第二章 研究原理和方法
2-1 毛細管電泳層析法的發展
電泳(electrophoresis)是指在電場的作用下,電解質中的荷電粒 子以不同的速率向荷電相反的電極遷移的現象。利用各荷電粒子所帶 荷電量與質量的不同,產生不同的遷移速率,藉此可達到分離的效 果,即稱為電泳分離法。
電泳技術發展至今,已有一百多年的歷史。最早發展是在 1886 年時,由 Lodge 在含有膠質的電解液中,兩端施以電壓,觀察出顏色 產生變化;1892 年 Picton 使用含有離子型色素,1899 年 Hardy 利用 含有膠質粒子的U 型管,都發現了電泳的現象,而且 Hardy 並於 1905 年利用有刻度的U 型管,記錄了膠質粒子的移動[35];1937 年 Tiselius 應用此方法分離出馬的三種血清蛋白,將它們分別命名為α、β、γ 球 蛋白[36],使電泳技術獲得重大突破,電泳分離技術漸漸受到重視,
並對此進一步更深入的探討,以期讓電泳成為簡便而有效率的分離技 巧,而他本人並因對電泳-吸附現象的研究及血清蛋白分離等相關的 研究,而獲得到1948 年諾貝爾獎化學獎。
由於電泳的分離效率和外加的電場大小成正比,早期的電泳分析 法是直接在電解質溶液中進行分離,但施加高電壓會產生焦耳熱
(Joule heating)而導致熱擴散及熱對流。這種熱效應會使介質產生 溫度梯度及速度梯度以致使譜帶加寬,降低分離效率,使電泳技術無
流的介質,如:聚丙醯胺(acrylamide)或瓊脂(agarose)凝膠等。
但此法還會延長電泳分離時間,並使再現性變差,而且凝膠介質製備 困難,故一直沒有受到廣泛的應用。直到 1965 年 Konstantinov 首度 嘗試利用毛細管進行電泳分析[37],1967 年,Hjerten 使用內徑 1-3 mm 的石英管,進行無機離子、核酸與蛋白質的分離[38],並以甲基纖維 素(methylcellulose)修飾石英管內部表面,然後將毛細管繞著縱軸 緩慢的旋轉,以消緩層帶擴散的影響,防止電滲流(electroosmosis)
的產生,進行區帶電泳的實驗。此一技術成功地將蛋白質與核酸完全 的分離。1979 年 Virtanen 和 Mikkers 利用內徑 200~500 µm 的玻璃材 質與聚四氟乙烯材質的毛細管進行電泳[39-40],由於管柱的表面積與 管柱體積比例大為提高,因此有效地控制了焦耳熱的產生。其後 Martin 和 Everaerts 的研究,進一步確立了毛細管電泳(Capillary Electrophoresis, CE)的理論基礎[41]。
目前大家普遍熟知的高效能毛細管電泳法(High Performance Capillary Electrophoresis, HPCE)則是由 Jorgenson 及 Lukacs 在 1981 年提出的(42-44)。他們發表以 75 µm 內徑的毛細管玻璃管柱,在 30 kV 的高電壓條件下,成功地分離胺基酸的衍生物。並且結合螢光 偵測器做線上偵測,將偵測靈敏度提高。且提出了毛細管區帶電泳分 析法(Capillary zone electrophoresis, CZE),其原理是利用離子分析物 在電埸作用下,由於質量和帶電量不同,產生不同的遷移速度,達到 分離效果。在電泳的實驗中,使用毛細管有許多優點,愈細的毛細管 所產生的焦耳熱愈少,毛細管能夠在高電場情況中產生很少的熱量,
而且毛細管電泳獨特的EOF(電滲流)所形成的流動界面也愈接近柱
狀層流(piston-like flow)(如圖 2-1 所示),因此可使得理論板數大 為提高。所以目前已有開發內徑細到5~10 µm 的毛細管,但太細的毛 細管會有校光、進樣及毛細管堵塞的問題存在,所以較為實用的毛細 管內徑約為25~75 µm,。
毛細管電泳在其發展主要 Jorgenson 等人提出的毛細管區帶電泳 層析法(CZE)。由於此法僅能分離質量和帶電量不同離子性分析物,
