國 立 交 通 大 學
分 子 科 學 研 究 所
碩士論文
二氧化鈦奈米管在可見光/紫外光分解水的效應
The Efficacy of TiO
2Nanotubes on Water-Splitting under Visible
and Ultraviolet Light Illumination
研究生 : 蕭世明
指導教授 : 林明璋 院士
指導教授 : 王念夏 博士
二氧化鈦奈米管在可見光/紫外光分解水的效應
The Efficacy of TiO
2Nanotubes on Water-Splitting
under Visible and Ultraviolet Light Illumination
研 究 生 : 蕭世明 Student : Shi-Ming Hsiao 指導教授 : 林明璋 Advisor : M. C. Lin
指導教授 : 王念夏 Advisor : Niann-Shiah Wang
國 立 交 通 大 學 分 子 科 學 研 究 所
碩 士 論 文
A Thesis
Submitted to the Department of Applied Chemistry National Chiao Tung University
In partial Fulfillment of the Requirements for the Degree of Master
in
Applied Chemistry
December 2009
Hsinchu, Taiwan
二氧化鈦奈米管在可見光/紫外光分解水的效應 學生: 蕭世明 指導教授 : 林明璋
指導教授 : 王念夏 國立交通大學分子科學研究所 摘要 本實驗主要研究利用電化學方法合成二氧化鈦奈米管,並摻雜了碳元素 在二氧化鈦奈米管中,經由水分解系統分別量測可見光及紫外光效率,發 現高密度排列的奈米管及參雜碳元素都可以明顯的增加反應效率,且紫外 光區電流及水分解效率都較可見光區好,而我們也經由SEM 圖譜、XRD 圖譜、及不同條件的二氧化鈦奈米管的水分解效率了解一些二氧化鈦奈米 管的物理特性,可以發現奈米管電解液組成為10%的 DI water + 0.5% NH4F + 89.5%的 glycerol,鍛燒溫度為 500℃,水分解的電解液為 1M KOH 效果最佳,水分解效率在紫外光區(320±70nm)可達 28%,但在可見光區 (520±30nm)卻只有 0.16%,並不如 Misra 在 J. Phys. Chem. C 2007[4]所發
表的結果,可是在可見光區的低效率卻與Murphy[44]最近的結果相似。最
後我們也利用XPS、IPCE 及反射式吸收光譜對可見光部分進行詳細討論。
The Efficacy of TiO2 Nanotubes on Water-Splitting
under Visible and Ultraviolet Light Illumination
Student : Shi-Ming Hsiao Advisor : M. C. Lin
Advisor : Niann-Shiah Wang
Department of Applied Chemistry National Chiao Tung University
ABSTRACT
In this research, we study TiO2 (TiO2-xCx) nanotubes synthesized by the
application of sonoelectrochemistry. We obtained vertically grown TiO2-xCx nanotubes arrays with high aspect ratios for water splitting in the
visible and ultraviolet light regions. The best electrolyte for anodization was found to be 10% DI water + 0.5%NH4F +89.5% glycerol with the annealing
temperature at 500℃. The observed efficiency results clearly from TiO2
nanotubes, and the ultraviolet light region showed a much greater efficiency than the visible region. The properties of the synthesized TiO2-xCx
nanotubes have been investigated with SEM, XRD and different electrolytes for water splitting. The best electrolyte for water splitting is 1M KOH solution. Finally we study in detail in the visible region using TiO2-xCx
characterized by XPS、IPCE and reflection-absorption spectroscopy. The low efficiency observed in the region disagrees with the 8.5% efficiency reported by Misra and coworkers(J. Phys. Chem. C 2007, 111, 8677-8685), but is consistent with the result of Murphy (Solar Energy Materials&Solar Cells, 92, 2008, 363-367).
誌 謝
在交大的研究生生涯終於到了結束的時候,真的投入研究後才發現原來 科學真的不是一件簡單的事,這時間受到很多人的照顧,有強者偉哥,馬 哥還有連話都說不清楚的老王,還有碩一時每天晚上都陪伴我們的學長, 還有可愛的學弟妹及王念夏實驗室的大家,感謝大家在這段時間的陪伴及 互相勉勵,當然家人的支持也是我可以走到這裡很大的原因,也感謝這幾 年林明璋教授對我實驗的指導,最後還是要說想感謝的人太多了,只有深 深的一句感謝大家,感謝大家的陪伴及對我的幫助。 iii目錄
中文摘要………i 英文摘要………ii 致謝………iii 目錄………iv 表目錄………vi 圖目錄………vii第一章 緒論
………1
1-1 前言 ………1 1-2 研究動機 ………3第二章
理論基礎 ………5
2-1 電化學拋光 ………5 2-2 二氧化鈦奈米管陽極化 ………8 2-2-1 二氧化鈦特性 ………8 2-2-2 二氧化鈦陽極化反應 ……….………… 10 2-3 水分解系統 ………13 2-3-1 原理 ……….………13 2-3-2 實驗裝置 ………15第三章
實驗方法與步驟………17
iv3-1 藥品與設備………17 3-2 電極製備………17 3-2-1 二氧化鈦奈米管製備………17 3-2-2 鉑電極製備………18 3-3 光電化學裝置與分析………18 3-4 分析儀器簡述………23 3-4-1 X 光繞射儀(XRD)………23 3-4-2 掃描式電子顯微鏡(SEM)………24 3-4-3 紫外光/可見光吸收光譜儀(UV/Visible Absorption Spectrometer)……….24 3-4-4 X 射線光電子能譜儀(XPS)………25 3-4-5 太陽光模擬器(Solar Simulator AM 1.5)………25
第四章
結果與討論………26
4-1 陽極化結果與分析………26 4-2 自製鉑電極結果………34 4 - 3 水 裂 解 效 率 結 果 … … … 3 6 4-4 TiO2-xCx奈米管對紫外光區與可見光區影響………46 4 - 5 I n N 對 二 氧 化 鈦 奈 米 管 影 響 … … … 5 0第五章
結論………58
第六章
參考文獻 ………60
v表目錄
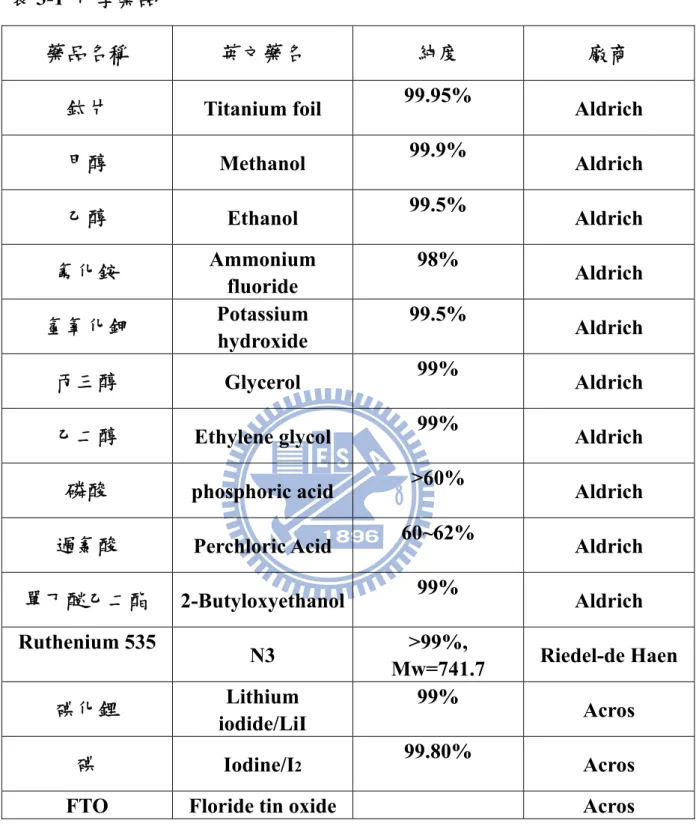
表(3-1) 化學藥品………19
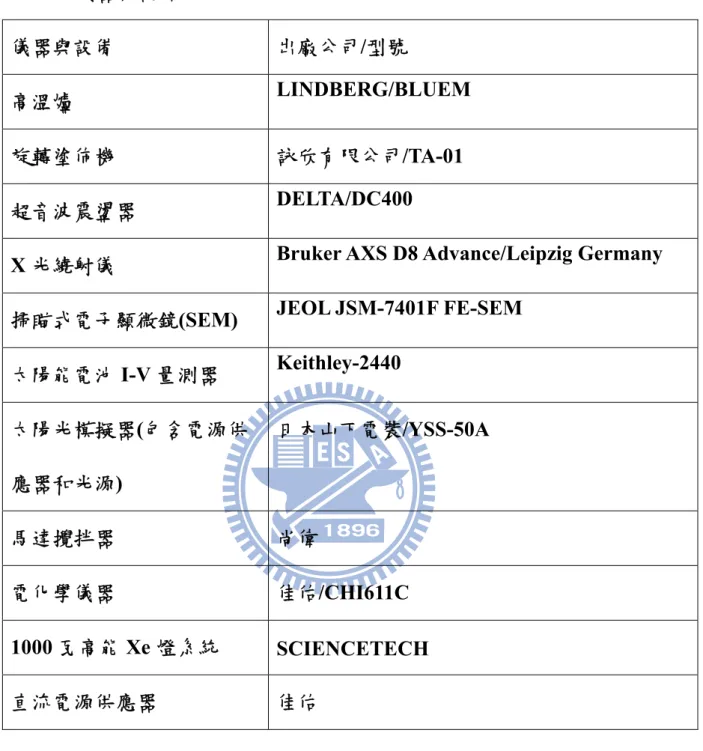
表(3-2) 儀器與設備………20
表(4-1) 拋光鈦片 EDS 元素分析………27
表(4-2) Pt/TiO2 的元素分析………35
表(4-3) TiO2-xCX nanotube/N3 dye 其光伏參數列表………54
表(4-4) 泡 TiCl4後的 TiO2-xCX nanotube/N3 dye 其光伏參數列表………55
表(4-5) TiCl4/InN/TiCl4的 TiO2-xCX nanotube/N3 dye 其光伏參數列表…57
圖目錄
