國立臺東大學應用科學系 碩士論文
Department of Applied Science National Taitung University
Master Thesis
單核與雙核二價錳、硫和磷錯合物的合 成及性質和其與氧氣反應的探討
Synthesis and Characterization of Mono- and Binuclear Phosphorus-Thiolate-Ligated Mn(II)
Complexes and the Reactivity Studies with Dioxygen
研 究 生: 吳汶諺 撰 指導教授: 李建明 博士
中華民國 106 年 5 月
國立臺東大學應用科學系 碩士論文
Department of Applied Science National Taitung University
Master Thesis
單核與雙核二價錳、硫和磷錯合物的合 成及性質和其與氧氣反應的探討
Synthesis and Characterization of Mono- and Binuclear Phosphorus-Thiolate-Ligated Mn(II)
Complexes and the Reactivity Studies with Dioxygen
研 究 生: 吳汶諺 撰 指導教授: 李建明 博士
中華民國 106 年 5 月
誌謝詞
至此我要感謝生活周遭的每個給予累積經驗知識的人,自大三到碩二在生
物無機實驗室這三年半的時間,首先我要感謝指導教授李建明老師給予需多知
識以及態度的教誨,另外讓我有機會參與許多的研討會見世面。實驗室中感謝
其和學長、任達學長在實驗儀器討論與教導,怡臻學姊、冠宇文獻搜尋的支
援,家偉、勇致、婇蕙、宥汯、守錞、明佑、家和、宣佑、詩柔和宛蓉等同學
學弟妹實驗陪伴以及交流豐富了生活。最後感謝口試委員廖文峯老師和陳以文
老師在應對之間給予意見並通過碩士學位考試。
今後學習旅途中或許會有許多挑戰,但這些際遇也將琢磨自身成就未來。
吳汶諺 撰
中文摘要
本研究合成一系列錳、硫和磷錯合物模擬生物酵素探討與氧氣的反應性。
其中雙硫橋接雙核錳的[PPN]2[MnII2(TMSPS3)2] (1)由二氯化錳、[PPN]Cl 和 Li3TM-
SPS3合成,其與過量 DABCO 反應轉變成單核[PPN][MnII(DABCO)(TMSPS3)]
(2)。利用 UV-vis 光譜儀探討變溫實驗指出雙核 1 在存有 DABCO 的環境與單
核 2 之間的平衡反應,其低溫傾向單核 2 的生成。針對 1 和 2 與雙邊鍵結的四
價過氧化物[PPN][MnIV(η2-O2)TMSPS3] (3)反應產生氧橋接雙錳的[PPN]2[MnIII2(μ-
O)(TMSPS3)2](4),化合物 4 能更進一步的被水分子質化生成[PPN][MnIII(OH)(TMS-
PS3)](5)。電化學顯示 1 和 2 的氧化電位(MnIII/MnII)之間的位移,並指出兩者皆 有被氧氣氧化的潛力;5 則突顯高還原電位更難以誘導催化反應的發生。在不
同的相態探討 1 和 2 與氧氣活化,晶體的 2 與乾燥的氧氣反應幾乎完全轉換成
四價過氧基的 3,這指出完整的固相轉換以及氧氣的兩電子的還原。在溶液狀
態下受氧氣濃度與擴散效應影響,導致 3 和 4 兩種產物生成比例不同。
Abstract
In this studies, we synthesized a series of manganese complex with sulfur and
phosphorus donor atoms. A thiolate-bridged binuclear [PPN]
2[(Mn
II(
TMSPS
3))
2] (1) prepared
from the reaction of MnCl
2/ [PPN]Cl and Li
3[
TMSPS
3] converts into a mononuclear [PPN]-
[Mn
II(
TMSPS
3)(DABCO)] (2) in the presence of excess amounts of DABCO. Variable
temperature studies of solution containing 1 and DABCO by UV–vis spectroscopy indicate
that 1 and 2 exist in significant amounts in equilibrium and mononuclear 2 is favored at low
temperature. Treatment of 1 and 2 with a O
2-side-on-bound [PPN][Mn
IV(η
2-O
2)
TMSPS
3] (3)
produces a mono-oxo-bridged dimeric [PPN]
2[Mn
III2(μ-O)(
TMSPS
3)
2](4). Reaction of 4 with
H
2O can generate complex [PPN][Mn
III(OH)(
TMSPS
3)](5). The electrochemistry of 1 and 2
reveal anodic peak(s) for Mn
III/Mn
IIredox couple at shifted potentials against Fc/Fc
+,
indicating that both complexes can be oxidize by dioxygen. The O
2activation mediated by 1
and 2 is investigated in both solution and the solid states. Crystals of 2 rapidly reacts with air
or dry O
2to generate the Mn(IV)-peroxo 3 in nearly quantitative yield, revealing a clean
solid-to-solid transformation and two-electron reduction of O
2. Oxygenation of 1 or 2 in
solution, however, is affected by diffusion and transient concentration of dioxygen in the two
different substrates, leading to generation of 3 and 4 in variable yield ratio
目錄
第一章 緒論
1-1 氧氣 ... 1
1-2 第二光系統與氧氣的釋放 ... 4
1-3 Ib 型核醣核苷酸還原酶與酪氨酸自由基誘導因子 ... 6
1-4 錳氧模擬化合物文獻探討 ... 9
1-5 研究方向 ... 13
第二章 實驗部分
2-1 一般實驗 ... 142-2 儀器 ... 14
2-3 藥品 ... 15
2-4 化合物的合成與性質鑑定 2-4-1 配位基Tris(2-thiol-3-trimethylsiylphenyl)phosphine, (TMSPS3H3)的合 成 ... 18
2-4-2 化合物[PPN]2[MnII2(TMSPS3)2] (1)的合成 ... 19
2-4-3 化合物[PPN][MnII(DABCO)(TMSPS3)] (2)的合成 ... 19
2-4-4 化合物[PPN][MnIV(η2-O2)(TMSPS3)] (3)的合成 ... 20
2-4-5 化合物[PPN]2[MnIII2(μ-O)(TMSPS3)2] (4)的合成 ... 20
2-4-6 化合物 1 與 3 的反應 ... 21
2-4-7 化合物 1 與 3 在含有 PPh3的反應 ... 21
2-4-8 化合物 2 與 PhIO 反應 ... 21
2-4-9 化合物 1 與氧氣反應 ... 22
2-4-10 化合物 2 與氧氣反應 ... 22
2-4-11 化合物 1 與 2 的固相氧化 ... 23
2-4-12 化合物[PPN][MnIII(OH)(TMSPS3)] (5)的合成 ... 23
2-4-13 化合物 5 與 2,6-di-tbu-4-R-PhOH(R=OMe)的反應 ... 24
2-4-14 平衡常數的測定 ... 24
第三章 結果與討論
3-1 含錳(II)化合物的合成、結構與性質鑑定 ... 253-2 含錳(III)化合物的合成、結構與性質鑑定 ... 31
3-3 含錳(II)化合物與氧氣的反應性 ... 39
3-4 錯合物的分子結構探討 ... 47
第四章 結論
... 55參考文獻
... 57Figure
Figure 1-1. Molecular orbital diagram of dioxygen. ... 2 Figure 1-2. Oxygen disproportionation reaction. ... 2 Figure 1-3. Binding modes for mono- and diatomic oxygen ligands. ... 2 Figure 1-4. Comparison of the O-O bond length and vibrational frequencies for O2、O2‧-and O22-. ... 2 Figure 1-5. X-ray Crystal structure of the Mn4O5Ca core of the oxygen evolving complex of Photosystem II at a resolution of 1.9 Å .The Kok cycle for oxygen
evolving reactions of PSII. ... 5 Figure 1-6. Possible mechanisms for the oxygen-evolving reaction. ... 5 Figure 1-7. Proposed mechanism of nucleotide reduction by ribonucleotide
reductases (RNRs). ... 6 Figure 1-8. Structures (left) and metallocofactors (right) of the class Ib RNR β2 (NrdF) subunits from E. coil. ... 7 Figure 1-9. The proposed biosynthetic and maintenance pathways for the
metallocofactors of the class Ib ribonucleotide reductases. ... 8 Figure 1-10. Side-on MnIII-peroxo compounds (a) [MnIII(O2)TPP]− (b)
[MnIII(HB(3,5-iPr2pz)3)(O2)( 3,5-iPr2pzH)] (c) [Mn(TMC)(O2)]+ (d)
[MnIII(O2)H2bupa]-. ... 9 Figure 1-11. Reaction scheme shows an intermolecular O2-Transfer between
[NiIII(12-TMC)(O2)]+ and [MnII(14-TMC)]2+. ... 10 Figure 1-12. Reaction scheme shows an intermolecular O2-Transfer from superoxo- [FeIII(TAML)(O2)]2- to [MnIII(TAML)]– that generate peroxo-[MnIV(TAML)(O2)]2–
and [FeIII(TAML)]-. ... 10 Figure 1-13. Reaction of five-coordinate [MnII(SMe2N4(6-Me-DPEN)]+ with O2 to
afford mono-oxo-bridged [[MnIII(SMe2N4(6-Me-DPEN)]2(μ-O)]2+ via metastable Mn−superoxo intermediate and Mn−peroxo intermediate [[MnIII(SMe2N4(6-Me-
