行政院國家科學委員會專題研究計畫 成果報告
登革病毒感染產生自體抗體對內皮細胞的影響及疫苗研發
的考量(3/3)
計畫類別: 個別型計畫 計畫編號: NSC93-3112-B-006-003- 執行期間: 93 年 05 月 01 日至 94 年 04 月 30 日 執行單位: 國立成功大學微生物免疫學研究所 計畫主持人: 林以行 計畫參與人員: 劉校生 教授, 莊偉哲 教授, 林秋烽, 鄭獻仁, 萬書 彣; 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 94 年 9 月 26 日
行政院國家科學委員會補助專題研究計畫
登革病毒感染產生自體抗體對內皮細胞的影響及疫苗研發的考量
(3/3)
計畫類別:■ 個別型計畫
□ 整合型計畫
計畫編號:NSC 93-3112-B-006-003
執行期間:九十三年五月一日至九十四年四月三十日
計畫主持人:林以行 教授
共同主持人:劉校生 教授 莊偉哲 教授
計畫參與人員:
博士後研究員:林秋烽
博士班研究生:鄭獻仁 (成大醫學院基礎醫學研究所)
萬書彣 (成大醫學院基礎醫學研究所)
成果報告類型(依經費核定清單規定繳交):□精簡報告
■完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
■出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:國立成功大學醫學院微生物及免疫學研究所
中
華
民
國
九十四
年
七
月
三十一
日
■
成果報告
□
期中進度報告
一、摘要 血管病變是出血性症狀 DHF/DSS 的 主要病徵。除了病毒本身會直接傷害宿主 細胞外,病毒引發宿主產生自體免疫反應 也可能參與登革病症的發生。我們認為DV 係利用”分子模擬”的特性引發與宿主相似 蛋白的交叉反應性抗體,即抗 DV NS1 抗 體 (anti-DV NS1)會交叉結合至內皮細胞 並且造成傷害。實驗發現anti-DV NS1 抗體 刺激內皮細胞 HMEC-1 所造成發炎性活化 作用,結果顯示抗體可引起內皮細胞內蛋 白質的磷酸化現象以及NF-B 蛋白的活化 作用。我們也觀察到內皮細胞的細胞激素 (IL-6)、化學趨化激素 (IL-8, MCP-1) 但不 包括RANTES,會受 anti-DV NS1 抗體的 刺激而增加其表現量。此調控情形可被DV NS1 蛋白質的先前競爭作用或 NF-B 抑制 劑的前處理所抑制。另外,anti-DV NS1 抗 體刺激生成細胞黏著分子 (ICAM-1) 的表 現,同時也促進週邊血液的單核球附著於 受刺激的內皮細胞表面。而這樣的現象則 可被預先處理中和抗體包括anti-MCP-1 及 anti-ICAM-1 所競爭抑制,顯示了 MCP-1 調控 ICAM-1 引發細胞黏著的可能機制。 根據本計畫的研究結果,我們認為內皮細 胞自體抗體的產生會造成細胞失去正常機 能,除了造成死亡的作用外亦會引起細胞 炎性活化作用。這樣的效應可能在 DV 感 染所引起的致病機制上扮演了一個重要的 角色。 關鍵詞:登革病毒、登革出血熱/登革休克 症候群、內皮細胞自體抗體、NS1 抗體、細胞凋亡、細胞激素、化 學趨化激素、細胞黏著分子 Abstract
Vascular dysfunction is a hallmark associated with disease onset in dengue hemorrhagic fever and dengue shock syndrome. In addition to direct viral damage, immune responses to dengue virus (DV) infection may also underlie the pathogenesis of disease. We have proposed a mechanism of molecular mimicry in which Abs directed against DV nonstructural protein 1 (NS1) cross-react with endothelial cells and induce damage. In this study, we demonstrated the inflammatory endothelial cell activation induced by anti-DV NS1 via the transcription factor NF-B-regulated pathway. Protein phosphorylation and NF-B activation were observed after anti-DV NS1 stimulation in a human microvascular endothelial cell line HMEC-1. The cytokine and chemokine
production, including IL-6, IL-8, and monocyte chemotactic protein-1 (MCP-1), but not RANTES, in endothelial cells increased after treatment with anti-DV NS1 Abs. The expression of IL-6, IL-8, and MCP-1 were blocked by the preabsorption of anti-DV NS1 with DV NS1 or by the inhibition of NF-B activation. Furthermore, the increases in both ICAM-1 expression and the ability of human PBMC to adhere to endothelial cells were also observed, and these effects were inhibited by pretreatment with anti-ICAM-1 or anti-MCP-1 Abs. Therefore, in addition to endothelial cell apoptosis, as previously reported, inflammatory activation occurs in endothelial cells after stimulation by anti-DV NS1 Abs. These results suggest the involvement of anti-DV NS1 Abs in the vasculopathy of DV infection.
Keywords:dengue virus; dengue
hemorrhagic fever/dengue shock syndrome; anti-endothelial cell autoantibody; anti-NS1; apoptosis; cytokine; chemokine; adhesion molecule
二、前言
Patients with dengue virus (DV) infection present a wide range of diseases
from mild dengue fever (DF) to
life-threatening dengue hemorrhagic fever and dengue shock syndrome (DHF/DSS) (1, 2). There is no vaccine against DHF/DSS because its pathogenic mechanisms are still not fully understood (3-6). In dengue pathogenesis, the involvement of viral and immune responses has been hypothesized (3-9). Virus variation and virus load involving Ab-dependent enhancement of infection have been suggested to be responsible for the progression and severity of dengue disease (10-17). Cell activation, impaired proliferation, cytokine and chemokine production, and apoptosis are caused after DV infection by a direct (18-32) or indirect route (33, 34). It is likely that different mechanisms are involved in the pathogenesis of DV infection.
complication caused by multiple symptoms, including thrombocytopenia, coagulopathy, and vasculopathy (7, 35-37). Plasma leakage, also called the hemorrhagic syndrome, occurs once vessel endothelium is disrupted and is followed by the loss of its barrier function. The pathogenesis of endothelial dysfunction resulting in vascular leakage remains unclear, however. In addition to direct viral damage, such as the induction of apoptosis in endothelial cells as observed in vitro (27), complement activation and cytokine and chemokine production induced by DV infection appear to be involved in the induction of endothelial cell damage (26, 27, 38, 39). DV can also indirectly modulate endothelial cell function through Ab-enhanced infection of DV in peripheral blood monocytes (33). Furthermore, the involvement of anti-endothelial cell autoAb in DV pathogenesis has been proposed (40-42). Endothelial cell apoptosis caused by autoAb may be related to the transient leakage syndrome in dengue vasculopathy.
Inflammatory immune responses
facilitate the progression of disease severity in DHF/DSS (7-9). Lymphocytes, monocytes and macrophages, mast cells, endothelial cells, liver cells, and dendritic cells are the targets of DV, and these cells could produce cytokines or chemokines, or both, after DV infection (20-22, 24-27, 33, 38, 39, 43, 44). Increased levels of cytokines, including IL-2, IL-6, IL-8, IL-10, IL-13, IL-18, IFN-, TNF-and monocyte chemotactic protein-1 (MCP-1), have been observed in patients with DV infections (8, 27, 45-52). The roles of various cytokines and chemokines in the pathogenesis of DHF/DSS need to be clarified, especially on endothelial cell activation, which is related to vasculopathy.
Previous studies in our laboratory showed the pathogenic role of Abs against nonstructural protein 1 (NS1) present in dengue patients or generated in mice (40, 41). Anti-DV NS1 Abs cross-reacted with endothelial cells and induced apoptosis via
an NO-mediated pathway (40, 42).
Pathogenic anti-DV NS1 may contribute to the progression of DHF/DSS (41). In the present study, we demonstrated protein tyrosine phosphorylation and NF-B activation after endothelial cells had been bound by anti-DV NS1 Abs. We also demonstrated that the increased levels of cytokine and chemokine expression were regulated by NF-B, and that anti-DV NS1 stimulation caused adhesion molecule
expression and PBMC adhesion to
endothelial cells.
