行政院國家科學委員會專題研究計畫 成果報告
兒童慢性 B 型肝炎病毒感染自然病程影響因子之長程研
究:病毒量及病毒全長基因變化之探討(3/3)
研究成果報告(完整版)
計 畫 類 別 : 個別型 計 畫 編 號 : NSC 95-2314-B-002-019- 執 行 期 間 : 95 年 08 月 01 日至 96 年 07 月 31 日 執 行 單 位 : 國立臺灣大學醫學院小兒科 計 畫 主 持 人 : 張美惠 計畫參與人員: 學士級-專任助理:李慧娟 報 告 附 件 : 出席國際會議研究心得報告及發表論文 處 理 方 式 : 本計畫可公開查詢中 華 民 國 96 年 09 月 03 日
行政院國家科學委員會補助專題研究計畫
☑ 成 果 報 告
□期中進度報告
(計畫名稱)
兒童慢性 B 型肝炎病毒感染自然病程影響因子之長程研究:
病毒量及病毒全長基因變化之探討(3/3)
計畫類別:
☑
個別型計畫 □ 整合型計畫
計畫編號:NSC 95-2314-B002-019
執行期間: 95 年 8 月 1 日至 96 年 7 月 31 日
計畫主持人:張美惠教授
共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告
☑
完整報告
本成果報告包括以下應繳交之附件:
☑
赴國外出差或研習心得報告一份 (附件一)
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:台大小兒科
中 華 民 國 96 年 8 月 31 日
2 中文摘要 第一部份:慢性B 型肝炎病毒感染兒童在 e 抗原抗體轉變自然史中之病毒量變化。 目的背景:本研究探討慢性B 型肝炎病毒(HBV)感染兒童在 e 抗原抗體轉變自然史中之血清 病毒量變化。方法:58 名慢性 B 型肝炎病毒感染兒童合乎下列標準:進入本研究時 ALT 值 正常,追蹤10 年以上,無抗病毒藥物治療,追蹤期間 e 抗原抗體自然轉變。依據其 e 抗原抗 體轉變後之HBV DNA 濃度將之分為二組:(1)低病毒血症組:一過性或者從未有>104個病毒 /ml(35 人),(2)浮動性高病毒血症組:至少有兩次相隔一年以上>104個病毒/ml(23 人)。腹部 超音波,ALT 值及 HBV DNA 值每年至少偵測一次。另有 14 名未發生 e 抗原抗體轉變者作為 對照組。HBV 核前區(nt1896)基因及基因型亦被測定。結果:58 名研究組兒童最初 HBV DNA 為108.4±1.0個/ml,最後降至 102.9±2.0個/ml。他們進入本研究時,最高 HBV DNA 值時,最高 ALT 值時,e 抗原抗體轉變時,及最後的年齡分別為 7.0±3.7,13.4±5.8,16.3±6.0,17.2± 5.8,及 23.7±4.1。HBV 核前區變異在浮動高病毒血症組高於低病毒血症組(45.5%對 7.1%, P=0.006)。B 型肝炎基因型對於病毒量無明顯影響。在 e 抗原抗體轉換後,平均最高 ALT 值
為26U/L,無人呈現持續 ALT 值續 ALT 值上昇現象。結論:一般而言,這些年輕時發生 e
抗原抗體轉變者病毒量會下降,ALT 值正常,轉變病程平順。未來更長程的追蹤研究將有助 於了解在兒童及青少年期e 抗原抗體轉變的意義。 第二部份:慢性B 型肝炎病毒感染及肝癌兒童所分離出之 B 型肝炎病毒全長基因之 研究。目的背景:過去對於長程追蹤之慢性B 型肝炎病毒感染兒童及兒童肝癌患者 B 型肝炎 病毒的全長基因變化並不瞭解。方法:21 名病程不同之 B 肝病毒慢性感染兒童及 7 名肝癌兒 童進入本研究。以PCR 及定序方法測定其 B 肝病毒全長基因序列。以建立基因樹的方法,我 們分析B 肝病毒序列在上述二組兒童之異同,並與國際文獻所報告的亞洲肝癌成人 B 肝病毒 序列作比較。結果:基因樹分析發現肝癌兒童與慢性B 型肝炎病童之 B 肝病毒序列分別聚集 在不同的羣落。並在表面抗原前二基因(Pre-S2)12 個核苷酸及其他多個特定區發現肝癌兒童特 有的B 肝病毒序列刪除的現象。結論:多數肝癌兒童的 B 肝病毒基因相當特別,含有核前二 區基因刪除等特別的序列,並與慢性B 肝帶原兒童的序列不同,值得注意。 關鍵詞:B 型肝炎病毒(HBV),HBV DNA,B 型肝炎 e 抗原抗體轉變,全長序列,種系演化 樹,慢性B 肝病毒感染,肝細胞癌
ABSTRACT
PART I : Viremia Profiles in Children with Chronic Hepatitis B Virus Infection and Spontaneous e Antigen Seroconversion
Aim/Background: This study investigated the viremia profiles in children with
chronic hepatitis B virus (HBV) infection and spontaneous hepatitis B e antigen (HBeAg) seroconversion. Methods: 58 children with chronic HBV infection met the following criteria: normal alanine aminotransferase (ALT) at enrollment, followed for >10 years, no antiviral treatment, and having undergone spontaneous HBeAg
seroconversion during follow-up. They were grouped according to the post-HBeAg seroconversion HBV DNA levels: (i) low viremia: transient or never ≥104 copies/ml (n=35) (ii) fluctuating high viremia: ≥104 copies/ml detected at least twice at intervals >1 year (n=23). Abdominal sonography, ALT, and HBV DNA levels were assessed annually. Another 14 non-seroconverted children served as controls. The precore mutant (nt1896) and genotypes were examined. Results: The initial HBV DNA level of the 58 seroconverters was 108.4±1.0 copies/ml and decreased to 102.9±2.0 copies/ml at the end of follow-up. Their mean ages at enrollment, peak HBV DNA, peak ALT, HBeAg seroconversion, and final follow-up were 7.0±3.7, 13.4±5.8, 16.3±6.0, 17.2±5.8, and 23.7±4.1 years, respectively. The precore mutant appeared more often in the fluctuating high viremia group than in the low viremia group (45.5% vs. 7.1%, p=0.006). HBV genotypes had no effect on the viremia profiles. After HBeAg seroconversion, the median peak ALT was 26 U/L and none had persistent abnormal ALT levels.
Conclusions: Generally, these young seroconverters had decreased viral loads,
normal ALT levels, and uneventful courses after HBeAg seroconversion during our follow-up. A longer follow-up is necessary to elucidate the significance of HBeAg seroconversion occurring in childhood and young adulthood.
Part II. Full-Length Hepatitis B virus genomes isolated from children with chronic hepatitis B virus infection and hepatocellular carcinoma
Aims & Background: The changes of the whole HBV genome associated with
different courses of childhood chronic hepatitis B (HBV) infection and HBV-related hepatocellular carcinoma (HCC) remain unclear and were under investigation in this study. Methods: We collected the serum samples from 21 children with different clinical courses of chronic HBV infection and from 7 children with HCC. The full-length HBV genomic sequences were obtained by PCR and sequencing method. Using phylogenetic tree construction, we analyzed the difference between HBV genomes from childhood HCC and those from other two groups of patients with either childhood chronic HBV infection or adult HCC. Results: Phylogenetic
4
analysis indicated that the majority of HBV genomes from children with HCC clustered in a specific class that was distinguishable from HBV genomes of either chronic HBV infection or adult HCC. Sequence analysis identified a consensus 12-bp pre-S2 deletion and other characteristic mutations throughout the genome.
Conclusion: The majority of HBV genomes isolated from children with HCC
contain unique mutations, such as a short pre-S2 deletion, that may be associated with early HCC development.
Key words: hepatitis B virus (HBV), HBV DNA, hepatitis B e antigen seroconversion,
full length sequence, phylogenetic tree, chronic hepatitis B vidrus infection, hepatocellular carcinoma
PART I. VIREMIC PROFILES IN CHILDREN WITH CHRONIC HEPATIITS B INFECTION AND SPONTANEOUS e ANTIGEN SEROCONVERSION
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a health problem worldwide.1 In hyperendemic areas such as Taiwan, most chronic HBV infection begins in infancy and early childhood.2, 3 It usually leads to a chronic infection,4 which may result in serious complications in adult life.5,6 After HBV acquisition in infancy or childhood, the virus replicates actively in the initial years, which is an immune-tolerant stage with high HBV DNA levels and normal alanine aminotransferase (ALT) levels in most cases. Whereas adults generally show a gradual loss of tolerance to HBV and enter an immune-clearance stage, 7 there are few viremia profile reports in children. The host-virus interaction leads to acute exacerbation and subsequent hepatitis B e antigen (HBeAg) seroconversion. Previously, we observed the mutations of the precore gene,8 basal core promoter,9 and core gene deletion10 in the process of HBeAg
seroconversion in children.
Current treatment modalities for chronic hepatitis B seek to achieve HBeAg seroconversion, normal ALT, and a sustained suppression of the HBV DNA
replication.11 These goals can occur spontaneously or achieved with antiviral therapy. Some adult HBeAg seroconverters would experience rebound viral replication, and are generally regarded as difficult to manage.12 This study was aimed to investigate whether HBeAg seroconversion in childhood, adolescence, or young adulthood might 5
lower the HBV replication early and lead to a relatively benign course of chronic HBV infection.