圖(1-1) 本多-藤嶋效應設備圖………3 圖(2-1) 電化學拋光反應電壓對電流關係圖 ………5 圖(2-2) 銅基材在硝酸電解液的拋光過程圖 ………6 圖(2-3) 金屬表面電化學拋光示意圖 ………7 圖(2-4) 二氧化鈦晶格結構 ………9 圖(2-5) 電鍍鈦基材的反應圖 ………11 圖(2-6) 在常壓下二氧化鈦奈米管形成的概略圖 ………12 圖(2-7) 常見的半導體能帶結構 ………14 圖(2-8) 水分解實驗原理圖 ………16 圖(2-9) 水分解實驗設計圖 ………16 圖(3-1) 光電化學反應裝置圖………21 圖(3-2)二氧化鈦奈米管實驗裝置圖………22 圖(4-1) 鈦片拋光圖………26 圖(4-2) 不同電壓奈米管的電流變化圖………27 圖(4-3) SEM 影像顯示定電壓 30V TiO2奈米管結構變化………29 圖(4-4) 不同電壓的 SEM 奈米管半徑圖………30 圖(4-5)電壓對(a)奈米管孔洞半徑(b)管璧厚度造成的變化………...30 圖(4-6)不同溫度 SEM 奈米管管璧圖………31 vii圖(4-7) 溫度對(a)奈米管孔洞半徑(b)管璧厚度造成的變化……….………31
圖(4-8) 奈米管表面清潔圖………...………32
圖(4-9) C1s XPS 圖……….………33
圖(4-10) 自製鉑電極 SEM 圖………...………34
圖(4-11)Pt vs Pt-TiO2紫外光區效率比較圖………35
圖(4-12)UV 與 VIS filter 波長穿透率圖………37
圖(4-13)含碳與不含碳的二氧化鈦奈米管光解水效率比較圖……...………39 圖(4-14) 1M KOH 不同溫度奈米管光解水效率圖………41 圖(4-15) 不同退火溫度二氧化鈦晶格 XRD 圖……...………42 圖(4-16) 紫外光分解水反應時間與效率圖……….………43 圖(4-17) 不同電解液對電壓的效率比較圖……….…………45 圖(4-18) 不同參雜元素的二氧化鈦奈米管 IPCE 效率圖……..………47 圖(4-19) 含碳的二氧化鈦奈米管反射式吸收光譜圖………….………47 圖(4-20) 參雜碳元素的能階圖……….………48 圖(4-21) 含碳二氧化碳奈米管理論計算結果圖……….………49 圖(4-22) InN 在不同奈米管半徑生成 SEM 圖……….………50 圖(4-23) 不同電解液對電壓的 InN 二氧化鈦奈米管效率比較圖……….…52
圖(4-24) TiO2-xCX nanotube/N3 dye I-V 曲線圖………..………54
圖(4-25) 泡 TiCl4後的TiO2-xCX nanotube/N3 dye I-V 曲線圖………..……55
ix
圖(4-26) InN/TiO2-xCX nanotube/N3 dye I-V 曲線圖………56
圖(4-27) TiCl4/InN/TiCl4的 TiO2-xCX nanotube/N3 dye I-V 曲線圖………57
第一章
緒論
1-1 前言
美國前總統布希於 2001 年 5 月發布「國家能源政策」,闡述美國國內能 源供應與國內能源需求之間嚴重不平衡乃其所面臨最根本之能源挑戰。美 國能源部於同年11 月在華府舉辦氫能會議,目標為尋找更安全與更乾淨的 能源,結果一致認為氫氣為未來重要能源之ㄧ。 氫氣具有含量豐富、乾淨、有效率與來源廣泛等優點,因此成為取代汽 油的新能源極佳選擇。宇宙約有75%由氫構成,故其於自然界含量豐富, 於自然界中氫常與其它元素化合,以化合物質存在而不以氫氣形式存在, 故具氫之化合物皆可視為氫燃料之來源,諸如硼氫化鈉水解、光觸媒分解 水,或以地熱、太陽能或風力電解水等。在幾種獲取氫來源的方法中,目 前以光觸媒分解水成氫氣和氧氣最受科學界之重視,原因主要是利用太陽 光做為分解水之能量來源,而據科學家估計,太陽的壽命約有50 億年之久, 故對於人類而言,可謂取之不盡、用之不竭。另外由於可見光佔太陽光組 成的45%[1],因此目前國際之光觸媒材料開發均朝能吸收可見光之研究方 向進行。 二氧化鈦因具有光催化活性、長期化學穩定性、低價格等優點,因此常 被應用於光催化相關領域。然而以二氧化鈦作為光觸媒應用於光化學氧化 1的技術上,由於很難將懸浮的二氧化鈦粒子從水溶液中分離,因此導致由 光產生的電子與電洞會快速結合,進而降低量子效率。為了解決此一問題, 有一些研究利用電化學的技術,於鈦基板上鍍上多孔性的二氧化鈦薄膜, 也有學者提出參雜不同元素(如碳、氮、硫等[2])後的二氧化鈦可使載子分離 較遠,且吸收光的波長範圍可由紫外光擴大到可見光區,期望可以將水分 解效率更提高一些。 2
1-2 研究動機
光觸媒於 1967 年被發現。當時還是研究生的東京大學教授藤嶋昭於某次 試驗中偶然發現,用二氧化鈦單晶及鉑做為電極放入水中形成迴路,當用 水銀燈照射紫外光,即使不通電,兩個電極上均有氣體產生。證實在二氧 化鈦電極及鉑電極上所產生之氣體分別為氧氣和氫氣。藤嶋昭於1972 年與 本多教授將此研究成果發表文章於Nature 雜誌上,此即為著名的本多-藤嶋 效應[3],而此處的二氧化鈦就是扮演光觸媒之角色。 光 白金對極 二氧化鈦電極 酵素 水素 圖(1-1) 本多-藤嶋效應設備圖[3] 3近年來二氧化鈦在太陽能電池上的研究非常多,主要是因為二氧化鈦具 有強大氧化還原能力、化學穩定性高及無毒的特性,且具有一般材料所沒 有的半導體性質,而太陽光電解水產氫之過程可完全避免化石能源之應 用,故符合氫能經濟之要求,此乃極佳之取氫方式。因此本研究主要著重 於利用電化學方法製作高密度排列的二氧化鈦奈米管,此法優點為控制實 驗中的電壓電流便可調節二氧化鈦奈米管的一些物理特性如奈米管半徑、 管壁厚度及長度,利用奈米管特殊的結構將可更有效的收集太陽光讓水裂 解效率變高。而我們知道太陽能電池在可見光波長的吸收效率一直都是極 差的,但在Misra 的研究中[4]卻說明了他的太陽能電池水分解系統的可見 光效率可以高達 8.5%,若他的研究屬實,太陽能電池效率將會有大大提 升,而這就是本研究實驗的最早動機,因此我們也摻雜了碳元素在奈米管 中,希望可以降低二氧化鈦本身的能階,讓二氧化鈦的吸收光譜可以延伸 到可見光區以增加效率,因此我們將會特別針對紫外光跟可見光區的水裂 解效率做深入的探討,以確定Misra 的結論是否正確的。 4
第二章 理論基礎
2-1 電化學拋光
電化學拋光(鈍化)為金相學常用之前處理,目的是移除機械拋光所造成之 缺陷。一般而言,可產生電化學拋光效果之電壓範圍相當廣,如圖(2-1)所 示[5]。當外加電壓較低時,幾乎無電流產生,此時金屬表面僅發生蝕刻 (etching)。當外加電壓持續增加時,金屬表面將產生電化學拋光反應,從圖 (2-1)可得知電化學拋光的範圍是相當大的。 Etching Polishing Current densitySingle electrode potential
圖(2-1) 電化學拋光反應電壓對電流關係圖 [5]
而拋光是一種金屬特殊的現象,它產生的現象是使原先易受腐蝕的陽極 金屬受到保護。有些金屬在特定的環境下,會有活性的轉變,也就是原先 活性很強的金屬,腐蝕速率應隨電極電位而增加,但是當陽極極化電位到 達某一電位時,腐蝕速率反而急遽下降,形成鈍化的現象,由蝕刻轉變為 電化學拋光過程中,存在一複雜之電壓—電流變化,如圖(2-2)所示[5]。在 AB 區間電流隨電壓增加,此時僅有部分金屬發生溶解反應且金屬表面將失 去光澤。經BC 之不穩定區後反應將過渡至 CD 穩定區,電化學拋光反應於 CD 區間開始發生(最佳化條件)。當反應過渡至 CD 或 EF 區時,金屬仍繼 續進行電化學拋光,然而可能伴隨部分氣泡產生並造成金屬發生孔蝕現 象。圖(2-2) [5]為一代表性說明,不同電解質則有不同之電壓電流特徵。 圖(2-2) 銅基材在硝酸電解液的拋光過程圖 [5] 6
電化學拋光反應主要可分為兩個過程:平坦化與亮光化。 1. 平坦化: 金屬經機械拋光後,表面仍存在粗糙度 1μm 左右的溝槽或 缺陷。當金屬置入電解液中時,表面會形成一層由電解液所組成的 拋光層,此拋光層的電阻較電解槽中溶液的電阻大,因此控制整個 電化學拋光的反應。由圖(2-3)可知,由於 A-B 兩點間之距離較 C-D 兩點小,因此在A-B 點之間的拋光層電阻較小。當施以外加電壓時, A-B 兩點間的電流會較 C-D 兩點間電流為大,換句話說,A-B 兩點 間金屬的溶解速度較大。藉由此金屬表面溶解速率的不同,可將表 面粗糙度較大的部分優先拋除,變成較平坦的平面[6,7]。 2. 亮光化:亮光化過程主要乃是去除表面尺寸 0.01μm 以下之缺陷。此 外,反應過程中金屬表面常產生一薄鈍化層並抑制金屬表面蝕刻反 應之發生。事實上,亮光化反應亦可能發生於金屬平坦化過程中[8]。 圖(2-3) 金屬表面電化學拋光示意圖[5] 7
2-2 二氧化鈦奈米管陽極化
本章節主要是描述二氧化鈦奈米管的一些物理特性,以及製作奈米管和 水裂解實驗的一些反應與原理。2-2-1 二氧化鈦特性
鈦金屬於自然環境中易生成一透明之二氧化鈦氧化膜,其膜厚可藉陽極 處理之外加電壓而增加,鈦之陽極處理條件若經特別的控制,則可製出高 品質或高附加價值之產品,如光觸媒感應器、奈米模板、太陽能電池、光 觸媒劑[9,10,11]。二氧化鈦是一陶瓷原料,在一般情況下與大部分化學試劑 不會發生反應,也不溶於水、脂肪酸和弱無機酸,但微溶於鹼,可視為相 當穩定的物質,二氧化鈦過去主要做為白色顏料,俗稱「鈦白」,因它具有 高折射率、高化學穩定性及不具毒性之物質,廣泛運用於塗料、化妝品、 瓷器原料等方面。以上產品均要求反應物具大表面積以達高反應效率。因 此,以陽極處理法控制鈦基材表面生成一多孔性之二氧化鈦氧化膜可達到 要 求 。 二 氧 化 鈦 具 有 三 種 結 晶 型 態[12] : 銳 鈦 礦 (Anatase) 、 金 紅 石(Rutile) 、板鈦礦(Brookite) ,其中結晶型態以 Rutile 最為常見,Anatase
