袁国亮 李爽 任申强 刘俊明
Excited charge-transfer organics with multiferroicity
Yuan Guo-Liang Li Shuang Ren Shen-Qiang Liu Jun-Ming
引用信息 Citation:
Acta Physica Sinica
, 67, 157509 (2018)
DOI: 10.7498/aps.67.20180759
在线阅读 View online:
http://dx.doi.org/10.7498/aps.67.20180759
当期内容 View table of contents:
http://wulixb.iphy.ac.cn/CN/Y2018/V67/I15
您可能感兴趣的其他文章
Articles you may be interested in
四方相多铁 BiMnO
3电控磁性的理论研究
Theoretical study on magnetoelectric effect in multiferroic tetragonal BiMnO
3物理学报.2018, 67(15): 157511
http://dx.doi.org/10.7498/aps.67.20180946
基于磁电耦合效应的基本电路元件和非易失性存储器
Fundamental circuit element and nonvolatile memory based on magnetoelectric effect
物理学报.2018, 67(12): 127501
http://dx.doi.org/10.7498/aps.67.127501
引入界面耦合系数的长片型磁电层状复合材料的等效电路模型
Equivalent circuit model for plate-type magnetoelectric laminate composite considering an interface
cou-pling factor
物理学报.2018, 67(2): 027501
http://dx.doi.org/10.7498/aps.67.20172080
基于能量转换原理的磁电层合材料低频磁电响应分析
Low frequency magnetoelectric response analysis of magnetoelectric laminate material based on energy
conversion principle
物理学报.2014, 63(20): 207501
http://dx.doi.org/10.7498/aps.63.207501
多铁材料 HoMnO
3中光学吸收和畸变驱动的第一性原理研究
Research on optical absorption and distortion driving in multiferroic HoMnO
3from the first principles
物理学报.2013, 62(12): 127502
http://dx.doi.org/10.7498/aps.62.127502
多铁性: 物理、材料及器件专题
激发态电荷转移有机体的多铁性研究
∗
袁国亮
1)
†
李爽
1)
任申强
2)
刘俊明
3)
‡
1) (南京理工大学材料科学与工程学院, 南京 210094) 2) (美国纽约州立大学布法罗校区机械和航天工程系, 美国纽约布法罗 14260) 3) (南京大学物理系, 南京 210046) ( 2018 年 4 月 20 日收到; 2018 年 6 月 13 日收到修改稿 )随着人们对多铁性的深入了解, 越来越多不同类型的有机多铁材料被合成出来. 激发态电荷转移有机体
的电荷转移网络是由一个提供电子的分子 (给体 donor, D
+) 和一个接受电子的分子 (受体 acceptor, A
−) 有
序排列后构成的. D
+A
−长程有序排列, 其激发态 (激子) 具有较长寿命和
±1/2 自旋, 这是产生室温铁电性和
铁磁性的根本原因. 激发态容易受外场刺激, 因此光照、磁场、电场、应力等能够很好地调控这类材料的铁电极
化、磁矩和相应的磁电耦合系数. 激发态电荷转移有机体不仅大大丰富了室温多铁材料体系, 而且可以为开发
新型多功能电子器件提供材料基础和技术储备.
关键词: 有机多铁, 磁电耦合, 电荷转移
PACS: 75.85.+t, 77.84.–s, 72.20.Jv
DOI: 10.7498/aps.67.20180759
1 引
言
多铁材料是指同时具备两种或者两种以上铁
性特征, 并且其间存在耦合的材料. 经典意义上的
多铁效应是指铁电、铁磁/反铁磁、铁弹性或铁涡
性之间的作用关系
[1]. 1894 年, Pierre Curie 首先
预言了低对称性下的特殊材料具备磁电耦合效应,
即外磁场调制材料的铁电性质或者外电场调制材
料的磁性. 这一效应随后在 Cr
2O
3中得到证实
[2,3].
近年来多铁材料因其在新功能磁电器件上的潜在
应用而受到越来越多的关注, 如超灵敏磁场传感
器
[4]、磁电数据存储
[5,6]、多态记忆存储
[7]. 目前而
言, 多铁材料可以分为单相多铁性材料和复合多铁
性材料, 单相多铁材料又可分为无机多铁和有机多
铁两类.
无 机 多 铁 材 料 中, 能 够 在 室 温 下 显 示 出 多
铁性的比较少, 而钙钛矿结构氧化物 BiMnO
3[8],
BiFeO
3[9]等能够在室温下同时具备自发的铁电性
和反铁磁性. BiFeO
3, PbFe
0.5Nb
0.5O
3等部分多铁
性材料的铁电性和磁性有各自独立的起源, 铁电
性源于晶格自发对称破缺, 磁性来自于离子 d 轨道
的部分填充, 它们的铁电极化和磁性往往都比较
强, 但是磁电耦合效应较弱, 这是第一类多铁性材
料
[10,11]. 此外还存在磁性引起的铁电性的多铁性
材料, 即第二类多铁性材料
[12]. 它们的磁电耦合很
强, 但是铁电性往往很弱. 近年来在 TbMnO
3[13],
GdMnO
3[14], MnWO
4[15], Ni
3V
2O
8[16]中发现了
磁电耦合效应, 从而诞生了一种新的多铁材料家
族 ——非共线多铁磁性材料. 这类材料中的磁电
效应是由自旋序同电子轨道的耦合作用导致的,
即逆 Dzyaloshinskii-Moriya 相互作用, 该作用与其
他交换作用相互竞争, 稳定了螺旋磁矩的存在, 螺
旋磁矩会引入电极化
[17−19]. 而对于共线多铁材
料, 如 HoMnO
3[11,20,21], YMnO
3[22,23]等, 自旋 -晶
格耦合作用通过超/双交换机制或对称交换收缩机
制诱发宏观铁电极化. 除了单相多铁材料外, 人
们也将目光放在了复合多铁异质结上, 如 BaTiO
3-CoFe
2O
4[24]构成的异质结薄膜结构. 将具备很强
∗ 国家自然科学基金 (批准号: 51790492, 51431006, 51472118) 和中央高校基本科研业务费专项资金 (批准号: 30916011104) 资助的课题. † 通信作者. E-mail:[email protected] ‡ 通信作者. E-mail:[email protected]铁电性的薄膜和具备很强磁性的薄膜交叠在一起,
从而引起这两者之间很强的磁电耦合效应. 一般而
言, 这种特殊结构往往只能在界面处很薄的区域引
起磁电耦合效应. 因此无论是无机单相多铁材料还
是无机复合多铁异质结, 在实际应用上仍存在诸多
障碍.