Most of the previous studies have several pitfalls: (i) the blood sampling was
done mostly in a cross-sectional manner when ALT levels were elevated, which often reflects the results of the virus-host interaction; (ii) the study subjects were a biased diseased population, not the community-based population; and (iii) previous pediatric reports did not use real-time polymerase chain reaction (PCR), the most sensitive method to measure the viral load. A prospective cohort study recruiting a large
community-based population starting from the initial phase of HBV infection, perhaps in early childhood, would be more revealing. Such a prospective study would allow us to observe the natural history and monitor the disease outcome in children,
adolescents, and young adults who undergo spontaneous HBeAg seroconversion.
6
Children with chronic HBV infection were recruited from a community-based general population through (i) four cross-sectional seroepidemiological surveys of HBV markers conducted in 1984, 1989, 1994, and 1999,13 (ii) a prospective screening program for children of HBsAg seropositive mothers, and (iii) the
outpatient clinic of the National Taiwan University Hospital as part of a prospective study that began 25 years ago. A physical examination, blood tests for ALT, HBV seromarkers (including HBsAg, anti-HBs, anti-HBc, HBeAg, anti-HBe), α-fetoprotein, and abdominal sonography were done at each visit at 6-month intervals.
Of these children with chronic HBV infection, 72 became the subjects of this study. Of these, 58 met the following criteria and were enrolled in this study: (i) HBeAg positive and normal ALT at enrollment, (ii) age at enrollment <15 years, (iii) follow-up duration >10 years, (iv) no antiviral treatment given, and (v) underwent spontaneous HBeAg seroconversion to anti-HBe during follow-up. The other 14 patients served as the control group. They met the same criteria except the last: they were persistently HBeAg positive and did not undergo HBeAg seroconversion. The sex ratio and follow-up duration of the control group were similar to those of the study group (Table 2). HBV DNA was quantified using real- time PCR at least once each year. The study protocol was approved by the Institutional Review Board of the National Taiwan University Hospital, and the patients themselves or their guardians signed the informed consent to collect serum samples.
We subdivided the 58 seroconverters into two groups based on their
post-seroconversion serum HBV DNA levels: (1) the low viremia group had viral loads of persistently <104 copies/ml (n=35) and (2) the fluctuating high viremia group had occasional episodes of viremia >104 copies/ml more than 6 months after HBeAg seroconversion (n=23). We used 104 copies/ml as the cut-off because this is the current indication for antiviral treatment for HBeAg-negative hepatitis patients.14 To ensure that the high viremia episodes were not insignificant, transient phenomena, we defined the fluctuating high viremia group as having two or more episodes of >104 copies/ml in an interval of more than 1 year.
HBV serological markers. The HBV seromarkers were measured using enzyme
immunoassays (EIA, Abbott Laboratories, North Chicago, IL, USA). ALT levels were determined by an autoanalyzer (Hitachi 7450, Tokyo, Japan).
HBV DNA quantification using real-time PCR. The detailed nucleotide sequences
and PCR procedures were described previously.15, 16 Briefly, HBV DNA was
extracted from 50 ul of serum and the nucleic acids were re-dissolved in 50 µl of H2O and subjected to PCR. The primers cover HBV nucleotide positions 1261-1279 and
1600-1580. The anchor probe is at nucleotide positions 1552-1576 and the sensor probe is at nucleotide positions 1533-1550 of HBV DNA. The real-time PCR measurement was performed by using LightCycler analysis software 3.5 (Roche Diagnostics Applied Science, Mannheim, Germany). The linear range of HBV DNA was 102-1011 copies/ml and the sensitivity of this method was 5x102 copies/ml of HBV in serum.
HBV genotyping. The latest serum samples of the 58 seroconverters were analyzed.
Briefly, the HBV genotypes were analyzed by using PCR with type-specific primers. The procedures were described previously.
Detection of the precore 1896 stop codon mutant. The latest serum samples of the
58 seroconverters were used to detect the precore 1896 stop codon mutant. Nested PCR was performed using two pairs of primers covering the HBV precore and core regions. The procedures were described previously.
Statistics. To compare the low viremia, fluctuating high viremia, and
nonseroconverter groups, the following statistical methods were used. The Chi-square test with Yates’ correction was used to analyze the variables gender and precore mutant. Fisher’s exact test was used to analyze the genotype. The Kruskal-Wallis test was used to compare the peak ALT levels with ALT levels expressed as the median and range. HBV DNA levels were log transformed and subjected to Student’s t test. A
p <0.05 was considered statistically significant. The data were expressed as the mean
± standard deviation.
RESULTS
The general profiles of the serum HBV DNA of the 58 HBeAg seroconverter
children are described in Table 1. The majority (77.6%, 45/58) of these patients were thought to have acquired the HBV infection from their mothers because their mothers were HBsAg positive. They initially had a high serum HBV DNA levels (108.4±1.0 copies/ml), which rose further to peak levels of 109.1±0.9 copies/ml, and then decreased to 102.9±2.0 copies/ml at the end of follow-up. Generally, the
seroconverters had a peak HBV DNA level at the mean age of 13.4 years, followed by a peak ALT level at 16.3 years, and then HBeAg seroconversion occurred at 17.2 years of age (Table 1 and Fig. 1). There were exceptions: (1) the peak ALT occurred before the peak HBV DNA in seven cases and the interval between these two events ranged from 0.2 to 6.8 years; (2) the peak HBV DNA occurred 6.6 and 1.0 years after HBeAg seroconversion in two cases; and (3) the peak ALT occurred after HBeAg
8
seroconversion in seven cases at intervals ranging from 0.2 to 15.1 years. No children underwent HBsAg seroconversion at the final follow-up. The mean age at enrollment and the age at the final follow-up of the non-seroconverted children in the control group were 8.7±4.5 and 25.2±3.5 years, respectively. Their peak ALT and highest and lowest HBV DNA levels did not differ from those of the seroconverters in the
pre-HBeAg seroconversion phase (Table 2). All of the children are still being
followed and none of them show any signs of decompensated liver diseases, or HCC on physical examination, blood tests, and abdominal ultrasound.
HBV DNA and ALT profiles before and after HBeAg seroconversion. HBV DNA
and ALT profiles before and after HBeAg seroconversion of these 58 seroconverter children are described in Table 2.
Post-HBeAg seroconversion low viremia group. A transient viremia of >104
copies/ml was found in 21 of the 35 cases (60%) in the low viremia group. Such events occurred an average of 2.0±1.5 years after HBeAg seroconversion.
Post-HBeAg seroconversion fluctuating high viremia group. In the post-HBeAg
seroconversion phase, the first up-surge viremia of >104 copies/ml occurred 2.1±2.2 years after HBeAg seroconversion; the second episode of high viremia came
4.1±2.8 years after HBeAg seroconversion. There were 3.3±1.3 episodes of high viremia during the 9.2±5.5 year follow-up period after HBeAg seroconversion in the fluctuating high viremia group. All of the viremia episodes fluctuated above 104 copies/ml, but never persisted for longer than 1 year.
Precore mutant. This mutant was detected in 38% of the seroconverters (22/58).
Fourteen seroconverters with the precore mutant were in the fluctuating high viremia group (14/23, 60.9%), which was much higher than the rate in the low viremia group (8/35, 23%; Table 3). Due to the longer follow-up duration after HBeAg
seroconversion in the former group, there was concern that the emergence of the precore mutant simply reflected the natural history, i.e., the longer the duration of infection after seroconversion, the more likely the precore mutant is to occur. A multiple logistic regression analysis was performed, and the precore mutation was found to be significantly associated with the fluctuating high viremia vs. the low viremia group (odds ratio=4.4, 95% confidence interval =1.3-14.3, p=0.016), but not the follow-up duration after HBeAg seroconversion ≥ 9 years vs. <9 years (odds ratio=2.3, 95% confidence interval=0.56-9.09, p=0.25). Nine years is the mean follow-up duration for the fluctuating high viremia group, and was used as the
cut-off. The viremia profile, rather than a longer follow-up duration, was the significant independent factor associated with the precore mutation.
In the fluctuating high viremia group, there were six cases with an abnormal ALT level after HBeAg seroconversion and four of them had the precore mutant (see below). Of the remaining 17 cases with a normal ALT in this group, 10 had the precore mutant. The prevalence of the precore mutant did not differ between those with normal and abnormal ALT levels in the fluctuating high viremia group (p=0.73, Fisher’s exact test with Yates’ correction). In the low viremia group, four of the six cases with an abnormal ALT had the precore mutant. Of the 29 cases with a normal ALT level in the low viremia group, only four cases had the precore mutant. The precore mutant was more prevalent in those with an abnormal ALT (Fisher’s exact test with Yates’ correction, p=0.005).
Abnormal ALT in fluctuating high viremia group after HBeAg seroconversion.
Six patients had these characteristics. In the four that had precore mutants, their ALT continued to fluctuate, but never exceeded 80 U/L after HBeAg seroconversion. Their peak HBV DNA levels were 104.6, 106.7, 105.7 and 107.4 copies/ml, respectively. The remaining two patients lacked the precore mutant and had mild ALT elevations after HBeAg seroconversion of 54 and 56 U/L, respectively. Their peak HBV DNA levels were 104.6 and 104.3 copies/ml, respectively.
Maternal HBsAg. Forty-five mothers of the 58 seroconverters were HBsAg positive
and the children were thus presumed to have acquired the HBV infection perinatally. There was no difference between the children infected perinatally or horizontally in terms of the age of HBeAg seroconversion (16.8±5.5 vs. 19.0±6.8 years, p=0.22). However, none of the horizontally infected children (0/13) had an ALT flare-up after HBeAg seroconversion as compared with nine of the perinatally transmitted children who had an abnormal ALT after HBeAg seroconversion (9/45; p=0.08, Fisher’s exact test).