次之,Brookite 最少見,其中只有 Anatase 晶相及 Rutile 晶相具有光催化 的能力,Anatase 晶相能隙(Energy Band Gap)為 3.2eV,Rutile 晶相則為 3.0eV,相對於此一能量躍遷,所需光能量之波長為 380nm。二氧化鈦在吸
收太陽光中紫外線或照明燈中的紫外光後會產生類似植物的光合作用原
理,只差異於生成物不同。 1991 年由 Iijima[13]先發現奈米碳管(Carbon Nanotubes),此後奈米碳管 便被視為極具潛力的新材料,學術界除了對奈米碳管持續研究外,同時也 將研究範圍延伸至其他材料組成之奈米管狀物,如二氧化矽(SiO2)[14] 、二 氧化鋁(Al2O3)[15] 、二氧化鈦(TiO2)[16,17]等,探討其特性結構及可應用之 領域,其中以二氧化鈦最具有強大的氧化還原能力、化學穩定性高及無毒 的特性最受到親睞,二氧化鈦具有一般材料所沒有的半導體性質,受到學 術界及科學家的重視,也是一種附加價值很高的功能性無機材料。由於近 來奈米製程的研發,使得材料尺寸降低,總表面積增加,進而增加材料介 面的反應效率,二氧化鈦為一價格低廉之原料,配合奈米技術的研發,將 使其應用範圍更為廣泛。奈米材料製程具有由小而大(bottom-up)與自我組 裝(self-assembly)的特性,其中 bottom-up 特性讓使用者可得到奈米級之材 料,而self-assembly 特性能讓研究者更方便的探討材料之反應機構。 圖(2-4) 二氧化鈦晶格結構 [18] 9
2-2-2 二氧化鈦陽極化反應
近十幾年來,陽極氧化鋁模版(AAO)的製備與應用已經發展的相當成熟 與多樣化,而近年來二氧化鈦奈米管的發展由初期的製備與改良,到後期 的應用價值探討與機制研究,已逐漸受到科學家的重視。由於二氧化鈦奈 米管有優良的光催化(photocatalytic)特性,是很好的光觸媒材料,具有自我 清潔環境汙染物的特性;二氧化鈦奈米管卓越的電催化(electrocatalyst)性質 可以將其組裝為光電極參與水的分解反應,或是應用於染料敏化太陽能電 池(dye-sensitized solar cell);亦可做為氫氣檢測器(H2 sensor)與生化測器(biosensor)或因組成穩定可應用在使用於人體的生醫材料(biomaterial)。 陽極處理法屬於電化學方法的一種,由於可藉由電化學條件控制來調整 二氧化鈦奈米管的型態與功能性,可藉由改變反應電位、電解液種類等參 數來控制管徑與生長速度及長度。而二氧化鈦陽極處理法主要分為四大 類,第一類為含有氫氟酸的水溶液[19,20] ,第二類為含氟離子與不同種類 的酸去搭配作為電解液,主要以硫酸、磷酸為主[21,22,23],第三類為含氟 離子的緩衝溶液[23,24],第四類為含氟離子的有機溶液[25],皆含有氟離子 是以上四種電解液的共同特性[26],這是由於鈦的材料特性,氟離子可與鈦 基板表面的二氧化鈦層反應形成錯合物而溶解至電解液中,從圖(2-5)可以 看到如電鍍時電解液沒有氟離子,鈦基材表面將會產生一個平坦的表面, 有氟離子時最後則會形成[TiF6]2-,然後離開鈦基材到電解液中,此化學溶 10
解使奈米管結構可在反應中生成,其他如氯、溴、碘離子卻無此效果[27]。 而整個反應過程可以以反應式(1)(2)(3)來表示[28,29],而反應式(1)(2)其實幾 乎是同時發生的,而當鈦基材表面形成一緻密層後,最後反應式(3)就會接 著產生,圖(2-6)就是詳細的管狀物產生過程[20]。 (1) Ti + 2H2O TiO2 + 4H++4e- (2) 6F- +TiO2 +4H+ [TiF6]2- + 2H2O
圖(2-5) 電鍍鈦基材的反應圖 (a) 無氟離子 (產生平坦表面) (b) 存在氟離 子 (管狀結構產生) [27] 11
圖(2-6) 在常壓下二氧化鈦奈米管形成的概略圖 (a) 緻密氧化層, (b) 孔洞 形成並慢慢加深, (c) 管狀物旁形成小孔洞, (d) 管狀層完全成形前,(e) 理想 的管狀層生成 [20]
2-3 水分解系統
太陽能是一種源源不絕且乾淨的能源,不會引起汙染更不會對環境造成 影響,長期以來,科學家一直希望能以太陽能來分解水製造氫氣。光分解 水製造氫氣的價值在於氫氣燃燒時只產生水,不會汙染環境,且相較於時 下使用的石化燃料,氫能蘊藏了較大的能量(119kJ/g) ,是極為理想的能 源。而光分解水製氫最大的問題就是轉換效率太低由於電子電動對的再結 合率太高。為了要克服這個問題,我們需先了解光觸媒半導體與水的氧化 還原電位的關係。2-3-1 原理
水分解成氫氣與氧氣的反應是伴隨著自由能正值的改變(大於零)的反 應。在反應式(4)中,製造一莫耳氫氣所需的自由能是 237.2KJ。又因為氫 氣與氧氣形成水的逆反應極易發生。使得光分解水的門檻較一般的光觸媒 催化反應高。 H2O H2+1/2O2 △G0=237.2kJ/mol(4) 圖(2-7)列出常見的半導體能帶結構圖[30,31]。當半導體材料吸收了一個 具有hν 大小能量的光子或者超過這個半導體能隙能量 Eg 的光子而激發 時,一個電子被從價帶激發到導帶,留下一個電洞在價帶中。當半導體的 能帶結構,導帶位置負於水的還原電位(0V,vs. NHE) ,而價帶位置正於水 的氧化電位(+1.23V, vs. NHE) 。所以光生電子由導帶還原水分子;光生電 13
洞則由價帶氧化水分子,而完成水的光分解。 半導體能帶的結構、能隙的寬度、導帶與價帶的位置及相對於水的氧化 還原電位程度,都是影響半導體的重要因素。然而,能帶的電位結構只是 熱力學的需求,其他如電荷分離、電子電洞對的再結合率、材料的結晶性、 表面積與其表面結構都是影響的重要因素之ㄧ。 圖(2-7) 常見的半導體能帶結構[30,31] 14
2-3-2 實驗裝置
而近年來許多科學家都致力於使用二氧化鈦奈米管當做光分解水的半導 體,管狀結構不只是增加了光的吸收性,他的高電子傳遞特性也顯示出了 較二氧化鈦薄膜高的轉換效率。由於二氧化鈦奈米管具有高密度且垂直於 表面的結構,且這種金屬氧化物的半導體在pH 值很強的電解液中有很高的 穩定性,因此是適合當光分解水的半導體結構。圖(2-8)可以把水裂解的反 應做一大概說明[32],當一光源照射在二氧化鈦奈米管激發產生電子電洞對 後,在價帶的電洞會與水溶液反應產生氫離子,此時氧氣也會產生,而在 導帶的電子會隨著導線傳到鉑電極上與溶液中的氫離子結合產生氫氣,理 論上一個穩定的水分解系統只要持續的照光反應將會一直持續的進行,電 流將不會隨時間改變。本實驗水分解系統主要是參考圖(2-9)[33,34]來設 計,與傳統太陽能電池不同的是水分解系統會多使用一個參考電極來當作 輔助,因為在水溶液中彼此電位是相對的,所以必須定義一個電極來當作 輔助,以幫助我們在之後可以換算我們的轉換效率。 15圖(2-8) 水分解實驗原理圖[32]
圖(2-9) 水分解實驗設計圖[33,34]
第三章
實驗方法與步驟
3-1 藥品與儀器
化學藥品與器材以及實驗設備分別列於表3-1 與表 3-2。3-2 電極製備
3-2-1 二氧化鈦奈米管製備 本實驗之高純度鈦片(99.95% Ti foil, 0.2mm),一開始需使用乙醇與丙酮 清洗表面,之後用氮氣吹乾,但是此時鈦片表面是粗糙不平的,所以我們 必須先經電解拋光後,可使鈦片表面具有光學級之平整度。鈦片的電解拋 光液主要成份為過氯酸(HClO4 )、甲醇(CH3OH) 、單丁醚乙二酯 (HOCH2CH2OC4H9)等三種溶液,本研究中純鈦之電解拋光液比例為 5 % 過氯酸 + 53 %單丁醚乙二酯 + 42 % 甲醇,外加電壓為 31V,拋光時間為 6min,實驗溫度控制在 1℃,拋光完成後鈦片表面就會有銀亮色。 拋光完鈦片將會開始第二階段的電化學實驗,而本研究中可以分為幾種 電解液,第一種電解液為實驗完後可以摻雜碳在奈米管中,電解液組成為 10wt%的 DI water+0.5wt%的 NH4F+89.5%的 ethylene glycol 或 10wt%的DI water+0.5wt%的 NH4F+89.5%的 glycerol[35,36],第二種為不含碳的奈
米管作法是將電解液改成體積比8:2 的 0.14M NaF:0.5M H3PO4[4],此種作
法奈米管中將會含有少量的磷。之後將要實驗的電解液置於超音波震盪器