DPEN)]2(μ-O)]2+. ... 11 Figure 1-14. Proposed mechanism for the formation of [MnIV(O2)P(C6H3-3-SiMe3-2- S)3]− and [MnII2(PS3)2]2-. ... 14 Figure 3-1. UV-vis spectra (in acetonitrile) for 1 and 2 at ambient temperature ... 26 Figure 3-2. (a) UV-vis spectra for 1 titrate 0 to 100 equivalent of DABCO in
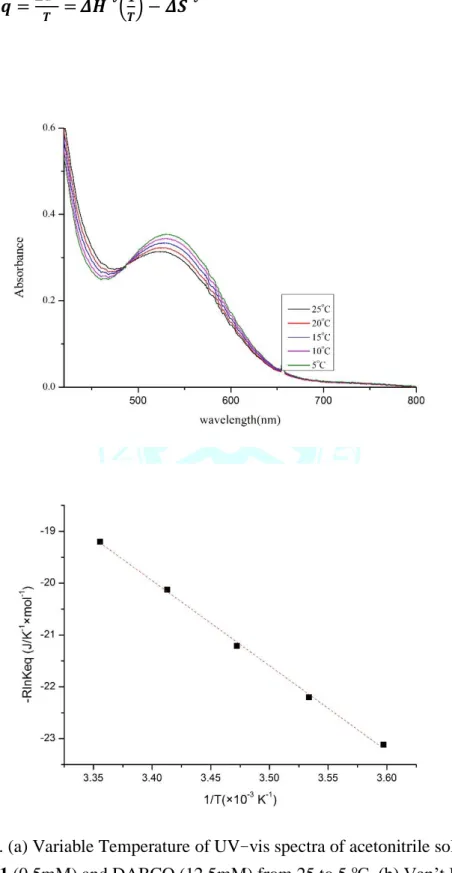
actontrile at room temperature. (b) The change of absorbance at 535 nm ... 27 Figure 3-3. Variable Temperature of UV-vis spectra of acetonitrile solution
containing 1 (0.5mM) and DABCO (12.5mM) from 25 to 5 oC. Inset: Van’t Hoff plot of equilibrium data based on eq 1 ... 28 Figure 3-4. Cylic voltammograms of 1 and 2 measured in a 1mM acetonitrile solution with 0.1M [n-Bu4N][PF6] as the supporting electrolyte at room temperature, scan rate 0.1 V/s ... 30 Figure 3-5. UV-vis spectra of 3 and 4 . Changes in UV-vis spectra (gray) after 3 with various equivalents of 2 in CH3CN at ambient temperature. Inset: absorbance changes at 670 nm upon addition of 2. ... 30 Figure 3-6. UV-vis spectrum (black line) of 4 generated from reaction of 1
(0.098mM) and 3 (0.065mM) based on eq :
(𝟑) +
32
(𝟏) → 2(𝟒)
; Red line shows the UV-vis spectrum of 4 generated from reaction of 2 (0.195mM) and 3 (0.065mM) based on eq :(𝟑) + 3 (𝟏) → 2 (𝟒)
... 33 Figure 3-7. GC-mass spectrum shows 3 reaction with 1 and PPh3 (10 equiv) that gerenated 279.1m/z (OPPh3+H+, black). 18O-labeled 3 confirms the source of O-atom that detected 281.1m/z (18OPPh3+H+, red) ... 33 Figure 3-8. UV-vis spectra of 4 with 10 equivalent H2O in acetonitrile that generate 2 equivalent 5 . Gray lines change show 4 convert to 5. The measured interval betweentwo curve is 1 min ... 34 Figure 3-9. Cylic voltammograms of 4 and 5 measured in a 1mM acetonitrile solution with 0.1M [n-Bu4N][PF6] as the supporting electrolyte at room temperature, scan rate 0.1 V/s. ... 35 Figure 3-10. UV-vis spectra of 5 with 100 equivalent 2,6-di-tbu-4-OMe-PhOH (black line) in acetonitrile. The measured interval between two curve is 20 min. ... 36 Figure 3-11. The microcrystals image of (a) 1, (b) after exposed dry O2 2 hours; (c) 2, (d) after exposed dry O2 2 hours. ... 37 Figure 3-12. Time-depended ATR-FTIR spectra of microcrystals of 1 and 2 after exposed to air. The measured interval between two curve is 1 min (top) and 5 min. 38 Figure 3-13. UV-vis spectra of solid-to-solid oxygenated 1 and 2. ... 39 Figure 3-14. UV-vis spectra of 1 (0.1mM) and 2 (0.2mM) reacts with excess oxygen by two different operation ... 40 Figure 3-15. UV-vis spectra of 2 reacts with excess oxygen by two different
operation. Oxygenated derivative generated was fitted by 3 and 4 extinction
coefficient ... 41 Figure 3-16. UV-vis spectra of 1 reacts with excess oxygen by two different
operation. According to 3 and 4 extinction coefficient, Fitted oxygenated derivative generated ... 42 Figure 3-17. UV-vis spectra of 2 (0.3 mM) layer above dry O2 in propionitrile at - 80 oC that shows 3 gradually generated ... 43 Figure 3-18. UV-vis spectra of 1 (0.17 mM) layer above dry O2 in propionitrile at -80 oC . ... 44 Figure 3-19. Geometry index for square pyramidal (τ5 = 0) and trigonal bipyramidal (τ5 = 1) are shown. ... 45
Figure 3-20. ORTEP diagram of dianion 1 with thermal ellipsoids at 50% probability,
hydrogen atoms and solvent of crystallization omitted for clarity. ... 47
Figure 3-21. ORTEP diagram of anion 2 with thermal ellipsoids at 50% probability, hydrogen atoms and solvent of crystallization omitted for clarity. ... 48
Figure 3-22. ORTEP diagram of dianion 4 with thermal ellipsoids at 50% probability, hydrogen atoms and solvent of crystallization omitted for clarity. ... 50
Figure 3-23. ORTEP diagram of anion 5 with thermal ellipsoids at 50% probability, 5hydrogen atoms and solvent of crystallization omitted for clarity. ... 51
Table
Table 1. Selected bond distances (Å ) and angles (deg) for 1. ... 47Table 2. Selected bond distances (Å ) and angles (deg) for 2. ... 49
Table 3. X-ray Crystallographic Data for 1 and 2. ... 50
Table 4. Selected bond distances (Å ) and angles (deg) for 4. ... 51
Table 5. Selected bond distances (Å ) and angles (deg) for 5. ... 52
Table 6. X-ray Crystallographic Data for 4 and 5. ... 53
第一章 緒論
1-1 氧氣
地球上絕大部分的氧氣來自於水分子受錳金屬的分解催化形成,其滋養了
世界成就鬱鬱蒼蒼的環境與生命。被視為最早行光合作用的藍菌使得地球上的
氧氣的含量逐步升高,這促使了嗜氧生物的演化。儲存在 O=O 鍵的能量提供
嗜氧生物的代謝、神經傳導以及 DNA 的合成,這些反應大多數由含錳、鐵和
銅的酶驅動,但這會導致代謝出活性氧物質(reactive oxygen species,ROS),例
如:單重態氧(1O2)、超氧化物(O2
‧-)、過氧化物(O22-)和氫氧自由基(‧OH),宛
如雙面刃的活性氧適用得宜促使生物反應,過量則破壞細胞及遺傳體。為了抗
衡上述物質的損毀,致使演化出抗氧化酶以及其他抗氧化代謝系統,達到活性
氧在生物系統中調節存量的平衡。
自然界中氧氣以三重態(triplet state oxygen, 3O2)穩定存在,安定的三重態氧
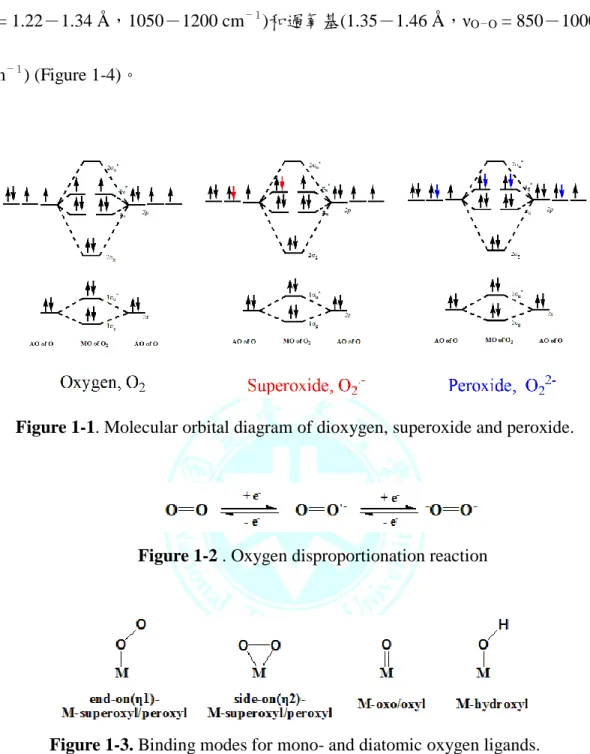
氣在不同環境能形成各種不同的活性氧物質。在分子軌域能得知氧氣在πg*有一
對未成對電子,超氧基有一個未成對電子,過氧基則呈現逆磁性(Figure 1-1)。
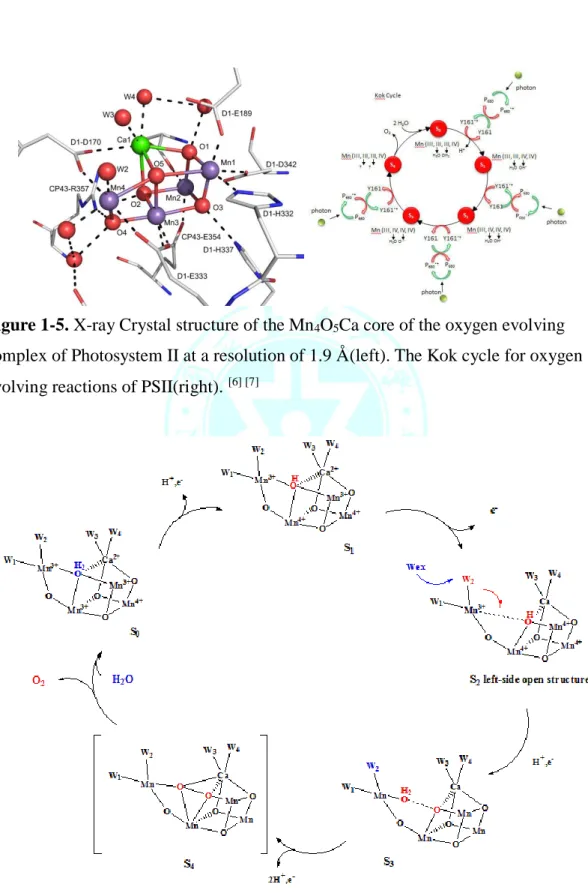
氧氣與其活性氧衍生物被視為自身氧化還原的配位基(redox non-innocent ligand)
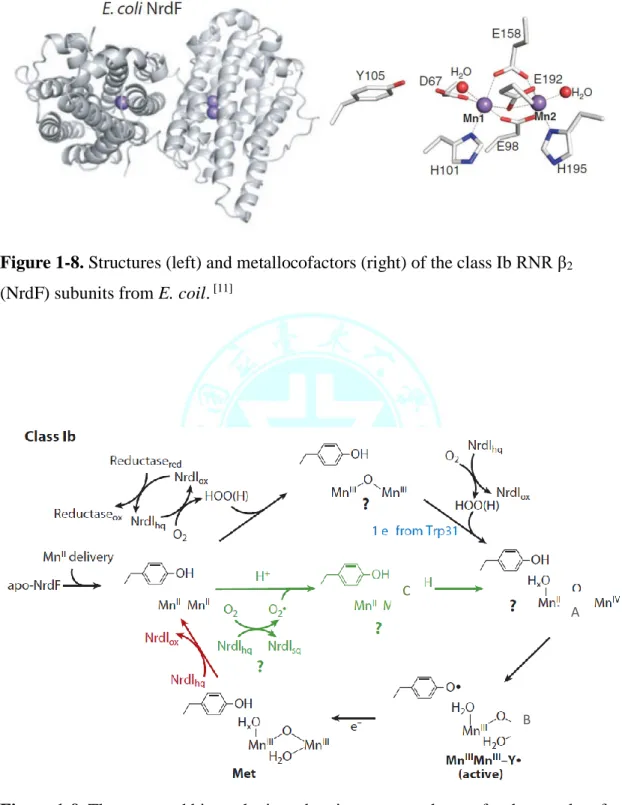
(Figure 1-2),與金屬交互作用後形成各種不同的結構,其氧氣鍵結形式大致可 分為 end-on(η1)和 side-on(η2)的超氧基(superoxyl)與過氧基(peroxyl),以及其他氧
基(oxo/oxyl)和羥基(hydroxyl) (Figure 1-3) [1]。隨著金屬氧化價數以及配位環境的
改變反映在鍵長、鍵角和鍵能,氧氣(1.2075 Å ,νO-O = 1580cm-1)、超氧基(νO-
O = 1.22-1.34 Å ,1050-1200 cm-1)和過氧基(1.35-1.46 Å ,νO-O = 850-1000 cm-1) (Figure 1-4)。
Figure 1-1. Molecular orbital diagram of dioxygen, superoxide and peroxide.