DISCUSSION
This prospective long-term follow-up study illustrated the natural course of serum HBV DNA levels and their relationship to ALT levels and HBeAg
seroconversion in children and young adults. A high HBV DNA viral load is found universally in the immune-tolerant phase in children with chronic HBV infection. The immune-clearance phase follows, during which ALT flare-ups occurred frequently and the peak ALT usually occurred after the peak HBV DNA.18We determined the average intervals between the successive peak HBV DNA levels, peak ALT levels,
10
and HBeAg seroconversion in children and adolescents in the natural history of chronic HBV infection (Fig. 1).
The current goals of antiviral treatments are to achieve sustained low viral replication after HBeAg seroconversion. How many subjects with spontaneous HBeAg seroconversion in childhood, adolescence, or young adulthood eventually develop a flare-up viremia (>104 copies/ml) and become HBeAg negative chronic hepatitis B victims?We found that 40% (23/58) of the seroconverters had more than one flare-up of HBV DNA levels and 20% (12/58) of them had ALT flare-ups after spontaneous HBeAg seroconversion. Nevertheless, all the peak ALT levels were less than two times the upper limit of normal. Therefore, none of these young
seroconverters, who might experience both persistent high viremia episodes and an occasional ALT flare-up, met the current indications for antiviral treatment as a case of HBeAg-negative chronic hepatitis. We need to continue monitoring these patients to see if they develop persistent liver damage later in life.
The precore 1896 stop codon mutant is an important factor for persistent viral replication after HBeAg seroconversion, and may prevail in one-half to two-thirds of HBeAg-negative children and adults with chronic HBV infection in Taiwan. 8,9,19An abnormal ALT after HBeAg seroconversion is associated with the emergence of the precore mutant in adults.20 As expected, this mutant appeared more frequently (60.9%) in the fluctuating high viremia group than in the low viremia group (22.9%). We recognized its association with an abnormal ALT level in the low viremia group but not in fluctuating high viremia group. It is likely that fluctuating high viremia itself is important enough in inducing an abnormal ALT and minimized the role of the precore mutant. Continuous monitoring of such patients, including the ALT and viremia profiles, can help to elucidate the role of the precore mutant in the post-HBeAg seroconversion phase.
In conclusion, children with chronic HBV infection carry a high viral load in the immune-tolerant phase, and it decreases dramatically after they undergo HBeAg seroconversion. Forty percent of these spontaneous seroconverters still had fluctuating high viremia (>104 copies/ml) after HBeAg seroconversion. The precore 1896 mutant after HBeAg seroconversion is more prominent in fluctuating high viremia group. None of them warranted treatment after HBeAg seroconversion. The outcome of these young HBeAg seroconverters appeared to be relatively benign during our follow-up, however, continuous follow-up is necessary to make a final conclusion.
1. Beasley RP, Stevens CE. Epidemiology of hepatitis B virus infection in Taiwan. In Sung JL, Yu JY, Wang TH, eds. “Proceedings of the International Symposium on Hepatitis in Taipei”. Taipei, Gastroenterologic Society of the Republic of China, pp. 1-10.
2. Hsu HY, Chang MH, Chen DS, Lee CY, Sung JL. Baseline seroepidemiology of hepatitis B virus in children in Taipei, 1984: A study just before mass hepatitis B vaccination program in Taiwan. J Med Virol 1986; 18: 301-307.
3. Stevens CE, Beasley RP, Tsui J, Lee WC. Vertical transmission of hepatitis B antigen in Taiwan. N Engl J Med 1975;292:771-774.
4. Chang MH. Chronic hepatitis virus infection in children. J Gastroenterol Hepatol 1998;13:541-548.
5. Chu CM. Toward control of hepatitis B in the Asia –Pacific Region- Natural history of chronic hepatitis B virus infection adults wit emphasis on the occurrence of cirrhosis and hepatocellular carcinoma. J Gastroenterol Hepatol 2000; 15: E25-30.
6. Yuen MF, Lai CL. Toward control of hepatitis B in the Asia –Pacific region- Natural history of chronic hepatitis B virus infection. J Gastroenterol Hepatol 2000; 15: E20-24.
7. Lee PI, Chang MH, Lee CY, Hsu HY, Chen JS, Chen PJ, Chen DS. Changes of serum hepatitis B virus DNA and aminotransferase levels during the course of chronic hepatitis B virus infection in children. Hepatology 1990; 12: 657-660. 8. Chang MH, Hsu HY, Ni YH, Tsai KS, Lee PI, Chen PJ, Hsu YL, Chen DS.
Preocore stop codon mutant in chronic hepatitis B virus infection in children: Its relation to hepatitis B seroconversion and maternal hepatitis B surface antigen. J Hepatol 1998;28: 915-922.
9. Ni YH, Chang MH, Hsu HY, Tsuei DJ. Longitudinal study on mutation profiles of core promoter and precore regions of hepatitis B virus genome in children. Pediatr Res 2004;56:396-399.
10. Ni YH, Chang MH, Hsu HY, Chen HL. Long-term follow-up study of core gene deletion mutants in children with chronic hepatitis B virus infection. Hepatology 2000;32:124-128.
11. Lok ASF, McMahon BJ. Chronic hepatitis B: update of recommendations. Hepatology 2004;39:857-861.
12. Hadziyannis SJ, Papatheodoridis GV. Hepatitis B e antigen-negative chronic hepatitis B: natural history and treatment. Semin Liver Dis 2006;26:130-141. 13. Ni YH, Chang MH, Huang LM, Chen HL, Hsu HY, Chiu TY, Tsai KS, Chen DS.
Hepatitis B virus infection in children and adolescents in a hyperendemic area: 15 years after universal hepatitis B vaccination. Ann Intern Med 2001; 135:796-800.
12
14. Liaw YF, Leung N, Guan R, Lau GKK, Merican I, McCaughan G, Gane E, Kao JH, Omata M for the Asian-Pacific consensus update working party on chronic hepatitis B. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2005 update. Liver Int 2005;25:472–489.
15. Ni YH, Chang MH, Wang KJ, Hsu HY, Chen HL, Kao JH, Yeh SH, Jeng YM, Tsai KS, Chen DS. Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology 2004;127:1733- 1738.
16. Yeh SH, Tsai CY, Kao JH, Liu CJ, Kuo TJ, Lin MW, Huang WL, Jih J, Chen DS, Chen PJ. Combined real time PCR quantification and signature single nucleotide polymorphism genotyping of hepatitis B virus in one-tube reaction. J Hepatol 2004;41:659-666.
17. Naito H, Hayashi S, Abe K. Rapid and specific genotyping system for hepatitis B virus corresponding to six major genotypes by PCR using type-specific primers. J Clin Microbiol 2001;39:362-364.
18. Liu CJ, Chen PJ, Lai MY, Kao JH, Chang CF, Wu HL, Shau WY, Chen DS. A prospective study characterizing full-length hepatitis B virus genomes during acute exacerbation. Gastroenterology 2003; 124:80-90.
19. Lin CL, Liao LY, Liu CJ, Chen PJ, Lai MY, Kao JH, Chen DS. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J Gastroenterol 2002;37:283-287.
20. Chu CM, Yeh CT, Lee CS, Sheen IS, Liaw YF. Precore stop mutant in HBeAg-positive patients with chronic hepatitis B: clinical characteristics and correlation with the course of HBeAg-to-anti-HBe seroconversion. J Clin Microbiol 2002;40:16-21.
PART II . UNIQUE HEPATITIS B GENEONS ISOLATED FROM
16
HEPATOCELLUALR CARCINOMA
Introduction
Chronic HBV infection is a grave issue of public health in hyperendemic regions such as Taiwan and other countries, since it can cause severe complications like liver cirrhosis and hepatocellular carcinoma (HCC) (1). Although human beings have known well the natural course of HBV infection, we still have limited
knowledge of the factors contributing to variable clinical course and outcome in different patients with chronic HBV infection. Since most chronic HBV infection begins during infancy or childhood (2) and tends to cause persistent infection (3) and more serious complications (4) than adults, the study in the pediatric patients is thus very important for identifying contributing factors of unfavorable outcome. The HBV genome is susceptible to mutation due to the spontaneous error of its reverse transcriptase (Gunther, 1999) and the lack of proofreading activity of the polymerase. As a consequence of a complex interaction of virus and host immune system, hot spots of mutation may gradually develop during the course of chronic infection. Of our special interest is the correlation between these hot spots of mutation and distinct clinical outcome of chronic HBV infection. In previous studies (5, 6, 7), it has been discovered that some specific HBV genotypes and mutations in the HBV genome have significant impact on the clinical course or outcome of chronic childhood HBV infection. For example, we have also found that genotype B dominates in children with both chronic HBV infection and HCC in Taiwan, while genotype C delays HBeAg seroconversion (6). It has also been found that children with earlier emergence of precore stop codon mutant (G1896A) tend to have more severe hepatocellular damage (5) and this mutant accounts for a half of childhood HBeAg seroconversion (7). In the core promoter region, higher rates of A1775G and G1799C mutations and a lower rate of A1752G mutation have been found in
childhood HBeAg seroconverters (7). In the core gene region, children with HCC have more mutations than those with chronic HBV infection; the former group has mutation hotspots at core gene codon 74, 87, and 159, while the later has mutation hotspots at core gene codon 21, 65, and 147 (8). However, previous studies have only focused on limited hot-spot domains, but a systemic analysis of the mutation status of the whole HBV genome and the establishment of its possible clinical relevance have not been achieved. Therefore, the purpose of this study aims at investigating whether a genome-wide comparison and phylogenetic analysis of HBV sequences isolated from children with different courses of chronic HBV infection, including those with HCC, can generate useful information pertaining to region or
sequence features that are specifically related to different consequences of chronic HBV infection in children. The knowledge obtained in this study will greatly
enhance our ability of predicting the outcome of chronic HBV infection and promote early therapeutic intervention for certain high-risk patients.