[37]或者馬達攪拌器中,定電壓為 30V,溫度控制在 20℃,實驗完成後以 去離水沖洗,再用氮氣吹乾,然後在500℃高溫爐中退火三小時即可完成, 退火完後的二氧化鈦奈米管需放在乙醇中以超音波震盪器震盪30 秒清除表 面上的一些多餘氧化物。 3-2-2 鉑電極製備 將拋光完後的鈦片使用體積比 2:8 的 0.14M NaF:0.5M H3PO4當作電解液 電鍍產生奈米管,反應條件與製作含碳奈米管相同,反應完成且退火過後 將此奈米管放置於稀釋過後的H2PtCl6中(8wt%在水中),以超音波震盪器 震盪三分鐘後,用去離子水稍微沖洗之後以氮氣吹乾,之後在高溫爐中加 入氫氣還原鉑並控制溫度在500℃退火兩小時即可完成,此時的二氧化鈦奈 米管中將會有許多的奈米等級鉑粒子。 3-3 光電化學裝置與分析 光電化學分析是利用 CHI611C 電化學儀器以三極式系統進行,分別以二 氧化鈦奈米管為工作電極,鉑電極為相對電極,Ag/AgCl 為參考電極;電 解液為1M KOH 水溶液,光照光源為 1000W 的高能 Xe 燈,並搭配使用 UV(330±70nm,光源強度到達二氧化鈦奈米管為 13.9 mW/cm2)與 Visible(520±46nm,光源強度到達二氧化鈦奈米管為 5.27 mW/cm2) filter, 光電化學反應槽以側面照光,照光面材質為石英玻璃,其餘為PYREX 玻 璃,實驗裝置為圖(3-1),圖(3-2)則為二氧化鈦奈米管實驗裝置圖。 18
表3-1 化學藥品
藥品名稱 英文藥名 純度 廠商
鈦片 Titanium foil 99.95% Aldrich
甲醇 Methanol 99.9% Aldrich 乙醇 Ethanol 99.5% Aldrich 氟化銨 Ammonium fluoride 98% Aldrich 氫氧化鉀 Potassium hydroxide 99.5% Aldrich 丙三醇 Glycerol 99% Aldrich
乙二醇 Ethylene glycol 99% Aldrich
磷酸 phosphoric acid >60% Aldrich
過氯酸 Perchloric Acid 60~62% Aldrich
單丁醚乙二酯 2-Butyloxyethanol 99% Aldrich Ruthenium 535 N3 >99%, Mw=741.7 Riedel-de Haen 碘化鋰 Lithium iodide/LiI 99% Acros 碘 Iodine/I2 99.80% Acros
FTO Floride tin oxide Acros
表3-2 儀器與設備
儀器與設備 出廠公司/型號
高溫爐 LINDBERG/BLUEM
旋轉塗佈機 詠欣有限公司/TA-01
超音波震盪器 DELTA/DC400
X 光繞射儀 Bruker AXS D8 Advance/Leipzig Germany
掃瞄式電子顯微鏡(SEM) JEOL JSM-7401F FE-SEM
太陽能電池I-V 量測器 Keithley-2440 太陽光模擬器(包含電源供 應器和光源) 日本山下電裝/YSS-50A 馬達攪拌器 尚偉 電化學儀器 佳佑/CHI611C 1000 瓦高能 Xe 燈系統 SCIENCETECH 直流電源供應器 佳佑 20
1 7 8 6 5 4 3 2 1. CHI611C 電化學儀器 2.1000 瓦高能 Xe 燈系統 3. 光電化學槽 4. 工作電極 5. 參考電極 Ag/AgCl 6. 相對電極(Pt 鉑薄片) 7. 光強度偵測器(Power meter) 8. 升降台(Support jack) 圖(3-1) 光電化學反應裝置圖 21
4
5
6
3
2
1
1.冷卻系統 2.直流電源供應器 3.馬達攪拌器 4.鉑電極 5.工作電極(二氧化鈦奈米管) 6.化學反應槽 圖(3-2)二氧化鈦奈米管實驗裝置圖 223-4 分析儀器簡述
3-4-1 X 光繞射儀(X-ray Diffraction Spectrometer,XRD)
X 射線是一種波長很短的電磁輻射,範圍介於 0.1~100Å 之間。當 X 射線 被晶體內有規則的環境散射時,散射的光線間會發生干涉現象(同時發生建 設性干涉與破壞性干涉),因此產生了繞射。我們可以利用布拉格方程式 (Bragg’s law),計算其晶格間的距離,同時也可以由繞射峰的半高寬計算出 結晶區塊的大小。 布拉格方程式(Bragg’s law): 2dsinθ=nλ 其中,n :為整數值 θ : 繞射波峰的布拉格角(Bragg Angle) λ: X 光的波長(1.5405Å) d : 結晶面間的距離 此外,XRD 亦可用來量測樣品結晶顆粒的大小,依據 Debye-Scherrer sequation 求出平均之二氧化鈦的結晶粒子大小,其計算公式如下: D=0.9λ/B cosθ 其中,D :平均晶粒大小 λ: X 光的波長(1.5405Å) B : 繞射峰的半高寬 θ : 繞射波峰的布拉格角(Bragg Angle) 23
3-4-2 掃描式電子顯微鏡(Scanning Electron Microscope,SEM) 利用電子槍產生電子束後,以約 0.2~40KV 電壓予以加速,經過柵極聚焦 及電磁透鏡,匯聚成電子束,接著聚焦於試片表面。在末端電磁透鏡上裝 有掃描線圈,可偏折電子束,使其在試片表面作二度空間掃描。當電子束 撞擊試片表面時,會產生向後散射的墊子及自材料中被激發射出的二次電 子(secondary electron) 。這些電子產生的狀態與材料表面的凹凸、物質種 類及電位有關。將這些電子訊號利用檢測器(detector)接收並經放大器放 大,再送到顯示螢幕上成像。由於掃描線圈上的電流與螢幕上的電流同步 變化,所以試片表面之任意點所產生的訊號與顯示螢幕的亮點會逐一對 應,因此亮點組合的成像可呈現該試樣的特徵與形貌。
3-4-3 紫外光/可見光吸收光譜儀(UV/Visible Absorption Spectrometer)
偵測光波長在 190~1100nm 範圍內化合物分子外層軌道電子躍遷的能
量,一般可做定性、定量分析、光的吸收量與偵測物物濃度之間的關係, 可用藍伯特-比爾定律(Lambert-Beer Law)表示,如下式 :
A= log Io/I=εbc
其中A 為吸收度,I0為入射光強度,I 為透射光(transmitted light)強度,ε
為吸收光係數(extinction coefficient),b 為光路徑長度(light path),通常以 cm 為單位,c 為待測物的莫耳濃度(molar concentration)。待測物的吸光係
數ε 在特定波長時為定值,因此根據藍伯特-比爾定律,可由吸收強度 A 推
測待測物的濃度。
3-4-4 X 射線光電子能譜儀(X-ray Photoelectron Spectrometer,XPS) 其原理為一束 X 光射線打到樣品上,藉由光電效應將某一能階的電子激 發出來成為光電子,然後再分析這些光電子的能量,可以獲得關於表面元 素組成以及元素的化學環境。當數個原子鍵結成一化合物時,其在化合物 中的結合能明顯與純原子時不同,其內核層電子的束縛能也會有不同的變 化,及一般的化學位移(Chemical Shift) ,分析束縛能的變化,便可獲得材 料中的電子結構與化學鍵結的訊息。其所造成的能譜位移可用來區來不同 氧化態之元素。偵測深度約為5nm(除氫元素外皆可偵測) 。 3-4-5 太陽光模擬器(Solar Simulator AM 1.5) 由模擬太陽光的入射光源,為 AM1.5 且入射光的日照強度為 100mw/cm2。一般測試環境上所採用標準測試條件為:在 AM1.5(Air Mass 1.5) ,且太陽能電池溫度為 25℃時日照量為 1kw/m2之情況。我們通常定 義一乾淨無污染的環境下,且太陽位於天頂時為AM1,而 AMX 約略近似 為AM1/cosθ,其中 θ 為太陽與天頂所夾的角度。在進行太陽能電池之 I-V 量測時,光源是置於電池上方約12 公分處,將待測之電池元件置於模擬光 源下且將兩電極利用外部線路外接至電位電流控制儀,經由儀器自動化量 測後再經計算,即可得到電池元件之操作表現,包含開路電壓、短路電流、 光電轉換效率等數值。 25
第四章 結果與討論
4-1 陽極化結果與分析
首先我們先探討電化學反應後的一些物理特性,圖(4-1)為電化學拋光完 後的鈦片表面,從EDS 的元素分析表(4-1)可以知道此時的鈦片表面已經有 一層平坦且緻密的氧化鈦了,而且因為拋光液中成分的關係也使得內部含 有一些碳的成分。 圖(4-1) 鈦片拋光圖 26表(4-1) 拋光鈦片 EDS 元素分析
Element Weight% Atomic%
C K 0.85 2.28 O K 23.6 47.22 TiK 75.55 50.50 接下來的奈米管形成反應,我們先使用不同的電壓並紀錄電流的變化 量,從圖(4-2)可以發現一開始的起始電壓會非常高,接下來會急據的下降, 再來就慢慢的趨於平緩,三個不同反應電壓的電流隨時間曲線非常的相 似,我們可以合理推論三個反應其實都是前面章節有提到的反應式1~3,唯 一差別就是電壓造成電流較大而已。 Time(min) 0 10 20 30 40 50 60 Curren t(mA) 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 (a) (b) (c) 圖(4-2) 不同電壓奈米管的電流變化圖(a)30V (b)50V (c)70V 27