三、研究目的
The onset of vascular leakage and hemorrhagic diathesis is one of the life-threatening complications that occurs in dengue patients, yet the pathogenic mechanisms are not well understood. We have proposed a mechanism of molecular mimicry in which antibodies directed against DV NS1 can cross-react with endothelial cells and induce these cells to undergo apoptosis. Immune responses to infectious agents are the critical defense mechanisms in a host, yet abnormal immunity may also be involved in the pathogenesis of disease. We hypothesize that the host-virus interactions that induce autoimmune responses may have implications in DHF/DSS pathogenesis. Gene expression profiling after anti-DV NS1 stimulation in endothelial cells are studied which may have implications in dengue disease pathogenesis. The epitopes in NS1 that may elicit autoimmune responses should
be avoided for safety in vaccine
development.
四、研究方法 Mice
BALB/cByJ breeder mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained on standard laboratory food and water ad libitum in our medical college laboratory animal center. Their 8-wk-old progeny were used for the
generation of Abs in the present study.
Abs and reagents
Both purified and FITC-conjugated mAbs against phosphotyrosine (clone PY20) were purchased from Oncogene Research Products (Boston, MA). Rabbit Abs specific for NF-B p65 were purchased from Chemicon (Temecula, CA). Monoclonal anti-human IL-6, IL-8, MCP-1, RANTES, and ICAM-1 Abs were from BD PharMingen (San Diego, CA). Neutralizing anti-human MCP-1 and ICAM-1 mAbs were also from BD PharMingen, and mouse mAb to -actin was from Sigma (St. Louis, MO). FITC- or HRP-conjugated goat anti-mouse or anti-rabbit IgG, NF-B inhibitor pyrrolidine dithiocarbamate (PDTC), protein synthesis inhibitor cycloheximide, caspase inhibitor benzyloxycarbonylvalylalanylaspartic acid fluoromethyl ketone (zVAD-fmk), and NO synthase (NOS) inhibitor N-nitro-L-arginine methyl ester (L-NAME) were obtained from Sigma.
Polyclonal Abs against DV or Japanese encephalitis virus (JEV) NS1 were obtained from BALB/c mice immunized i.p. with purified recombinant DV-2 (New Guinea C strain) or JEV (NT109 strain) NS1 proteins as described previously (40). The IgG fractions from hyperimmunized mouse sera were purified with a protein G-Sepharose affinity chromatography column (Amersham Pharmacia, Uppsala, Sweden) and recovered with HCl-glycine. The reactivity of purified
IgG against NS1 was confirmed by
SDS-PAGE and Western blot. The control IgG was eluted from a protein G column loaded with normal mouse sera. The endotoxin concentration of each of these preparations was < 0.03 EU/g, as determined by a Limulus amebocyte lysate assay (Associates of Cape Cod, Falmouth, MA).
Patient sera
Dengue patient sera were obtained from Dr. N. T. Hung (Department of Dengue
HemorrhagicFever,Children’sHospitalNo.1, Ho Chi Minh City, Vietnam). Seventeen serum samples were collected from patients with DHF disease severity-grades I-III. Diagnosis of DHF was based on the clinical criteria established by the World Health Organization. Sera from five healthy volunteers were used as the normal controls.
Cell and virus culture
Human microvascular endothelial cell line-1 (HMEC-1) was passed in culture plates containing endothelial cell growth medium (EGM; Clonetics, Walkersville, MD)
composed of 2% FBS, 1 g/ml
hydrocortisone, 10 ng/ml epidermal growth factor, and antibiotics (40). Cells were detached using 1000 U/ml trypsin and 0.5 mM EDTA. Only those cultures from three to five passages with a viability of over 95% by eosin-Y staining were used for experiments. Baby hamster kidney cell line (BHK) and C6/36 cells were cultured in DMEM medium containing 10% FBS and antibiotics. PBMC were isolated from normal volunteer blood using Ficoll-Paque isolation (Amersham Pharmacia) according to the standard procedures and were used for adhesion assay.
Dengue-2 virus (PL046, Taiwan isolated) was maintained in the C6/36 cells. Briefly, monolayers of C6/36 were incubated with DV at a multiplicity of infection (MOI) of 0.01 and incubated at 26°C in 5% CO2 for 5 days. The cultured medium was harvested
and cell debris was removed by
centrifugation at 900 × g for 10 min. After further centrifugation at 16,000 × g for 10 min, the virus supernatant was collected and stored at 70°C until use. Virus titer was determined by plaque assay using the BHK-21 cell line as described previously (26).
Western blotting and flow cytometry for detection of protein tyrosine phosphorylation
HMEC-1 cells underwent starvation in serum-free EGM for 1 h before the
experiment. Next, the cells were treated with mouse anti-DV NS1, anti-JEV NS1, or control IgG for various time intervals. Cells were then harvested and lysed with a buffer containing 1% Triton X-100, 50 mM Tris (pH 7.5), 10 mM EDTA, 0.02% NaN3, and a protease inhibitor cocktail (Boehringer Mannheim, Mannheim, Germany). After being freeze-thawed once, cell lysates were centrifuged at 12,000 rpm for 20 min at 4°C. The supernatants were collected and boiled in sample buffer for 5 min. Following SDS-PAGE, proteins were transferred to PVDF membrane (Millipore, Bedford, MA), blocked overnight at 4°C in PBS-T (PBS plus 0.1% Tween-20) containing 5% skim milk, and probed with Abs against phosphotyrosine at 4°C overnight. After being washed with PBS-T, blots were incubated with a 1:5000 dilution of HRP-conjugated goat anti-rabbit IgG for 1 h at 4°C. The protein bands were developed with an AEC substrate kit (Zymed Laboratories, San Francisco, CA).
For immunostaining followed by flow cytometric analysis, cells were washed in PBS, fixed with 1% paraformaldehyde in PBS at room temperature for 10 min, and permeabilized with 70% ethanol. After being washed with PBS three times, cells were incubated with FITC-conjugated mAbs against phosphotyrosine for 1 h at 4°C and analyzed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA) with excitation set at 488 nm.
Western blotting and EMSA for detection of NF-B activation
Nuclear translocation of NF-B was detected by Western blotting. Harvested cells were lysed with buffer A (10 mM HEPES (pH 7.8), 5 mM MgCl2, 10 mM KCl, 1 mM ZnCl2, 0.2 mM EDTA, 1 mM Na3VO4, 10 mM NaF, 0.5 mM DTT, and 0.5 mM PMSF), incubated on ice for 10 min, and centrifuged at 12,000 rpm for 20 min at 4°C. The supernatants were collected as the cytosolic lysates, and the nuclear extracts were obtained from pellets lysed in buffer B (20
mM HEPES (pH 7.8), 5 mM MgCl2, 300 mM NaCl, 1 mM ZnCl2, 0.2 mM EDTA, 25% glycerol, 1 mM Na3VO4, 10 mM NaF, 0.5 mM DTT, and 0.5 mM PMSF) and incubated for 15 min on ice with occasional mixing. Following SDS-PAGE, proteins were transferred to PVDF membrane, blocked overnight at 4°C in PBS-T containing 5% skim milk, and probed with Abs against NF-B p65 subunit at 4°C overnight. After being washed with PBS-T, blots were incubated with a 1:5000 dilution of HRP-conjugated goat anti-rabbit IgG for 1 h at 4°C. The protein bands were developed with an AEC substrate kit.
For EMSA, nuclear extracts were collected as described above and separated by 6% acrylamide gel. The DIG Gel Shift kit (Roche Diagnostics, Penzberg, Germany) was used according to the manufacturer’s instructions. The detection probe specific for NF-B consisted of a digoxigenin-labeled
double-strand oligonucleotide
(5'-AGTTGAGGGGACTTTCCCAGGC-3'). Specificity of protein-DNA complexes was detected by immunoreactivity with sheep anti-digoxigenin conjugated with alkaline
phosphatase and developed using
chemiluminescent substrate CSPD (Roche). The generated chemiluminescent signals
were recorded on X-ray film by
autoradiography.
Immunohistochemistry and flow cytometry for detection of cytokine, chemokine, and adhesion molecule
Monolayers of HMEC-1 cells were cultured on sterile glass slides followed by treatment with mouse anti-DV NS1, anti-JEV NS1, or control IgG for various time intervals. For flow cytometric analysis, cells were detached using 1000 U/ml trypsin and 0.5
mM EDTA. Cells were fixed and
permeabilized with Cytofix/Cytoperm kit (BD PharMingen). For immunohistochemical staining, fixed cells were incubated with 0.3% H2O2 in PBS for 5 min to quench endogenous peroxidase activity and then
washed again with PBS. Abs specific for IL-6, IL-8, RANTES, MCP-1, and ICAM-1 were added to cells and incubated for 1 h at 4°C. After being washed with PBS, cells were incubated with HRP- or FITC-conjugated anti-primary Ab for 1 h at 4°C. After being washed with PBS, cells were developed using the AEC substrate kit (Zymed), counterstained with hematoxylin (Sigma), and viewed with light microscopy or analyzed by flow cytometry with excitation set at 488 nm.
