寡聚蒽衍生物的合成及其光電特性
187
0
0
全文
(2) 謝誌 就要離開這個待了四年半的有機光電材料實驗室,心情其實是很矛盾 的,謝謝當初 郭文章老師讓我加入這間實驗室當專題生,老師帶領實驗 室的方式以及做人做事的客觀態度都是讓我留在這間實驗室最大的驕傲 和動力;感謝老師對於 group meeting 的堅持,讓我在這之中扎扎實實學 到了如何上台報告、如何客觀的去看待和解讀文獻、如何準備一份好的 投影片、如何好好安排時間等,也感謝老師總是全力支持這幾年來我所 做的決定,給予我很多具體的建議和幫助,無時無刻都很樂意和學生討 論,並教導和建議我許多研究上的概念和方法,給我很多彈性空間,讓 我每次在研究上遇到瓶頸時,可以盡快解決問題。郭老師,謝謝您! 有機光電材料實驗室這個大家庭:謝謝當初帶我的學長昭霖,讓我從 一個一無所知的混亂製造者進入軌道,進而對這些實驗有了興趣,還有 後來在其他事情的鼎力相助,謝謝你!看起來像流氓的學長長勳、一起 打球一起做實驗的學姐羽婷,謝謝你們的照顧,讓這間實驗室這麼有趣 讓人想一直待在這裡!人又帥又好的學長柏志、同是網球愛好者的學長 佳慶,謝謝你們永無止盡的(鬼)話讓實驗室充滿歡笑!接著是豪哥和阿 捲,非常謝謝你們一起盡責的分擔實驗室所有大小事情、一起討論實驗 上的瓶頸,每件爆笑恐怖白痴驚人的事我都會一直放在心裡!遊戲王哈.
(3) 比,謝謝你這麼好笑讓我釋放許多壓力!惟嘉,實驗加油!專題生們: 角哥、圈圈、阿尼、小菜、舜仔你們都很懂事也都很優秀;阿緯、當年 的小助手何政霖、人稱王太太的翰翰,謝謝你們的幫忙,不管是蒸 DCM 還是幫忙切水果,都讓我印象深刻;洞洞腦黑黑、很有想法又很可愛的 張偉榤與沈俞均夫婦,你們的天真話語和時不時的驚人舉動,在我壓力 最大的時候給我很多關心和笑料,以後記得不要再用小畫家畫 NMR 圖! 謝謝我的男友 ELVA,從一起當專題生做實驗到現在,不管快樂、難 過還是實驗低潮你總是陪著我支持我、給我很多建議和想法,陪我一起 走過壓力最大的那段日子、不斷鼓勵我,謝謝你! 謝謝我的研究所好同學、好朋友:小酸和歪歪,這些日子一起討論課 業、一起打球釋放壓力和燃燒脂肪、一起聊天排解很多研究上和生活上 的不悅,認識妳們是我研究所遇到最幸運的一件事,謝謝妳們! 謝謝其他實驗室曾經幫過我的人:謝謝汪達學姐都在我最低潮時給我 很多人生的建議和正面的能量;謝謝 Ricky 在質譜方面的傾力相助以及 好吃的下午茶點心,少吃宵夜才能瘦回來喔;195 同學,謝謝你總是沒有 第二句話的幫我蒸甲苯、使用手套箱以及借我玻璃儀器,東西我應該都 還清了吧;我最貼心的直屬學妹小小聯,謝謝妳的貼心和懂事總能給我. II.
(4) 最真誠的建議,還有,妳永遠是我籃底下最可敬的對手;menu,謝謝妳 在我壓力最大的幾個月用妳特有的風格分享好笑的故事。 謝謝 詹立行老師、 陳俐吟老師與 黃榮宗老師百忙之中擔任我的口 試委員,給我許多建議與指導而讓這本論文能夠更加完善,謝謝你們! 特別謝謝 黃榮宗老師,把有機化學這門課教得很好,奠定我有機化 學的基礎與興趣,並在生活上與研究上給我許多寶貴的經驗和鼓勵;謝 謝 何永皓老師,質譜學這門課您教得很好讓我扎扎實實學了很多,也傾 力幫我解決質譜方面的問題,更在我最茫然的時候撥空開示我、鼓勵我, 讓我了解出社會重要的課題;謝謝 李頂瑜老師,曾經極力鼓勵我追求夢 想,雖然計畫總趕不上變化;謝謝 莊曜遠老師理論計算上的協助;還有 其他應化系的老師,沒有您們的指導不會有這份論文和現在的我! 謝謝我的父母,總是全力支持我所有決定,謝謝你們體諒這段做研究 的日子讓我很少回家陪伴你們,但你們讓我知道家是永遠的避風港!現 在我畢業了,可以好好陪你們了! 特別謝謝中山大學貴儀中心的何妱璉小姐與中興大學貴儀中心的許 麗梅小姐,謝謝妳們辛苦加班幫我趕出急件 data,讓我得以順利完成論 文,謝謝妳們的幫忙! 中華民國 102 年 4 月 19 日 李唯甄 謹致. III.
