國立臺灣大學電機資訊學院光電工程學研究所 碩士論文
Graduate Institute of Photonics and Optoelectronics College of Electrical Engineering and Computer Science
National Taiwan University Master Thesis
氮化鋁鎵/氮化鎵金氧半高電子遷移率場效 電晶體之製作與特性研究
Fabrication and Characterization of AlGaN/GaN Metal-Oxide-Semiconductor High Electron
Mobility Field Effect Transistors
洪瑞宏 Jui-Hung Hung
指導教授:彭隆瀚 博士 Advisor: Lung-Han Peng, Ph.D.
中華民國 102 年 7 月
July, 2013
i
誌謝
完成這篇論文的此刻,心中充滿了許多的喜悅以及感謝。本論文得以完成,
首先要感謝的是我的指導教授彭隆瀚博士,您的悉心指導,讓我在這碩士班生涯 中成長許多,與您相處的這兩年中,除了您以身作則的研究態度,您高度的道德 操守也深深地影響了我,讓我在碩士生涯中,在智識以及人格方面皆有所提升,
我將銘記在心。
感謝口試委員王維新博士,您和藹的待人處事態度,與您的交談、討論,每 每讓我覺得如沐春風;感謝口試委員胡振國博士,您對我的論文提出了最為關鍵 的問題,使我對於我的研究主題有更深刻的認識;感謝口試委員賴志明博士,無 論是碩一的去光斑實驗或是碩二的電晶體研究,與您的討論,都能帶給我更加明 確的方向。在此由衷地感謝三位口試委員。
感謝王德豐先生,在碩士班的兩年中,你變魔術般的生出了許多機台,讓我 的實驗有許許多多的變化,而你即時的維修服務,真的是「揪感心ㄟ」;另外,
是你帶我進入咖啡的世界,讓只知道壹咖啡以及星巴克的我,漸漸能品嘗出咖啡 的各種風味,讓我活得更加的精彩、更加的有滋味。
感謝 No 哥學長,在我實驗、報賬兩頭燒時,你為我接下一些報賬的事務,你 默默的付出,讓我在最忙的時刻中順利的撐下去。感謝伯淳學長,你出手救援我 碩一的光學實驗,讓不熟悉光學的我最後能順利完成計畫,碩二的電晶體研究,
也是靠著與你不時的討論,才能有所突破,達到本實驗室碩士論文的標準,以你 的研究能力,相信你能順利畢業。此外,感謝永銘學長,在一開始進入實驗室時 給予指導及協助,讓我得以快速上手;並感謝世豪、思德、濟民學長一年多的照 顧,尤其是大便學長陪我做實驗到凌晨的義氣,讓我能在實驗室中感受到許多溫 暖,在此致上十分的感謝。
感謝同屆一起奮鬥兩年的夥伴:LN 組同學恩綺、翰棕以及同為 GN 組的耀德;
感謝恩綺,在我為光斑計畫奮鬥時,讓出光學桌以及光學室的使用時間,使我能 有充足的時間做實驗,而與妳、葉伯、凱勛的晚餐時刻,總能讓我舒緩一整天下 來所累積的壓力並回復繼續做實驗的動力;感謝翰棕,為實驗室的生活帶來了許
ii
多的歡笑以及娛樂,實驗室許多的活動都是由你主辦,辛苦你了;最後,感謝同 組的耀德,在我最初進來的半年內,感謝有你的幫忙我才能順利度過一開始的適 應期,而後來的光斑元件,感謝你的大力支援,我們才能完成老師交代的各種奇 妙的元件。感謝三位共度碩士生涯的同學,我們從一開始的互不相識,到後來成 為患難與共的夥伴,期待未來的日子裡,我們仍能保持聯繫,維持這得來不易的 緣份。
感謝小我一屆的三位學弟:凱勛、鈞皓及柏廷;感謝在工作態度上與我最相 近的凱勛,你接下我的報賬業務,交接上因為你的一點就通,使我毋須多耗費精 力在雜務上,讓我能專心於研究,完成畢業論文,相信你也能順利完成碩士學位;
感謝處理了實驗室許多雜務的鈞皓,有你處理這些業務,相信這些任務都能順利 完成;感謝陪我打球的柏廷,因為有你,使我碩二的生活增加了許多打網球的樂 趣,每次早晨揮灑汗水的時刻,總是讓我期待不已,另外,感謝你在我低潮聽我 發牢騷,並且鼓勵我忘掉不愉快的經驗,面對現實,你給我的建議總是十分務實 的。感謝新進來的學弟們,實驗室有你們這些新血加入,相信能帶來新的氣象。
最後要感謝一直無怨無悔支持我的家人、父親、母親,有你們的支持,我才 能毫無後顧之憂的全力衝刺碩士學程的研究,順利取得碩士學位,我今日的成就,
都是你們所造成,僅以此學位論文獻給我最親愛的家人。
iii
中文摘要
本研究主要探討兩大部分:1. AlGaN/GaN MOS-HEMT 以及 2. 閘極掘入式 AlGaN/GaN MOS-HEMT 兩種元件,除了對於製作過程中所需的各項技術進行確認,
更對於製程中光致電化學氧化法對於元件特性的影響提出一些觀察。
在 AlGaN/GaN MOS-HEMT 方面,我們發現到薄層的 PEC 閘極氧化層對於元 件特性(電流開關比、閘極漏電流)有所提升,使電流開關比由 5 個數量級提升至 6 個數量級、閘極漏電流下降 1 個數量級,而閾值電壓往負偏移約 2 V,可能與 PEC 氧化層/AlGaN 介面狀態有關、或與 AlGaN 受到氧化層的應力有關。在 PEC 閘極 氧化層增厚的情況下,比起薄層的 PEC 閘極氧化,閾值電壓再往正電壓偏移 2.2 V 左右。然而,元件特性卻大幅下降,在 VG=0 V 時,飽和電流由 410 mA/mm 下降 至 144.5 mA/mm,轉導峰值則由 63 mS/mm 下降至 30.67 mS/mm,說明以 PEC 氧 化法削減 AlGaN 層厚度並非製作增強型元件的優良手段。
閘極掘入式 AlGaN/GaN MOS-HEMT 證明了閘極掘入可製作出增強型元件,
而 PEC 氧化層鈍化表面可以進一步提升元件的各種電特性,電流開關比由 5 個數 量級提升至 5.5 個數量級,轉導峰值由 95 mS/mm 上升至 120 mS/mm,調整 PEC 氧化時間以及原子層沉積的氧化層厚度等,可望做出特性更優良的電晶體。
關鍵字:MOS-HEMT、光致電化學氧化法(PEC Oxidation)、閘極掘入
iv
ABSTRACT
This work is focused on the effects of the photoelectrochemical oxidation (PEC oxidation) method on both AlGaN/GaN Metal-Oxide-Semiconductor High Electron Mobility Transistors (MOS-HEMTs) and gate recessed AlGaN/GaN MOS-HEMTs.
The measurement of AlGaN/GaN MOS-HEMTs with PEC oxide gate shows that with lightly PEC oxidation, the current on/off ratio has increased from 5 to 6 order of magnitude and the gate leakage current decreased almost 1 order of magnitude.
However with short-time PEC oxidation, the threshold voltage has shifted -2 V. This phenomenon is related to the interface state of the PEC oxide and the AlGaN layer or the strain of the AlGaN layer induced by the PEC oxide. Moreover, with long-time PEC oxidation, the threshold voltage has shifted 2.2 V, but the device performance is greatly decreased.
The measurement of gate recessed AlGaN/GaN MOS-HEMTs shows that the gate recess method can effectively shift threshold voltage from -5 V to -1 V. The PEC oxidized passivation layer further increases the threshold voltage to nearly zero volts.
with moderate PEC oxidation, the transconductance can be increased from 95 mS/mm to 120 mS/mm, though the recessed gate structure is suffered from the poor gate leakage current.
In a nutshell, we propose a useful method to fabricate an enhancement mode MOS-HEMT, but we still need some calibrations and optimizations to further increase our device performance.