部 分 金 属 -有 机 骨 架 材 料 的 单 相 化 合 物 也
受到越来越多的关注
[25−28]. 在 [(CH
3)
2NH
2]Fe-(COOH)
3中发现了很强的磁电耦合效应
[29,30], 铁
离子和二甲基阳离子通过氢键结合在一起形成
Fe-O-C-(O
· · · HN)-Fe, 其磁性源自于两个 Fe 离子间的
远距离超 -超交换作用
[31]. 在高温下, 氢键由于很
高的内在动能而呈无序态, 当温度降到居里温度
(T
C) 以下, 氢键变得有序排列. 施加外电场时, 氢
键连接的偶极子会随着外电场排列, 引起铁电极化
翻转. 当氢键沿着同一方向排列时, Fe 离子间的远
距离超 -超交换作用会被氢键阻碍, 因此磁性能够
被外加电场调控.
无机多铁材料的发展瓶颈使得更多的学者将
目光转投于有机多铁材料. 近年来在有机电荷转移
化合物中, 人们相继发现了铁电性、铁磁性以及磁
电耦合性质
[32−34]. 电荷转移化合物是由一个提供
电子的分子 (给体 donor, D
+) 和一个接受电子的分
子 (受体 acceptor, A
−) 有序排列后构成的有机电
荷转移化合物, 因其超分子结构 D
+A
−D
+A
−· · ·
而在室温下表现出很好的铁电性. 例如由均苯四
甲酸二酰亚胺作为电子受体和嵌二萘衍生物作为
电子给体产生的电荷转移晶体在 10 kV/cm 的电场
下电极化强度可以达到 6 µC/cm
2 [32]. 而电荷转移
晶体化合物中未成对电子自旋间的交换耦合作用也
导致了室温铁磁性
[35−39]. 本文将介绍几种电荷转
移化合物的铁电性、磁性和磁电耦合性质.
2 激发态电荷转移有机体
2.1
四硫戊烯 -对溴电荷转移体的多铁性
2010 年, Kagawa 等
[40]报道了一维有机电荷
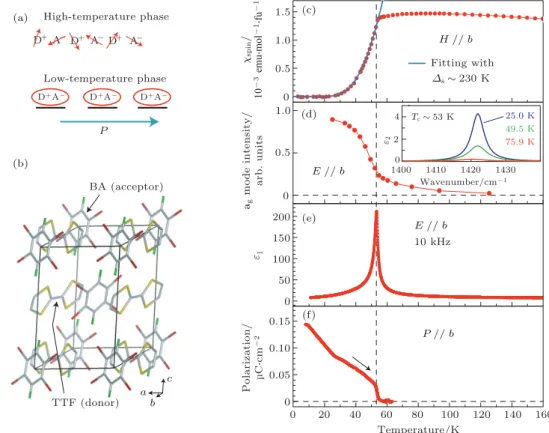
E ⊳⊳ b E ⊳⊳ b 10 kHz P ⊳⊳ b Po la ri z a ti o n / µ C Sc m -2 P High–temperature phase Low–temperature phase b c Temperature/K ag mo d e i n te n si ty/ a rb . u n it s Fitting with Wavenumber/cm-1 25.0 K 49.5 K 75.9 K H ⊳⊳ b ∆s ~230 K Tc ~53 K a ε2 4 2 0 BA (acceptor) TTF (donor) 0 1.5 0.5 1.0 0 0.5 1.0 200 100 150 0 50 0.05 0 0.15 0.10 60 80 40 20 100 120 140 160 0 ε1 χsp in / 10 -3 e m u S m ol -1S fu -1 D+ D+A- D+A- D+A -D+ D+ A- A -A -1410 1420 1430 1400 (a) (b) (c) (d) (e) (f)图 1 (a) TTF-BA 的给体 TTF (donor/D+) 和受体 BA (acceptor/A−) 分别在高温和低温下的结构示意图; 箭头表示−1/2 自 旋, 下划线表示二聚合物, 椭圆表示单重态, P 表示电极化; (b) TTF-BA 的晶格结构; (c) 不同温度下 TTF-BA 的磁化率; (d) 标准 化后的 ag模 (1422 cm−1) 光谱强度; (e) 10 kHz 下的介电常数 ε1; (f) 沿 b 轴的自发铁电极化强度[40]
Fig. 1. (a) Schematic structure of ionic donor (D+) and acceptor (A−) mixed stacks in high- and low-temperature phases of TTF-BA. The arrow, underline and ellipsoid represent spin−1/2, a dimer and a singlet state, respectively. P denotes
electric polarization. (b) Crystal structure of TTF-BA. (c) Temperature dependence of spin susceptibility. (d) Normalized spectral weight of the agmode at 1422 cm−1as a measure of local D+A−dimerization. (e) Dielectric constant at 10 kHz.
