國立臺灣大學理學院化學系 碩士論文
Department of Chemistry College of Science
National Taiwan University Master Thesis
立障萘啶酮配位基之三核金屬串合成與研究 Synthesis and Studies of Triunclear Metal String
Complexes with Bulky Ligand (7-methyl-1,8-naphthyridin-2(1H)-one)
郭世英 Shih-Ying Kuo
指導教授:彭旭明 博士 Advisor: Shie-Ming Peng, Ph.D.
中華民國 108 年 7 月
July, 2019
i
中文摘要
本 篇 論 文 使 用 帶 有 甲 基 的 立 障 不 對 稱 型 萘 啶 酮 (7-methyl-1,8-naphthyridin-2(1H)-one, Hmnpo)配基來合成出第二周期過渡金屬非螺 旋性金屬串錯合物,並透過 X-ray 單晶繞射、循環伏安法、紫外光-可見光光譜、
核磁共振儀來鑑定及探討其性質。在合成上,將立障萘啶酮配基分別加入釕 (ruthenium)以及鉬(molybdenum)的相關前驅物並以萘(naphthalene)作為溶劑,於 220
oC 含氬氣的條件下進行萘燒反應 8 個小時,透過分層養晶(bilayer)的方式可得到第 二 周 期 過 渡 金 屬 的 三 核 金 屬 串 錯 合 物 Ru3(mnpo)4Cl(PF6) (1) 以 及 RuMoRu(mnpo)4Cl(PF6) (2)。錯合物(2)是第一個 RuMoRu 的三核金屬串錯合物被合 成出來。透過單晶繞射的結構可以發現,四片配位基皆有脫氫的情形,並以非螺 旋的形式和金屬串配位。因為配基的立障因素,其中一片配基有移位的現象發生,
造成萘啶上的一個氮原子沒有和金屬配位,此金屬串並沒有失序(disorder)的情形,
因此有利於結構的探討。由第二周期過渡金屬三核金屬串的分子軌域可以判斷,
錯合物(1)和(2)的相近金屬離子之間的鍵級均為 1.5 鍵,在磁性上均為逆磁。從單 晶結構可以發現,錯合物(1)及錯合物(2)的相鄰金屬離子鍵長均在 2.25Å ~ 2.27Å 之 間,屬於1.5 鍵的範圍。錯合物(2)的核磁共振圖譜以及順磁共振圖譜更顯示其逆磁 的特性,此外由循環伏安法可以看出錯合物(1) 在+0.21 伏特時有一個不可逆的氧 化反應,而錯合物(2)在+1.10 伏特以及-0.68 伏特各有一個可逆的氧化還原對。
Abstract
In recent years, scientists has fascinated with the concept of single-molecole conducting wire with the advancement of nanotechnology. In this thesis, we focus on extended metal-atom chain about trinuclear second-row transition metal string complexes with bulky ligand Hmnpo (7-methyl-1,8-naphthyridin-2(1H)-one).
[Ru3(mnpo)4Cl](PF6) (1) and [RuMoRu(mnpo)4Cl](PF6) (2) have been successfully synthesized. Complex 2 is the first synthesized RuMoRu trinuclear string complex. The molecular structure shows that the metal-atoms chains in these complexes are nearly non-helically coordinated by four deprotonated ligands. Due to the steric effect of Hmnpo, the displacement of one of the ligands is inevitable, leaving a nitrogen atom on naphthyridine remain uncoordinated. According to the molecular orbital model of tri-metal-atom chain described previously1, the electronic configurations of both Complex 1 and 2 results in the diamagnetism and the bond order is 1.5 between adjacent metal ions. The crystal structures show that the Ru–Ru and Ru–Mo bond lengths is 2.2699(14), 2.2753(14) Å in (1) and 2.2552(12), 2.2694(13) Å in (2), respectively, which is corresponding to a bond order of 1.5 between adjacent metal as expected. NMR and EPR spectrum of (2) reveals the diamagnetism of the complexes.
Cyclic voltammetry shows that Complex 1 undergoes an irreversible oxidative process at E1/2 = 0.22V, while Complex 2 exhibits two reversible redox couples.
iii
目錄
中文摘要 ... i
Abstract ... ii
目錄 ... iii
圖目錄 ... v
表目錄 ... vii
第一章 緒論 ... 1
1-1 前言 ... 1
1-2 金屬串分子導線 ... 2
1-2-1 無架橋配基之分子金屬串錯合物 ... 4
1-2-2 有架橋配位基之分子金屬導線 ... 6
1-3 金屬-金屬鍵結理論 ... 9
1-3-1 雙核金屬錯合物之金屬-金屬鍵結 ... 10
1-3-2 直線型三核過渡金屬錯合物之金屬-金屬鍵結 ... 13
1-4 金屬串錯合物之多氮配位基 ... 16
1-4-1 多吡啶胺配位基 ... 17
1-4-2 多萘啶胺配位基 ... 19
1-4-3 多萘啶吡啶胺配基 ... 21
1-5 金屬串錯合物之探討 ... 22
1-5-1 同核金屬串錯合物 ... 22
1-5-2 三核異金屬串錯合物 ... 23
1-5-3 不對稱配位基金屬串錯合物 ... 27
1-5-4 以非氮原子配位金屬串錯合物 ... 29
1-6 研究動機 ... 30
第二章:實驗部分 ... 31
2-1 試藥與儀器 ... 31
2-1-1 試藥與溶劑 ... 31
2-1-2 實驗儀器 ... 33
2-2 化合物合成 ... 34
2-2-1 金屬前驅物的合成 ... 34
2-2-2 配位基的合成 ... 35
2-2-3 金屬串錯合物的合成 ... 37
2-3 晶體數據之收集與整理 ... 40
2-3-1 [Ru3(mnpo)4Cl](PF6) (1)... 40
2-3-2 [RuMoRu(mnpo)4Cl](PF6) (2) ... 41
第三章:結果與討論 ... 42
3-1 晶體結構解析 ... 42
3-1-1 [Ru3(mnpo)4Cl](PF6)(1)晶體結構解析 ... 42
