The cells were then spun down at 3200 rpm, 4°C for 5 minutes. supernatant was then discarded.. The. The cell pellets was then suspended. in 0.3 ml RNA Isolation Buffer (50mM NaCl, 20mM Tris-HCl, pH7.6, 1% EDTA and 1% SDS) with vortex.. One third volume of glass beads. (Sigma G-9268, 425-600 microns unwashed glass beads, washed by acetic acid before use, Step B.2.) was then added to the cell suspension before being vortexed for 5 minutes to break the cell wall. A volume of 0.3 ml of phenol was then added and the mixture was vortexed for 5 minutes.. A volume of 0.5 ml of RNA Isolation Buffer. was added before being vortexed for 5 minutes.. The whole mixture. was then centrifuged at 14000 rpm, 4°C for 5 minutes. supernatant. was. transferred. to. new. tubes.. In. transferring, the interface should not be taken.. the. process. The of. Equal volume of. phenol was then added to the supernatant prior to vortex for 30 seconds followed by spinning at 14000 rpm for 5 minutes.. The. supernatant was transferred to new tubes with care to avoid the interface.. The steps of 6 and 7 were repeated for one more time.. The supernatant was transferred to new tubes. In the process of 10.
(18) transferring, one should avoid the interface.. One eighth volume of 2.5. M NaOAc (pH 7) and 2.5 volume of 100% cold ethanol were added to the supernatant and kept on ice for 20 minutes prior to centrifugation at 3000 rpm for 1 min. The supernatant was discarded carefully and then the pellets were washed with 75% cold ethanol prior to centrifugation at 3000 rpm, 4°C for 1 minute.. The supernatant was discarded.. The RNA pellets were. then dissolved in 1X TE before measurement of the OD260/280 to estimate the purity and concentration of RNA.. To assess the integrity. of RNA, 2 µg of isolated RNA were electrophoresed in 1% agarose gel. 2. Preparation of Glass Beads Glass beads were poured into a 500ml-beaker, and acetic acid was added covering every glass beads.. The beaker was covered with. aluminum foil, and left in the laminar flow overnight. poured out very carefully.. Liquid was. The glass beads were washed with ddH2O. for 5-6 times and then dried in the oven.. The glass beads were. divided into 1.5ml eppendorf tubes and then autoclaved.. The. eppendorf tubes containing glass beads were dried in the oven. C. Subtractive hybridization a. cDNA Synthesis Ten micromoles of both CDS (cDNA Synthesis) primer and SmartII SMART™ primer (Switch Mechanism At 5' end of the RNA Transcript) were used for every 3µg of RNA and H2O was added to total volume of 5 µl. The tubes were incubated in hot water at 70°C for 5 minutes and then cooled on ice before brief centrifugation. The following mixture was added to each tube and the tubes were vortexed and spun. First-strand buffer, 5X, 2µl (250mM Tris-HCl, pH 8.5, 40 mM MgCl2, 150 mM KCl, and 5 mM Dithiothreitol), DTT (100mM) 1µl, dNTP (10mM) 1µl, and Superscript II/MMLV RT (200u/µl) 1µl. 11.
(19) The tubes were incubated in a water bath at 42°C for 2 hours and then 40µl of TE was added into each tube.. The reaction was. inactivated by 7 minutes’ hot water bath at 72°C. b. cDNA Amplification by LD PCR The master mix, containing 1050µl of H2O, 126µl of 10X Advantage 2 PCR buffer (Clontech Laboratory, Inc.), 24µl of dNTP (10mM), 24µl of PCR primer (10µM), and 24µl of 50X Advantage 2 Polymerase Mix was prepared for 4 samples (12 tubes). Each tube contains 312µl of master mix. Three microliter of cDNA template from the previous step, the synthesized cDNA, was added to each tube. The tubes were vortexed and spun.. The mixture in tubes was divided into 3 tubes.. The PCR. condition was as following: 65°C’s annealing, 68°C’s elongation for 6 minutes, and totally 25 cycles. cycle.. The program was interrupted at 15th. Two tubes of each sample were taken out and carefully kept.. Eight microliters of mixture from the other tube was transferred into a new tube. other cycle.. PCR continued and 8 µl of sample was taken out every Agarose gel (1%) at 250 Volt for 20 minutes was used to. determine the optimal cycle for each sample.. The 2 previous. taken-away tubes were then continued with the rest of cycles.. The 2. tubes of each sample were combined and then 4 µl of 0.5M EDTA was added to stop the reaction.. Eight microliters from each sample was. kept in a new tube for further analysis (Sample 1). c. Column Chromatography Samples from step b were mixed with equal volume of DNA phenol, vortexed for 30 seconds and then centrifuged at 12000g for 10 minutes. The water phase in the tube was transferred into the upper part of the Centricon 100 containing 1.8 ml of H2O.. The Centricon tube was. centrifuged at 3000 rpm for 30 minutes. After centrifugation, the lower. 12.
(20) part solution was discarded, 2 ml of H2O was added and this step was repeated 3 times.. The solution in the column was mixed by inverting. and then centrifuged at 3000 rpm for 5 minutes.. The solution in the. cap was collected. The column was inverted several times to re-suspend the gel matrix. The top and bottom plug of the column were removed and the inner buffer flowing through.. TNE buffer (1.5ml, 10 mM Tris-HCl, pH 8,. 10mM NaCl and 0.1mM EDTA) was added and after it flowing through, the buffer was collected and discarded.. The sample was added in the. center of gel in the column and followed 25 µl of TNE.. After TNE buffer. drained out, 150 µl of TNE was added and drained out.. In the tube,. 320 µl of TNE was added and the solution flowed through was collected. The sample 1 was run on 1.0% agarose gel to check the quantity of DNA.. If the cDNA fragments on the gel were not able to be seen,. another 75 µl of TNE should be added and the solution flowed through collected.. The sample was confirmed on 1.0% agarose gel.. Eight. microliters of each sample was transferred into a clean tube for further analysis (Sample 2). d. RsaI Digestion The following procedure was performed to both wild type and mutant cDNA.. The shorter, blunt-ended double-stranded cDNA and optimal. fragments will be prepared in this step.. The cDNA from Step c. was. taken by 300 µl and mixed with 33.5 µl of 10X RsaI restriction buffer attached to the restriction enzyme and 20 units of RsaI.. The mixture. was incubated in 37°C of water bath overnight. The sample was run in 1.0% agarose gel and checked for the cDNA fragments.. Each sample. was added with 8 µl of 0.5M EDTA to stop the reaction and 8 µl of samples were transferred for further analysis (Sample 3). DNA phenol was added and mixed by vortex and then centrifuged at 12000g for 10 minutes.. The water phase was transferred into the. 13.
(21) upper part of Centricon 100 containing 1.7 ml of H2O.. The. centrifugation step was repeated at 3000 rpm for 30 minutes and the Two milliliters of H2O was added. lower part of solution was discarded.. into the tube for 4 times. The column was inverted several times for mixing and centrifuged at 3000 rpm for 10 minutes.. The solution was. collected in the cap. The samples was transferred into new tubes and concentrated by an evaporator (SpeedVac). The RsaI digested cDNA fragments were dissolved in 10 µl of H2O.. From every sample, 1 µl was taken and. mixed with 2 µl of dye and 7 µl of H2O.. Agarose gel (1%) was used to. check Sample 3. e. Adaptor Ligation Every sample from Step d. was diluted with 5 times of H2O; prepared for Tester cDNA T, and C. The Master Mix (4 µl of H2O, 1 µl of 10X ligation buffer, and 1 µl of T4 DNA ligase for each reaction) was prepared for 2 samples (4 tubes).. See the table below for recipes.. The tubes were incubated at 16°C overnight and after incubation, 1 µl of EDTA/glycogen mix was added.. The reaction was stopped in a. thermal cycler at 72°C for 5 minutes.. The fragments were stored at. -20°C.. Sample. WT-Ac. Mut-Ad. WT-Be. Mut-Bf. Diluted tester cDNA. WT, 2µl. Mut, 2µl. WT, 2µl. Mut, 2µl. Adaptor 1 (10 µM). 2. 2. -. -. Adaptor 2R (10 µM). -. -. 2. 2. Master Mix. 6. 6. 6. 6. 10. 10. 10. 10. Final Volume. cWT-A: wild type DNA ligased with A adaptor;. 14.