ELISA
The concentrations of cytokines and chemokines, including IL-6, IL-8, and MCP-1, in culture supernatants and DHF patient sera were determined using ELISA kits (R&D Systems, Minneapolis, MN). The manufacturer’s instructions were followed and the concentrations were determined by spectrophotometry at 450 nm (Molecular Devices, Sunnyvale, CA).
RT-PCR
The expression of MCP-1 and RANTES mRNA was determined using RT-PCR analysis. Total cellular RNA from endothelial cells was extracted using TRIzol Reagent (Life Technologies, Gaithersburg, MD) according to themanufacturer’sinstructions. The concentration of RNA was quantified by spectrophotometry at 260 nm (U-2000; Hitachi, Tokyo, Japan). The cDNA was prepared by reverse transcription as previously described (40) and PCR was performed using a PCR controller (GeneAmp PCR System 2400; PerkinElmer, Wellesley, MA). PCR was carried out in 50 l of the reaction mixture (1.5 mM MgCl2 and 0.2 mM each of dATP, dGTP, dCTP, and dTTP) containing primers at 1.5M each, 0.2 g/ml of RNase A (Sigma), and 1 U of Taq DNA polymerase (Promega, Madison, WI) as follows: 94°C for 1 min; 30 thermal cycles at 95°C for 1 min, 55°C for 1 min, and 72°C for 2 min; and a final cycle at 72C for 5 min. The oligonucleotide primers for human
MCP-1 (sense:
5'-CAAACTGAAGCTCGCACTCTCGCC-3
' and antisense:
5'-ATTCTTGGGTTGTGGAGTGAGTGTTC
A-3'), RANTES (sense:
5'-TGCCTCCCCATATTCCTCGG-3' and antisense: 5'-TCATGTTTGCCAGTAAGC-3'), and -actin (sense: 5'-AGCGGGAAATCGTGCGTG-3' and antisense:
5'-CAGGGTACAT-GGTGGTGGTGCC-3') were used according to previously published sequences (27, 40).
DV primer set (sense:
5'-GATATGGGTTATTGGATAGA-3' and
antisense: 5'-CTGATTTCCATCCCGTA-3') was used as an infected control (26). The PCR products were analyzed by 1.5% agarose gel electrophoresis, stained with ethidium bromide, and viewed with UV light.
Transient transfection
In experiments using dominant-negative mutant, HMEC-1 cells were co-transfected with the dominant negative IKK-(the kind gift of Dr. C. C. Chen, Department of Pharmacology, National Taiwan University, Taipei, Taiwan) or the empty vector pcDNA3.1 (the kind gift of Dr. M. D. Lai, Department of Biochemistry, National Cheng Kung University, Tainan, Taiwan). Briefly, HMEC-1 cells were grown to 50% confluent in six-well plate, then transfected with the dominant negative IKK- (0.5 g) or the empty vector (1.0 µg) using Lipofectamine 2000 reagent (Invitrogen, San Diego, CA). After 24 h of transfection, the cells were used for experiments.
Adhesion assay
HMEC-1 cells (5 × 104 cells/well) were plated into 8-well glass chamber slides (Nalge Nunc International, Naperville, IL). When monolayers were confluent, the cells were stimulated with anti-DV NS1, anti-JEV NS1, or control IgG in serum-free culture medium. After 24 h of incubation, the cells were washed once with medium and incubated for 2 h at 37C with isolated PBMC (1 × 105 cells/well) in a total volume
of 250 l/well. At the end of the incubation period, the nonadherent cells were removed by washing twice with 0.1% BSA in PBS. Adherent cells were stained with Liu’sstain (TONYAR Biotech, Taipei, Taiwan) and viewed with light microscopy. The adherent cells were counted on three consecutive microscopic fields (53, 54).
Statistical analysis
Comparisons between various
treatments were performed by Student’s t test with SigmaPlot version 4.0 for Windows (Cytel Software Corporation, Cambridge, MA). Values were considered statistically significant at p < 0.05.
五、結果
Protein tyrosine phosphorylation and NF-B activation induced by anti-DV NS1 Abs in human endothelial cells
In a previous study (40), we showed that anti-DV NS1 cross-reacted with endothelial cells. To assess the signal transduction induced by anti-DV NS1 in endothelial cells, we investigated protein phosphorylation and NF-B activation in the present study. We used Western blotting to assay the levels of protein tyrosine phosphorylation from Abs-treated HMEC-1 cells (Fig. 1A, upper
panel). We also determined the relative
optical densities of bands (Fig. 1A, lower
panel). Results showed tyrosine phosphorylation of proteins in HMEC-1 after binding with anti-DV NS1, but not with anti-JEV NS1 or control IgG. Endothelial cell growth medium (EGM)-treated cells were used as the positive control for protein phosphorylation induced by growth factors. Protein tyrosine phosphorylation was further analyzed by flow cytometry as presented by the percentages of fluorescence-positive cells (Fig. 1B, upper panel). The time-kinetic changes of phosphorylated cells after Abs stimulation are shown in Fig. 1B (lower
panel). Results indicated that protein tyrosine
phosphorylation reached its highest level 30 min after anti-DV NS1 treatment. The
specificity of Abs stimulation was demonstrated by the blockage of protein tyrosine phosphorylation when anti-DV NS1 Abs were pretreated with recombinant DV NS1 proteins (data not shown). Tyrosine phosphorylation of cellular proteins was also observed in HMEC-1 cells after they had been treated with DHF patient sera (data not shown).
We next investigated the involvement of NF-B in the anti-DV NS1-triggered signaling pathways. After Abs treatment for 3 or 24 h, endothelial cell extracts were separated for cytoplasmic and nuclear
fractions, and then followed by
immunoblotting for NF-B. The expression of NF-B decreased in the cytoplasmic fraction and increased in the nuclear fraction in cells with anti-DV NS1 treatment compared to those with anti-JEV NS1 or control IgG treatment (Fig. 2A). Activation of NF-B as indicated by its translocation from cytoplasm to nucleus was also observed by immunohistochemical staining (data not shown). The DNA-binding ability of NF-B was determined by EMSA, which showed positive signals in the anti-DV NS1-treated cells compared to the untreated, anti-JEV NS1, or control IgG group (Fig. 2B). The DNA-binding ability of NF-B was inhibited when anti-DV NS1 was preabsorbed with
recombinant DV NS1 proteins. The
specificity of the cross-reactivity of anti-NS1 Abs was thus confirmed. To further demonstrate whether NF-B activation is requisite for its DNA-binding ability mediated by anti-DV NS1, the effect of NF-B inhibitor pyrrolidine dithiocarbamate (PDTC) was examined. Results showed that the addition of PDTC to cell cultures caused an inhibition of the DNA-binding activity of NF-B in anti-DV NS1-treated cells (data not shown).
Production of cytokines and chemokines in endothelial cells after anti-DV NS1 stimulation
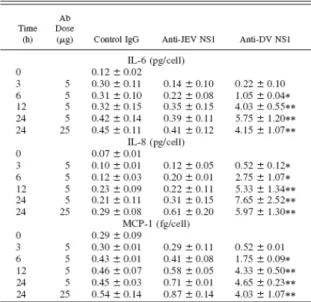
along with the induction of proinflammatory responses underlie dengue vascular disorders (7, 9). We therefore examined whether anti-DV NS1 Abs could induce inflammatory factors in endothelial cells. Results from immunohistochemical staining revealed the expression of IL-6, IL-8, and MCP-1 by HMEC-1 cells after they had been treated with anti-DV NS1 for 24 h (Fig. 3A). The expression of RANTES, however, could not be detected in anti-DV NS1-treated cells. Expression of IL-6, IL-8, and MCP-1 was further quantified using both flow cytometric analysis (Fig. 3B) and ELISA (Table I). Treatment with anti-JEV NS1 Abs showed only basal levels of IL-6, IL-8, MCP-1, and RANTES, similar to those with control IgG.
