國 立 交 通 大 學
材料科學與工程研究所
碩士論文
以
Dithienothiophene 及 2,7-Carbazole 單元為基礎之新
穎型共軛高分子於有機光伏電池應用之合成與鑑定
Synthesis and Characterization of Novel Dithienothiophene- and
2,7-Carbazole-Based Conjugated Polymers for
Organic Photovoltaic Cells Applications
研 究 生:林家瑋
指導教授:林宏洲 博士
以
Dithienothiophene 及 2,7-Carbazole 單元為基礎之新穎型
共軛高分子於有機光伏電池應用之合成與鑑定
Synthesis and Characterization of Novel Dithienothiophene- and
2,7-Carbazole-Based Conjugated Polymers for
Organic Photovoltaic Cells Applications
研 究 生: 林家瑋 Student: Chia-Wei Lin
指導教授: 林宏洲 博士 Advisor: Dr. Hong-Cheu Lin
國 立 交 通 大 學
材料科學與工程研究所
碩 士 論 文
A Thesis Submitted to Department of
Materials Science and Engineering College of Engineering National Chiao Tung University
In partial Fulfillment of the Requirements For the Degree of Master of Science In Materials Science and Engineering
July 2009 Hsinchu, Taiwan
摘要
本論文中,我們利用 Suzuki coupling reaction 合成了以 2,7-carbazole 及 fused dithienothiophene 單元為分子主鍊的共軛型高分子,此系列高分子的吸 收範圍約在300 nm-550 nm;另一方面,我們也引入了 benzothiadiazole 單 元作為電子受體,而藉由2,7-carbazole 施體與 benzothiadiazole 受體之間的 作用力,有效的將吸光範圍延長至紅光區,其吸收範圍約在300 nm-630 nm,但此高分子之光轉換效率偏低,我們推估是因為在分子主鍊上引入的 長碳鏈較多,使得分子間的π-π stacking 較差,而影響其效率。 此兩類型共軛高分子因不具結晶態,因此可以利用旋轉塗佈的方式製 成薄膜,當與PCBM 1:1 混摻時,在 AM 1.5 的標準太陽光照射下,最佳 元件效率可達到η=0.49%,Jsc=1.82 mA/cm2、Voc=0.82 V、FF=0.37 %。
Abstract
In this reaserch, we synthesized new kinds of low band gap polymers by Suzuki coupling reaction with 2,7-carbazole and fused dithienothiophene rings as the main-chain conjugated polymers. The absorption spectra of polymers in this series showed an absorption range at 300 nm to 550 nm. Moreover, we have also introduced benzothiadiazole molecule as an electron acceptor, thus extending the absorption range at about 300 nm to 630 nm by the donor-acceptor effect between 2,7-carbazole and benzothiadiazole. However, the conversion efficiency of this polymer was low. This could be further explained by the introduction of the long alkyl chain along the polymer backbone, in which the inter-molecular π-π stacking decreased and consequently limited its conversion efficiency.
These two types of conjugated polymers were non-crystalline, so we investigated the potential use of polymers P1-P4 in bulk heterojunction solar cells by spin coating. The active layer where the polymers P1-P4 blended with PCBM in a weight ratio of 1:1. Under the standard AM 1.5G, the power conversion efficiency has been up to 0.49% (Jsc = 1.82 mA/cm2, Voc = 0.82 V,
目錄
中文摘要... i 英文摘要...ii 目錄...iii 圖目錄...vii 表目錄... ix 第一章 緒論... 1 1-1 前言... 2 1-2 太陽能電池簡介... 3 1-3 太陽能電池種類概述 ... 5 1-3-1 單結晶矽、非結晶矽、多結晶矽太陽能電池... 5 1. 單結晶矽太陽電池... 5 2. 多結晶矽太陽電池... 6 3. 非結晶矽太陽電池... 6 1-3-2 薄膜太陽能電池 ... 61-3-3 有機太陽能電池 ... 7
1-4 太陽光譜介紹... 8
第二章 文獻回顧及研究動機... 10
2-1 高分子有機太陽能電池發展概況 ... 11
2-2 高分子太陽能電池原理 ... 12
2-3 Low Bandgap 材料與 Tandem Cell ... 17
2-4 受體-給體系統(Donor-Acceptor System) ... 23 2-5 研究動機... 25 第三章 實驗部分... 28 3-1 實驗儀器... 29 3-2 實驗藥品及溶劑... 31 3-3 合成流程... 33 3-3-1 化合物 2 的合成 ... 33 3-3-2 化合物 4 的合成 ... 34 3-3-3 化合物 Monomer 1 的合成,代號 M1... 35
3-3-4 化合物的 Monomer 2 合成,代號 M2... 37 3-3-5 化合物 13 的合成 ... 40 3-3-6 化合物 Monomer 3 的合成,代號 M3... 43 3-3-7 化合物 Monomer 4 的合成,代號 M4... 46 3-4 高分子之聚合... 49 3-4-1 高分子 P1-P2 之聚合 ... 50 3-4-2 高分子 P3 之聚合... 50 3-4-3 高分子 P4 之製備... 51 第四章 結果與討論... 52 4-1 合成機制探討... 53
4-1-1 Stille coupling reaction... 53
4-1-2 Suzuki coupling reaction ... 54
4-2 高分子基礎性質... 55
4-3 高分子光物理性質之探討 ... 56
4-4 高分子電化學性質之探討 ... 60
參考文獻... 69
圖目錄
圖1 太陽能電池構造... 4 圖2 太陽電池發電原理... 4 圖3 各種類太陽能電池外觀... 5 圖4 太陽能電池之種類分類及效率... 7 圖5 太陽光能量光譜圖... 9 圖6 高分子太陽能電池的(a)發電原理(b)元件結構 ... 13 圖7 高分子太陽能電池結構示意圖及其 SEM 圖 ... 14 圖8 太陽能電池於照光下的電流-電壓(I-V)特性曲線 ... 15 圖9 高分子太陽能電池元件發電機制... 16 圖10 P3HT、MDMO-PPV、PCBM 與 AM 1.5G 太陽光譜比較圖... 18圖11 各類高效能 low band gap 材料結構圖 ... 20
圖12 (a) Poly-TPT、P3HT 及 PCBM 能階比較圖(b) poly-TPT:PC71BM 混摻 主動層TEM 圖(c) poly-TPT:PC71BM 混摻元件之光電轉換效率圖... 22 圖13 PCPDTBT/P3HT tandem cell 示意圖 ... 23 圖14 施體(D)與受體(A)的分子軌域交互作用... 25 圖15 本論文所合成出之單體... 27 圖19 各個高分子熱重分析圖... 56 圖20 各個高分子在 chlorobenzene 中的吸收光譜圖(10-6 M)... 58
圖21 各個高分子薄膜狀態吸收光譜圖... 59 圖22 各個高分子薄膜狀態放光光譜圖... 59 圖23 各個高分子的 HOMO 和 LUMO 能階示意圖... 61 圖25 太陽能電池元件結構圖... 63 圖26 各高分子 J-V 曲線圖 ... 65 圖27 各高分子之暗電流 J-V 曲線圖... 65
表目錄
表1 高分子基礎性質(GPC、DSC、TGA) ... 55 表2 高分子紫外-可見光(UV-Vis)吸收光譜、放光光譜(PL)數據 ... 58 表3 各個高分子電化學性質數據... 62 表4 高分子和 PCBM 經過 1:1 混摻後之元件量測數據 ... 63 表5 高分子 P1 和 PCBM 在 1:1(1 wt%:1 wt%),800 rpm 條件下之元件量測 數據... 661-1 前言
隨著人類文明迅速地發展,對能源之需求與依賴也與日俱增,傳統的 石油、天然氣及燃煤等石化能源,因為大量開採之故,不但很快地將面臨 枯竭的危機,近來價格更是劇烈波動。此外,燃燒石化能源後將產生二氧 化碳等廢棄物而造成溫室效應及酸雨等環境污染,因此目前全球對於再生 性(renewable)能源的重視也日漸提高,像是利用太陽能、生質能、地熱能、 海洋能或者風力...等項。其中太陽光電,是一種利用太陽電池將照射入太陽 的光能直接轉變成電能輸出的發電方式,由於它不易枯竭、潔淨無噪音、 較少或無排放二氧化碳、幾乎無地域限制,如此多的重要優勢使得科學家 們注意到太陽能利用的重要性 1。根據相關研究報告 2,推論目前地球石油 蘊藏量只能維持約 30 年、天然氣約 60 年、煤礦約 200 年,一般估計,太 陽每年照射到地球的能量約為目前人類所需電力的五十萬倍,因此如果能 夠充分利用太陽能,相信往後的能源問題皆可輕易解決。而最直接可將太 陽能轉換成能源的方式就是使用太陽能電池(solar cells),也稱為光伏打電池 (photovoltaic cells)。目前多數商品化的太陽能電池均以矽半導體材料製作。 依矽的晶體型態又可分為單晶、多晶及非晶矽等種類。單晶矽太陽能電池 的能量轉換效率很高且穩定,但成本十分昂貴;非晶矽元件效率則較低, 壽命也較短。在價格方面,雖然非晶矽元件比單晶矽太陽能電池便宜,但 比起一般的家庭用電成本,還是貴了許多。因此各國也積極地發展高效率和低成本的太陽能應用科技,期望藉由增加太陽能源的利用來減低對化石 能源的依賴性。