(22) d Mut-A: mutant DNA ligased with A adaptor; eWT-B: wild type DNA ligased with B adaptor; f Mut-B: mutant DNA ligased with B adaptor.. f. First Hybridization Samples were prepared according the following recipe, and incubated in thermo cycler at 98°C for 2 minutes then 68°C for 6 hours. incubation, step g was immediately performed. Sample. WT-A-Mutc. Driver cDNA. Mut, 1.5 µl. WT, 1.5 µl. Tester cDNA. WT-A, 1.5µl. Mut-A, 1.5µl. 1 µl. 1 µl. WT-B-Mute. Mut-B-WTf. Driver cDNA. Mut, 1.5 µl. WT, 1.5 µl. Tester cDNA. WT-B, 1.5µl. 4X hybridization buffer Sample. 4X hybridization buffer. Mut-A-WTd. Mut-B, 1.5µl. 1 µl. 1 µl. cWT-A-Mut: mix WT-A DNA with mutant DNA; dMut-A-WT: mix Mut-A DNA with wild type DNA; e WT-B-Mut: mix WT-B DNA with mutant DNA; f Mut-B-WT: mix Mut-B DNA with wild type DNA.. g. Second Hybridization Samples were prepared as follows: WT. Mut. Driver cDNA (µl). 1. 1. 4X Hybridization buffer (µl). 1. 1. H2O (µl). 2. 1. Total volume (µl). 4. 3. 15. After.
(23) Tubes were spun and 1 µl of solution in each tube was transferred to new tubes.. DNA fragments were denatured in thermal cycler at 98°C. for 2 minutes.. The denatured driver cDNA and T-DB were mixed and. T-DA was then added. The mixture was thoroughly mixed and spun. Then the steps 4-6 were repeated and the mixture was incubated at 68°C overnight.. One hundred of dilution buffer was added, then was. mixed by vortex and spun down.. The reaction was inactivated in. thermal cycler at 68°C for 7 minutes. Samples were transferred to a new tube and stored at -20°C. h. PCR Amplification First PCR solution was prepared as follows and run the PCR program1Ж. H2O. 19.5 µl. 10X Advantage 2 PCR buffer. 2.5 µl. dNTP mix, 10mM. 0.5 µl. PCR primer 1, 10µM. 1.0 µl. 50X Advantage 2 polymerase mix. 0.5 µl. Subtracted cDNA fragments from Step g-11. 1.0 µl. Three microliter of PCR product was transferred and diluted with 27 µl of H2O.. The PCR product from first PCR was the template of second. PCR. Ж. PCR program1: 75°C. 5 min. 94°C. 25 sec. 94°C. 15 sec. 66°C. 30 sec. 72°C. 110 sec. 27 cycles. 16.
(24) Second PCR solution was prepared as follows and run the PCR program2 Є.. Agarose gel (1%) was used to check the PCR products.. H2O. 18.5 µl. 10X Advantage 2 PCR buffer. 2.5 µl. dNTP mix, 10mM. 0.5 µl. Nested PCR primer 1, 10µM. 1.0 µl. Nested PCR primer 2R, 10µM. 1.0 µl. 50X Advantage 2 polymerase mix. 0.5 µl. Є. PCR program2: 94°C. 10 sec. 68°C. 30 sec. 72°C. 10/12/14 cycles. 110 sec. i. Final PCR cycles First step PCR solution was prepared as follows and 1 µl of subtracted cDNA fragments from step g-11 was added to run the PCR program‡. The PCR product was transferred and diluted with 4 times of H2O. H2O. 19.5 µl. 10X Advantage 2 PCR buffer. 2.5 µl. dNTP mix, 10mM. 0.5 µl. PCR primer 1, 10µM. 1.0 µl. 50X Advantage 2 polymerase mix. 0.5 µl. PCR program‡: 75°C. 5 min. 94°C. 25 sec. 94°C. 15 sec. 66°C. 30 sec. 72°C. 110 sec. 20 cycles. 17.
(25) Second step PCR solution was prepared as follows and 1 µl of diluted first PCR product was added as templates (2 for each sample). The. PCR program†. H2O. 41 µl. 10X PCR buffer. 5 µl. dNTP mix, 10mM. 1 µl. Nested PCR primer 1, 10µM. 1 µl. Nested PCR primer 2R, 10µM. 1 µl. 50X Advantage cDNA polymerase. 1 µl. PCR program† 94°C. 10 sec. 68°C. 30 sec. 72°C. 110 sec. optimized cycles. Two of each sample was combined and taken out 8 µl for 1.0% agarose gel. The PCR products were purified and ligated (TA cloning).. The water. phase of PCR products from previous step were transferred into the upper part of Centricon 100 containing 1.8 ml of H2O. centrifuged at 3000 rpm for 30 minutes.. Tubes were. The lower part of the solution. was removed and 2 ml of H2O was added for 4 times. The column was inverted for mixing and centrifuged at 3000 rpm for 5 minutes.. The solution in the cap was collected.. Taq DNA. polymerase, 10X PCR buffer and 25 mM dNTP were added.. The. mixture was incubated at 72°C in a thermal cycler for 10 minutes. Two hundred microliters of DNA phenol and 150 µl of H2O were added and mixed by vortex, and then centrifuged at 12000g for 10 minutes. The water phase was transferred into the upper part of Centricon 100 containing 1.8 ml of H2O. minutes.. Tubes were centrifuged at 3000 rpm for 30. The lower part of the solution was removed and 2 ml of H2O 18.
(26) was added for 4 times.. The column was inverted for mixing and. centrifuged at 3000 rpm for 5 minutes. collected. hours.. The solution in the cap was. Samples were concentrated by evaporator (SpeedVac) for 3. The cDNA fragments were dissolved in 12 µl H2O.. Small. amount of the cDNA sample was taken for 1.0% agarose gel. The cDNA fragments were taken by 6 µl to be ligated with master mix (vector 2 µl, buffer 1 µl and ligase 1 µl) and spun down.. Samples. were incubated at 14.5°C in thermal cycler overnight. j. Transformation (Invitrogen TA Cloning One Shot competent cell) A tube containing frozen competent cell DH5α (50µl) was placed on ice and 2 µl of DNA was added slowly and mixed gently with the pipette tip.. The tube was tapped gently and then kept on ice for 30 minutes.. The tube was incubated at 37°C for 20 seconds; shaking should be avoided. After incubation, it was kept on ice for 2 minutes and then 0.45 ml of room temperature SOC medium was added.. The mixture. was transferred into a new centrifugation tube and then centrifuged at 225 rpm at 37°C for 50 minutes.. The solution was dispersed averagely. 70µl on medium plate containing ampicillin. D. Plasmid DNA Isolation Cells were cultured in 24-well-plates with LB medium (Scharlau) containing ampicillin (10mg/ml) and incubated at 37°C overnight with shaking. Plates containing cells were spun at 3000 rpm for 15 minutes and the supernatant was poured out.. Cell pellets were re-suspended. with 200 µl of P1 buffer (re-suspension buffer, 50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 100 µg/ml RNase A, QIAGEN) and rocked for 5 minutes. The solution was incubated at room temperature for 8 minutes with 300 µl of P2 buffer (lysis buffer, 200mM NaOH/ 1% SDS, QIAGEN) added, and then on ice for 2 minutes.. Cold P3 buffer (neutralizing. buffer, 3M potassium acetate, pH 5.5) was added and placed on ice for. 19.