(5) 寡聚蒽衍生物的合成及其光電特性 指導教授:郭文章 博士 (副教授) 國立高雄大學應用化學系碩士班. 學生:李唯甄 國立高雄大學應用化學系. 摘要. 自從白川英樹教授發現有機高分子可以導電後,隨即讓有機分子之導電特性開始 受到材料學家的注目,使得有機光電材料成為另一重要學門。迄今,不管是有機太陽 能電池、有機發光二極體甚至是有機場效電晶體等研究,雖然仍無法追上傳統太陽能 電池、發光二極體及矽半導體的表現,但有機電子元件之魅力在於能夠讓光電元件應 用在更多不同的基板上,製成可撓式的軟性電子元件,讓電子產品能夠更人性化、讓 人類的生活更便利。 本研究開發一系列以蒽為主體的多環芳香烴材料,在蒽的不同化學位置導入甲氧 基,並在分子的二號和(或)六號位置進行延伸,得到蒽之雙聚物及三聚物,其中,係 以不同的合成方法得到單體蒽衍生物,再以 Suzuki-Miyaura 交叉耦合反應,合成出十 個蒽衍生物分子,對於合成之中間物以及最後產物是藉由核磁共振光譜儀鑑定其結構 的變化及形成,此類分子皆可溶於二氯甲烷溶劑中,其中以甲氧基導入在一號及四號 位置之分子擁有最佳的溶解度。 為了瞭解合成出來的蒽衍生物在有機薄膜電晶體方面的應用潛力,本研究利用 UV 吸收光譜及 FL 放射光譜來研究蒽衍生物分子的光學特性及其 HOMO/LUMO 能. IV.
(6) 階,研究結果顯示當這些分子的共軛長度增加,其吸收波長皆有明顯的紅位移,取代 基位置的改變亦影響著分子的紅位移程度;材料電學性質係利用循環電位儀進行測 量,所有分子皆能進行氧化及還原反應,即同時具有傳遞電子及電洞之特性,另外, 分子的氧化及還原電位隨著取代基位置的差異而有規律性的改變,利用氧化及還原電 位計算可知分子的 HOMO 能階座落在 5.2~5.45 eV 之間,LUMO 能階則在 2.73~2.9 eV 之間;在材料的熱性質部分則以 TGA 及 DSC 進行研究,延伸共軛長度後的蒽衍生物 (雙體及三體)分子,在 300℃以內分子幾乎無明顯的型態變化(未見熔化溫度及玻璃轉 移溫度),且其熱裂解溫度皆在 300℃之上,顯示這系列分子具有良好的熱穩定性。 綜合以上結果,此系列之蒽衍生物分子應有機會應用在電晶體材料中。. 關鍵字:蒽、雙體蒽、三體蒽、有機薄膜電晶體材料. V.
(7) Synthesis of Oligo-Anthracene and its Electro-Optical Characteristics Advisor(s): Dr.(Professor) Wen-Jang Kuo Institute of Applied Chemistry National University of Kaohsiung Student: Wei-Chen, Lee Institute of Applied Chemistry National University of Kaohsiung ABSTRACT. Since Professor Hideki Shirakawa discovered conjugated organic molecule can be conductive, the conductive properties of organic molecules began to obtain great attention of material scientists immediately and became another vital discipline. So far, regardless of the organic solar cells, organic light-emitting diodes or even organic field effect transistors still unable to catch up the performance with the traditional solar cells, light-emitting diodes and silicon semiconductor, the charm is the ability of the organic electronic components could be applied to flexible substrate. This way allows electronic products can be more humane, to make human life more convenient. In this study, a series of the methoxy-substituted anthracene at different chemical position was synthesized. Incorporating the substitued-anthracene with one or two substitued-anthracene at 2,6-position, the anthracene dimers and trimers were obtained. Of these, we use different method to acquire the substituded anthracene monomers, then. VI.