從圖(4-2)得知電流會圍著時間呈現階段性的下降,因此我們改為固定電 壓30V,電解液使用 10wt%的 DI water+0.5wt%的 NH4F+89.5%的 glycerol,溫度控制在 20℃,我們可以從 SEM 影像圖(4-3)觀察到隨著陽極 化時間增加,鈦片表面開始產生奈米孔洞的變化,可以發現在25 分鐘後才 有明顯的奈米管陣列出現,當陽極化時間持續增加,奈米管表面其實並不 會有太大變化而只是會增加奈米管長度。因此我們也針對陽極化電壓對奈 米管孔洞半徑以及管璧厚度做一系列的測試與紀錄。測試時我們使用固定 的電解液為10wt%的 DI water+0.5wt%的 NH4F+89.5%的 glycerol,溫度 控制在20℃,從圖(4-4)可以明顯發現當電壓伏特數加大時,孔洞半徑會隨 著增大但是管璧厚度卻沒有影響[38],圖(4-5)的紀錄就有很明顯的趨勢顯示 出來。為此我們另外改將電壓控制為30V,電解液一樣為 10wt%的 DI water+0.5wt%的 NH4F+89.5%的 glycerol,從圖(4-7)可以看到溫度才是影 響管璧厚度的主要因素[39],但是增加差異不大。而我們也可以發現改變實 驗溫度半徑也會增加,但是觀察圖(4-6)可以看到奈米管表面並不太一樣, 因為溫度會影響反應的生成速度,因此對於實驗溫度我們主要是著重於能 長出最好品質的奈米管為主,對於控制半徑大小我們還是會利用外加電 壓。根據上述的數據表示,可以長出最整齊乾淨的奈米管的條件為控制溫 度在20~25℃,長度隨著時間而半徑隨著電壓而呈正比成長。 28
29
(a) (b)
(c) (d)
(e) (f)
圖(4-3) SEM 影像顯示定電壓 30V TiO2奈米管結構變化;(a)1 min (b)5 min
30 (a) (b) (c) (d) 圖(4-4) 不同電壓的 SEM 奈米管半徑圖(a)50V (b)70V (c)90V (d)110V Anodization Potenial(V) 20 40 60 80 100 120 A v e rage Tub e Dia m ete r and w a ll Th ickness (nm) 0 20 40 60 80 100 120 140 160 180 200 (a) (b) 圖(4-5) 電壓對(a)奈米管孔洞半徑(b)管璧厚度造成的變化
31 (a) (b) (c) (d) 圖(4-6)不同溫度 SEM 奈米管管璧圖 (a)8℃ (b)13℃ (c)18℃ (d)23℃ Av era ge Tube Diame ter and w a ll Th ickness (nm) 10 20 30 40 50 60 Temperature(oC) (a) (b) 0 5 10 15 20 25 30 圖(4-7)溫度對(a)奈米管孔洞半徑(b)管璧厚度造成的變化
在章節 4-1 所看到的二氧化鈦奈米管 SEM 圖,都是已經使用乙醇在超音 波震盪器中清洗過後的結果,圖(4-8)可以看到電化學製備完的奈米管表面 其實都會有滿多氧化物分布,若不清除掉會嚴重影響我們水分解的效率, 若只用去離子水清洗奈米管表面,發現表面的氧化物並沒有辦法完全清除 掉,因此我們多使用乙醇再做第二次的清潔,可以看出使用乙醇在超音波 震盪器中震盪30 秒後就將表面氧化物都清除掉了。 32 (a) (b) (c) 圖(4-8) 奈米管表面清潔圖 (a)清洗前 (b)去離子水清洗後 (c)乙醇清洗後
再來我們將製作完成後的二氧化鈦奈米管做 XPS 資料分析,圖(4-9)為
C1s 的 XPS 圖,我們將它積分後可以發現有兩根 peak,我們從 XPS
database[40]先列出幾根 peaks,C-C(285.3ev)、C-O(286.5ev)、
C-O-O(289ev)、C-Ti(281.9ev) ,再對照我們的 XPS 圖可發現 peak1 是
graphitized carbon(C-C),peak2 應為 doped carbon(C-O),相對含量比例
接近2:1,而在 281ev 的位置卻無 C-Ti 的 peak 出現。
292 290 288 286 284 282 280 0 500 1000 1500 2000 Coun ts Binging Energy(ev) Pk1=285.4 69.6% Pk2=286.7 30.4%
圖(4-9) C1s XPS 圖 Pk1 is for graphitized carbon, and Pk2 is for doped carbon
4-2 自製鉑電極結果
使用自製鉑電極當作陰極是為了希望可以降低實驗上使用的經費,因為 若是直接買鉑金屬價錢將會差10 倍以上,圖(4-10)為自製後的成果,表(4-2) 為EDS 顯示出有鉑的粒子存在,圖(4-11)則是比較使用 100%鉑電極與使用 自製鉑電極當作陰極是否有差別,從圖中可以看出自製鉑電極測量出來的 結果會比100%的鉑電極小一些,因此若是就價錢方面考量的話,也可以使 用自製的二氧化鈦奈米管參雜鉑粒子當作陰極。 圖(4-10) 自製鉑電極 SEM 圖 34表(4-2)Pt/TiO2的元素分析
Element Weight% Atomic%
C K 1.9 4.32
O K 36.12 61.72
P K 0.17 0.15
Ti K 58.39 33.33
Pt K 3.42 0.48
Applied Potential V(Ag/AgCl)
-1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6
Photocurrent(mA/cm
2)
0 1 2 3 4 5 6 Pt-TiO2vsC-TiO2 Pt vsC-TiO2 圖(4-11) Pt vs Pt-TiO2紫外光區效率比較圖 354-3 水裂解效率結果
本實驗所做的水裂解效率實驗主要是參考 Misra 的論文[4], The photoconversion efficiency (
η
) of the photoanodeη (%) = [ (total power output − electrical power input) / light power input ] × 100= Jp [ E0rev − Eapp ) / I0 ] × 100
Jp = photocurrent density (mA/cm2)
JpE0rev = total power output JpEapp = electrical power input
I0 = power density of incident light (mW/cm2)
η (%) = Jp [E0rev − Eapp ) / I0 ] × 100 E0rev = 1.23 VNHE, standard rev. potential Eapp = Emeas − Eaoc
Emeas = electrode potential ( vs Ag/AgCl) of the working electrode Eaoc = electrode potential ( vs Ag/AgCl) of the working electrode at open circuit condition 而在水裂解實驗中,因為實際上全光譜光源的水分解效率仍然偏低,所以 我們分為UV 區與 VIS 區來量測,UV 區是大家已知二氧化鈦本來就會吸收 的區域,而VIS 區是因為要測試含碳的二氧化鈦是否真的會紅位移至可見 光區以增加我們水分解效率,圖(4-12)為我們選擇的 UV(330±70)與 VIS(520±46) filter 的穿透率,實驗的光源為 1000W 的 Xe 燈,在通過 UV
filter 後光強度為 9.28mw/cm2,VIS filter 為 5.36 mw/cm2。
Wavelength(nm) 100 200 300 400 500 600 700 800 900 T% -20 0 20 40 60 80 100 UV filter Wavelength(nm) 100 200 300 400 500 600 700 800 900 T% -20 0 20 40 60 80 100 VIS filter 圖(4-12) UV 與 VIS filter 波長穿透率圖 37
當有了一些基本資料後,我們先比較當二氧化鈦奈米管有含碳跟沒有含 碳時的電壓跟效率的關係圖,含碳的二氧化碳奈米管電解液主要為Glycerol or Ethylene Glycol,不含碳的主要為 H3PO4,實驗溫度控制在 23℃,時間 為一小時,實驗完成後以去離子水沖洗並退火3 小時,從圖(4-13)可以知道 含有碳的二氧化鈦奈米管在分解水系統中不管哪個反應條件的電壓下UV 區效率都大於不含碳的,而且都有趨勢為電壓越高效率會往下降,當然含 碳奈米管效率變高可知道是因為碳與二氧化鈦晶格會產生一些鍵結,使得 二氧化鈦本身能階變的更小,讓電子更容易傳遞出去,而電壓越高其實在 最底層的緻密氧化鈦會變的較厚,會讓電子傳遞不易反而造成效率變低, 因此若二氧化鈦奈米管表面沒有要再另外反應如生成一些半導體,其實並 不需要做大半徑的二氧化鈦奈米管,而可見光區沒有什麼變化,且幾乎可 以說是沒有什麼吸收,之後我們會對這部份再做另外討論。 38