(27) at least 15 minutes (Sheibani, 1997).. The mixture was spun at 3000. rpm for 15 minutes and the supernatant was carefully poured to new 24-well plates.. Equal volume of iso-propanol was added and spun at. 3000 rpm for 15 minutes.. The supernatant was poured out.. Plates. were washed with 1 ml of 70% ethanol and spun at 3000 rpm for 10 minutes.. The supernatant was poured out and the plate was placed on. the bench until it dries.. The pellets were resuspended in ddH2O and. stored at –20°C.. E. Restriction Mapping DNA samples and pCRII-TOPO vector (Invitrogen, Figure 4) were digested with HaeIII and Sau3AI endonucleases (Gibco), respectively. Plasmids were isolated in 24-well microplate. The enzyme solution was prepared as follows and incubated at 37°C for 2 hours (Sambrook et al, 1989). Hae III (10 unit/µl). 0.2µl / Sau 3AI (10 unit/µl). 0.2µl. REACT® 2 Buffer. 1.5µl / REACT® 4 Buffer. 1.5µl. H2O. 3.3µl. Plasmid DNA. 10.0µl. Total. 15.0 µl/ Reaction. F. Acrylamide gel for endonuclease digested DNA fragments The gel electrophoresis cassette, Mini-PROTEAN 3 (Bio-rad), was set up to cast a polyacrylamide gel.. The gel size was 8.3 cm wide, 7.3 cm. height and 0.075 cm thick. The solution was prepared by mixing 1.2 ml of 30% acrylamide/bis acrylamide ( 29:1 ), 0.6 ml of 10X TBE, 30 µl of 10% ammonium persulfate and 4.2 ml of H2O.. 20. The polymerization.
(28) stated when 3 µl of TEMED added.. The mixture was immediately. poured in between the glass plates and then the 10-well comb was inserted.. The gel was completely polymerized at room temperature. for 25-30 minutes.. The comb was removed and the cassettes were. transferred to the electrode assembly.. The electrode assembly was. placed in the clamping frame and the tank was filled with running buffer (0.5X TBE Buffer, Gibco BRL). The first lane was always loaded with size marker, 1kb Plus (Gibco BRL) and the tenth was vector sequence digested by restriction enzyme, HaeIII or Sau3AI.. The gel was run at 100 volts for 50 minutes.. G. Visualization of the Gel The gels were stained with Ethidium bromide (0.5 µg/ml, Sambrook et al, 1989) for 30 seconds and destained in H2O for another 30 seconds. The bands were visualized and stored with Gel Doc 2000 (Bio-rad).. H. Classification Restriction Maps The molecular weight of each band of samples can be determined according to the 1kb size marker and the enzyme digested vector fragments on the same gel.. First, samples were classified by the. number of bands and then similar band patterns.. Finally, samples. within the same group carrying the same molecular weights of bands are regarded as the same fragment of DNA.. 21.
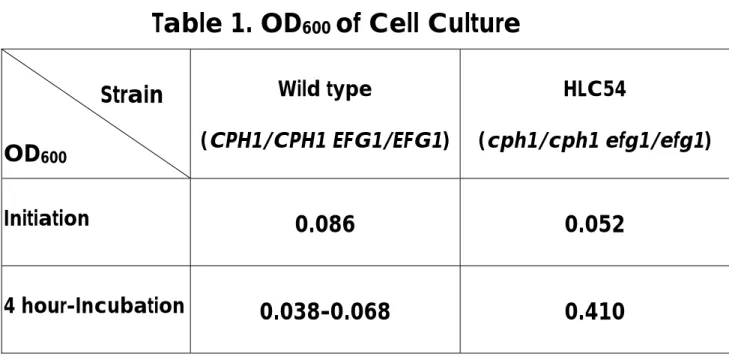
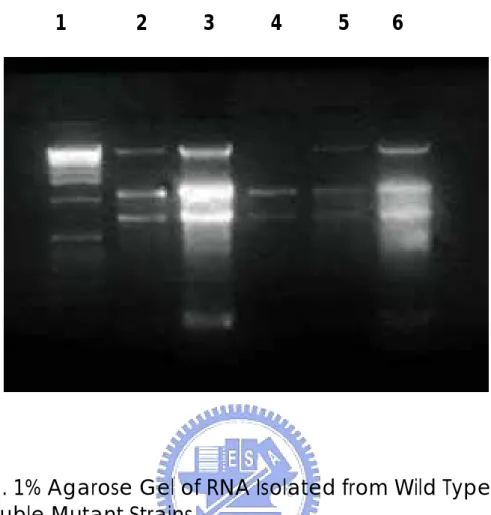
(29) Results and Discussion A. Cell Culture C. albicans were inoculated into fresh media and incubated with shaking at 37°C overnight. The cell density was estimated by spectrophotometer at 600nm. The optical density measured at 600nm of mutant cells reached 0.41 (Table 1) at the 4th hour and then cells were harvested.. Wild-type C.. albicans cells tended to aggregate at the bottom of tubes and the absorbance was varying and difficult to determine, so they were harvested at the same time as mutant cells.. The double mutant strain cells were. suspending in the media (Figure 1). The morphological expression of wild type and that of mutant strain were corresponding to the referred publication (Lo et al, 1997). B. RNA Isolation The RNA isolation steps followed the protocol in Chapter 7 of Molecular Cloning (Sambrook et al, 1989). spectrophotometer at 260nm.. The quantity of RNA was estimated by. The OD260 of isolated RNA of the wild type. was 0.296 and that of the double mutant was 0.598.. From 100 ml of cell. culture media, 118.4 µg of total RNA were isolated from wild-type cells, while. 239.2. µg. of. total. RNA. from. double. mutant. strain.. The. electrophoresis results were shown in Figure 2. Theoretically, the whole process of RNA isolation should be kept under the circumstance of 4°C to ensure the stability of RNA.. In this study, RNA. isolation procedure was performed five times following the same protocol. Among them, three times were at room temperature and two in cold room at 4°C.. There were different degrees of degradation in some samples, the. bottom bands are the degradation products.. Lane 3 has the strongest. degradation band, Land 2 and 6 also have these bands (Figure 2). C. Subtractive Hybridization There were 991 transformants resulted from subtractive hybridization 22.