1-2 太陽能電池簡介
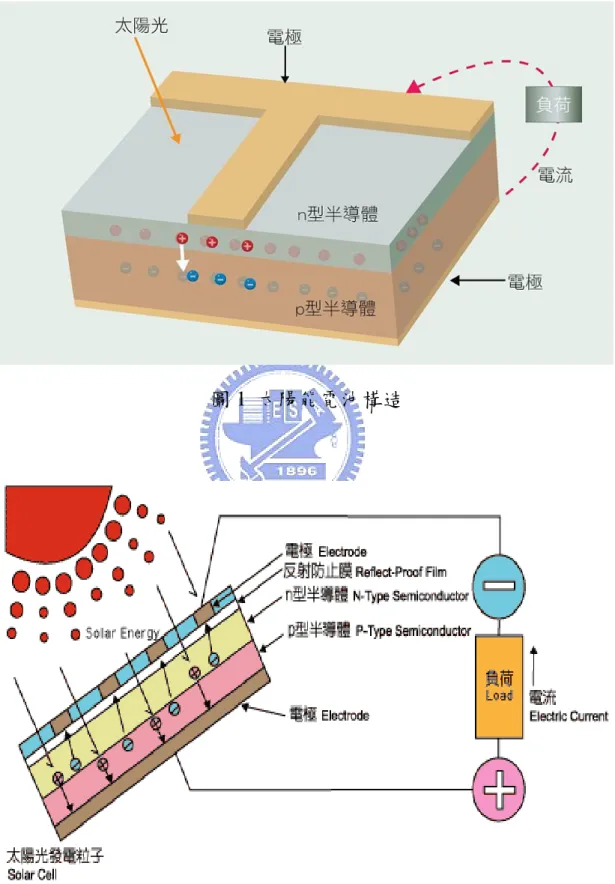
太陽電池(solar cell)是一種可以將能量轉換的光電元件,其基本構造是 運用P型與N型半導體接合而成的。半導體最基本的材料是「矽」,它是不導 電的,但如果在半導體中摻入不同的元素,就可以做成P型與N型半導體, 當P型及N型半導體互相接觸時,N型半導體內的電子會湧入P型半導體中, 以填補其內的電洞。在P-N接面附近,因電子-電洞的結合形成一個載子 空乏區(depletion area),而P 型及N 型半導體中也因而分別帶有正、負電 荷,因此形成一個內建電場。當太陽光照射到此P-N結構時,光子所提供的 能量可能會把半導體中的電子激發出來,產生電子—電洞對,電子與電洞 均會受到內建電位的影響,電洞往電場的方向移動,而電子則往相反的方 向移動,這整個程序稱為光伏效應(photovoltaic effect)3。若經由電極把電流 引出,形成一個迴路,這就是太陽能電池發電的原理(圖1)、(圖2)。圖1 太陽能電池構造
1-3 太陽能電池種類概述
1-3-1 單結晶矽、非結晶矽、多結晶矽太陽能電池 目前市場應用上大多為單結晶矽及非結晶矽,較多且廣泛,其原因有: 1.單結晶矽效率最高;2.非結晶矽價格最便宜,且無需封裝,生產也最快; 3.多結晶矽的切割及下游再加工較不易,而前述兩種都較易於再切割及加 工。 圖3 各種類太陽能電池外觀 1. 單結晶矽太陽電池4 單晶矽電池最普遍,多用於發電廠、充電系統、道路照明系統及交通 號誌等,所發電力與電壓範圍廣,轉換效率高,使用年限長,世界主要大 廠,如德國西門子、英國石油公司及日本夏普公司均以生產此類單晶矽太 陽能電池為主要,市場佔有率約五成,單晶矽電池效率從11%~24%,太空級(蒸鍍式) 晶片從16%~24%,當然效率愈高其價格也就愈貴。 2. 多結晶矽太陽電池 多晶矽電池的效率較單晶矽低,但因製程步驟較簡單,成本亦低廉, 較單晶矽電池便宜20%,因此一些低功率的電力應用系統均採用多晶矽太陽 電池。 3. 非結晶矽太陽電池 非晶矽電池為目前成本最低的商業化太陽能電池,且無需封裝,生產 也最快,產品種類多,使用廣泛,多用於消費性電子產品,且新的應用產 品不斷在開發中,其中以砷化鉀電池是最高效率的電池,但成本也最高。 1-3-2薄膜太陽能電池5 薄膜太陽能電池,乃是在塑膠、玻璃或是金屬基板上形成可產生光電 效應的薄膜,厚度僅需數μm,因此在同一受光面積之下可較矽晶圓太陽能 電池大幅減少原料的用量,厚度可低於矽晶圓太陽能電池90%以上,目前 轉換效率最高以可達13%。薄膜太陽能電池除了平面之外,也因為具有可 撓性可以製作成非平面構造其應用範圍大,可與建築物結合或是變成建築 體的一部份,在薄膜太陽電池製造上,則可使用各式各樣的沈積(deposition) 技術,一層又一層地把P型或N型材料長上去,常見的薄膜太陽電池有非晶 矽、CuInSe2 (CIS)、CuInGaSe2 (CIGS)、和CdTe..等。
1-3-3 有機太陽能電池 有機太陽能電池發展的潛力在於成本低廉、製程簡便、易於大面積化 以及重量輕可具撓曲性(flexibility),並且耐摔、耐衝擊、低成本,也可製作 在軟性塑膠或薄金屬基板上,然而此系列的太陽能電池至今尚未量產之主 因是在於其轉換效率太低以及穩定性的問題,因此,提高轉換效率則成為 目前研究的首要目標。有機太陽能電池大致可分為以下四大類:有機高分 子太陽能電池、有機⁄無機奈米複合材料太陽能電池、有機C60奈米複合材料 太陽能電池、染料敏化太陽能電池。本論文中所開發出的高分子太陽能電 池材料即屬於有機太陽能電池種類的一種,較深入的說明將於後段文章內 容詳敘。 圖4 太陽能電池之種類分類及效率6
1-4 太陽光譜介紹
太陽能電池的功用是將太陽光能轉換成電能,所以太陽光譜對太陽能 電池的效率有相當大的影響,也因此在這裡也需要簡單的介紹一下太陽光 譜。太陽表面溫度接近6000 K,因此其放射光譜幾乎等同於該溫度下的黑 體輻射,並且光譜照射並無方向性,地球與太陽相距約1億5千萬公里遠, 能到達地球表面的光子,幾乎只有正向入射至地球表面的光譜所貢獻,到 達地球大氣圈表面的光譜輻射能量定義為太陽常數(solar constant),其數值 大約1.353 kW/m2,因此大氣圈外的太陽光譜定義為AM 0,其中大氣質量(air mass, AM)用來估量因為大氣層吸收後,所導致影響太陽光譜表現與總體能 量值,而這些能量值亦是地球表面應用的太陽電池元件所能運用的,而大 氣質量的計算方法是使用air mass = 1/cosθ 來計算,其中θ = 0所代表的是太 陽光線從頭頂上方直射下來,而由上述的計算式中可知,地球表面用以衡 量太陽光譜的大氣質量值是大於等於1,目前被慣以使用的太陽光譜AM 1.5,即是太陽光入射角偏離頭頂46.8度,當太陽光照射到地球表面時,由 於大氣層與地表景物的散射與折射的因素,會多增加20% 的太陽光入射 量,抵達地表上所使用的太陽電池表面,其中這些能量稱之為擴散部份 (diffusion component),因此針對地表上的太陽光譜能量有AM 1.5G (Global) 與AM 1.5D (Direct)之分,其中AM 1.5G 即是有包含擴散部分的太陽光能 量,而AM 1.5D則沒有。圖五表示的即是大氣圈外(AM 0)與地表上(AM 1.5)太陽光能量光譜。太空用的太陽電池元件電性量測所使用的標準光譜是採 AM 0,而地面上應用的太陽電池元件電性量測所使用的標準光譜,依其應 用性之不同,可採用AM 1.5G 或是AM 1.5D,其中AM 1.5G光譜的總照度 為963.75W/m2,而AM 1.5D光譜的總照度為768.31W/m2,在量測計算應用 上方便,常會將此二值做normalize至1000 W/m27。 太陽輻射的光譜主要是以可見光為中心,波長從0.3 μm 的紫外光到數 微米的紅外光是主要的分布範圍。對於太陽光的模擬,本研究採用汞燈(Hg lamp)做為光源作為模擬太陽光之光源。 圖5 太陽光能量光譜圖
2-1 高分子有機太陽能電池發展概況
在1977年MacDiarmid與A. J. Heeger利用電化學和化學法摻雜聚乙炔
(poly -acetylene)而使其導電度提升109倍8。因而開啟了各種不同共軛導電高
分子的研究,如:poly p-phenylene vinylene (PPV)、polythiophene (PT)、 polypyrrole (PPy)、polyaniline (PAn)等。
導電高分子太陽能電池首先沿襲LED製程,最初以雙層(donor-acceptor bilayer)有機化合物建構donor-acceptor活性層(active layer)。1979年,由美國 柯達公司的C. W. Tang 研究團隊,利用copper PC及perylene衍生物所組成
9。其主要貢獻在於將異相體(heterojuction)之觀念導入電池製程,為現階段
有機小分子太陽能電池之始祖,然當時的元件效率太低,因而商業價值較 低 。 而 第 一 個 具 備 較 高 效 率 的 高 分 子 有 機 太 陽 能 電 池 為University of California, Santa Barbara Alan Heeger 研究團隊所發表,其使用MEHPPV/C60
bilayer hetrojucton 的概念,製作出第一個高分子碳材太陽能電池10。接著 1995 年 在 Science 期 刊 發 表 bulk hetero-junction (BHJ) 為 主 要 製 程 的 MEH-PPV/C60 太陽能電池11後,此類型的太陽能電池才真正受到重視並發
展其高分子/碳材太陽能電池原型。迄今高分子有機太陽能電池仍沿襲此 BHJ觀念,以搭配在導電高分子/碳材上的變化來提昇效率。
2-2 高分子太陽能電池原理
高分子太陽能電池的特點為,光主要由donor材料conjugated polymer吸 收,由於共軛高分子材料具高的吸收係數,因此其元件的厚度為100 nm (polycrystalline CuInSe, CdTe:1 μm、crystalline silcon: 100 μm),為最輕薄 的太陽能電池。光電轉換詳細作用機制如圖6所示,利用donor-type材料與 acceptor-type材料進行混摻,藉由太陽光的照射,以產生電子與電洞對 (electron/hole pair),最後電子與電洞分離,並分別經由電子與電洞傳導材 料,傳輸至陰陽電極而形成電壓降,以產生電能。由於有機半導體材料的 exciton有較高的束縛能(binding energy)約在0.2~1.0 eV,而無機材料的 binding energy約在0.015 eV,其束縛能與有機半導體材料相比,約大上一兩 個order,故於室溫條件下,有機材料無法形成自由的電子或電洞,必須藉 由N型 與P型材料界面的勢能差,才能達 到電子與電洞分離的效果。
圖6 高分子太陽能電池的(a)發電原理(b)元件結構
目前最常見的有機混成太陽光電系統,主要採A. J. Heeger 與F. Wudl 所設計的BHJ結構,元件結構如圖6(b)所示。圖7為高分子碳材太陽能電池 之元件結構的SEM圖,由圖中可以更清楚的了解到其元件構造。以高分 poly(3-hexylthiophene) (P3HT)為p-type而[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) 為 n-type 材 料 所 組 成 的 主 動 層 , 配 合 ITO 基 材 與 poly(3,4-ethylenedioxythiophene)poly(styrenesulfonate) (PEDOT: PSS)組成的 陽極及以Al陰極所構成。其結構看似簡單,然而不同層材料的選擇,皆有 其限制與功用。
圖 7 高分子太陽能電池結構示意圖及其 SEM 圖