Keywords: MOS-HEMT, PEC Oxidation, Gate Recessed MOS-HEMT
v
目錄
口試委員會審定書 ... #
誌謝 ...i
中文摘要 ... iii
ABSTRACT ...iv
目錄 ... v
圖目錄 ... viii
表目錄 ...xi
第 1 章 緒論... 1
1.1 簡介 ... 1
1.2 研究動機 ... 5
1.3 論文架構 ... 7
第 2 章 AlGaN/GaN MOS-HEMT 理論介紹 ... 8
2.1 AlGaN/GaN 極化現象與二維電子氣形成機制 ... 8
2.1.1 自發極化效應 ... 8
2.1.2 壓電極化效應 ... 10
2.1.3 二維電子氣形成機制 ... 11
2.2 晶格面蝕刻法 ... 15
2.3 金屬接觸原理 ... 16
2.3.1 歐姆接觸原理 ... 16
2.3.2 傳輸線模型原理 ... 19
vi
2.4 光致電化學氧化法原理 ... 21
2.5 原子層沉積[30] ... 23
2.6 電性量測架構 ... 25
第 3 章 AlGaN/GaN MOS-HEMT 的製程技術 ... 26
3.1 表面清潔 ... 27
3.2 元件隔離 ... 28
3.3 晶格面蝕刻法 ... 29
3.4 歐姆接觸(傳輸線模型量測用) ... 30
3.5 光致化學氧化法 ... 31
3.6 AlGaN/GaN MOS-HEMT 製作流程 ... 33
3.6.1 元件隔離 ... 33
3.6.2 PEC 網格電極製作 ... 35
3.6.3 PEC 閘極氧化層及表面鈍化層製作 ... 37
3.6.4 原子層沉積 ... 39
3.6.5 歐姆接觸 ... 40
3.6.6 閘極金屬沉積 ... 42
3.7 閘極掘入式 AlGaN/GaN MOS-HEMT 製作流程 ... 44
第 4 章 AlGaN/GaN MOS-HEMT 直流量測 ... 45
4.1 傳輸線模型量測 ... 45
4.2 PEC 光致電化學氧化法之研究 ... 47
4.3 MOS-HEMT IDS-VDS與 IDS-VG特性 ... 50
4.3.1 閾值電壓變化(Threshold Voltage Shift) ... 54
vii
4.3.2 電流開關比(On/Off Current Ratio) ... 55
4.3.3 閘極漏電流(Gate Leakage Current) ... 56
4.4 閘極掘入式 MOS-HEMT IDS-VDS與 IDS-VG特性 ... 57
4.4.1 閾值電壓變化(Threshold Voltage Shift) ... 62
4.4.2 電流開關比(On/Off Current Ratio) ... 63
4.4.3 閘極漏電流(Gate Leakage Current) ... 64
4.4.4 轉導(Transconductance) ... 65
第 5 章 結論與未來展望 ... 66
5.1 結論 ... 66
5.2 未來展望 ... 69
參考文獻 ... 70
viii
圖目錄
圖 1.1 More Moore and More than Moore[2] ... 2
圖 1.2 三五族材料電子速率[5] ... 3
圖 1.3 不同閘極絕緣層與沒有絕緣層之 HEMT 元件的 IG-VG特性 ... 6
圖 2.1 烏采結構之氮化鎵及鎵原子終結面與氮原子終結面示意圖[17] ... 9
圖 2.2 自發極化與晶格常數[19] ... 9
圖 2.3 AlGaN/GaN 磊晶示意圖 (a)應變前 (b)應變後[17] ... 10
圖 2.4 AlGaN/GaN 接面與 AlGaAs/GaAs 接面比較[21] ... 11
圖 2.5 AlGaN/GaN 結構中極化效應之方向(Ga 面) ... 12
圖 2.6 AlGaN/GaN 接面量子井與極化現象之關係(Au/AlGaN 接觸)[22] ... 12
圖 2.7 2DEG 面密度與 AlGaN 厚度關係(內插圖為理論計算)[23, 24] ... 13
圖 2.8 能帶結構隨著 AlGaN 厚度變化的示意圖[23] ... 14
圖 2.9 能帶結構隨著外加閘極偏壓的變化[25] ... 14
圖 2.10 金屬-n 型半導體接面在接觸後之熱平衡歐姆接觸能帶圖 ... 16
圖 2.11 Ti/Al/Ti/Au 與 GaN 反應示意圖 ... 18
圖 2.12 傳輸線模型示意圖 ... 19
圖 2.13 電阻 RT與長度 T 之函數圖 ... 20
圖 2.14 光致電氧化法中 n 型氮化鎵與電解液接觸後能帶示意圖 ... 21
圖 2.15 利用 ALD 沉積 Al2O3之成膜原理[30] ... 24
圖 2.16 直流量側架構 ... 25
圖 3.1 氮化鋁鎵/氮化鎵高電子遷移率電晶體磊晶結構 ... 26
ix
圖 3.2 光致電化學氧化法實驗架構圖[33] ... 31
圖 3.3 元件隔離製程示意圖 ... 34
圖 3.4 PEC 金屬網格製作示意圖 ... 36
圖 3.5 PEC 氧化層與表面鈍化層示意圖 ... 38
圖 3.6 ALD 沉積氧化鋁示意圖 ... 39
圖 3.7 歐姆接觸製作流程示意圖 ... 41
圖 3.8 閘極金屬沉積示意圖 ... 43
圖 4.1 相同間距(d=34 um)下的不同條件下歐姆接觸之優劣 ... 46
圖 4.2 PEC 氧化層高度(上)與深度(下):1 分鐘 ... 48
圖 4.3 PEC 氧化層高度(上)與深度(下):2 分鐘 ... 48
圖 4.4 PEC 氧化層高度(上)與深度(下):4 分鐘 ... 49
圖 4.5 閘極氧化層 PEC 時間為 0 分鐘之電晶體飽和電流特性圖 ... 51
圖 4.6 閘極氧化層 PEC 時間為 0 分鐘之電晶體轉換特性圖 ... 51
圖 4.7 閘極氧化層 PEC 時間為 1 分鐘之電晶體飽和電流特性圖 ... 52
圖 4.8 閘極氧化層 PEC 時間為 1 分鐘之電晶體轉換特性圖 ... 52
圖 4.9 閘極氧化層 PEC 時間為 4 分鐘之電晶體飽和電流特性圖 ... 53
圖 4.10 閘極氧化層 PEC 時間為 4 分鐘之電晶體轉換特性圖 ... 53
圖 4.11 閘極氧化時間對於閾值電壓之影響 ... 54
圖 4.12 閘極氧化時間對於電流開關比之影響 ... 55
圖 4.13 閘極氧化時間對於閘極漏電流之影響 ... 56
圖 4.14 非閘極掘入式且 PEC 較少的電晶體飽和電流特性圖 ... 58
圖 4.15 非閘極掘入式且 PEC 較少的電晶體轉換特性圖 ... 58
x
圖 4.16 非閘極掘入式且 PEC 較多的電晶體飽和電流特性圖 ... 59
圖 4.17 非閘極掘入式且 PEC 較多的電晶體轉換特性圖 ... 59
圖 4.18 閘極掘入式且 PEC 較少的電晶體飽和電流特性圖 ... 60
圖 4.19 閘極掘入式且 PEC 較少的電晶體轉換特性圖 ... 60
圖 4.20 閘極掘入式且 PEC 較多的電晶體飽和電流特性圖 ... 61
圖 4.21 閘極掘入式且 PEC 較多的電晶體轉換特性圖 ... 61
圖 4.22 PEC 氧化程度與閘極掘入程度對於閾值電壓之影響 ... 62
圖 4.23 PEC 氧化程度與閘極掘入程度對於電流開關比之影響 ... 63
圖 4.24 PEC 氧化程度與閘極掘入程度對於閘極漏電流的影響 ... 64
圖 4.25 PEC 氧化程度與閘極掘入程度對於轉導之影響 ... 65
圖 5.1 閘極 PEC 氧化層厚度對(a)閘極漏電流及(b)轉換特性之影響[38] ... 67
圖 5.2 閘極掘入對於轉導特性之影響[39] ... 67
xi
表目錄
表 1.1 不同半導體材料的物理參數[3] ... 3
表 2.1 GaN 在不同溶液中的橫切面蝕刻形貌示意圖[26] ... 15
表 2.2 金屬功函數表[28] ... 17
表 4.1 PEC 氧化層測試 ... 47
1
緒論 第 1 章
1.1 簡介