转 移 材 料 四 硫 戊 烯 -对 溴
(tetrathiafulvalene-p-bromanil, TTF-BA) 的磁性, 这是自旋引起铁电性
质并导致多铁性质的典型例子. 图
1
(a) 和图
1
(b)
是 TTF-BA 的 结 构 示 意 图, 其 中 电 荷 转 移 网 络
(donor-acceptor charge-transfer, DACT) 是由一个
给体 D
+和一个接受电子的受体 A
−有序排列后构
成的. 在 84 K 以下, TTF 分子和 BA 分子总是以
离子态形式组合成 TTF-BA 系统
[41], 离子化率约
0.95, 因此每一个 D
+A
−D
+A
−· · · 链条可以看成具
有
−1/2 自旋的一维海森伯链. 在 T
C(53 K) 温度
以下时, 自旋 -晶格耦合作用会引入电极化,
TTF-BA 同时具备自发的铁磁性和铁电性, 并表现出磁
电耦合效应, 如图
1
(c)—(f) 所示. D
+A
−D
+A
−· · ·
的激发态 (激子) 具有较长寿命和
±1/2 自旋, 这些
激子是由电子 -空穴同周围晶格耦合在一起构成的,
每一个都可看成单独的电偶极子, 这是激发态电荷
转移有机体产生铁电性和铁磁性的根本原因. 单
重态激子是由两个自旋相反的电荷组成的, 当施加
一个具备特定方向的外加磁场时, 相反于磁场方向
的电荷自旋就会被抑制, 从而限制了单重态的产
生, 更多的多重态将因为系统内部的交互作用而产
生
[42]. 在 50 和 52 K 时, 电极化强度在 56 T 的外加
磁场下几乎消失, 这是因为高磁场抑制了单重态激
子的产生, 从而使得这类给体 -受体共聚物的铁电
性消失. 因此, 电荷给体 -受体 (D
+A
−) 共聚物的多
铁性是来源于自旋 - 晶格耦合作用的不稳定性
[43].
2.2
聚 (3-己基噻吩)-苯基 C
61丁酸甲酯电
荷转移体的室温多铁性
聚 (3-己 基 噻 吩)-苯 基 C
61丁 酸 甲 酯
(poly(3-hexylthiophene)-phenyl C
61butyric acid methy
lester, P3HT-PCBM) 电 荷转移有机体在室温下具
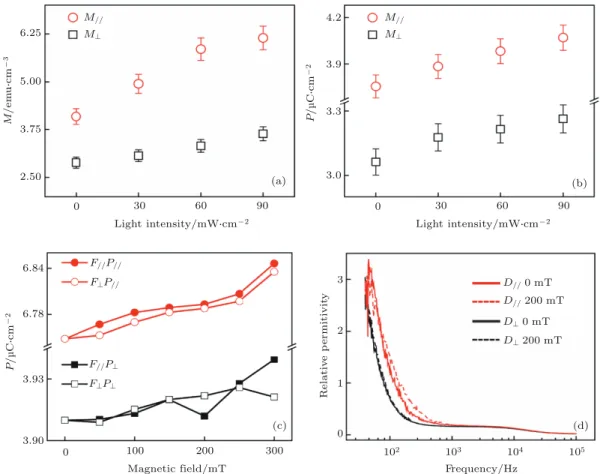
6.25 5.00 3.75 2.50 4.2 3.9 3.3 3.0 0 30 60 90 M / e m u Sc m -3 Light intensity/mWScm-2 0 30 60 90 Light intensity/mWScm-2 P / m C Sc m -2 M// Mu M// Mu (a) (b) 6.84 6.78 3.93 3.90 3 2 1 0 102 103 104 105 0 100 200 300Magnetic field/mT Frequency/Hz
R e la ti v e p e rmi ti v it y P / m C Sc m -2 F//P// FuP// F//Pu FuPu D// mT D// mT Du mT Du mT (c) (d)
图 2 (a) 室温下定向 P3HT0.75:PCBM0.25纤维薄膜在不同强度光照下的饱和磁化强度 (1 emu/cm3= 103 A/m); (b) 室温下定向
P3HT0.75:PCBM0.25纤维薄膜在不同强度光照下的饱和铁电极化强度; (c) 施加平行于纳米线 (F//) 和垂直于纳米线 (F⊥) 的磁场时, 饱和铁电极化 P//和 P⊥随磁场的变化; (d) 不同方向的相对介电常数在外加磁场下的变化[45]
Fig. 2. (a) Illumination-dependent saturation magnetization of anisotropic P3HT0.75:PCBM0.25complex at room temperature
(1 emu/cm3 = 103 A/m); (b) illumination-dependent saturation polarization of anisotropic P3HT
0.75:PCBM0.25 complex at
room temperature; (c) magnetic field-dependent saturation polarization; increasing magnetic fields of two directions, parallel (F//) and perpendicular (F⊥) to the fiber axis, were applied to the sample to measure two in-plane polarizations (P//and P⊥);
有多铁性质. P3HT 和 PCBM 组成电荷转移有机
多铁体系中, P3HT 为电子给体, PCBM 为电子受
体, 它们以特定有序的方式自组装形成 DACT
[44].
在室温下 P3HT
0.75:PCBM
0.25定向纤维薄膜具有
磁序和电序各向 异性. 面内沿着纳米线轴线方
向 的 饱 和 磁 化 强 度 M
//远 远 超 出 了 平 面 内 垂 直
于轴线的磁化强度 M
⊥和平面外垂直于轴线的磁
化强度 M
OP. 而相对于磁序的各向异性, 铁电性
也显示出明显的 各向异性. 通过外场可以调控
P3HT
0.75:PCBM
0.25薄膜的磁性、铁电性、微波性
质和磁电耦合效应 (图
2
), 受磁场调控的多重态
激子是导致电荷转移多铁性的根本原因. 首先随
着光照强度的增大, 更多的多重态能够直接从能
量基态中被激发出来, 也就是说光照能够同时引
起磁序和电序的增大. 沿着纳米线方向, 增加的
饱和磁化强度 M
//和饱和铁电极化强度 P
//分别
为 0.5 和 0.085, 比垂直于纳米线方向 ∆M
⊥= 0.26
和 ∆P
⊥= 0.065 要高很多. 这说明沿着纳米线方
向产生了更多的多重态激子和更窄的自旋锥 (spin
cone). 面内沿着纳米线轴线方向的极化强度 P
//和
平面内垂直于轴线的 P
⊥也能随着外加磁场增加而
逐渐增大, 但是磁场方向的影响却并不明显. 介电
常数随着外加磁场的变化也进一步证明了其磁电
耦合的各向异性. 而当施加外加电场时, 电场会注
入更多的自由电荷, 因而引入了一个更大的电荷转
移密度, 导致更多的电荷 -晶格耦合、多重态激子和
更强的磁电耦合效应
[45].