The ELISA (Table I) and
immunohistochemical staining (data not shown) showed IL-6, IL-8, and MCP-1 production after anti-DV NS1 treatment for as short as 3 to 6 h. The specificity of the cross-reactivity of anti-NS1 Abs was further confirmed by the inhibitory effect of preabsorption of anti-DV NS1 with recombinant DV NS1 but not JEV NS1 proteins (Fig. 3B). Further studies showed that the mRNA expression of MCP-1, but not RANTES, was elevated after anti-DV NS1 stimulation in HMEC-1 cells, as detected via RT-PCR (Fig. 4). By contrast, cells infected
with DV showed increased mRNA
expression in RANTES but not in MCP-1. The ELISA quantification demonstrated high levels of MCP-1 in DHF patient sera at different disease grades compared to healthy controls (Table II), similar to results in a study of DSS patients (27).
Requirement for NF-B in anti-DV NS1-induced cytokine and chemokine production
Anti-DV NS1 induced NF-B activation (Fig. 2). We further examined the involvement of NF-B modulation on the expression of IL-6, IL-8, and MCP-1. After treatment with anti-DV NS1 in HMEC-1 cultures for 24 h, the protein levels of IL-6, IL-8, and MCP-1 increased. NF-B inhibitor
PDTC, but not NOS inhibitor L-NAME or caspase inhibitor zVAD-fmk, was able to inhibit the anti-DV NS1-mediated elevations of IL-6, IL-8, and MCP-1 expression (Fig. 5,
left). Further confirming the requirement for
NF-B activation, HMEC-1 cells transfected with dominant negative IKK- showed a blockage in IL-6, IL-8, and MCP-1 production compared with the vector control (Fig. 5, right). These results revealed that NF-B in anti-DV NS1-induced endothelial cell activation upregulates cytokine and chemokine expression.
MCP-1 facilitates the expression of ICAM-1 induced by anti-DV NS1 in endothelial cells
The activation of endothelial cells involves not only the induction of cytokines and chemokines; our data showed that the expression of adhesion molecules was also augmented. After anti-DV NS1 stimulation, an increase in the expression of ICAM-1 was detected with immunohistochemical staining (Fig. 6A). The increase in ICAM-1 expression was confirmed using flow cytometric analysis (see Fig. 6B for time kinetics and dose-response data). To further investigate the regulation of ICAM-1 expression by NF-B in anti-DV NS1-treated cells, PDTC was used. Results showed a reduction of ICAM-1 expression once cells were pretreated with PDTC (data not shown). MCP-1 elevates ICAM-1 expression (55-57). We therefore investigated the relationship between MCP-1 and ICAM-1 expression after anti-DV NS1 stimulation. Pretreatment with MCP-1-neutralizing Abs reduced the expression of ICAM-1, suggesting that the ICAM-1 expressed was regulated, at least in part, by MCP-1 in anti-DV NS1-treated endothelial cells. Similar results were observed using both immunohistochemical staining (Fig. 6A) and flow cytometric analysis (Fig. 6B). Further studies using anti-DV NS1 F(ab')2 confirmed that the activation of endothelial cells, as detected by ICAM-1 expression, was not via an Fc-mediated signaling pathway (data not
shown).
Regulation of ICAM-1 and MCP-1 on the adherence of PBMC to anti-DV NS1-treated endothelial cells
The ability of immune cells to adhere to DV-infected endothelial cells was investigated next. Based on the findings shown in Fig. 6, anti-DV NS1 Abs-induced expression of ICAM-1 was downregulated by anti-MCP-1. To explore the effects of ICAM-1 upregulation on endothelial cell and immune cell interaction, the adhesion of PBMC to anti-DV NS1-treated HMEC-1 cells was assessed. Higher levels of PBMC adhering to endothelial cells could be observed in cells treated with anti-DV NS1 for 24 h, compared to those with anti-JEV NS1 or control IgG (Fig. 7A). The increase in PBMC adherence was quantified by counting the numbers of adherent cells under a microscope (see Fig. 7B for time kinetics and dose-response data). We further investigated whether the increase in ICAM-1 expression was related to the ability of PBMC to adhere to endothelial cells. Pretreatment with ICAM-1-neutralizing Abs inhibited the adherence of PBMC, suggesting that ICAM-1 expressed on endothelial cells was involved in this binding effect. We also tested the potential regulatory role of MCP-1 on ICAM-1-mediated PBMC adherence to endothelial cells. A reduction in PBMC
adherence was demonstrated when
endothelial cells were pretreated with anti-MCP-1 Abs (Fig. 7). These results thus suggested that the ICAM-1-mediated ability of PBMC to adhere to endothelial cells was at least partially regulated by MCP-1. To further investigate whether the upregulation of cell adherence was dependent on new protein synthesis, HMEC-1 cells were pretreated with protein synthesis inhibitor cycloheximide. Results showed that cycloheximide treatment inhibited PBMC adhesion, suggesting a requirement for protein synthesis of ICAM-1 or MCP-1 to increase PBMC adherence to HMEC-1 cells (Fig. 7B).
六、討論
Dengue disease is an emerging public health problem, and an increasing number of cases are being reported (5, 6, 58). Vascular leakage, liver abnormality, and hemorrhagic diathesis are the life-threatening complications that occur in dengue patients
with DHF/DSS. Their pathogenic
mechanisms, however, are not well understood. We have proposed a mechanism of molecular mimicry in which Abs directed against DV NS1 would cross-react with endothelial cells and cause damage. Our studies showed anti-endothelial cell Abs (AECA) in dengue patient sera (41). Moreover, endothelial cell damage induced by anti-DV NS1 by the production of NO may play a role in the disruption of vessel endothelium and contribute to the AECA-induced pathogenesis of vasculopathy (40, 42). When taken together with previous findings that anti-DV NS1 Abs also cross-reacted with platelets (59) and that transient thrombocytopenia in DV-infected mice was associated with the generation of anti-platelet Abs (60), our results suggest that the onset of autoimmune responses in DV infection may have implications in DHF immunopathogenesis (9). Several other reports have also shown the autoimmune responses in DV infection (61, 62).
In addition to cell apoptosis (40), we showed in this study that immune activation in endothelial cells also occurred after anti-DV NS1 stimulation. Protein tyrosine phosphorylation and NF-B activation in HMEC-1 cells were observed. There was also an increase in the expression of cytokines and chemokines, including IL-6, IL-8, and MCP-1, but not RANTES. The involvement of NF-B in the expression of IL-6, IL-8, and MCP-1 was also demonstrated. Upregulation
of ICAM-1 expression and
ICAM-1-mediated adherence of PBMC to endothelial cells were observed after anti-DV NS1 stimulation. The expression of ICAM-1
was modulated, at least in part, by MCP-1 and by NF-B. Taken together, these facts indicate that endothelial cell activation that involves NF-B-regulated cytokine and chemokine production and adhesion molecule expression, as well as adherence of immune cells to endothelial cells, may represent the inflammatory mechanisms mediated by anti-DV NS1 Abs. The signaling pathways induced by anti-DV NS1 in endothelial cells leading to immune activation and inflammation are shown in Fig. 8. Although our previous study showed an NO-mediated mechanism of apoptosis induced by anti-DV NS1, immune activation
is NF-B-dependent but NO- and
apoptosis-independent.
Several in vitro studies have shown that endothelial cells are the targets of DV and that induction of proinflammatory responses and apoptotic cell death are directly caused by DV (26, 27, 63). We have shown somewhat similar effects mediated by anti-DV NS1 Abs, specifically that endothelial cells underwent apoptosis (40-42) and, in the present study, induced proinflammatory effects. The combined effects of DV and anti-NS1 Abs on endothelial cells related to dengue pathogenesis remain to be investigated. Recently, DV NS1 has been reported to express in a GPI-linked form that results in signal transduction, as evidenced by tyrosine phosphorylation of cellular proteins (64). In
this study, we showed tyrosine
phosphorylation and NF-B activation after anti-DV NS1 stimulation in endothelial cells without DV infection. The role of protein
tyrosine phosphorylation in the
NF-B-mediated immune activation requires further clarification. To explore the signaling pathways mediated by anti-DV NS1, the surface molecules of endothelial cells, GPI-linked or not, recognized by these autoAbs need to be identified. Studies on potential autoantigens using anti-DV NS1 Abs and DHF/DSS patient sera are in progress.