3-1-2 [RuMoRu(mnpo)4Cl](PF6)(2)晶體結構解析 ... 46
3-2 磁性探討 ... 51
3-3 電子吸收光譜分析 ... 54
3-4 電化學分析 ... 56
第四章:結論 ... 60
參考文獻 ... 61
附錄 光譜和晶體數據 ... 65
v
圖目錄
圖1-1 金屬導線和微小化後的分子導線示意圖4b ... 3
圖1-2 分子導線示意圖4c ... 3
圖1-3 n[Pt(CN)4]2-透過部分氧化而成為[Pt(CN)4]n1.7- ... 5
圖1-4 (a)一維無限延長銠金屬串錯合物[Rh(MeCN)4(BF4)1.5]x的晶體結構 (b)由 C 軸方向觀察其結構堆積情形7 ... 5
圖1-5 [Pd10(β-carotene)2][B(ArF)4]之晶體結構圖8 ... 6
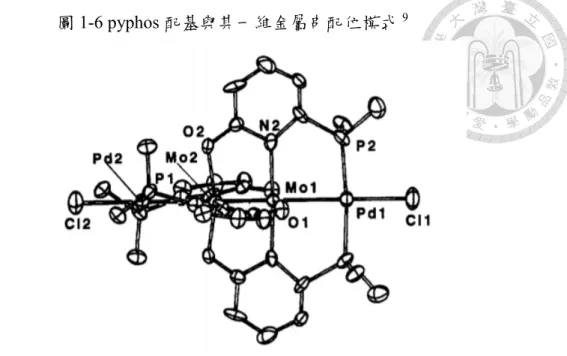
圖1-6 pyphos 配基與其一維金屬串配位模式9 ... 6
圖1-7 PdMoMoPd(pyphos)4Cl2之晶體結構圖9 ... 7
圖1-8 各式各樣的配位基與不同金屬組合出多樣的金屬串錯合物16 ... 7
圖1-9 三個鉬為鹵素(X)所連結16 ... 9
圖1-10 [Re2Cl8]2-錯合物晶體結構圖2 ... 9
圖1-11 兩個金屬離子透過五種 d 軌域所生成的鍵結軌域 ... 10
圖1-12 雙核金屬錯合物之中的金屬金屬四重鍵作用力 ... 12
圖1-13 雙核金屬串電子數與鍵級關係圖 ... 12
圖1-14 直線型三核金屬錯合物之鍵結、反鍵結和非鍵結軌域圖 ... 13
圖1-15 M3(dpa)4(NCS)2 中M=Cr, Ru, Co 能階示意圖18 ... 14
圖1-16 軸向配位基對三核釕金屬串能階影響示意圖1... 14
圖1-17 二吡啶胺氮原子配位的三種構型 ... 16
圖1-18 五吡啶四胺之九核純鎳串錯合物單晶結構圖13a ... 17
圖1-19 多吡啶胺配位基 ... 18
圖1-20 多吡啶胺系統之中已得到單晶結構的純串錯合物 ... 18
圖1-21 三吡啶二胺中三個吡啶環上的氮以全反向型和鐵離子配位21 ... 18
圖1-22 [Ni2(ny)4Br2][BPh4]的單晶結構圖23 ... 19
圖1-23 多吡啶胺配位基 ... 19
圖1-24 [Ni11(tentra)4Cl2]4+之單晶結構圖14 ... 20
圖1-25 多萘啶胺系統之中已得到單晶結構的純串錯合物 ... 20
圖1-26 部分多萘啶吡啶胺配位基結構 ... 21
圖1-27 [Ru3(dpa)4Cl2](左)及[Rh3(dpa)4Cl2](右)單晶結構圖10a ... 22
圖1-28 在異三核金屬串系統中金屬的排列情形示意圖 ... 23
圖1-29 (a) [CuPdCu(dpa)4Cl2];(b) [CuPtCu(dpa)4Cl2]之單晶結構圖27-28 ... 24
圖1-30 (a) [MnNiMn(dpa)4Cl2];(b) [MnPtMn(dpa)4Cl2]之單晶結構圖29 ... 24
圖1-31 Ru2Cu(dpa)4Cl2金屬串錯合物30 ... 25
圖1-32 MoWCr(dpa)4Cl2的合成途徑與單晶結構31 ... 25
圖1-33 NiCoRh(dpa)4Cl2合成中金屬離子置換示意圖32 ... 26
圖1-34 NiCoRh(dpa)4Cl2單晶結構圖32 ... 26
圖1-35 不對稱配基所產生出的四種結構異構物36 ... 27
圖1-36 金屬串錯合物之異構物分布機率36 ... 27
圖1-37 Ni4(phdpda)4之單晶結構圖33 ... 28
圖1-38 Mo2(mnpo)4(左)以及 Ru2(mnpo)4(右)之單晶結構圖34 ... 28
圖1-39 (a)吡啶噻唑胺 (b)萘啶酮 (c)萘啶硫酮之結構圖35-37 ... 29
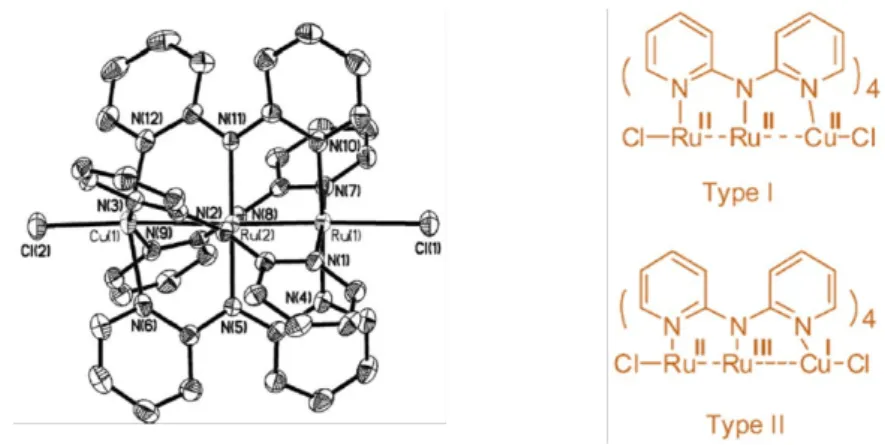
圖3-1 [Ru3(mnpo)4Cl](PF6) (1)之單晶結構圖 ... 42
圖3-2 [Ru3(npo)4Cl2]-之金屬串錯合物結構圖36 ... 43
圖3-3 錯合物
1 部分鍵長(Å) ... 44
圖3-4 [RuMoRu(mnpo)4Cl](PF6) (2)之單晶結構圖 ... 46
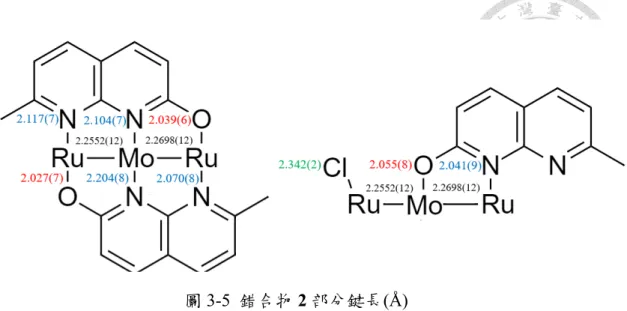
圖3-5 錯合物
2 部分鍵長(Å) ... 48
圖3-6 (a)順磁性 (b)鐵磁性(c) 反鐵磁性(d) 陶鐵磁性36 ... 51
圖3-7 電子能階分裂圖 ... 52
圖3-8 錯合物
2 之電子順磁共振光譜 ... 53
vii
圖3-9 [Ru3(mnpo)4Cl](PF6) (1)、[RuMoRu(mnpo)4Cl](PF6) (2)以及配位(Hmnpo)在
CH2Cl2之中的紫外光-可見光吸收光譜圖 ... 55
圖3-10 錯合物
1 之循環伏安圖 ... 57
圖3-11 錯合物
2 之循環伏安圖 ... 58
表目錄
表1-1 M(II) [Pt(CN)4]晶體分子間鉑鉑距離和晶體顏色6 ... 4表1-2 [Ru3(dpa)4C12] M=Ru, Rh 之金屬鍵比較10a ... 15
表3-1 錯合物
1 與其他釕三核金屬串錯合物之部分鍵長(Å)及鍵角(°)比較 ... 46
表3-2 含有釕及鉬金屬離子三核金屬串之金屬氮鍵比較 ... 49
表3-3 直線型三核金屬串之金屬-金屬鍵結比較... 50
表3-4 配位基、錯合物
1 及錯合物 2 之最大吸收波長和消光係數 ... 56
表3-5 錯合物
1 和 2 及其他金屬串錯合物之 d-d transition 最大吸收波長 ... 56
表3-6 錯合物
1 和 2 及其他釕三核金屬串錯合物之氧化還原電位 ... 59
第一章 緒論
1-1 前言
化學經歷了長時間的發展,它是一門研究物質的性質、組成、結構、以及變 化規律的基礎自然科學,自所謂科技始終來自人性,生活中的每一部分都因化學 而有所改變,對於人類的進步有極大的貢獻。奈米科技的發展隨著時代的變遷、
產業的轉型,逐漸成為科學領域中最具潛力的技術之一。
奈米是一個全新的研究領域,研究一維尺寸在約 1~100 奈米範圍內的材料、
結構、性質的科學,我們稱之為奈米科學。而奈米科技起源於各類型電子顯微鏡 技術逐漸成熟,乃至近年來x-ray 單晶繞射儀相關技術快速發展,對於奈米量級物 質得以清楚的觀察解析,因此原子級或分子級的材料得以迅速發展,使得所有電 子元件開始趨於微型化,也開始有分子導線的概念被提出。
奈米技術的概念最早是由美國的物理學家理察·費曼(Richard Feynman)在一次 的演講中所提出。樣品的製程可利用單分子由下而上的的自組裝來完成(bottom up),
因此人類可以隨心所欲地利用小尺寸材料並呈現出嶄新的應用,將對人類的科技 與生活創造出新的世界。
一般而言,奈米材料的製作方法可以依照其尺度的變化而分成兩種:ㄧ種是 由上到下(top-down)的方式來製成,透過外力作用或化學反應來將塊材(bulk)逐步 地 分 解 , 使 其 大 小 逐 漸 的 下 降 至 奈 米 尺 度 的 範 圍 ; 另 外 一 種 則 是 由 下 到 上 (bottom-up) 的方式,透過化學反應或物理的作用力,來將原子或分子組裝至奈米 尺度的大小。本實驗室的合成方法即是屬於bottom-up 的方式之一,我們使用以氮 為主的配位基來和金屬離子產生鍵結,組裝成長達數奈米的直線型金屬串錯合物。
在除了探討金屬與金屬之間的作用力之外,也使用掃描穿隧式電子顯微鏡技術研 究其導電度,期望發展成分子導線(molecular wires)或分子開關,希望在未來能 應用在奈米電子元件上。