無法對中性分子加以分析,乃缺點之一。在 1984 年,Terabe 利用微 胞液相層析(micellar liquid chromatography)的原理,發展出微胞電 動毛細管層析法(Micellar Electrokinetic Chromatography 或 Micellar Electrokinetic Capillary Electrophoresis,簡稱 MEKC 或 MECC),在緩 衝溶液中添加界面活性劑而形成微胞,使分離系統中如同有兩相的存 在,一是由緩衝溶液組成的水相、一是由界面活性劑形成的微胞相,
分析物在兩相間分配係數不同,電泳時產生不同的遷移速率,可以成 功分離不帶電的中性物質[45-46]。事實上,界面活性劑的添加,對於 荷電分子的分離也極具效率,使毛細管電泳的發展更為廣泛[47]。隨 後Hjerten 在 1985 年將傳統的等電聚焦過程移到毛細管進行,進而提 出毛細管等電聚焦電泳法(Capillary isoelectric focusing, CIEF)
[48-49],使用不同 pH 梯度的溶液,利用分析物等電點的差異進行分 離蛋白質。1987 年 Cohen 與 Karger 亦將傳統的凝膠電泳法應用到毛 細管中,將十二烷基硫酸鈉-聚丙烯醯氯在毛細管內形成凝膠,直接 應 用 在 蛋 白 質 的 分 離 與 分 子 量 的 決 定 , 發 展 成 毛 細 管 凝 膠 電 泳
(Capillary gel electrophoresis, CGE)[50]。其間,還有其他的毛細管 電泳技術被發展出來,如:利用不連續的緩衝溶液系統,造成電場差
異,使分析物依據不同電場下電泳速率的不同,而形成樣品區帶的分 離機制,稱為毛細管等速電泳法(Capillary isotachophoresis, CITP)
[51-53]。此外,Tusda 結合液相層析與毛細管電泳的優點,使用填充 物於毛細管中,利用分析物在這種特殊填充物與溶液間的分佈平衡,
而 產 生 的 一 種 分 離 機 制 , 稱 為 毛 細 管 電 層 析 法 (Capillary electrochromatography, CEC)[54-56]。這些技術使得毛細管電泳法應 用日益廣泛。在毛細管區帶電泳中,理論板數一般可達數十萬,高的 可達數百萬,同時由於儀器的自動化,使得操作更為簡便。還有目前 新興的微晶片毛細管電泳分析[57-61],使毛細管電泳的技術更趨成 熟,成為分析技術的主流。
由於毛細管電泳分析法具有許多優點,包含樣品及溶劑使用量 少,不會造成污染,而且分離時間短且效率佳,可搭配不同的電泳模 式,使分離物種類多樣化,加上商業自動化儀器的普及,使CE 技術 被廣泛應用在刑事鑑定、藥物分析、環境檢測、食品品管、生化醫藥 上,成為簡便且有效率的分析方法[62-71]。
圖 2-1 毛細管柱內流動的示意圖及相對溶質的區帶
泳動方向
EOF
泳動方向
Laminar flow
a b
泳動方向
EOF
泳動方向
Laminar flow
a b
a b
2-2 毛細管電泳層析法的分離原理
毛細管電泳的基本組件如圖2-2 所示,包含毛細管(capillary)、
電壓電源(HV)、緩衝溶液儲存槽(buffer solution)、樣品儲存槽(sample solution)、偵測器(detector)。先在毛細管中注滿緩衝溶液或背景溶 液(background solution),再將適量的分析物進樣至毛細管內,然後 把毛細管兩端放入緩衝溶液儲存槽中,利用白金電極通以高壓直流 電,進行電泳分析。
2-2.1 電泳的分離與遷移率
電泳的分離機制係以帶電粒子在電場中的速度差異為基準,當一 個帶電粒子在自由溶液中,外加電場作用下所受到的靜電力為Fi[72]:
Fi=qE
qe=粒子帶電量(單位為庫倫)
Ee=電場強度(單位為 V/cm)
此靜電力會使得此荷電粒子加速運動,直到和反方向的黏滯力 Fd相等而達成平衡。
Fi = Fd
這個黏滯力與粒子的移動速度vi、半徑ri、介質的黏度 η 成正比,
可表示為:Fd
= 6 πη ×
ri×vi當這兩個作用力互相平衡時,在電場 E 下,粒子的移動能達到一 個穩定狀態的速度vi為: E
r v q
i e
i ×
= ×
πη
6又 vi =µi E