(8) synthesized the oligo-anthracene derivatives by means of Suzuki-Miyaura cross-coupling reaction. These intermediates and final molecules were soluble in dichloromethane, was characterized by NMR. The OFET application potential of the anthracene derivatives was assessed in the aspects of photopysical properties, thermal stabilities and electrochemical behaviors. Photophysical properties and the HOMO/LUMO energy levels of the molecules were characterized from UV- visible and fluorescence spectra. The results show that the molecules with more extensive conjugation ha ve an obvious red-shift. It suggested that the presence of increasing electron delocalization along the main chain. The positions of methoxy group also influence the range of red-shift. CV was employed to investigate the electrochemical behaviors of these molecules. All of them have occurred both oxidation and reduction reactions, means that it would transport electron and hole simultaneously. The HOMO levels of these molecules were estimated to be around 5.2~5.45 eV and the LUMO level were estimated to be around 2.73~2.9 eV. Thermal properties of the molecules were investigated by DSC and TGA. The anthracene dimers and trimers were almost no apparent morphological change within 300℃(no melting temperature and glass transition temperature) and the thermal decomposition temperature were up to 300 ℃. These results indicated that these series of anthracene derivatives possessed high thermal stability.. Key words: anthracene, di-anthracene, tri-anthracene, OFET. VII.
(9) 目錄 中文摘要……………………………………………………………………IV 英文摘要……………………………………………………………………VI 第一章、緒論............................................................................................ 1 1.1 前言................................................................................................. 1 1.2 OTFT 元件簡介................................................................................ 2 1.3 有機材料導電原理與堆疊方式........................................................ 2 1.4 好的 OFET 材料之必要條件 ........................................................... 4 第二章、文獻回顧 .................................................................................... 7 2.1 多環芳香烴:五環素及其衍生物.................................................... 7 2.2 多環芳香烴:六環素 .................................................................... 13 2.3 多環芳香烴:四環素 .................................................................... 15 2.4 多環芳香烴:三環素 .................................................................... 17 第三章、研究動機 .................................................................................. 25 3.1 本計畫選擇蒽分子的理由 ............................................................. 25 3.2 分子設計藍圖................................................................................ 28 第四章、結果與討論 .............................................................................. 30. VIII.
(10) 4.1 目標分子的合成討論 .................................................................... 30 4.2 分子溶解度比較 ............................................................................ 41 4.3 光學性質分析................................................................................ 42 4.4 電化學性質分析 ............................................................................ 50 4.5 熱穩定性質分析 ............................................................................ 55 第五章、結論.......................................................................................... 62 第六章、實驗方法 .................................................................................. 64 6.1 化學藥品及溶劑 ............................................................................ 64 6.1.1 化學藥品 ................................................................................. 64 6.1.2 溶劑及其除水方式 .................................................................. 67 6.2 測量儀器 ....................................................................................... 68 6.3 合成步驟 ....................................................................................... 71 6.3.1 單體蒽分子的合成 ................................................................. 71 6.3.1.1 單體 1,4-dimethoxyanthracene 的合成 .............................. 71 6.3.1.2 單體 9,10-dimethoxyanthracene 的合成 ............................ 75 6.3.1.3 單體 5,8-dimethoxyanthracene 的合成 .............................. 79 6.3.1.4 單體 9,10-bisalkynylanthrylene 的合成 ............................. 85. IX.
(11) 6.3.1.5 單體 2,3-dimethoxyanthracene 的合成 .............................. 89 6.3.2 雙體蒽分子的合成 ................................................................. 96 6.3.2.1 1,1',4,4'-tetramethoxy-2,2'-bianthracene <di-1,4DMA>的合成 ..................................................................................................... 96 6.3.2.2 9,9',10,10'-tetramethoxy-2,2'-bianthracene <di-9,10DMA>的 合成 ............................................................................................. 97 6.3.2.2 5,5',8,8'-tetramethoxy-2,2'-bianthracene <di-5,8DMA>的合成 ..................................................................................................... 98 6.3.3 三體蒽分子的合成 ................................................................100 6.3.3.1 9',10’-bis(octyl-1-ynyl)-1,4,1’’,4’’-tetramethoxy- teranthrylene <tri-1,4DMA>合成 ......................................................................100 6.3.3.2 9',10’-bis(octyl-1-ynyl)-9,10,9’’,10’’-tetramethoxyteranthrylene <tri-9,10DMA>合成................................................101 6.3.3.3 9',10’-bis(octyl-1-ynyl)-5,8,5’’,8’’-tetramethoxy- teranthrylene <tri-5,8DMA>合成 ......................................................................103 6.3.3.3 9',10’-bis(octyl-1-ynyl)-6,7,6’’,7’’-tetramethoxy- teranthrylene <tri-6,7DMA>合成 ......................................................................104 第七章、參考資料 .................................................................................106. X.