Voltage(V) 20 30 40 50 60 70 80 90 100 Ef ficien cy (% ) 0 5 10 15 20 25 UV(Glycerol) VIS(Glycerol) UV(H3PO4) VIS(H3PO4) 圖(4-13)含碳(Glycerol)與不含碳(H3PO4)的二氧化鈦奈米管光解水效率比較 圖 當知道含碳二氧化鈦奈米管的確對水分解效率有幫助後,我們便測試不 同溫度鍛燒的奈米管效率,希望可以比較不同晶形的二氧化鈦與碳鍵結反 應後是否會有更好的效率。所以我們便將電壓控制在30V,電解液主體一
樣為Glycerol or Ethylene Glycol,實驗溫度控制在 23℃,實驗完成以去離
子水沖洗後退火3 小時,從圖(4-14)可以發現,不管實驗長度多久的 UV 區
效率遠遠高於VIS 區,這結果與圖(4-13)效率相符合,不同溫度鍛燒的 VIS
區,效率幾乎集中在0.14~0.16%,而在 UV 區就可以明顯看出退火 500oC
效率比較高,從圖(4-15)的 XRD 圖可以知道二氧化鈦在不同退火溫度時的
晶格變化,在475℃時二氧化鈦的晶格還不明顯所以效率在幾個實驗條件中 相對偏低,500℃時 anaste 晶格明顯出現後效率就提高的非常多,之後隨著 溫度再升高rutile 的晶格也慢慢增加,可以發現當二氧化鈦結構有較多 anaste 時,效率是會比 Rutile 好許多,另外可以從圖(4-14)得知長成二氧化 鈦奈米管的時間在超過50 分鐘後普遍效率都是下降的,而我們知道反應時 間越長奈米管長度也越長,對於是否是因為長度關係造成效率下降我們也 將反應時間變的更長,實驗結果記錄為圖(4-16) ,對於這個結果我們知道 一般半導體在水分解系統中會吸收低於能階波長的光進而產生電子電洞 對,電洞會被擴散到近奈米管表面跟水反應,而這擴散的距離是很小的(在 二氧化鈦中不超過200nm) [41,42,43],所以可以說光子幾乎都是在接近表 面附近就被吸收了,我們可以推論為奈米管越長在水分解系統中反而會增 加電子電洞對重新結合的機會,且從水分解原理可以得知我們並不需要像 敏化染料太陽能電池一樣需要大面積來吸附染料,所以我們並不需要做太 長的二氧化鈦奈米管,因此我們可以說水分解實驗中最適合的反應時間為 40~50 分鐘。 40
Time(min) 20 30 40 50 60 70 Effi ci enc y (% ) 0 5 10 15 20 25 30 475oC 500oC 525oC 550oC 575oC 600oC 625oC (a) Time(min) 20 30 40 50 60 70 Efficiency (% ) 0.10 0.12 0.14 0.16 0.18 0.20 475oC 500oC 525oC 550oC 575oC 600oC 625oC (b) 圖(4-14) 1M KOH 不同溫度奈米管光解水效率圖(a)UV 區(320±70nm)效率 (b)Vis 區 (520±30nm) 效率 41
20
30
40
50
60
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
2 - t h e t a
A R
475
oC
500
oC
550
oC
575
oC
600
oC
625
oC
圖(4-15) 不同退火溫度二氧化鈦晶格 XRD 圖 42Time(min) 0 50 100 150 200 250 300 Efficiency(% ) 0 5 10 15 20 25 30 35 圖(4-16) 紫外光分解水反應時間與效率圖 從圖(4-14)可知在水分解中有最好效率的退火溫度為 500℃,奈米管生成 時間為40~50 分鐘之間,而這些測試所使用的電解液全都是 1M 的 KOH, 所以最後我們改變另外新的電解液看看是否會有更高的效率。而我們測試 的電解液主要為1M 的 KOH、0.24M Na2S.XH2O+0.35MNa2SO3及1M KCl。圖(4-17)從三張效率圖中可以發現說使用 1M 的 KOH 當電解液效率 會優於其他兩種,合理推論為1M KOH PH 值為 12.6,電解液中離子性較 強使得反應較好,而三種電解液也都有低電壓條件做出來的奈米管效率都 會比高電壓的高,主要原因是當使用較高電壓條件做出來的二氧化鈦奈米 管底部的氧化層會比低電壓來的厚,而底部氧化層太厚會降低我們傳導電 43
子的效率,三種電解液都明顯的呈現出這種特性出來。因此單純就二氧化 鈦奈米管在水裂解的反應效應而言,最好的條件為使用低電壓為條件做出 來的二氧化鈦奈米管,鍛燒溫度為500℃,水裂解實驗中的電解液為 1M KOH 會有最好效率。 (a) 1M KOH Voltage(V) 20 30 40 50 60 70 80 90 100 E ffi ci en cy (% ) 0 5 10 15 20 25 44
(b) 0.24M Na2S.XH2O+0.35MNa2SO3 Voltage(V) 20 30 40 50 60 70 80 90 100 Eff ici e ncy (% ) 0 2 4 6 8 10 12 14 16 18 20 (c)1M KCl Voltage(V) 20 30 40 50 60 70 80 90 100 Effi ci en cy (% ) 0 2 4 6 8 圖(4-17) 不同電解液對電壓的效率比較圖。圓圈為 UV 區效率﹔三角為 VIS 區效率 45
4-4 TiO
2-xC
x奈米管對紫外光區與可見光區影響
在 4-3 節中我們根據 Misra 的結果來做一系列測試,而他們 UV 區效率為 13.3%,VIS 區效率為 8.5%。我們測試的 UV 區效率則為 28%,VIS 區效 率為0.15%左右,對於 UV 區水分解效率增加,但是 VIS 區卻降低的非常 多,這樣異常的結果與我們一開始的實驗動機相差非常多,因此我們將會 提出一些合理的解釋。首先我們根據Murphy[44]也對可見光效率提出的質 疑中說明應該要多一些證據如IPCE 或反射式吸收的效率來應證,從圖 (4-18)IPCE 效率可以看到當二氧化鈦奈米管參雜碳元素後,在紫外光區 IPCE 效率的確會有明顯的增加,可以增加到 40%左右,而其他不同的奈 米管可以發現光電轉換效率最高的波長大概都在320nm,在 400nm 後幾乎 都降到零了,此時就可以發現Misra 的水分解轉換效率公式就有點瑕疵, 因為他並沒有把IPCE 測試出來,所以我們合理假設他把紫外光區與可見 光區的光電轉換效率都當成100%了,但實際上就測試出來的結果並不是如 此,因此我們若使用Misra 的水分解轉換效率公式的話,最後應該要再乘 上我們的光電轉換效率,此時的可見光區水分解轉換效率將會非常的低, 正好符合我們自己的實驗結果,也代表了參雜碳元素並沒有辦法將吸收波 長延伸到可見光區,為此我們也測試了反射式吸收光譜,圖(4-19)也可以看 到在接近400nm 時吸收就會快速下降了。 46Wavelength(nm) 300 350 400 450 500 550 600 IPCE(% ) 0 10 20 30 40 50 P doped tube N doped tube
C deoped tube(Eehylene Glycol) C deped tube(Glycerol) 圖(4-18) 不同參雜元素的二氧化鈦奈米管 IPCE 效率圖 Wavelength(nm) 200 400 600 800 Abs o rp ti on(a.u .) 0.2 0.4 0.6 0.8 1.0 1.2
C doped TiO2 nanotbe TiO2 nanotube
圖(4-19) 含碳的二氧化鈦奈米管反射式吸收光譜圖
從圖(4-9)的 XPS 可得知的確是有不同型態的碳元素參雜在二氧化碳奈米 管中,而就水分解效率來看在可見光區的確是沒有明顯增加的吸收,因此 本實驗室也對含碳的二氧化鈦奈米管進行了一系列的理論計算,根據實驗 室雯妃學姊的計算結果與實驗結果的結合得知,圖(4-20)為摻雜碳元素後的 不同能階變化,但是(a)的能階圖為可見光應該會有吸收的結果,這與我們 的實驗效率不符合,而(b)為可見光區不會有影響但紫外光區會有吸收的增 加,因此(b)應該才是我們實驗後的能階變化圖。此外再根據圖(4-21)實驗室 的理論計算得知,我們摻雜的碳原子是屬於interstitial 的碳原子,因為這 樣的鍵結屬於能量最低且最穩定,而圖(4-21)的(b)可以看出當碳與氧與鈦原 子鍵結時,有一小根peak 出現,這表示碳原子對紫外光的確有紅位移的影 響。 (a) (b) 圖(4-20) 參雜碳元素的能階圖[18] 48
-12 -11 -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 0 1 2 pσ pσ #3O pπ pπ t2g (d) C #1Ti, #2T Energy (eV) total DOS 1 3 2 圖(4-21) 含碳二氧化碳奈米管理論計算結果圖[45] 49
4-5 InN 對二氧化鈦奈米管影響
從上述結果討論我們已經說明了含碳的二氧化鈦奈米管在水分解系統中 並無法有效吸收可見光,因此我們嘗試了另一個方法為使用PECVD 系統 在二氧化鈦奈米管上長成InN,希望可以利用 InN 的半導體特性增加我們 的水分解效率,首先我們先觀察到了圖(4-22) ,當我們奈米管半徑越大時 卻發現InN 的粒子一樣只會吸附在管徑旁邊,似乎並無法有效的滲入到管 徑內。 50 (a) (b) (c)