半導體科技的發展使得上個世紀人類的發展有了翻天覆地的變化,其重要性 及影響力一直延續至今,她將人類社會帶向前所未有的「大數據」時代,改變了 人類社會的生產、生活、溝通以及思維方式。半導體材料在半導體科技的成長過 程中始終扮演著重要的角色。世界上第一顆的電晶體是以鍺(Ge,室溫下能隙寬度 為 0.66 eV)為材料,然而帶動過去 60 年的半導體發展的始終以矽(Si,室溫下能隙 寬度為 1.12 eV)為主,除了地表上含量豐富外,直接從塊材生長出的高品質氧化層 也是其它半導體材料所望塵莫及的[1],直到今天,矽仍然是微電子領域中主要的 半導體材料;為了滿足超高速、大功率微波元件及電路的需求,1970 年代開始了 砷化鎵(GaAs,室溫下能隙寬度為 1.42 eV)以及磷化銦(InP,室溫下能隙寬度為 1.35 eV)材料的研究;直至 1990 年代,隨著長晶科技的進步,碳化矽(SiC,室溫下能隙 寬度為 3.25eV)以及三五族化合物半導體材料(III-V)漸漸受到重視,其優良的通道 特性以及寬能隙的優勢,使它們成為下一代電晶體的發展重點。
對於未來電晶體的發展,ITRS 技術路程規格[2]定義出現今電晶體的兩大發展 方向(圖 1.1):微縮化(Miniaturazation)以及多樣化(Diversification),微縮化指的是 在現有的矽材料下,對電晶體元件大小進行微縮以增加電路密度且提升運算速度,
另外降低閾值電壓(Threshold Voltage)以降低功耗亦是設計元件時須考量的面向,
主要是對邏輯電路而言;而多樣化則多指將電晶體用於類比的領域,諸如微波元
2
件、高功率元件、感應器、生醫用晶片…等。微縮化的發展目前以業界為主力,
並已成功導入鰭狀場效電晶體(FinFET)架構;而多樣化方面則有多元的內容,各種 半導體材料因其特性不同,對於不同的領域有不同的優勢,研究內容可說是百家 爭鳴,本實驗室躬逢其盛,以氮化鎵(GaN)為實驗發展的一大方向。
圖 1.1 More Moore and More than Moore[2]
由表 1.1[3]可知,氮化鎵除了在電子遷移率方面不如砷化鎵以外,其他各項特 性皆優於其餘三者,其中 BFOM Ratio 為表示半導體材料在高頻、大功率元件的應 用潛力上的常用指標[4],因此以氮化鎵發展大功率或是高頻電晶體是相當合理的 選擇。另外,雖然電子遷移率不如砷化鎵,但是由理論計算[5](圖 1.2)可發現,事 實上氮化鎵的電子速度峰值優於砷化鎵,只是峰值的位置不同,且砷化鎵於中高 電場下其飽和速度遠遜於氮化鎵,在大功率元件上沒有優勢;而相較之下,碳化 矽在能隙寬度、耐受溫度等條件均能與氮化鎵匹敵,然而電子遷移率較差的缺點
3
使得其在高頻方面的應用潛力上不如氮化鎵系列的電晶體結構。
表 1.1 不同半導體材料的物理參數[3]
BFOM=Baliga figure of Merit for power transistor performance (**EG3
)
圖 1.2 三五族材料電子速率[5]
材料
電子遷移率
(cm2/V-s)
介電系數
能隙寬度 EG (eV)
耐受溫度 Tmax(oC)
BFOM Ratio
氮化鎵 1500 9.5 3.4 700 24.6
砷化鎵 5000 13.1 1.4 300 9.6
碳化矽 260 9.7 2.9 600 3.1
矽 1300 11.4 1.1 300 1.0
4
氮化鎵的電晶體主要以氮化鎵異質接面電晶體(GaN Heterojunction Field Effect Transistor,GaN HFET)為主,在氮化鋁鎵/氮化鎵(AlGaN/GaN)或氮化銦鎵/
氮化鎵(InGaN/GaN)異質接面形成時,由於氮化物材料的自發極化(Spontaneous Polarization)及壓電極化(Piezoelectric Polarization)效應,在沒有雜質摻雜下,在 GaN 側即有高濃度的二維電子氣(2DEG)或二維電洞氣(2DHG)產生,且因通道處未有摻 雜,不受傳導時雜質散射之影響,同時具有高電子遷移率的特性,也因此 GaN HFET 常稱為氮化鎵高電子遷移率電晶體 (GaN High Electron Mobility Transistor,
GaN HEMT)。
然而 GaN HEMT 的發展仍面臨了許多的挑戰:首先,異質磊晶使得氮化物材 料缺陷密度高(Defect Density),其次,氮化物材料本身強大的極化效應與介面缺陷 狀態(Interface States)不易控制,另外,GaN HEMT 在高工作電壓下的元件漏流或 逆壓電效應(High-voltage induced defect formation through the inverse piezoelectric effect)所造成元件可靠性不佳…等更是商業化的一大障礙[6-9]。因此,關於氮化物 半導體尚有許多課題值得進一步研究,例如:設計不同的磊晶結構以降低缺陷並 藉由磊晶結構對於極化場量進行控制、對於介面缺陷的模擬以及特性研究、增強 型元件(Enhancement Mode Device)的實現…等。
5
1.2 研究動機
傳統上 GaN HEMT 的以蕭特基接觸做為閘極,其缺點是閘極漏電流大(圖 1.3)[10],相較之下,GaN 金屬-氧化層-半導體 HEMT(GaN MOS-HEMT)有較佳的 電性表現,閘極絕緣層材料的研究從早期電漿輔助化學氣相沉積(Plasma Enhanced Chemical Vapor Deposition,PECVD)的二氧化矽(SiO2)及氮化矽(SiNx)漸漸轉變為 原子層沉積(Atomic Layer Deposition,ALD)的高介電係數材料,如氮化鋁(AlN)[11]、
氧化鋁(Al2O3)[12]、氧化鉿(HfO2)[10, 13]…等,然而其絕緣層/半導體介面缺陷密 度仍無法與二氧化矽/矽介面相比,所以如何在氮化鋁鎵/氮化鎵表面成長高品質且 低表面缺陷的絕緣層為目前重要的研究目標,由於氮化鋁鎵/氮化鎵材料因異質磊 晶所形成的表面缺陷,為影響電晶體效能的關鍵因素,本實驗室所研究的光致電 化學氧化(Photoelectrochemical Oxidation,PEC Oxidation)裝置能於氮化鎵上成長出 高品質的氧化鎵(Ga2O3)[14],搭配本實驗室的 ALD 所生長的氧化鋁,以製作高品 質的 AlGaN/GaN MOS-HEMT;最後,筆者嘗試以閘極掘入(Gate Recess)的方法,
製作出接近增強型的 AlGaN/GaN MOS-HEMT;量測方面,將以探討 PEC 對於不 同元件的直流操作之影響做為主軸。
6
圖 1.3 不同閘極絕緣層與沒有絕緣層之 HEMT 元件的 IG-VG特性
7
1.3 論文架構
本論文的撰寫架構可分為 5 個章節,依序為緒論、AlGaN/GaN MOS-HEMT 理論介紹、AlGaN/GaN MOS-HEMT 製程技術、AlGaN/GaN MOS-HEMT 直流量 測與討論、最後一章則為結論與未來展望。
第 1 章說明此研究主題之簡介與動機,並提出論文架構。
第 2 章首先介紹 AlGaN/GaN MOS-HEMT 的工作原理,接著介紹晶格面蝕刻 法、金屬接觸原理、光致電化學氧化法理論以及原子層沉積(ALD)機台介紹,最後 是電性量測架構。
第 3 章為 AlGaN/GaN MOS-HEMT 的製程技術,包含表面清潔、元件隔離、
晶格面蝕刻、PEC 氧化法、歐姆接觸(以傳輸線模型測試),最後是 AlGaN/GaN MOS-HEMT 以及閘極掘入式 AlGaN/GaN MOS-HEMT 的製作流程。
第 4 章首先敘述歐姆接觸(以傳輸線模型測試)條件測試,接著為 PEC 氧化法 速率測試,然後使用 Agilent 4155C 探討不同厚度的 PEC 氧化層對 AlGaN/GaN MOS-HEMT 的影響,最後是閘極掘入式 AlGaN/GaN MOS-HEMT 的直流量測。
第 5 章總結了整篇論文並給出與研究的未來展望及發展方向。
8
AlGaN/GaN MOS-HEMT 理論介紹 第 2 章
本章將介紹 AlGaN/GaN 材料性質、極化現象,以及二維電子氣(2DEG)形成的 理論基礎,接著介紹製程關鍵步驟的理論基礎,分別是:晶格面蝕刻法及其對後 續製程必要性,金屬接觸原理及歐姆接觸的測試方法,光致電化學氧化法原理和 原子層沉積(ALD)的成膜原理,最後則是本實驗室電性量測架構。
2.1 AlGaN/GaN 極化現象與二維電子氣形成機制
III 族氮化物半導體(AlN、GaN、InN)存在兩種晶體結構,分別是六方晶系的 烏采結構(Wurtzite)以及立方晶系的閃鋅礦結構(Zinc Blende)。烏采結構是氮化物的 熱力學穩態結構也是氮化物最常見的結構。烏采結構由兩個六方最密堆積結構相 互交疊組成,最密堆積面為(0001),每個鎵原子被四個相鄰的氮原子包圍,反之亦 然,每個單位晶胞有 12 個原子,鎵原子與氮原子各占一半[15];因閃鋅礦結構較 少見且不穩定,在此不多做介紹。
2.1.1 自發極化效應
III 族氮化物磊晶時會沿著特定軸向交互堆疊兩種不同的原子,烏采結構 III 族氮化物的軸向為 c 軸,即晶體沿著[0001]或是[000-1]方向生長,若是沿著[0001]