1-2 金屬串分子導線
隨著奈米科技的進步,電子元件逐漸微小化,分子級的導線發展的潛力也逐 漸顯現,將擔任在奈米級的電子元件之間傳遞電荷及能量的功能。
分 子 導 線 的 研 究 可 以 追 溯 至 於 1964 年具有金屬-金屬四重鍵的錯合物 [Re2Cl8]2-由Cotton 團隊所合成出來並確認2,開始有了金屬離子之間多重鍵的概念,
錯合物中金屬離子之間的鍵結使金屬之間可以有直接的交互作用,引發了科學家 的興趣,從此錯合物的研究不在侷限於單一金屬,目前已經由雙金屬錯合物擴張 至三核,乃至多核的金屬串分子導線。本實驗室於1994 年開始進行直線型多核金 屬串錯合物的合成與研究,及具有金屬-金屬鍵錯合物分子的探討4。從結構上來 看本實驗室所合成出來的金屬串錯合物,其中的金屬離子彼此排列成直線,形成 直線型的金屬骨架,由四個脫氫的有機含氮配位基所圍繞配位,其圍繞在金屬離 子外的有機配基可視為絕緣體,而中間金屬離子則視為導體。
微觀尺度上的金屬串錯合物可以視為巨觀尺度上生活中常見的電線,電線一 般以銅線作為導線,外為塑膠絕緣體所包覆,金屬串中金屬原子之間可以通導電 子,經過理論計算,金屬串錯合物在施以電流時,電荷幾乎僅侷限在中間金屬離 子中流動,配位基則鮮少有電子流經。故金屬串錯合物的金屬骨架相當於電線中 的導線,其導線的直徑僅為一個金屬原子,配位基則相當於電線中絕緣體的部份,
沒有電流流經而作為支撐金屬導線的功能。
直線型的金屬串錯合物在做為奈米級的導線有很大的潛力,因此許多科學家 以及研究團隊開始致力於金屬串錯合物的合成與研究。在眾多的直線型金屬串錯 合物中可以分為兩大類:(1) 無架橋配基之金屬串錯合物 (2) 有架橋配基之金屬串 錯合物,將在之後章節加以討論。
圖1-1 金屬導線和微小化後的分子導線示意圖4b
圖1-2 分子導線示意圖3
1-2-1 無架橋配基之分子金屬串錯合物
無架橋配基金屬錯合物是由金屬金屬離子之間的鍵結所排列而成
,
這類的金 屬串錯合物不需要透過配位基來支撐分子的架構,而是由金屬金屬間直接的作用 力去支持整個金屬串的結構。從 1960 年代起,開始有金屬之間的鍵結相關的文獻報告,其中在 1969 年由 K. Krogmann 研究團隊所發表的文章6,[Pt(CN)4]2-中鉑離子以方形平面的配位模式,
和四個 CN-的碳端進行鍵結配位。對於在水溶液中無色的錯合物[Pt(CN)4]2-,形成 晶體時,會因為晶體中陽離子的不同以及結晶水數量的差異改變晶體中分子的排 列,造成分子間鉑鉑距離的不同,進而使鉑金屬之間的作用力有所差異,導致晶 體顏色的不同(如表 1-1 所示),文獻中也透過部分氧化來將各個平面分子串連成一 維延伸的分子導線 (如圖 1-3 所示)。在形成一維鍊狀結構後,[Pt(CN)4]2-單體中鉑 之間透過dz2軌域來生成作用力,以σ 鍵的型式產生鍵結,形成一維直線型金屬串 的模式,其中鉑和鉑之間不具有架橋,因為擁有相對於一般的錯合物較好的導電 能力,對於分子金屬導線具有相當高的潛能,因此在奈米材料的製程上備受矚目。
表1-1 M(II) [Pt(CN)4]晶體分子間鉑鉑距離和晶體顏色6
圖1-3 n[Pt(CN)4]2-透過部分氧化而成為[Pt(CN)4]n1.7-
原本無架橋配基的金屬錯合物皆為有限長度,直到 1999 年 Dunbar 教授實驗 室嘗試將銠的雙核錯合物(Rh2II,II)分別進行氧化以及還原的反應7,在氧化的反應中 得到單核的銠三價錯合物,在還原的過程卻得到一維無限延長的銠金屬串錯合物,
其中銠的金屬平均價數為1.5,為混價的型態。
圖1-4 (a)一維無限延長銠金屬串錯合物[Rh(MeCN)4(BF4)1.5]x的晶體結構7 (b)由 C 軸方向觀察其結構堆積情形7
(a) (b)
1-2-2 有架橋配位基之分子金屬導線
部份分子導線是透過具有架橋的配位基合成的,這類的分子導線的長度會受 到配位基的限制,因此選用較長的配位基較有合成較長的金屬導線的潛力。這一 類的金屬串錯合物因為存在架橋配位基支撐整個分子,因此金屬和金屬離子之間 可以不存在鍵級或作用力。
其中於2015 年由 T. Murahashi 團隊發表在 Nature Chem 的論文8,以β-胡蘿蔔 素分子作為架橋配位基,鈀金屬與β-胡蘿蔔素分子上的碳配位形成有機金屬錯合 物,十個鈀金屬呈直鏈排列作為金屬串的骨架,由2 個 β-胡蘿蔔素分子配位,如 圖1-5 所示,此為現今最長的鈀金屬串錯合物。
圖1-5 [Pd10(β-carotene)2][B(ArF)4]之晶體結構圖8
1993 年 , Mashima 教 授 在 J. Am. Chem. Soc. 發 表 以 6-(Diphenylphosphino)-2-pyridone (pyphos)作為架橋配位基9,配位基以三種不同的 元素進行配位,利用鉬金屬雙核錯合物加入鈀金屬,得到四核異金屬串錯合物,
如圖1 和圖 1-6 所示。除了鉬-鉬四重鍵之外,更探討金屬串中鉬和鈀之間的作用 力。
圖1-6 pyphos 配基與其一維金屬串配位模式9
圖1-7:PdMoMoPd(pyphos)4Cl2之晶體結構圖9
本 實 驗 室 所 合 成 的 金 屬 串 錯 合 物 也 屬 於 此 一 類 型 , 常 見 使 用 多 吡 啶 胺 (oligo-α-pyridylamine) 作為架橋配位基,透過配位基上的氮原子來和金屬離子進行 鍵結,以形成多核金屬串錯合物,故配基上的氮原子數目愈多,就更有合成出長 串錯合物的潛力。使用多吡啶胺所合成出來的金屬串分子,大多是以直線型排列 的金屬離子作為中心骨幹,四個配基脫氫以螺旋型式圍繞,配位基會以螺旋性圍 繞的原因是相鄰吡啶環的氫原子互相排斥,促使可塑性高的配位基藉由單鍵旋轉 排開立障所致。選用不同種類的金屬以及配位基來進行合成,其所產生的配位模 式、電子組態都將會有所不同,進而可以針對金屬金屬鍵進行探討,在本實驗室 近20 年來致力於金屬串的合成及研究得到的成果,我們已經合成出一系列三核5,10、 五核 11、七核 12、九核 13甚至十一核 14及十三核 15之金屬串錯合物,且除了同核 金屬外,近幾年更是加入其他種類的金屬形成異核金屬串錯合物,不同種類的金 屬串骨架與各式各樣的配位基排列組合,可以產生非常多樣的金屬串錯合物(圖 1-7),藉著研究這些錯合物的特性,我們可以逐漸了解金屬串中金屬離子之間的相 互作用力。多年的研究以來,我們針對金屬離子之間的作用力進行探討,主要是
金屬串,都得到了相當豐富的結果。
對於有架橋配位基的金屬串錯合物而言,架橋配位基可以粗分為三大類,分 別為多吡啶胺配位基、多萘啶胺配位基以及多萘啶吡啶胺配位基,將在1-4 章節介 紹,三者的研究在本實驗室均有相當多的成果。
圖1-8 各式各樣的配位基與不同金屬組合出多樣的金屬串錯合物16
1-3 金屬-金屬鍵結理論
配位錯合物的存在早已為科學家們所知,但是在1893 年由 Alfred Werner 提出 過渡金屬錯合物六配位八面體的模型後才逐漸趨於完善,奠定了配位化學發展的 基礎。Alfred Werner 同時也了解到多核金屬錯合物的存在,但僅解釋為多個單核 金屬的錯合物透過架橋配位基結合,金屬彼此之間並不存在任何形式的作用力(如 圖1-8),金屬-金屬之間鍵結理論在這個時期仍未被討論16。
圖1-9 三個鉬為鹵素(X)所連結,當時認為鉬鉬之間不存在作用力16
直到1964 年,F. A. Cotton 針對[Re2Cl8]2-中Re2之金屬-金屬多重鍵進行討論(圖 1-9),Re-Re 鍵長為 2.24 Å,Re 金屬的氧化數是+3,價電子之電子組態為 5d4,所 以兩個錸金屬之間可以有8 個 d 電子作鍵結,形成 4 個鍵,即鍵序(bond order)為 4;