2.3
噻吩 -富勒烯电荷转移体的室温多铁性
另外一种电荷引起的电极化的有机多铁材料
是噻吩 -富勒烯化合物 (thiophene fullerene). 在这
类超高分子混合物中, 噻吩作为电子给体和富勒烯
作为电子受体共结晶形成给体 -受体电荷转移网络
结构 DACT, 其室温下的磁性和自发电极化特性都
已被观察到
[35,36]. 噻吩纳米线向富勒烯移动, 结晶
的噻吩纳米线会产生排列好的非配对自旋并引起
铁磁性. 此外, 电荷转移能够分解成自由电荷, 光
照能够通过在噻吩纳米线内直接引入非配对自旋
而提高其磁性. 为了证明自旋引起的电极化, 在噻
吩 -富勒烯化合物上施加磁场来调控单重态和多重
态比例的关系
[46−49]. 特定方向的外加磁场能够减
少自旋的随机分布, 因而增大了多重态激子的产
生
[46,47]. 这些激子是由电子 -空穴同周围晶格耦合
在一起构成的, 每一个都可看成单独的电偶极子.
当大量的多重激子产生后, 因其具有微秒级的寿
命
[50,51], 可以导致宏观的电极化. 因此, 噻吩 -富勒
烯化合物的铁电极化能够被外加磁场调控.
噻吩 -富勒烯超分子共晶体在室温同样具有
多铁性. 结晶化的噻吩纳米线给体和聚合化的碳
基 受 体 共 同 结 晶, 形 成 三 维 的 超 分 子 电 荷 转 移
结 构 (supramolecular charge-transfer co-crystals,
SCTCs). 通 过 调 控 非 共 价 键, 能 够 使 得 这 类 超
分子共晶体的生长表现出各向异性. 电子自旋共
振 (electron spin resonance, ESR) 技术可以用于观
察这类规则排列的超分子共晶体的非配对自旋
态. 发现其磁化率在 100 和 200 K 时具有异常变
化
[52]. 通过调控电荷转移密度, 可以实现铁电性和
铁磁性在室温下的共存. 外部因素如光照、磁/电
场, 能够调控电偶极子和自旋序, 从而实现对磁电
耦合效应的控制, 这意味着 SCTCs 在室温下存在
多铁性.
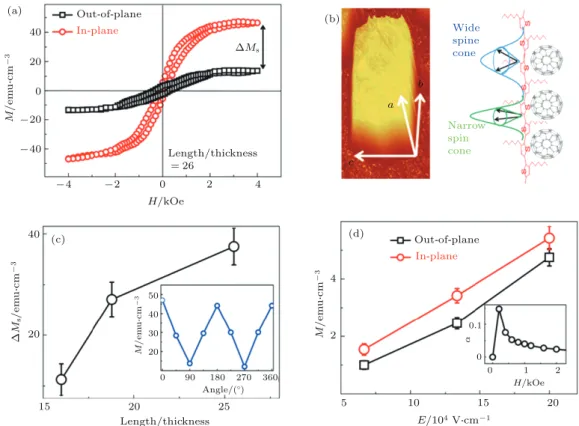
图
3
(a) 所示的磁滞回线证明了 SCTCs 沿着面
内和面外方向时的磁各向异性. 在结晶的噻吩纳米
线中, 电荷和周围晶体的耦合会产生自旋密度波,
从而引发 SCTCs 中的自发磁性. 当电荷同周围晶
格作用力很强时, 能够引入一个很窄的自旋锥, 并
迫使自旋序沿着易磁化轴, 因此沿着面外磁性更
弱, 而沿着面内磁性更强. 图
3
(b) 显示同一方向排
列的自旋能够增大易磁化轴方向的磁性
[53]. 反之,
较宽的自旋锥会分散自旋, 并有一定自旋沿着难磁
化轴, 因而一定程度上降低了磁性, 自旋锥的方向
和宽度取决于电荷 -晶格耦合和自旋方向. 为了进
一步证明自旋锥在磁性各向异性中的作用, 我们进
一步研究电荷密度和角度的关系. SCTCs 有着很
大的长/厚比例, 并能够引入很高的电荷密度, 因而
引起了电荷晶格之间很强的作用力并导致晶格微
微扭曲. 这会使得沿着易轴方向的自旋锥变窄并引
起更高的磁性, 最终导致饱和磁化强度 ∆M
s的巨
大差别, 可以看到 ∆M
s随着长/厚比的增大而增大
(图
3
(c)). 此外, 改变外加磁场的角度, 磁场在面内
方向角度 θ 从 0
◦变到 180
◦时, 可以看到饱和磁化
强度随着角度变化而明显变化, 证明了在共晶体低
维态的情况下自旋锥取向同外加磁场角度的紧密
关系
[54]. 此外, 图
3
(d) 中外加电场调控饱和磁矩
实验证明了室温下的逆磁电耦合效应, 插图显示了
在 200 Oe (1 Oe = 10
3/(4π) A/m) 下磁电耦合系
数达到 0.148 V/(cm
·Oe).