當中ITO為照光面的透明電極材料,必需具備高導電度(<20 Ω/□)及高穿透 度功能(於可見區域穿透度T% > 85%)。而PEDOT:PSS 主要功能,為修 飾ITO的work function(減少hole injection barrier,使電洞傳導效率提昇), 並 使 基 板 平 坦 化 , 另 外 亦 扮 演 電 子 阻 檔(electron blocking) 的 角 色 。 ITO/PEDOT及Al電極的選擇,亦是在能階考量下所搭配出,如圖6(a)所示, ITO/PEDOT 的 work function 必 須 配 合 P3HT 的 HOMO (highest occupied molecular orbital)能階,才能有效的將電洞引導出來,而Al的選擇也是符合 由PCBM的LUMO (lowest unoccupied molecular orbital)萃取出電子需求的緣 故。另外如圖7所示,一般亦會於主動層(active layer)上,鍍上LiF或Ca,而 藉由這層材料的導入,可以增進electron injection,並幫助效率的提昇。 太陽能電池的特性與二極體的特性相似,一般稱為IV曲線特性,如圖
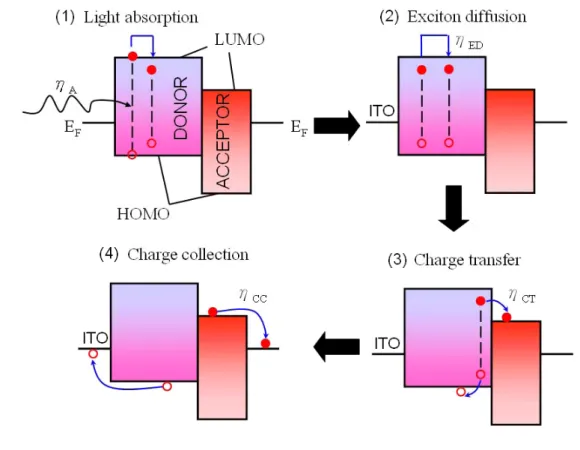
8所示,決定太陽電池能源轉換效率的基本特性參數:短路電流(Isc)、開路 電壓(Voc)、充填因子(FF),及最大功率(Pmpp),此IV曲線會因光照度及環境 溫度影響它的發電量。一般標準的測試條件為1000 W/m2 照度、1.5G 空氣 質量、25°C 室溫,若日照度越強,則發電量越大12。一般說來,元件的光 電轉換效率可以下式(1)表示: Efficiency η = VOC * ISC * FF / Pinput (1) FF = (I * V)max/(ISC * VOC) 其中, Voc:開路電壓、 Isc:短路電流、FF:Fill Factor 。 圖8 太陽能電池於照光下的電流-電壓(I-V)特性曲線 為達到元件應用的基本需求,其小型元件之光電轉換效率需達10% , 以此為目標,對BHJ元件的深入檢視是必需的。若細分發電過程,主要可分 四個部份13(圖9):(1)主動層材料吸收太陽光,將電子從HOMO能階位置, 激發至LUMO能階形成excitons;(2)激子(excitons)傳導(excitons朝donor及
acceptor界面擴散);(3)在donor及acceptor界面產生電荷分離(光致電荷轉 移(photo-induced charge transfer));(4)主動層中,charge carriers路徑選擇, donor負責傳送電洞,而acceptor負責傳送電子,以確保電子/電洞持續的被 傳導至電極,並維持電子/電洞傳輸路徑管道暢通與減少電子/電洞再結合的 機率。
圖9 高分子太陽能電池元件發電機制
(1)在第一個步驟,可改進的方向為推升主動層材料的吸光效率與吸光範 圍,如使用low bandgap材料及tandem cell概念的導入,皆能有效的提昇光電 流密度(Jsc);(2)由於exciton擴散距離(diffuse length)為10 nm左右,於第二步
離,如此將有助於提昇有效的exciton濃度,繼而提昇整體元件的效率;(3) exciton遇到界面時,其電荷轉移速度(~p sec)遠短於其它的競爭機制(~μ sec),故此光致電荷轉移過程,並不會伴隨能量損失,故只要exciton能遇到 D/A界面,便能有效使exciton游離成自由的電子與電洞;(4)最後一個步驟, 即自由離子的傳導,亦是最為複雜的過程,首先材料必須擁有高且平衡的 mobility,主動層材料的電子/電洞mobility需達到平衡,才能避免space charge 的累積,然一般而言donor (polymer)與acceptor (PCBM)其mobility相比,約 差1~2個order,因此提昇donor材料的hole mobility,亦為一重要課題。此外 產生的自由離子,亦須沿著連續的路徑傳遞(continued pathway),避免自由 離子於傳送過程,再結合(recombination)機率的上昇,通常這過程可由混摻 溶劑選擇、退火(annealing)等製程步驟來控制。以整體元件而言,各層材料 間介面的關係,亦是必須掌握的問題,不良的介面除了使元件的series resistivity提高,也將使fill factor及光電流降低。綜觀上述要達到元件的極致 表現,必須要對這些層次的問題有更深度的瞭解。
2-3 Low Bandgap 材料與 Tandem Cell
Low bandgap導電高分子材料開發,一直是高分子太陽能電池所致力的 目 標 , 如 圖 10 所 示 , 目 前 最 好 的 高 分 子 材 料 為 P3HT 及 MDMO-PPV(poly[2-methoxy-5-3(3,7-dimethyloctyloxy)-1-4-phenylene vinyle
部分的光無法被吸收,若能將主動層材料的bandgap縮小,將可幫助吸收更 多太陽能光譜,繼而產生高的光電轉換效能。
圖 10 P3HT、MDMO-PPV、PCBM 與 AM 1.5G 太陽光譜比較圖
然而並非所有low bandgap 材料皆有較好的光電轉換效率,這些材料必須兼 顧優良物性,如高的吸光係數(Film>105cm-1 at λmax)及高的hole mobility
(>10-4cm2 V-1s-1 (field effect transitor)),加上能階(HOMO與LUMO須符合碳 材與電極的work function系統)及元件製程配合下(即導電高分子和碳材混 摻的morphology控制)),才能擁有優良的元件效率。在上述因素缺一不可情 形下,絕大部份發展的low bandgap導電高分子,在元件效率表現皆不盡理 想(<<1%)。瑞典Linkoping大學Inganas教授研究群最早發表具較高光電轉換 效 率 的l o w b a n d g a p 材 料 1 8 , 利 用 混 合 溶 劑 系 統 成 功 地 將 APFO-3(poly(2,7-(9,9-dioctyl-fluorene)-alt-5,5-(4’,7’-di-2thienyl-2’,1’3’-benzo
thiadiazole))與PCBM混合,製作出效率為2.8%的low bandgap太陽能電池, 此效率雖距離P3HT/PCBM系統5%效率還有段差距,但已打破以往low band gap導電高分子效率低於1%的記錄,同時使low bandgap導電高分子材 料在太陽能電池中樹立了一個新的里程碑。圖11整理目前文獻所報導效能 與P3HT/PCBM相似的low bandgap材料19-23,如Alan J. Heeger團隊所開發的 PCDBTBT材料,效率有5.5%的表現,PCDBTBT的bandgap約為1.55 eV,但 可有效的吸收太陽光至800 nm的波長,藉由形態的控制,其元件的Jsc可提
高至16 mA/cm2 的水準。新材料的開發必須兼具整體光電流與電壓之間的 Off-set考量,不因電流的提昇而過度犧牲 Voc; 且donor-acceptor的 band
structure (HOMO-LUMO)乃至所對應的金屬電極的功函數(work function), 都需要仔細調整才能提昇元件的開路電壓。
S S * N S N * n N S N S S Pt * P(C4H9)3 P(C4H9)3 n S S C6H13 C6H13 S C6H13 C6H13 N S N S * * m n S N S N S * * n APFO-3 Pt-metallopolyyn PCPDTBT P-(TPT)
圖 11 各類高效能 low band gap 材料結構圖
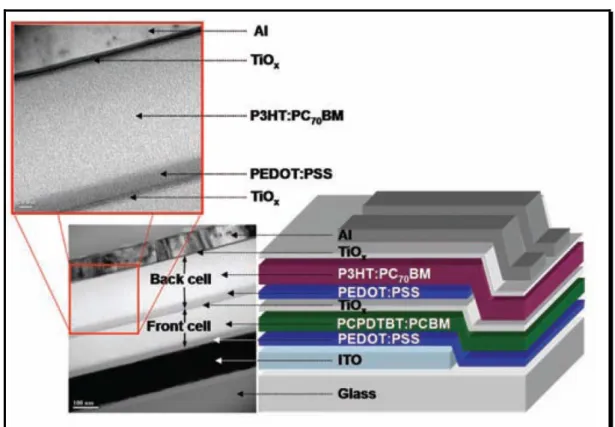
以 工 研 院 材 化 所(MCL/ITRI) 發 表 的 poly-TPT 為 例圖12,poly-TPT 材 料 bandgap約為1.7 eV,利用循環伏安法,可從氧化還原電位,推算出其對應 HOMO 與 LUMO 值 , 並 與 PCBM 的 HOMO/LUMO 相 比 , 可 判 定 poly-TPT/PCBM在高分子太陽能電池的適用性。如圖12(a)所示poly-TPT其 HOMO與LUMO分別為-5.4與-3.7eV,其LUMO與PCBM的LUMO相比約有 0.6 eV的能階差,能有效的達成exciton解離。觀察poly-TPT的HOMO值,
與P3HT相比,具備較低之HOMO值,故poly-TPT材料擁有較P3HT穩定之能 階,理論上來說,poly-TPT半導體材料,較不易受氧氣的doping,繼而失去 半導體特性,並使元件效率下降。另外較低的HOMO亦有提高元件開路電 壓的優點,從光激發(photoluminescence)光譜中,觀察poly-TPT/PCBM混摻 薄膜,poly-TPT的發光會被PCBM所淬息(quenching),即材料間產生 光致電荷轉程現象,由上文所介紹的發電原理中,此行為即是材料吸光後 不再產生會發光的exciton,而是產生有效的自由電子與電洞。一般元件的 製程可藉由溶劑的選擇、spin-coating轉速及annealing溫度/時間調控元件主 動層的形態(morphology),使用原子力顯微鏡(AFM)或穿透式電子顯微 鏡(TEM),可觀察主動層形態的變化,理想的主動層形態為polymer與PCBM 形成優良的微觀相分離,使donor與acceptor能有足夠的面積,產生大量的光 致電荷轉程(自由電洞/電子),且在donor與acceptor材料能形成網狀互穿 結構情形下,使電子與電洞沿著各自管道,傳送到對應電極。圖12(b)為混 摻 後Poly-TPT 材 料 的 TEM 圖 , 黑 色 區 塊 為 PCBM-rich , 而 白 色 為 poly-TPT-rich 的 區 塊 , 由 圖 可 以 觀 察 到 PCBM 均 勻 分 散 在 poly-TPT 的 matrix,其domain大小約在20 nm左右,此形態為不錯的主動層材料形態。
圖12(c) Poly-TPT : PC71BM混摻元件之光電轉換效率,效率為4.4%,當中
圖 12 (a) Poly-TPT、P3HT 及 PCBM 能階比較圖(b) poly-TPT:PC71BM 混摻
主動層TEM 圖(c) poly-TPT:PC71BM 混摻元件之光電轉換效率圖
除了使用新材料來提昇元件效率外,學者亦從製程角度來改變元件的效 率,如利用tandem結構,以multiple device的stacked結構設計,來達成Voc及
Isc增加的目的,配合sping coating時所用的溶劑不同,而達到保護下層薄膜
的目的,如UCLA的Prof. Yang Yang研究團隊24選用MEH-PPV/PCBM,以串 聯形成製作的stacking cell,可將電池之Voc 提升到1.3 V以上。Heeger等人更
TiOx與材料系統能階配合,在ITO/PEDOT/PCPDTBT/TiOx/PEDOT/
P3HT/TiOx/Al的元件結構下,利用PCPDTBT 吸收紅外光,而P3HT吸收可 見光,達到高的光電轉換效率。
圖13 PCPDTBT/P3HT tandem cell 示意圖