Patients and Methods
Collection and grouping of patients
a) Children with chronic HBV infection
We followed up 415 HBsAg carrier children who were less than 15 years longitudinally for liver function profiles and HBV markers. If their alanine
aminotransferase (ALT) levels increased up to more than 2 times of the upper limit of the normal value, monthly determination of HBeAg/anti-HBe and ALT levels were performed until normalization of ALT was reached. We performed full-length HBV sequencing using the blood from 21 age- and sex-matched children who were divided into 3 groups. They were selected according to the following criteria. For group 1, 7 children were randomly selected among HBeAg seropositive patients with acute exacerbation and a peak ALT > 400 IU/L followed by a subsequent HBeAg seroconversion. For group 2, 7 children will be randomly selected from initially HBeAg seropositive patients with a peak ALT level of < 200 IU/L, followed by subsequent HBeAg seroconversion. For group 3, 7 children were randomly selected from patients remaining HBeAg positive without seroconversion during follow-up. For patients in group 1 and group 2, three time points of blood
(enrollment, peak ALT levels, and final serum) were analyzed for full-length HBV DNA sequences (totally 21 blood samples for both groups). For group 3, only the initial and final blood samples were subjected to analysis for full-length HBV DNA sequences.
b) Children with HCC
From our HCC blood bank, we randomly selected 7 children with HBV-related HCC whose first diagnoses were made before the age of 15 years. The follow-up period ranged from 1 month to 24.5 years. All except one blood sample, which was obtained 9 years after the diagnosis of HCC, were withdrawn after the diagnostic confirmation of HCC.
18
The serum samples at enrollment and the latest serum sample available were submitted for HBV genotype analysis. The determination of HBV genotypes has been prescribed previously (6). Briefly, The HBV genotypes were analyzed by using polymerase chain reaction (PCR) with type-specific primers (9). For those samples in which viral loads were so low that HBV genotype could not be determined experimentally, we employed the viral genotyping tool in the National Center for
Biotechnology Information(NCBI) web site
(http://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi) to determine the
genotype.
Extraction of DNA from sera
Serum DNA was extracted from 200μl serum by using QIAamp DNA mini kit (Qiagen Inc, Valencia, CA) by following the instructions from the manufacturer. The DNA was dissolved in 100μl buffer TE.
Amplification of full-length HBV genome.
To amplify the full-length HBV genome, we performed the PCR with 3 different primer pairs as shown in Table1 to yield 3 overlapping sub-genomic fragments that covered the full length of HBV genome. The HBV nucleotides were numbered from the EcoRI site. The 50μl PCR mixture contained 10X buffer, 5X enhancer, 50mM MgCl2 , and 50XdNTP. The polymerase for PCR was the
Combizyme DNA polymerase (InViTek GmbH, Berlin, Germany). The PCR condition was performed as following: 96 for 2 min; 75 for 5 min; 94 for 40 ℃ ℃ ℃ sec, 57 for 40 sec, 72 for 120 sec, for 10 cycles; 94 for ℃ ℃ ℃ 40 sec, 55 for 40 ℃ sec, 72 for 120 sec, for 10 cycles; 94 for 40 sec, 53 for 40 sec, 72 for 120 ℃ ℃ ℃ ℃ sec, for 10 cycles; 94 for 40 sec, 51 for 40 sec, 72 for 120 sec, for 10 cycles; ℃ ℃ ℃ and finally 72 for 10 min. However, the primer pairs described above co℃ uld not amplify some samples with very low viral loads. Thus, we designed other primers as shown in Table 2 for low viral load samples and the same PCR condition to amplify the full-length HBV sequence. The PCR products were isolated by the
electrophoresis and were purified from the 1.5% agarose gel by Gel/PCR DNA Fragments Extraction Kit (Geneaid biotech Ltd. Taiwan).
Cloning of PCR products and isolation of plasmid DNA
The purified PCR products were subjected to cloning using the TOPO XL Cloning Kit (Invitrogen/Life Technologies, Carlsbad, CA) and transformed into
recombinant clones were selected and the plasmid DNA was extracted with Mini-M Plasmid DNA Extraction System (Viogene Biotek corp. Taiwan). The DNA samples were dissolved in the 50μl elution buffer. Finally, each plasmid DNA was confirmed by enzyme digestion with EcoR1 (New England Biolabs GmbH, Frankfurt,
Germany).
Sequencing and assembling of the full-length of HBV genome, sources of adult HCC full-length sequences, construction of consensus sequences, and
phylogenetic analysis
We performed DNA sequencing using M13 forward and reverse primers by ABI DNA sequencer (model3730, version 3.2, Applied Biosystems). We performed the assembling and analysis of HBV genome using the software SeqWeb Version 3.1 (Accelrys Software Inc.). We obtained 48 full-length HBV nucleotide sequences from three groups of sequences submitted to GenBank database. The accession numbers of the first group were AB014360-14390 (10); the numbers of the second group were AB241109, AB241113, AB241114, AB241116, and AB241117 (11); the numbers of the third group were AB113875, AB113876, and AB113877 (published only in GenBank database by Michitaka et al., 2004). The consensus sequences of HBV genomes from patients with either chronic HBV infection or adult HCC were constructed using the software SeqWeb 3.1 supplied by National Health Research Institute (Miaoli County, Taiwan). We carried out phylogenetic analysis by using Phylip version 3.6 provided by National Health Research Institutes (Miaoli County, Taiwan). We used Neighbor-Joining method to do phylogenetic analysis. To evaluate the statistical support, we performed 1,000 bootstrap replications.
Ethical considerations
The blood samples of patients in this study were withdrawn after we obtained the written informed consents from parents of the patients or patients themselves. The Ethical Committee of the National Taiwan University Hospital approved the protocol of study. All sera in this study were processed to protect personal information.
Statistic analysis
The frequency of mutation for each sub-genomic region was calculated by dividing the number of mutation by the number of base pairs of the region. The
20
frequencies of the highest one and the second highest one were then compared by a chi square test with Yates’ correction.
RESULTS
Clinical and laboratory data of patients
The clinical and laboratory profile of three groups (HBeAg seroconverters with high or low peaking ALT levels, and HBeAg non-seroconverters) of patients with chronic HBV infection and 7 children with HCC are listed in Table 3 and Table 4,
respectively. Totally 63 HBV full-length sequences were amplified, cloned, and sequenced successfully. Among them, 7 were from children with HCC and 56 were from 21 HBV chronically infected children whose blood samples were withdrawn 2 (enrollment and final follow-up) or 3 (enrollment, peak ALT, and final follow-up) times. In addition, we also retrieved 48 HBV genomes from sera of adult HCC published in GenBank for comparison.
Phylogenetic features of HBV genomes from different clinical settings
In the phylogenetic tree (Fig. 1) based on 111 full-length HBV nucleotide sequences, the HBV genomes from children HCC (H1-H7) were clearly diverged from those from patients with chronic HBV infection and were related to those from adult HCC. Moreover, HBV genomes from patient H1, H2, H3, H6, and H7 were more closely related to each other than to those from H4, and H5. The genomes of H1, H2, H3, and H7 were clustered on a branch that was separated from those of adult HCC patients with a significant bootstrap value (86 %). Although H6 sequence was classified as a neighboring branch to the one consisting of H1, H2, H3, and H7, indeed it was classified in a same cluster as H1, H2, H3, and H7 when only adult HCC isolates were included with 7 childhood HCC isolates to construct the phylogenetic tree (data not shown). To further minimize the region responsible for this classification, we constructed other phylogenetic trees (data not shown or supplementary data) using nucleotide sequences from 7 sub-genomic regions of HBV alone, including those encoding pre-S1 region, pre-S2 region, S region, and the region encoding the large S protein (including the pre-S1, pre-S2, and S), the P gene, the precore/core gene, and the X gene. A similar topology with high
relatedness of H1, H2, H3, and H7 was maintained within the regions of pre-S1, pre-S2, and the large S protein, but not in the regions of core and X genes. For the S region and the P gene regions, the topology was loosely maintained, with H1, H2, and H3 still clustered together, but their relationship with H4, H5, H6, and H7 changed. On the other hand, isolates of different groups of children with chronic HBV infection sampled at different time points did not form any cluster with
The nucleotide and amino acid characteristics of HBV genomes of H1, H2, H3, H6, and H7
Mutations of the pre-S and the S genes
Since HBV genomes of H1, H2, H3, H6, and H7 share more relatedness, we first focused our analysis on the sequence characteristics of these 5 isolates. By aligning these 5 genomes with the consensus sequence deduced from 56 HBV isolates of chronic HBV infection and with the consensus sequence deduced from 48 adult HCC, we identified some unique nucleotide positions with non-synonymous mutations or deletions (Table 5) in these 5 isolates. These mutations were chosen only when they caused amino acid substitutions in both consensus sequences from chronic HBV infection and adult HCC. In the pre-S1 region, we detected an early stop codon mutation (W4STOP) at nucleotide 2859 position in H1 sequence, which might abolish the synthesis of most of the pre-S1 domain. A similar stop codon mutation located more distally (nucleotide 3055) in the pre-S1 gene has been reported in patients with exacerbation of chronic HBV infection (Minami et al, 1993). The other pre-S1 mutation was found in the amino half of all 5 childhood HCC isolates. In the pre-S2 region, notably, the 4 isolates from childhood HCC (H1, H2, H3, and H7) had a same 12 base-pair deletion (nucleotide 44-55, pre-S2 amino acid, aa 19-22) in the pre-S2 region (Fig. 2). The range of deletion was smaller than those reported in a largest series of study (13). This deletion affected both the epitopes of B cells (aa 1-26) (14) and T cells (aa 21-30) (15) in the pre-S2 region. Other 6 non-synonymous mutations were found in the pre-S2 region. In the S gene, 4 patients (H1, H2, H3, and H7) shared 4 common non-synonymous mutations (G44E, P46Q, and T/V47K, which was generated by mutations of both nucleotide 293 and 294) located within a cytotoxic T lymphocyte (CTL) epitope region (aa 28-51) (16). Another 3 amino acid substitutions (S34L, F41S, T/V47A) also occurred in this region (Table 5), but only in one single patient individually. As for the major hydrophilic region (aa 99-169) (17) of HBsAg, 2 non-synonymous mutations (C121Y and S154P) were found in H7 and H2 sequences, respectively; neither of them was located in the region of immunodominant “a” determinant (18) of HBsAg.