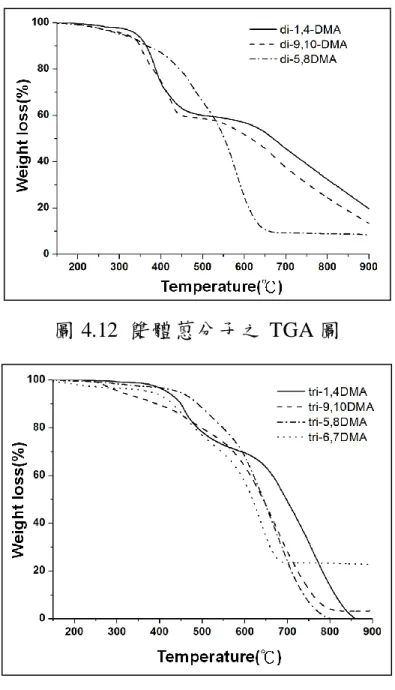
(12) 圖目錄 圖 1.1 有機薄膜電晶體之元件結構 ........................................................... 2 圖 1.2 電荷以跳躍的方式在有機分子間傳遞 ........................................... 3 圖 1.3 有機分子固態堆疊方式(A)EDGE-TO-FACE 魚骨狀; (B)FACE-TOFACE 魚骨狀; (C)一維片狀; (D)磚牆式; (E)二維磚牆式 4......................... 4 圖 2.1 五環素分子結構示意圖及其分子堆疊 7......................................... 7 圖 2.2 五環素的氧化過程 ......................................................................... 8 圖 2.3 五環素副產物 ................................................................................ 9 圖 2.4 分子單晶結構圖(A) TESPEN; (B) TIPSPEN; (C) TPSPEN; (D) TMSMSPEN............................................................................................ 10 圖 2.5 含四氯化苯之五環素前驅物結構示意圖 ..................................... 11 圖 2.6 (A) N-磺醯胺基五環素;(B)具感光性 N-磺醯胺基五環素前驅物 .. 12 圖 2.7 醌基型五環素結構示意圖及其分子排列 ..................................... 12 圖 2.8 六環素的合成演進 ....................................................................... 14 圖 2.9 六環素分子 .................................................................................. 15 圖 2.10 四環素及其衍生物之分子堆疊示意圖 16.................................... 16. XI.
(13) 圖 2.11 四環素衍生物分子示意圖.......................................................... 16 圖 2.12 蒽的 PERI-與 END-POSITION 示意圖 ...................................... 17 圖 2.13 寡聚蒽衍生物結構示意圖與其電荷遷移率比較 ........................ 18 圖 2.14 DTANT 與 DHTANT 分子單晶結構與半導體電性比較 ............. 19 圖 2.15 五環素(左圖)與 DPPVANT(右圖)之分子排列方式比較 ............ 20 圖 2.16 TIPSANT 分子單晶結構圖 ......................................................... 21 圖 2.17 (A)(B)TIPSANTHT 與(C)(D)TIPSANTPV 分子單晶結構圖 ....... 22 圖 2.18 TIPSANTT、TIPSANTBET 與 TIPSANTPT 分子排列方式示意圖 ................................................................................................................ 23 圖 2.19 OA-5A 與 OA-5B 分子示意圖及 OA-1 與其單晶結構圖 ............ 24 圖 3.1 多環芳香烴之稠環數量與其特性比較.............................................25 圖 3.2 延伸蒽的構想 ............................................................................... 26 圖 3.3 蒽分子之官能基修飾示意圖 ........................................................ 27 圖 4.1 蒽單體示意圖 .............................................................................. 30 圖 4.2 蒽單體分子之 UV 吸收光譜圖 .................................................... 44 圖 4.3 蒽雙體分子之 UV 吸收光譜圖 .................................................... 45. XII.