2-4 受體-給體系統(Donor-Acceptor System)
能隙(band gap energy:Eg)是一種介於最高填滿的分子軌域(highest occupied molecular orbital : HOMO) 和 最 低 未 填 滿 的 分 子 軌 域 (lowest unoccupied molecular orbital:LUMO)之間的能量間隔,其決定共軛高分子
的 導 電 性 。 若 降 低 高 分 子 的 能 隙 將 會 增 進 傳 導 帶 的 熱 量 總 數(thermal
metals)。另一方面,窄能隙有較低的氧化電位時,將會導致參雜狀態穩定, 因此藉由化學修飾高分子來控制HOMO-LUMO 之間的間隙,對於窄能隙共 軛高分子來說是極其重要的。 共軛高分子的電性和光學性質是來自位於 HOMO 和 LUMO 周圍的能 階所影響的。根據能帶理論來說,最高填滿帶來自於每個單體的 HOMO, 也就是所謂的價帶;而最低未填滿帶則是來自於每個單體的的 LUMO,可 當成傳導帶。在共軛高分子中,能帶的形成是來自於聚合時每個單體的參 與 26。在吸收光譜裡若要得到共軛高分子的 Eg,則是將吸收峰進行外插法 而得,因此要得到窄能隙的共軛高分子,其吸收邊緣(absorbtion edge)應在 近紅外光區(near infrared region),也就是吸收長波長。
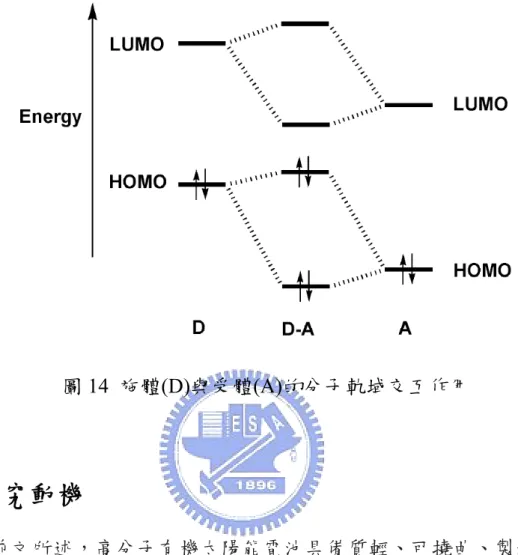
到目前為止,許多分子工程策略用來設計窄能隙的共軛高分子已發展 多年 27,本論文中的所合成之共軛高分子之一就是選用施體-受體的系統 (donor-acceptor)的概念來降低能隙。而 donor-acceptor 系統中合成策略是利 用強推電子部分和強拉電子部分進行結合並以一種連續的單元形成共軛高 分子,使得單元和單元之間的鍵結扭轉程度降到最低。由於donor 和 acceptor 之間的作用力,使得單元之間的鍵結更具有雙鍵的特性,讓窄能隙共軛高 分子的主鏈上形成類似醌環的穩定結構。 近年來的分子軌域計算呈現 donor 和 acceptor 之間的能階經過混成後,
和acceptor 部分的 LUMO 靠的很近的話,就會形成窄能隙,如圖 14:
圖14 施體(D)與受體(A)的分子軌域交互作用
2-5 研究動機
如前文所述,高分子有機太陽能電池具備質輕、可撓曲、製程環保、 低成本及應用性佳等優點,因此成為具有潛力的新興太陽能電池。Dr. Serdar Sariciftci 研 究 團 隊 於 2001 年 所 發 表 的 MDMO - PPV : PCBM blend
(polymer/C60)27,也成為近幾年有機高分子C60奈米複合材料太陽能電池
的技術基礎。由於polymer/fulleren複合物有很高的電子遷移率,同時可透
過自組裝(self-assembly)的方式,形成PPV-PS(C60) copolymer奈米薄膜,不
須經過燒結程序,更適合製作在塑膠基板上,不過,由於發展的歷史並不 長,因此發電效率偏低。
在本論文中,實驗主要目的在於合成出具有電子予體功能之共軛高分 子,以及具有電子予體與受體混成軌域之窄能隙共軛高分子,進一步與 PCBM混摻,期望在太陽能電池轉化效率上能夠突破以往的有機高分子太 陽能電池。在電子予體的部分,我們引入了2,7-carbazole取代以往共軛性不 錯的fluorene分子,原因除了carbazole分子上的N原子具有良好的電子捐贈
(electron-donating)能力28之外,近幾年Dr. Mullen以及Dr. Leclerc等研究團隊
也發表了一系列以2,7-carbazole為基礎之高分子延伸物,在太陽能電池轉換 效率方面皆有不錯的表現29-30;而與fluorene相比,carbazole分子則為fully aromatic,也因此可以提供較佳的穩定性。另一方面,林宏洲老師實驗室在 2009年所發表的文獻31中,包含一了系列含有融合環(fused ring)的窄能隙共 軛高分子,其中含有thiophene片段的雜環,因其有共平面的結構較易於堆 疊,因此能夠增加π−π stacking,進而使其化合物擁有較高的電洞遷移率
(hole mobility)32,除此之外,較低的能隙(band gap),加上應用於太陽能電
池 有 不 錯 的 元 件 轉 換 效 率 , 近 幾 年 為 科 學 家 們 所 喜 愛 , 諸 如 : cyclopentadithiophene33、thienothiophene34...等,可見此類化合物確實能夠成 為 良 好 的 有 機 太 陽 能 電 池 材 料 ; 而 本 論 文 中 引 入 的 片 段 為fused dithienothiophene,除了上述的優點之外,此化合物在n-doping及p-doping 狀態時,具有良好的穩定性35;加上其片段在高極性的溶劑之中有較低的 自由能36,也因此這類化合物在光伏效應產生的同時,擁有較佳的再生能
力。 而在電子受體方面,本論文引入的是benzothiadiazole 片段,希望能夠 藉由施體與受體之間的作用力,使得單體與單體之間的鍵結更具有雙鍵的 特性,而不容易扭轉,將有利於載體的傳導,而此D-A effect 當然也能夠使 吸收光譜範圍更為廣泛。同時,我們也引入了八個碳以及六個碳的長烷基 鏈於 2,7-carbazole、dithienothiophene 單體,以增進分子的溶解度以及成膜 性,避免因為有機薄膜不平整,在元件製作時容易發生的短路現象。 N C8H17 C8H17 O O B O O B Br Br N O C C6H12
M2
O O S S S C6H13 C6H13 S S Br BrM3
S S S C6H13 C6H13 S S S C6H13 C6H13 N S N Br BrM4
M1
圖15 本論文所合成出之單體3-1 實驗儀器
1、真空系統 (Vacuum Line & Schlenk Line)
2、核磁共振光譜儀 (Nuclear Megnetic Resonance):Burker AC-300 型。將
sample 溶於 d-solvent 中,利用所測得 1H 與 13C 光譜判斷化合物之結構
與純度。化學位移單位為 ppm,偶合常數單位為 Hz,並以 d-solvent 值為 內標 (d-CHCl3 1H: δ = 7.24 ppm, 13C: δ = 77 ppm)。s 代表 singlet, d 代表
doublet, t 代表 triplet, m 代表 multiplet。
3. 紫 外 光 - 可 見 光 - 近 紅 外 光 光 譜 儀 (Ultraviolet-Visible-Near Infrared Spectrophotometer):Lambda 950型。紫外光-可見光吸收光譜可以用來偵測 軌域中之電子被激發而產生躍遷的情況,所以放射光的波長和物質內的電 子結構有關。操作方法為先將溶劑放入方形的石英管且放入儀器的樣品槽 中,並設定好儀器參數作基線掃描,再將溶劑倒掉,放入樣品溶液進行掃 描。 4. 螢光光譜儀 (Fluorescence Spectormeter):Hitachi F-4500 型。螢光光譜可 以用來偵測發光團分子之電子被激發後以發光的型式回到基態的情況,所以 發射光的波長和物質內的電子結構有關。操作方法為配置適當濃度並設定其 激發波長,再放入螢光光譜儀掃描。 3.循環伏安電化學儀 (Cyclic Voltammeter,CV):AutoLab型。先配置電解質 溶液為0.1M tetra-n-butylammonium hexafluorophosphate的無水乙睛溶液,再
將各個高分子配置成10 mg/1 ml的氯苯溶液。本次實驗所用到的CV是屬於 三電極式,工作電極是Pt薄膜,參考電極是Ag/AgCl參考電極,輔助電極是 Pt線,而量測時先將高分子溶液滴加在Pt薄膜上,等自然風乾成薄膜後,再 放入電解質溶液裡,並用100 mV/s的掃描速率去進行電化學的量測。我們 可以由循環伏安法來得知高分子的氧化還原電位、最高填滿分子軌域 (HOMO)、最低未填滿分子軌域(LUMO),並利用HOMO和LUMO的電位相 減進而得知電化學能隙。
5.示差掃描熱量計 (Differential Scanning Calorimeter, DSC):Perkin Elmer Pyris 7 型。DSC 是分析熱向性液晶之熱力學性質之有效利器。使用儀器前 先作儀器校正,接著將待測之樣品稱重在 1.5~5.0 mg 之間,將其裝在金屬 鋁盤中加蓋密閉後便可進行量測,並從吸熱或放熱的熱分析圖形,得其熱 焓值大小,及相轉變時的溫度。
6.凝膠滲透層析儀 (Gel Permeation Chromatography,GPC):Waters 2414型。 7.紅外線光譜儀 (Infrared Spectrometer, IR):Perkin-Elmer Spectrum 100 型。
8. 質譜儀 (Mass Spectrometer):Micromass Trio-2000型。用FAB (Fast Atom
Bombardment)或 EI (Electron Ionization)來將樣品離子化。此外,由於分子量 較大的分子非常難以被離子化,在其離子化的過程中很容易就分散掉了,因
此必須使用MALDI-TOF (Matrix-Assisted Laser Desorption/Ionization-Time
9.元素分析儀 (Elemental Analyzer):Perkin-Elmer 240C 型。由交通大學貴重 儀器中心代測樣品。
3-2 實驗藥品及溶劑
藥品名稱 廠牌 Bromine MERCK Butyllithium 2.5M Chemetall Benzo[c][1,2,5]thiadiazole ACROS 4-Dimethylaminopyridine Aldrich 4,4′-Dibromobiphenyl ACROS 1,6-Dibromohexane Aldrich Dimethylsulfoxide GRANDMagnesium sulfate anhydrous SHOWA
2-Methylpropan-2-ol ACROS NH4Cl Aldrich N-Bromosuccinimide Fluka N,N′-Dicyclohexylcarbodiimide Fluka Triphenylphosphine ACROS TsCl Aldrich Thiophene Aldrich Tributyl(thiophen-2-yl)stannane ACROS Triethylamine GRAND
Tri-n-butyltin chloride ACROS
Tetrakis(triphenylphosphine)palladi um(0)
Ultra Fine Chemical Technology Corp.