(1) 虹吸進樣時,將毛細管兩端分別置於 sample solution 及 buffer solution 中,並將 sample solution 抬高,利用兩端壓 力差將樣品注入毛細管內。
(2) 進樣完成後,將毛細管兩端分別放入 buffer solution 中,
通入高壓直流電,進行電泳分析
圖 2-2 CE 儀器裝置簡圖
HV Detector
buffer
buffer sample
capillary
HV Detector
buffer
buffer sample
capillary
其中,vi為電泳速度,E 為電場強度,µi為帶電粒子的遷移率。
這種帶電粒子的遷移行為即為電泳。我們可定義電泳遷移率為單位電 場下的電泳速率 µi:
i i
r q E
V
= ×
= 6 πη µ
i2-2.2 Zeta 電位與電滲流
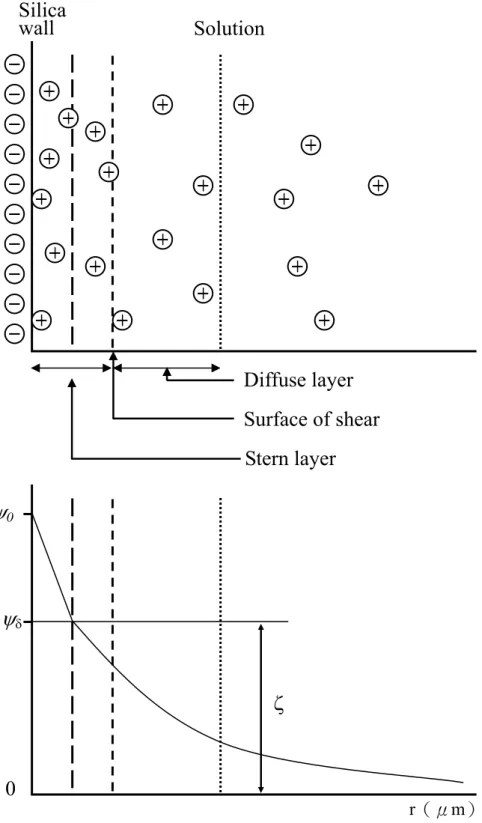
電滲流(electroosmotic flow, EOF)為毛細管的自然現象,是在 十九世紀末由Helmholtz 將電場應用至含鹽類水溶液之水平玻璃管中 時發現的[73]。主要是指毛細管內壁表面電荷所引起管內液體的整體 流動。主要形成的來源為外加電場對管壁溶液電雙層的作用。
如圖 2-3 所示,毛細管壁之離子化矽醇基(SiO-)會因緩衝溶液 的pH 值不同而有不同比例的解離,進而吸引緩衝溶液中的陽離子,
形成了所謂的電雙層。根據 Stern 的電雙層理論,溶液的正離子在經 活化後的毛細管壁上,將因靜電吸引力而吸附在管壁表面上,形成不 可逆吸附的第一層吸附,稱為固定層(stern layer)。當稍遠離於毛細 管表面時,也會有過多的正電荷被靜電力吸引,此時吸附的程度則沒 有第一層來的強,其電荷隨遠離管壁程度呈指數趨勢急遽下降,此層 吸附稱為擴散層(diffuse layer)。由於溶液在毛細管內電荷分佈的不 均勻,因此溶液與管壁間會有電位差的存在,此稱為 zeta 電位(ζ potential)。
若將溶液的中央部分(bulk solution)定為零電位,則管壁的電 位為
ψ
0,此時在固定層中的電位由ψ
0降至ψ
δ,而在擴散層中電位則由圖 2-3 毛細管壁因帶電荷而形成電雙層及zeta電位示意圖 Solution
Silica wall
Stern layer Surface of shear Diffuse layer
ψ
0ψ
δ0
r(μm)
ζ
ψ
δ降至零。用公式則表示為:ε ζ = 4 πδ ×
e其中,δ為電雙層的厚度;e 為每單位表面積的電荷;ε為溶液的 介電常數。因為zeta 電位的存在,在高壓電場的作用下,使得毛細管 中的液體會有泳動的現象,即為電滲流(electroosmotic flow, EOF),
如圖 2-4。將電滲流遷移率定義為單位電場下的電滲流速率 µeof,而 毛細管中各部位介質的電滲流與該點的zeta 電位成正比,可表示為:
η ζ
µ
eof= ε ×
其中,η 為溶液的黏度。由於帶電粒子在緩衝溶液中,藉著靜電 力和本身質量的影響,有其電泳遷移率(µep),而溶液本身因pH 值、
離子強度、濃度的控制亦存在著電滲流遷移率(µeof),因此帶電粒子 整體的遷移率則為這兩者向量的加成;電滲流遷移率之作用如一種泵 機制,可帶動所有粒子朝向偵測器,測定各分析物在電泳遷移中的差 異而達分離目的。當通以正的高壓電時,陽離子移動方向與電滲流µeof 相同,其單位電場的遷移速率為µep+µeof,陰離子移動則與電滲流相 反,其單位電場的遷移速率為︱µep-µeof︱。
電泳分離的機制就是靠著各種分析物在電解質溶液中電泳遷移 率的差異來達成,如圖2-5。表 2-1 列出了控制 EOF 常見的方法,由 此可藉由改變電解質的條件,如組成、濃度、pH 值、有機修飾劑、
電解質修飾劑、管柱修飾劑等,來達成不同的分離效果。
圖 2-4 EOF的形成及流動 (a) 毛細管壁的解離 (b) 電雙層的形成
(c) 通電後,電滲流的形成
a
b
c
Si Si Si Si Si Si Si Si Si