從下往上的排列為氮原子面在下,III 族原子面在上,則材料表面呈現 III 族原子面 極性;若是沿著[000-1] 從下往上的排列為 III 族原子面在下,氮原子面在上,則 材料表面呈現氮原子面極性,圖 2.1 以氮化鎵為例說明。不同的極性面的物理性
9
質與化學性質相當不同,因此兩種極性面並非等價[16, 17]。文獻指出自發極化的 方向為[000-1][18, 19],即由鎵原子面指向氮原子面,文獻[19]指出:烏采結構的晶 體屬於非中心對稱晶體,且鎵原子與氮原子並非對稱性分布,導致正負電荷的中 心無法重合,從而在沿 c 軸方向造成自發極化,自發極化的強度值可由現代極化 理論計算而得(圖 2.2)[20]。
圖 2.1 烏采結構之氮化鎵及鎵原子終結面與氮原子終結面示意圖[17]
圖 2.2 自發極化與晶格常數[19]
10
2.1.2 壓電極化效應
壓電效應為電介質材料中電能與機械能互換之現象,而晶體在外力作用下產 生的內部電子重新分布則稱為壓電極化。在磊晶過程中,由於異質接面上下材料 晶格常數不匹配,會有應變產生,造成電荷隨著應變重新分布。圖 2.3 為
AlGaN/GaN 異質接面,一般磊晶狀況下,GaN 的厚度遠大於 AlGaN,故可假設應 變都落在較薄的 AlGaN 上,由於 AlGaN 之晶格常數小於 GaN,因此若要形成假形 晶體(Pseudomorphic structure)或是應變層(Strain layer),AlGaN 須受到一伸張應變,
而沿著 c 軸的壓電極化在應變層受到張應變時,極化方向為[000-1],正好與自發 極化方向相同[18]。
圖 2.3 AlGaN/GaN 磊晶示意圖 (a)應變前 (b)應變後[17]
11
2.1.3 二維電子氣形成機制
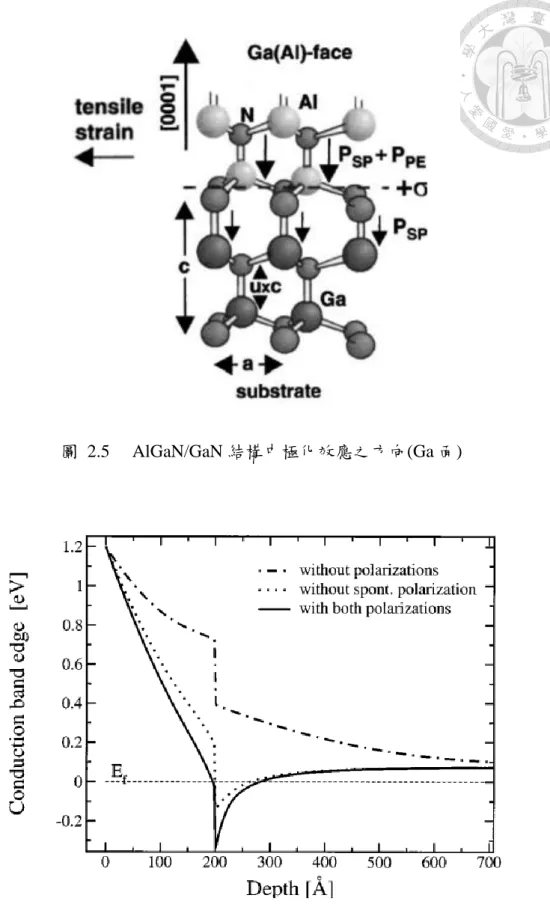
氮化物材料中,二維電子氣的形成機制除了異質接面造成的能帶不連續外,
更需要考慮極化場量的梯度效應。傳統的 AlGaAs/GaA 材料中,二維電子氣(2DEG) 的來源是 AlGaAs 和 GaAs 的調變摻雜,然而 AlGaN/GaN 結構中,在沒有人為摻 雜的情況下仍能產生極高密度的二維電子氣[21](圖 2.4),這是因為極化效應為異 質接面提供了很強的內建電場(又,對於 Ga 面之磊晶結構,AlGaN 的自發極化與 壓電極化同向,有加成的效果。如圖 2.5),使得異質接面 GaN 側形成既深且窄的 量子井結構,侷限了電子於此二維結構傳輸,文獻[22]對於調變摻雜 AlGaN 的 AlGaN/GaN 接面模擬其傳導帶結構(圖 2.6),可看出極化效應是形成 2DEG 相當重 要的因素。
圖 2.4 AlGaN/GaN 接面與 AlGaAs/GaAs 接面比較[21]
12
圖 2.5 AlGaN/GaN 結構中極化效應之方向(Ga 面)
圖 2.6 AlGaN/GaN 接面量子井與極化現象之關係(Au/AlGaN 接觸)[22]
13
文獻[23]及[24]分別以實驗及理論計算出 2DEG 須在 AlGaN 超過某一關鍵厚度 後,才會大量產生,之後隨著厚度增加漸漸達到峰值,然後開始緩慢減少(圖 2.7)。
他們提出 2DEG 主要來源於 AlGaN 表面的類施體態(surface donor-like states),如圖 2.8 所示,2DEG 形成前,隨著 AlGaN 厚度增加,極化電場令表面電位(eΦB,表面 導帶底相對於費米能階 EF的高度)逐步升高,相當於類施體態漸漸接近費米能階,
一旦費米能階與類施體態切齊,類施體態開始釋放電子,此時,2DEG 在介面處開 始形成,AlGaN 的內建電場也會被減弱;若 AlGaN 厚度繼續增加,由於類施體態 與費米能階切齊,則 2DEG 密度趨向飽和,接近於 AlGaN/GaN 介面極化電荷的密 度;另外,雖然 AlGaN 中 Al 成分增加亦會令 2DEG 濃度上升[16],但以異質磊晶 的角度而言,也使得 AlGaN 處於更強的張應變狀態,為了避免應變鬆弛(strain relaxation)的產生,AlGaN 的厚度應隨著 Al 成分上升而減少。
圖 2.9 [25]以理論計算表明了能帶結構隨著閘極偏壓的變化,說明了當負偏壓 時,產生 2DEG 的量子井結構變淺且變寬,使得 2DEG 濃度降低,2DEG 漸漸呈現 空乏狀態。
圖 2.7 2DEG 面密度與 AlGaN 厚度關係(內插圖為理論計算)[23, 24]
14
圖 2.8 能帶結構隨著 AlGaN 厚度變化的示意圖[23]
圖 2.9 能帶結構隨著外加閘極偏壓的變化[25]
15
2.2 晶格面蝕刻法
晶格面蝕刻法為先利用氫氧化鉀(KOH)溶液搭配紫外光的光致電(PEC)蝕刻法 來蝕刻氮化鎵材料,之後再泡入不同溶液中以蝕刻出不同晶格面的兩段式蝕刻架 構,第二段的濕蝕刻稱為晶格面蝕刻法(wet chemical crystallographic)[26],由於 PEC 蝕刻法無法直接蝕刻出平滑側壁,需要搭配後續的溶液處理始可將氮化鎵蝕刻出 平滑側壁。文獻[26]整理出利用熔融氫氧化鉀(molten KOH)、磷酸(H3PO4)或氫氧化 鉀溶液(KOH solution)來蝕刻氮化鎵,如表 2.1,而本研究是以晶格面蝕刻法修復 因乾蝕刻(Reactive Ion Etching,RIE)造成氮化鎵側壁殘留的電漿損傷與不規則側壁,
減少元件因不規則側壁所產生漏流,表 2.1 最下方為本研究蝕刻之條件。
表 2.1 GaN 在不同溶液中的橫切面蝕刻形貌示意圖[26]
16
2.3 金屬接觸原理
2.3.1 歐姆接觸原理
歐姆接觸提供金屬與半導體接面雙向傳導的低電阻接觸[27]。為了降低金屬與 半導體接面的損耗,並提升元件操作效率,須選擇功函數較低的金屬與半導體形 成接觸面,當金屬、半導體接觸後,為達到熱平衡,半導體側的能帶將會彎曲(圖 2.10),使得費米能階與導帶能隙變小,因此,由於不存在位障,在金屬上外加正 偏壓,電子容易由半導體流向金屬;反之,在金屬上外加負偏壓,電子由金屬流 向半導體所受到的位障約為 qΦBn,若慎選金屬,則 qΦBn可以是非常小的位障。總 之,無論在金屬上外加正或負偏壓,電子均可輕易流動,有很好的導電性,是決 定元件特性的基本要件。
圖 2.10 金屬-n 型半導體接面在接觸後之熱平衡歐姆接觸能帶圖 EC
EF
EV
qV
nq
BnEvac
s
m
q
q
17
為了判斷歐姆接觸的好壞,通常是以接觸電阻值 Rc(contact resistance)做為指 標,由接觸面積 A 及接觸電阻 Rc 的乘積可得特徵電阻值 ρc(Specific contact resistance)
2
1
V cm J
C
(2.1)
其中 J 為電流密度(A/cm2),V 為電壓(Voltage,V)。
表 2.2 金屬功函數表[28]
金屬 功函數 金屬 功函數 金屬 功函數 金屬 功函數 金屬 功函數 金屬 功函數
Ag 4.26 Al 4.28 As 3.75 Au 5.1 B 4.45 Ba 2.7 Be 4.98 Bi 4.22 C 5 Ca 2.87 Cd 4.22 Ce 2.9 Co 5 Cr 4.5 Cs 2.14 Cu 4.65 Eu 2.5 Fe 4.5 Ga 4.2 Gd 3.1 Hf 3.9 Hg 4.49 In 4.12 Ir 5.27
K 2.3 La 3.5 Li 2.9 Lu 3.3 Mg 3.66 Mn 4.1 Mo 4.6 Na 2.75 Nb 4.3 Nd 3.2 Ni 5.15 Os 4.83
Pb 4.25 Pt 5.65 Rb 2.16 Re 4.96 Rh 4.98 Ru 4.71 Sb 4.55 Sc 3.5 Se 5.9 Si 4.85 Sm 2.7 Sn 4.42 Sr 2.59 Ta 4.25 Tb 3 Te 4.95 Th 3.4 Ti 4.33 Tl 3.84 U 3.63 V 4.3 W 4.55 Zn 4.33 Zr 4.05
18
表 2.2 為金屬功函數總表,去除掉活性過高、電性不佳、太少見等元素厚,
適合與 GaN 形成歐姆接觸的金屬只有 Ti 或 Al,一般選用 Ti/Al 為基礎的金屬組合,
然而因為 Al 在後續快速熱退火(Rapid Thermal Anneal,RTA)製程中容易氧化形成 Al2O3,且 Al 熔點較低,使得單純 Ti/Al 不易形成具有高度熱穩定佳的歐姆接觸。
有鑑於此,一般多以 Ti/Al/X/Au 的多層金屬作為 GaN 的歐姆接觸,X 可以是 Ti、
Mo、Ni 等[29],Ti 與 GaN 在熱退火過程中可能產生些許的固相反應而形成 TiN,
同時 GaN 表面 N 空缺濃度上升,相當於重摻雜的 n-GaN,有利於形成歐姆接觸;