Figure 1-2 . Oxygen disproportionation reaction
Figure 1-3. Binding modes for mono- and diatomic oxygen ligands.
Oxygen Superoxide peroxide νO-O (cm-1) 1580 1050-1200 850-1000 Bond length (Å ) 1.2075 Å 1.22-1.34 Å 1.35-1.46Å
Figure 1-4 . Comparision of the O-O bond length and vibrational frequencies for O2、O2‧-and O22-.
作為一個有力的氧化劑,氧氣提供酵素基質電子受體,能接受四電子四質
子自身還原成水。過渡金屬在其中扮演著基質與氧氣的媒介,因擁有豐富的氧
化價、低能量障礙和蛋白質構形的調控,驅動電子與基質在酵素中流動,承載
氧氣到細胞各處以及形成活性氧進行催化反應。舉例來說好氧生物細胞必須仰
賴粒線體呼吸作用生成能量來源 ATP,高等生物攜帶氧氣的血紅素(hemoglobin)
是由血基質(heme)紫質二價攜帶,將氧氣傳遞氧氣至細胞組織之中,給予其他
酵素催化受質的能量來源。這些傳遞進細胞組織中的氧氣被應用於數種酵素當
中,例如廣泛分佈在肝臟及腸道的細胞色素 P450 (Cytochrome P450)單加氧酶,
是一群含三價鐵離子並附著於細胞內質網,其結構類似鐵蛋白結構及功能類似
血紅素,參與類固醇的生物合成、膽固醇的降解、維生素的活化以及外來物質
的代謝。[2]
然而好氧生物代謝反應會產生微量的活性氧自由基,這些自由基極易與體
內其他分子發生連鎖反應,甚使較不具活性的自由基也會因交互產生活性更強
的自由基。為了平衡過多活性氧自由基的生成,則由超氧歧化酶(superoxide
dismutase)及過氧化酶(catalase)等氧化還原酶代謝,相較其他酵素兩者皆是最佳 轉換效率之一,前者利用鎳、錳、鐵和銅/鋅,提供超氧離子自由基高效的清除
速率(約 7×109 M-1s-1) [3],進而預防氧中毒;後者則是以血基質,經過高價鐵
氧基(FeIV-oxo-por.+or FeV-oxo-por)催化過氧化氫生成水與氧氣。
相較於鐵,錳作為活性中心的氧化還原酶,這些被確立的例子佔相對少數。
錳跟鐵一樣可以在催化反應中作為路易士酸,在特定環境能提供氧氣預反應。
通常錳在酵素輔酶中是二價或三價,但它可以在團簇中瞬間轉換成四價,如第
二光系統 Mn4O5Ca 釋氧錯合物[4][5]。二價錳離子電子密度比鐵更加的扎實,能
跟羧基有更好的親和力,除了上述的錳超氧歧化酶之外,還有 Ib 型核醣核酸還
原酶等[8]。
1-2 第二光系統與氧氣的釋放
而地球上的氧氣來自於光合作用第二光系統(Photosystem II),這是由複雜
蛋白質在類囊體上組成,其中輔酶釋氧錯合物(oxygen-evolving complex,OEC)
以 Mn4O5Ca 作為活性中心(Figure 1-5) [6]。類囊體間其他的作用角色如葉綠素
P680吸收 680 nm 光能激發電子驅使酵素電子傳遞,β-胡蘿蔔素淬滅過量激發 光,質醌(plastoquinone, Pq, QA and QB)傳遞電子至 cytb6f 以及第一光系統,但
至今尚未有完整的催化機制被證實。從 Kok-Cycle (Figure 1-5)能得知從 S0到 S4
的光電子傳遞,P680吸收光激發電子使得電子從醌汲取,導致氧化態醌氧化 D1-
Tyr161(Yz),被氧化的 Tyr161.+從錳金屬提取,然後錳進一步地將水氧化。然 而水 S4到 S0釋放氧氣的途徑並沒有在結構以及物理性測定上證實 O-O 鍵是如
何形成,近年來文獻提出是由 S0-state 立方體上的 O5 水分子被 Yz 奪去質子和
電子[7],循序漸進地形成高價錳氧基(MnIV-oxo),並與鍵結在立方體外部的錳的
水分子進行反應並生成 O-O 鍵結,最後釋放氧氣分子完成催化水分子裂解循
環,過程總失去四電子四質子(Figure 1-6)。
Figure 1-5. X-ray Crystal structure of the Mn4O5Ca core of the oxygen evolving complex of Photosystem II at a resolution of 1.9 Å (left). The Kok cycle for oxygen evolving reactions of PSII(right). [6] [7]
Figure 1-6. Possible mechanisms for the oxygen-evolving reaction. [7] [8]
1-3 Ib 型核醣核苷酸還原酶與酪氨酸自由基誘導因子
核醣核苷酸還原酶(ribonucleotide reductase,RNR)在生物體裡的功能是將基
質核苷三磷酸(nucleoside triphosphate,NTPs)還原成去氧核苷三磷酸(deoxy
nucleoside triphosphate,dNTPs)提供 DNA 合成的素材,也是扮演調控 DNA 合 成速率的角色,使細胞分裂及 DNA 複製時質量比例維持恆定比例。而文獻上
提出 RNR 被分類成三種,這三種都擁有類似的 α 亞基以及催化機制(Figure 1-
7),不同的是使用金屬輔酶的形式。其中第一型 RNR 發現在原核與真核生物並 使用雙金屬驅使酪胺酸自由基進行催化反應,而又被分類成兩個亞種 Ia 、Ib
和 Ic,分別使用雙鐵、雙鐵/雙錳以及錳鐵催化酪胺酸自由基的生成[9]。
Figure 1-7. Proposed mechanism of nucleotide reduction by ribonucleotide reduct- ases (RNRs). [9]
舉例來說 class I RNR 催化 NTPs 的機制是以酪胺酸自由基(β2-Tyr-O·)機制
調控。其催化循環先由β2-Tyr-O·將基質五碳醣 3’-H 奪走,導致自由基出現在
3’-C。接著 α2-Cys-SH 質子化 2’-OH,2’-C-O+H2脫水形成 2’-C+,質子再由附 近的α2-Cys-SH 傳輸。然後氫原子轉移至還原 2’-C+,形成雙硫鍵α2-Cys-S-S-
Cys 以及 2’-C-H。最後 3’-C·再從 β2-Tyr-OH 轉移氫原子,而氧化形成雙硫鍵的 硫氧還原蛋白再被 NADPH 提供兩質子還原,致使催化循環回歸原點(Figure 1-
7)。[10]
在此我們以文獻上大腸桿菌 Ib RNR 探討 β2 雙錳活性中心(Figure 1-8),催
化酪氨酸自由基的生成,以及誘導 NTPs 還原成 dNTPs。Ib RNR-β2 是大約由三
至四百個胺基酸組成,金屬輔酶由 Asp67、His101 以及水分子鍵結 Mn1,
His195 與水分子鍵結 Mn2,Glu158、Glu192 和 Glu98 橋接雙錳以及外部配位環 境的 Tyr105 作為自由基活化處(Figure 1-9)。然而雙錳的 Ib RNR 的催化機制尚
未被完整的定義出,目前被接受的的路徑被分成兩種:(1)氧氣分子氧化 hydro-
quinone-Nrdl (Nrdlhq)形成過氧化氫,然後第一當量過氧化氫在氧化雙錳中心成 氧橋接三價錳中間體,第二當量過氧化氫在與 Trp31 提供單電子形成羥基橋接
和氧橋接雙錳的 A;(2) 氧氣氧化 Nrdlhq形成 Nrdlsq和超氧離子,超氧在質子化
氧化其中一個錳形成 end-on 的過氧基 C,進一步的生成 A。而 A 的橋接羥基奪
取 Tyr105 的氫原子形成活性的 B,以提供核糖核酸苷的還原。這些被推測的中
間體錳氧錯合物的可能性還需要更多實驗驗證,而模擬化合物提供重要性。[10]
Figure 1-8. Structures (left) and metallocofactors (right) of the class Ib RNR β2
(NrdF) subunits from E. coil. [11]
Figure 1-9. The proposed biosynthetic and maintenance pathways for the metalocofa- ctors of the class Ib ribonucleotide reductases. The steps shown in blue highlight the requirement for the extra reducing equivalent, and in red, the maintenance path-way.