Various autoimmune diseases associated with vascular dysfunction may be related to the generation of AECA (42). Vasculopathy in autoimmune diseases involves both cytotoxicity and inflammation facilitated by AECA via a direct or an indirect route. The elevated expression of inflammatory mediators, including cytokines (IL-1 and IL-6), chemokines (IL-8 and MCP-1), and adhesion molecules (ICAM-1, VCAM-1, and
E-selectin) has been shown in
AECA-stimulated endothelial cells (65-68). However, the molecular mechanisms of AECA-mediated endothelial cell activation remain poorly defined. Studies on the mechanisms of endothelial activation have shown that degradation of I-B followed by NF-B activation might contribute to the induction of proinflammatory responses (67). Activation of NF-B was also detected when anti-DV NS1 cross-reacted with uninfected endothelial cells. A recent study provided a possible molecular mechanism of AECA’s regulation of ICAM-1 expression through ERK1/2 activation in Behcet’s disease (69).
In dengue pathogenesis, elevated levels of inflammatory IL-6 and IL-8 were correlated with clinical presentation of DHF (46-48, 52). Also, endothelial cells infected with DV induced IL-6 and IL-8 production (26, 27). In the present study, we showed that anti-DV NS1 promoted proinflammatory endothelial cell activation causing increases in IL-6 and IL-8 protein expression and secretion. Furthermore, anti-DV NS1 induces the generation of MCP-1 in endothelial cells. A previous study showed MCP-1 in DSS patient plasma and pleural fluid, but not in DV-infected endothelial cells (27). We found, in the present study, MCP-1 production by endothelial cells after anti-DV NS1 stimulation; MCP-1 in DHF patient sera was also confirmed. In addition to the two chemokines IL-8 and MCP-1, RANTES was also checked in this study and was found not induced by anti-DV NS1 stimulation, although it was produced in endothelial cells
after DV infection (27). MCP-1-mediated elevation of ICAM-1 expression and facilitation of leukocyte transmigration has been suggested (53-55). Results in this study showed that MCP-1 upregulated ICAM-1 expression in anti-DV NS1-treated HMEC-1 cells. Endothelial cell activation followed by elevated expression of ICAM-1 may contribute to the adherence of immune cells to endothelial cells in the inflammatory responses associated with dengue disease pathogenesis. The involvement of other cytokines, chemokines, and adhesion molecules is currently under investigation. In the present study, we showed the inflammatory initiator role of anti-DV NS1 resulting in endothelial cell activation. In addition to the increased expression of cytokines and chemokines, a possible chemotactic effect on PBMC adherence may contribute to the indirect damage in endothelial cells. Activation of endothelial cells via DV-infected peripheral blood monocytes has been reported (33). Endothelial cell activation and apoptosis might play a role in the fulminant but short-lived vascular leakage of DHF pathogenesis (27). The clinical features indicate that vascular endothelia in serosal tissues are preferentially affected by dengue pathogenetic mechanisms, resulting in ascites and pleural effusion for most of the plasma leakage in dengue shock syndrome (70). Tissue culture fluid from DV-infected monocytes has activated endothelial cells (33). Serum inflammatory cytokines may relate to the development of plasma leakage and disease severity (8, 52). Given all these data, endothelial vascular dysfunction in dengue pathogenesis involves multiple factors, including the dengue virus itself and anti-DV NS1, direct and indirect effects on endothelial cells, and immune activation and apoptosis. 七、計畫成果自評 登革病毒 (DV) 感染造成致病的發生 機制是多因的,我們的研究顯示 DV 感染 產生自體免疫反應生成抗內皮細胞自體抗 體,係源自於病毒利用其NS1 蛋白所引發 的分子模擬自體免疫反應,進而產生與宿 主細胞有交叉結合作用的病理效應。本計 畫初始的目標為發展以登革病毒非結構性 蛋白 1 (NS1) 做為基礎抗原蛋白,研發中 和性登革疫苗抗體。先前的研究發現,NS1 在登革病毒感染時參與了自體免疫抗體生 成的機制。而根據 DV 感染引起種種的病 理反應都顯示除了病毒的直接傷害外,愈 來 愈 多 的 證 據 顯 示 宿 主 因 子 可 能 參 與 DHF/DSS 的病程發展與機制。我們的研究 顯示DV NS1 抗體 (anti-DV NS1) 就是一 種自體免疫抗體,對血管內皮細胞均有結 合作用並造成對細胞功能上的影響。我們 認為來自病毒的直接感染和抗體的合併效 應,亦或是抗體直接的影響都有可能是造 成內皮細胞死亡進而導致血管內皮系統失 去正常機能進而參與出血性病症的發生。 抗體與內皮細胞的結合作用會造成細胞死 亡的結果,除此之外,相關激素的生成如 IL-6、IL-8 及 MCP-1 亦會受到抗體的作用。 換句話說,除了病毒感染的細胞活化反 應,抗DV NS1 抗體似乎也可刺激內皮細 胞造成細胞不正當的活化反應生成激素, 而是否與 DV 的致病機制有關是必須再進 一步釐清。基於這些結果的發現,警示我 們在研發疫苗之前,針對抗體所可能參與 的自體免疫反應,有必要去釐清它所隱藏 的潛在性致病角色。我們以NS1 抗體為研 究對象,希冀瞭解抗體其自體免疫反應可 能會對宿主細胞造成的影響,並研究自體 免疫反應在登革病毒引發嚴重出血性及休 克併發症 DHF/DSS 之致病機制中可能扮 演的角色。研究結果希望能提供我們重要 的訊息,特別是找出NS1 蛋白引發自體免 疫反應的抗原決定位。計劃將此決定位去 除或予以修飾的方式,排除NS1 所引發的 cross-reactive 自體免疫抗體。如此一來,才 能研發具保護性而無致病性的 candidate vaccines。 本計畫「登革病毒感染產生自體抗體 對內皮細胞的影響及疫苗研發的考量」為 三年期計畫,計畫執行之目的為了解登革 病毒感染產生抗登革病毒非結構性蛋白 1 抗體(anti-DV NS1)對內皮細胞的自體免疫 現象,除了進一步探討自體抗體的細胞病 理效應,鑑定引發自體抗體的抗原分子亦 是本計畫的既定目標。第三年的研究重點 除了繼續研究可能的自體抗原之外,並以 基因剔除及重組的方式將可能引發自體抗 體的NS1 某些抗原序列做為修飾蛋白並評 估其產生的抗體對內皮細胞的作用。過去 三年的研究成果除基因體計畫辦公室定期 排定之進度報告之外,計畫執行期間亦參 與或受邀國內多次生物醫學會議,發表與 本計畫相關之研究成果。國外的會議則包 括 2003 年 12 月在美國舊金山舉行的 43rd
Annual Meeting of The American Society for Cell Biology、以及將於 2004 年 7 月在加拿 大蒙特婁舉行的12th International Congress of Immunology and 4th Annual Conference of FOCIS 進行口頭報告及張貼 poster。另 外,計畫期間業已彙整部分研究成果論文
發表於國際期刊,計有 2002 年刊登於
Journal of Immunology (40)、2003 年刊登於 Journal of Medical Virology (41)、以及 2004
年應邀撰寫 review paper 刊登於 Current
Pharmaceutical Design (42),而且令人興奮 的是我們所整理製作的圖被選為該期的封 面。此外與越南胡志明市第一兒童醫院合
作的臨床檢體研究結果刊登於 2004 年
Journal of Infectious Diseases (52)。另有一 篇 與 cytokine 、 chemokine 、 adhesion molecule expression 相關的 paper 於今年發 表於Journal of Immunology (71)。另外,亦 受 WHO 之邀請撰寫論文發表於 2004 年 Dengue Bulletin (72),還有一篇在實驗動物 的研究目前正在撰寫中。 目前登革疫苗的臨床效果並不如預期 或仍在改良階段。本計畫的執行目標即找 出引發自體免疫效應的抗原決定位,在未 來修飾疫苗的研發上提供重要的考量依 據。本計畫近期利用基因體計畫核心實驗 室蛋白質體學工具的輔助下,從事自體抗 體結合抗原的搜尋與鑑定,初步結果顯示 了數個內皮細胞細胞膜上蛋白質與自體抗 體的交叉結合作用。我們將針對這些標的 蛋白質做基因序列上或結構上的比對,進 一步了解其與NS1 蛋白質及 anti-NS1 抗體 的作用關係。另外,我們根據人類基因庫
序列的比對分析發現Mucin 5AC 及 Mucin
5B 蛋白質與登革病毒 NS1 蛋白質有部分 序列相似,除了製備將此相似序列剔除修 飾後的NS1 重組蛋白質,我們將進一步評 估修飾蛋白質引發抗體的免疫特性。另一 方面,我們持續研究自體抗體的細胞病理 效應,利用基因體計畫核心實驗室核酸微 陣列的技術廣泛地研究自體抗體對內皮細 胞的基因調控。未來,擬以小鼠模式評估 研究重組核酸或蛋白質疫苗對登革病毒的 保護性及降低引發自體免疫的現象。 八、參考文獻
1. Henchal, E. A., and J. R. Putnak. 1990. The dengue viruses. Clin. Microbiol. Rev. 3:376. 2. Gubler, D. J. 1998. Dengue and dengue
hemorrhagic fever. Clin. Microbiol. Rev. 11:480. 3. Bhakdi, S., and M. D. Kazatchkine. 1990.