Potassium hydroxide SHOWA
Potassium carbonate SHOWA
Potassium iodide SHOWA
Pyridinium chlorochromate Aldrich
Phenylbronic acid Landcaster
Sodiun bisulphite SHOWA
4-Hydroxybenzoic acid ACROS
Heptanal Alfa Aesar
Hydrochloric acid 37% Fisher Scientific
Hydrobromic acid ACROS
Ethyl 2-sulfanylacetate TCI
Ethyl formate Aldrich
Copper powder MERCK
Chloroform GRAND
Octylmagnesium bromide Aldrich
o-Dichlorobenzene TEDIA
Me3N‧HCl SHOWA
Lithiun hydroxide Aldrich
Fuming nitric acid 聯工化學
Quinoline ACROS 2-Isopropoxy-4,4,5,5-tetramethyl-1,
3,2- dioxaborolane Aldrich
Iodobenzen ACROS
反應用的溶劑,如:acetone、dichloromethane、hexane、methanol、toluene、
N,N-dimethylformamide(DMF)、tetrahydrofuran(THF)、ethyl acetate (EA)
3-3 合成流程
3-3-1 化合物 2 的合成 Br Br HNO3 HOAc, 100℃ Br Br NO2 PPh3 o-DCB, reflux Br Br N H (1) (2) a. 4,4′-Dibromo-2-nitrobiphenyl (1)將4,4′-dibromobiphenyl (5.0 g, 16.1 mmol) 溶於 75 mL 的 HOAc 中加
熱至100 ℃,待起始物完全溶解後,緩慢滴入 HNO3 (23.2 mL)持續約兩小 時,降溫至室溫,加入大量的水會有淡黃色固體產生,以抽氣過濾裝置過 濾,再以 EtOH 再結晶,析出後即可得到淡黃色色固體化合物 4.0 g, 產率 70%。 1H NMR(CDCl 3, 300 MHz): δ 8.01 (d, J = 2.0 Hz, 1H), 7.74 (dd, J = 8.2、 2.0 Hz, 1H), 7.54 (d, J = 8.6 Hz, 2H), 7.27 (d, J = 8.2 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H)。 b. 2,7-dibromo-carbazole (2)
將4,4′-Dibromo-2-nitrobiphenyl (13.0 g, 36.8 mmol)、PPh3 (24.1 g, 2.5 eq)
置入雙頸反應瓶中,真空抽換氮氣數次,加入 o-DCB (5.6 mL, 每 mmL
4,4′-Dibromo-2-nitrobiphenyl, 2mL)溶解,加熱回流 12 小時。待反應完全後
冷卻至室溫,以 CH2Cl2和大量水萃取數次,收集有機層,經無水硫酸鎂除
hexane:CH2Cl2 (3:1)之混合液為沖堤液,得到白色固體化合物 9.4 g,產率為 79%。 1H NMR(CDCl 3, 300 MHz): δ 8.02 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.54 (s, 2H), 7.33 (dd, J = 8.4、 1.5 Hz, 2H)。 3-3-2 化合物 4 的合成 O EtO H C8H17MgBr THF OH C8H17 C8H17 TsCl MeNHCl , Et3N CH2Cl2 OTs C8H17 C8H17 (3) (4) c. Heptadecan-9-ol (3)
將ethyl formate (2.17 mL, 34.4 mmol)置入雙頸反應瓶中,真空抽換氮氣
數次,加入適量溶劑THF (55.0 mL),在-78 ℃下預冷 10 分鐘,octylmagnesium
bromide (52.0 mL, 2.0 M in diethyl ether, 3.0 eq) 緩慢加入反應瓶中,在室溫
下攪拌24 小時後,會有白色固體產生,在冰浴下緩慢加入飽和 NH4Cl 水溶 液,接著以CH2Cl2 和水萃取數次,收集有機層,經無水硫酸鎂除水,過濾 後濾液以迴旋濃縮儀抽除有機溶劑,可得白色固體的產物6.8 g, 產率 77%。 1H NMR(CDCl 3, 300 MHz): δ 3.57-3.54 (m, 1H), 1.47-1.23 (m, 28H), 0.86 (t, J = 6.5 Hz, 6H)。
d. Heptadecan-9-yl 4-methylbenzenesulfonate (4)
將Heptadecan-9-ol (1.0 mL, 3.9 mmol)與 Me3N‧HCl (0.37 g, 1 eq)置入
三頸反應瓶中,TsCl (0.93g, 1.3 eq) 以彎管放置,真空抽換氮氣數次,加入 適量溶劑CH2Cl2 (10.0 mL)後,再加入 Et3N (1.36mL, 2.5 eq),冰浴下將 TsCl 緩慢加入,在室溫下攪拌3 小時後,以 CH2Cl2 和水萃取數次,收集有機層, 粗產物再以矽膠管柱層析純化,以hexane:EA (10:1)之混合液為沖堤液,得 到無色油狀產物1.12 g,產率為 70%。 1H NMR(CDCl 3, 300 MHz): δ 7.80 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 4.58-4.50 (m, 1H), 2.43 (s, 3H), 1.58-1.42 (m, 4H), 1.28-0.82 (m, 24H), 0.94-0.81 (m, 6H)。 3-3-3 化合物 Monomer 1 的合成,代號 M1 Br Br N C8H17 C8H17 (5) 1) BuLi, THF, -78℃ 2) O O B O N C8H17 C8H17 O O B O O B (M1) e. 2,7-Dibromo-9-(heptadecan-9-yl)-9H-carbazole (5)
將2,7-dibromo-carbazole (0.59 g, 1.8 mmol)與 KOH (0.51 g, 5 eq) 放入
圓底燒瓶中,真空抽換氮氣數次,加入DMSO (5mL),再將 heptadecan-9-yl
4-methylbenzenesulfonate (1.11 g, 1.5 eq)溶於 DMSO (5mL)慢慢滴入反應瓶
大 量 水 萃 取 數 次 , 收 集 有 機 層 , 粗 產 物 再 以 矽 膠 管 柱 層 析 純 化 , 以 hexane:CH2Cl2 (2:1)之混合液為沖堤液,得到白色固體產物 0.68 g,產率為 67%。 1H NMR(CDCl 3, 300 MHz): δ 7.88 (d, J =7.9 Hz, 2H), 7.67 (s, 1H), 7.52 (s, 1H), 7.31 (d, J =7.8 Hz), 4.43-4.36 (m, 1H), 2.18-2.16 (m, 2H), 1.94-1.87 (m, 2H), 1.31-1.12 (m, 22H), 0.88-0.79 (m, 8H)。 f. 9-(Heptadecan-9-yl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan- 2-yl)-9H-carbazole (M1) 將2,7-dibromo-9-(heptadecan-9-yl)-9H-carbazole (3.62 g, 6.42 mmol)置 入雙頸反應瓶中,真空抽換氮氣數次,加入適量溶劑THF (40.0 mL),在-78
℃下預冷10 分鐘,加入 n-BuLi (5.65 mL, 2.5 M in hexane, 2.2 eq)緩慢加入反
應 瓶 中 , 繼 續 在 -78 ℃ 持 續 保 持 60 分 鐘 後 , 將
2-isopropoxy-4,4,5,5-tetramethyl-1,3,2- dioxaborolane (3.15 mL, 2.4 eq)注入反
應瓶中,慢慢回到室溫後至12 小時,以乙醚 和飽和食鹽水萃取數次,收集 有機層,經無水硫酸鎂除水,過濾後濾液以迴旋濃縮儀抽除有機溶劑,粗 產物再以acetone:MeOH (1:10)再結晶,得到白色固體產物 2.69 g,產率 64 %。 1H NMR(CDCl 3, 300 MHz):
δ 8.12 (d, J = 8.0 Hz, 2H), 8.02 (s, 1H), 7.89 (s, 1H), 7.66 (d, J = 8.1 Hz, 2H), 4.73-4.66 (m, 1H), 2.35-2.30 (m, 2H), 1.98-1.90 (m, 2H), 1.39-1.12 (m, 48H), 0.82 (t, J = 7.0 Hz, 6H)。MS (FAB): m/z 544 (calcd [M]+). 3-3-4 化合物的 Monomer 2 合成,代號 M2 HO C OH O + HO DCC, DMAP THF HO C O O (6) HO C O O (6) + Br Br K2CO3, KI acetone O C O O (7) BrC6H12 + O C O O (7) BrC6H12 NaH THF Br Br N H (2) Br Br N O C C6H12 (M2) O O g. Tert-butyl 4-hydroxybenzoate (6)
將4-hydroxybenzoic acid (7.0 g, 50.7 mmol)與4-dimethylaminopyridine (0.62 g, 0.1 eq) 放入雙頸圓底燒瓶中,真空抽換氮氣數次,加入THF (60mL),再加入2-methylpropan-2-ol (60 mL, 2-methylpropan-2-ol為反應試劑
N,N′-dicyclohexylcarbodiimide (12.6 g, 1.2 eq)慢慢滴入反應瓶中,室溫下反 應至隔天,待反應完全後,以CH2Cl2和飽和食鹽水萃取數次,收集有機層, 粗產物再以矽膠管柱層析純化,以hexane:ethyl acetate (4:1)之混合液為沖堤 液,得到白色固體產物3.95 g,產率為40%。 1H NMR(CDCl 3, 300 MHz): δ 7.87 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 6.44 (s, 1H), 1.59 (s, 9H)。 h. Tert-butyl 4-(6-bromohexyloxy)benzoate (7)
將tert-butyl 4-hydroxybenzoate (3.95 g, 20.3 mmol)、K2CO3 (8.43 g, 3 eq)
與KI (0.2 g, 0.06 eq) 放入雙頸圓底燒瓶中,真空抽換氮氣數次,加入 acetone
(150 mL),再將 1,6-dibromohexane (29.18 g, 6 eq)加入反應瓶中,加熱至迴 流反應至隔天,待反應完全後回到室溫,先以減壓蒸餾移除溶劑,再以
CH2Cl2 和飽和食鹽水萃取數次,收集有機層,粗產物再以矽膠管柱層析純
化,以hexane: ethyl acetate (30:1)之混合液為沖堤液,得到無色油狀產物 6.48
g,產率為 89%。
1H NMR(CDCl
3, 300 MHz):
δ 7.89 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.98 (t, J = 6.4 Hz, 2H ), 3.40 (t, J = 6.8 Hz, 2H ), 1.90-1.77 (m, 4H), 1.56 (s, 9H), 1.51-1.46 (m, 4H)。
i. Tert-butyl 4-(6-(2,7-dibromo-9H-carbazol-9-yl)hexyloxy)benzoate (M2)
將NaH (0.36 g, 1.8 eq) 放入雙頸圓底燒瓶中,真空抽換氮氣數次,加
入 THF (50 mL),2,7-dibromo-carbazole (2.71 g, 8.34 mmol)先溶於適量
THF(10 mL),慢慢滴入反應瓶中,加熱迴流一個小時後,再將溶於 THF(10 mL)的 tert-butyl 4-(6-bromohexyloxy)benzoate (3.7 g, 1.3 eq)慢慢滴入反應瓶
中,加熱迴流反應至隔天,待反應完全後回到室溫,以 CH2Cl2和飽和食鹽 水萃取數次,收集有機層,粗產物再以矽膠管柱層析純化,以hexane: ethyl acetate (1:7)之混合液為沖堤液,得到白色固體產物 2.90 g,產率為 60%。 1H NMR(CDCl 3, 300 MHz): δ 7.90 (m, 4H), 7.53 (d, J = 1.2 Hz, 2H), 7.34 (dd, J = 9.0、1.6 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 4.22 (t, J = 7.2 Hz, 2H ), 3.96 (t, J = 6.29 Hz, 2H ), 1.91-1.75 (m, 4H), 1.59-1.44 (m, 13H)。MS (EI): m/z 601 (calcd [M]+).