[8]
A C
B
1-4 錳氧模擬化合物文獻探討
這些金屬與氧結合的中間體是瞭解酵素是如何作用的重要指標,但這些中
間體往往是難以觀察的不穩定存在,這促使合成應用在酵素模擬及鑑定的依
據。這些錳氧配位基的模擬化合物近年陸續地被廣泛地探討著,像是 Valentine
研究團隊利用二價錳錯合物與雙氧水反應得到三價[Mn(O2)TPP]− (TPP= meso-
tetraphenylporphyrin),晶體結構顯示氧到氧的鍵長分別為 1.421 Å[12]。Kitajima 研究團隊利用雙核錳二價化合物[MnII(HB(3,5-iPr2pz)3)]2(OH)2與兩當量 3,5-
iPr
2pzH 及過量雙氧水反應得到三價錳過氧基化合物[MnIII(HB(3,5-iPr2pz)3)(O2)( 3,5-iPr2pzH)],晶體結構顯示氧到氧的鍵長分別為 1.428 Å[13]。Nam 研究團 隊則發表兩個單核三價錳過氧基化合物[Mn(13-TMC)(O2)]+和[Mn(14-
TMC)(O2)]+(TMC= tetramethyl-cyclam),晶體結構顯示氧到氧的鍵長分別為
1.410(4)和 1.403(4) Å[15]。Borovik 研究團隊利用錳二價[MnIIH2bupa]−化合物與 氧氣分子反應,由 FTIR 得知氧氧震動能量在 885 cm-1,及其他光譜結果,證
實產生雙邊鍵結錳三價過氧基化合物[MnIII(O2)H2bupa]-(Figure 1-10) [14]。
Figure 1-10. Side-on MnIII-peroxo compounds (a) [MnIII(O2)TPP]− (b) [MnIII(HB- (3,5-iPr2pz)3)(O2)( 3,5-iPr2pzH)] (c) [Mn(TMC)(O2)]+ (d) [MnIII(O2)H2bupa]-. [12] [13]
[14] [15]
Nam 研究團隊探討一系列過渡金屬鍵結氧分子錳的催化活性,其中三價錳
過氧基化合物[MnIII(14-TMC)(O2)]+被探討能夠與醛類進行去甲醛化反應,將環
己烷甲醛去甲醛化成環己烷,藉由調控反應物對位取代基(para-Y-Ph-CHO;
Y=OMe, Me, F, H, Cl),以及控制軸向配位基的提供電子能力(N3
-, CF3CO2-,
NCS-, CN-),得知其反應為親核性反應[15]。此外錳錯合物也能作為其他金 屬 O2分此外錳錯合物也能作為其他金屬 O2分子的受體,如[MnII(14-TMC)]2+能
提供三價鎳過氧化物[NiIII(12-TMC)(O2)]+的 O2分子轉移的受體形成[MnIII(14-
TMC)(O2)]+(Figure 1-11) [15]。相似的例子還有三價鐵超氧化物
[FeIII(TAML)(O2)]2–將 O2分子轉移至[MnIII(TAML)]–產生四價錳雙邊鍵結過氧基 的[MnIV(TAML)(O2)]2–(Figure 1-12) [16]。
Figure 1-11. Reaction scheme shows an intermolecular O2-Transfer between [NiIII(12-TMC)(O2)]+ and [MnII(14-TMC)]2+.[15]
Figure 1-12. Reaction scheme shows an intermolecular O2-Transfer from superoxo- [FeIII(TAML)(O2)]2– to [MnIII(TAML)]– that generate peroxo-[MnIV(TAML)(O2)]2–
and [FeIII(TAML)]–. [16]
Kovacs 研究團隊更進一步的探討二價錳與氧氣的反應,並提出了在低溫反
應時其中一個 pyridine 上 N 配位基會離開並空出一個位置而產生單核錳三價超
氧基中間物,此中間體在與另一當量的二價錳離子反應生成過氧橋接三價雙錳
錯合物[[MnIII(SMe2N4(6-Me-DPEN)]2(trans-μ-1,2-O2)]2+,並在低溫得出晶體結構
顯示氧到氧的鍵長為 1.452(5) Å 以及低溫拉曼共振光譜測得氧-氧震動能量約為
819 cm-1,這些數據傾向於過氧基的形式鍵結在雙錳之間。然而這過氧橋接三 價雙錳錯合物會熱不穩定最終形成氧橋接三價雙核錳錯合物[[MnIII(SMe2N4(6-
Me-DPEN)]2(μ-O)]2+(Figure 1-13)。[17] [18]
Figure 1-13. Reaction of five-coordinate [MnII(SMe2N4(6-Me-DPEN)]+ with O2 to afford mono-oxobridged [[MnIII(SMe2N4(6-Me-DPEN)]2(μ-O)]2+ via metastable Mn
−superoxo intermediate and Mn−peroxo intermediate [[MnIII(SMe2N4(6-Me-DPE- N)]2(μ-O)]2+.[17] [18]
本團隊過去研究成果提出錳金屬與氧氣分子反應時配位基本身的立體障礙
影響最終的產物。當 cis-[PPN][Mn(CO)4(SPh)2]分別與一當量配位 P(C6H5SH)3和
P(C6H3-3-SiMe3-2-SH)3反應會生成化合物[MnI (CO)3P(C6H5S)2(C6H5SH)]−和[MnI-
(CO)3P(C6H3-3-SiMe3-2-S)2(C6H3-3-SiMe3-2-SH)]−,上述兩個幾何結構最大的特 色就是有一個未參與鍵結的硫醇基落在第二層的配位環境。當錳一價的錳化合
物分別與氧氣分子發生反應時,我們認為過程經歷一個單核四配位二價錳中間
體。立體障礙較小的化合物會轉換成雙核錳二價化合物,若長時間的氧氣反應
化合物會分解成不溶的黃色固體;相反地,當立體障礙較大的配位基的錳化合
物與過量氧氣分子反應時會得到雙邊鍵結錳四價過氧基化合物[MnIV(O2)P(C6H3-
3-SiMe3-2-S)3]− (Figure 1-14),這第一個具有晶體結構的雙邊鍵結錳四價過氧基 化合物,氧到氧的鍵長為 1.379(5)Å 。另外氧氧鍵的震動能量(vO-O)約為 903 cm-
1,參數均支持化合物的雙氧是屬於過氧基[19]。
Figure 1-14. Proposed mechanism for the formation of [MnIV(O2)P(C6H3-3-SiMe3-2- S)3]- and [MnII2(PS3)2]2-.