O - OH O - OH O - O - OH OH OH
KSiOH
H+ H+
H+ H+
H+
H+
H+ Electrolyte solution
Silica wall
+
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
+ + + + + + + + + + + +
+ + + + +
+
+ + + +
+ + + + + + +
+ + +
+ + +
+
+ + + + + + +
+ + + + +
+ +
+
+ +
+
+ + +
+ + +
+ +
+ +
+ +
+ + + + +
+ +
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
EOF
a
b
c a
b
c
Si Si Si Si Si Si Si Si Si
O - OH O - OH O - O - OH OH OH
KSiOH
H+ H+
H+ H+
H+
H+
H+ Electrolyte solution
Silica wall Si Si Si Si Si Si Si Si Si
O - OH O - OH O - O - OH OH OH
Si Si Si Si Si Si Si Si Si
O - OH O - OH O - O - OH OH OH
KSiOH
H+ H+
H+ H+
H+
H+
H+ Electrolyte solution
Silica wall
+
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
+ + + + + + + + + + + +
+ + + + +
+
+ + + +
+ + + + + + +
+ + +
+ + +
+
+ + + + + + +
+ + + + +
+ +
+
+ +
+
+ + +
+ + +
+ +
+ +
+ +
+ + + + +
+ +
+
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
+ + + + + + + + + + + +
+ + + + +
+
+ + + +
+ + + + + + +
+ + +
+ + +
+
+ + + + + + +
+ + + + +
+ +
+
+ +
+
+ + +
+ + +
+ +
+ +
+ +
+ + + + +
+ +
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
EOF
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
EOF
表2-1 常見控制 EOF 的方法
影響因素 電場強度
緩衝溶液 pH
離子強度或緩 衝溶液濃度
溫度
有機修飾劑
表面活性劑
中性親水高分 子聚合物 共價鍵結
結果
●電滲流呈比例變化
●低 pH,EOF 降低
●高 pH,EOF 增加
●增加離子強度,zata 下降,
EOF 下降
●每℃黏度變化 2~3%
●改變 zata 電位及黏度,一般 使 EOF 下降
●透過疏水性或離子相互作 用吸附於毛細管壁
●透過疏水性相互作用吸附 於毛細管壁
●化學鍵結於毛細管壁
說明
●場強降低可能引起分離效率和 解析度下降
●場強增加可能引起焦耳熱
●改變 EOF 最方便有用的方法
●可能改變溶質的電荷或結構
●高離子強度產生較大電流引起 焦耳熱
●低離子強度產生樣品吸附問題
●樣品導電度不同引起譜峰變形
●低離子強度降低樣品堆積效果
●若由儀器控溫,改變溫度是有效 的方法
●變化複雜,需透過實驗確認
●可能改變選擇性
●陰離子表面活性劑增加 EOF
●陽離子表面活性劑減少 EOF 或 使之改變方向
●改變選擇性
●通過覆蓋表面電荷和黏度增加 降低 EOF
●多種可能影響(親水性或帶電性)
●穩定性常有問題
圖 2-5 在CZE的分離模式下,不同粒子的遷移
(a)分離示意圖
(b)層析圖譜示意圖
t=0
t t t
Nt t
Migration time
b