Mutations of Precore/Core Genes
The analysis in the precore region showed that 2 reported mutations (G1862T and G1896A) (19-21) coexisted in 4 childhood HCC isolates (H1, H2, H3, and H7). Both G1862T (19) and G1896A (19, 21) mutations are associated with reduced capacity of HBV to produce hepatitis B e antigen (HBeAg) from its precore protein
22
precursor. The G1896A mutation is also associated with severe liver damage in patients with chronic hepatitis B (22). In the core gene, it is remarkable that all 5 childhood HCC patients (H1-3, H6, and H7) shared 2 mutations at nucleotide 2048 (P50A) and 2073 (A58E) in a region of CD4+T cell epitope (aa 48-69) (23). The mutation at nucleotide 2048 (core gene codon 147) is one of the 3 most frequently occurred mutation in chronic HBV infected children (8). For other known epitopes, there was 1 non-synonymous mutation (V13A) in the HLA-A2-restricted CD8+ CTL epitope (aa 18-27) (24-5) in isolates from patient H1, H2, H3, and H7, while there were 2 non-synonymous mutations (T147A and R151Q) in
HLA-Aw68-restricted CTL epitope (aa 141-151) (25) in patient H1, H3, H6, and H7. Within the hot-spot mutational domain (aa 80-120) reported to be related to severe liver diseases (26-28), we found 3 non-synonymous mutations (A80T, L84A, and Y118F) in patient H1, H3, H6, and H7.
Mutations of polymerase and X genes
The analysis of the polymerase (pol) gene of 5 isolates showed that the majority of non-synonymous mutations (Table 5) were located in the reverse
transcriptase (RT) (nucleotide 130-1161) and spacer domains (nucleotide 2838-129) (29). As in the pre-S2 region, there was a 4-amino acid deletion in the spacer domain of isolated from patient H1, H2, H3, and H7. In the RT domain of isolates from patient H1 and H2, there was a mutation at nucleotide position 880 (aa 251 of the RT gene) that changed glycine to a stop codon (G251STOP) and generated a truncated RT lacking both the c-terminal region of the RT domain and the RNase-H domain (nucleotide 1152-1623) (29). In the RNase-H domain, the isolate from patient H6 carried a non-synonymous mutation at nucleotide 1613 (rhR240K or R841K), which also cause a synonymous mutation in X gene open reading frame known to have a higher relative risk of HCC development (30). In addition, there were 2 mutations located in 2 HLA class I restricted T-cell epitopes of the pol gene (31): nucleotide 760 (A557S, patient H6 and H7) of YMDDVVLGA (aa 549-557), and nucleotide 828 (I579M, patient H1) of FLLSLGIHL (aa 573-581). Our 5 isolated did not carry the well-known rtM204I/V mutation in the YMDD motif (32) or other mutations in the pol gene associated with antiviral therapy (33-34). The analysis of X gene region revealed only 4 non-synonymous mutations in isolated from patient H1, H2, H3, and H7 (Table 5). We did not find other mutations or deletions in the X gene reported previously in patients with severe liver diseases (35) or with HCC (30, 36). Since parts of sequences in this region also function as core promoter and other regulatory elements, the mutations within them will be discussed in the following paragraphs. Mutational frequencies of different regions of the HBV genome
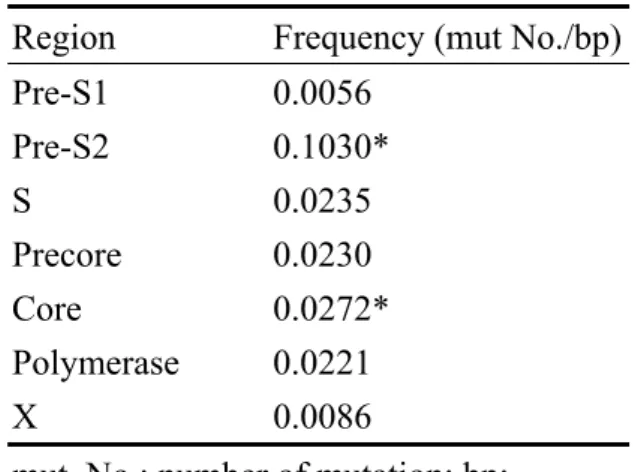
We compared the frequencies of non-synonymous mutations in different regions of HBV (pre-S1, pre-S2, S, precore, core, pol, and X genes) (Table 6) and found that the pre-S2 region had the highest mutation frequency (compared with that of the second highest one, the core gene, X2 = 16.677, P < 0.0001).
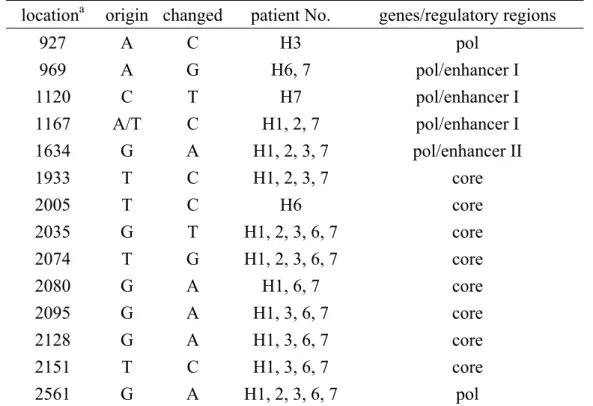
Mutations of the regulatory regions of HBV
In addition to the non-synonymous mutations, we also found 14 synonymous mutations in HBV genomes of the isolates from 5 children with HCC (Table 7). The analysis of both groups of mutations in the regulatory sequences of HBV showed that some of them might change the important regulatory sites. In the core
promoter/enhancer II region, the clustered mutations at nucleotide 1632, 1633, and 1634 (Table 8) of isolates from patient H1, H2, H3, and H7 were immediately next to a Sp1 site (nucleotide 1622-1631) (37). In addition, the HBV genomes of these 4 patients also had 2 mutations (nucleotide 1482 and 1566, Table 8) located in the negative regulatory element gamma and alpha (38), respectively. In the basal core promoter (BCP), the well-known BCP (A1762T, G1764A) mutation that increases that risk of HCC in HBV carries (36) existed in isolates from patient H4, H5, H6, and the majority of HCC adults, but not in those from patient H1, H2, H3, and H7. In the region of S1 and S2 promoters, 4 mutations (Table 8) were noted but were not close to any known transcription binding sites. In the enhancer/X promoter regions, we found 7 mutations (Table 8). Notably, 3 of them (nucleotide 1120, 1123, and 1128, all from patient H7) were located in the hepatocyte nuclear factor 3 (HNF3) binding site (nucleotide 1120-1129) (39). In addition, the retinoic acid responsive element (RARE, nucleotide 1136-1148) (40) in enhancer I of the isolate from patient H7 harbored one mutation (nucleotide 1138).
Discussion
Our phylogenetic analysis of the HBV genomes from children either with HCC or chronic HBV infection at different clinical settings, and from adult HCC available in GenBank indicated the emergence of a unique and childhood HCC-related cluster of HBV genomes. It suggested that selection pressure might be great enough in these patients to cause the genetic differentiation of HBV virus. In addition, the direct sequence comparison among these 3 groups of HBV genomes revealed several characteristic mutations/deletions specific to childhood HCC group. The most
striking one may be the consensus 12-bp in-frame pre-S2 deletion in 4 out of 7 cases. Recently several studies have shown significant association of pre-S deletions of HBV genomes with severe liver diseases, including HCC (13, 41-45). However, the reported pre-S deletions involve both the pre-S1 and pre-S2 regions but only the pre-S2 deletion was found in our cases. In HBV genomes from adult HCC, Chen et
24
al. (13) have identified multiple patterns of pre-S deletion. Our deletion, which was located in the N-half of the pre-S2 gene, can be classified as type V deletion
according to their system. Type V deletion comprises 29.3% of pre-S deletions found in adult HCC. Interestingly, 75% of their type V deletions are larger in length than our deletion and 58.3% of type V deletions contain our 12-bp deletion region. Therefore, it is probable that the deletion we found defines a smaller critical region, which when deleted may be linked to HCC pathogenesis. How does the internal pre-S2 deletion in HBV contribute to the development of HCC? One possibility is that it may compromise both B- and T-cell epitopes of the pre-S2 region (14, 15) and escape the immune-mediated viral clearance. Other plausible mechanisms include that the large surface protein containing the pre-S2 deletion (so called Delta S2-LHBs) may induce endoplasmic reticulum stress (46) that exerts genotoxic effects leading to cancer formation and that the Delta S2-LHBs can upregulate cyclinA expression and hepatocyte proliferation (47). It remains to be determined whether pre-S2 deletion is sufficient to generate real liver cancers in vivo. For confirming the role of pre-S2 deletion in HCC oncogenesis, it may be also necessary to screen the pre-S2 deletion in a larger cohort with chronic HBV infection and determine the relative risk of HCC development prospectively in those whom harbor these deletions.