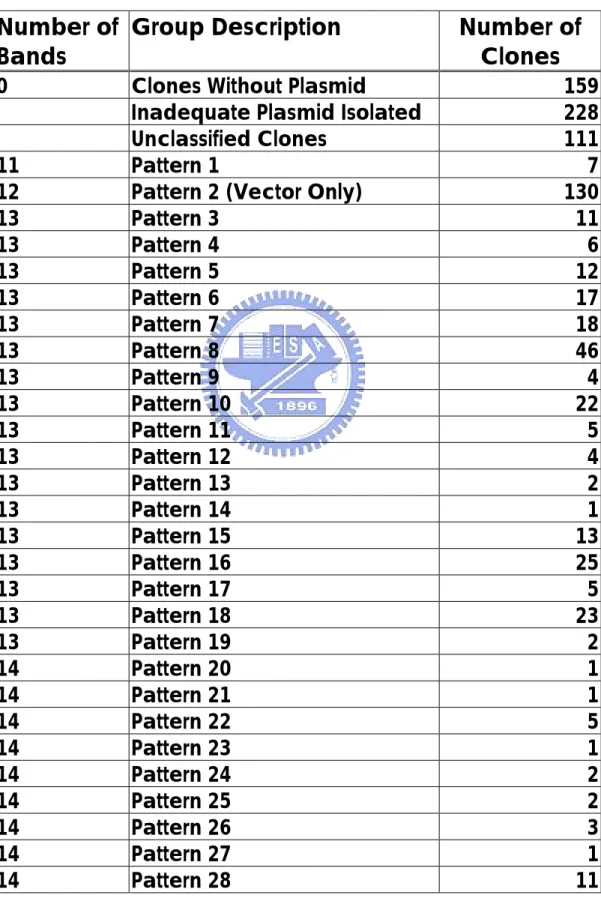
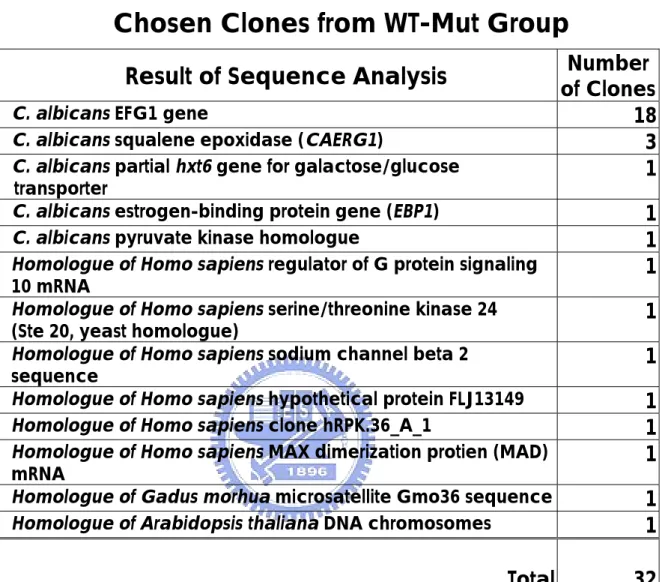
(30) using wild type as testers and mutant strain as drivers (WT-Mut group). On the other hand, 340 clones were isolated when mutant RNA was used as testers and wild-type RNA as drivers (Mut-WT group). C. albicans cph1/cph1 efg1/efg1 double mutant is unable to form filaments, and it is avirulent in a mouse model (Lo et al, 1997).. Genes. regulated by CPH1 and EFG1 genes are related to morphology and the pathogenic ability, and each gene represents a separate pathway of control (Braun et al, 2000).. By comparing the gene expression in wild. type and mutant cells, genes from both pathways were found.. Further. studies will be necessary to determine which pathway, morphology,or virulence, these genes are involved in.. Of course, some genes may be. involved in both. Among the 32 randomly picked and sequenced clones, 18 of them were different restriction DNA fragment of the EFG1 gene and 3 of them CAERG1 gene (Table3). Genes in the WT-Mut group are expressed only in wild type C. albicans or expressed at higher level in wild-type than in the mutant strain. Virulence genes could be in this group.. The discovery of EFG1 gene in. this group demonstrated that subtractive hybridization can actually single out the specifically expressed genes in WT-Mut group.. Since EFG1 gene. was included in the WT-Mut group as expected, this technique has been proven reliable in this case.. According to the results of the restriction. mapping patterns of Hae III digested DNA fragments in WT-Mut group, there were 130 clones carrying no insert and only the vector.. The. efficiency was 86.9%. Avirulence genes are supposed to be found in the Mut-WT group, which means the genes were expressed at a higher level in a mutant cell than in a wild-type cell or expressed only in the mutant strain. has been found in the Mut-WT group.. So far, TUP1 gene. TUP1 was reported to repress. genes responsible for the filamentous growth in C. albicans (Braun et al, 2000).. Many genes with unknown function were found in this group,. 23.
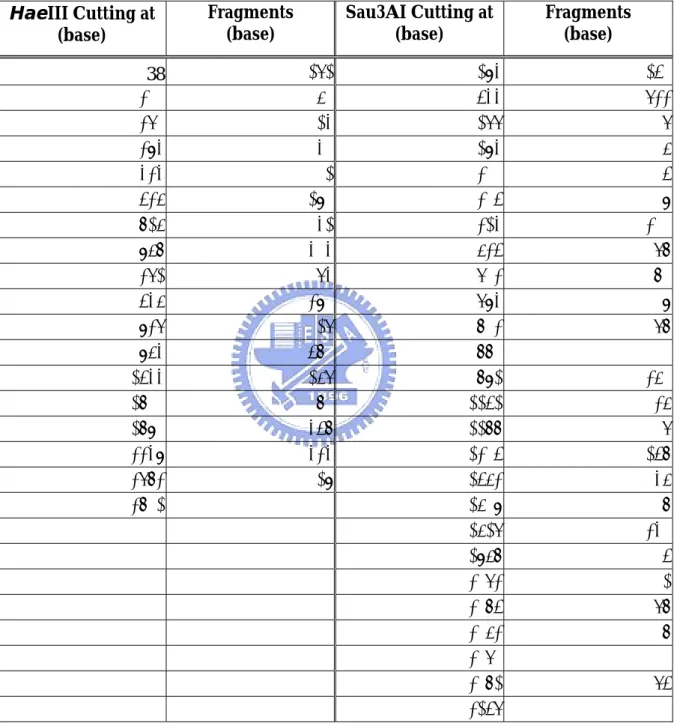
(31) which could be potential therapeutic targets.. There were 66 clones. carrying vector only in the Mut-WT group digested by Sau3AI and 29 in the group digested by HaeIII.. The efficiency was 80.6% and 91.5%.. D. Restriction Mapping There are four groups of results, cDNA clones from WT-Mut digested by HaeIII, WT-Mut digested by Sau3AI, Mut-WT digested by HaeIII and Mut-WT digested by Sau3AI. Many lanes on the polyacrylamide gel showed no DNA at all. some reasons of the absence of the DNA fragments. contaminant was mistakenly identified as a clone.. There are First, the. Second, the plasmid. was lost in the process of isolation. Some lanes on the gel were too weak to be identified. Quantifying the plasmid before incubation with the restriction enzyme helps to determine how much plasmid to be added and would have a better result for classification.. The quantity of the restriction enzyme added to each well. should also depend on the quantity of the plasmid extracted.. If the. quantification has been done in this research, the results of restriction mapping and classification may have been improved. The clones with vector only (no insert) should have been eliminated by using agarose gel to identify whether the clone carries an insert or not. After elimination of the empty clones, inadequate plasmid ones, vector only ones and unclassified ones, the remaining 363 clones in the WT-Mut digested by HaeIII group were classified into 61 different patterns (Table 2).. In the Mut-WT group, the restriction mapping results were classified. into 47 different patterns (Table 5). The pCRII-TOPO vector used for cloning the cDNA fragments was digested into 17 fragments by HaeIII and 25 fragments by Sau3AI. cutting positions and lengths of fragments were shown in Table 4.. 24. The.