1-5 研究方向
目前發現生物以錳金屬作為酵素輔酶的物種日益劇增,然而這些酵素作用
機制尚未完整的被驗證,如第二光系統(PSII)和 Ib 型核醣核苷酸還原酶(class Ib
RNR )。近年來探討錳金屬的模擬化合物逐步地被提出,過去本團隊提出一價 錳化合物與氧氣反應生成四價過氧基化合物,並推測過程可能有單核的四配位
錳二價中間體,進而被氧氣氧化至四價過氧基化合物[PPN][MnIV(O2)P(C6H3-3-
SiMe3-2-S)3]。在此我們認為能藉由其他方法合成出推測的二價錳中間體並嘗試 與氧氣反應,提供過去的推論佐證以及其中是否參與不同的反應,並進一步探
索金屬-氧氣之間的反應關係。
第二章 實驗部分
2-3 一般實驗
凡是對於空氣或水氣敏感的化合物,其操作、反應與轉移的過程皆在氮氣
環境下完成以 Schlenk techniques 或氮氣手套箱(glove box)中進行。使用之溶劑
皆經過適當的除水步驟,並在氮氣環境下蒸餾收集。Acetonitrile 以 CaH2/P2O5
二重除水;n-hexane 與 diethyl ether 以及 tetrahydrofuran (THF)皆以 sodium
benzophenone 除水,並保存在充滿氮氣且置有分子篩(4 Å molecular sieves)乾燥 的燒瓶中,並通入氮氣去除額外的空氣。
2-2 儀器
電子吸收光譜儀(UV-Visible electronic spectrometer, UV-vis):SCINCO S-
4100 spectrophotometer 以及 Agilent Cary 8454 UV/Vis,使用光徑(path
length)為 1.0cm 的石英槽。
紅外線光譜儀(Infrared spectrometer, IR):
SHIMADZU FTIR-8400S,使用 KBr 壓片;全反射式 PerkinElmer FTIR
電子順磁共振光譜儀(Electron paramagnetic resonance, EPR):委託科技部中
興大學貴儀中心 Bruker E580 CW/Pulse EPR。
電化學分析儀(Electrochemical analyser):
CH Instruments, Inc. CHI729E,三電極系統,工作電極為玻璃碳電極,參考
電極為非水溶液 Ag/AgCl 電極,輔助電極為 Pt。
核磁共振儀(Nuclear Magnetic Resonance spectrometer, NMR):400-MR
DD2。
X-ray 單晶繞射解析:
委託科技部台灣大學貴儀中心,Bruker SMART APEXCCD four-circle
diffractometer。
2-3 藥品
1. 正丁基鋰 n-Butyllithium n-BuLi 2.5M Chemetall
分子量:64.05 g/mol,密度:0.68 g/cm3,沸點:分解,熔點:-76 ℃。
2. 四甲基乙二胺 N,N,N´,N´-Tetramethylethylenediamine TMEDA ACROS 分子量:116.24 g/mol,密度:0.87 g/cm3,沸點:120-122 ℃,熔點:-
76℃。
3. 硫化酚 Thiophenol C6H5SH Merck
分子量:110.19 g/mol,密度:1.08 g/cm3,沸點:169 ℃,熔點:-15 ℃。
4. 正己烷 n-Hexane C6H14 ACROS
分子量:86.18 g/mol,密度:0.65 g/cm3,沸點:69 ℃,熔點:-95 ℃,
5. 四氫呋喃 Tetrahydrofuran THF ACROS
分子量:72.11 g/mol,密度:0.89 g/cm3,沸點:66 ℃,熔點:-108.4 ℃,
6. 三甲基氯矽烷 Trimethylsilyl chloride Me3SiCl ACROS
分子量:108.94 g/mol,密度:0.86 g/cm3,沸點:57 ℃,熔點:-40 ℃。
7. 濃鹽酸 Hydrogen chloride HCl ACROS
純度 37%,分子量:36.46 g/mol,密度:1.18 g/cm3,沸點:48 ℃,熔點:
-27℃。
8. 二氯甲烷 Dichloromethane CH2Cl2 ACROS
分子量:84.93 g/mol,密度:1.33 g/cm3,沸點:39.6 ℃,熔點:-96.7
℃。
9. 無水硫酸鎂 Magnesium sulfate MgSO4 ACROS
分子量:120.42 g/mol,密度:2.66 g/cm3,熔點:1124 ℃分解。
10. 三氯化磷 Phosphorus trichloride PCl3 ACROS
分子量:137.33 g/mol,密度:1.57 g/cm3,沸點:76.1 ℃,熔點:-93.6
℃。
11. 異丙醇 isopropyl alcohol C3H8O ACROS
分子量:60.1 g/mol,密度:0.786 g/cm3,沸點:82.6 ℃,熔點:-89 ℃
12. Bis(triphenylphosphine)iminium chloride [PPN]Cl SIGMA-ALDRICH 分子量:574.03 g/mol,熔點:260-262 ℃。
13. 無水氯化錳 Manganese(II) chloride MnCl2 SIGMA-ALDRICH
分子量:125.84 g/mol,密度:2.98 g/cm3,沸點:1225℃,熔點:654℃。
14. 乙腈 Acetonitrile CH3CN J.T.Baker
分子量:41.05 g/mol,密度:0.786 g/cm3,沸點:82℃,熔點:-45℃。
15. 乙醚 Diethyl ether Ether ACROS
分子量:74.12 g/mol,密度:0.713 g/cm3,沸點:34.6 ℃,熔點:-
116℃,在氮氣下加入切碎的鈉金屬,用 benzophenone 為指示劑,迴流 24 小時,顏色呈紫色時,收集並保存於氮氣下。
16. 三亞乙基二胺 1,4-diazabicyclo[2,2,2]octane DABCO ACROS
分子量:112.17 g/mol,密度:1.14 g/cm3,沸點:174℃,熔點:158-160
℃。
17. 三苯基磷 Triphenylphosphine P(C6H5)3 ACROS
分子量:262.29 g/mol,密度:1.1g/cm3,沸點:377 ℃,熔點:80 ℃。
18. Dimanganesedecacarbonyl, Mn2(CO)10 ACROS
分子量:389.98 g/mol,密度:1.750 g/cm3,沸點:昇華 60 ℃,熔點:154
℃。
19. 氫氧化鉀 Potassium Hydroxide KOH J.T.Baker
分子量:56.105 g/mol,密度:1.1 g/cm3,沸點:1327 ℃,熔點:360.4
℃。
20. 二茂鐵 Ferrocene Fe(C5H5)2 ACROS
分子量:186.04 g/mol,密度:1.107 g/cm3,沸點:249 ℃,熔點:172.5 ℃。
2-4 化合物的合成與性質鑑定
2-4-1 配位基Tris(2-thiol-3-trimethylsiylphenyl)phosphine, (TMSPS3)的合成[20]
定量n-butyllithium溶液(120 mL, 2.5 M, 0.3 mol)轉移進N2(g)下的加料漏斗與
Schlenk flask組合瓶,之後加入N,N,N',N'-tetramethylethane-1,2-diamine (40 mL,
0.3mol)形成亮黃色液體,在冰浴下滴加thiophenol (14.4 mL, 0.15 mol)反應,回 溫後反應隔夜逐漸產生白色固體,靜置分層後將上層液移除並用THF溶解固
體,而後浸入-78℃丙酮浴中滴加入trimethylsilyl chloride(18.7 mL, 0.3 mol),回
溫後反應隔夜形成荔枝色溶液,加入40毫升二次蒸餾水去除多餘反應物,並減
壓濃縮去除THF,而後用7% HCl酸化至pH值2-3,以CH2Cl2萃取後,加入無水
MgSO4除水並減壓蒸餾得到透明無色液體為2-trimethylsiylthiophenol。
以同樣方法鋰化2-trimethylsiylthiophenol,隔夜逐漸產生白色固體,靜置分
層後將上層液移除並用THF溶解固體,而後浸入-78℃丙酮浴中滴加入1/3當量
的phosphorus trichloride回溫後反應隔夜形成亮橘色溶液,加入40毫升二次蒸餾
水去除多餘反應物,並減壓濃縮去除THF,而後用7%鹽酸酸化至pH值為2-3,
產生乳白色固體,以CH2Cl2萃取後,加入無水MgSO4除水一天,以異丙醇室溫
長晶,一周後得到白色晶體,結晶產率為88%,1H NMR (400 MHz):δ4.55 (d, J
= 11.9 Hz, 3H),δ7.48 (dd, J = 7.3, 1.4 Hz, 3H),δ7.09 (t, J = 7.4 Hz, 3H),δ6.79
(ddd, J = 7.0, 5.5 Hz, 3H)。
2-4-2 化合物[PPN]2[MnII2(TMSPS3)2] (1)的合成
在氮氣手套箱中將去質子化Li3TMSPS3的THF溶液(0.1 M, 5 mL, 0.5 mmol)轉
移至已秤取MnCl2(0.063 g, 0.5 mmol)與[PPN]Cl (0.2875 g, 0.5 mmol)的Schlenk
flask中,並加入2 mL乙腈助溶解,反應三十分鐘後呈現帶點橘的紅色液體,在 室溫下擴散乙醚進溶液中並重長晶兩次,生成暗紅色塊狀晶體,並以X-ray單晶
繞射儀鑑定結構,其結晶產率為85-95%。紫外線可見光光譜(THF:acetonitrile
=1:1v/v) [nm, λmax(M-1cm-1, ε)]:420(650)。元素分析C126H132N2P6S6Si6Mn2: 理論值(C,64.92; H,5.71; N,1.20); 實驗值(C,63.92; H,5.94; N,1.56)。
2-4-3 化合物[PPN][MnII(DABCO)(TMSPS3)] (2)的合成
在氮氣手套箱中秤取[PPN]2[MnII2(TMSPS3)2] (0.046 g, 0.02 mmol)置於Schlenk
flask中以2 mL THF:乙腈=1:1(v/v)溶解,而後加入過量DABCO(1,4-
diazabicyclo[2.2.2]octane)直到呈現紫色,在室溫下layer一層過量DABCO的
diethyl ether:n-hexane =1:1(v/v)長晶,生成紫色針狀晶體,並以X-ray單晶繞 射儀鑑定結構,其結晶產率為90-95%。紫外線可見光光譜為(acetonitrile) [nm,
λmax(M-1cm-1, ε)]:370(2450)、535(540)。元素分析C69H78N3P3S3Si3Mn:理論值
(C,64.86; H,6.15; N,3.29); 實驗值(C,63.76; H,6.30; N,3.37)。
2-4-4 化合物[PPN][MnIV(η2-O2)(TMSPS3)] (3)的合成
在氮氣手套箱中秤取[PPN][Mn(CO)5] (0.073 g, 0.1 mmol)與Li3TMSPS3(0.0575
g, 0.1 mmol)以及diphenyl disulfide(0.0218 g,0.1 mmol)並以5 mL THF溶解攪拌三 十分鐘後溶液呈現褐色,以n-hexane沉降出固體並移除上層液後抽乾,將固體
回溶THF並加入N,N-diisopropylethylamine後以針筒抽純氧反應兩小時,溶液轉