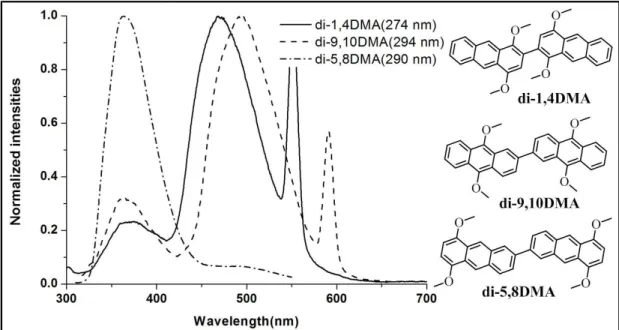
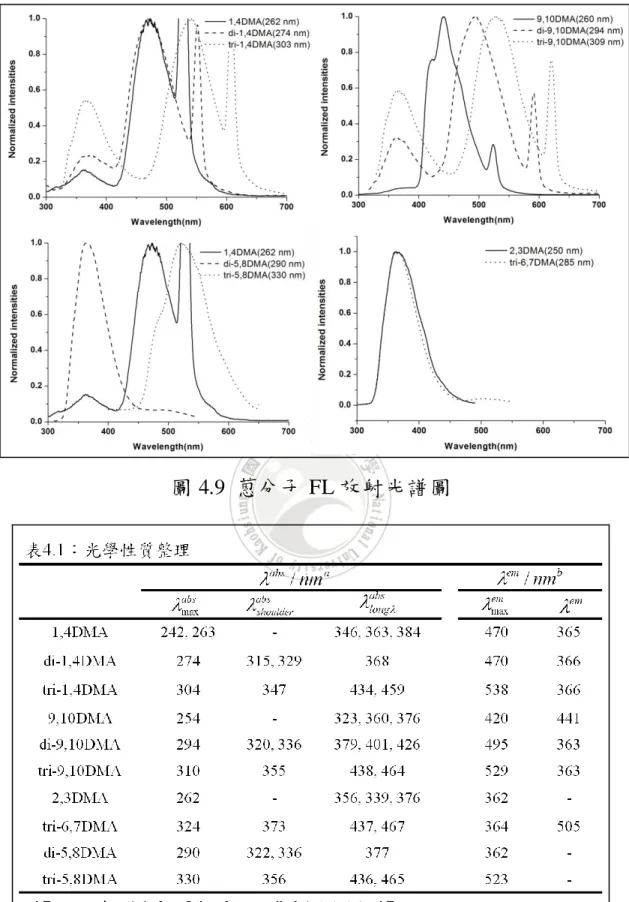
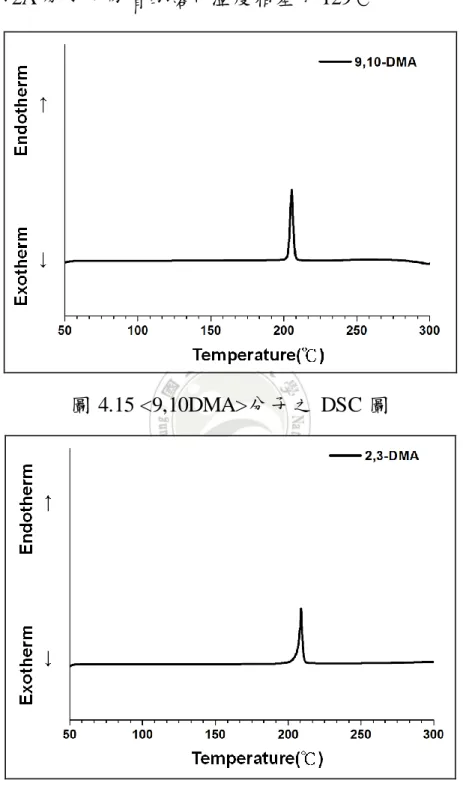
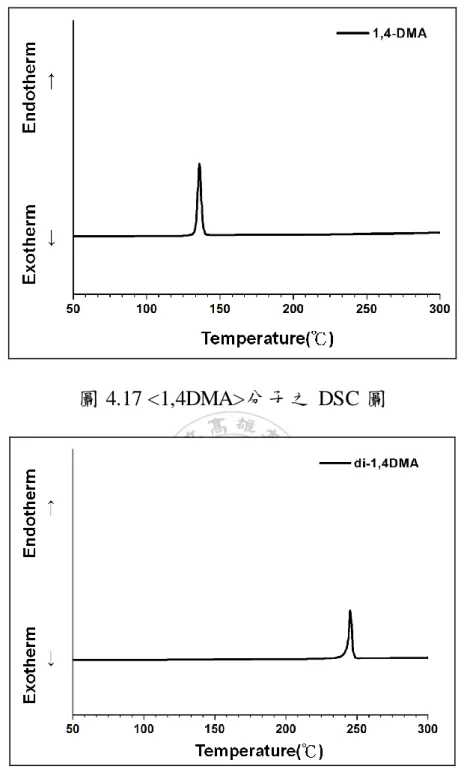
(14) 圖 4.4 蒽三體分子之 UV 吸收光譜圖 .................................................... 45 圖 4.5 蒽分子 UV 吸收光譜圖................................................................ 46 圖 4.6 蒽單體分子之 FL 放射光譜圖 ..................................................... 47 圖 4.7 蒽雙體分子之 FL 放射光譜圖 ..................................................... 48 圖 4.8 蒽三體分子之 FL 放射光譜圖 ..................................................... 48 圖 4.9 蒽分子 FL 放射光譜圖................................................................. 49 圖 4.10 蒽分子之循環電位 ..................................................................... 54 圖 4.11 單體蒽分子之 TGA 圖 ............................................................... 56 圖 4.12 雙體蒽分子之 TGA 圖 ............................................................... 57 圖 4.13 三體蒽分子之 TGA 圖 ............................................................... 57 圖 4.14 <DI-1,4DMA>分子之第二次升溫 DSC 圖 .................................. 58 圖 4.15 <9,10DMA>分子之 DSC 圖 ........................................................ 59 圖 4.16 <2,3DMA>分子之 DSC 圖 .......................................................... 59 圖 4.17 <1,4DMA>分子之 DSC 圖 .......................................................... 60 圖 4.18 <DI-1,4DMA>分子之 DSC 圖 ..................................................... 60. XIII.
(15) 表目錄 表 1.1 有機及無機半導體電荷傳遞速率比較 ........................................... 6 表 4.1 光學性質整理.....................................................................................49 表 4.2 電學性質與分子能階整理.................................................................55 表 4.3 熱學性質整理.....................................................................................61. XIV.
(16) 流程目錄 流程 4.1 <D2>之合成路徑....................................................................... 31 流程 4.2 <B2>之合成路徑....................................................................... 31 流程 4.3 <9,10DMA>之合成路徑............................................................ 32 流程 4.4 <1,4DMA>之合成路徑.............................................................. 