(32) E. Sequence Analysis Figure 3 shows different EFG1 fragments discovered in this study and how they matched to the EFG1 sequence.. The repeated presence of. EFG1 DNA fragments may imply the abundance of its mRNA in wild type C. albicans. On the other hand, there was not CPH1 gene found which may stand for the expression level of CPH1 was much lower than that of EFG1 in wild type. In addition to EFG1 and CAERG1, there were C. albicans partial hxt6 gene for galactose/glucose transporter, C. albicans estrogen-binding protein gene (EBP1), and pyruvate kinase homologue.. Other included. homologues of Homo sapiens regulator of G protein signaling 10 mRNA, Homo sapiens serine/threonine kinase 24 (Ste 20, yeast homologue), Gadus morhua microsatellite Gmo36 sequence, Homo sapiens sodium channel beta 2 sequence, Arabidopsis thaliana DNA chromosomes, Homo sapiens hypothetical protein FLJ13149, Homo sapiens clone hRPK.36_A_1, and Homo sapiens MAX dimerization protien (MAD) mRNA (Table 3). These may contribute to the regulation of the virulence.. 25.
(33) Reference 1. Diatchenko, L., S. Lukyanov, Y. F. Lau, P. D. Siebert. “Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes.” Methods Enzymol, 303, pp.349-80, 1999.. 2. Diatchenko, L., Y. F. C. Lau, A. P. Campbell, A. Chenchik, F. Moqadam, B. Huang, S. Lukyanov, K. Lukyanov, N. Gurskaya, E. D. Sverdlov, P. D. Siebert. “Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries.” Proc Natl Acad Sci USA, 93, Issue 12, pp.6025-6030, Jun 1996.. 3. Hedrick, S. M., D. I. Cohen, E. L. Nielsen, and M. M. Davis. “Isolation of cDNA clones encoding T cell-specific membrane-associated proteins” Nature 308, pp.149-153. Mar 1984. 4. Hara, E., T. Kato, S. Nakada, S. Sekiya, and K. Oda. “Subtractive cDNA cloning using oligo(dT)30-latex and PCR: isolation of cDNA clones specific to undifferentiated human embryonal carcinoma cells.” Nucleic Acids Res 19, pp. 7097-104. Dec 1991.. 5. Duguid, J. R., and M. C. Dinauer. “Library subtraction of in vitro cDNA libraries to identify differentially expressed genes in scrapie infection.” Nucleic Acids Res 18, pp. 2789-92. May 1990.. 6. Sargent, T. D., and I. B. Dawid. “Differential gene expression in the gastrula of Xenopus laevis.” Science 222, pp.135-9. Oct 1983.. 7. Davis, M. M.. “Cell-type-specific cDNA probes and the murine I region: 26.
(34) the localization and orientation of Ad alpha.” Proc Natl Acad Sci USA, 81, pp.2194-8, April 1984.. 8. Lisitsyn, N., N. Lisitsyn, and M. Wigler. “Cloning the differences between two complex genomes.“ Science, 259, pp. 946-51, Feb 1993.. 9. Hubank, M., and D. G. Schatz. “Identifying differences in mRNA expression by representational difference analysis of cDNA.” Nucleic Acids Res, 22, pp. 5640-8, Dec 1994.. 10. Liang, P., and A. B. Pardee. “Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction.” Science, 257, pp. 967-71, Aug 1992.. 11. Welsh, J., K. Chada, S. S. Dalal, R. Cheng, D. Ralph, and M. McClelland. “Arbitrarily primed PCR fingerprinting of RNA.” Nucleic Acids Res, 20, pp. 4965-70, Oct 1992.. 12. Bauer, D., et al., PCR Methods and Applications, Supplement, pp. S97-S108, Cold Spring Harbor Lab. Press, Plainview, New York, 1994.. 13. Sompayrac, L., S. Jane, T. C. Burn, D. G. Tenen, K. J. Danna. “Overcoming limitations of the mRNA differential display technique.” Nucleic Acids Res, 23, pp. 4738-9, Nov 1995.. 14. Bertioli, D. J., U. H. Schlichter, M. J. Adams, P. R. Burrows, H. H. Steinbiss, J. F. Antoniw. “An analysis of differential display shows a 27.
(35) strong bias towards high copy number mRNAs.” Nucleic Acids Res, 23, pp. 4520-3, Nov 1995. 15. Lukyanov, K. A., G. A. Launer, V. S. Tarabykin, A. G. Zaraisky, S. A. Lukyanov. “Inverted terminal repeats permit the average length of amplified DNA fragments to be regulated during preparation of cDNA libraries by polymerase chain reaction.” Anal Biochem, 229, pp. 198-202, Aug 1995.. 16. Chenchik, A., L. Diachenko, F. Moqadam, V. Tarabykin, S. Lukyanov, P. D. Siebert. “Full-length cDNA cloning and determination of mRNA 5' and 3' ends by amplification of adaptor-ligated cDNA.” Biotechniques, 21, pp. 526-34, Sep 1996.. 17. Siebert P. D., A. Chenchik, D. E. Kellogg, K. A. Lukyanov, S. A. Lukyanov. “An improved PCR method for walking in uncloned genomic DNA” Nucleic Acids Res, 23, pp. 1087-8, Mar 1995.. 18. Chenchik, A., et al, P. Krieg ed., In A Laboratory Guide to RNA: Isolation, Analysis, and Synthesis, pp. 273-321, Wiley, New York, 1996.. 19. Sheibani, N., and W. A. Frazier. “Miniprep DNA Isolation for Automated Sequencing of Multiple Samples” Ana Biochem, 250, pp. 117-119, July 1997. 20. Sambrook, J. et al., Molecular Cloning, Second Edition, pp. 5.3-5.32, Cold Spring Harbor Laboratory Press, 1989.. 28.
(36) 21. Sambrook, J. et al, Molecular Cloning, Second Edition, pp. 6.16-6.17, Cold Spring Harbor Laboratory Press, 1989.. 22. Cutler, J. E.. “Putative virulence factors of Candida albicans” Annu Rev Microbiol, 45, pp. 187-218, 1991.. 23. Ganesan, K., A. Banerjee, A. Datta. “Molecular cloning of the secretory acid. proteinase. gene. from. Candida. albicans. and. its. use. as. a. species-specific probe” Infect Immun, 59, pp. 2972-7, Sep 1991. 24. Gillum, A. M., E. Y. Tsay, D. R. Kirsch. “Isolation of the Candida albicans. gene. for. orotidine-5'-phosphate. decarboxylase. by. complementation of S. cerevisiae ura3 and E. coli pyrF mutations” Mol Gen Genet, 198, pp. 179-82, 1984. 25. Lo, H. J., J. R. Kohler, B. DiDomenico, D. Loebenberg, A. Cacciapuoti, G. R. Fink. “Nonfilamentous C. albicans mutants are avirulent” Cell, 90, pp. 939-49, Sep 1997.. 26. Braun, B. R., and A.D. Johnson. “TUP1, CPH1 and EFG1 make independent contributions to filamentation in Candida albicans.” Genetics, 155, pp. 57-67, May 2000.. 27. Tsarfaty, I., H. Sandovsky-Losica, L. Mittelman, I. Berdicevsky, E. Segal. “Cellular actin is affected by interaction with Candida albicans.” 29.