The coexistence of both G1862T and G1896A precore (PC) mutations found in HBV genomes from 4 of 7 childhood HCC is consistent with previous reports showing that both mutations were associated with severe liver diseases and HCC (19-21). Interestingly, HBV genomes of these 4 patients also harbored the consensus 12-bp pre-S2 internal deletion. In contrast, we did not detect the well-known BCP (A1762T, G1764A) mutations (36) in these 4 cases, although BCP mutations were present in the majority of HBV genomes from adult HCC we obtained from GenBank and in one out of 7 isolates from group 1 chronic HBV-infected patients who had higher peak ALT. Recently it has been reported (13) that in HBV genomes of adult HCC, pre-S2 deletions rarely exist alone and coexist frequently with PC rather than with BCP mutations. Our results are in agreement with it and we did not find co-existence of BCP mutations and pre-S2 deletions in any case.
In other regions, we also found unique mutations of HBV genomes from childhood HCC that are rarely reported. Some of them may alter the immune
response of the host against the virus, since they were located in the epitopes of B or T cells. For example, in the of the majority of childhood HCC-related isolates, we found G44E, P46Q, and T/V47K in a CTL region (aa 28-51) (16) of the small surface protein, P50A and A58E in a CD4+ T cell epitope (aa 48-69) (23) of the core protein, and T147A and R151Q in a HLA-Aw68-restricted CTL epitope (aa 141-151)
(25) in the core protein. Other less frequently occurred mutations include A557S and I579M (Table 5) in class I restricted T-cell epitopes of polymerase (31). In addition, the childhood HCC-related HBV genomes also contained mutations in previously reported regions where mutations are associated with severe liver diseases and HCC (26-28, 30), such as aa 88 (R841K) (30) in the polymerase and the hot-spot
mutational domain (aa 80-120) (26-28) of the core protein (Table 5). Finally, in the P gene, we found several mutations in the domains of polymerase that interacts heat shock protein 90 (Hsp90) (48), including one common mutation (A137T, all 5 patients) in the C terminal domain (after aa 97) of the terminal protein (TP), and a premature stop codon mutation (G597STOP, patient H1 and H2) that abolishes the expression of the thumb region of RT domain (aa 616-680). By interacting with these 2 domains of polymerase, Hsp90 can associate with RT and facilitate the folding of RT into an active form. However, it remains to be determined to what extent the mutations change the interaction and what the biological consequences of the change are.
HBV mutations accumulate gradually during long-term chronic infection. What cause the early emergence of HBV mutants that possibly linked to HCC
development in these children? So far the answer is still vague, although the
interplay of genetic, immune, and environmental factors may be responsible for the pathogenesis. Most of these patients came to our clinics with initial manifestation of HCC without regular follow up in hospitals. However, we clearly know that they were likely infected vertically since all the mothers were HBV carriers. All patients were HBeAg seroconverters before the detection of HCC. No significant difference was seen in their HBV genotypes (B: C = 3: 4) although the cohort is small.
Interestingly, among these 7 patients, we found one whose blood analyzed was withdrawn before he developed detectable HCC, suggesting that these unique genomes may exist in the liver previously and may possibly participate in the process of carcinogenesis instead of merely being a biomarker of HCC. To confirm this hypothesis may require incorporation of more HBV genomes from both children and adults into our databank for phylogenetic analysis and a well-designed
prospective study that compares the clinical outcome between patients with distinct HBV genomes.
In conclusion, we identified unique HBV genomes in children with HCC. These HBV genomes share a 12-bp pre-S2 deletion and several characteristic
mutations that when in combination may be possibly related to early development of HCC. Moreover, our results indicated that analysis of the whole genome of HBV in more patients with chronic HBV infection may be helpful, especially when
26
mild and advanced liver diseases, since this comprehensive approach may identify unique HCC-related HBV genomes and those who harbor them may benefit greatly from early detection and therapeutic intervention of advanced diseases.
REFERENCES
1. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet. 1981;2:1129-33.
2. Hsu HY, Chang MH, Chen DS, et al. Baseline seroepidemiology of hepatitis B virus in children in Taipei, 1984 : A study just before mass hepatitis B vaccination program in Taiwan. J Med Virol 1986; 18: 301-7.
3. Stevens CE, Beasley RP, Tsui J, et al. Vertical transmission of hepatitis B antigen in Taiwan. N Engl J Med 1975; 292:771-4.
4. Chu CM. Toward control of hepatitis B in the Asia –Pacific Region- Natural history of chronic hepatitis B virus infection adults wit emphasis on the occurrence of cirrhosis and hepatocellular carcinoma. J Gastroenterol Hepatol 2000; 15: E25-30.
5. Chang MH, Hsu HY, Ni YH, et al. Preocore stop codon mutant in chronic hepatitis B virus infection in children : Its relation to hepatitis B seroconversion and maternal hepatitis B surface antigen. J Hepatol 1998; 28: 915-22.
6. Ni YH, Chang MH, Wang KJ, et al., Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology. 2004;127:1733-8.
7. Ni YH, Chang MH, Hsu HY, Tsuei DJ. Longitudinal study on mutation profiles of core promoter and precore regions of the hepatitis B virus genome in children. Pediatr Res. 2004;56:396-9.
8. Ni YH, Chang MH, Hsu HY, Tsuei DJ. Different hepatitis B virus core gene
mutations in children with chronic infection and hepatocellular carcinoma. Gut. 2003;52:122-5.
9. Naito H, Hayashi S, Abe K. Rapid and specific genotyping system for hepatitis B virus corresponding to six major genotypes by PCR using type-specific primers. J Clin Microbiol. 2001;39:362-4.
10. Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full-length isolates. Arch Virol. 1998;143:2313-26.
11. Sakamoto T, Tanaka Y, Orito E, Co J, Clavio J, Sugauchi F, Ito K, Ozasa A, Quino A, Ueda R, Sollano J, Mizokami M.Novel subtypes (subgenotypes) of hepatitis B virus genotypes B and C among chronic liver disease patients in the Philippines. J Gen Virol. 2006 Jul;87:1873-82.
12. Minami M, Okanoue T, Nakajima E, Yasui K, Kagawa K, Kashima K. Significance of pre-S region-defective hepatitis B virus that emerged during exacerbation of chronic type B hepatitis. Hepatology. 1993;17:558-63.
28
13. Chen BF, Liu CJ, Jow GM, Chen PJ, Kao JH, Chen DS. High prevalence and
mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology. 2006;130:1153-68.
14. Milich DR, Thornton GB, Neurath AR, Kent SB, Michel ML, Tiollais P, Chisari FV. Enhanced immunogenicity of the pre-S region of hepatitis B surface antigen. Science. 1985;228:1195-9.
15. Milich DR, McLachlan A, Chisari FV, Thornton GB. Nonoverlapping T and B cell determinants on an hepatitis B surface antigen pre-S(2) region synthetic peptide. J Exp Med. 1986;164:532-47.
16. Tai PC, Banik D, Lin GI, Pai S, Pai K, Lin MH, Yuoh G, Che S, Hsu SH, Chen TC, Kuo TT, Lee CS, Yang CS, Shih C. Novel and frequent mutations of hepatitis B virus coincide with a major histocompatibility complex class I-restricted T-cell epitope of the surface antigen. J Virol. 1997;71:4852-6. 17. Carman WF. The clinical significance of surface antigen variants of hepatitis B
virus. J Viral Hepat. 1997;4 Suppl 1:11-20.
18. Brown SE, Howard CR, Zuckerman AJ, Steward MW. Affinity of anti-peptides. Lancet 1984;2:184-187.
19. Parvez MK, Thakur V, Kazim SN, Guptan RC, Hasnain SE, Sarin SK Base-pair alterations in the epsilon-lower stem due to a novel double substitution in the precore gene of HBV-e negative variant were recovered by secondary
mutations.Virus Genes. 2001;23:315-20.
20. Hou J, Lin Y, Waters J, Wang Z, Min J, Liao H, Jiang J, Chen J, Luo K, Karayiannis P.Detection and significance of a G1862T variant of hepatitis B virus in Chinese patients with fulminant hepatitis. J Gen Virol. 2002;83:2291-8. 21. Lin CL, Liao LY, Liu CJ, Chen PJ, Lai MY, Kao JH, Chen DS. Hepatitis B
genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J Gastroenterol. 2002;37:283-7.
22. Rezende RE, Fonseca BA, Ramalho LN, Zucoloto S, Pinho JR, Bertolini DA, Martinelli AL. The precore mutation is associated with severity of liver damage in Brazilian patients with chronic hepatitis B. J Clin Virol. 2005 Jan;32(1):53-9. 23. Ferrari C, Bertoletti A, Penna A, Cavalli A, Valli A, Missale G, Pilli M, Fowler P,
Giuberti T, Chisari FV, et al. Identification of immunodominant T cell epitopes of the hepatitis B virus nucleocapsid antigen. J Clin Invest. 1991;88:214-22. 24. Bertoletti A, Chisari FV, Penna A, Guilhot S, Galati L, Missale G, Fowler P,
Schlicht HJ, Vitiello A, Chesnut RC, et al. Definition of a minimal optimal cytotoxic T-cell epitope within the hepatitis B virus nucleocapsid protein. J Virol. 1993;67:2376-80.