圖(4-22) InN 在不同奈米管半徑生成 SEM 圖 (a)30V(D=50nm) (b)50V(D=70nm) (c)70V(D=110nm)
而這三種不同半徑生成的InN 二氧化鈦奈米管我們也在三種電解液中測 試水分解效率,其結果如圖(4-23)。可以發現效率並沒有增加甚至還會比不
長成InN 時還小一點,推論其原因為因為使用 PECVD 系統所生成的 InN
為氣相沉積,在二氧化鈦奈米管上只是吸附並沒有很強的鍵結,因此在離 子性很高的電解液中會很容易被分解反應成In(OH)3,當我們測試完成後從 電解液取出就可以發現表面從有InN 的咖啡色變成一般二氧化鈦奈米管的 銀色了,所以在水分解系統中InN 並無法達到我們想要的半導體特性之前 就被分解掉了,因此效率並無增加。 (a) 1M KOH Voltage(V) 20 30 40 50 60 70 80 Efficiency (%) 0 5 10 15 20 25 51
(B) 0.24M Na2S.XH2O+0.35MNa2SO3 Voltage(V) 20 30 40 50 60 70 80 Efficiency (% ) 0 2 4 6 8 10 12 14 16 18 20 (c)1M KCl Voltage(V) 20 30 40 50 60 70 80 Ef ficiency( %) 0 1 2 3 4 5 6 7 圖(4-23) 不同電解液對電壓的 InN 二氧化鈦奈米管效率比較圖。圓圈為 UV 區效率﹔三角為 VIS 區效率 52
此外我們也利用含碳的二氧化碳奈米管吸附染料(N3)後測量光電轉換效 率,從前面的實驗可知當增加長成二氧化鈦奈米管的時間,其奈米管長度 也會隨之增加,而目前我們測量的是電化學長成二氧化鈦奈米管時間為1 到5 小時,因為目前當實驗時間超過 5 小時時,奈米管會呈現斷裂的情形, 經由SEM 得知是因為奈米管的長度可能太長但強度不夠導致斷裂,使得表 面會形成一些氧化物堆積造成電子傳導不佳使得整體效率下降。圖(4-24)為 不同實驗成長時間的含碳奈米管在泡N3 dye 24 小時後的 I-V 曲線圖,可以 發現效率隨著二氧化鈦奈米管的實驗時間增加也隨之上升,而目前測到的 最高效率為實驗時間五小時的條件,效率有 3.8%左右。 之後我們試著在泡染料之前先浸泡在 TiCl4的溶液裡,實驗條件為先泡在 0.2M TiCl4溶液裡,溫度控制在 50℃一小時,之後退火溫度為 300℃ 30 分 鐘,然後再泡一次0.1M TiCl4溶液,溫度控制在 50℃ 30 分鐘,退火溫度為 450℃ 30 分鐘,最後就是再經由泡 N3 dye 24 小時後測量光電轉換效率,從 圖(4-25)可以得知浸泡 TiCl4溶液對效率的確有明顯的效率增加,主要是因 為在退火時附著在奈米管表面的TiCl4會氧化變成小顆粒的TiO2,如此一 來會增加染料吸附的表面積,因此效率就提高了,而從表(4-3)與表(4-4)比 較得知效率可以比單純泡N3 dye 的結果增加到 0.5%左右。 53
表(4-3) TiO2-xCX nanotube/N3 dye 其光伏參數列表 Time(hr)* 1h 2h 3h 4h 5h Jsc(mA/ cm2) 7.95 9.58 10.4 10.9 11.9 Voc(volt) 0.707 0.691 0.682 0.674 0.645 η(%) 2.71 3.21 3.43 3.57 3.80 * Time: 二氧化鈦奈米管的實驗時間 Voltage(V) 0.0 0.2 0.4 0.6 0.8 I(m A /c m 2 ) 0 2 4 6 8 10 12 14 1h 2h 3h 4h 5h
圖(4-24) TiO2-xCX nanotube/N3 dye I-V 曲線圖
*Time: 二氧化鈦奈米管的實驗時間
表(4-4) 泡 TiCl4後的TiO2-xCX nanotube/N3 dye 其光伏參數列表 Time(hr)* 1h 2h 3h 4h 5h Jsc(mA/ cm2) 9.3 11.4 12.0 12.6 12.9 Voc(volt) 0.712 0.680 0.683 0.676 0.649 η(%) 3.18 3.73 3.98 4.12 4.32 *Time: 二氧化鈦奈米管的實驗時間 Voltage(V) 0.0 0.2 0.4 0.6 0.8 I( m A /c m 2 ) 0 2 4 6 8 10 12 14 1h 2h 3h 4h 5h
圖(4-25) 泡 TiCl4後的 TiO2-xCX nanotube/N3 dye I-V 曲線圖
*Time: 二氧化鈦奈米管的實驗時間
從圖(4-25)可知在泡染料之前經過 TiCl4的前處理會增加效率,之後我們 希望使用PECVD 系統在二氧化鈦奈米管上長成 InN,使我們的效率更增 加,因此二氧化鈦奈米管在經過TiCl4的前處理後,會在PECVD 系統裡長 成InN,實驗溫度為 85℃,時間為 20 分鐘,濃度比則是 NH3 :20 sccm、 In(CH3)3 : 6sccm,最後泡在 N3 dye 24 小時,圖(4-26)為實驗條件 5 小時的 二氧化鈦奈米管結果,顯示結果當長成InN 時卻沒有明顯的變化。 Voltage(V) 0.0 0.2 0.4 0.6 0.8 I( mA /cm 2 ) 0 2 4 6 8 10 12 14 TiO 2 nanotube InN/TiO2 nanotube
圖(4-26) InN/TiO2-xCX nanotube/N3 dye I-V 曲線圖
最後我們發現將長成 InN 的二氧化鈦奈米管在泡染料之前再經過一次
TiCl4的處理後,效率就有一些提升,條件為將樣品泡在 0.1M TiCl4 後放入
冰箱5~7 個小時,取出後放入烤箱退火溫度為 150℃ 30 分鐘,圖(4-27)為實
驗結果,可以發現電流大約增加0.5~1 mA,電壓會增加 0.03 V,效率可再
提高0.3%。
表(4-5) TiCl4/InN/TiCl4的TiO2-xCX nanotube/N3 dye 其光伏參數列表
Time(hr)* 1h 2h 3h 4h 5h Jsc(mA/ cm2) 9.8 11.978 12.72 13.25 14.459 Voc(volt) 0.742 0.704 0.714 0.6993 0.68 η(%) 3.54 3.95 4.30 4.36 4.60 *Time: 二氧化鈦奈米管的實驗時間 Voltage(V) 0.0 0.2 0.4 0.6 0.8 I(mA/c m 2 ) 0 2 4 6 8 10 12 14 1h 2h 3h 4h 5h 57
圖(4-27) TiCl4/InN/TiCl4的 TiO2-xCX nanotube/N3 dye I-V 曲線圖
第五章 結論
在許多生成二氧化鈦奈米管的方法中,電化學製程的確有許多優點,當 電解液的成分比例確定後,只要控制我們實驗的電壓、溫度與時間便可隨 時調整我們要的物理變化(奈米管半徑、管璧厚度、長度) ,實驗的步驟相 對其他方法簡單許多,但是由於電解液的成分需控制得宜才能成長出好的 二氧化鈦奈米管,這也造成了一個缺點就是在同一種電解液裡我無法改變 奈米管含碳比例,因此如要比較碳含量的話可能需要就必須要另外的電解 液才行。 而參雜碳元素的二氧化鈦奈米管的確在紫外光區會有很明顯的效率增 加,但是對可見光區還是沒有幫助,從IPCE 與反射式吸收圖譜可以看到 雖然含碳的二氧化鈦奈米管的確會有紅位移,但是在400nm 之後就幾乎為 零了,因此造成的效果並不明顯,從可見光的水分解效率圖就可看出目前 的含碳奈米管還是無法達到我們要的目的,此外理論計算也支持了我們的 實驗結果,我們的含碳二氧化鈦奈米管是屬於interstitial 的碳原子,因為 這樣的鍵結能量最低且最穩定,而這種結構就是在可見光區沒有吸收的。 所以就Misra 在可見光區的分解水效率可達 8.5%,我們可以說是他計算效 率的一些人為錯誤,此論點也與Murphy 的結果一致。 所以我們試著利用 PECVD 系統在奈米管上生成 InN,但是氣相沉積的 58InN 在水分解系統中的電解液無法有效存活,當電解液效率圖可以得知 pH 值越高離子性越強則水分解效率會越好,但是pH 值越高電解液會越容易跟 In 生成 In (OH)3,如此一來我們想要的 InN 半導體特性也被破壞了,所以 並不建議利用PECVD 系統生成的 InN 在水分解系統中測試。 而我們也試著將長有 InN 的二氧化鈦奈米管運用在 N3 dye 的測試,就整 個實驗而言效率雖然有增加但是InN 的影響目前似乎還沒有很明顯,就這 部份可以提出幾個想法,首先從SEM 圖可以看出 InN 都只散布在二氧化鈦 奈米管的表面,由於InN 可能與二氧化鈦奈米管沒有產生很好的鍵結,因 此有可能在泡染料時InN 也被反應掉了,導致最後實驗效率沒有增加,再 者有可能二氧化鈦奈米管本身的品質還不夠好,並無法有效反應出InN 的 效用,應該試著其他的方法提高二氧化鈦奈米管的長度,若是本身的效率 可以先大幅增加,也許就可看出InN 的作用了,而從圖(4-27)的效率增加得 知浸泡低溫的TiCl4溶液可能使二氧化鈦奈米管與InN 的連結變好,因此最 後的總結論就是若是想要N3 dye 的效率增加,需先提高二氧化鈦奈米管本 身的品質(強度與長度),再來就是讓二氧化鈦奈米管與 InN 的連結變好,相 信這樣整體效率應該就會有明顯增加了。 59