3-3-5 化合物 13 的合成 S Br2 CHCl3 S Br Br Br Br 1) BuLi, THF, -78℃ 2) C6H13CHO S Br Br O C6H13 C6H13 O pcc CH2Cl2 HSCH2COOEt, K2CO3 DMF, 60℃ S S S C6H13 C6H13 EtOOC COOEt LiOH THF, reflux S S S C6H13 C6H13
HOOC COOH quinoline, 260℃
Cu S S S C6H13 C6H13 (8) (13) S Br Br OH C6H13 C6H13 HO (9) (10) (11) (12) j. Perbromothiophene (8) 將thiophene (10.0 mL, 0.2 mol)和 CHCl3 (12 mL)置入反應瓶中,緩慢滴 入Br2 (46.0 mL, 0.9 mole, 4.5 eq),在室溫下攪拌 24 小時後,使用亞硫酸氫 鈉水溶液除去多於的 Br2,加入 CH2Cl2萃取後,收集有機層,經無水硫酸 鎂除水,過濾後濾液以迴旋濃縮儀抽除有機溶劑,可得白色固體的產物(72.0 g, 90%)。 k. 1,1′-(3,4-Bromo-2,5-thienyl)diheptanol (9) 將perbromothiophene (6.17 g, 5.4 mmol) 置入雙頸反應瓶中,真空抽灌 dry THF(50 mL),在-78 ℃下預冷 10 分鐘,n-BuLi (12.35
mL, 2.5 M in hexane, 2.0 eq)緩慢加入反應瓶中,打入時間大約 30 分鐘,繼 續在-78 ℃持續保持 60 分鐘後,heptanal (4.41 mL, 2.05 eq),緩慢加入反應 瓶中,慢慢回到室溫後攪拌24 小時,以 CH2Cl2和飽和食鹽水萃取數次,收 集 有 機 層 , 經 無 水 硫 酸 鎂 除 水 。 粗 產 物 再 以 矽 膠 管 柱 層 析 純 化 , 以 hexane:ethyl acetate (4:1)之混合液為沖堤液,得到白色固體化合物 3.72 g, 產率為51%。 1H NMR(CDCl 3, 300 MHz): δ 4.97 (t, J = 6.36 Hz, 2H), 3.49 (s, 1H), 1.74-1.67 (m, 4H), 1.33-1.23 (m, 16H), 0.87 (t, J = 6.54 Hz, 6H)。 l. 1,1′-(3,4-Bromo-2,5-thienyl)diheptanone (10) 將 1,1′-(3,4-bromo-2,5-thienyl)diheptanol (7.77 g, 16.5 mmol) 溶於適量 溶劑CH2Cl2 (30 mL)中,待起始物完全溶解後,將 pyridinium chlorochromate (8.9 g, 2.5 eq)慢慢加入反應瓶中,室溫下反應至隔天,會有黑色固體產生, 以抽氣過濾裝置過濾固體,收集有機溶液,經無水硫酸鎂除水後過濾,再 以減壓蒸餾揮發溶劑。粗產物以矽膠管柱層析純化,以hexane:ethyl acetate (10:1)之混合液為沖堤液,得到白色固體化合物 6.0 g,產率為 78%。 1H NMR(CDCl 3, 300 MHz): δ 3.02 (t, J = 7.20 Hz, 4H), 1.73-1.66 (m, 4H), 1.38-1.28 (m, 12H), 0.87 (t, J = 6.7 Hz, 6H)。
m. 2,6,-Dicarboethoxy- 3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (11)
將 1,1′-(3,4-bromo-2,5-thienyl)diheptanone (7.51 g, 16.1 mmol)、ethyl 2-sulfanylacetate (3.70 mL, 2.1 eq)與碳酸鉀(9.35 g, 4.2eq)置入反應瓶中,加
入DMF (40 mL),加熱至 60 ℃反應 48 小時,待反應完全後冷卻至室溫, 以ethyl acetate 和大量水萃取數次,收集有機層,經無水硫酸鎂除水後過濾, 並以減壓蒸餾揮發溶劑。粗產物再以矽膠管柱層析純化,以 hexane:ethyl acetate (15:1)之混合液為沖堤液,得到液體產物 3.5 g,產率為 43%。 1H NMR(CDCl 3, 300 MHz): δ 4.38-4.31 (m, 4H), 3.12 (t, J = 7.8 Hz, 4H), 1.75-1.65 (m, 4H), 1.40-1.22 (m, 18H), 0.88-0.84 (m, 6H)。 n. 2,6,-Dicarboxylate acid-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (12) 取2,6,-Dicarboethoxy-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (0.67g, 1.32 mmol)置於反應瓶中,再加入1 M LiOH水溶液(11 mL)與THF (11 mL)。 加熱迴流12小時後,再以過量的2M 鹽酸水溶液中和,以濾紙過濾水溶液, 收集固體,用蒸餾水沖洗,再以真空系統移除殘留溶劑,可得黃色固體的 產物0.53 g,產率89%。 1H NMR(DMSO-d 6, 300 MHz): δ 3.11-3.06 (m, 4H), 1.33-1.26 (m, 16H), 0.85-0.83 (m, 6H)。
o. 3,5-Dihexyldithieno[3,2-b:2′3′-d]thiophene (13)
在 反 應 瓶 中 置2 , 6 , - d i c a r b o x y l a t e a c i d - 3 , 5 - d i h e x y l d i t h i e n o [3,2-b:2′3′-d]thiophene (4.68 g, 10.3 mmol) 與1.58 g (1.18 g, 1.8 eq)的Cu粉 末,加入quinoline (70 mL),加熱至迴流,持續12小時。反應降溫至室溫, 加入乙醚(50 mL)使Cu沉澱後,以濾紙過濾Cu,反覆用1 M的鹽酸水溶液除 去quinoline,再用乙醚萃取,收集有機層,經無水硫酸鎂除水,過濾後濾液 以迴旋濃縮儀抽除有機溶劑,殘餘物經silica管柱層析純化,以hexane為沖 堤液,可得產物1.77 g,產率35%。 1H NMR(CDCl 3, 300 MHz): δ 6.95 (s, 2H), 2.74 (t, J = 7.5 Hz, 4H), 1.80-1.73 (m, 4H), 1.35-1.28 (m, 12H), 0.91 (t, J = 6.7 Hz, 6H)。 3-3-6 化合物 Monomer 3 的合成,代號 M3 S S S C6H13 C6H13 (13) NBS DMF S S S C6H13 C6H13 Br Br (14) + S SnBu3 Pd(PPh3)4 toluene, reflux S S S C6H13 C6H13 S S NBS DMF S S S C6H13 C6H13 S S Br Br (15) (M3) p. 2,6-Dibromo-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (14) 將3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (1.96 g, 5.38 mmol)放入圓
底燒瓶中,加入DMF (15mL),再將 NBS (2.39 g, 2.5 eq)溶於 DMF (5mL)慢 慢滴入反應瓶中,在室溫下反應至隔天,以ethyl acetate 和大量水萃取數次, 收集有機層,經無水硫酸鎂除水後過濾,並以減壓蒸餾揮發溶劑。粗產物 再以矽膠管柱層析純化,以hexane 為沖堤液,得到固體產物 2.51 g,產率 為89%。 1H NMR(CDCl 3, 300 MHz): δ 2.72 (t, J = 7.5 Hz, 4H), 1.75-1.65 (m, 4H), 1.42-1.32 (m, 12H), 0.92-0.88 (m, 6H)。 q. 2,6-Dithienyl-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (15) 將 3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (2.46g, 4.71 mmol),以及 Pd(PPh3)4 (0.32g, 0.06eq),置於反應瓶中,用真空系統抽灌氮氣數次,加入
tributyl(thiophen-2-yl)stannane (3.29mL , 2.2 eq)以及 toluene (25 mL),加熱迴
流反應12 小時,反應後冷卻至室溫,先以減壓蒸餾揮發溶劑,粗產物再以 矽膠管柱層析純化,以hexane 為沖堤液,得到黃色固體產物 2.12 g,產率 為86%。 1H NMR(CDCl 3, 300 MHz): δ 7.35 (dd, J = 5.1、1.0 Hz, 2H ), 7.18 (t, J = 1.0 Hz, 2H), 7.09 (dd, J = 5.1、3.6 Hz, 2H), 2.91 (t, J = 8.1 Hz, 4H), 1.67-1.61 (m, 4H), 1.42-1.27 (m, 12H), 0.94-0.89 (m, 6H)。
r. 2,6-Bis(2′-bromothien-5′-yl) 3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (M3) 將 2,6-dithienyl-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (2.12 g, 4.01 mmol)放入圓底燒瓶中,加入 DMF (15mL),再將 NBS (2.1 g, 2.1 eq)溶於 DMF (5mL)慢慢滴入反應瓶中,在室溫下反應至隔天,以 ethyl acetate 和大 量水萃取數次,收集有機層,經無水硫酸鎂除水後過濾,並以減壓蒸餾揮 發溶劑。粗產物再以矽膠管柱層析純化,以 hexane 為沖堤液,得到固體產 物2.0 g,產率為 73%。 1H NMR(CDCl 3, 300 MHz): δ 7.04 (d, J = 3.5 Hz, 2H ), 6.9 (d, J = 3.5 Hz, 2H), 2.85 (t, J = 7.3 Hz, 4H), 1.75-1.73 (m, 4H), 1.41-1.26 (m, 12H), 0.92-0.89 (m, 6H)。MS (EI): m/z 685.4 (calcd [M]+).