(37) FEMS Microbiology Letters, 189, pp. 225-232, 2000.. 28. MaCullough, M. J., B. C. Ross, P. C. Reade. “Candida albicans: a reviw of its history, taxonomy, epidemiology, virulence attributes, and methods of strain differentiation.” Int J Oral Maxillofac Surg 25, pp.136-144, 1996. 29. Asakura, K., S. Iwaguchi, M. Momma, T. Sukai, K. Higashide, K. Tanakak. “Electrophoretic karyotypes of clinically isolated yeast of Candida albicans and C. glabrata.” J Gen Microbiol 137, pp.2531-8, 1991. 30. Yang, Y. L.. “Virulence factors of Candida species.” J. Microbiol Immunol Infect, 36, pp. 223-228, 2003. 31. Ghannoum, M, K. Asu-Elteen. “Correlative relationship between proteinase production, adherence and pathogenicity of various strains of Candida albicans.” J Med Vet Mycol, 24, pp.407-13, 1986. 32. Ghezzi, M. C., M. Trancassini, P. Cipriant, C. Mancini, M. I. Brenciaglia. “Comparison between adherence of C. albicans and Candida spp. to human epithelial cells.” Boll Ist Sieroter Milan, 65, pp.436-9, 1986. 33. Liu, H., J. Köhler, G. R. Fink. “Suppression of hyphal formation in Candida albicans by mutation of a STE12 homolog.” Science, 266, pp.1723-6.1994 34. Kron, S. J., N. A. Gow. “Budding yeast morphogenesis: signalling, cytoskeleton and cell cycle.” Curr Opin Cell Biol, 7, pp.845-55, 1995. 35. Hazen, K. C.. “Participation of yeast cell surface hydrophobicity in adherence of Candida albicans to human epithelial cells.” Infect Immun, 57, pp.1894-1900, 1989. 30.
(38) 36. Leberer, E., K. Ziegelbauer,A. Schmidt,D. Harcus,D. Dignard,J. Ash,L. Johnson,D. Y. Thomas “Virulence and hyphal formation of Candida albicans require the Ste20p-like protein kinase CaCla4p.” Curr Biol, 8, pp.539-46, 1997. 37. Phan, O. T., P. H. Belanger, S. G. Filler. “Role of hyphal formation in interactions of Candida albicans with endothelial cells.” Infect Immun, 68, pp.3485-90, 2000. 38. Antley, P. P., K. C. Hazen. “Role of yeast cell growth temperature on Candida albicans virulence in mice. Infect Immun, 11, pp.2884-90, 1988. 39. Hazen, B. W., K. C. Hazen. “Dynamic expression of cell surface hydrophobicity during initial yeast cell growth and before germ tube formation of Candida albicans.” Infect Immun, 9, pp.2521-5, 1988.. 40. Hazen, K. C., J. G. Lay, B. W. Hazen, R. C. Fu, S. Murthy. “Partial biochemical. characterization. of. cell. surface. hydrophobicity. and. hydrophilicity of Candida albicans.” Infect Immun, 11, pp.3469-76, 1991. 41. Calderone, R. A., P. C. Brawn. “Adherence and receptor relationships of Candida albicans.” Microbiol Rev, 55, pp.1-20, 1991. 42. Filler, S. G., A. S. Pfunder, B. J. Spellberg, J. P. Spellberg, J. E. Edwards, JR. “Candida albicans stimulates cytokine production and leukocyte adhesion molecule expression by endothelial cells.” Infect Immun 64: 2609-17, 1996. 43. Gale, C. A., C. M. Bendel, M. McClellan, M. Hauser, J. M. Becker, J. Berman, M. K. Hostetter. “Linkage of adhesion, filamentous growth, and. 31.
(39) virulence in Candida albicans to a single gene, INT1.” Science, 279, pp.1355-8, 1998. 44. Macdonald, F., F. C. Odds. “Inducible proteinase of Candida albicans in diagnostic serology and in the pathogenesis of systemic candidosis.” J Med Microbiol, 129, pp.431-8, 1980.. 45. Portillo, F., C. Gancedo. “Purification and properties of three intracellular proteinases from Candida albicans. Biochim Biophys Acta, 881, pp.229-35, 1986. 46. Remold, H., H. Fasold, F. Staib. “Purification and characterization of a proteolytic enzyme from Candida albicans.” Biochim Biophys Acta, 167, pp.399-406, 1968. 47. Ruchel, R.. “Properties of a purified proteinase from the yeast Candida albicans.” Biochim Biophys Acta. 659, pp.99-113, 1981. 48. De Bernardis, F., S. Arancia, L. Morelli,B. Hube,D. Sanglard,W. Schafer,A. Cassone. “Evidence that members of the secretory aspartyl proteinase gene family, in particular SAP2, are virulence factors for Candida vaginitis.” J Infect Dis, 179, pp.201-8, 1999. 49. Kerridge, D., R. O. Nicholas. “Drug resistance in the opportunistic pathogens. Candida. albicans. and. Candida. glabrata.”. J. Antimicrob. Chemother, 18, Suppl B, pp.39-49, 1986. 50. Ernst, J. F., 2000 “Transcription factors in Candida albicans – environmental control of morphogenesis” Microbiology (2000), 146, 1763–1774 51. Clontech “Clontech PCR-Select cDNA Subtraction Kit User Manual”. 32.
(40) Table 1. OD600 of Cell Culture Strain OD600 Initiation. 4 hour-Incubation. Wild type. HLC54. (CPH1/CPH1 EFG1/EFG1). (cph1/cph1 efg1/efg1). 0.086. 0.052. 0.038-0.068. 0.410. 33.
(41) Table 2. Groups of Restriction Mapping Pattern In WT-Mut Group Digested by Hae III Number of Group Description Bands 0. 11 12 13 13 13 13 13 13 13 13 13 13 13 13 13 13 13 13 13 14 14 14 14 14 14 14 14 14. Clones Without Plasmid Inadequate Plasmid Isolated Unclassified Clones Pattern 1 Pattern 2 (Vector Only) Pattern 3 Pattern 4 Pattern 5 Pattern 6 Pattern 7 Pattern 8 Pattern 9 Pattern 10 Pattern 11 Pattern 12 Pattern 13 Pattern 14 Pattern 15 Pattern 16 Pattern 17 Pattern 18 Pattern 19 Pattern 20 Pattern 21 Pattern 22 Pattern 23 Pattern 24 Pattern 25 Pattern 26 Pattern 27 Pattern 28 34. Number of Clones 159 228 111 7 130 11 6 12 17 18 46 4 22 5 4 2 1 13 25 5 23 2 1 1 5 1 2 2 3 1 11.
(42) 14 14 14 14 14 14 14 14 14 14 14 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 16 16 16 17 17 17. Pattern 29 Pattern 30 Pattern 31 Pattern 32 Pattern 33 Pattern 34 Pattern 35 Pattern 36 Pattern 37 Pattern 38 Pattern 39 Pattern 40 Pattern 41 Pattern 42 Pattern 43 Pattern 44 Pattern 45 Pattern 46 Pattern 47 Pattern 48 Pattern 49 Pattern 50 Pattern 51 Pattern 52 Pattern 53 Pattern 54 Pattern 55 Pattern 56 Pattern 57 Pattern 58 Pattern 59 Pattern 60 Pattern 61 Total. 12 9 1 5 2 2 5 6 1 1 12 25 1 1 3 2 1 1 1 1 1 2 3 1 2 1 3 2 1 2 1 1 1 991. 35.
(43) Table 3. DNA Sequencing Results of Randomly Chosen Clones from WT-Mut Group Result of Sequence Analysis C. albicans EFG1 gene C. albicans squalene epoxidase (CAERG1) C. albicans partial hxt6 gene for galactose/glucose transporter C. albicans estrogen-binding protein gene (EBP1) C. albicans pyruvate kinase homologue Homologue of Homo sapiens regulator of G protein signaling 10 mRNA Homologue of Homo sapiens serine/threonine kinase 24 (Ste 20, yeast homologue) Homologue of Homo sapiens sodium channel beta 2 sequence Homologue of Homo sapiens hypothetical protein FLJ13149 Homologue of Homo sapiens clone hRPK.36_A_1 Homologue of Homo sapiens MAX dimerization protien (MAD) mRNA Homologue of Gadus morhua microsatellite Gmo36 sequence Homologue of Arabidopsis thaliana DNA chromosomes. Total. 36. Number of Clones 18 3 1 1 1 1 1 1 1 1 1 1 1 32.