Al 則是作為催化劑促進 Ti 與 GaN 的固相反應,並與 Ti 形成功函數低且緻密的合 金(TiAl);X 是為了在 RTA 時,阻擋上方金屬 Au 往下方 GaN 表面擴散,避免高 功函數的 Au 降低歐姆接觸的特性;而 Au 除了為下方 Ti/Al/X 提供保護,在 RTA 時,避免下方金屬氧化之外,更提供了與外部系統良好的接觸(圖 2.11)。在本研究 中,以 Ti 做為 X 層金屬。
圖 2.11 Ti/Al/Ti/Au 與 GaN 反應示意圖
19
2.3.2 傳輸線模型原理
量測特徵接觸電阻的方法有很多種,本研究利用傳輸線模型(Transmission Line Model,TLM)結構來測量歐姆接觸之電阻值。大部分半導體元件皆為平面式結構,
因此可利用傳輸線模型測量其特徵接觸電阻值ρc來判斷歐姆接觸特性的好壞,圖 2.12 為傳輸線模型示意圖。
圖 2.12 傳輸線模型示意圖
由圖 2.12 可知,任意相鄰之二金屬墊(metal pad)的電阻值為
(2.2) 其特徵電阻值,可表示為
ρ (2.3)
其中 Rs 為金屬間距 L 的電阻值,而 Rc 為金屬與半導體接面的電阻,由式(2.3)中 可得知電阻 R 為長度 L 的函數,因此可經由量測不同長度 L 的電阻值 R,作 R-L 直線逼近,如圖 2.13 所示,在 L=0 處之 R 軸截距可求得 R=2Rc,在 R=0 處之 L
L1 L2 L3 L4
W
t
20
截距可求得 L=2LT,再將電極寬度 W 代入式(2.3)中,如此即可求得特徵電阻值 ρc。
圖 2.13 電阻 RT與長度 T 之函數圖
L(μm) R(Ω)
2R
c2L
T0
21
2.4 光致電化學氧化法原理
光致電化學反應(Photoelectrochemical,PEC)是利用能量大於或等於半導體能 帶的光源照射半導體表面,使其表面產生大量電子電洞對,光源所激發的電洞會 因內建電場漂移至表面與電解液形成氧化反應,而電子則與電極產生還原反應,
完成光致電化學反應,針對氮化鎵的 PEC 反應為本實驗室發展成熟之技術,以下 介紹此技術之原理。
圖 2.14 光致電氧化法中 n 型氮化鎵與電解液接觸後能帶示意圖
圖 2.14 為 PEC 氧化法中 n 型氮化鎵置於電解液中的能帶示意圖,在熱平衡狀 態下,須滿足半導體費米能階與電解液的氧化還原電位(redox potential)相等,即讓 電荷移動達到穩態平衡,電荷須在半導體與電解液的介面處上移動來平衡兩者能
紫外光 hν>Eg
電解液面 半導體表面
22
量,由於電荷的移動,在半導體表面形成內建電場,使得氮化鎵的價電帶和導電 帶向下彎曲。因此,若再利用能量高於半導體能階的紫外光激發氮化鎵的電子電 洞對,激發出來的高能量電子、電洞受到表面電場驅動,電洞會移動至電解液與 半導體介面處,累積於半導體表面,如果此時具有適當的氧化基(Oxident)時,將使 高能量的電洞越過介面而產生氧化作用;而電子則藉由電極導引至電解液中,與 液體中的還原基(Reductant)產生還原反應,形成封閉的氧化還原電流迴路,其反應 式如式(2.4)與式(2.5)所示。
Cathode: 2H2O + 2e- → 2OH- + H2 (2.4) Anode: 2GaN + 6OH- +6h+→ Ga2O3 + N2 + 3H2O (2.5)
於本研究中,氮化鎵會因氧化反應從表面開始消耗,又電解液刻意挑選弱酸 性溶液,不會將氧化鎵迅速溶解,因而在表面處形成氧化鎵薄膜,成為電晶體之 閘極絕緣層及鈍化層。
23
2.5 原子層沉積[30]
原子層沉積(Atomic Layer Deposition,ALD )是將物質以逐層堆疊原子級厚度 薄膜的方式的一種化學氣相沉積(Chemical Vapor Deposition,CVD)技術,ALD 利 用氣態金屬有機前驅物(Metal Organic Precursor)在基板表面的選擇性化學吸附性,
在表面吸附層達到飽和狀態後,即不再吸附前驅物,在成膜過程中,因為自我限 制的反應特性,可以達成原子級厚度的薄膜成長,具有優異的覆蓋率與順應性。
圖 2.15 為利用 Al(CH3)3 (亦稱 TMA)與 H2O 作為反應物以成長 Al2O3的步驟示意 圖:
1. 適當地加熱基板,使基板表面產生氫氧基(OH 基);
2. 將 TMA 前驅物通入反應腔,TMA 與 OH 表面官能基發生反應,生成物甲 烷(CH4),氣態的 CH4會在高溫狀態下脫離表面;
3. 當表面層的 OH 基完全反應完畢,基材表面便不再吸附 TMA;
4. 利用氬氣(Ar)(或是 N2)流過表面,帶走殘留在表面的雜質及多餘的反應 物;
5. 通入水氣,由於 Al-O 的鍵結會比 Al-CH3更強,所以-CH3鍵結很容易因為 H2O 的存在就斷開,換成 Al-O 的鍵結,因此,H2O 與表面上的甲基(CH3) 反應生成 OH 表面官能基與 CH4氣體分子,而兩個相鄰的 OH 基發生脫水 反應,鋁原子終於與氧原子形成化學鍵結,
6. 當表面所有的 CH3基全被 OH 基取代,氧化鋁第一層原子層於是產生,
7. 接著通入 Ar (或是 N2),帶走殘留在表面的雜質及多餘的反應物。
24
如此便完成一個循環,由於表面上已經成長出了一層非常均勻的原子級厚 度的薄膜生成物,要增加薄膜厚度,只須重覆前述的循環步驟,即可達成接 近原子級平整的薄膜。
圖 2.15 利用 ALD 沉積 Al2O3之成膜原理[30]
25
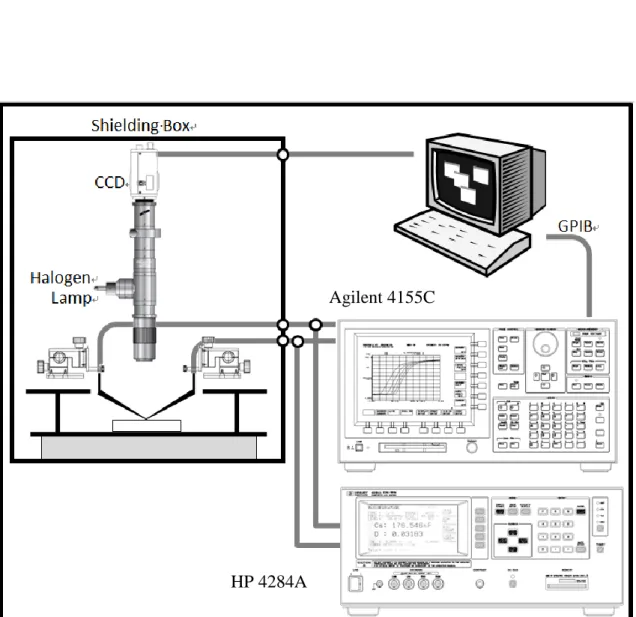
2.6 電性量測架構
本實驗室採用 Agilent 4155C 半導體參數分析儀來量測氮化鋁鎵/氮化鎵金氧 半高電子遷移率電晶體(AlGaN/GaN MOS-HEMT)以及閘極掘入式氮化鋁鎵/氮化 鎵金氧半高電子遷移率電晶體(Gate Recessed AlGaN/GaN MOS-HEMT)的直流特性,
包括 VDS-IDS、VG-IDS、漏電流 VG-IG,量測架構如圖 2.16 所示。
圖 2.16 直流量側架構 Agilent 4155C
HP 4284A
26
AlGaN/GaN MOS-HEMT 的製程技術 第 3 章
本章介紹表面清潔、元件隔離、晶格面蝕刻、PEC 氧化法、歐姆接觸等製作 AlGaN/GaN MOS-HEMT 的技術,最後是 AlGaN/GaN MOS-HEMT 以及閘極掘入 式 AlGaN/GaN MOS-HEMT 的製作流程。
圖 3.1 為本研究所用的 AlGaN/GaN 異質磊晶結構圖,由 Hitachi Cable 所生產 製造,使用藍寶石(sapphire)作為基板,基板上首先沉積 GaN 做為緩衝層,厚度約 為 1 μm,其上為約 25 nm 的 AlGaN,因此二維電子氣位能井在 GaN 與 AlGaN 接 面處會形成一平面通道,侷限了電子於此平面上傳輸,最後在最頂層在沉積厚度 1 nm 的 n 型氮化鎵,增強金屬與半導體間的歐姆接觸。
圖 3.1 氮化鋁鎵/氮化鎵高電子遷移率電晶體磊晶結構 1 m 25 nm
1 nm n-GaN Cap Layer
Sapphire Substrate Undoped GaN Buffer Layer
Undoped AlGaN 2DEG
27
3.1 表面清潔
元件製作前的表面清潔是整個製作過程中最重要的部份之一,由於有機金屬 化學氣相沉積法(Metal-organic Chemical Vapor Deposition,MOCVD)成長完後表面 仍有部分反應後的殘留物,多為金屬或有機物,且當樣品暴露在空氣中過久後,
表面會生成自然氧化層,這些殘留物,對於元件特性會造成莫大的傷害。
不同的清潔方法會對元件特性造成不同影響[31],因此選擇適當的清潔流程是 必要的。文獻[32]中提出各種清潔樣品的方法,考量到實驗室現有設備,筆者採行 的清潔方法如下:
1. 將樣品浸在 80 oC 丙酮中,以超音波震盪器震洗 5 分鐘,去除有機雜質;
2. 再將樣品浸在甲醇中,以超音波震盪器震洗 5 分鐘,去除丙酮殘留;
3. 接著以去離子水(Deionized Water,DI Water)沖洗一分鐘以去除甲醇殘留,
再以氮氣槍吹乾,以上完成對有機物的清潔;
4. 完成有機物清潔後,將樣品浸入 36.5 %的鹽酸(HCl)溶液中 10 分鐘,以去 除金屬殘留物並減少氧化物殘餘量,再以去離子水沖洗,以氮氣槍吹乾;
5. 最後將樣品置入氫氟酸(HF)中 1 分鐘,再以大量的去離子水沖洗,以減少 HF 在樣品表面的殘留,避免長時間下對樣品表面造成損傷。
28
3.2 元件隔離
製作 MOS HEMT 時,元件隔離製程會影響其特性,若元件隔離製程不完善,