Pathogenesis of dengue: an alternative hypothesis. Southeast Asian J. Trop. Med. Pub. Hlth. 21:652. 4. Bielefeldt-Ohmann, H. 1997. Pathogenesis of
dengue virus diseases: missing pieces in the jigsaw. Trends Microbiol. 5:409.
5. Pancharoen, C., U. Thisyakorn, and C. Thisyakorn. 2001. Dengue infection. J. Infect. Dis. Antimicrob. Agents 18:115.
6. Halstead, S. B. 2002. Dengue. Curr. Opin. Infect. Dis. 15:471.
7. Rothman, A. L., and F. A. Ennis. 1999. Immunopathogenesis of dengue hemorrhagic fever. Virology 257:1.
8. Green, S., D. W. Vaughn, S. Kalayanarooj, S. Nimmannitya, S. Suntayakorn, A. Nisalak, R. Lew, B. L. Innis, I. Kurane, A. L. Rothman, and F. A. Ennis. 1999. Early immune activation in acute dengue illness is related to development of plasma leakage and disease severity. J. Infect. Dis. 179:755.
9. Lei, H. Y., T. M. Yeh, H. S. Liu, Y. S. Lin, S. H. Chen, and C. C. Liu. 2001. Immunopathogenesis of dengue virus infection. J. Biomed. Sci. 8:377. 10. Halstead, S. B., C. N. Venkateshan, M. K. Gentry,
and L. K. Larsen. 1984. Heterogeneity of infection enhancement of dengue 2 strains by monoclonal antibodies. J. Immunol. 132:1529. 11. Littaua, R., I. Kurane, and F. A. Ennis. 1990.
Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. J. Immunol. 144:3183.
12. Mady, B. J., D. V. Erbe, I. Kurane, M. W. Fanger, and F. A. Ennis. 1991. Antibody-dependent enhancement of dengue virus infection mediated by bispecific antibodies against cell surface molecules other than Fcreceptors. J. Immunol. 147:3139.
13. Morens, D. M. 1994. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clin. Infect. Dis. 19:500.
14. Rico-Hesse, R., L. M. Harrison, R. A. Salas, D. Tovar, A. Nisalak, C. Ramos, J. Boshell, M. T. R. de Mesa, R. M. R. Nogueira, and A. Travassos da Rosa. 1997. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 230:244.
15. Leitmeyer, K. C., D. W. Vaughn, D. M. Watts, R. Salas, I. Villalobos de Chacon, C. Ramos, and R. Rico-Hesse. 1999. Dengue virus structural differences that correlate with pathogenesis. J. Virol. 73:4738.
16. Diamond, M. S., D. Edgil, T. G. Roberts, B. Lu, and E. Harris. 2000. Infection of human cells by dengue virus is modulated by different cell types and viral strains. J. Virol. 74:7814.
17. Vaughn, D. W., S. Green, S. Kalayanarooj, B. L. Innis, S. Nimmannitya, S. Suntayakorn, T. P. Endy, B. Raengsakulrach, A. L. Rothman, F. A. Ennis, and A. Nisalak. 2000. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181:2.
18. Kurane, I., B. L. Innis, A. Nisalak, C. Hoke, S. Nimmannitya, A. Meager, and F. A. Ennis. 1989.
Human T cell responses to dengue virus antigens: proliferative responses and interferon gamma production. J. Clin. Invest. 83:506.
19. Mathew, A., I. Kurane, S. Green, D. W. Vaughn, S. Kalayanarooj, S. Suntayakorn, F. A. Ennis, and A. L. Rothman. 1999. Impaired T cell proliferation in acute dengue infection. J. Immunol. 162:5609. 20. Lin, Y. W., K. J. Wang, H. Y. Lei, Y. S. Lin, T. M.
Yeh, H. S. Liu, C. C. Liu, and S. H. Chen. 2002. Virus replication and cytokine production in dengue virus-infected human B lymphocytes. J. Virol. 76:12242.
21. King, C. A., J. S. Marshall, H. Alshurafa, and R. Anderson. 2000. Release of vasoactive cytokines by antibody-enhanced dengue virus infection of a human mast cell/basophil line. J. Virol. 74:7146. 22. Marianneau, P., A. M. Steffan, C. Royer, M. T.
Drouet, D. Jaeck, A. Kirn, and V. Deubel. 1999. Infection of primary cultures of human Kupffer cells by Dengue virus: no viral progeny synthesis, but cytokine production is evident. J. Virol. 73:5201.
23. Wu, S. J., G. Grouard-Vogel, W. Sun, J. R. Mascola, E. Brachtel, R. Putvatana, M. K. Louder, L. Filgueira, M. A. Marovich, H. K. Wong, A. Blauvelt, G. S. Murphy, M. L. Robb, B. L. Innes, D. L. Birx, C. G. Hayes, and S. S. Frankel. 2000. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 6:816.
24. Ho, L. J., J. J. Wang, M. F. Shaio, C. L. Kao, D. M. Chang, S. W. Han, and J. H. Lai. 2001. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J. Immunol. 166:1499.
25. Libraty, D. H., S. Pichyangkul, C. Ajariyakhajorn, T. P. Endy, and F. A. Ennis. 2001. Human dendritic cells are activated by dengue virus infection: enhancement by gamma interferon and implications for disease pathogenesis. J. Virol. 75:3501.
26. Huang, Y. H., H. Y. Lei, H. S. Liu, Y. S. Lin, C. C. Liu, and T. M. Yeh. 2000. Dengue virus infects human endothelial cells and induces IL-6 and IL-8 production. Am. J. Trop. Med. Hyg. 63:71. 27. Avirutnan, P., P. Malasit, B. Seliger, S. Bhakdi,
and M. Husmann. 1998. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J. Immunol. 161:6338.
28. Despres, P., M. Flamand, P.-E. Ceccaldi, and V. Deubel. 1996. Human isolates of dengue type 1 virus induce apoptosis in mouse neuroblastoma cells. J. Virol. 70:4090.
29. Marianneau, P., A. Cardona, L. Edelman, V. Deubel, and P. Despres. 1997. Dengue virus replication in human hepatoma cells activates NF-B which in turn induces apoptotic cell death. J. Virol. 71:3244.
30. Despres, P., M. P. Frenkiel, P. E. Ceccaldi, C.
Duarte dos Santos, and V. Deubel. 1998. Apoptosis in the mouse central nervous system in response to infection with mouse-neurovirulent dengue viruses. J. Virol. 72:823.
31. Marianneau, P., M. Flamand, V. Deubel, and P. Despres. 1998. Apoptotic cell death in response to dengue virus infection: the pathogenesis of dengue haemorrhagic fever revisited. Clin. Diagn. Virol. 10:113.
32. Jan, J. T., B. H. Chen, S. H. Ma, C. I. Liu, H. P. Tsai, H. C. Wu, S. Y. Jiang, K. D. Yang, and M. F. Shaio. 2000. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: arachidonic acid, superoxide anion, and NF-B are sequentially involved. J. Virol. 74:8680. 33. Anderson, R., S. Wang, C. Osiowy, and A. C.
Issekutz. 1997. Activation of endothelial cells via antibody-enhanced dengue virus infection of peripheral blood monocytes. J. Virol. 71:4226. 34. Gagnon, S. J., F. A. Ennis, and A. L. Rothman.
1999. Bystander target cell lysis and cytokine production by dengue virus-specific human CD4+
cytotoxic T-lymphocyte clones. J. Virol. 73:3623. 35. Nimmannitya, S. 1987. Clinical spectrum and
management of dengue haemorrhagic fever. Southeast Asian J. Trop. Med. Pub. Hlth. 18:392. 36. Bhamarapravati, N. 1989. Hemostatic defects in
dengue hemorrhagic fever. Rev. Infect. Dis. 11:S826.