(44) Table 4. pCRII-TOPO Vector Fragments Digested by HaeIII and Sau3AI HaeIII Cutting at (base) 38 310 370 394 434 536 826 968 1372 1546 1937 1964 2544 2811 2891 3349 3783 3812. Fragments (base). Sau3AI Cutting at (base) 272 60 24 40 102 290 142 404 174 391 27 580 267 80 458 434 29. 37. 294 544 1277 1294 1300 1305 1324 1635 1713 1794 1803 1881 1892 2252 2288 2305 2563 2609 2627 2968 3073 3085 3163 3171 3182 3257. Fragments (base) 250 733 17 6 5 19 311 78 81 9 78 11 360 36 17 258 46 18 341 105 12 78 8 11 75.
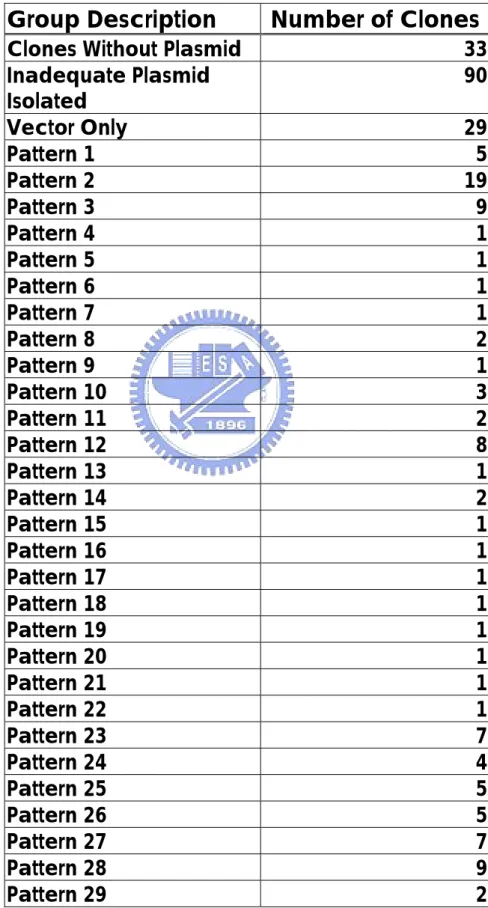
(45) Table 5. Groups of Restriction Mapping Pattern In Mut-WT Group Digested by Hae III Group Description. Number of Clones. Clones Without Plasmid Inadequate Plasmid Isolated Vector Only Pattern 1 Pattern 2 Pattern 3 Pattern 4 Pattern 5 Pattern 6 Pattern 7 Pattern 8 Pattern 9 Pattern 10 Pattern 11 Pattern 12 Pattern 13 Pattern 14 Pattern 15 Pattern 16 Pattern 17 Pattern 18 Pattern 19 Pattern 20 Pattern 21 Pattern 22 Pattern 23 Pattern 24 Pattern 25 Pattern 26 Pattern 27 Pattern 28 Pattern 29 38. 33 90 29 5 19 9 1 1 1 1 2 1 3 2 8 1 2 1 1 1 1 1 1 1 1 7 4 5 5 7 9 2.
(46) Pattern 30 Pattern 31 Pattern 32 Pattern 33 Pattern 34 Pattern 35 Pattern 36 Pattern 37 Pattern 38 Pattern 39 Pattern 40 Pattern 41 Pattern 42 Pattern 43 Pattern 44 Pattern 45 Pattern 46 Pattern 47 Total. 13 6 8 3 34 4 4 2 1 1 2 1 1 1 1 1 1 1 340. 39.
(47) Wild type cell. Double mutant cph1/cph1. CPH1/CPH1 EFG1/EFG1. efg1/efg1. Figure 1. Wild Type and cph1/cph1 efg1/efg1 Candida albicans grown in the media. 40.
(48) 1. 2. 3. 4. 5. 6. Figure 2. 1% Agarose Gel of RNA Isolated from Wild Type and Double Mutant Strains Lane 1: 1 kb PLUS DNA Ladder (Gibco BRL), 1µg; Lane 2: Wild Type Strain, Dec. 2 1999, RNA concentration undetermined; Lane 3: Double Mutant Strain, Dec. 2 1999, RNA concentration undetermined; Lane 4: The Same as Lane 2 Lane 5: Wild Type Strain, Nov. 19 1999, RNA 2 µg (sample used in this study); Lane 6: Double Mutant Strain, Nov. 19 1999, RNA 2 µg (sample used in this study).. 41.
(49) 1 agatct//tttggg gggaggaaat tttaacttta agtttttgcc tactggaagc. 25:. 3860-4018. 3901 tatatatttg atttagtgta ttacatccag ccaacccact taacttacaa ttgaagagac. A8 3859-4019 3961 aagcaaaaaa cgaccaaatt atacaaaatt taagaagcca gaatattaaa gaaatagaat. 4021 tttatttttt tgtttttgtt ttttcgcttt ttttttggtt aacccctttg tgtcccttgc 4081 atacttttac attggaaaca tacatacact aacattcaca ctcaatacac tcatattatt. 4141 taccattttt gttgtgaaga tacacgtatt tattgagtat tccttcataa catttaattt. 25:. 4057-4328. A8 4094-4322. 4201 atattccaag agttaattga ttaaacaact tggtccaaga attcattacc aggcgtgttt. 4261 tattaaattc ctttttttaa ttagcctttt ttgcctccca cattagttgc tcaggtcacg. 4321 ttatttaata tatttctttt ttttttctta cttgctgaaa aaaaaaaaaa aaaacaacca. 25:. 4377-4550 4611-4671. 4381 accaaccctt aacccattaa cgaattaaga tttgttctat ttgactacca agaatataac. 4728-4869. 4441 ccatattaat gtcaacgtat tctataccct attacaatca aatgaacgga aattacaata 4501 acggtatgcc ccagcaaaca actgcagcca atcaacaggc ttttcctcag caacaacaac 4561 caacaacaac aggcaatgct agccaacagc aacagcaagc agctgctacg gcagctgcag 4621 tccaacagcc ttataactac atgttctatc aacaacaagg acaaccaggt caacagactg 4681 gacagacagc aggacaacaa caacaacagc agcagcagca gcaacaatat gattacaata 4741 cctacaacag atatcagtat cctgccgcaa catctcaagg aaactattac cagcaaacaa. 19:. 4771-4949. 4801 ttcctaatca attgtcacag ccacaacctc agcattacaa tggatctaat cgtaattaca. 4861 caagtgctcc tagtggtgcc cccatacctt ccaattctac cagtggacct tcacaacagc. 4921 caccactacc aggtcaacaa gcagtaccta tcccaccaca tgtatcgaca atgcaacaac. 40 & 50: 4951-5463. 41: 4944-5625 4981 caactcctgt tcaggatacg ttgaacgcct cgagcacttc cactgtgggg caattccaac. 181: 5012-5556 42.