不僅會造成嚴重的漏電流與影響元件電壓-電流特性外,甚至會影響到元件操作的 穩定性。在元件隔離製程時,首要重點是蝕刻深度必須超過二維電子氣所在之位 置,如此才可達到元件隔離的效果,使元件間不會互相影響。在本研究中使用感 應式耦合電漿離子蝕刻系統(Inductively Coupled Plasma Reactive Ion Etching,
ICP-RIE)來進行元件隔離製程。
ICP-RIE 的蝕刻條件對元件隔離製程相當重要,由於 AlGaN 的厚度十分地薄,
故蝕刻速率的掌握變得十分重要,而 RF 功率大小與蝕刻氣體之選擇,則是條件中 最重要的參數。本實驗室蝕刻氣體選用 Ar 與 Cl,其原因為離子反應蝕刻中包含了 化學反應,與蝕刻表面之原子產生反應後,會產生揮發性物質,可藉由機台中真 空設備將其抽走,因此不會再次沉積於表面,而造成表面蝕刻速率不一的情況產 生。本研究中蝕刻氣體條件選用 Ar=20 sccm、Cl=20 sccm,操作功率為 RF Power=200 W、Bias=100 W,蝕刻壓力則為 0.6 Pa。
29
3.3 晶格面蝕刻法
RIE 高台蝕刻後,須以晶格面蝕刻法蝕刻隔離高台的側壁,利用氫氧化鉀(KOH) 或是磷酸(H3PO4)溶液來修復乾蝕刻所造成 1)表面殘留的電漿損傷與 2)不規則側壁,
讓側壁呈現平滑的狀態,如表 2.1 所示,本研究利用 KOH 溶液,降低 KOH 的濃 度,以控制其對氮化鎵蝕刻速度,減少 KOH 溶液對高台表面的損傷,並蝕刻出類 似梯型的斜邊側壁;需要平滑側壁的原因為元件操作時,如果高台的側壁不規則 排列或是存在電漿損傷時,可能會增加元件漏流路徑,產生不必要的損耗,因此,
藉由晶格面蝕刻,蝕刻出平滑斜邊側壁,可減少不必要的漏電流產生。另外,根 據本實驗室傳承,若不做晶格面修復蝕刻,則無法以 PEC 法成長高品質的氧化鎵。
晶格面蝕刻法的製程步驟如下:
1. 配置氫氧化鉀溶液,以 2.8 克(g)氫氧化鉀錠與 50 毫升(mL)的去離子水充 分攪拌混合,再放置於加熱台(Hot Plate)上加熱到 120℃。
2. 將主動區高台蝕刻完之樣品,放置於 120 ℃的氫氧化鉀中,浸泡 10 分鐘,
當實驗完成後,用去離子水沖洗 1 分鐘,完成晶格面蝕刻製程。
30
3.4 歐姆接觸(傳輸線模型量測用)
使用完成元件隔離與晶格面蝕刻後的樣品,製作傳輸線模型之歐姆接觸金屬,
以便測試歐姆金屬在不同條件下的電性特性,測出達到最低接觸電阻所需之條件,
以下為傳輸線模型製程步驟:
1. 將完成元件隔離與晶格面修復蝕刻後之樣品先滴上 LOR5B,以每分鐘 5000 轉 (5000 r.p.m)旋轉塗佈 40 秒,使 LOR5B 均勻的分佈在樣品上,將樣品放置於加 熱台上以 115 度加熱 120 秒,除去 LOR5B 中的水分,樣品塗佈完 LOR5B 之後,
再滴上光阻 S1813,以每分鐘 5000 轉旋轉塗佈 40 秒,再將樣品放置於加熱台 上以 115 度加熱 90 秒,增強光阻鍵結,以利曝光顯影。
2. 曝光 22 秒,使用 MF319 顯影 18 秒定義出歐姆接觸區域,之後再以去離子水 清洗,使用氮氣槍吹乾。
3. 將樣品迅速的放置於電子束蒸鍍機(E-Gun)中,鍍上金屬鈦(Ti)/鋁(Al)/鈦(Ti)/金 (Au),厚度約為 20 nm/100 nm/55 nm/100 nm。
4. 將樣品放置於丙酮中,以光阻掀離法(Lift off) 去除遮罩區域之外的 S1813 光阻,
再以甲醇、去離子水清洗樣品,使用氮氣槍吹乾,再將樣品置入 MF319 中約 5 秒,去除 LOR5B,以去離子水清洗樣品,再以氮氣槍將樣品吹乾。
5. 將樣品放置於快速熱退火爐(Rapid thermal anneal,RTA)之腔體內,以 925 oC 烘烤 70 秒做退火處理,完成歐姆接觸的傳輸線模型。
31
3.5 光致化學氧化法
圖 3.2 為光致電化學(PEC)氧化實驗架構圖[33],先將氮化鎵樣品用黃臘黏在 載玻片上,再放置到鐵氟龍(Teflon)載台上,利用長條狀的鉑(Pt)金屬片做為電極與 樣品上的金屬電極緊緊接觸,鉑電極經由外部電路連接至另一片同樣浸泡在電解 液中的鉑電極,將載台、樣品與電極沉浸於適當的電解液中,兩白金電極間加一 電流計,當實驗進行時可利用其電流計觀察電流之變動得知反應是否正常進行。
圖 3.2 光致電化學氧化法實驗架構圖[33]
PEC 氧化實驗中使用電解液為醋酸銨(CH3COONH4)與醋酸(CH3COOH)的混和 溶液,調配出 pH 值為 6.4 的電解液來進行氧化反應,利用光源為 254 nm 之汞燈 直接照射樣品,光源的發光強度約 20 mW/cm2,使用低轉速攪拌子的目的在於確 保電解液於實驗過程中均勻分散,並利用外加的安培計觀察反應時的電流,進而
A
紫外光
攪拌子
載台 樣品
白金片
32
得知實驗過程是否正常進行。
PEC 氧化法實驗過程如下:
1. 將樣品固定於載台上,以 2.5 g 醋酸銨與 500 ml 的去離子水混合後,將醋酸 以 1:5 的比例與去離子水適當稀釋後,加入稀釋後的醋酸,調整電解溶液 pH 值至 6.4,完成電解液的調製。
2. 將載台放入 pH 值為 6.4 的電解液中,開啟汞燈源樣品表面產生氧化還原反 應,實驗結束後,立刻以去離子水沖洗 1 分鐘,避免酸性電解液在結束氧 化反應後產生的蝕刻氧化層的效果。
3. 將樣品放置於高溫爐中,通入氧氣 15 分鐘後,當氧氣充滿整個高溫爐爐體 後,再以 400 度燒烤 2 小時,去除其表面所殘餘之水分,通入氧氣可以增 強自生氧化層之鍵結,完成光致電化學氧化法之氧化物的生成。
33
3.6 AlGaN/GaN MOS-HEMT 製作流程
3.6.1 元件隔離
1. 氮化鋁鎵/氮化鎵晶圓先以切割機(Dicing Saw)切出所需要的樣品大小;
2. 使用 3.1 節所提出的方法清潔樣品表面;
3. 因本實驗室樣品的氫氟酸(HF)為公用溶液,為減少 HF 中的雜質汙染樣品,將 樣品再次浸在丙酮中,以超音波震盪器震洗 1 分鐘,再浸入甲醇中,以超音波 震盪器再震洗 1 分鐘,之後以去離子水沖洗,去除樣品上之甲醇溶液,最後再 用氮氣將樣品吹乾,如圖 3.3 (a)所示。
4. 將清洗好之樣品滴上 S1813 光阻(Photoresist,PR),以每分鐘 5000 轉(5000 r.p.m) 旋轉塗佈 40 秒,使光阻均勻分佈於樣品上,再將樣品放置於加熱台上以 115 度加熱 100 秒,去除光阻水分並增強鍵結,將樣品曝光 22 秒,再以 MF319 顯 影 19 秒定義出元件有效區域,之後以去離子水清洗 1 分鐘,以氮氣吹乾,如 圖 3.3 (b)所示。
5. 使用感應式耦合電漿離子反應蝕刻機(ICP-RIE),蝕刻出深度約為 90 nm 的高台 (Mesa),如圖 3.3 (c)所示。
6. 將蝕刻完的樣品放置於丙酮中,除去蝕刻過後的 S1813 光阻,之後以甲醇與去 離子水清洗樣品,再以氮氣吹乾樣品,如圖 3.3 (d)所示。
7. 使用 3.3 節的晶格面蝕刻法修復 ICP-RIE 造成的損傷。
34
圖 3.3 元件隔離製程示意圖 Sapphire Substrate
i-GaN i-AlGaN n-GaN
Sapphire Substrate i-GaN
i-AlGaN n-GaN
Sapphire Substrate i-GaN
i-AlGaN n-GaN
(a) (b)
(d) (c)
PR
Sapphire Substrate i-GaN
i-AlGaN n-GaN
35
3.6.2 PEC 網格電極製作
1. 將製作好隔離高台之樣品浸入氫氟酸(HF)溶液中,時間為 1 分鐘,以大量的去 離子水沖洗 1 分鐘,再以氮氣槍將樣品吹乾,如圖 3.4(a)所示。
2. 將處理過後之樣品滴上 LOR5B,以 5000 rpm 轉速旋轉塗佈 40 秒,放置於加熱 台上以 115 度加熱 120 秒,再滴上 S1813 光阻,以 5000 rpm 轉速旋轉塗佈 40 秒,再次將樣品放置於加熱台上以 115 度加熱 100 秒,將樣品曝光 22 秒,再 以 MF319 顯影 20 秒定義出金屬網格區域,之後再以去離子水清洗,並以氮氣 槍吹乾,如圖 3.4(b)所示。
3. 將樣品迅速放置於電子鎗真空蒸鍍系統 (E-gun System)中,鍍上金屬鈦(Ti),厚 度約為 100nm,如圖 3.4(c)所示。
4. 將鍍完金屬之樣品置入於丙酮中,以光阻掀離法(Lift off)去除遮罩區域之外的 S1813 光阻,再以甲醇、去離子水清洗樣品,並以氮氣槍吹乾,再將樣品置入 MF319 中,去除 LOR5B,以去離子水清洗樣品,並將樣品吹乾,如圖 3.4(d) 所 示。完成 PEC 氧化時所需的金屬網格。