3-3-7 化合物 Monomer 4 的合成,代號 M4 S S S C6H13 C6H13 1. n-BuLi, THF, -78℃ 2. SnBu3Cl S S S C6H13 C6H13 SnBu3 N S N Br 2 HBr, reflux N S N Br Br (17) S S S C6H13 C6H13 SnBu3 + N S N Br Br Pd(PPh3)4 toluene, reflux S S S C6H13 C6H13 S S S C6H13 C6H13 N S N S S S C6H13 C6H13 S S S C6H13 C6H13 N S N NBS DMF Br Br (M4) (13) (16) (16) (17) (18) s. 2-Tributylstannyl-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (16) 將3,5-dihexyldithieno[3,2-b:2′3′-d]thiophen (2.0 g, 5.49 mmol)置於反應 瓶中,利用真空系統抽灌氮氣數次,加入dry THF (30 mL),在-78 ℃下預冷
10 分鐘,將 n-BuLi (2.41 mL, 2.5 M in hexane, 1.1 eq)緩慢加入反應瓶中,持
續保持180 分鐘。隨後將 Bu3SnCl (1.64 mL, 1.0 eq)注入反應瓶中,在低溫
下維持20 分鐘,慢慢回到室溫後,以 ethyl ether 萃取,收集有機層,經無
乾得無色油狀物3.81 g。 t. 4,7-Dibromobenzo[c][1,2,5]thiadiazole (17) 將 benzo[c][1,2,5]thiadiazole (5.0 g, 36.7 mmol)置入反應瓶中,加入 HBr (100 mL),加熱至迴流後,將 Br2 (5.65 mL, 3.0 eq)溶於 50 mL HBr,慢慢滴 入反應瓶中,滴入時間約為兩小時,再持續迴流 6 小時後,冷卻至室溫, 加入大量飽和NaHSO3水溶液約 200 mL,會有白色固體產生,以抽氣過濾 裝置過濾,並且使用 EtOH 再結晶,可得到白色固體化合物 8.42 g 產率為 78%。 1H NMR(CDCl 3, 300 MHz): δ 7.69 (s, 2H )。 u. 4,7-Bis(3′,5′-dihexyldithieno[3,2-b:2′3′-d]thiophene-2′-yl) benzo[c][1,2,5] thiadiazole (18) 將 4,7-dibromobenzo[c][1,2,5]thiadiazole (0.83 g, 2.65 mmol) , 以 及 2-tributylstannyl-3,5-dihexyldithieno[3,2-b:2′3′-d]thiophene (4.06 g, 2.2 eq )、 Pd(PPh3)4 (98 mg, 0.03eq),置於反應瓶中,用真空系統抽灌氮氣數次,加入 toluene (45 mL),加熱迴流反應 12 小時,反應後冷卻至室溫,先以減壓蒸 餾揮發溶劑,粗產物再以矽膠管柱層析純化,以hexane 為沖堤液,得到紅 色液體產物2.31 g,產率為 95%。
1H NMR(CDCl 3, 300 MHz): δ 7.69 (s, 2H ), 6.70 (s, 2H), 2.90-2.75 (m, 8H), 1.84-1.56 (m, 8H), 1.39-1.25 (m, 24H), 0.96-0.83 (m, 12H)。 v. 4,7-Bis(6′-bromo-3′,5′-dihexyldithieno[3,2-b:2′3′-d]thiophene-2′-yl) benzo[c] [1,2,5]thiadiazole (M4) 將4,7-bis(3′,5′-dihexyldithieno[3,2-b:2′3′-d]thiophene-2′-yl) benzo[c][1,2,5] thiadiazole (0.94 g, 1.09 mmol)放入圓底燒瓶中,加入 DMF (10mL),再將 NBS (0.78 g, 2.5 eq)溶於 DMF (5mL)慢慢滴入反應瓶中,在室溫下反應至隔天, 以ethyl acetate 和大量水萃取數次,收集有機層,經無水硫酸鎂除水後過濾, 並以減壓蒸餾揮發溶劑。粗產物再以矽膠管柱層析純化,以hexane 為沖堤 液,得到紅色固體產物0.71 g,產率為 64%。 1H NMR(CDCl 3, 300 MHz): δ 7.74 (s, 2H ), 2.87-2.76 (m, 8H), 1.80-1.57 (m, 8H), 1.36-1.22 (m, 24H), 0.93-0.80 (m, 12H)。MS (EI): m/z 1019 (calcd [M]+).
3-4 高分子之聚合
S S S C6H13 C6H13 S S N O C O C6H12 * * y z P3: x = 0.5, y = 0.05, z = 0.45 P4: x = 0.5, y= 0.05, z = 0.45 O C8H17 C8H17 N S S S C6H13 C6H13 S S N O COOH C6H12 * * C8H17 C8H17 N N S N S S S C6H13 C6H13 S S S C6H13 C6H13 C8H17 C8H17 N * x y P2: x = 0.5, y= 0.5 * x x y z C8H17 C8H17 N S S S C6H13 C6H13 S S * * x y P1: x = 0.5, y= 0.5 圖 16 各高分子結構圖3-4-1 高分子 P1-P2 之聚合
本實驗所使用的聚合方法是Suzuki coupling,將0.5 mmol之M1單體以及
0.5 mmol之雙溴單體M3或M4、Pd(PPh4)4 (1 mol%)放入25mL雙頸圓底瓶
中,用真空系統抽換氮氣數次,加入無水toluene (5 mL),使反應溶液濃度
為0.2M,加溫至90 ℃,待瓶內溫度到達90 ℃後,加入2M的K2CO3水溶液(3.3
mL),在90 ℃下反應48小時後,加入過量iodobenzen繼續反應4小時後,再 加入過量phenylbronic acid以90 ℃繼續反應至隔天,結束end-capping。反應 後先降溫至約40 ℃,接著將濃稠的高分子溶液慢慢滴入甲醇中,讓其在甲 醇裡沉澱,以抽氣過濾裝置過濾後再用適當的溶劑去進行清洗動作,以減 壓蒸餾揮發溶劑後即可得到純的高分子固體。
3-4-2 高分子 P3 之聚合
本實驗所使用的聚合方法是Suzuki coupling,將0.5 mmol之M1單體以及
0.45 mmol之雙溴單體M2、0.05 mmol之雙溴單體M3、Pd(PPh4)4 (1 mol%)
放入25mL雙頸圓底瓶中,用真空系統抽換氮氣數次,加入無水toluene (5 mL),使反應溶液濃度為0.2M,加溫至90 ℃,待瓶內溫度到達90 ℃後,加
入2M 的 K2CO3水 溶 液(3.3 mL) , 在 90 ℃ 下 反 應 48 小 時 後 , 加 入 過 量
iodobenzen繼續反應4小時後,再加入過量phenylbronic acid以90 ℃繼續反應 至隔天,結束end-capping。反應後先降溫至約40 ℃,接著將濃稠的高分子
當的溶劑去進行清洗動作,以減壓蒸餾揮發溶劑後即可得到純的高分子固 體。 3-4-3 高分子 P4 之製備 取200 mg 高分子P3溶於約20 mL toluene中,慢慢滴入過量之HCl水溶 液,加溫至80 ℃反應48小時,接著將高分子溶液慢慢滴入甲醇中,讓其在 甲醇裡沉澱,以抽氣過濾裝置過濾後再用適當的溶劑去進行清洗動作,以 減壓蒸餾揮發溶劑後即可得到純的高分子固體。
4-1 合成機制探討
4-1-1 Stille coupling reaction
Stille 反應是一個利用鈀金屬催化有機錫化合物與有機鹵化物,而形成 C-C 鍵化學偶合的反應,反應式如下所示: R1-Sn(Bu)3 + R2-X R1-R2 + X-Sn(Bu)3 本文中,使用的鹵化物是以溴為主,故必須先使用NBS (N-bromosuccinimide) 或bromine 將化合物進行溴化反應(bromination)後,和有機錫化合物再進一 步行偶合反應,可能反應機構如圖 17。但由於金屬錫毒性甚大,除非必要
應避免使用,可以其他有機反應可取代之,如 Suzuki reaction34、Negishi
reaction35等。 PdⅡ Pd0Ln XSnBu3 R2SnBu3 R1-PdⅡLm-R 2 R1-PdⅡLm-X R1-R2 1 2 3 4 5 6 7 8 R1-X 圖17 Stille coupling 可能之反應機構
4-1-2 Suzuki coupling reaction 此反應乃藉 Pd 催化結合有機硼酸與鹵化物,而形成 C-C 鍵化學偶合 的反應,反應式如下所示:
R
1-B(OH)
2+ R
2-X
R
1-R
2+ B(OH)
4 -近年來催化劑與反應條件開發擴展運用可能性,以致於作用領域不再限制 在芳香族羥基,也包含脂肪族烷基、烯基與炔基化合物。Suzuki 偶合反應 最大特點在硼酸必須具有反應活性,使有機配位基增強極性,有利於 transmetallation 進行。本文中,使用的鹵化物是以溴為主,故必須先使用 NBS (N-bromosuccinimide)或 bromine 將化合物進行溴化反應(bromination)後,和有機硼化合物再進一步行偶合反應,可能反應機構如圖18 所示:
Ar-Ar'
Pd(0)
ArX
Ar'-Pd(II)-Ar
Ar'-Pd(II)-X
Ar-Pd(II)-OH
NaOH
NaX
B(OH)
4 -B OH OH Ar' OH NaOHAr'B(OH)
2X=Br,I
圖18 Suzuki coupling 可能之反應機構4-2 高分子基礎性質
表 1 高分子基礎性質(GPC、DSC、TGA) Polymer Mna Mwb PDIc Tgd (0C) Tde (0C) Yield (%) P1 16674 19786 1.19 130 421 71.4 P2 16625 18209 1.10 160 404 76.4 P3 14396 16297 1.13 135 366 79.5 P4 - - - 143 408 - a平均數量分子量(number average molecular weight);b平均重量分子量(weight average molecular weight);c polydispersity indices (PDI=Mw/Mn);d玻璃轉換溫度(glass transition
temperature);e熱分解溫度(decomposition temperature (0C) at 5% weight loss.)