40 20 0 -20 -40 Out-of-plane In-plane Out-of-plane In-plane -4 -2 0 2 4 DMs 40 4 2 20 15 20 0 90 50 40 30 20 180 270 360 25 Length/thickness H/kOe H/kOe 5 10 0 0 0.1 1 2 15 20 E/104 VScm-1 Length/thickness =26 Wide spine cone Narrow spin cone M / e m u Sc m -3 M / e m u Sc m -3 M / e m u Sc m -3 D M s / e m u Sc m -3 c a b Angle/(O) α (a) (b) (c) (d) 图 3 超分子共晶体的磁性和磁电耦合各向异性 (a) SCTCs 在面内和面外方向上的磁滞回线; (b) 原子力显微镜图像和 电荷 -晶格耦合产生的自旋锥的示意图, 自旋锥的方向和宽度取决于电荷 -晶格耦合和自旋方向; (c) 面内 (易磁化轴) 和面 外 (难磁化轴) 方向上长厚比对饱和磁化强度各向异性的影响, 插图显示了饱和磁化强度随外加磁场方向的变化 (0◦, 180◦, 360◦时磁场沿着易轴); (d) 外电场调控磁矩从而产生磁电耦合, 插图则是磁场影响下的磁电耦合系数[55]
Fig. 3. Anisotropy of magnetization and magnetoelectric coupling in SCTCs: (a) The in-plane and out-of-plane magnetic hysteresis (M -H) loops of SCTCs; (b) the atomic force microscope image of one SCTC, and the scheme of spin cone distribution due to the exciton-lattice coupling; the width and orientation of spin cone would be different based on the exciton-lattice coupling extent and spin direction; (c) the length/thickness of SCTC dependent anisotropy of magnetization (∆M ) between in-plane (easy axis) and out-of-plane (hard axis) directions; the inset shows the angle dependent saturation magnetization M (the 0◦, 180◦, and 360◦means magnetic field parallel to in-plane direction); (d) electric field dependent magnetization (magnetoelectric coupling) of SCTC devices; the inset shows the magnetic field dependent magnetoelectric coupling coefficient[55].
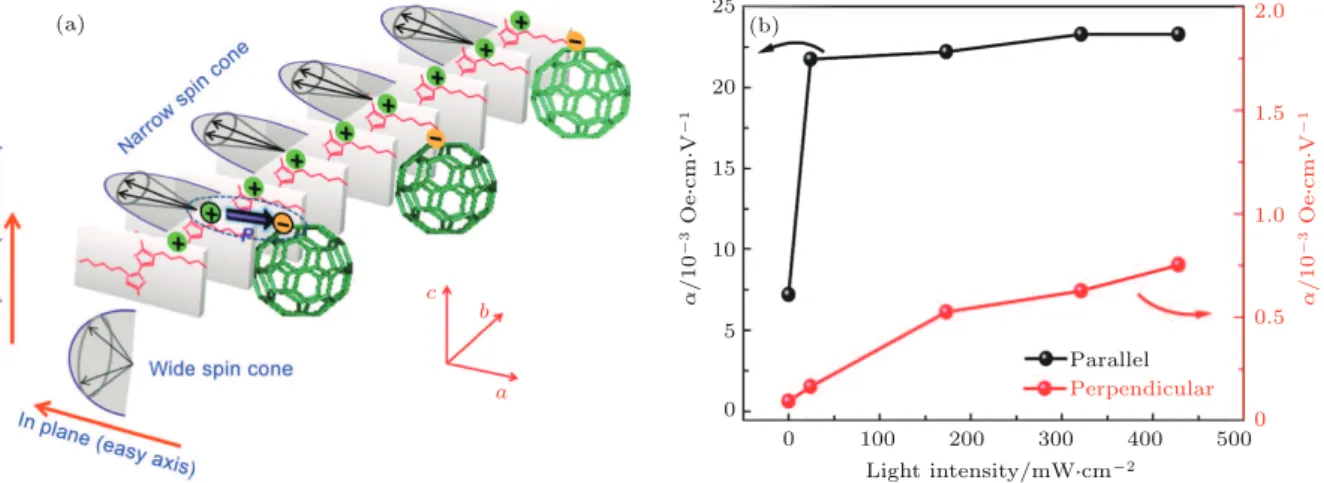
2.4
聚噻吩 -C
60电荷转移体的室温多铁性
由 聚 噻 吩 和 C
60分 别 作 为 电 荷 给 体 和 受 体
的 电 荷 转 移 有 机 体 (polythiophene-C
60) 在 室 温
具 有 较 大 铁 电 极 化、 饱 和 磁 矩 和 磁 电 耦 合 系
数. 如图
4
所示, 与噻吩 -富勒烯超分子共晶体类
似, polythiophene-C
60共晶体中的电荷转移及其
激子诱导了铁电极化和铁磁性.
polythiophene-C
60的 铁 电 极 化 随 外 加 电 场 的 增 大 而 增 强, 当
外 电 场 与 磁 场 都 沿 b 轴 时 其 磁 电 耦 合 系 数 可
达 到 0.0072 Oe
·cm/V, 在 光 照 下 可 以 增 大 到 约
0.022 Oe
·cm/V.
2.5
电荷转移有机薄膜 -铁电薄膜异质结的
室温多铁性
为了更进一步提高磁电耦合效应, 构建了由电
荷转移有机多铁材料和 P(VDF-TrFE) 铁电薄膜组
成的异质结, 并研究了其多铁性. 由电荷给体的
单壁纳米碳管富勒烯 (SWCNTs) 和作为电荷受体
的 C
60组成的电荷转移有机体 (SWCNTs-C
60) 同
样具有室温多铁性. 图
5
(a) 显示了 SWCNTs-C
60与 P(VDF-TrFE) 铁电薄膜构成的异质结. 在生
长 SWCNTs-C
60之前, 先用外加电场极化
P(VDF-TrFE) 层, 使得电偶极子沿着同一方向排列
[57,58].