25. Missale G, Redeker A, Person J, Fowler P, Guilhot S, Schlicht HJ, Ferrari C, Chisari FV. HLA-A31- and HLA-Aw68-restricted cytotoxic T cell responses to a
single hepatitis B virus nucleocapsid epitope during acute viral hepatitis. J Exp Med. 1993;177:751-62.
26. Ehata T, Omata M, Chuang WL, Yokosuka O, Ito Y, Hosada K, et al. Mutations in core nucleotide sequence of hepatitis B virus correlate with fulminant and severe hepatitis. J Clin Invest 1993; 91: 1206-1213
27. Akarca US, Lok AS. Naturally occurring hepatitis B virus core gene mutations. Hepatology 1995; 22: 50-60
28. Stuyver L, De Gengt R, Cadranel JF, Van Geyt C, Van Reybroeck G, Dorent R, et al. Three cases of severe subfulminant hepatitis in heart-transplanted patients after nosocomial transmission of a mutant hepatitis B virus. Hepatology 1999; 29: 1876-1883.
29. Lanford RE, Kim YH, Lee H, Notvall L, Beames B. Mapping of the hepatitis B
virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by protein-protein interaction. J Virol. 1999;7:1885-93. 30. Chen GG, Li MY, Ho RL, Chak EC, Lau WY, Lai PB. Identification of hepatitis
B virus X gene mutation in Hong Kong patients with hepatocellular carcinoma. J Clin Virol. 2005;34:7-12.
31. Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M, Moss B, Sette A, Chisari FV.The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med. 1995;181:1047-58.
32. Ling R, Mutimer D, Ahmed M, Boxall EH, Elias E, Dusheiko GM, Harrison TJ. Selection of mutations in the hepatitis B virus polymerase during therapy of transplant recipients with lamivudine. Hepatology. 1996;24:711-3.
33. Niesters HG, Honkoop P, Haagsma EB, de Man RA, Schalm SW, Osterhaus AD. Identification of more than one mutation in the hepatitis B virus polymerase gene arising during prolonged lamivudine treatment. J Infect Dis.
1998;177:1382-5.
34. Aye TT, Bartholomeusz A, Shaw T, Bowden S, Breschkin A, McMillan J, Angus P, Locarnini S.Hepatitis B virus polymerase mutations during antiviral therapy in a patient following liver transplantation. J Hepatol. 1997 May;26(5):1148-53. 35. Fukuda R, Nguyen XT, Ishimura N, Ishihara S, Chowdhury A, Kohge N, Akagi
S, Watanabe M, Fukumoto S.X gene and precore region mutations in the
hepatitis B virus genome in persons positive for antibody to hepatitis B e antigen: comparison between asymptomatic "healthy" carriers and patients with severe chronic active hepatitis. J Infect Dis. 1995;172:1191-7.
30
36. Kao JH, Chen PJ, Lai MY, Chen DS.Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327-34.
37. Li J, Ou JH.Differential regulation of hepatitis B virus gene expression by the Sp1 transcription factor. J Virol. 2001;75:8400-6.
38. Chen M, Qu JH. Cell type-dependent regulation of the activity of the negative regulatory element of the hepatitis B virus core promoter. Virology.
1995;214:198-206.
39. Chen M, Hieng S, Qian X, Costa R, Ou JH.Regulation of hepatitis B virus ENI enhancer activity by hepatocyte-enriched transcription factor HNF3. Virology. 1994;205:127-32.
40. Huan B, Kosovsky MJ, Siddiqui A. Retinoid X receptor alpha transactivates the
hepatitis B virus enhancer 1 element by forming a heterodimeric complex with the peroxisome proliferator-activated receptor. J Virol. 1995;69:547-51.
41. Fan YF, Lu CC, Chang YC, Chang TT, Lin PW, Lei HY, Su IJ. Identification of a pre-S2 mutant in hepatocytes expressing a novel marginal pattern of surface antigen in advanced diseases of chronic hepatitis B virus infection.
J Gastroenterol Hepatol. 2000;15:519-28.
42. Huy TT, Ushijima H, Win KM, Luengrojanakul P, Shrestha PK, Zhong ZH, Smirnov AV, Taltavull TC, Sata T, Abe K. High prevalence of hepatitis B virus
pre-s mutant in countries where it is endemic and its relationship with genotype and chronicity. J Clin Microbiol. 2003;41:5449-55.
43. Sugauchi F, Ohno T, Orito E, Sakugawa H, Ichida T, Komatsu M, Kuramitsu T, Ueda R, Miyakawa Y, Mizokami M. Influence of hepatitis B virus genotypes on the development of preS deletions and advanced liver disease. J Med Virol. 2003;70:537-44.
44. Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, Chang WT, Huang W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage.Carcinogenesis. 2004;25:2023-32. 45. Choi MS, Kim DY, Lee DH, Lee JH, Koh KC, Paik SW, Rhee JC, Yoo BC.
Clinical significance of pre-S mutations in patients with genotype C hepatitis B virus infection. J Viral Hepat. 2007;14:161-8.
46. Wang HC, Wu HC, Chen CF, Fausto N, Lei HY, Su IJ.Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol.
47. Wang HC, Chang WT, Chang WW, Wu HC, Huang W, Lei HY, Lai MD, Fausto N, Su IJ. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology. 2005;41:761-70. 48. Cho G, Park SG, Jung G.. Localization of HSP90 binding sites in the human
hepatitis B virus polymerase. Biochem Biophys Res Commun. 2000;269:191-6. Table 1 The primers used for PCR
Fragment 1 (HBV nt 1821-2516)
P1 (forward, nt 1821-1844) 5’ –TTTTTCACCTCTGCCTAATCATCT– 3’ NP111038A (reverse, nt 2516-2497) 5’ –AAAGACAGGTACAGTAGAAG– 3’ Fragment 2 (HBV nt 427-1825)
NP210101A (forward, nt 427-448) 5’ –CATCTTCTTGTTGGTTCTTCTG– 3’ P2 (reverse, nt 1825-1804) 5’ –AAAAAGTTGCATGGTGCTGGTG– 3’ Fragment 3 (HBV nt 2268-479)
NP3 (forward, nt 2268-2291) 5’ –GAGTGTGGATTCGCACTCCTCCAG– 3’ NP4 (reverse, nt 479-456) 5’ –GAGGACAAACGGGCAACATACCTT– 3’ Table 2 The primers used for PCR with low viral-load samples
Fragment 1 (HBV nt 1573-2516)
NP1SF (forward, nt1573-1594) 5’ – GACCGTGTGCACTTCGCTTCAC – 3’ NP111038A (reverse, nt 2516-2497) 5’ – AAAGACAGGTACAGTAGAAG – 3’ Fragment 2 (HBV nt 542-1741) NP2SF (forward, nt 542-564) 5’ –GGAAACTCTATGTTTCCCTCATG– 3’ NP2SR (reverse, nt1741-1719) 5’ –AACTCCTCCCACTCAKTAAACAC– 3’ Fragment 2-1 (HBV nt 488-1188) HCC 2-1F ( forward, nt 488 - 507) 5’ – GAAACATCAACTACCAGCAC– 3’ HCC 2-1R (reverse, nt 1188-1169) 5’ – AGCAAACACTTGACAGAGAC– 3’ Fragment 2-2 (HBV nt 1136-2098) HCC 2-2F (forward, nt 1136-1155) 5’ – TGAACCTTTACCCCGTTGCC– 3’ HCC 2-2R ( reverse, nt 2098-2078) 5’ –CATCAACTCACCCCAACACAC– 3’ Fragment 3 (HBV nt 2528-704) HCC 3F (forward, nt 2528-2549) 5’ – GCAAACTCCCTCCTTTCCTCAC– 3’ HCC 3R ( reverse, nt 704-683) 5’ – CGAACCACTGAACAAATGGCAC–3’
32
Fragment 3 (HBV nt 2268-690)
NP3 (forward, nt 2268-2291) 5’ –GAGTGTGGATTCGCACTCCTCCAG –3’ NP410004C (reverse, nt 690-671) 5’ –AATGGCACTAGTAAACTGAG– 3’
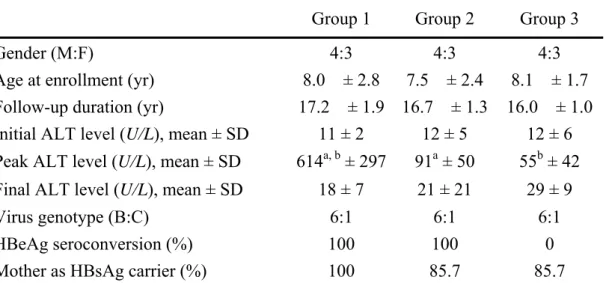
Table 3. Basic data of three groups of patients with chronic HBV infection Group 1 Group 2 Group 3
Gender (M:F) 4:3 4:3 4:3
Age at enrollment (yr) 8.0 ± 2.8 7.5 ± 2.4 8.1 ± 1.7 Follow-up duration (yr) 17.2 ± 1.9 16.7 ± 1.3 16.0 ± 1.0 Initial ALT level (U/L), mean ± SD 11 ± 2 12 ± 5 12 ± 6 Peak ALT level (U/L), mean ± SD 614a, b ± 297 91a ± 50 55b ± 42 Final ALT level (U/L), mean ± SD 18 ± 7 21 ± 21 29 ± 9
Virus genotype (B:C) 6:1 6:1 6:1
HBeAg seroconversion (%) 100 100 0
Mother as HBsAg carrier (%) 100 85.7 85.7
a: The peak ALT levels between group 1 and 2 showed significant difference (P <
0.001), Mann-Whitney test; b: The peak ALT levels between group 1 and 3 showed significant difference (P < 0.001), Mann-Whitney test.