這證實了金屬具有四重鍵的存在。此後金屬-金屬多重鍵理論慢慢受到重視,並引 發科學家們對於探討金屬間的作用力的興趣,關於多核金屬錯合物的研究才開始 蓬勃發展2。
圖1-10 [Re2Cl8]2-錯合物晶體結構圖2
1-3-1 雙核金屬錯合物之金屬-金屬鍵結
再生成金屬金屬鍵時,填滿了電子的(n-1)s 與(n-1)p 軌域不予考慮,多半得依 靠金屬中的(n-1)d 軌域來進行混層,而金屬的 ns 和 np 軌域會因的能量較高提供空 得軌域而去和配位基生成配位鍵,若不和配基形成鍵結時,則往往因為能量高為 空軌域而不參與金屬間的鍵結。當要透過 d 軌域來生成金屬鍵時,和一般軌域之 間要形成鍵結時的條件類似,兩個d 軌域彼此之間的能量要相近、對稱性要相同,
並且其軌域重疊(overlap)要達到一定的程度,當這三要素都符合時,兩個軌域才可 以進行鍵結。對於過渡金屬而言有5 個 d 軌域,可鍵結形成一個 σ 鍵、二個 π 鍵、
二個δ 鍵,以及其相對的反鍵結(anti-bonding orbital):σ*、π*、δ*,如圖 1-11。
圖1-11 兩個金屬離子透過五種 d 軌域所生成的鍵結軌域 (bonding orbital)
根據基本分子軌域觀念,其鍵結強度與軌域的重疊度成正比,軌域的重疊度 為σ>π>>δ,鍵結強度越強的軌域其反鍵結能量通常越高。而鍵結能階的穩定度和 軌域形成鍵結時產生的節點(node)數目為負相關,σ 的節點數為 0,所以最穩定,
π 的節點數為 1,δ 的節點數為 2,所以能量較高,因此在定性上各能階依照能量 的大小,排列如下:
σ < π << d < δ* << π* < σ*
若以通過金屬-金屬鍵中心軸為 Z 軸時,會受到來自 x 以及 y 方向配位基的作 用力,金屬離子的dx2
-y2軌域往往會與配位基配位形成σ 鍵,所以 dx2
-y2 軌域並不 參與金屬-金屬鍵結,因此參與金屬-金屬鍵結軌域的為其他 4 個 d 軌域,即 dxy、 dyz、dxz 及 dz2軌域。
根據分子軌域理論定義,其鍵級 (bond order) 的定義如下:
bond order =(n
b-n
ab)/2
nb : 鍵結(bonding)軌域電子數 nab : 反鍵結(anti-bonding)軌域電子數
一般而言針對鍵級以及鍵長(bond length)的關係為負相關,對於特定的金屬金 屬鍵的特定鍵級,其鍵長會在一定的範圍內。除了鍵級以外,金屬串錯合物中金 屬-金屬之間距離還會受到很多其他因素影響,如:架橋配位基帶來結構上的扭力、
軸向配基的電負度強度以及配位能力等。
當錯合物是以方型平面 (square planar) 的形式和配基鍵結時,金屬離子會以 dx2-y2軌域來和配基生成配位鍵,而剩下四個未使用的d 軌域。此時若兩個金屬的 d 軌域能量相當,即可以透過軌域的重疊產生鍵結,分別形成一個σ、兩個 π、一個 δ 軌域和其各自的反鍵結軌域。
在 [Re2Cl8]2-之中 2,雙核錸金屬離子均為正三價,各自具有四個 d 電子,總 計共有八個 d 電子,若將其依照能階由低至高填入分子軌域當中,可以得到電子
(quadruple bond) 作用力。
圖1-12 雙核金屬錯合物之中的金屬金屬四重鍵作用力
將雙核金屬錯合物各種金屬離子含有的 d 電子數,由較低的分子能階依序向 上填入於分子軌域中,可以得到d 電子數和鍵級的關係如 1-12 所示,當電子組態 為 d4時,此時鍵級達到最大值 4,意味著金屬之間的四重鍵。當金屬離子是鎳二 價時,電子組態為d8,此時鍵序為0,雙核金屬間不具有金屬鍵存在。
圖1-13 雙核金屬串電子數與鍵級關係圖
1-3-2 直線型三核過渡金屬錯合物之金屬-金屬鍵結
從雙核金屬錯合物的金屬-金屬鍵結理論,可以進一步延伸到直線型三核過渡 金屬金屬之間的鍵結,可以視為雙核的金屬-金屬鍵於 Z 方向再加入一個金屬參與 鍵結,和雙核金屬錯合物一樣,其dx2-y2軌域往往會與配位基以σ 鍵的形式進行配 位。三個金屬的dz2可以形成三個σ 分子軌域,一個為鍵結(bonding orbital)軌域(σ),
一個為未鍵結軌域(non-bonding orbital) (σnb),一個為反鍵結軌域(anti-bonding orbital) (σ*),同樣形成分子軌域的方式,dyz 及 dxz 軌域可以產生兩個
π
鍵結軌域,兩個
π
nb未鍵結軌域,以及兩個π*
反鍵結軌域,dxy 軌域則可以產生一個δ
鍵結軌域,一個
δ
nb未鍵結軌域,以及一個δ*
反鍵結軌域,其鍵結模式如圖1-13 所示。圖1-14 直線型三核金屬錯合物之鍵結、反鍵結和非鍵結軌域圖
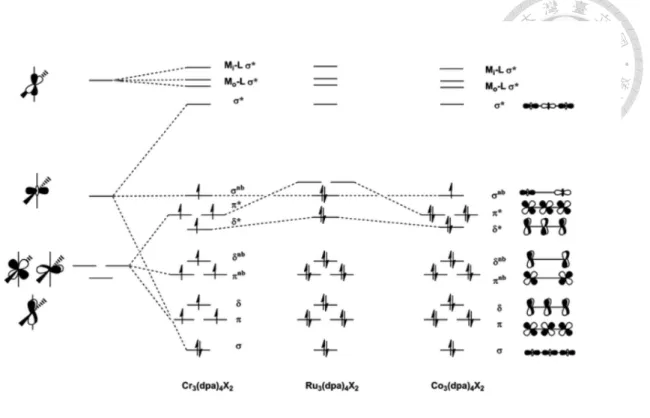
近年來透過直線型三核金屬串錯合物x-ray 單晶結構的解析,以及各種性質的 研究和理論計算可以得知,此分子軌域系統各能階高低會受到金屬種類18以及軸 向配位基(axail ligand)的陰電性和在光化學序列 (spectrochemical series) 1的排序等 因素而有所影響,如圖1-14、1-15 所示。
圖1-15 M3(dpa)4(NCS)2 中M=Cr, Ru, Co 能階示意圖18
圖1-16 軸向配位基對三核釕金屬串能階影響示意圖1
若將電子由低能量之能階依序往上填,可以得到三核金屬串的電子組態,透 過先前所提及計算鍵級的公式bond order = (nb-na) / 2,可以得到三核金屬串的總鍵 級,唯三核金屬串含有兩個金屬-金屬鍵,若電子在三個金屬離子之間為未定域 (delocolize),鍵級被平均分配於兩個金屬鍵,因此在計算三核金屬串中鄰近兩個金 屬離子之間的鍵級,須將原始公式進一步再除以二,修正如下:
bond order = (n
b-n
ab)/4
n
b: 鍵結軌域電子數 n
ab: 反鍵結軌域電子數
經由以上公式計算可以得知三核金屬串中鄰近兩個金屬之間的鍵級,進而了 解三核金屬串中,金屬與金屬之間的鍵結模式。
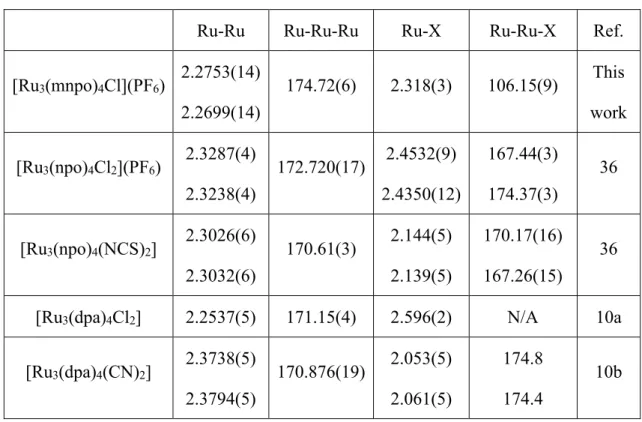
在[Ru3(dpa)4C12]及[Rh3(dpa)4C12]中10a,因為釕金屬以及銠金屬均為第二週期 過渡元素金屬,而且軸向配位基氯是弱場,因此推斷金屬串分子軌域如圖1-15,
Ru36+三個釕總計有18 個價電子,由能量低的能階依序往上填入電子,按照鍵級公 式去計算,[Ru3(dpa)4C12]之三個釕總鍵級為 3,相鄰兩個釕為 1.5 鍵,同樣的計算 方式可得[Rh3(dpa)4C12]中 Rh36+總鍵級為1.5,相鄰兩個銠之間為 0.75 鍵,實驗結 果也顯示鍵長以及鍵級是呈負相關的趨勢(表 1-2),與理論是相符的。
表1-2 [Ru3(dpa)4C12] M=Ru, Rh 之金屬鍵比較10a
當三核釕錯合物使用強場做為軸向配位基,會使
π*
穩定化(stabilized),導致電 子優先填入反鍵結軌域,使得nab增加,進而減少三核金屬串的鍵級,從金屬金屬 鍵的鍵長以及磁性上的分析都有觀察到相關現象,如[Ru3(dpa)4C12]中 Ru 之間為 1.5 鍵,錯合物為逆磁,[Ru3(dpa)4(CN)2]中 Ru 之間為 1.0 鍵,錯合物為順磁10b。1-4 金屬串錯合物之多氮配位基
自 1893 年由 Alfred Werner 提出配位化學相關理論開始,在金屬的研究發展自 今以有100 多年的歷史,研究的方向從單核金屬離子的配位,擴展自雙核金屬-金 屬鍵,延伸至今日的多核金屬錯合物甚至是無限延伸的聚合物,同時配位基也不 斷的推陳出新。
在1968 年二吡啶胺的三核鎳串錯合物首次被合成出來,但當時未成功預測其 結構19,直到1991 年 Thomas J. Hurley 等人發表於 Dalton 的文獻20中,他們重複 之前的實驗,並且利用X-ray 單晶繞射證實此錯合物為直線型三核金屬串。本實驗 室自1994 年開始對多吡啶胺系統進行探討,率先以二吡啶胺核出了三核純鈷串錯 合物5,除了多吡啶胺系統之外,實驗室同樣也針對多萘啶胺系統進行廣泛金屬串 的合成及探討,並發現其和傳統的多吡啶胺系統之間,在性質上存有些許的差異,