33 流程 4.5 萘醌還原為羥基萘之測試 ........................................................ 33 流程 4.6 <C5>與<A2>的合成差異 .......................................................... 33 流程 4.7 <C1>合成之條件測試及<C2>之合成方法 ................................ 34 流程 4.8 以 FRIDEL-CRAFTS ACYLATION 合成<C5>之反應測試與流程 ................................................................................................................ 36 流程 4.9 以 DIELS-ALDER REACTION 合成<C5>之流程 .................... 37 流程 4.10 <2,3DMA>與<E5>之合成 ....................................................... 38 流程 4.11 硼酯衍生物的合成及產率整理............................................... 39 流程 4.12 二聚體的合成及產率整理 ...................................................... 40 流程 4.13 三聚體的合成及產率整理 ...................................................... 41. XV.
(17) 附錄目錄 圖 S1 <A1>之 1H-NMR ......................................................................... 111 圖 S2 <A1>之 13C-NMR ........................................................................ 112 圖 S3 <1,4DMA>之 1H-NMR................................................................. 113 圖 S4 <1,4DMA>之 13C-NMR................................................................ 114 圖 S5 <A2>之 1H-NMR ......................................................................... 115 圖 S5 <A2>之 13C-NMR ........................................................................ 116 圖 S7 <A3>之 1H-NMR ......................................................................... 117 圖 S8 <A3>之 13C-NMR ........................................................................ 118 圖 S9 <9,10DMA>之 1H-NMR............................................................... 119 圖 S10 <9,10DMA >之 13C-NMR ........................................................... 120 圖 S11 <B1>之 1H-NMR........................................................................ 121 圖 S12 <B1>之 13C-NMR....................................................................... 122 圖 S13 <B2>之 1H-NMR........................................................................ 123 圖 S14 <B2>之 13C-NMR....................................................................... 124 圖 S15 <B3>之 1H-NMR........................................................................ 125 XVI.