detector
EOF
N
N N
N N
N
a
2-2.3 電分散作用
樣品區帶和緩衝液之間導電度的差異會引起電分散作用,而使譜 峰產生fronting 與 tailing 的現象。如圖 2-6(a)所示,當溶質區帶的 電泳遷移率較緩衝溶液快時(即高電導、低電阻),則溶質區帶的譜 峰前沿將會擴散而後尖銳(fronting)。相反地,當溶質區帶具有比緩 衝溶液低的電泳遷移率時(即低電導、高電阻),則溶質區帶的譜峰 前沿尖銳而後沿擴散(tailing),如圖 2-6(c)。當溶質區帶與緩衝溶 液導電度相同時,則不會出現譜峰畸變現象,而譜峰以高斯曲線或標 準偏差曲線的形式出現,為理想峰形,如圖2-6(b)[74-76]。
上述情形的發生必須待測樣品本身帶有電荷,亦即樣品本身具有 導電度才會有影響。對於電中性的物質,則不受導電度差異的影響。
儘管這些譜峰畸變的情形經常發生,若導致解析度降低,則必需透過 分析樣品及緩衝溶液的導電度調整,加以改善,若並不影響分離效 率,則譜峰的扭曲變形,往往可以忽略。
圖 2-6 緩衝溶液與樣品的導電度不同,導致溶質的電分散作用
(a)分析物區帶的導電度較高
(b)分析物與緩衝液的導電度相近
(c)分析物區帶的導電度較低
buffer
buffer
sample zone
低電導 高電導 低電導
a b c
buffer buffer
sample zone
高電導 低電導 高電導
buffer buffer
sample zone
等電導
t=0 t=0 t=0
2-2.4 分離效率及解析度
理論板數(N)是反應物質在固定相和流動相中動力學的重要層 析參數,代表層析管柱分離效能的指標,毛細管電泳的分離效率與一 般層析方法一樣,可用理論板數(N)當作指標:
2
2 / 1
54 .
5 ⎟⎟ ⎠
⎜⎜ ⎞
⎝
× ⎛
= W
N t
其中t 為物質的遷移時間;W1/2為吸收峰半波寬。一般而言,毛 細管電泳的理論板數在 105~106之間,比 HPLC 來得高。但實際上,
常因縱向擴散、焦耳熱所造成的溫度梯度、樣品吸附管壁、樣品進樣 長度、溶質及緩衝液間導電度的差異等不同的因素而造成譜帶加寬和 分離效率降低。一般常見可以提高分離效率的方法有︰將電壓調高、
使用內徑較小或較長的毛細管、降低分析樣品的濃度、降低毛細管的 溫度、添加修飾劑減少管壁吸附現象…等。而解析度(RS)可用來描 述兩譜峰的分離情形
1 2
1
2
)
( 2
W W
t R
st
−
= −
其中,t1及t2為溶質1 及 2 的遷移時間;W1及W2則為各別的吸 收峰底部的寬度。在定量方法中,通常 RS大於 1.5 表示兩譜峰以達基 線分離。因此,在毛細管電泳的分離過程中,必須特別注意上述影響 分離效率的因素,才能成功地分離樣品、增加分離效率及解析度。
2-3 毛細管電泳層析法的分離模式
毛細管電泳層析法的分離模式很多,但由於本實驗主要以添加界 面活性劑的微胞電動毛細管電泳(MEKC)分離法為主,因此本處只 簡述毛細管電泳層析法中最基本的毛細管區帶電泳層析法(CZE)及 本研究所使用的 MEKC 法。並將其他毛細管電泳模式,列於表 2-2 中。
2-3.1 毛細管區帶電泳層析法(CZE)
CZE(capillary zone electrophoresis)是毛細管電泳法中最基本也 是應用最廣的一種操作模式。其分離原理是在外加電場的存在下,分 析物在毛細管內緩衝溶液中,因質量與有效電荷的差別,造成電泳遷 移率的差異而達到分離的效果,故又稱自由溶液電泳(free zone electrophoresis)。如圖 2-5 所示,在通正高壓電場的情況下,正離子 分析物運動方向和電滲流一致,會最先通過檢測器;中性物質本身在 電場中不移動,則隨電滲流遷移但彼此不能分離;而負離子因其運動 方向和電滲流相反,則在中性物質之後才被偵測到。
在CZE 中,可藉由改變電壓、溫度、緩衝溶液組成、pH 值、濃 度及添加有機修飾劑等方法來改善分離效率。因為這個方法很簡便,
所以應用的範圍很廣,包括胺基酸的分析、蛋白質的純度分析、陰陽 離子分析等等[77-80]。