此外還針對多吡啶胺系統和多萘啶胺系統進行組合,形成多萘啶吡啶胺系統,希 望可以結合兩者的優點,發展出更加多元的金屬串錯合物。
以二吡啶胺為研究的出發點,按照配位基上的氮原子之配位位向進行區分,
則可以有三種配位形式,分別為:I. 全反向型 、II. 反向-順向型、III. 全順向型 等三種類型(
圖1-16),金屬串的合成必須是全順向式的配位,但此時因為吡啶環之間存在 彼此交錯的氫原子產生立障因此不利於合成,配位基較容易以反向的構型存在。
圖1-17 二吡啶胺氮原子配位的三種構型
1-4-1 多吡啶胺配位基
在研究的初期,我們是以二吡啶胺來合成金屬串錯合物,使用不同的金屬以 探討金屬金屬之間的作用力,及測量分子導線的導電度等等,但因為有架橋配位 基的分子導線長度受到配位基的限制,因此所得到的成果終究有限。若要得到進 一步的發展,我們必須試圖去延長配基本身的長度,以期能夠得到更多核的金屬 串錯合物,而將二吡啶胺延展成三吡啶二胺 (tripyridyldiamine, Htpda),金屬串錯 合物的長度由三核延展到五核,但是要再進一步合成出更長的配位基時,在合成 上遭遇到了困難。直到 1997 年,我們在有機配位基的合成上加入鈀金屬催化劑 Pd2(dba)3以便合成出更長的多吡啶胺配位基19。最終我們在多吡啶胺系統之中合成 出最長的配位基十一吡啶十胺,在金屬串錯合物的合成上最長則為九核純鎳串(圖 1-17)13a以及純鉻串13b,並且透過金屬串中金屬離子的置換,在純串中加入其他種 類的異金屬,多吡啶胺配位基系統的金屬串已經從同核擴展到異核,甚至是異三 核的金屬串錯合物,為各式各樣金屬-金屬鍵的探討帶來了嶄新的潛力。
圖1-18 五吡啶四胺之九核純鎳串錯合物單晶結構圖13a
利用有機合成方法,本實驗室已經合成出一系列的多吡啶胺配基,如圖 1-18。
其組成是由吡啶與胺基互相間隔組合而成,並藉由氮原子上的孤對電子對,可以 與金屬形成良好的配位鍵。
N N
H N N
H n N
n = 0,1, 2, ..., 9
圖1-19 多吡啶胺配位基
在多吡啶胺系統之中,目前最長的配位基為十一吡啶十胺,若可以形成金屬 串錯合物其長度將會達到二十一核,但金屬串錯合物的部分只停留於九核的長度 (如圖 1-19),原因主要在於配基本身具有一定程度的的彈性 (flexible),隨著配位 基的延長,配位基容易透過單鍵的旋轉而產生彎折的現象,而以類似髮夾的形式 反向型去和金屬離子進行配位,形成單核或是核數較少的團簇金屬錯合物,如1-20 所示。而為了改善配位基剛性不足的缺點,我們開發出了多萘啶胺系統,希望藉 由萘啶環來提高配基整體的剛性,以期能以全順向式和金屬離子配位,形成核數 較多的金屬串錯合物。
. 圖1-20 多吡啶胺系統之中已得到單晶結構的純串錯合物
圖1-21 三吡啶二胺中三個吡啶環上的氮以全反向型和鐵離子配位21
1-4-2 多萘啶胺配位基
為了解決吡啶環缺乏剛性以致於在合成長鏈金屬串錯合物上的困難,因此我 們開發出了萘啶胺配位基(1,8-naphthyridine, ny)。萘啶胺配位基系統在 1973 年時,
Sacconi Luigi 研究團隊以萘啶做為配基和溴化鎳進行反應,順利得到以四片萘啶夾 住兩個鎳離子的鎳雙核錯合物[Ni2(ny)4Br2][BPh4] 23,單晶結構如圖 1-21。在電荷 上可以發現,兩個鎳離子發生了還原反應而僅帶正三價,為正一、二價混價 (mixed-valence)的形式,推測萘啶配位基系統在還原金屬離子的同時,具有穩定低 氧化態金屬的功能,在此雙核鎳金屬離子之間電子完全未定域化,未成對電子在 兩個鎳金屬離子之間快速的傳遞。
圖1-22 [Ni2(ny)4Br2][BPh4]的單晶結構圖23
多吡啶胺配位基不容易合成出高核數的金屬串錯合物,因此我們開始開發多 萘啶系統,同樣在以鈀金屬做為催化劑的幫助下,我們合成出了最長為十四氮的 純萘啶胺配基(圖 1-22),在金屬串方面,二萘啶胺的鎳五核金屬串錯合物於 2007 年率先被合成出來24,隨後合成出鎳八核25至最長為十一核的純鎳金屬串14。
在四萘啶三胺(tentra)之十一核純鎳串中(圖 1-23),和之前 Sacconi Luigi 的研究 有類似的發現,其分子式為[Ni11(tentra)4Cl2](PF6)4,在電荷上十一個鎳只帶正18價,
被還原了四個電子,且萘啶胺中的萘啶環上的兩個氮原子所配位的兩個鎳距離稍 短,因此可以判斷兩個鎳之間有部份金屬鍵,共有四個鎳雙核鎳單元,鎳雙核單 元中有發生還原的現象,兩個鎳由正四價被還原成正三價。和先前的研究一樣,
多萘啶胺配位基系統具有穩定低氧化態金屬離子的能力。
圖1-24 [Ni11(tentra)4Cl2]4+之單晶結構圖14
在萘啶環單元的剛性幫助下,配位基的剛性增加了,減少了反向型配位發生 的情形,金屬串的長度也順利從九核拓展到十一核,並且具有更為豐富的電化學 特性,但配基本身在脫氫之後所帶的負電荷相對較少,如[Ni11(tentra)4Cl2](PF6)4中 金屬串即帶有四個正電,在金屬串的合成上有一定程度的困難,故我們將多吡啶 胺系統和多萘啶胺系統進行結合,希望可以互相補足彼此的缺點,創造出更加豐 富的多萘啶吡啶胺系統。
圖1-25 多萘啶胺系統之中已得到單晶結構的純串錯合物
1-4-3 多萘啶吡啶胺配基
在多吡啶胺系統之中,配位基容易發生反向式配位的情形,但在脫氫之後能 夠攜帶足夠的負電荷,在多萘啶胺系統之中,配基雖然較為剛性,而且能夠穩定 氧化數較低的金屬離子,豐富了電化學的探討,但本身脫氫後所攜帶的負電荷則 相對不足。有鑑於此,我們嘗試將兩種系統結合,建構出多萘啶吡啶胺系統,希 望能夠互相補足彼此的缺點,往更長鏈的金屬串錯合物發展。
在多萘啶吡啶胺系統之中,吡啶環和萘啶環透過不同的排列組合可以產生出 許多的配位基,其中最簡單的對稱型配位基如圖 1-25 (A)所示,本實驗室於 2005 年成功合成其六核純鎳串錯合物26,開啟了對於多萘啶吡啶胺配位基系統的研究,
近年來更將金屬串錯合物的長度由多萘啶胺系統的十一核延長到進一步的延長到 十三核,配位基結構如1-25 (B)所示,其金屬串的長度達到 27.28 Å,為現今文獻 中最長的分子導線15。
此外,多萘啶吡啶胺系統也衍生出了非對稱性配位基,最簡單為四核配位基,
其結構如圖1-25 (C)所示,相關金屬串錯合物在本實驗室也有成功合成27,非對稱 性配位基會因為在金屬串中的正反排列而產生結構異構物,為金屬串錯合物在異 構物方面更增添了討論的空間。
1-5 金屬串錯合物之探討
在金屬串錯合物的合成上,本實驗室主要以萘當作溶劑在 220 度下反應,利 用高溫來解決起始物溶解度的問題。本實驗室與國外知名化學家F. A. Cotton 皆致 力於金屬串錯合物的合成與研究,得到了許多的成果並且發表在文獻中。本章節 將針對各類型的金屬串錯合物進行探討。
1-5-1 同核金屬串錯合物
在同核合金屬串錯合物中,以純鎳串研究成果最為豐富,因為鎳二價本身在 空氣中相對穩定,在合成操作上相對容易,同時鎳金屬離子與四片配基具有良好 的配位能力,可以使金屬串錯合物穩定存在。除了鎳金屬串以外,其他還有鈷、
鉻、銅、釕及銠等純金屬串。由於這類金屬對於水氣及空氣敏感,因此在合成上 較為困難。以二吡啶胺(Hdpa)配位基之三核純串錯合物來說,[Ni3(dpa)4Cl2]的結構 在1991 年被確認20,本實驗室則於1994 年首先合成出[Co3(dpa)4Cl2]5,隨後於1997 年本實驗室也發表[Ru3(dpa)4Cl2]及[Rh3(dpa)4Cl2]10(a) (圖 1-26),接著 2000 年 F. A.