(50) 5041 caccaggaat cagaccacga gtaacaacta ccatgtggga agatgaaaaa actttgtgct. 5:. 4991-5464. 76: 5071-5464 5101 atcaagttga tgccaataat gtgtcggttg tcagaagagc agataataat atgatcaacg. 464 & 526: 4998-5464. 5161 gaaccaaatt gctcaatgtg gcccaaatga cacgtggtag aagagatggg attttgaaat. 3: 5113-5464. 5221 cagaaaaggt gagacacgtt gtgaaaatcg gatcaatgca tttgaaagga gtctggattc. 55 & 47: 4944-546. 5281 catttgaaag agcattggcc atggctcaac gtgaacaaat tgtggatatg ttgtatcctt. 5341 tgtttgtcag agatattaaa cgagtgattc aaaccggagt aactcctaat gcagctgctg. 55: 5401 caacggccgc cgccgctgcc actgccactt ctgcttcggc tcctccacct ccacctccac. 43. 5497-5625.
(51) 5461 ccgttgctgc tgctactact actgctgcta ctgctatttc caaaagttct agcggtaatg. 5:. 5497-5543. 3 & 76: 5521 ggaacagtat atctgctacc agtggtggca gtaatgtgtc tggtgcttct ggtgcaggtt. 5497-5625. 464: 5581 ccaccactag tccggtaaat accaaggctg ccactgctgc tggtatccct caaggtaatt. 5641 attatcaaac ttacaaccaa cagcagtatc ctcaacagta tggtcagtat aatgctcctg. 5497-5625. 17: 5622-577. 5701 gtaagaacca aaatacacct gcatcacaac caggttctac aaccaatgat caatatttac. 5761 aacaacaaca gcaacaacaa caacaaatgt atgggtatca actgaattat taccagggtg. 5821 gtgctgctaa tagtagttac tatccaaatt attatcaaca acaacaacca aattatgcat. 45: 5874-5900. 5881 catcatatcc ataccaacag caacaacaaa agcaacaaca acaacaacca aatcaacaac. 5963-6125 5941 aacagtcaga tcaacaacaa acttctacac caagtggtgg tgcaggaact agatctgtgc. 17: 5963-612 6001 accaatcacc ccaagttcag tcattgactc aaggttcagt tcacccttca ccccaacaac. 45: 5963-6125 6061 atcaagctaa tcaatcagct agcactgttg ccaaagaaga aaagtaataa atatcattcg. 44.
(52) 6121 tgtacatcac cttctgcttt ctgccataaa ttccaaatta gattatagca tatatttcat. 46: 6122-6352 6181 taagaaatga ttggaaagtt tccttcattc aaaattagag agaaaataat aaaaaaagaa. 6392-6417. 6241 atagtataga tcataatctt ctcccaaatc cattgacatg aacgaattta tactatttgg. 6301 gacaaattta aattggattt attattttta ttttctttca gttctatcat agaaacaaac. 6361 ataaacataa acaaaaacgt tttctttttt tgcataatat ctatctatgt atatgtatat. 45: 6405-6424. 6421 atatgtgtgt aagtcattgt cttttccatt ttcttttcca ttttcttttt tttttagttt. 46: 6481 tgttttcaag tgtgtaataa taataatatt aattgta//acccaagaat tc. 6490-6512. Figure 3. Different DNA Fragments of Candida albicans EFG1 Gene Example: 46:. 6490-6512. 46:the number of the clone. 6490 and 6512 mean the base position in the sequence of C. albicans EFG1. After sequencing and alignment, the sequences of colne 46 match those of EFG1 base 6490-6512 and the color of the frame corresponds to that of the undelines.. 45.
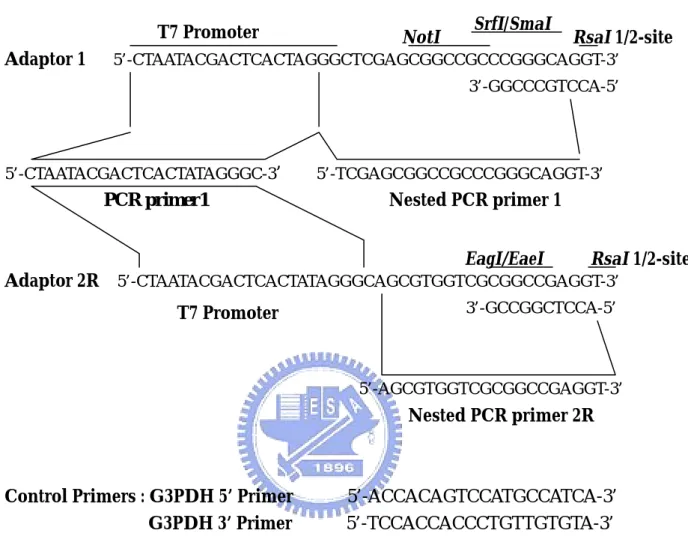
(53) cDNA synthesis primer. RsaI. Hind Ⅲ. 5’-TTTTGTACAAGCTT30N1N-3’. T7 Promoter Adaptor 1. NotI. SrfI/SmaI. RsaI 1/2-site. 5’-CTAATACGACTCACTAGGGCTCGAGCGGCCGCCCGGGCAGGT-3’ 3’-GGCCCGTCCA-5’. 5’-CTAATACGACTCACTATAGGGC-3’. 5’-TCGAGCGGCCGCCCGGGCAGGT-3’. Nested PCR primer 1. PCR primer1. EagI/EaeI. RsaI 1/2-site. Adaptor 2R 5’-CTAATACGACTCACTATAGGGCAGCGTGGTCGCGGCCGAGGT-3’ 3’-GCCGGCTCCA-5’. T7 Promoter. 5’-AGCGTGGTCGCGGCCGAGGT-3’. Nested PCR primer 2R. Control Primers : G3PDH 5’ Primer G3PDH 3’ Primer. 5’-ACCACAGTCCATGCCATCA-3’ 5’-TCCACCACCCTGTTGTGTA-3’. Figure 4. Sequences of the PCR-Select cDNA Synthesis Primer, Adaptors, and PCR Primers When the adaptors are ligaed to Rsa-digested cDNA, the site is restored. Adaptor 2R was reformulated in December 1996. Before reformulation, this adaptor was called Adaptor 2; the sequence of the old Adaptor 2 was different. However, please note that in some lots of the PCR-Select Kit, the reformulated Adaptor 2R was still called Adaptor 2. If you received version PR6Y996 of the Use Manual with your PCR-Select Kit, your Adaptor 2 has the same sequence as Adaptor 2R shown here. (From : CLONTECH PCR-Select cDNA Subtraction Kit Use Manual). 46. TM.
(54) ®. Figure 5. Map of pCR II-TOPO®. 47.
(55)
數據
+6


相關文件
目前全世界生產結構存在全球供應鍊的整合體系,在 這種環境下,土國相信保護主義的形成亦對實施保護
Krajcik, Czerniak, & Berger (1999) 大力倡導以「專題」為基礎,教導學生學習科 學探究的方法,這種稱之為專題導向的科學學習(Project-Based Science,
彼所依體由二種因增上力故。從自種子即於是處中有異熟無間得生。死生同時如秤兩
下手傳球是接發球, 後排防守, 保護的主要技術. 正面下手傳球是最 基本的傳球方法, 是各種傳球技術的基礎, 適合於接各種發球, 扣球 等.. ──
黑木耳 (Auricularia polytricha) 是台灣普遍的食用 菌之一,為一種低熱量的食品,亦是一種富含食用纖維及
1.補充影片:BBC《 Wild New World》 (中 文 名:BBC 野性新世界) ---介紹冰河時期的各式物種及環境
烏克蘭的車諾比於 1986 年發生核能災變,讓原本繁榮的小鎮瞬間成為人煙罕至的 鬼城。因為核能災變產生的輻射持續汙染環境,政府曾經警告該地在未來
9 大老虎亞種之中,峇裏虎、爪哇虎及波斯虎早已絕種,另一種野生華南虎亦瀕臨絕