2-3.2 微胞電動毛細管層析法(MEKC)
MEKC(Micellar Electrokinetic Capillary Chromatography)技術是 由Terabe 等人在 1984 年首先提出。此一技術最大的特點在於兼具電 泳與層析的特性,使毛細管電泳技術可同時分離離子型化合物及中性 物質。
MEKC 的分離通常是在緩衝溶液中加入濃度大於臨界微胞濃度
(critical micelles concentration, CMC)的界面活性劑。如表 2-3 為常 用的界面活性劑及其臨界濃度(74)。當一定數量的界面活性劑單體 分子會聚集在一起,形成一個球體,稱為微胞 (micelles)圖 2-7。
在水溶液中的微胞,其單體聚集的方式為疏水性的一端聚合而形成微 胞的中心,親水性的一端則朝向水相。由於在緩衝溶液中有水相及微 胞相同時存在,因此分析物可在水相和微胞相中有不同的分配比,而 將分析物分離。如圖2-8 所示,在電泳的過程中,由於緩衝溶液在靠 近管壁處會形成的正電荷,導致強大的電滲流產生而向負極移動,對 於陰離子微胞(如 SDS 微膠粒)而言,因其外殼帶很大的負電荷,
使之往正極遷移,這與電滲流方向相反,因此陰離子微胞會最慢到達 偵測器。分析物也會隨著本身疏水或親水的性質,以及與帶電荷微胞 的靜電作用力,在水相與微胞相之間有不同程度的分佈(partition)。
在 MEKC 中,界面活性劑的選擇對分離的影響很大,因為不同 的界面活性劑會產生不同的交互作用力、不同的溶解度以及不同的聚 合數(aggregation number)和形狀,這些因素都會影響分離的選擇性。
圖 2-7 陽離子和陰離子微胞的示意圖
表 2-2 毛細管電泳層析法的分離模式
分離模式 分離機制
毛細管區帶電泳法(CZE) Capillary zone electrophoresis
●分析物緩衝溶液中的電泳 遷移率不同
微胞電動層析法(MEKC)
Micellar electrokinetic chromatography
●分析物與微胞間的相互作 用力
毛細管凝膠電泳法(CGE) Capillary gel electrophoresis
●大小及電荷不同的分析物 通過分子篩的難易度 毛細管等電聚焦法(CIEF)
Capillary isoelectric focusing
●蛋白質分析物會在等電點 的位置被聚焦
毛細管等速電泳法(ITP) Isotachophoresis
●分析物在兩種緩衝液造成 的分離區帶中等速移動 毛細管電色析層法 (CEC)
Capillary electrochromatography
●毛細管填充的靜相物與分 析物間的交互作用
表 2-3 微胞電動毛細管層析法常用的界面活性劑
Biological detergents CMC
(mM)
臨界濃度
Aggregation number 分子聚集數
Anionic SDS DOSS
8.2
─
62
─ Cationic DTAB
CTAB
14 1.3
50 78 Non ionic Octylglucoside
n-Dodecyl-β-D-maltoside Triton X-100
─ 0.16 0.24
─
─ 140 Zwitterionic CHAPS
CHAPSO
8 8
10 11 Bile Salt Cholic acid
Deoxycholic acid Taurocholic acid
14 5 10-15
2-4 4-10
4
圖 2-8 MEKC的分離模式
S1及S2分別代表不同的溶質粒子,因與微胞產生作用力的不同,
而有不同的遷移速率(a)分離示意圖(b)層析圖譜示意圖
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
µ
eofµ
mcS
2S1
t=0
t
eoft
S1t
S2t
mcb
a Detector
mc
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
µ
eofµ
mcS
2S1
t=0
t
eoft
S1t
S2t
mcb