Cotton 團隊則發表[Cr3(dpa)4Cl2]金屬串26,接著合成更長鏈的金屬串錯合物是我們 實驗室期望突破的目標,於是我們嘗試使用更長的配位基,也得到了不少的成果(圖 1-19、1-24),但因為較長的金屬串在合成上不容易,因此以純鎳串為主。
圖1-27 [Ru3(dpa)4Cl2](左)及[Rh3(dpa)4Cl2](右)單晶結構圖10a
1-5-2 三核異金屬串錯合物
多核金屬串錯合物中,中心金屬串骨架為兩種以上的金屬離子即為多核異金 屬串錯合物,由於金屬種類的不同,使得金屬串的金屬離子之間的作用力的探討 更為複雜,磁性其他等化學性質也就更有多變性。但是在異金屬串的合成上會因 為金屬的離子排列而使反應更為雜亂,因此產率普遍都不高。近年來本實驗室團 隊研究主要利用二吡啶胺(Hdpa, dipyridylamide)作為架橋配位基,也合成出了各式 各樣的三核異金屬串,得到不錯的研究成果。
若不考慮金屬的種類,以對稱型配位基來講,三核異金屬串錯合物的排列可 以有以下三種:(1)MAMAMB (2)MAMBMA (3) MAMBMC,二吡啶胺為例,如圖1。
圖1-28 在異三核金屬串系統中金屬的排列情形示意圖
若我們使用了三種不同的金屬離子欲合成三核異金屬串錯合物,即使使用對 稱型架橋配位基,包含同核金屬串錯合物在內,也會產生 21 種不同的組合模式,
故多種產物的生成是是此類型金屬串在合成時所需要面對的一大挑戰。本實驗室 於2007 年時發表了第一個三核異金屬串錯合物27,以二吡啶胺分別加入鈷以及鈀 的相關前驅物成功合成出了[CuPdCu(dpa)4Cl2],接著在同年也成功合成並發表 [CuPdCu(dpa)4Cl2]以及[CuPtCu(dpa)4Cl2]28兩種異金屬串錯合物,發現Cu2+為順磁,
兩者均有兩個磁性中心,被中間逆磁的Pd2+、Pt2+金屬離子所隔開。經過對於這一 系列的對稱型三核異金屬串的研究後,我們發現Pd 以及 Pt 易以方型平面的模式配 位,且似乎傾向於二吡啶胺中amido 的位置。如圖 1-28 所示
圖1-29 (a) [CuPdCu(dpa)4Cl2];(b) [CuPtCu(dpa)4Cl2]之單晶結構圖27-28
錳三核純串錯合物還沒有被成功合成,但本實驗室於2016 年發表了一系列 [MnMMn(dpa)4Cl2]之三核異金屬串錯合物,其中 M 為第十族的二價金屬離子,即 Ni2+, Pd2+, Pt2+,如圖1-29 所示,其中 M 也同樣偏好於中間 amido 的位置,同時他 們也發現金屬串中兩個終端之Mn2+之間具有反鐵磁作用(antiferromagnetic
interactions)。29
圖1-30 (a) [MnNiMn(dpa)4Cl2];(b) [MnPtMn(dpa)4Cl2]之單晶結構圖29
異金屬串錯合物也可以以雙核釕金屬錯合物Ru2(OAc)4Cl 做為反應前驅物,並 加入第三個金屬離子以合成不對稱異核金屬串錯合物[Ru2M(dpa)4Cl2],本實驗室首 先發表 M 為 Ni 及 Cu 的金屬串錯合物 30,如圖 1-30,並且透過氧化反應而形成 [Ru2M(dpa)4Cl2]+(PF6)1-。經由磁性測定研判,[Ru2M(dpa)4Cl2]中兩個釕金屬離子視 為二、三混價單元(Ru25+,S = 3/2),鈷以及鎳則被還原為正一價的金屬離子,(圖 1-30,type II)。
圖1-31 Ru2Cu(dpa)4Cl2金屬串錯合物30
2009 年 J. F. Berry 實驗室團隊先以二吡啶胺(Hdpa)配位基合成 MoW(dpa)4之雙 核金屬串作為前驅物,加入第三種金屬離子CrCl2,以萘作為溶劑在220 度高溫下 反應,經過金屬離子與配位基之間的重排即可形成MoWCr(dpa)4Cl2的異三核金屬 串31,合成途徑以及單晶結構如所示。
1-32 MoWCr(dpa) Cl 31
本實驗室於2013 年也發表了三種不同金屬的異金屬串錯合物,利用金屬配位 能力不同的特性,將[CoCoRh(dpa)4Cl2]中的一個的鈷金屬離子取代成配位能力較好 的鎳金屬離子(圖 1-32),成功合成出[NiCoRh(dpa)4Cl2]32(圖 1-33(A)),並且透過氧 化反應得到[NiCoRh(dpa)4Cl2]+(PF6)- (圖 1-33(B)),以及透過 Ag+容易與Cl-產生沉 澱的特性,使用銀離子成功拔除一個氯離子而成功得到[NiCoRh(dpa)4Cl]+(PF6)-(圖 1-33(C))。
圖1-33 NiCoRh(dpa)4Cl2合成中金屬離子置換示意圖32
圖1-34 NiCoRh(dpa)4Cl2單晶結構圖32
1-5-3 不對稱配位基金屬串錯合物
在研究初期時,我們所使用的多吡啶胺以及多萘啶胺配位基均為對稱型結構,
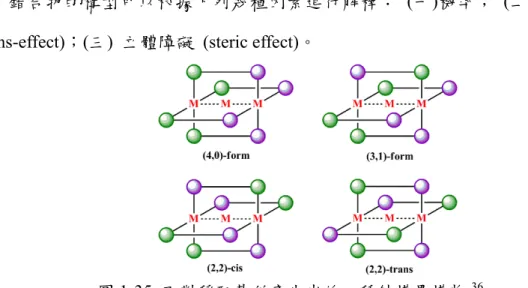
因此其金屬串錯合物並不會產生異構物。在嘗試對架橋配位基進行修飾之後,所 產生出來的配位基開始具有不對稱的特性,因此在形成金屬串錯合物時,會根據 配位基的正反排列而產生出不同的構型,根據不對稱配基排列的方向可分為下列 四種構型:(4,0)-form、(3,1)-form、(2,2)-cis 以及(2,2)-trans (如圖 1-4 所示)。而金 屬串錯合物的構型可以根據下列幾種因素進行解釋: (一)機率; (二)對位效應 (trans-effect);(三) 立體障礙 (steric effect)。
圖1-35 不對稱配基所產生出的四種結構異構物36
若考慮機率因素,則可以以排列組合來進行探討,可以發現(3,1)-form 的構型 具有最大的可能性,其機率為 1/2,其次為(2,2)-cis 的構型機率為 1/4,(4,0)-form 以及 (2,2)-trans 的構型則以 1/8 的機率為最低(圖 1-35)。單一金屬串錯合物可能同 時能夠產生一種或多種的構型,機率因素無法控制,可以藉由調控後面兩個因素,
來使金屬串錯合物的單一種構型有較大的產率。
1-36 金屬串錯合物之異構物分布機率36
對位效應因素則以 1999 年本實驗室所發表的苯基二吡啶二胺(H2phdpda)的四 核鎳串錯合物來探討33,金屬串錯合物分子式為Ni4(phdpda)4(圖 1-36),因為對位 效應,amido 位置的氮(Namido)以及 pyridine 位置的氮(Npyridine)配位能力不同,錯合
物中所有Namido其對位皆為Npyridine,四個鎳均被兩個相鄰排列的Namido以及兩個相
鄰排列的Npyridine所配位,造成金屬串錯合物的構型為(2,2)-cis。
圖1-37 Ni4(phdpda)4之單晶結構圖33
若以立體障礙因素進行探討,則對於不對稱型的架橋配位基來說,其兩端的 立體障礙效應存在差異,在形成金屬串錯合物時,配位基中具有高立障的一端傾 向於互相排開,彼此交錯排列,而使金屬串錯合物以(2,2)-trans 的構型為主產物。
如在1994 年所發表的鉬雙核以及釕雙核甲基萘啶酮(Hmnpo)錯合物中,因存在具 有立障的甲基使金屬串錯合物皆為(2,2)-trans 的構型 (圖 1-37) 34。
圖1-38 Mo2(mnpo)4(左)以及 Ru2(mnpo)4(右)之單晶結構圖34
1-5-4 以非氮原子配位金屬串錯合物
在實驗室長期的努力下,以傳統二吡啶胺為配位基所合成的各式各樣同核及 異核金屬串錯合物已經有相當多的成果,為了能夠有更進一步拓展金屬串錯合物 的可能性,開始對配基進行各種的修飾,但是實驗室一直以氮作為配位基的主要 配位元素,較少加入其它種類元素來合成配位基與金屬串錯合物。2007 年本實驗 室開發出了吡啶噻唑胺配位基(圖 1-38(a))35,配位基中雖然含有硫元素,但仍然是 以氮原子來與金屬串中的金屬離子配位,直到近年我們設計出含氧之萘啶酮配基 (Hnpo)(圖 1-38(b))36,可以用氮、氮、氧原子進行配位,並以鉬雙核作為主體,成 功合成出一系列[Mo2M(npo)4Cl2] (M = Cr, Mn, Fe, Co, Ni, Cu, Zn, Pd, Ru, Rh, Ir)之 三核金屬串錯合物,為本實驗室第一個以非氮配位的異金屬串錯合物。
萘啶酮上的氧原子也可以透過有機反應轉換為硫,形成萘啶硫酮(Hnpt)(圖 1-38(c))37,以氮、氮、硫原子進行配位,並功合成出鎳三核的金屬串錯合物 [Ni3(npt)4Cl2]。不同的原子其原子半徑以及電負度等因素存在差異,配位在各種金 屬上所形成的金屬串錯合物,相信將有更加多元又有趣的性質可以探討。
圖1-39 (a)吡啶噻唑胺 (b)萘啶酮 (c)萘啶硫酮之結構圖35-37
1-6 研究動機
近年來本實驗室致力於異核金屬串錯合物的合成與研究,因為在結構、磁性、
或是導電度上都比以往同核金屬串具有更加多元的性質,並期望金屬串錯合物能 朝更實用方向邁進,所以異核金屬串的合成與研究引起本實驗室的興趣。
實驗室傳統的二吡啶胺配位基在長期的研究下已經有相當豐富的研究成果,
但是以非氮原子配位金屬串錯合物到近年來才逐漸發展,以新穎的配位基萘啶酮 合成出本實驗室第一個以非氮原子配位的金屬串錯合物36。我們在含氧之萘啶配基 上引入一個甲基,經過修飾形成甲基萘啶酮(7-methyl-1,8-naphthyridin-2(1H)-one, Hmnpo),使配位基的立體障礙提高,希望能夠藉此改善晶體失序(disorder)的現象,