(18) 圖 S16 <B3>之 13C-NMR....................................................................... 126 圖 S17 <C1>之 1H-NMR........................................................................ 127 圖 S18 <C1>之 13C-NMR....................................................................... 128 圖 S19 <C2>之 1H-NMR........................................................................ 129 圖 S20 <C2>之 13C-NMR....................................................................... 130 圖 S21 <C3>之 1H-NMR........................................................................ 131 圖 S22 <C3>之 13C-NMR....................................................................... 132 圖 S23 <C4>之 1H-NMR........................................................................ 133 圖 S24 <C4>之 13C-NMR....................................................................... 134 圖 S25 <C5>之 1H-NMR........................................................................ 135 圖 S26 <C5>之 13C-NMR....................................................................... 136 圖 S27 <C6>之 1H-NMR........................................................................ 137 圖 S28 <C6>之 13C-NMR....................................................................... 138 圖 S29 <D1>之 1H-NMR ....................................................................... 139 圖 S30 <D1>之 13C-NMR ...................................................................... 140 圖 S31 <D2>之 1H-NMR ....................................................................... 141 XVII.
(19) 圖 S32 <D2>之 13C-NMR ...................................................................... 142 圖 S33 <D3>之 1H-NMR ....................................................................... 143 圖 S34 <D3>之 13C-NMR ...................................................................... 144 圖 S35 <E2>之 1H-NMR ........................................................................ 145 圖 S36 <E2>之 13C-NMR ....................................................................... 146 圖 S37 <2,3DMA>之 1H-NMR............................................................... 147 圖 S38 <2,3DMA>之 13C-NMR.............................................................. 148 圖 S39 <E4>之 1H-NMR ........................................................................ 149 圖 S40 <E4>之 13C-NMR ....................................................................... 150 圖 S41 <E5>之 1H-NMR ........................................................................ 151 圖 S42 <E5>之 13C-NMR ....................................................................... 152 圖 S43 <E6>之 1H-NMR ........................................................................ 153 圖 S44 <E6>之 13C-NMR ....................................................................... 154 圖 S45 <DI-1,4DMA>之 1H-NMR ......................................................... 155 圖 S46 <DI-1,4DMA>之 13C-NMR ........................................................ 156 圖 S47 <DI-9,10DMA>之 1H-NMR........................................................ 157 XVIII.
(20) 圖 S48 <DI-9,10DMA>之 13C-NMR....................................................... 158 圖 S49 <DI-5,8DMA>之 1H-NMR ......................................................... 159 圖 S50 <DI-5,8DMA>之 13C-NMR ........................................................ 160 圖 S51<TRI-1,4DMA>之 1H-NMR ........................................................ 161 圖 S52 <TRI-1,4DMA>之 13C-NMR ...................................................... 162 圖 S53 <TRI-9,10DMA>之 1H-NMR...................................................... 163 圖 S54 <TRI-9,10DMA>之 13C-NMR..................................................... 164 圖 S55 <TRI-5,8DMA>之 1H-NMR ....................................................... 165 圖 S56 <TRI-5,8DMA>之 13C-NMR ...................................................... 166 圖 S57 <TRI-2,3DMA>之 1H -NMR ...................................................... 167. XIX.
(21) 第一章、緒論 1.1 前言 有機半導體的發展可以追溯至 1963 年,Pope 等人發現將蒽 (anthracene)做成晶體元件後,施以高外加電壓後可觀察到電激發光的 現象,接著在 1977 年,Shirakawa、Heeger 以及 MacDiarmid 等人發 現聚乙炔(polyacetylene)的薄膜經過碘蒸氣氧化後,其導電度增加了 十億倍之多,使有機高分子的導電度接近金屬的導電度 1。而有機場 效電晶體最早發表於 1986 年,由 Tsumura 等人將有機分子聚噻吩 (polythiophene)做為電晶體的載子傳輸層,雖然當時電荷遷移率僅 10-5 cm2/Vs,卻開啟了有機場效電晶體(Organic Field Effect Transistor,簡 稱 OFET)的相關研究 2。 迄今,雖然有機電晶體的電荷遷移率仍不及以矽晶圓為主體之無 機半導體,但有別於傳統無機半導體的製程方式,當有機分子應用於 電晶體材料時,由於有機分子的特性,半導體元件可在低溫環境下以 溶液製程進行大面積的塗佈,以製作大尺寸的顯示器,另外,低溫製 程讓基板材料有更多的選擇,例如以塑膠基板取代以往的金屬導體, 而可做出可撓曲性的產品,這是一般無機半導體無法達成的。. 1.