變成紫紅色。紫外線可見光光譜為(THF)[nm, λmax(M-1cm-1, ε)]:490(2450)、
550(2760)、780 (1200)。 利用diethyl ether在-20°C擴散長晶,兩周後得出暗紫 色片狀晶體,結晶產率為52%。
2-4-5 化合物[PPN]2[MnIII2(μ-O)(TMSPS3)2] (4)的合成
在氮氣手套箱中秤取 2 (0.038 g, 0.015 mmol)置於Schlenk flask中以2 mL乙
腈溶解,而後加入過量DABCO (1.68 g, 15 mmol)直到溶液呈現紫色,而後將溶
液轉移進秤有 3 (0.012 g, 0.01 mmol)的Schlenk flask中反應一小時,溶液顏色轉
變成綠色,溶液加入30mL乙醚析出綠色固體([PPN]2[MnIII2(μ-O)(TMSPS3)2](4),
0.044 g, 93%)。紫外線可見光光譜為(乙腈)[nm, λmax(M-1cm-1, ε)]:455(5380)、
675(3780)。將翠綠色溶液在-20°C下擴散乙醚長晶,一週後得到針狀晶體,並 以X-ray單晶繞射儀鑑定結。元素分析C126H132N2OP6S6Si6Mn2:理論值(C,64.48;
H,5.67; N,1.19); 實驗值(C,64.30; H,5.79; N,1.37)。
2-4-6 化合物 1 與 3 的反應
在氮氣環境下秤取 3 (0.08 g, 0.067 mmol)溶解在10mL乙腈,並將溶液加入
秤有 3 (0.233 g, 0.1 mmol)的乙腈溶液中,室溫下攪拌反應三十分鐘。結果轉變
成暗綠色溶液,並在真空下去除溶劑至5 mL,加入30 mL乙醚析出綠色固體
([PPN]2[MnIII2(μ-O)(TMSPS3)2](4), 0.220 g, 70%)。
2-4-7 化合物 1 與 3 在含有PPh3的反應
在氮氣環境下秤取 3 (0.12 g, 0.1 mmol)與PPh3(0.27 g, 1.0 mmol)溶解在10
mL乙腈,並將溶液加入秤有 3 (0.350 g, 0.15 mmol)的乙腈溶液中,室溫下攪拌 反應三十分鐘。溶液轉變成暗綠色,並在真空下去除溶劑至5 mL,加入30 mL
乙醚析出綠色固體([PPN]2[MnIII2(μ-O)(TMSPS3)2](4), 0.250 g, 64%)。殘餘溶液利
用氣相層析儀分析(GC/MS:279 m/z (16OPPh3+H+),281 m/z (18OPPh3+H+)18O標
記 3 作為反應物。
2-4-8 化合物2 與PhIO反應
在氮氣手套箱中秤取 2 (0.038g, 0.03mmol)置於Schlenk flask中以2mL乙腈溶
解,加入過量DABCO(0.28 g, 2.5 mmol)直到溶液呈現紫色。將溶液轉移至
PhIO(0.035 g, 0.16 mmol)容器中室溫攪拌反應一小時。紫外線可見光光譜分析
生成 4。溶液加入30 mL乙醚沉降出綠色固體 4,產率為96%。
2-4-9 化合物 1 與氧氣反應
在氮氣環境下秤取 1 (0.023 g, 0.01 mmol)並溶於25 mL乙腈,以不同方式與
過量氧氣反應一小時。
(1) 將過量氧氣通入液面下:溶液由亮橘色轉變為帶黃的綠色,分析UV-vis混 合光譜計算生成 3 (基於550nm消光係數,佔混合光譜比例51%),4 (49%,
基於ε 670nm)
(2) 緩慢擴散氧氣至液面上:溶液由亮橘色轉變為亮綠色,分析UV-vis混合光 譜計算生成 3 (基於550nm消光係數,佔混合光譜比例27%),4 (72%,基於ε
670nm)
2-4-10 化合物 2 與氧氣反應
在氮氣環境下秤取 1 (0.023 g, 0.01 mmol)與過量DABCO(0.28 g, 2.5 mmol)
並溶於25 mL乙腈,以不同方式與過量氧氣反應一小時。
(3) 將過量氧氣通入液面下:溶液由亮紫色轉變為紫紅色,分析UV-vis混合光 譜計算生成 3 (基於550 nm消光係數,佔混合光譜比例95%),4 (5%,基於ε
670nm)
(4) 緩慢擴散氧氣至液面上:溶液由亮紫色轉變為褐色,分析UV-vis混合光譜
計算生成 3 (基於550 nm消光係數,佔混合光譜比例80%),4 (20%,基於ε
670nm)
2-4-11 化合物 1 與 2 的固相氧化
(1) 將橘紅色 1 微晶體(0.055 g, 0.02 mmol)置於小瓶中,然後暴露乾燥氧氣兩小 時顏色由橘紅色轉變成黃褐色。FTIR (soild, cm-1):3060(w), 2950(w),
2890(w), 1588(w), 1554(w), 1532(w), 1482(w), 1438(m), 1350(m), 1238(m),
1192(w), 1114(m), 1044(w), 998(w), 848(s), 830(s), 799(w), 781(w), 750(m),
722(s), 690(s)。生成的黃色固體以UV-vis(THF)有500nm特徵峰,但並未成功 辨明此物種。
(2) 將紫色 2 微晶體(0.064 g, 0.05 mmol)置於小瓶中,然後暴露乾燥氧氣兩小時 顏色由橘紅色轉變成黃褐色。FTIR (KBr, νO-O=903(w) cm-1),生成固體回
溶乙腈並由UV-vis分析,其特徵峰(490、550及755nm)證實 3 生成(產率
99%)
2-4-12 化合物[PPN][MnIII(OH)(TMSPS3)] (5)的合成
在氮氣手套箱中秤取[PPN]2[MnIII2(μ-O)(TMSPS3)2] (0.023 g, 0.01 mmol)於
Schlenk flask中,並以3 mL 乙腈溶解,滴入一滴二次水,顏色逐漸由亮綠色轉 變為翠綠色。紫外線可見光光譜為(THF)[nm, λmax(M-1cm-1, ε)]:480(1880)、
670(2330)。利用diethyl ether:n-hexane=1:1(v/v)擴散長晶,兩週後得到綠色晶 體,並以X-ray單晶繞射儀鑑定結構,結晶產率為48%。FTIR (KBr, cm-1):3626
(m)。
2-4-13 化合物 5 與2,6-di-tbu-4-R-PhOH(R=OMe)的反應
秤取 5 (0.018 g, 0.01 mmol)與2,6-di-tbu-4-OMe-PhOH (0.263 g, 1 mmol)溶於
10 mL乙腈反應,UV-vis隨時間監測反應光譜變化,出現尖細的特徵峰在403
nm (1065),在真空下去除溶劑,加入10 mL正己烷萃取,由UV-vis光譜計算自 由基產率為7-8%。
2-4-14 平衡常數的測定
將固體的1 (0.061g, 0.25mmol)和DABCO (0.070 g, 0.625 mmol)置於燒瓶,加
入10 mL乙腈溶解混合直到色澤不改變,已知初始濃度[1]0 = 0.5 mM和
[DABCO]0 = 12.5 mM,紫外線可見光光譜儀測定535 nm特徵峰吸收度隨溫度改 變變化。平衡常數根據以下公式計算
Keq = [ 𝟐 ]2
([ 𝟏 ]0)([DABCO]0− 2[ 𝟐 ])2)
第三章 結果與討論
金屬含氧類官能基的仿生模擬化合物已被廣泛的研究,其中以鐵、銅、錳
居多,但關於錳金屬與氧氣的反應則較少被研究,在文獻中 Kovacs 的團隊提出
由錳(II)錯合物([MnII(SMe2N4(6-Me-DPEN)]+)與氧氣反應並在低溫(-40/-80oC)能
觀測並結晶出帶過氧基橋接的中間體化合物[[MnIII(SMe2N4(6-Me-DPEN)]2(trans- μ-1,2-O2)]2+,其在室溫逐漸轉換成氧基橋接錳(III)錯合物[[MnIII(SMe2N4(6-Me-
DPEN)]2(μ-O)]2+[17];本實驗室過去提出由錳(I)錯合物與氧氣反應最終得到單核 錳(IV)[PPN][MnIV(η2-O2)TMSPS3]過氧基錯合物,推測其中反應機制中參與錳(II)
中間體的形成,其進一步與氧氣反應而得之[19]。因此由推測中間體出發合成出
TMSPS3螯合錳(II)等錯合物並探討與氧氣的反應性。
3-1 含錳(II)化合物的合成、結構與性質鑑定
將 MnCl2、[PPN]Cl 以及 Li3 TMSPS3 THF 溶液以 1:1:1 混合並滴加入乙腈,
溶液逐漸轉換成紅帶橘色(scheme 1),電子吸收光譜約在 500 nm 後沒有特徵峰
(Figure 3-1),在氮氣環境室溫下以 diethyl ether : n-hexane=1:1(v/v) 擴散長晶,
最終形成二聚體[PPN]2[MnII2(TMSPS3)2] (1)其結晶產率 95%,X-ray 單晶繞射結構
性質(Figure 3-20),其結構為硫橋接雙核五配位;將(1)溶解在乙腈並加入過量
DABCO(~100 equiv),溶液立刻轉換成紫色,電子吸收光譜在 535 nm 有一特徵 峰(Figure 3-1),以 diethyl ether : n-hexane=1:1(v/v)與過量 DABCO 混合,在氮
氣環境室溫下 double layered 長晶,形成[PPN][MnII(DABCO)(TMSPS3)] (2),其結
晶產率 90%,X-ray 單晶繞射結構性質(Figure 3-21),其結構為單核五配位。
雙核 1 反應形成單核 2 為平衡反應,在滴定實驗中,利用紫外線可見光光
譜儀觀測 535 nm 的吸收值隨著外加 DABCO 當量數變化,觀測到加入約 50 當
量時,535 nm 特徵峰有明顯的升高並接近飽和(Figure 3-2)。
在變溫實驗中,將 1 與 25 當量 DABCO 混合在乙腈中,並藉由紫外線可見
光光譜儀觀測特徵峰有明顯的差異,證實平衡反應中 1 與 2 生成量的改變,基
於 eq 1.計算平衡常數(Keq)在 25oC 乙腈溶劑下為 10(1),基於 eq 2.利用 Van’t
Hoff plot 分析 298 K 到 278 K 平衡常數的變化求出反應熱力學參數(ΔGex°= -5.7
kJ·mol-1,ΔH°= -16.4(1) kJ·mol-1,ΔS°= -35.8(1) J·K-1mol) (Figure 3-3),得知在低 溫下更傾向 2 的生成,與文獻[CuIII(TMSPS3)(Cl)]-與 CuIII(TMSPS3) (DABCO)置換
平衡反應有相同的趨勢(Keq=8(1),ΔGex°= -5.2 kJ·mol-1) [21]
Figure 3-1. UV-vis spectra (in acetonitrile) for 1(orange) and 2(purple) at ambient temperature.
Scheme 1
Figure 3-2. (a) UV-vis spectra for 1 titrate 0 to 100 equivalent of DABCO in acetoni- trile at room temperature. (b) The change of absorbance at 535 nm.
(a)
(b)
(1) (2)
[(MnII(TMSPS3)2)]2- + 2 DABCO ⇌ 2 [MnII(DABCO)(TMSPS3)]- eq 1.
Figure 3-3. (a) Variable Temperature of UV-vis spectra of acetonitrile solution containing 1 (0.5mM) and DABCO (12.5mM) from 25 to 5 oC. (b) Van’t Hoff plot of equilibrium data based on eq 1 and eq 2.
− 𝐑𝐥𝐧 𝑲𝒆𝒒 =𝜟𝑮°
𝑻 = 𝜟𝑯° (𝟏
𝑻) − 𝜟𝑺° eq 2.