37. Kalayanarooj, S., D. W. Vaughn, S. Nimmannitya, S, Green, S. Suntayakorn, N. Kunentrasai, W. Viramitrachai, S. Ratanachueke, S. Kiatpolpoj, B. L. Innis, A. L. Rothman, A. Nisalak, and F. A. Ennis. 1997. Early clinical and laboratory indictors of acute dengue illness. J. Infect. Dis. 176:313.
38. Bosch I, K. Xhaja, L. Estevez, G. Raines, H. Melichar, R. V. Warke, M. V. Fournier, F. A. Ennis, and A. L. Rothman. 2002. Increased production of interleukin-8 in primary human monocytes and in human epithelial and endothelial cell lines after dengue virus challenge. J. Virol. 76:5588.
39. Warke R. V., K. Xhaja, K. J. Martin, M. F. Fournier, S. K. Shaw, N. Brizuela, N. De Bosch, D. Lapointe, F. A. Ennis, A. L. Rothman, and I. Bosch. 2003. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J Virol. 77:11822.
40. Lin, C. F., H. Y. Lei, A. L. Shiau, H. S. Liu, T. M. Yeh, S. H. Chen, C. C. Liu, S. C. Chiu, and Y. S. Lin. 2002. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J. Immunol. 169:657.
41. Lin, C. F., H. Y. Lei, A. L. Shiau, C. C. Liu, H. S. Liu, T. M. Yeh, S. H. Chen, and Y. S. Lin. 2003. Antibodies from dengue patient sera cross-react with endothelial cells and induce damage. J. Med. Virol. 69:82.
42. Lin, Y. S., C. F. Lin, C. C. Liu, H. S. Liu, T. M. Yeh, S. H. Chen, and H. Y. Lei. 2004. Antibody-mediated endothelial cell damage via nitric oxide. Curr. Pharm. Design 10:213.
43. Lin, Y. L., C. C. Liu, J. I. Chuang, H. Y. Lei, T. M. Yeh, Y. S. Lin, Y. H. Huang, and H. S. Liu. 2000. Involvement of oxidative stress, NF-IL-6, and RANTES expression in dengue-2-virus-infected human liver cells. Virology 276:114.
44. King, C. A., R. Anderson, and J. S. Marshall. 2002. Dengue virus selectively induces human mast cell chemokine production. J. Virol. 76:8408.
45. Kurane, I., B. L. Innis, S. Nimmannitya, A. Nisalak, A. Meager, J. Janus, and F. A. Ennis. 1991. Activation of T lymphocytes in dengue virus infections. High levels of soluble interleukin 2 receptor, soluble CD4, soluble CD8, interleukin 2, and interferon-gamma in sera of children with dengue. J. Clin. Invest. 88:1473.
46. Hober, D., L. Poli, B. Roblin, P. Gestas, E. Chungue, G. Granic, P. Imbert, J. Pecarere, R. Vergez-Pascal, P. Wattre, and M. Maniez-Montreuil. 1993. Serum levels of tumor necrosis factor- (TNF-), interleukin-6 (IL-6), and interleukin-1 (IL-1) in dengue-infected patients. Am. J. Trop. Med. Hyg. 48:324.
47. Raghupathy, R., U. C. Chaturvedi, H. Al-Sayer, E. A. Elbishbishi, R. Agarwal, R. Nagar, S. Kapoor, A. Misra, A. Mathur, H. Nusrat, F. Azizieh, M. A. Y. Khan, and A. S. Mustafa. 1998. Elevated levels of IL-8 in dengue hemorrhagic fever. J. Med. Virol. 56:280.
48. Juffrie, M., G. M. van Der Meer, C. E. Hack, K. Haasnoot, K, Sutaryo, A. J. Veerman, and L. G. Thijs. 2000. Inflammatory mediators in dengue virus infection in children: interleukin-8 and its relationship to neutrophil degranulation. Infect. Immun. 68:702.
49. Mustafa, A. S., E. A. Elibishbishi, R. Agarwal, and U. C. Chaturvedi. 2001. Elevated levels of interleukin-13 and IL-18 in patients with dengue hemorrhagic fever. FEMS Immunol. Med. Microbiol. 30:229.
50. Gagnon, S. J., M. Mori, I. Kurane, S. Green, D. W. Vaughn, S. Kalayanarooj, S. Suntayakorn, F. A. Ennis, and A. L. Rothman. 2002. Cytokine gene expression and protein production in peripheral blood mononuclear cells of children with acute dengue virus infections. J. Med. Virol. 67:41. 51. Suharti, C., E. C. van Gorp, W. M. Dolmans, T. E.
Setiati, C. E. Hack, R. Djokomoeljanto, and J. W. van der Meer. 2003. Cytokine patterns during dengue shock syndrome. Eur. Cytokine Netw. 14:172.
52. Hung, N. T., H. Y. Lei, N. T. Lan, Y. S. Lin, K. J. Huang, B. Lien le, C. F. Lin, T. M. Yeh, Q. Ha do, V. T. Huong, L. C. Chen, J. H. Huang, L. T. My, C. C. Liu, and S. B. Halstead. 2004. Dengue
hemorrhagic fever in infants: a study of clinical and cytokine profiles. J. Infect. Dis. 189:221. 53. Frostegard, J., J. Nilsson, A. Haegerstrand, A.
Hamsten, H. Wigzell, and M. Gidlund. 1990. Oxidized low density lipoprotein induces differentiation and adhesion of human monocytes and the monocytic cell line U937. Proc. Natl. Acad. Sci. USA 87:904.
54. Adams, M. R., W. Jessup, D. Hailstones, and D. S. Celermajer. 1997. L-Arginine reduces human monocyte adhesion to vascular endothelium and endothelial expression of cell adhesion molecules. Circulation 95:662.
55. Weiss, J. M., S. A. Downie, W. D. Lyman, and J. W. Berman. 1998. Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J. Immunol. 161:6896. 56. Tekstra, J., H. Beekhuizen, J. S. Van De Gevel, I. J. Van Benten, C. W. Tuk, and R. H. Beelen. 1999. Infection of human endothelial cells with Staphylococcus aureus induces the production of monocyte chemotactic protein-1 (MCP-1) and monocyte chemotaxis. Clin. Exp. Immunol. 117:489.
57. Viedt, C., R. Dechend, J. Fei, G. M. Hansch, J. Kreuzer, and S. R. Orth. 2002. MCP-1 induces inflammatory activation of human tubular epithelial cells: involvement of the transcription factors, nuclear factor-kappaB and activating protein-1. J. Am. Soc. Nephrol. 13:1534.
58. Monath, T. P. 1994. Dengue: the risk to developed and developing countries. Proc. Natl. Acad. Sci. USA 91:2395.
59. Lin, C. F., H. Y. Lei, C. C. Liu, H. S. Liu, T. M. Yeh, S. T. Wang, T. I. Yang, F. C. Sheu, C. F. Kuo, and Y. S. Lin. 2001. Generation of IgM anti-platelet autoantibody in dengue patients. J. Med. Virol. 63:143.
60. Huang, K. J., S. Y. J. Li, S. C. Chen, H. S. Liu, Y. S. Lin, T. M. Yeh, C. C. Liu, and H. Y. Lei. 2000. Manifestation of thrombocytopenia in dengue-2-virus-infected mice. J. Gen. Virol. 81:217.
61. Falconar, A. K. I. 1997. The dengue virus nonstructural-1 protein (NS1) generates antibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to human endothelial cells: potential implications in haemorrhagic fever pathogenesis. Arch. Virol. 142:897.
62. Oishi, K., S. Inoue, M. T. Cinco, E. M. Dimaano, M. T. Alera, J. A. Alfon, F. Abanes, D. J. Cruz, R. R. Matias, H. Matsuura, F. Hasebe, S. Tanimura, A. Kumatori, K. Morita, F. F. Natividad, and T. Nagatake. 2003. Correlation between increased platelet-associated IgG and thrombocytopenia in secondary dengue virus infections. J. Med. Virol. 71:259.
63. Andraws, B. S., A. N. Theofilopoulos, C. J. Peters, D. J. Loskutoff, W. E. Brandt, and F. J. Dixon. 1978. Replication of dengue and junin viruses in cultured rabbit and human endothelial cells. Infect. Immun. 20:776.