当电偶极子方向同外加电场一致时, 电偶极子中
产生的电场能够增加 SWCNTs-C
60的有效电场, 从
而增大了电荷转移密度. 此外, P(VDF-TrFE) 层
中规则排列的电偶极子也能在 SWCNTs-C
60层中
直接引入电偶极子. 如图
5
(b) 所示, 外加电场为
1.8
× 10
5V/cm 时, 拥有 P(VDF-TrFE) 层的纳米
碳合物器件比单纯的纳米碳合物有着更强的磁电
耦合效应. 外加电场能够调控磁性主要通过两个方
a b c 25 20 15 10 5 0 0 100 200 300 400 Parallel 500 2.0 1.5 1.0 0.5 Perpendicular 0 α / 1 0 -3 Oe Sc m SV -1 α / 1 0 -3 Oe Sc m SV -1 Light intensity/mWScm-2 (b) (a) 图 4 (a) 聚噻吩 -C60电荷转移共晶体中自旋锥沿长轴 (b 轴) 的分布, 其中聚噻吩供体向 C60受体转移电子, 其界面电荷有 序导致铁电极化, 自旋锥的易/难轴方向和宽度取决于自旋方向及其电荷 -晶格耦合; 正负电荷只用来说明电荷转移和偶极子 的情况, 不表示真实电荷分布; (b) 外电场平行/垂直于共晶体长轴 (b 轴) 时, 磁电耦合系数及其与光强的关系[56]
Fig. 4. (a) Spin cone distribution along the long axis (b axis) of the charge-transfer cocrystal superstructures (CTCCs) and the polarization induced by charge ordering and charge-transfer at the interface. The direction and width of the spin cone depend on the spin direction and the charge-lattice coupling extent. The positive and negative charges in polythiophene and C60are used for the illustration of charge-transfer and dipoles, which do not represent
the real charge distribution in the cocrystals. (b) Light intensity-dependent magnetoelectric coupling of CTCCs[56].
12 8 4 0 40 80 120 5 0 -5 0 100 200 300 400 0.5 0 1.0 1.5 E/105 VScm-1 P(VDF-TRFE) thickness/nm Time/s M / e m u Sc m -3 12 8 4 M / e m u Sc m -3 M / e m u Sc m -3 With P(VDF-TrFE) Without P(VDF-TrFE) P(VDF-TrFE) on top P(VDF-TrFE) on bottom
OFF ONOFF ON OFF ON OFF ONOFF
(a)
(b)
(c) (d)
图 5 (a) 纳米碳合物器件示意图; (b) 外加电场开 (ON) 关 (OFF) 下调控具有和不具有铁电层 P(VDF-TrFE) 时的 纳米碳合物磁性; (c) 铁电层 P(VDF-TrFE) 分别在纳米碳合物层底部 (蓝线) 和顶部 (红线) 时磁性与 P(VDF-TrFE) 层 厚度的关系, 外加电场 1.8× 105 V/cm, SWCNT 为 2 wt% 时, 最优 P(VDF-TrFE) 层厚度为 45 nm; (d) 45 nm 厚度
P(VDF-TrFE) 层在电场影响下的磁化强度[39]
Fig. 5. (a) The scheme of the nanocarbon device structure; (b) the tunability of magnetization by electric field with and without the ferroelectric P(VDF-TrFE) layer, ON (OFF) means applied electric field (1.8× 105 V/cm)
is tuned on (off); (c) the P(VDF-TrFE) thickness-dependent tunability of magnetization in both bottom (circles) and top locations (squares), where the optimized thickness of P(VDF-TrFE) is 45 nm, applied electric field is 1.8× 105 V/cm and SWCNT loading ratio is 2 wt%; (d) the electric-field-dependent magnetoelectric coupling with
面: 1) 调控电荷转移密度; 2) 调节 P(VDF-TrFE)
层 的 电 极 化. 从图
5
(c) 可 见, 通 过 控 制
P(VDF-TrFE) 层的厚度, 电荷转移和铁电极化影响的磁
性可以达到平衡状态并得到最好的磁电耦合效
应. 另一方面, 器件的形貌结构也对磁电耦合有
着重要影响. 当把 P(VDF-TrFE) 层从底部位置切
换到顶部位置并且外加电场为 1.8
× 10
5V/cm 时,
磁电耦合系数从 5.03
× 10
−4Oe
·cm/V 增加到了
9.75
× 10
−4Oe
·cm/V, 这是来自于钝化作用对纳米
碳合物层的影响. 从图
5
(d) 可以看出, 磁性可以通
过电场调控, 即具有逆磁电耦合效应. 通过 (
1
) 式
的计算证明了电场调控磁性的耦合机制, 发现理论
计算出来的结果同实验结果非常符合.
M =
µµ
2 B(k
BT )
2
√
2π
~
3(m
eeEl)
3/2exp
(
−
2
3eEl
βφ
)
×
[(
π
2β
)
1/2−
1
β
+
π
(2β)
2]
H + bH
2,
(1)
其中 µ 是磁导率, µ
B是玻尔磁子, k
B是玻尔兹曼
常数, T 是温度,
~ 是普朗克常数, m
e是电子质量,
e 是电子电荷, E 是电场, l 是碳基层厚度, φ 是界
面处的势垒高度, β =
4
√
2m
eπ
l
~
φ
1/2, H 是磁场,
b 是 瑞 利 常 数. 可 见, 外 加 电 场 可 以 显 著 地 调 控
SWCNTs-C
60有机体的磁电耦合效应. 此外, 改变
单重态激子和多重态激子的比例可以控制其磁 -介
电/电流的耦合关系. 在加入了额外的有机铁电层
P(VDF-TrFE) 后, 纳米碳合物层和有机铁电层之
间的耦合作用加强了器件的磁电耦合作用.
3 结论与展望
激发态电荷转移有机多铁材料是一个全新的
材料体系. 电荷转移有机体由一个供给电子的分
子 (给体 D
+) 和一个接受电子的分子 (受体 A
−) 构
成电荷转移网络, D
+A
−长程有序排列, 其激发产
生的多重态激子是产生室温多铁性的根本原因,
这完全不同于传统的氧化物多铁材料和其他高分
子多铁材料. 通过光照、施加应力等可以实现对
电荷转移有机体中磁电耦合效应的调控, 证明了
电荷转移及其激发态在这类材料的多铁性中具有
关键作用.