以期能夠透過單晶結構更加精確的分析金屬金屬之間的作用力,以及期望透過立 體障礙能夠將金屬串錯合物控制在單一構型,並探討具有立體障礙的配位基對金 屬串錯合物的結構以及其他性質所造成的影響。
此甲基萘啶酮配位基早在1994 年成功合成出鉬雙核以及釕雙核的金屬錯合物 並得到單晶解構(圖 1-37)34,判斷此配位基有形成三核金屬串錯合物的潛力,但至 今為止並沒有成功合成出具有三核金屬串的相關文獻,於是引起了我們極大的興 趣,想藉此合成出同樣以非氮原子配位且異金屬的金屬串,相信其金屬串一定具 有非常豐富有趣的性質。
我們選用具有立障的甲基萘啶酮作為配位基,並且為了和以萘啶酮作為配位 基的金屬串錯合物比較,選用第二周期的過渡金屬,希望可以合成出一系列的同 核以及異核的金屬串錯合物,並且透過晶體結構的解析以及磁性的探討,研究直 線型三核過渡金屬串錯合物之金屬-金屬鍵結。
第二章:實驗部分
2-1 試藥與儀器 2-1-1 試藥與溶劑
(1) 反應溶劑
試藥名稱 藥品廠商
Acetone TEDIA (試藥級,光譜級)
Dichloromethane TEDIA (試藥級,光譜級)
Ether TEDIA (試藥級,光譜級)
n-Hexane TEDIA (試藥級,光譜級)
Methanol TEDIA (試藥級,光譜級)
n-PrOH ACROS(試藥級)
t-BuOH ACROS (試藥級)
Acetic acid
Acetic anhydride
ACROS (試藥級) ACROS (試藥級)
Ethyl acetate TEDIA (試藥級,光譜級)
DMSO-d6 Merck (光譜級)
CDCl3-d1 Merck (光譜級)
Naphthalene Phosphoric acid Ammonia Solution Sulfuric acid
ACROS (試藥級) ACROS (試藥級) ACROS (試藥級) ACROS (試藥級)
(2) 反應試藥
試藥名稱 藥品廠商
2,6-Diaminopyridine ACROS (試藥級) 4,4-Dimethoxy-2-butanone
o-Dichlorobenzene 1,5-Cyclooctadiene NaNO2
ACROS (試藥級) ACROS (試藥級) ACROS (試藥級) ACROS (試藥級)
t-BuOK ACROS (試藥級)
RuCl3·3H2O FeCl2·4H2O
Heraeus (試藥級) ACROS (試藥級)
Mo(CO)6 ACROS (試藥級)
LiCl ACROS (試藥級)
KPF6 ACROS (試藥級)
2-1-2 實驗儀器
1. X-ray 單晶繞射儀:Bruker D8 VENTURE 委託科技部臺大貴重儀器中心李錦祥
先生代測。2. 核磁共振光譜儀:
1H NMR 光譜使用 Varian 400 MHz NMR 測量,委託台大化 學系蘇仁寬先生代測。化學位移則相對於CHCl3 (7.24 ppm)、DMSO (2.48 ppm) 做為參考。3. 基質輔助雷射游離質譜儀(MALDI-MS)及電噴灑游離質譜儀(ESI-MS):委託中研
院化學所質譜中心、台大化學系代測。4. 紅外線光譜儀 (infrared spectroscopy, IR):Nicolet MAGNA-IR 550 型,樣品均以
KBr 打片,自行操作。5. 循環伏安法 (cyclic voltammetry, CV) : 使用三電極電位測定儀(CH Instruments,
Model 750A)進行電位掃描實驗。三電極:工作電極 (working electrode) 為 BAS 面積0.02 cm2的白金電極,輔助電極 (auxiliary electrode) 為直徑 0.25 mm 的白 金絲,參考電極 (reference electrode) 則為自製 Ag/AgCl (sat’d KCl) 電極。參考 電極則以飽合甘汞電極 (saturated calomel electrode, SCE) 校正,誤差在± 3mV 內,電解質溶液為0.1 M TBAP,自行操作。
6. 電 子 吸 收 光 譜 儀 (Ultraviolet-Visible Spectrometer) : Hitichi (U-3310)
spectrophotometer,以 dichloromethane 為溶劑配置 10 uM 的溶液自行操作測量。7.
電子順磁共振光譜儀(Electron Paramagnetic Resonance, EPR): Bruker 廠牌 EMXmicro-6/1/S/L 型,委託臺師大 EPR 實驗室操作員邢凱捷小姐代測。2-2 化合物合成
2-2-1 金屬前驅物的合成 2-2-1a Ru
3(COD)Cl
2之合成
取RuCl3·3H2O (4.64g, 22.3 mmole)於 250 mL 單頸瓶中,加入約 100 mL 之 n-PrOH 作為溶劑,接著加入 FeCl2·4H2O (15.51g, 66.2 mmole)及 1,5-cyclooctadiene (COD,9.0 mL, d=0.880, 66.7 mmole),以液態氮將溶劑凝固並上真空抽氣約 10 分鐘,
灌入氬氣氣球,以97oC 油浴迴流 24 小時,靜置降溫後減壓過濾,所得固體以 methanol 沖洗,即得橘紅色固體,產率 98% (6.14g, 21.9 mmole)。
2-2-1b Mo
2(OAC)
4之合成
38取 Mo(CO)6 (3.93g, 14.9 mmole) 於 250 mL 單頸瓶中,分別加入 CH3COOH (35 mL)、acetic anhydride (3.5 mL)、o-dichlorobenzene (25 mL)、n-hexane (4 mL),以 150oC 油浴迴流 2 天,靜置降溫後減壓過濾,所得固體以 methanol 沖洗,即得金 黃色固體,產率57% (1.84g, 4.29 mmole)。
2-2-2 配位基的合成
2-2-2a: 7-methyl-2-amino-1,8-naphthyridine 之合成
39取 2,6-diaminopyridine (6.50 g, 59.6 mmol),放入 250 mL 單頸瓶中,再利用約 100 mL 之 磷 酸 室 溫 下 攪 拌 至 溶 解 。 接 著 以 液 體 添 加 器 緩 慢 加 入 4,4-dimethoxy-2-butanone (7.60 ml, d=0.996, 57.7 mmol),加入後以 90 oC 油浴迴流 24 小時。靜置降溫後,在冰浴中以加入 NH4OH 中和溶液直到 pH 為 7-8 左右,接 著 減 壓 過 濾 , 濾 液 以 ethyl acetate 萃 取 8 次 , 即 可 得 到 咖 啡 色 的 7-methyl-2-amino-1,8-naphthyridine 固體,產率為 51 % (4.64 g, 29.6 mmole)。
鑑定結果
MS(ESI) m/z:160.0871 ([M+H]+).
1H NMR (400 MHz, [d6]DMSO, 25 °C): δ = 8.62 (dd, 1H, J = 4.4 Hz, J = 2 Hz), 8.00 (dd, 1H, J = 7.8 Hz, J = 2.2 Hz), 7.89 (d, 1H, J = 8.8 Hz ), 7.11 (dd, 1H, J = 8 Hz, J = 4.4 Hz), 5.090 (s, 2H), 6.77 (d, 1H, J = 8.8 Hz) ppm
IR(KBr,cm-1):3324 (s) (N-H)、3149 (s) (N-H)、1604 (vs) (C=O)、1550 (vs)、1514 (vs)、1441 (s)、1384 (vs)、1346 (s)、1211 (m)、1138 (m)、1033(m)、844(m)、808(m)、
795(m)、779(m)、688(m)、634(m).
2-2-2b: 7-methyl-1,8-naphthyridin-2(1H)-one (Hmnpo)之合成
34取7-methyl-2-amino-1,8-naphthyridine (1.60 g 10.0 mmol),在冰浴下加入 2 M 的硫酸溶液(25 ml),接著加入 NaNO2 (1.01g, 14.3 mmole)水溶液 25ml,攪拌 30 分 鐘後在冰浴中以NH4OH 中和溶液至 pH 為 7-8 左右,接著減壓過濾,濾液以 CH2Cl2
萃取5 次,最後使用管柱層析以 acetone/dichloromethane (1:6)的比例做為沖堤液進 行純化,得到白色固體7-methyl-1,8-naphthyridin-2(1H)-one,產率 52% (0.84g, 5.2 mmole )。
鑑定結果
MS(ESI) m/z:161.0628 [Hmnpo + H]+
1H NMR (400 MHz, [d1]chloroform, 25 °C): δ = 2.56-2.60 (s, 3 H), 6.61-6.63 (d, 1 H), 7.01-7.03 (d, 1 H), 7.64-7.66 (d, 1 H), 7.73-7.75 (d, 1 H), 9.53 (s, 1 H) ppm.
IR(KBr,cm-1):3432 (N-H)、1647 (C=O)、1611 (m)、1601(m)、1540(m)、1400 (m)、
1358 (m)、1330 (m)、1228 (m)、1147(m)、887 (m)、852 (m)、784 (m).