(a)
(b)
在 1 的觀測中-1.10V 為 MnIIMnII/MnIIIMnII的氧化還原峰並在電化學上生成
MnIIMnIII的物種,此外在-0.77V 能夠更進一步地被氧化成 MnIIIMnIII;而在 2 的可逆氧化還原過程中 E1/2=-1.16V 為 MnII/MnIII氧化還原峰。相較於 1 (E1/2=-
1.10V)第一個氧化還原峰,2 (E1/2=-1.16V)擁有一個更負的電位峰值,這推測可 能為配位基 DABCO 擁有比硫橋接配位基還強的推電子能力造成。雖然在循環
伏安法指出在乙腈下(a weaker donor than DABCO)依然與固相結構中觀測 1 皆為
雙核狀態,但這不排除形成低濃度的溶劑鍵結單核錳([MnII(NCCH3)(TMSPS3)]-)
的生成。
近期文獻提出一硫橋接雙核[MnII2(LS)(LSH)]ClO4在 Epa=-0.01V(vs Fc/Fc+)
有一個不可逆的氧化峰,其被定義為 MnIIMnII至 MnIIIMnIII兩電子的氧化[24],
不同於 1 的電化學性質。推測其電位的下降可能是因TMSPS33-硫醇基(S-)擁有
比橋接硫(μ-S)更強的電子給予能力,導致錳(II)中心帶有更高的電子豐富度。觀
測到穩定混合價數 MnIIMnIII物種,可歸咎於相對剛性的四芽基及具自身氧化還
原能力的配位基TMSPS33-。另一個硫醇基配位錳(II)的例子能提供 2 擁有更低電
位的解釋,例如[MnII(SMe2N4-(6-Me-DPEN)](BF4)有個可逆的 MnIII/MnII氧化還原
峰在 E1/2=0.035V (vs Fc/Fc+)。此外較低的 MnIII/II氧化還原電位突顯 1 和 2 更容
易被氧氣氧化。[18]
Figure 3-4. Cylic voltammograms of 1 (orange) and 2 (purple) measured in a 1mM acetonitrile solution with 0.1M [n-Bu4N][PF6] as the supporting electrolyte at room temperature, scan rate 0.1 V/s.
Figure 3-5. UV-vis spectra of 3 (black) and 4 (purle). Changes in UV-vis spectra (gray) after 3 with various equivalents of 2 in CH3CN at ambient temperature. Inset:
absorbance changes at 670 nm upon addition of 2
3-2 含錳(III)化合物的合成、結構、性質鑑定與反應性
在室溫氮氣環境下,2 乙腈溶液當中滴入[PPN][MnIV(η2-O2)TMSPS3] (3) 觀
測到綠色的特徵峰在 450(ε=5410)nm 與 670(ε=3760)nm 快速生成。暗綠色的晶
體藉由反應生成的綠色乙腈溶液 double layer 乙醚長晶獲得,X-ray 單晶繞射結
構性質(Figure 3-22),其結構為單氧橋接雙核五配位[PPN]2[MnIII2(μ-O)(TMSPS3)-
2](4),其產率為 95% (scheme 2)。
Scheme 2
在滴定實驗中,利用 UV-vis 光譜儀觀測需要約 3.0 當量 2 才能導致 3 完全
地生成 4,4 生成的量基於加入的 3 決定(Figure 3-5)。近期一個氧橋接雙錳的例
子{[MnIII(SMe2N4(6-Me-DPEN)]2(μ-O)}2+被 Kovas 團隊提出,其化合物來自於次
穩定的{[MnIII(SMe2N4(6-Me-DPEN)]2(1,2-trans-μ-O2)}2+熱不穩定生成而至,過氧
橋接雙錳(III)化合物的生成機制參與了單核錳(III)超氧中間體與第二當量的
[MnII(SMe2N4(6-Me-DPEN)]+反應的過程[17];上述例子以及其他被廣為接受的例
子解釋μ-peroxo 與 μ-oxo 轉變機制探討,我們從中推測本實驗單核 Mn(II) 2 與
Mn(IV)-peroxo 3 生成雙核 [(TMSPS3)MnIII (μ-O2) MnIII (TMSPS3)]2- (intermediate
A, Scheme 2)。在文獻上提出利用低溫 UV-vis 光譜儀偵測,並在較高可見光區
(3) (4)
觀測到 peroxo-to-manganese charge transfer band,但是在-80oC 丙腈下混合 2 與
3 並未觀察到任何中間體的產生,想必不存在穩定中間體是因為缺乏分子內交
互作用,例如π-stacking 穩定過氧橋接雙錳的物種。[17]
在 Figure 3-1 我們可以得知計量上每當量的 3 需要三當量 2 反應,並基於
eq 2.;獨立實驗中 2 能夠與氧原子轉移試劑 iodosylbenzene (PhIO)進行反應,並 產生接近理想產率的 4。這意味著 4 的生成牽扯到中間體 A O-O 鍵的斷裂,進
一步生成兩當量高價 Mn-oxo 過渡態,然後產生的 Mn-oxo 再與另一當量的 2 反
應形成μ-oxo 4。基於 eq 3.和 eq 4.在競爭反應中比較 2 存在三苯基磷(PPh3)的環
境,是否會影響氧原子轉移生成 Mn-oxo 物種。UV-vis 光譜儀觀測 4 的生成量
不受過量三苯基磷的影響(Figure 3-6),至此假設過渡態 Mn-oxo 與單核 2 快速反
應,使得其中並未觀測到 OPPh3的形成。
這實驗支持著中間體化合物並沒有親電子性的氧,換句話說其擁有相對的
電子豐富度。此外雙核 1 與過氧基 3 在相同環境(10 equiv PPh3)也會形成 4,其
產量下降至 75%,稟明了 PPh3參與 1 氧化的競爭反應。 GC/MS 分析產物
OPPh3生成,18O 同位數標定證實氧原子來自 3 (Figure 3-7)。則關於 Mn(II)
TMSPS3系列與氧氣結合反應見下章節探討。
[MnIV(O2)(TMSPS3)]-+3
2 [MnII2 (TMSPS3)2]2-→ 2 [MnII2 (μ-O)(TMSPS3)2]2-eq 3.
[MnIV(O2)(TMSPS3)]-+3 [MnII(DABCO) (TMSPS3)2]-→ 2 [MnII2 (μ-O)(TMS-
PS3)2]2-+3 DABCO eq 4.
Figure 3-6. UV-vis spectrum (black line) of 4 generated from reaction of 1 (0.098mM) and 3 (0.065mM) based on eq :
(𝟑) +
32
(𝟏) → 2(𝟒)
; Red line shows the UV-vis spectrum of 4 generated from reaction of 2 (0.195mM) and 3 (0.065mM) based on eq :(𝟑) + 3 (𝟏) → 2 (𝟒)
Figure 3-7. GC-mass spectrum shows 3 reaction with 1 and PPh3 (10 equiv) that gerenated 279.1m/z (OPPh3+H+, black). 18O-labeled 3 confirms the source of O-atom that detected 281.1m/z (18OPPh3+H+, red).
3 reaction with monomeric 2 and 10 equiv PPh
3.
3 reaction with dimeric 1 and 10 equiv PPh3.
室溫下將 4 乙腈溶液滴入 H2O(~10 equiv),觀測色澤由亮綠色迅速轉變為
青綠色,UV-vis 光譜儀監測特徵峰 480 (ε=1880)nm、670 (ε=2330)nm 增消長以
及 490nm 出現一等吸收點(Figure 3-8)。暗綠色晶體由反應產物在室溫下擴散乙
醚進入溶液,X-ray 單晶繞射結構性質(Figure 3-23),其結構為羥基鍵結單核錳
五配位[PPN][MnIII(OH)(TMSPS3)](5),其產率為 90% (scheme 3)。
利用循環伏安法檢測,在氮氣環境下以 0.1M [n-Bu4N][PF6]作為電解質測量
4 (1 mM)與 5 (2 mM)乙腈溶液的電化學性質(Figure 3-9)。其中 4 觀測到一組清
Scheme 3
Figure 3-8. UV-vis spectra of 4 (black line) with 10 equivalent H2O in acetonitrile that generate 2 equivalent 5 (green line). Gray lines change show 4 convert to 5. The measured interval between two curve is 1 min.
(4) (5)
晰的可逆氧化還原峰在E1/2=0.00V (vs Fc/Fc+)為MnIIIMnIV/MnIIIMnIII特徵峰,在
與Mn(II) TMSPS3系統相同視窗中並未觀測到任何電位更負的氧化還原峰,先前
文獻提出{[MnIII(SMe2N4(6-Me-DPEN)]2(μ-O)}(BF4)也有一個MnIII/MnIV可逆氧化
還原峰在E1/2= +0.465V (vs SCE;0.09V vs Fc/Fc+)[27];5觀測到一組清晰的可逆
氧化還原峰在E1/2= -1.18V (vs Fc/Fc+)為MnIII/MnII特徵峰,不同於氧橋接4並未
觀測到更高電位的可逆氧化還原峰(MnIV/MnIII)。這電位值介於[MnIII(OH)(PY-
5)]2+ (E1/2=+0.17V vs Fc/Fc+)[82]以及[MnIII(OH)(SMe2N4(tren))]+ (E1/2= -0.6V vs
Fc/Fc+)-[29],[MnIII(OH)(OAc)(Me2EBC)]+ (E1/2= -1.33V vs Fc/Fc+)[30]以及[MnIII-
(buea)(O-H)]- (E1/2=-1.51V vs Fc/Fc+)之間[31],在文獻上得知隨著MnIII/MnII的 電位越低,錳羥基化合物更難以進行氫原子轉移反應。
Figure 3-9. Cylic voltammograms of 4 (black) and 5 (green) measured in a 1mM acetonitrile solution with 0.1M [n-Bu4N][PF6] as the supporting electrolyte at room temperature, scan rate 0.1 V/s.