目前, 激发态电荷转移有机多铁材料还有很多
问题亟待解决. 寻找新的电荷转移体系并制备超分
子共晶体, 其规则排列的分子链结构可望提高磁电
性能. 其次, 发展异质结体系, 在激发态有机薄膜
上外延生长有机铁电或者铁磁体系, 可望大幅度提
高其磁电耦合效应. 此外, 需要发展新的电荷转移
有机多铁器件, 促进新型多铁器件在实际工业生产
中的应用.
参考文献
[1] Gao W X, Brennan R, Hu Y, Wuttig M, Yuan G L, Quandt E, Ren S Q2018 Mater. Today (In Press) DOI: 101016/j.mattod.201801032
[2] Astrov D1960 Sov. Phys. JETP 11 708
[3] Dzyaloshinskii I E1960 Sov. Phys. JETP 10 628
[4] Greve H, Woltermann E, Quenzer H J, Wagner B, Quandt E2010 Appl. Phys. Lett. 96 182501
[5] Fiebig M2005 J. Phys. D: Appl. Phys. 38 R123
[6] Martins P, Lanceros-Mendez S2013 Adv. Funct. Mater.
23 3371
[7] Zavaliche F, Zhao T, Zheng H, Straub F, Cruz M P, Yang P L, Hao D, Ramesh R2007 Nano Lett. 7 1586
[8] Chou C C, Taran S, Her J L, Sun C P, Huang C L, Sakurai H, Belik A A, Takayama-Muromachi E, Yang H D2008 Phys. Rev. B 78 092404
[9] Wang J, Neaton J B, Zheng H, Nagarajan V, Ogale S B, Liu B, Viehland D, Vaithyanathan V, Schlom D G, Waghmare U V2003 Science 34 1719
[10] Ratcliff W, Lynn J W, Kiryukhin V, Jain P, Fitzsim-mons M R2016 npj Quantum Mater. 1 16003
[11] Liu J M, Nan C W2014 Physics 43 88(in Chinese) [刘 俊明, 南策文 2014 物理 43 88]
[12] Wang F, Shen S P, Sun Y2016 Chin. Phys. B 25 087503
[13] Kimura T, Goto T, Shintani H, Ishizaka K, Arima T, Tokura Y2003 Nature 426 55
[14] Al Qahtani M S, Alshammari M S, Blythe H J, Fox A M, Gehring G A, Andreev N, Chichkov V, Mukovskii Y
2012 J. Phys.: Conf. Ser. 391 012083
[15] Arkenbout A H, Palstra T T M, Siegrist T, Kimura T
2006 Phys. Rev. B 74 184431
[16] Lawes G, Kenzelmann M, Rogado N, Kim K H, Jorge G A, Cava R J, Aharony A, Entin-Wohlman O, Harris A B, Yildirim T, Huang Q Z, Park S, Broholm C, Ramirez A P2004 Phys. Rev. Lett. 93 247201
[17] Wang K F, Liu J M, Ren Z F2009 Adv. Phys. 58 321
[18] Cheong S W, Talbayev D, Kiryukhin V, Saxena A2018
npj Quantum Mater. 3 19
[19] Sergienko I A, Dagotto E2006 Phys. Rev. B 73 094434
[20] Hur N, Jeong I K, Hundley M F, Kim S B, Cheong S W
2009 Phys. Rev. B 79 134120
[21] Bukhari S H, Ahmad J2017 Chin. Phys. B 26 018103
[22] Choi T, Horibe Y, Yi H T, Choi Y J, Wu W D, Cheong S W2010 Nat. Mater. 9 253
[23] Chatterji T, Ouladdiaf B, Henry P F, Bhattacharya D
2012 J. Phys.: Condens. Matter 24 336003
[24] Zheng H, Wang J, Lofland S E, Ma Z, Mohaddes-Ardabili L, Zhao T, Salamanca-Riba L, Shinde S R, Ogale S B, Bai F2004 Science 303 661
[25] Pato-Doldan B, Gomez-Aguirre L C, Bermudez-Garcia J M, Sanchez-Andujar M, Fondado A, Mira J, Castro-Garcia S, Senaris-Rodriguez M A 2013 RSC Adv. 3 22404
[26] Xu G C, Zhang W, Ma X M, Chen Y H, Zhang L, Cai H L, Wang Z M, Xiong R G, Gao S2011 J. Am. Chem.
Soc. 133 14948
[27] Fu D W, Zhang W, Cai H L, Zhang Y, Ge J Z, Xiong R G, Huang S D, Nakamura T2011 Angew. Chem. Int.
Ed. Engl. 50 11947
[28] Jain P, Stroppa A, Nabok D, Marino A, Rubano A, Pa-paro D, Matsubara M, Nakotte H, Fiebig M, Picozzi S, Choi E S, Cheetham A K, Draxl C, Dalal N S, Zapf V S2016 npj Quantum Mater. 1 16012
[29] Tian Y, Cong J Z, Shen S P, Chai Y S, Yan L Q, Wang S G, Sun Y2014 Phys. Status Solidi RRL 8 91
[30] Tian Y, Stroppa A, Chai Y S, Yan L Q, Wang S G, Barone P, Picozzi S, Sun Y2014 Sci. Rep. 4 6062
[31] Tian Y, Wang W, Chai Y S, Cong J Z, Shen S P, Yan L Q, Wang S G, Han X F, Sun Y2014 Phys. Rev. Lett.
112 017202
[32] Tayi A S, Shveyd A K, Sue A C, Szarko J M, Rolczyn-ski B S, Cao D, Kennedy T J, Sarjeant A A, Stern C L, Paxton W F2012 Nature 488 485
[33] Wang Y, Liu J L, Tran H D, Mecklenburg M, Guan X N, Stieg A Z, Regan B C, Martin D C, Kaner R B2012
J. Am. Chem. Soc. 134 9251
[34] Zhang Z L, Li H S, Luo Z P, Chang S Q, Li Z, Guan M M, Zhou Z Y, Liu M, Grossman J C, Ren S Q2017
Chem. Mater. 29 9851
[35] Qin W, Jasion D, Chen X M, Wuttig M, Ren S Q2014
ACS Nano 8 3671
[36] Ren S Q, Wuttig M2012 Adv. Mater. 24 724
[37] Lohrman J, Liu Y Y, Duan S F, Zhao X Y, Wuttig M, Ren S Q2013 Adv. Mater. 25 783
[38] Qin W, Lohrman J, Ren S Q2014 Angew. Chem. Int.