36
圖 3.4 PEC 金屬網格製作示意圖 PR
(a)
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(b)
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(c)
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
Ti Ti
Ti
(d)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
37
3.6.3 PEC 閘極氧化層及表面鈍化層製作
1. 將製作好 PEC 網格電極的樣品浸在丙酮中,以超音波震盪器震洗 1 分鐘,再浸 入甲醇中,以超音波震盪器再震洗 1 分鐘,之後以去離子水沖洗,去除樣品上 之甲醇溶液,最後再用氮氣槍將樣品吹乾,如圖 3.5 (a)所示。
2. 將處理過後之樣品滴上 S1813,以 5000 rpm 轉速旋轉塗佈 40 秒,放置於加熱 台上以 115 度加熱 120 秒,將樣品曝光 22 秒,再以 MF319 顯影 17 秒定義出 閘極區域,之後再以去離子水清洗,並以氮氣槍吹乾,如 圖 3.5 (b)所示。
3. 以載波片固定樣品,將樣品固定在鐵氟龍載台上,並放入溶液 pH 值為 6.4 的 電解液中,照入汞燈源,使其產生氧化還原反應,當反應時間結束,立即以去 離子水沖洗 1 分鐘,降低酸性電解液蝕刻反應所產生之自氧化層,如 圖 3.5 (c) 所示。。
4. 將製作好 PEC 閘極氧化層的樣品浸在丙酮中,以超音波震盪器震洗,以去除光 阻遮擋層,再浸入甲醇中,以超音波震盪器再震洗 1 分鐘,之後以去離子水沖 洗,去除樣品上之甲醇溶液,最後再用氮氣槍將樣品吹乾,如圖 3.5 (d)所示。
5. 再次將樣品固定於載玻片上,將樣品固定在鐵氟龍載台上,並放入溶液 pH 值 為 6.4 的電解液中,照入汞燈源,使其產生氧化還原反應,當反應時間結束,
立即以去離子水沖洗 1 分鐘,降低酸性電解液蝕刻反應所產生之自氧化層,此 PEC 氧化層為表面以及側壁鈍化層,如圖 3.5 (e)所示。
6. 將樣品放置於高溫爐中,通入氧氣 15 分鐘後,當氧氣充滿整個高溫爐爐體後,
再以 400 度燒烤 2 小時,去除其表面所殘餘之水分,通入氧氣可以增強自生氧 化層之鍵結,完成光致電化學氧化法之氧化物的生成,如圖 3.5 (f)所示。
38
圖 3.5 PEC 氧化層與表面鈍化層示意圖 PR
PEC Oxide
(a)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(b)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(c)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(d)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(e)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
(f)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
39
3.6.4 原子層沉積
1. 將完成 PEC 自氧化層的樣品浸在丙酮中,以超音波震盪器震洗 1 分鐘,再浸入 甲醇中,以超音波震盪器再震洗 1 分鐘,之後以去離子水沖洗,去除樣品上之 甲醇溶液,最後再用氮氣槍將樣品吹乾,如 圖 3.6(a)所示。。
2. 將清潔過後之樣品置入原子層沉積機台(ALD)腔體內,沉積約 4 nm 厚度的氧化 鋁,作為 PEC 自氧化層的保護層。
3. 將樣品放置於高溫爐中,通入氮氣 15 分鐘後,當氮氣充滿整個高溫爐爐體後,
再以 400 度燒烤 30 分鐘,去除其表面所殘餘之水分, 圖 3.6(b)所示。
圖 3.6 ALD 沉積氧化鋁示意圖 PR
PEC Oxide ALD Oxide
Ti Ti
Sapphire Substrate i-GaN i-AlGaN
n-GaN
(a)
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
(b)
40
3.6.5 歐姆接觸
1. 將完成氧化層生長的樣品浸在丙酮中,以超音波震盪器震洗 1 分鐘,再浸入甲 醇中,以超音波震盪器再震洗 1 分鐘,之後以去離子水沖洗,去除樣品上之甲 醇溶液,最後再用氮氣槍將樣品吹乾,如圖 3.7 (a)所示。。
2. 將清潔過後之樣品滴上 LOR5B,以 5000 rpm 轉速旋轉塗佈 40 秒,放置於加熱 台上以 115 度加熱 120 秒,再滴上 S1813 光阻,以 5000 rpm 轉速旋轉塗佈 40 秒,再次將樣品放置於加熱台上以 115 度加熱 100 秒,將樣品曝光 22 秒,再 以 MF319 顯影 20 秒定義出源極(Source)及汲極(Drain)的歐姆接觸區域,之後再 以去離子水清洗,並以氮氣槍吹乾,如圖 3.7 (b)所示。
3. 將樣品迅速送入反應式離子蝕刻機(Reactive Ion Etching,RIE)內進行氧化層蝕 刻,之後將樣品浸入二氧化矽蝕刻液(Buffer Oxide Etchant,BOE)約 20 秒,避 免氧化鎵殘餘,如圖 3.7 (c)(d)。
4. 將樣品迅速放置於電子鎗真空蒸鍍系統 (E-gun System)中,鍍上金屬鈦(Ti)/鋁 (Al)/鈦(Ti)/金(Au),厚度約為 20 nm/100 nm/55 nm/100 nm,如 圖 3.7 (e)所示。
5. 將鍍完金屬之樣品置入於丙酮中,以光阻掀離法(Lift off)去除遮罩區域之外的 S1813 光阻,再以甲醇、去離子水清洗樣品,並以氮氣槍吹乾,再將樣品置入 MF319 中,去除 LOR5B,以去離子水清洗樣品,並將樣品吹乾,如圖 3.7 (f) 所示。
6. 將樣品放置於快速熱退火爐(RTA)腔體內,以 925 oC 烘烤 70 秒做退火處理,完 成源極(Source)及汲極(Drain)的歐姆接觸。
41
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-GaN
PR
PEC Oxide ALD Oxide
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
Ti/Al/Ti/Au Ti/Al/Ti/ Ti/Al/Ti/Au Au
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
(a)
(b)
(d)
(c)
(e) (f)
圖 3.7 歐姆接觸製作流程示意圖
42
3.6.6 閘極金屬沉積
1. 將完成歐姆接觸源極和汲極金屬的樣品浸在丙酮中,以超音波震盪器震洗 1 分 鐘,再浸入甲醇中,以超音波震盪器再震洗 1 分鐘,之後以去離子水沖洗,去 除樣品上之甲醇溶液,最後再用氮氣槍將樣品吹乾,如圖 3.8 (a)所示。。
2. 將清潔過後之樣品滴上 LOR5B,以 5000 rpm 轉速旋轉塗佈 40 秒,放置於加熱 台上以 115 度加熱 120 秒,再滴上 S1813 光阻,以 5000 rpm 轉速旋轉塗佈 40 秒,再次將樣品放置於加熱台上以 115 度加熱 100 秒,將樣品曝光 22 秒,再 以 MF319 顯影 20 秒定義出閘極金屬區域,之後再以去離子水清洗,並以氮氣 槍吹乾,如圖 3.8 (b)所示。
3. 將樣品迅速放置於電子鎗真空蒸鍍系統 (E-gun System)中,鍍上金屬鎳(Ni)/金 (Au),厚度約為 30 nm/100 nm,如 圖 3.8 (c)所示。
4. 將鍍完金屬之樣品置入於丙酮中,以光阻掀離法(Lift off)去除遮罩區域之外的 S1813 光阻,再以甲醇、去離子水清洗樣品,並以氮氣槍吹乾,再將樣品置入 MF319 中,去除 LOR5B,以去離子水清洗樣品,並將樣品吹乾,如圖 3.8(d) 所示。
43
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
PR
PEC Oxide
ALD Oxide Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
G
Ni/Au Ni/Au
Ti Ti
Sapphire Substrate i-GaN
i-AlGaN
n-aN
S D
G
圖 3.8 閘極金屬沉積示意圖
(d) (c)
(b) (a)
44