近期研究中許多錳羥基化物被提出,其結構性質以及擁有氫原子轉移 (hy-
drogen atom transfer,HAT)的能力,例如[MnIII(OH)(dpaq)](OTf)、[MnIII(OH)(S-
Me2N4(tren))](PF6)·H2O以及[MnIII(OH)(PY5)]2+,能與1-hydroxy-2,2,6,6-tetramethy-
lpiperidine(TEMPOH)或酚類進行氫原子轉移,參考其方法作以下反應性測試。
5與100當量2,6-di-tbu-4-OMe-PhOH反應(Figure 3-10),發現並沒有觀測到5有如 同其他文獻例子單純還原成Mn(II),觀測到405 nm (1350)尖細的特徵峰增長,
這表明反應中有2,6-di-tbu-4-OMe-PhO.自由基的生成[25]。萃取出自由基產率為
7-8%,值得注意的是光譜有些許紅位移至700 nm,似乎反應中並不只牽扯到氫 原子轉移使金屬還原至Mn(II),但未能分離出其結構與性質。從電化學上的比
較能提出5有較低的電位使得金屬難以還原,這與難以進行氫原子轉移有所契
合。
Figure 3-10. UV-vis spectra of 5 with 100 equivalent 2,6-di-tbu-4-OMe-PhOH (black line) in acetonitrile. The measured interval between two curve is 20 min.
3-3 含錳(II)化合物與氧氣的反應性
電化學分析中 1、2 的低電位支持著兩者皆能夠被氧氣氧化,其在固相以及
溶液相皆對於氧氣有高反應性,然而在溶液相與氧氣氧化反應較為複雜,故先
討論固相反應。將 2 微晶體置於乾燥氧氣中兩小時,表層色澤由紫色氧化後轉
為紫紅色(Figure 3-12),氧化後在氮氣環境回溶乙腈,觀測到三個特徵峰分別在
490、550、755 nm,這指出形成錳(IV)過氧化物 3 (基於 550nm 消光係數,產率
99%)。在 ATR-FTIR 光譜儀時間追蹤實驗中觀測到 νperoxo在 903 cm-1,這支持 著固相氧化中形成 3[19]
(Figure 3-11)。相較於 2,1 在固相氧化沒有觀測到 ATR-
FTIR 有 νo-o
=903cm
-1特徵峰增消長(Figure 3-12),顯示著並未 Mn(IV)-peroxo 3 的生成。橘紅色的 1 微晶體在室溫與過量乾燥氧氣反應兩小時,其逐漸轉變為黃棕色固體(Figure 3-11)。生成的固體能夠被 THF 溶解以及 UV-vis 光譜儀觀測
到約 500nm 有特徵峰,試圖分離衍生物但並未成功。如同(Figure 3-13)。兩者
皆在真空環境下去除空氣,其氧氣鍵結是不可逆的反應。
Figure 3-11. The microcrystals image of (a) 2, (b) after exposed dry O2 2 hours; (c) 1, (d) after exposed dry O2 2 hours.
Figure 3-12. Time-depended ATR-FTIR spectra of microcrystals of 1 (bottom) and 2 (top) after exposed to air. The measured interval between two curve is 1 min (top) and 5 min (bottom).
Figure 3-13. UV-vis spectra of solid-to-solid oxygenated 1 (black) and 2 (red) disso- lveed in acetonitrile at room temperature. Inset plot shows 500 nm feature peak.
在乙腈溶液中,針對 1 及 2 與過量的氧氣在室溫下反應,在此發現不同於
固相氧化的結果,不論 1 或 2 兩者皆生成類似的產物,但其生成比例相異。藉
由 UV-vis 光譜儀監測其結果為混合產物,擬合混合光譜證實主要生成 3 與 4
(Figure 3-14)。為了證實 3 和 4 生成比例與瞬間參與反應的氧氣濃度有關,以下 做了兩種操作(1)將氧氣灌入溶液;(2)將氧氣通入液面上層並逐間擴散進溶液。
在此發現將氧氣大量的通入單核 2 乙腈溶液內,產生的 3 產率(~95 %)遠高
於擴散操作的生成量(~80%)(Figure 3-15)。這證實了氧氣的擴散速率遠大於 2 的
氧化速率時,絕大部分的 2 皆被氧化成 3,殘餘微量的 2 決定了進一步反應衍
生物 4 的產率。相較於固態反應中擴散效應沒有溶劑的支撐,導致氧化完整地
生成 3。
雙核 1 乙腈溶液不同於 2。其與氧氣反應其生成比例與加入氧氣的方式並
沒有太顯著的關係(Figure 3-16)。氧氣氧化 1 乙腈溶液觀測到 3 化合物的生成,
但在固相氧化反應並沒有偵測到,這大概是因為雙核 1 在乙腈中發生溶劑鍵結
形成單核[MnII(NCCH3)(TMSPS3)]-而造成的。藉由 1 與 DABCO 平衡反應的熱力
學參數可以推測與乙腈鍵結的單核體生成。雖然難以控制通入氧氣流量與濃
度,但理應隨著溫度降低而濃度升高,3 產量進一步地提高。
Figure 3-14. UV-vis spectra of 1 (0.1mM, bottom) and 2 (0.2mM, top) reacts with excess oxygen by two different operation.
Purge O2 to solution of 2
Layer O2 above solution of 2
Purge O2 to solution of 1
Layer O2 above solution of 1
Figure 3-15. UV-vis spectra of 2 reacts with excess oxygen by two different
operation. Oxygenated derivative generated was fitted by 3 (blue line) and 4 (red line) extinction coefficient.
Figure 3-16. UV-vis spectra of 1 reacts with excess oxygen by two different operation. According to 3 (blue line) and 4 (red line) extinction coefficient, Fitted oxygenated derivative generated.
此外針對 2 在-80 oC 丙腈溶劑下緩慢擴散氧氣進溶液,其結果與固相氧 化類似,能夠得到出純粹的 3 光譜生成(Figure 3-17)。並沒有觀測到穩定存在的
中間體生成,推測歸咎於中間體的溶解度以及擴散速率受溫度下降的影響。而
1 在-80 oC 丙腈溶劑下與氧氣氧化與室溫相同逐漸生成 3 與 4 生成的混合光
譜。值得注意的是在低溫還存在著另一個未知產物其特徵峰在 950 nm,但又熱
不穩定隨著時間而消長(Figure 3-18)。雙核的 1 似乎存在著其他的反應,這可能
與固態氧化黃褐色產物有關聯性。
Figure 3-17. UV-vis spectra of 2 (0.3 mM) layer above dry O2 in propionitrile at - 80 oC that shows 3 gradually generated.
Figure 3-18. UV-vis spectra of 1 (0.17 mM) layer above dry O2 in propionitrile at - 80 oC . Black line and blue line show the initial spectrum and final spectrum,
respectively. Red line shows the spectrum in which highest peak of absorbance of 950 nm. Inset: absorbance changes at 950 nm upon diffusion of dry O2. The measured interval between two curve is 2 min.
3-4 錯合物的分子結構探討
配位基TMSPS3為主體合成出一系列螯合錳金屬錯合物,在此描述其結構性
質。錯合物 1、2、4 以及 5 的 ORTEP 圖呈現在 Figure 3-20 至 Figure 3-23,並
附上篩選後的鍵長和鍵角。這一系列的配位化合物皆是五配位,得以用
geometry index (τ, degree of trigonality)描述其結構性質。τ5能區分配位中心是四 角錐體又或是雙三角錐幾何結構,其中τ5 = 0 時為標準四角錐體,τ5 =1 時為標
準雙三角錐體,計算公式τ5 = (β-α)/60,其中 β 與 α 分別為 Lbasal-M-Lbasal中的最
大角與次大角(Figure 3-19)[26]。
Figure 3-19. Geometry index for square pyramidal (τ5 = 0) and trigonal bipyramidal (τ5 = 1) are shown. [26]
擁有兩個對等錳中心的 1,其中 S1MnS1A 與 S1MnAS1A 的二面角為零,
呈現了 Mn2S2菱形共平面。兩個錳(II)離子之間的距離長達 3.0055(13) Å ,這遠
大於有金屬-金屬交互作用的距離。幾何結構上呈現兩個扭曲的雙三角錐(τ5 =
0.67),相對於磷有三個硫坐落在赤道向,第四個硫呈現反式並落於軸向。Mn-
Sbridge和 Mn-Sterminal平均鍵長分別為 2.5242(14)和 2.4441(13) Å ,與文獻硫橋接
雙錳(II) [MnII2(LS)(LSH)]ClO4 的 Mn-Sbridge和 Mn-Sterminal鍵長分別為 2.6462 與

![Figure 1-10. Side-on Mn III -peroxo compounds (a) [Mn III (O 2 )TPP] − (b) [Mn III (HB- (HB-(3,5-iPr 2 pz) 3 )(O 2 )( 3,5-iPr 2 pzH)] (c) [Mn(TMC)(O 2 )] + (d) [Mn III (O 2 )H 2 bupa] -](https://thumb-ap.123doks.com/thumbv2/9libinfo/9563968.617730/23.893.129.762.394.1103/figure-side-iii-peroxo-compounds-iii-tpp-iii.webp)
![Figure 1-11. Reaction scheme shows an intermolecular O 2 -Transfer between [Ni III (12-TMC)(O 2 )] + and [Mn II (14-TMC)] 2+](https://thumb-ap.123doks.com/thumbv2/9libinfo/9563968.617730/24.893.129.760.478.1118/figure-reaction-scheme-shows-intermolecular-transfer-iii-tmc.webp)
![Figure 1-13. Reaction of five-coordinate [Mn II (S Me2 N 4 (6-Me-DPEN)] + with O 2 to afford mono-oxobridged [[Mn III (S Me2 N 4 (6-Me-DPEN)] 2 (μ-O)] 2+ via metastable Mn](https://thumb-ap.123doks.com/thumbv2/9libinfo/9563968.617730/25.893.174.727.502.938/figure-reaction-coordinate-dpen-afford-oxobridged-dpen-metastable.webp)
![Figure 1-14. Proposed mechanism for the formation of [Mn IV (O 2 )P(C 6 H 3 -3-SiMe 3 -2- -2-S) 3 ] - and [Mn II 2 (PS 3 ) 2 ] 2 -](https://thumb-ap.123doks.com/thumbv2/9libinfo/9563968.617730/26.893.137.757.530.1106/figure-proposed-mechanism-formation-mn-iv-sime-mn.webp)