a Detector Detector
mc
2-4 毛細管電泳線上濃縮技術
毛細管電泳發展初期所使用的偵測器仍然是傳統層析技術的紫 外光吸收法,但毛細管內徑比HPLC 管柱小很多,所以在樣品偵測極 限上反而比較不好。因此有許多的科學家,致力於改善偵測極限的研 究,常用的方法有下列幾種︰(一)將毛細管的光徑加大(81),例如使 用 Z 形毛細管、直角毛細管,可增加吸收度;(二)利用多次反射槽 (multi-reflection cell)來增加光徑的距離,提高偵測靈敏度[82];(三) 利用液相萃取或固相萃取的樣品前處理步驟,濃縮樣品的濃度[83];
(四)使用功率較大的偵測器,例如使用雷射誘導螢光偵測器(Laser Induced Fluoresence, LIF)[84],其偵測極限可達到 10-10M 以下,但本 身具有螢光性質的分析物並不多,通常分析物需經過衍生的處理步驟 才能偵測;(五)或是採用複雜的使用技術,如電化學偵測法、質譜 分析法,但這些方法都有較多的困難存在。
因此有人提出了另一種增加CE 靈敏度且較簡單的方法為線上樣 品堆積技術[85-86],用來降低偵測極限,此方法既有效又經濟。樣品 堆積的方法是先增加樣品的進樣時間,使樣品的總進樣量增加,則樣 品區間亦隨之變長,再透過樣品區間的聚集濃縮,提高樣品的濃度。
由於不需預先於實驗前進行樣品濃縮,而是在整個實驗過程中,同時 達成樣品堆積與分離,稱此技術為線上濃縮(on-line concentration)。
樣品堆積技術可根據緩衝溶液間形成之界面移動方式歸納成兩大系 統:(1)移動界面堆積(moving boundary staking):所有樣品區帶是 以相同速度移動與(2)靜相界面堆積(stationary boundary staking):
由 Chien 與 Burgi[87]在 CZE 模式中對帶電粒子所導入的電場放大樣
品堆積技術(field amplified sample staking),其中樣品區濃度較低,
緩衝溶液濃度較高,兩者之間形成的界面在電泳分析期間是保持不動 的。一旦施加高電壓,帶電粒子受到電場增強效應而速度增加,之後 在樣品與緩衝溶液的交接處堆積。這是因為緩衝溶液導電度大、電場 較小,帶電粒子至此速度驟減而堆積。
後來 Terabe 以及許多科學家努力研究[88-97],提出中性分析物 的樣品濃縮方法-正向堆積模式,並開發出在 MEKC 下數種不同的 線上濃縮模式,如表 2-4 所示,以下僅介紹本實驗過程中所採用的毛 細管電泳掃集法(sweeping-MEKC)
表 2-4 各種毛細管電泳線上濃縮的模式
正向堆積模式
(Normal Stacking Mode, NSM)
反向電極極性堆積模式
(Reversed Electrode Polarity Mode, REPSM)
反向遷移微胞堆積
(Stacking with Reverse Migration Micells, SRMM)
場強放大樣品注射
(Field-Enhanced Sample Injection, FESI)
反向遷移微胞之場強放大樣品注射
(Field-Enhance Sample Injection with Reverse Migration Micells, FESI-RMM)
反向遷移微胞與水之堆積
(Stacking with Reverse Migration Micells and Water Plug, SRW)
掃集堆積模式
(Sweeping-MEKC)
陽離子選擇性注入掃集堆積模式
(CSEI-sweeping-MEKC)
2-4.1 毛細管電泳掃集法(sweeping)
1998 年 Quirino 與 Terabe 消除了 EOF 存在時可能帶來的影響,
發展出一種稱為掃集法的 MEKC 線上樣品堆積技術。該堆積模式特 色在於樣品基質與分離緩衝溶液間維持相等的導電度。機制為先在毛 細管中注滿低pH 值,含有微胞的緩衝溶液(micellar backgrand solution, BGS)以降低電滲流值幾近於零,然後進樣一段樣品基質,再將毛細 管柱兩端置於BGS 溶液中,通高壓電後,來自 BGS 的微胞在電壓作 用下會進入樣品區,當微胞經過樣品基質時,會與分析物結合,並將 分析物掃集至一個狹窄的濃縮區。此過程就稱為掃集(sweeping),