3.7 閘極掘入式 AlGaN/GaN MOS-HEMT 製作流程
對於氮化物電晶體元件,一般在不施加偏壓下,通道為自然導通,是為空乏 型(Depletion mode)元件。在電力電子的應用上,增強型(Enhancement mode)元件對 於降低驅動電路設計的複雜度以及電路在開關切換所產生的功率損耗是相當重要 的。因此,筆者嘗試以閘極掘入的作法實現接近增強型的 AlGaN/GaN
MOS-HEMT。
閘極掘入式 AlGaN/GaN MOS-HEMT 的作法大致與 AlGaN/GaN MOS-HEMT 相當,不同點在於
1. 將完成元件隔離的樣品再次置入感應式耦合電漿離子反應蝕刻機中,將 閘極區域下方蝕刻出約 20nm 深的溝槽,之後跟隨 AlGaN/GaN
MOS-HEMT 製程類似步驟。
2. PEC 的自氧化層並沒有用光阻定義出閘極下方的絕緣層,僅作為表面以 及側壁的鈍化層,PEC 時間為 2 分鐘。
3. 另外,出於製程需求以及時間考量,沒有鍍上 PEC 網格電極。
4. 同樣出於製程考量,做完原子層沉積的樣品,沒有經過高溫爐的燒烤。
45
AlGaN/GaN MOS-HEMT 直流量測 第 4 章
本章對於上章節所完成的實驗進行分析,首先是對於傳輸線模型量測分析,
接著是 PEC 光致氧化法的研究,最後是不同氧化層厚度的 AlGaN/GaN MOS-HEMT 的直流量測,最後是不同 PEC 覆蓋層厚度的閘極掘入式 AlGaN/GaN MOS-HEMT 的直流量測及分析。
4.1 傳輸線模型量測
如本研究利用鈦(Ti)/鋁(Al)/鈦(Ti)/金(Au)金屬堆疊層作為歐姆接觸電阻值測 試,參考過去的研究,將堆疊層的厚度選定為 20 nm/100 nm/55 nm/100 nm,主要 以固定時間 70 秒,調變熱退火的溫度。
本研究所使用的快速熱退火(Rapid thermal anneal,RTA)機台為冷壁式(Cold Wall)RTA,是藉由石英爐管外的燈管對爐管內的樣品進行輻射加熱,並以爐管內 的熱電偶計(Thermal Coupler)進行溫度控制,然而因為熱電偶計與樣品對於輻射熱 的吸收能力有所不同,因此監控到的溫度通常並不是樣品所受到的溫度;根據前 期的實驗(對於非本研究所用的晶片的歐姆接觸研究),為了讓樣品溫度與觀測值稍 微接近,且避免退火時間過長導致樣品其他裸露部分的損傷,退火時間以 70 秒為 佳(一般文獻多為 30 秒[34-36])。
由圖 4.1 可知,選用 925 oC 作為熱退火溫度是最佳的,沒有繼續往上增加溫 度測試的原因為擔心樣品表面的 GaN 會因此解離;經過計算後,最佳歐姆特徵電 阻值為 7.08 x 10-6 Ω- cm2,此特徵電阻值已符合一般製作電晶體之要求。
46
圖 4.1 相同間距(d=34 um)下的不同條件下歐姆接觸之優劣
47
4.2 PEC 光致電化學氧化法之研究
於此研究中,先於清潔完的樣品上鍍上 PEC 用金屬網格,在以光阻定義出 PEC 氧化之位置(本研究使用的是閘極光罩),再使用 PEC 氧化法製作氧化層,經過高 溫爐燒烤後,再以原子力顯微鏡(Atomic Force Microscopy,AFM)量測氧化區域與 光阻遮擋區域的落差,最後再以鹽酸蝕刻 PEC 氧化層,再次以 AFM 量測落差,
以此確定氧化層之深度。
原實驗設計有 5 個時間條件,分別為 1、2、4、8、12 分鐘,然而光阻遮擋層 於 8、12 分鐘條件下有邊緣殘留的問題,無法使用測量此兩條件之高度與深度,
因此本實驗的數據略有不足,相關數據整理於表 4.1,可注意到 D/T(深厚比)在不 同條件下接近相等,證明 PEC 氧化、高溫爐燒烤製程對於氧化層的影響是相同的。
然而,本實驗數據無法與電晶體製程相對應,主要原因為製作電晶體時,PEC 氧化前已經做了元件隔離,樣品表面特性與本實驗不同,目視結果,製作電晶體 時,相同條件下,速率小於本實驗。
表 4.1 PEC 氧化層測試
PEC 時間 (分鐘)
氧化層高度(H) (nm)
氧化層深度(D) (nm)
氧化層厚度 (T=H+D)(nm)
D/T
1 7~9 13~15 20~24 0.6364
2 ~14 20~22 34~36 0.6286
4 ~16 28~30 44~46 0.6444
48
圖 4.2 PEC 氧化層高度(上)與深度(下):1 分鐘
圖 4.3 PEC 氧化層高度(上)與深度(下):2 分鐘
49
圖 4.4 PEC 氧化層高度(上)與深度(下):4 分鐘
50
4.3 MOS-HEMT I
DS-V
DS與 I
DS-V
G特性
在本小節中,探討在穩定製程下,調變閘極氧化層的生長時間,依序為 0 分 鐘、1 分鐘、4 分鐘,藉以改變閘極氧化層的厚度,觀察閘極氧化層厚度對於 AlGaN/GaN MOS-HEMT 電晶體,值得注意的點是本製程中對於閘極 PEC 氧化、
鈍化層以及 ALD 鈍化層各分別做了熱退火(Post Oxide Anneal,POA)。
本研究中,電晶體的幾何參數如下:源極與汲極的間距(LDS)為 15 m,閘極 寬度(WG)為 200 m,閘極長度(LG)為 2 m。
圖 4.5 顯示 IDS-VDS的直流特性,電晶體閘極氧化層 PEC 時間為 0 分鐘,閘極 偏壓以 2 V 的間格從-10 V 到 0 V。在閘極電壓為 0 V 下,其汲極飽和電流約為 363 mA/mm。圖 4.6 顯示電晶體在固定汲極偏壓為 8 V 下,對不同閘極偏壓的轉導以 及汲極電流。VDS=8 V 和 VG=-4.12 V 時,其轉導峰值為 64 mS/mm。
圖 4.7 顯示 IDS-VDS的直流特性,電晶體閘極氧化層 PEC 時間為 1 分鐘,閘極 偏壓以 2 V 的間格從-10 V 到 0 V。在閘極電壓為 0 V 下,其汲極飽和電流約為 410 mA/mm。圖 4.8 顯示電晶體在固定汲極偏壓為 8 V 下,對不同閘極偏壓的轉導以 及汲極電流。VDS=8 V 和 VG=-6.6 V 時,其轉導峰值為 63 mS/mm。
圖 4.9 顯示 IDS-VDS的直流特性,電晶體閘極氧化層 PEC 時間為 4 分鐘,閘極 偏壓以 2 V 的間格從-10 V 到 0 V。在閘極電壓為 0 V 下,其汲極飽和電流約為 144.5 mA/mm。圖 4.10 顯示電晶體在固定汲極偏壓為 8 V 下,對不同閘極偏壓的轉導以 及汲極電流。VDS=8 V 和 VG=-3.04 V 時,其轉導峰值為 30.67 mS/mm。
51
圖 4.5 閘極氧化層 PEC 時間為 0 分鐘之電晶體飽和電流特性圖
圖 4.6 閘極氧化層 PEC 時間為 0 分鐘之電晶體轉換特性圖
![表 1.1 不同半導體材料的物理參數[3]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/16.892.118.781.265.1072/表11不同半導體材料的物理參數3.webp)

![圖 2.3 AlGaN/GaN 磊晶示意圖 (a)應變前 (b)應變後[17]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/23.892.151.770.95.1003/圖23AlGaNGaN磊晶示意圖a應變前b應變後17.webp)
![圖 2.7 2DEG 面密度與 AlGaN 厚度關係(內插圖為理論計算)[23, 24]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/26.892.126.792.113.313/圖272DEG面密度與AlGaN厚度關係內插圖為理論計算2324.webp)
![圖 2.8 能帶結構隨著 AlGaN 厚度變化的示意圖[23]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/27.892.254.681.117.984/圖28能帶結構隨著AlGaN厚度變化的示意圖23.webp)
![表 2.1 GaN 在不同溶液中的橫切面蝕刻形貌示意圖[26]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/28.892.196.698.654.1027/表21GaN在不同溶液中的橫切面蝕刻形貌示意圖26.webp)

![圖 2.15 利用 ALD 沉積 Al 2 O 3 之成膜原理[30]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9607122.632933/37.892.161.755.349.931/圖215利用ALD沉積Al2O3之成膜原理3.webp)