2-2-3 金屬串錯合物的合成
2-2-3a: [Ru
3(mnpo)
4Cl](PF
6) (1) 的合成
取Hmnpo (0.2024 g, 1.264 mmol)以及 Ru(COD)Cl2 (0.3228 g, 1.153 mmol)並加 入約25 g naphthalene作為溶劑於250 mL 萘燒錐型瓶中,同時配置t-BuOK/ t-BuOH 溶液:取過量t-BuOK (0.1711g, 1.525 mmole)溶於適量 t-BuOH(約 7 mL)中,並適度 加熱使t-BuOK 溶解。將錐形瓶架上卜型管及緩衝瓶真空抽氣 10 分鐘,灌入含氬 氣氣球,加熱至220 ℃並攪拌進行反應,30 分鐘後以針筒逐滴加入事先配置好的 t-BuOK/ t-BuOH 溶液,持續在 220 ℃下反應 6 小時後靜置降溫冷卻,加入約 250 mL 之n-hexane 將 naphthalene 融化並抽氣過濾,得深棕色粉末。再用 CH2Cl2萃取,即 可得紅棕色溶液,加入過量 KPF6於 CH2Cl2溶液中攪拌約 2 小時,再使用管柱層 析以acetone/dichloromethane (1:3)的比例做為沖堤液進行純化,可得黃褐色溶液,
純化後的溶液以CH2Cl2 / Hexane 分層養晶,靜置約 2 天後即可得橘色針狀晶體,
產率1.1 % (4.8 mg, 0.0035 mmole)。
鑑定結果
MS (ESI-TOF): m/z = 975.9112 [M + H]+
2-2-3b: [RuMoRu(mnpo)
4Cl](PF
6) (2)的合成
秤取Hmnpo (0.2038 g, 1.266 mmol)、Mo2(OAc)4 (0.1749 g, 0.1619 mmol)、
Ru(COD)Cl2 (0.3334 g, 1.191 mmol) 並加入約 25 g naphthalene 作為溶劑於 250 mL 萘燒錐型瓶中,直接在空氣中以220 ℃加熱約 16 小時之後靜置降溫冷卻,加入約 250 mL 之 n-hexane 將 naphthalene 融化並抽氣過濾,得深褐色粉末。再用 CH2Cl2
萃取,即可得黃褐色溶液,加入過量 KPF6於 CH2Cl2溶液中攪拌約 2 小時,再使 用管柱層析以acetone/dichloromethane (1:3)的比例做為沖堤液進行純化,得黃褐色 溶液,純化後的溶液以CH2Cl2 / Hexane 分層養晶,靜置 1 天後即可得紅棕色針狀 晶體,產率27 % (116.5 mg, 0.08497 mmole)。
鑑定結果
MS (ESI-TOF): m/z = 971.9079 [M + H]+.
1H NMR (400 MHz, [d6]DMSO, 25 °C): δ = 8.46 (d, 2H), 8.36 (d, 1H), 8.30 (d, 1H), 8.23 (d, 2H), 8.17 (d, 1H), 8.10 (d, 1H) 7.63 (d, 2H), 8.58 (d, 1H), 7.50 (d, 1H) 7.21 (d, 2H), 7.05 (d, 1H), 6.95 (d, 1H) 5.67 (CH2Cl2) , 3.38 (ether), 3.14 (ss,6H+3H), 2.69 (s,3H), 1.08 (ether)
2-2-3c: [Mo
2Pd(mnpo)
4] (3)的合成
秤取Hmnpo (0.1005 g, 0.6278 mmol)、Mo2(OAc)4 (0.0693 g, 0.162 mmol)、PdCl2
(0.0564 g, 0.318 mmol) 並加入約 25 g naphthalene 作為溶劑於 250 mL 萘燒錐型瓶 中,將錐形瓶架上卜型管及緩衝瓶真空抽氣10 分鐘,灌入含氬氣氣球,加熱至 150
℃並攪拌進行反應 10 分鐘後靜置降溫冷卻,加入約 250 mL 之 n-hexane 將 naphthalene 融化並抽氣過濾,得深棕色粉末。再用 CH2Cl2萃取,即可得黃褐色溶 液。至此產率已過低無法近一步做純化並養晶,因此僅由質譜作為合成的證據。(附 錄圖A-12)
鑑定結果
MS (MALDI-TOF): m/z = 936.2 [M + H]+.
2-3 晶體數據之收集與整理
晶體之X-ray 繞射資料的收集,是利用 Bruker D8 VENTURE 繞射儀,以 MoKα (λ = 0.71073 Å)為光源,進行 X-ray 單晶結構解析。晶體結構中的所有非氫原子位 置,利用SHELXTL program 以直接法 (direct methods) 解出,所有氫原子位置則 經 由 SHELXTL 軟體 計算而得其位置,吸收校正(absorption correction)使用 semi-empirical from equivalents。所有繞射數據的最小平方差計算是依據 full-matrix least-squares on F2 ( SHELXTL-2018/3 )。
2-3-1 [Ru
3(mnpo)
4Cl](PF
6) (1)
將所得之橘色針狀晶體(0.470 × 0.084 × 0.022 mm3),在 150(2) K 的溫度下,以 MoKα (0.71073 Å)為光源,利用 Bruker D8 VENTURE 繞射儀進行 X 光單晶繞射數 據收集。所求得化合物的結晶系統為單斜晶系(monoclinic),空間群為 P21/m,晶胞 參數a = 11.9887(8) Å、b = 15.8513(11) Å、c = 13.7188(9) Å,α = 90°、β = 92.033(3)°、
γ = 90°,V = 2605.4(3) Å3,Z = 2。繞射點收集範圍(limiting indices):-14 ≦h≦ 12,
-18 ≦k≦ 18,-16 ≦l≦ 16;θ 角收集範圍為 2.563-24.996°,共收集了 15438 個 繞射點。經數據統計平均後有 4750 個獨立繞射點(independent reflections),Rint = 0.0591;穿透係數(transmission coefficient) Tmin = 0.4577、Tmax = 0.7456。其收斂值 為:R1 = 0.0861,wR2 = 0.2135 [I≧2σ(I)],R1 = 0.1186,wR2 = 0.2454 (all data)。詳 細晶體數據參數如附表
2-3-2 [RuMoRu(mnpo)
4Cl](PF
6) (2)
將所得之紅棕色針狀晶體(0.457 × 0.046 × 0.044 mm3),在 150(2) K 的溫度下,
以MoKα(0.71073Å)為光源,利用 Bruker D8 VENTURE 繞射儀進行 X 光單晶繞射 數據收集。所求得化合物的結晶系統為單斜晶系(monoclinic),空間群為 P21/m,晶 胞參數a = 11.9526(8) Å、b = 15.9326(10) Å、c = 13.7853(9) Å,α = 90°、β = 91.924(2)°、
γ = 90°,V = 2623.7(3) Å3,Z = 2。繞射點收集範圍(limiting indices):-14 ≦h≦ 12,
-19 ≦k≦ 19,-16 ≦l≦ 16;θ 角收集範圍為 2.561-25.719°,共收集了 14165 個 繞射點。經數據統計平均後有 5174 個獨立繞射點(independent reflections),Rint = 0.0543;穿透係數(transmission coefficient) Tmin = 0.5680、Tmax = 0.7453。其收斂值 為:R1 = 0.0738,wR2 = 0.1779 [I≧2σ(I)],R1 = 0.0936,wR2 = 0.1900 (all data)。詳 細晶體數據參數如附表。
第三章:結果與討論
3-1 晶體結構解析
3-1-1 [Ru
3(mnpo)
4Cl](PF
6) (1)晶體結構解析
圖3-1 [Ru3(mnpo)4Cl](PF6) (1)之單晶結構圖
[Ru3(mnpo)4Cl](PF6)之單晶結構圖如圖 3-1 所示,晶體為橘色針狀晶體,經由 X-ray 單晶繞射儀器進行鑑定解析,此晶體屬於單斜(monoclinic)晶系,空間群為 P21/m,金屬串錯合物陽離子以及 PF6陰離子皆位於鏡面(mirror plane)上,圖 3-1 中 可以看出平行於紙面有一個對稱面。解晶體解1/2 個金屬串錯合物以及 1/2 個 PF6
陰離子,溶劑的部分為1/2 個正己烷以及一個二氯甲烷,透過對稱面的對稱操作得 到 完 整 的 金 屬 串 錯 合 物 分 子 。 晶 體 的 實 驗 式 (empirical formula) 為 [Ru3(mnpo)4Cl](PF6).(C6H14).2(CH2Cl2)。Z 值為 2,即每一個單位晶格中存在著兩個 金屬串錯合物分子。

及[Rh 3 (dpa) 4 Cl 2 ](右)單晶結構圖 10a](https://thumb-ap.123doks.com/thumbv2/9libinfo/9603588.629922/31.892.145.745.823.1119/圖127Ru3dpa4Cl2左及Rh3dpa4Cl2右單晶結構圖1a.webp)
![圖 1-30 (a) [MnNiMn(dpa) 4 Cl 2 ];(b) [MnPtMn(dpa) 4 Cl 2 ]之單晶結構圖 29](https://thumb-ap.123doks.com/thumbv2/9libinfo/9603588.629922/33.892.192.729.759.1047/圖13aMnNiMndpa4Cl2bMnPtMndpa4Cl2之單晶結構圖29.webp)