Ed. Engl. 53 7316
[39] Wei Q, Gong M G, Chen X M, Shastry T A, Sakidja R, Yuan G L, Hersam M C, Wuttig M, Ren S Q2015 Adv.
Mater. 27 734
[40] Kagawa F, Horiuchi S, Tokunaga M, Fujioka J, Tokura Y2010 Nat. Phys. 6 169
[41] Torrance J B, Girlando A, Mayerle J J, Crowley J I, Lee V Y, Batail P, Laplaca S J 1981 Phys. Rev. Lett. 47 1747
[42] Lamola A A, Hammond G S 1965 J. Chem. Phys. 43 2129
[43] Ding L J, Yao K L, Fu H H2011 J. Mater. Chem. 21 449
[44] Yu G, Gao J, Hummelen J C, Wudl F, Heeger A J1995
Science 270 1789
[45] Brédas J L, Beljonne D, Coropceanu V, Cornil J2004
Chem. Rev. 104 4971
[46] Hu B, Wu Y2007 Nat. Mater. 6 985
[47] Qin W, Gao K, Yin S, Xie S J2013 J. Appl. Phys. 113 193301
[48] Janssen P, Cox M, Wouters S H W, Kemerink M, Wienk M M, Koopmans B2013 Nat. Commun. 4 2286
[49] Majumdar S, Majumdar H S, Aarnio H, Vanderzande D, Laiho R, Osterbacka R2009 Phys. Rev. B 79 201202
[50] Baldo M A, O’Brien D F, You Y, Shoustikov A, Sibley S, Thompson M E, Forrest S R1998 Nature 395 151
[51] Jariwala D, Sangwan V K, Lauhon L J, Marks T J, Her-sam M C2013 Chem. Soc. Rev. 42 2824
[52] Chen X M2016 Ph. D. Dissertation (Nanjing: Nanjing University of Science and Technology)(in Chinese) [陈 孝敏 2016 博士学位论文 (南京: 南京理工大学)]
[53] Armstrong J N, Hua S Z, Chopra H D2013 Phys. Status
Solidi B 250 387
[54] Callen E R1960 J. Appl. Phys. 31 S149
[55] Qin W, Chen X M, Li H S, Gong M G, Yuan G L, Gross-man J C, Wuttig M, Ren S Q2015 ACS Nano 9 9373
[56] Xu B B, Li H S, Hall A, Gao W X, Gong M G, Yuan G L, Grossman J, Ren S Q2015 Sci. Adv. 1 e1501264
[57] Jin J Z, Lu S G, Chanthad C, Zhang Q M, Haque M A, Wang Q2011 Adv. Mater. 23 3853
[58] Carvell J, Cheng R H, Dowben P A, Yang Q2013 Appl.
SPECIAL TOPIC — Multiferroicity: Physics, materials, and devices
Excited charge-transfer organics with multiferroicity
∗
Yuan Guo-Liang
1)†Li Shuang
1)Ren Shen-Qiang
2)Liu Jun-Ming
3)‡1) (School of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China) 2) (Department of Mechanical and Aerospace Engineering, University at Buffalo, The State University of
New York, Buffalo, New York 14260, USA)
3) (Department of Physics, Nanjing University, Nanjing 210046, China) ( Received 20 April 2018; revised manuscript received 13 June 2018 )
Abstract
Multiferroics, showing simultaneous electric and magnetic degree of freedom, has aroused increasing interest due
to tailored multiferroic properties and magneto-electric coupling for shaping the development of energy-efficient
mul-tifunctional devices. Now, the multiferroics can be classified as two groups: 1) inorganic multiferroics, which can be
single-phase, multi-phases oxide multiferroic or multiferroic heterojunction and 2) organic counterpart, which is mostly
determined by instinct charge-transfer behavior. But it is difficult to find the polarization and the magnetization
co-exist in a single-phase oxide multiferroic material, and their coupling range in the multiferroic heterojunction is only
several atomic layers, which limits the applications. As a result, more and more different types of organic
multifer-roics have been studied. Some organic complexes can display dual ferroelectric and ferromagnetic properties at ambient
temperature, e.g. thiophene-fullerene donor-acceptor charge-transfer networks. The organic charge-transfer complex
is based on electron donor (D
+) and acceptor (A
−) assembly. D
+A
−are long-range ordering, the excitons have µs
lifetime and
±1/2 spin, which contributes to the room temperature ferroelectricity and ferromagnetism. The excitons
can be excited by external magnetic field, electric field, illumination and stress, and eventually influence the
polariza-tion, magnetization and magnetoelectric coupling coefficient. However, there are still many problems to be solved, i.e.,
searching for new charge-transfer systems and preparing supramolecular co-crystal with ordered molecular chain, further
improving magnetoelectric properties; developing the heterojunction technology and epitaxial growth of organic
ferro-electric or ferromagnetic systems on excited organic films, which is expected to greatly improve their magnetoferro-electric
coupling effects; inventing more new charge transport organic multiferroic devices to extend the application scope of new
multiferroic devices in actual industrial production. Generally speaking, the organic charge-transfer complexes not only
greatly enrich the room temperature multiferroics materials, but also provide the technical basis for developing the new
multifunctional electronic devices.
Keywords: organic multiferroics, magneto-electric coupling, charge-transfer
PACS: 75.85.+t, 77.84.–s, 72.20.Jv
DOI: 10.7498/aps.67.20180759
* Project supported by the National Natural Science Foundation of China (Grant Nos. 51790492, 51431006, 51472118) and the Fundamental Research Funds for the Central Universities of Ministry of Education of China (Grant No. 30916011104). † Corresponding author. E-mail:[email protected]