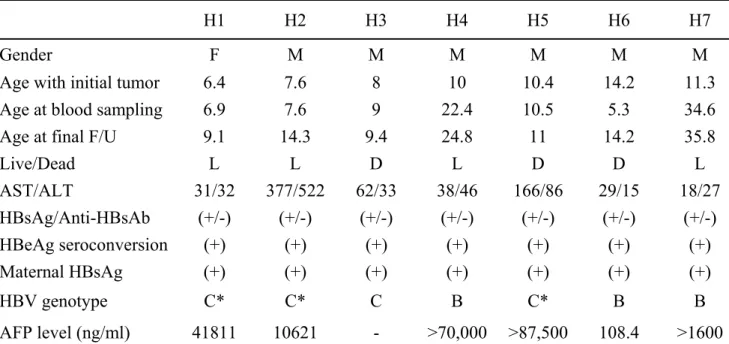
Table 4. Clinical profiles of 7 children with HCC
H1 H2 H3 H4 H5 H6 H7
Gender F M M M M M M
Age with initial tumor 6.4 7.6 8 10 10.4 14.2 11.3
Age at blood sampling 6.9 7.6 9 22.4 10.5 5.3 34.6
Age at final F/U 9.1 14.3 9.4 24.8 11 14.2 35.8
Live/Dead L L D L D D L AST/ALT 31/32 377/522 62/33 38/46 166/86 29/15 18/27 HBsAg/Anti-HBsAb (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) HBeAg seroconversion (+) (+) (+) (+) (+) (+) (+) Maternal HBsAg (+) (+) (+) (+) (+) (+) (+) HBV genotype C* C* C B C* B B AFP level (ng/ml) 41811 10621 - >70,000 >87,500 108.4 >1600 H1-7: HCC patients No.1-No.7; F/U: follow-up; *: cases whose viral genotypes
determined by sequence analysis only due to insufficient blood amount; AFP: alpha-fetopprotein
34
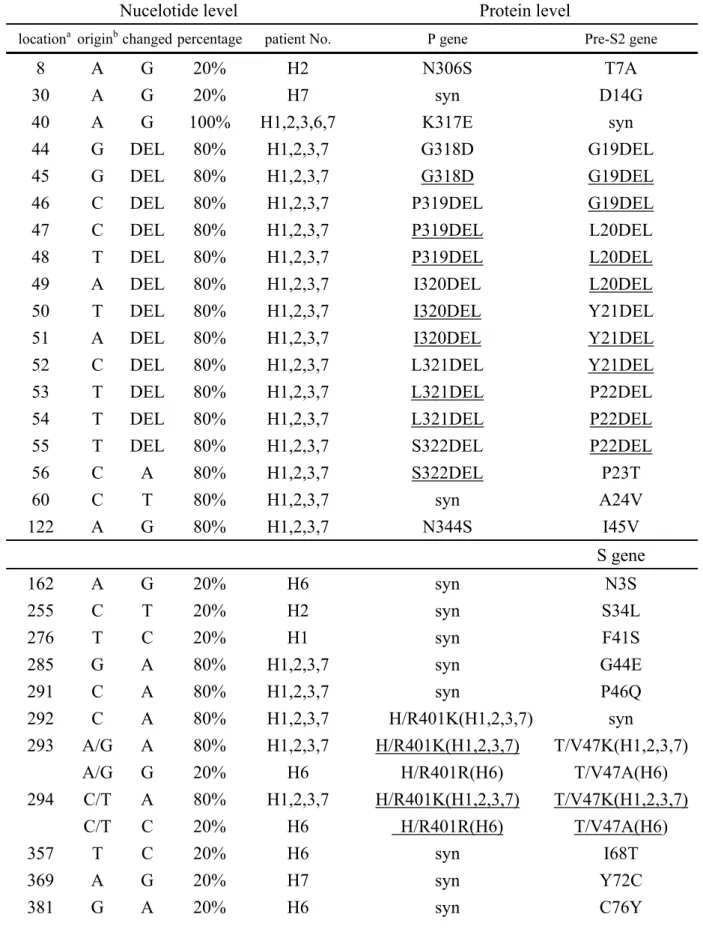
Table 5: Unique non-synomonous mutations found in 5 childhood HCC full length HBV sequences
Nucelotide level Protein level
locationa originb changed percentage patient No. P gene Pre-S2 gene
8 A G 20% H2 N306S T7A
30 A G 20% H7 syn D14G
40 A G 100% H1,2,3,6,7 K317E syn
44 G DEL 80% H1,2,3,7 G318D G19DEL
45 G DEL 80% H1,2,3,7 G318D G19DEL
46 C DEL 80% H1,2,3,7 P319DEL G19DEL
47 C DEL 80% H1,2,3,7 P319DEL L20DEL
48 T DEL 80% H1,2,3,7 P319DEL L20DEL
49 A DEL 80% H1,2,3,7 I320DEL L20DEL
50 T DEL 80% H1,2,3,7 I320DEL Y21DEL
51 A DEL 80% H1,2,3,7 I320DEL Y21DEL
52 C DEL 80% H1,2,3,7 L321DEL Y21DEL
53 T DEL 80% H1,2,3,7 L321DEL P22DEL
54 T DEL 80% H1,2,3,7 L321DEL P22DEL
55 T DEL 80% H1,2,3,7 S322DEL P22DEL
56 C A 80% H1,2,3,7 S322DEL P23T 60 C T 80% H1,2,3,7 syn A24V 122 A G 80% H1,2,3,7 N344S I45V S gene 162 A G 20% H6 syn N3S 255 C T 20% H2 syn S34L 276 T C 20% H1 syn F41S 285 G A 80% H1,2,3,7 syn G44E 291 C A 80% H1,2,3,7 syn P46Q 292 C A 80% H1,2,3,7 H/R401K(H1,2,3,7) syn 293 A/G A 80% H1,2,3,7 H/R401K(H1,2,3,7) T/V47K(H1,2,3,7) A/G G 20% H6 H/R401R(H6) T/V47A(H6) 294 C/T A 80% H1,2,3,7 H/R401K(H1,2,3,7) T/V47K(H1,2,3,7) C/T C 20% H6 H/R401R(H6) T/V47A(H6) 357 T C 20% H6 syn I68T 369 A G 20% H7 syn Y72C 381 G A 20% H6 syn C76Y
429 T C 80% H1,2,3,7 syn I92T 445 T C 20% H1 S452P syn 499 T C 60% H1,2,7 Y470H syn 511 A G 80% H1,2,3,7 T474A syn 516 G A 20% H7 M475I C121Y 517 C T 20% H3 Q476T syn 532 T C 20% H1 S481P syn 617 T C 20% H2 I509T S154P 709 G A 20% H3 A540T syn 760 G T 40% H6,7 A557S syn 828 A G 20% H1 I579M Y225C 880 G T 40% H1,2 G597STOP 898 T G 20% H3 W603F 934 A C 60% H1,2,3 I615L 936 C T 20% H3 I615L 1083 G C 20% H6 K664N 1123 T/A C 20% H7 C/S678R 1128 A C 20% H7 K679N 1138 A C 20% H7 N683H X gene 1482 C G 80% H1, 2, 3, 7 L797 L37V 1566 T C 80% H1,2,3,7 H825R S65P 1613 G A 20% H6 R841K syn 1632 C G 80% H1, 2, 3, 7 Q/A87G 1633 A/G G 80% H1, 2, 3, 7 Q/A87G Precore gene 1862 G T 80% H1, 2, 3, 7 V17F 1896 G A 80% H1, 2, 3, 7 W28Stop Core gene 1938 T C 80% H1, 2, 3, 7 V13A 2001 C T 20% H6 A34V 2003 T A 80% H1, 2, 3, 7 S35T T C 20% H6 S35L 2004 C T 20% H6 S35L 2007 C A 20% H6 A36D 2010 T G 20% H6 L37R 2048 C G 100% H1, 2, 3, 6, 7 P50A 2073 C A 100% H1, 2, 3, 6, 7 A58E
36 2138 G A 80% H1, 3, 6, 7 A80T 2150 T G 80% H1, 3, 6, 7 L84A 2253 A T 80% H1, 3, 6, 7 Y118F P gene 2339 A G 80% H1, 3, 6, 7 syn T147A 2352 G A 80% H1, 3, 6, 7 D16K R151Q 2366 C G 80% H1, 3, 6, 7 syn P156A 2423 A C 80% H1, 3, 6, 7 E39D syn 2525 G/A T 20% H2 E73D 2547 C/G A 40% H6,7 H81N 2563 A G 100% H1,2,3,6,7 E86G 2715 G A 100% H1,2,3,6,7 A137T Pre-S1 gene 2859 G A 20% H1 V185I W4STOP 2991 C A 100% H1,2,3,6,7 Q229K N48K 3063 C/G A 100% H1,2,3,6,7 R/G253R syn 3165 T C 20% H2 S287P syn
a: nucleotide position counted from the EcoRI site; b: when two characters shown
and separated by a slash, meaning that the former was from adult HCC group and the later was from children with chronic HBV infection; syn: synonymous mutations; DEL: deletion; underlined characters: meaning that the amino acid mutation was generated together by the one underlined and previous 1 or 2 nucleotide positions.
Table 6: Frequencies of non-synonymous mutations of different HBV regions Region Frequency (mut No./bp) Pre-S1 0.0056 Pre-S2 0.1030* S 0.0235 Precore 0.0230 Core 0.0272* Polymerase 0.0221 X 0.0086 mut. No.: number of mutation; bp: number of base pair; * P < 0.0001 (chi square test with Yates’ correction