當微胞通過樣品基質區以後,就以一般的 MEKC 模式,完成電泳分 析。
分析物依據和微胞分布係數的不同與微胞結合,與微胞結合度越 強的分析物,掃集的效果就越好,相對的,與微胞結合度差的分析物,
所堆積的譜帶較寬,分離效率較差。
一般而言,利用毛細管電泳掃集法可降低大約 3~4 個級數的偵 測極限,可用來改善紫外光偵測器在低濃度的樣品分析時,無法偵測 到訊號的缺點。
micelles >0 micelles=0
sample
concentration zone micelles=0
ab
c
injection
detector
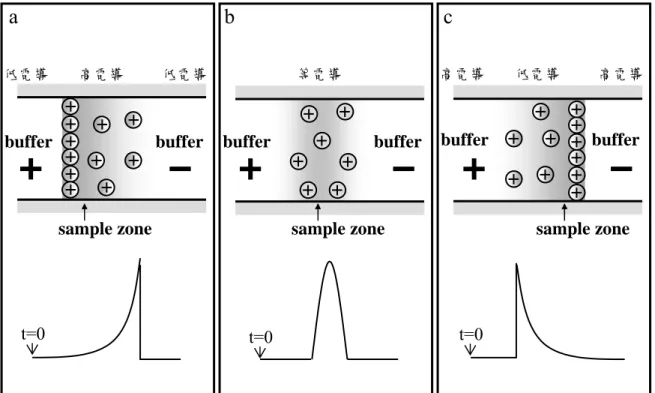
圖 2-9 毛細管電泳掃集模式
(a)毛細管內充滿酸性含有微胞的緩衝溶液(BGS),將樣品配在 導電度與緩衝溶液相同的基質中,進樣至毛細管中
(b)通負電進行電泳,微胞進入毛細管,與分析物結合開始掃集
(c)微胞通過樣品區帶後,以一般MEKC模式,完成電泳分析
2-5 77 K 低溫螢光光譜分析法
1960 年,Shpol’skii[98-99]等研究者首先發現利用長鏈的烷類(如 n-alkane)當作溶劑來測量有機化合物時,尤其是多環芳香族化合物
(PAHs),由於長鏈烷基在低溫時會形成 glasses(晶格狀),恰可將 PAHs 包住,使 PAHs 的分子與分子之間無法相互作用,電子的能量 無法傳遞,只能利用螢光或磷光的形式放出,因此可增強螢光或磷光 的強度,而可觀測到相當窄(高解析度)的譜線。並可由每個譜線間 的差異得知分子各個振動能階的量值。所以,他們把這種在低溫下以 長鏈烷類為基質的螢光光譜稱為 Shpol’skii 低溫光譜。因為室溫環境 下螢光光譜振動譜寬太大(~50nm),僅能提供相當有限的振電躍遷 的光譜資訊,而在液態氮(77 K)的溫度下,光譜的頻寬為 10-30 cm-1, 在液態氦(4.2 K)的溫度下,光譜頻寬更窄,可降至 1-10 cm-1左右。
因此在低溫下,光譜的解析可以相當的窄(又稱 quasi-lines)[99],
可以清楚的獲知分子的各個振動能態。圖2-10 為分子在室溫(如圖 a)
及0 K(如圖 b)時的振動能階示意圖。當電子吸收固定能量後,會 躍遷至激發態,再經由緩解作用,以不同的方式放出螢光、磷光,甚 至於以熱能形式放出。但由於在室溫下,分子的量子分佈呈現波茲曼 分佈,並非全部都處於基態,所以當分子激發至激發態而放出螢光 時,是以帶狀光譜的形式呈現,根本無法解析出分子的細微能階。但 是,若將溫度降低至液態氮的溫度(77 K)甚至液態氦的溫度(4.2 K)
時,且溶於長鏈烷類基質或在低溫之下足以形成較佳晶格的溶液中,
則分子在基態的電子分佈範圍可較為一致,因此放出的螢光譜線較 窄,可以得到較細微的電子振動能階資訊。
圖2-11 為 trans-resveratrol 溶於乙醇的室溫及 77 K 低溫螢光光譜 之比較。在室溫下測得的螢光光譜譜峰相當寬廣,且螢光量子產率
(ΦF)僅有0.05,為弱螢光性物質。但是將溫度降到液態氮的溫度時
(77 K),螢光量子產率驟增為 0.75,螢光強度明顯的增強,且容易 觀測到清晰的譜峰。由於降低溫度可使化合物展現其特定的譜峰,因 此對於相似異構物而言,藉由譜峰位置及強度的相對關係,可供給最 佳的光譜指紋比對作為精確定性分析之用。
本研究結合毛細管電泳sweeping-MECK 以及 Shpol’skii 的低溫螢 光光譜法的好處,發展出獨創的毛細管電泳掃集濃縮/77 K 低溫螢 光光譜分析法,以電泳濃縮分離、瞬間冷卻、on-line 取得低溫螢光光 譜進行分析為實驗步驟,針對葡萄酒中 trans-resveratrol 化合物進行分 析研究,不僅可使分析物螢光強度增強,更可獲知更多更明顯的譜峰 以供光譜指紋(spectral fingerprint)比對。