參考文獻
[1] http://www.solar-i.com/know.html#12
[2] A. R. Chourasia, D. R. Chopia, Thin Solid Films, 266, 1995, 298~301
[3] A. Fujishima, K. Honda, Nature, 238, 1972, 37-38
[4] S.K. Mohapatra, M. Misra, V.K. Mahajan, K.S. Raja, J. Phys. Chem. C, 111, 2007, 8677-8685
[5] George F. , Metallography and microstructures, American Society of Metal, 9, 1985
[6] P. A. Jacquet, Electrolytic polishing of metallic surface, 5, 1949, 48
[7] P. A. Jacquet, Electrolytic polishing of metallic surface, 2, 1950, 55
[8] P. V. Schigolev, Electrolytic and chemical polishing of metal, Freund , Holon , Israel , 1970.
[9] M. Adachi, Y. Murata and M. Harada M and S. Yoshikawa., Chem. Lett., 29, 2000, 942
[10] S. Z. Chu, S. Inoue, K. Wada, D. Li, H.Haneda, J. Phys. Chem. B, 107, 2003, 6586
[11] G.K. Mor, K. Shankar, M. Paulose, O.K. Varghese, C.A. Grimes, Nano Lett., 5, 2005, 191-195
[12] R. L. Kurtz, R. L. Stockbauer, T. E. Madey, E. Roman, J. L. deSegovia, Surf. Sci, 218, 1989, 178
[13] Iijima S., Nature, 354, 1991, 56
[14] H. Nakamura and Y. Matsui, J. Am. Chem. Soc., 117, 1995, 2651-2651
[15] C. N. R. Rao, B. C. Satishkumar, A. Govindaraj, E. M. Vogl and L. Basumallick, J. Mater. Res., 12, 1997, 604
[16] B.B. Lakshmi, C.J. Patrissi and C.R. Martin, Chem. Mater., 9, 1997, 2544
[17] S. Kobayashi, K. Hanabusa, N. Hamasaki, M. Kimura and H. Shirai, Chem. Mater., 12, 2000, 1523
[18] C.D. Valentin, G. Pacchioni, A. Selloni, Chem. Mater,17, 2005, 6656-6665
[19] G.K. Mor, Oomman K. Varghese J. Mater. Res., 18, 2003, 2588~2593
[20] J. Bai, B. Zhou, L. Li, Y. Liu, J. Mater Sci, 43, 2008, 1880-1884
[21] Q. Cai, M. Paulose, O.K. Varghese, C.A. Grimes, J. Mater. Res., 20, 2005, 230-236
[22] G.K. Mor, K. Shankar, M. Paulose, O.K. Varghese, C.A. Grimes, Nano Lett., 5, 2005, 191-195
[23] Q. Cai, L. Yang, Y. Yu, Thin Solid Films, 515, 2006, 1802-1806
[24] H. Wang, C.T. Yip, K.Y. Cheung, A.B. Djuriic, M.H. Xie, Appl. Phys. Lett., 89, 2006, 1-3
[25] K. Zhu, N.R. Neale, A. Miedaner, A.J. Frank, Nano Lett., 7, 2007, 69-74
[26] K.S. Raja, M. Misra, K. Paramguru, Electrochimica Acta, 51, 2005, 154-165
[27] J.M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer, P. Schmuki, Current Opinion in Solid State and Materials Science,11, 2007,
3-18
[28] J. Tao, J. Zhao, C. Tang, Y. Kang, Y. Li, New J. Chem., 32, 2008, 2164-2168
[29] S. Yoriya, M. Paulose, O.K. Varghese, G.K. Mor, C.A. Grimes, J. Phys. Chem. C, 111, 2007, 13770-13776
[30] M. Gratzel, Nature, 414, 2001, 338~344
[31] A.J. Nozik, R. Memming, J. Phys. Chem., 100, 1996, 100, 13061-12078
[32] S.K. Mohapatra, V.K. Mahajan, M. Misra, Nanotechnology, 18, 2007, 1-6
[33] G.K. Mor, O.K. Varghese, M. Paulose, K. Shankar, C.A. Grimes, Solar Energy Materials&Solar Cells, 90, 2006, 2011-2075
[34] J.H. Park, S. Kim, A.J. Bard, Nano Lett., 6, 2006, 24-28
[35] A.Valota, D.J. LeClere, T. Hashimoto, P. Skeldon, G.E. Thompson, S. Berger, J. Kunze, P. Schmuki, Nanotechnology, 19, 2008, 1-7
[36] J.M. Macak, P. Schmuki, Electrochimica Acta, 52, 2006, 1258-1264
[37] S.K. Mohapatra, M. Misra, V.K. Mahajan, K.S. Raja, Journal of Catalysis, 246, 2007, 362-369
[38] Q. Cai, M. Paulose, O. K. Varghese, C. A. Grimes, J. Mater., 20, 2005, 230-236
[39] G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese, C. A. Grimes, Nano Lett., 5, 2005, 191-195
[40] K. Shankar, G. K. Mor, H. E. Prakasam, S. Yoriya, M. Paulose, O. K. Varghese, C. A. Grimes, Solar Energy Material & Solar Cells, 90, 2006,
63
2011-2075
[41] A. B. Murphy, P.R.F. Barnes, L.K. Randeniya, I.C. Plumb, M.D. Horne, Journal of Hydrogen Energy, 31, 2006, 1999
[42] M. F. Weber, M.J. Dignam, J. Electrochem Soc., 131, 1984, 1258-1265
[43] J. R. Bolton, S.J. Strickler, J.S. Connolly, Nature, 316, 1985, 495-500
[44] A. B. Murphy, Solar Energy Materials&Solar Cells, 92, 2008, 363-367