64. Jacobs, M. G., P. J. Robinson, C. Bletchly, J. M. Mackenzie, and P. R. Young. 2000. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 14:1603. 65. Carvalho, D., C. O. S. Savage, C. M. Black, and J.
D. Pearson. 1996. IgG antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelial-derived cytokines. J. Clin. Invest. 97:111.
66. Del Papa, N., E. Raschi, G. Moroni, P. Panzeri, M. O. Borghi, C. Ponticelli, A. Tincani, G. Balestrieri, and P. L. Meroni. 1999. Anti-endothelial cell IgG fractions from systemic lupus erythematosus patients bind to human endothelial cells and induce a pro-adhesive and a pro-inflammatory phenotype in vitro. Lupus 8:423.
67. Yazici, Z. A., E. Raschi, A. Patel, C. Testoni, M. O. Borghi, A. M. Graham, P. L. Meroni, and N. Lindsey. 2001. Human monoclonal anti-endothelial cell IgG-derived from a systemic lupus erythematosus patient binds and activates human endothelium in vitro. Int. Immunol. 13:349.
68. Praprotnik, S., M. Blank, Y. Levy, S. Tavor, M. C. Boffa, B. Weksler, A. Eldor, and Y. Shoenfeld. 2001. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int. Immunol. 13:203.
69. Lee, K. H., H. J. Cho, H. S. Kim, W. J. Lee, S. Lee, and D. Bang. 2002. Activation of extracellular signal regulated kinase 1/2 in human dermal microvascular endothelial cells stimulated by anti-endothelial cell antibodies in sera of patients with Behçet's disease. J. Dermatol. Sci. 30:63.
70. Innis, B. L. 1995. Dengue and dengue hemorrhagic fever. In Kass Handbook of Infectious Diseases: Exotic Viral Diseases, J. S. Porterfield, ed. Chapman and Hall, London, p. 103.
71. Lin, C. F., S. C. Chiu, Y. L. Hsiao, S. W. Wan, H. Y. Lei, A. L. Shiau, H. S. Liu, T. M. Yeh, S. H. Chen, C. C. Liu, and Y. S. Lin. 2005. Expression of cytokine, chemokine, and adhesion molecules during endothelial cell activation induced by antibodies against dengue virus nonstructural protein 1. Journal of Immunology 174:395. 72. Lin, C. F., H. Y. Lei, C. C. Liu, H. S. Liu, T. M.
Yeh, S. H. Chen, and Y. S. Lin. 2004.
Autoimmunity in dengue virus infection. Dengue Bulletin 28:51. Geneva: World Health Organization (invited paper)
Figure legend
Figure 1. Tyrosine phosphorylation of cellular proteins after treatment with anti-DV NS1 Abs in endothelial cells. After serum-free starvation for 1 h, HMEC-1 cells were treated with 5 g of anti-DV NS1, anti-JEV NS1, or control IgG, or replaced with endothelial cell growth medium (EGM, used as positive controls for cell signaling) composed of 2% FBS for 30 min. The tyrosine phosphorylated protein levels were analyzed by Western blotting (A, upper panel) and quantified by measuring optical density (A,
lower panel). The cells were stained with
FITC-conjugated phosphorylated tyrosine-specific IgG and analyzed using flow cytometry. A histogram and the percentage of positive cells are shown (B, upper panel). Time-kinetic changes of protein tyrosine phosphorylation induced by control IgG (●), anti-DV NS1 (▲), and EGM () are shown, respectively. The untreated cells () were used as negative controls (B, lower panel).
Figure 2. NF-B activation induced by anti-DV NS1 Abs in endothelial cells. A, HMEC-1 cells were untreated or treated with 5 g of anti-DV NS1, anti-JEV NS1, or control IgG for 3 or 24 h, and the expression of NF-B in cytoplasm and nucleus was determined using Western blotting. -actin was used as an internal control of total cell lysate before cytoplasmic and nuclear extraction. B, the DNA-binding ability of NF-B in HMEC-1 cells after treatment with anti-DV NS1 (5 or 25 g), anti-JEV NS1 (25 g), or control IgG (25 g) for 3 h was detected by EMSA using NF-B specific probes. Cells treated with anti-DV NS1 preabsorbed with 25 g of recombinant DV NS1 protein were tested for the specificity of Ab stimulation. NF-B p65 fragment was used as a positive control. Free probe was used as a loading control.
Figure 3. Production of IL-6, IL-8, and MCP-1, but not RANTES, by endothelial cells after treatment with anti-DV NS1 Abs. HMEC-1 cells were treated with 5g of anti-DV NS1, anti-JEV NS1, or control IgG for 24 h. The expression of IL-6, IL-8, MCP-1, and RANTES were detected with immunohistochemical staining using specific Abs as described in Materials and
Methods (A, the positive cells are indicated by arrowheads) and with flow cytometry analysis
individual cultures were performed and the averages of the percentages of positive cells are shown (mean SD). Cells treated with anti-DV NS1 preabsorbed with 5 g of recombinant DV or JEV NS1 protein were tested for the specificity of Ab stimulation.
Figure 4. The expression of MCP-1 but not RANTES mRNA by endothelial cells after anti-DV NS1 Abs treatment. Total cellular mRNA was extracted at 24 h from HMEC-1 cells after treatment with 5 g of anti-DV NS1, anti-JEV NS1, or control IgG or with 100 MOI of dengue-2 virus, and detected using RT-PCR. -actin was used as an internal control.
Figure 5. The involvement of NF-B in the expression of IL-6, IL-8, and MCP-1 in endothelial cells after anti-DV NS1 stimulation. In the presence or absence of PDTC (100 M), L-NAME (10 M), or zVAD-fmk (10 M) for 1 h pretreatment or transfected with dominant negative IKK- ( Δ IKK-) or empty vector, HMEC-1 cells were treated with 5g of anti-DV NS1, anti-JEV NS1, or control IgG for 24 h. The expression of IL-6, IL-8, and MCP-1 was detected using ELISA as described in Materials
and Methods. Three individual cultures were
performed in ELISA and the averages are shown (meanSD).
Figure 6. MCP-1 regulates the expression of ICAM-1 in endothelial cells after anti-DV NS1 Abs stimulation. A, HMEC-1 cells were treated with 5 g of anti-DV NS1, anti-JEV NS1, or control IgG for 24 h. Cells were labeled with mouse anti-human ICAM-1 Abs followed by HRP- or FITC-conjugated goat anti-mouse IgG. Cells treated with anti-DV NS1 in the presence of 5g neutralizing anti-MCP-1 were also tested. The ICAM-1 expression was detected using immunohistochemical staining. B, HMEC-1 cells were treated with 1, 5, or 25g of anti-DV NS1, anti-JEV NS1, or control IgG for 6, 12, or 24 h. The ICAM-1 expression was detected using flow cytometry analysis. Three individual cultures were performed and the percentages of positive cells are shown (meanSD).
Figure 7. The ability of PBMC to adhere to anti-DV NS1-treated endothelial cells is blocked by pretreatment with anti-ICAM-1 and anti-MCP-1. A, HMEC-1 cells were cultured in 8-well glass chamber slides and treated with 5g of anti-DV NS1, anti-JEV NS1, or control IgG for 24 h. In anti-DV NS1-treated groups, cells cultured with 5 g neutralizing anti-ICAM-1 or anti-MCP-1 were also tested. Cells were washed with PBS, and then co-cultured with fresh human
PBMC for 2 h. Cell adhesion was observed using Liu's stain. B, time kinetics and dose response of anti-DV NS1 on the PBMC adhesion to endothelial cells in the presence or absence of 100 M cycloheximide or 5 g anti-ICAM-1 or anti-MCP-1. The numbers of adherent cells were quantified in each tested culture.
Figure 8. The signaling pathways mediated by anti-DV NS1 Abs which lead to endothelial cell activation. Signal transduction in endothelial cells after anti-NS1 stimulation involves protein tyrosine phosphorylation and NF-B activation. NF-B is required for the expression of cytokines (e.g., IL-6) and chemokines (e.g., IL-8 and MCP-1). Expression of ICAM-1 induced by anti-NS1 is, at least in part, regulated by NF-B and by MCP-1. Furthermore, the ability of PBMC to adhere to endothelial cells is mediated by ICAM-1. The endothelial cell activation-induced by anti-DV NS1 Abs leading to inflammatory responses may be related to DHF pathogenesis.
Figure 2
Figure 3
Figure 4
Figure 5
Figure 7