高分子基礎性質的部分,我們利用凝膠滲透層析法 (Gel Permeation Chromatography,GPC) 分別得知四個高分子的分子量,而 P4 因為含有 COOH 關能基,不能進行 GPC 量測,因此分子量則以 P3 (P4 水解之前)為
參考值。四個高分子對於 tetrahydrofuran (THF)、chloroform (CHCl3)、
ortho-dichlorobenzene (o-DCB)、chlorobenzene (CB)皆有不錯的溶解度。
在熱性質方面,P1-P4 皆可以利用 Differential Scanning Calorimetry
(DSC) 以每分鐘升高十度的速率掃描得到化合物的 Tg,四個高分子的 Tg皆
在130 0C 以上,而 P2 的 Tg值較其他三個高分子高,推測原因可能為 P2 分
子主鍊上引入較多的fused dithienothiophene 環使得結構較為剛硬,進而導
致其 Tg 值上升。再者,我們利用 Thermogravimetric Analyses (TGA)得到
上,其中P3 的 Td較低,推測原因為 monomer 2 分子末端的 tert-butyl 官能 基較不穩定,而使得Td下降。 100 200 300 400 500 600 700 800 40 50 60 70 80 90 100 Weight perc e nt age Temperature (oC) P1 P2 P3 P4 圖19 各個高分子熱重分析圖
4-3 高分子光物理性質之探討
圖20-22 和表 2,為 P1-P4 測得之紫外-可見光(UV-Vis)吸收光譜、放光 光譜(PL)與其數據。由溶液狀態吸收光譜圖(圖 20)中得知,P1、P3、P4 因 為結構相似,其最大吸收峰值皆落在440 nm 附近,我們歸諸為化合物的π -π* 電子躍遷,而吸收範圍則落在 300 nm 到 550 nm 左右。以 P2 而言, 因為在高分子主鍊上引入了含有拉電子能力的 benzothiadiazole 片段,吸收 光譜上除了在短波長處仍然有化合物的π-π* 電子躍遷之外,在長波長區 (donor)、受體(acceptor)之間作用而產生的intramolecular charge transfer(ICT)吸收峰,而其最大吸收峰值落在 488 nm; 同 時 我 們 也 在 P2 分 子 主 鍊 引 入 了 含 有 較 多 有 效 共 軛 長 度 的 fused dithienothiophene 環,進而使得其吸收範圍更為廣泛,約延伸至 630 nm。 由薄膜狀態吸收光譜圖(圖 21)中,我們可以看到 P1-P4 的吸收波型與在 溶液狀態時非常相似;其中P1 的最大吸收峰由 448 nm 位移到 470 nm,其 他三個高分子的最大吸收峰值也分別都有紅位移的現象,我們歸因於薄膜 狀態的分子較易產生堆疊作用(aggregation),造成 interchain association 以及 π-πstacking,因此比溶液狀態吸收光譜還要紅位移,範圍約在 22-24 nm 之間。而四個高分子的光學能隙(optical band gap)皆以此薄膜狀態吸收光譜
(absorption edge)而求得,其中 P2 之 Eg為 1.98 eV,是所有高分子中能隙最
窄的。
而由薄膜狀態放光光譜圖(圖 22)中,我們可以看到四個高分子的放光
強度皆非常的弱,其中 P2、P4 幾乎不放光,此現象或許能夠間接解釋 P2
分子擁有比 P1 分子強的 photoinduced charge transfer 以及 intermolecular
energy transfer 能力之外;其中 P4 分子放光較 P3 分子弱的原因,我們推測
是因為P4 分子當中含有的少部分 COOH 官能基可能形成二聚物(dimer),使
表 2 高分子紫外-可見光(UV-Vis)吸收光譜、放光光譜(PL)數據 polymer λabs, sola (nm) λabs, filmb (nm) λPL,filmb (nm)
P1 448 470 546
P2 378, 488 388, 510 -
P3 442 465 543
P4 441 465 -
a The absorption spectra were recorded in dilute chlorobenzene solution at room temperature. bThe absorption and PL films were spin-coated from 10 mg/1 mL chlorobenzene solution.
300 400 500 600 0.0 0.2 0.4 0.6 0.8 1.0 Normali z e d abso rban ce Wavelength (nm) P1 P2 P3 P4 圖20 各個高分子在 chlorobenzene 中的吸收光譜圖(10-6 M)
300 400 500 600 0.0 0.2 0.4 0.6 0.8 1.0 Normalized absorbance Wavelength (nm) P1 P2 P3 P4 圖21 各個高分子薄膜狀態吸收光譜圖 500 550 600 650 700 750 0 100 200 300 400 500 600 P h ot o lum in ese n c e int e nsit y Wavelength (nm) P1 P2 P3 P4 圖22 各個高分子薄膜狀態放光光譜圖
4-4 高分子電化學性質之探討
我們利用循環伏安法(cyclic voltammetry,CV) ,量測各高分子之電化
學 性 質 。 進 行 CV 量 測 時 , 先 配 置 電 解 質 溶 液 為 0.1 M 的
tetra-n-butylammonium hexafluorophosphate (Bu4NPF6)無水 acetonenitrile 溶
液,各個高分子配置成10 mg/1 ml 的 chlorobenzene 溶液。本次實驗所使用 的CV 是屬於三電極式,工作電極是 Pt 薄膜,參考電極是 Ag/AgCl 參考電 極,輔助電極是Pt 線。量測時先將高分子溶液滴加在 Pt 薄膜上,等自然風 乾成薄膜後,再放入電解質溶液裡,並用100 mV/s 的掃描速率去進行電化 學的量測。在本文中,P1-P4 高分子在還原電位皆測不出其氧化還原的特 性,因此主要可利用 CV 測得高分子之氧化電位求得最高填滿分子軌域 (HOMO),配合吸收光譜也可得到化合物之最低未填滿分子軌域(LUMO)。 詳細的電化學資料則列於表 3 和圖 23。由於本次 CV 量測的是薄膜狀態的 氧化還原,其膜厚較難逐一控制,因此電流範圍較廣;而因為濃度較高的 關係,其電流值也較一般在溶液狀態時要大許多。 由圖 23 可得知,P1-P4 高分子在氧化電位皆呈現半可逆(qusi-reversible) 的氧化還原對,而在還原電位方面,我們並未觀測到其氧化還原的特性, 此結果也顯示出 P1-P4 高分子可能是較適合被應用在 p-type 的材料。其中 P1、P3、P4 化合物,因為結構組成相似,其 HOMO、LUMO 以及光學能 隙也較為相近。其中P2 分子因為分子主鍊上π共軛長度的延伸,以及引入
了含有拉電子能力的 benzothiadiazole 片段受體,而有強拉電子能力的影 響,使得LUMO 電位下降(約 0.21 eV),光學能隙變窄。若要應用在與 PCBM 混摻而製成的太陽能電池元件時,其高分子的LUMO 電位最好要比 PCBM 的LUMO 電位高 0.3 eV 以上37,以確保高分子能有效地將電子傳遞到PCBM 上。而在本實驗中,四個高分子的 LUMO 位電皆大於 PCBM 的 LUMO 電 位0.3 eV 以上,此數據雖然可以預測高分子的電流傳遞可能沒有問題;但 就理論上的電子電洞分離能力來探討,若高分子的HOMO 電位和 PCBM 的 LUMO 電位差距越大,其分離效果則越好。詳細的元件量測結果在下一節 詳述。 圖23 各個高分子的 HOMO 和 LUMO 能階示意圖 P1 P2 P3 P4 PCBM ITO -5.58 eV -5.54 eV -5.60 eV -5.60 eV -6.1 eV -4.1 eV -3.35 eV -3.35 eV -3.56 eV -3.34 eV -4.7 eV
表3 各個高分子電化學性質數據
polymer λonset, abs (nm)
Ega (eV) E1/2b (ox) HOMO c LUMOd P1 553 2.24 0.89 -5.58 -3.34 P2 626 1.98 0.85 -5.54 -3.56 P3 550 2.25 0.91 -5.60 -3.35 P4 551 2.25 0.91 -5.60 -3.35
a The optical band gap was obtained from the equation E
gopt = 1240/λonset
bE
1/2 was the average value of oxidation and reduction potential.
c E
HOMO = [-(E1/2 - 0.11) - 4.8] eV where 0.11 V is the value for ferrocene vs. Ag/Ag+ and 4.8
eV is the energy level of ferrocene below the vacuum.
dLUMO = HOMO - E g 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 -0.0003 -0.0002 -0.0001 0.0000 0.0001 0.0002 Cu rr en t (A ) Potential (V) P1 P2 P3 P4 圖 24 各個高分子在氧化電位下之循環伏安圖
4-5 太陽能電池之性質
各高分子的混摻異質接面太陽能電池(bulk heterojunction solar cells)之
短路電流 (Jsc)、開環電壓(Voc)、填充因子(FF)、轉換效率(PCE)如表 4 所示。 本 論 文 元 件 中 的 主 動 層 (active layer) 所 選 用 的 溶 劑 是 高 沸 點 的 o-dichlorobenzene,而高分子是以 polymer(2 wt%):PCBM(2 wt%) = 1:1 去混合,再用固定轉速 600 rpm 塗佈在 PEDOT:PSS 層上,並鍍上電極 Ca/Al , 而 所 選 用 的 基 材 是 ITO 玻 璃 。 其 元 件 結 構 為 ITO/PEDOT:PSS/Polymer(2wt%):PCBM(2wt%)/Ca(50nm)/Al(100nm) , 如 圖 25 所示。 圖25 太陽能電池元件結構圖 表4 高分子和 PCBM 經過 1:1 混摻後之元件量測數據 active layer (polymer:PCBM=1:1) Voc (V) Jsc (mA/cm2) FF (%) PCE (%) p.s. P1 0.82 1.82 0.37 0.49 - P2 0.87 0.12 0.26 0.03 - P3 0.49 0.84 0.28 0.12 broken film P4 0.90 1.08 0.27 0.26 - substrate ITO PEDOT:PSS polymer:PCBM (1:1) Ca Ca Ca Al Al Al