(22) 1.2 OTFT 元件簡介 有機薄膜電晶體(Organic Thin Film Transistor,簡稱 OTFT)是 OFET 的一種形式,由於有機半導體層通常是薄膜的形式,因此常被 稱為 OTFT 3。 OTFT 的元件結構可概略分成三個部分(如圖 1.1):有機半導體 (organic semiconductor)、三個電極以及絕緣層(dielectric insulator)。有 機半導體層是以有機分子所組成,在有機半導體層的兩端有兩個電 極,分別是源極(source)與汲極(drain),可由調整兩者偏壓來進行電晶 體之電性研究;而整個 OTFT 的開關則是閘極(gate)所控制,藉由在 閘極通入正或負偏壓,使電晶體得以運作。在有機半導體層與閘極之 間須以一絕緣層阻隔,以避免短路 4。. 圖 1.1 有機薄膜電晶體之元件結構. 1.3 有機材料導電原理與堆疊方式 在一般的無機半導體材料中,原子之間藉由較強的作用力(如: 共價鍵或離子鍵)形成高度結晶的三維結構,因此電荷能夠在非定域 2.
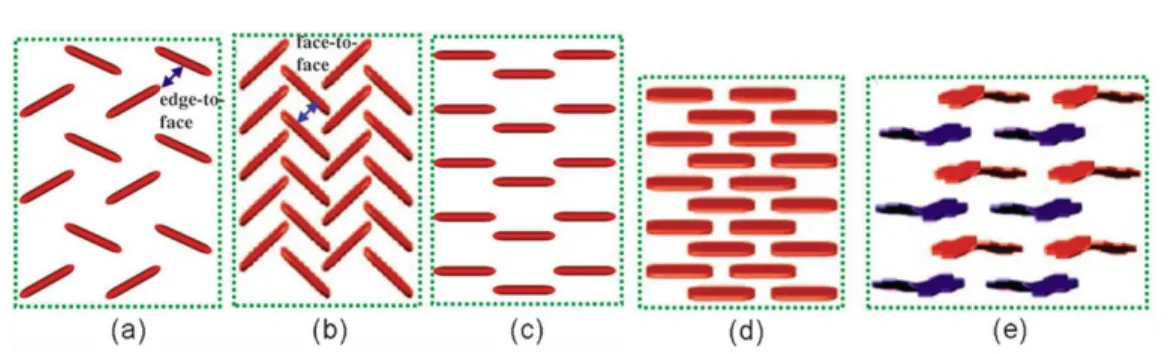
(23) 化軌域中進行電荷傳遞;而有機半導體是由許多獨立的有機分子組 成,這些分子通常以凡得瓦力(van der Waals force)、氫鍵(hydrogen bonding)、π 軌域間的交互作用(π-π interaction)等這些較弱的方式形成 分子堆疊,因此在單一有機分子中,電荷以非定域化(delocalization) 的方式沿著分子的共軛骨架進行傳遞;若在相鄰兩分子之間,則須藉 由 π 軌域重疊部分以跳躍(hopping)的方式進行電荷遷移(圖 1.2),也就 是在有機材料中,電荷傳遞被認為是藉由電荷在定域化軌域間跳躍, 從一帶電荷分子轉移至鄰近中性分子。 因此,若要有機半導體分子具有高導電性,有機分子的 π 共軛系 統就扮演了重要的角色,也就是有機半導體分子必須具有共振結構, 且分子間的 π 軌域重疊部分越多且分子間的間距越小,都將有助於電 子在有機分子間中的傳遞能力。. 圖 1.2 電荷以跳躍的方式在有機分子間傳遞 分子除了須具有良好的共軛 π 系統之外,分子間的堆疊方式影響 著半導體的電性,一般而言有機分子之堆疊型態可分為分子以單一方 向進行堆疊的單晶相(single crystalline)、分子無特定堆疊方向的非晶 相(amorphous)以及具有多種堆疊方向的多晶相(polycrystalline)。. 3.
(24) 在單晶相堆疊的方式中,又可細分成數種排列方式(如圖 1.3), 這五種方式是已在文獻中被發表過的排列方式,在文獻回顧中會做分 子結構及其排列方式的詳細探討。. 圖 1.3 有機分子固態堆疊方式(a)edge-to-face 魚骨狀; (b)face-to- face 魚骨狀; (c)一維片狀; (d)磚牆式; (e)二維磚牆式. 4. 1.4 好的 OFET 材料之必要條件 欲設計一個高效率的有機半導體材料,必須考慮到幾個層面:元 件角度的考量、材料導電性的考量以及元件製程的考量。 從元件角度的考量,衡量一個半導體的好壞可從三個數據去判 斷:起始電壓(threshold voltage)、電荷遷移率(charge mobility)以及電 流開關比(on/off ratio)。起始電壓就是驅動元件所需的最小閘極電 壓,也就是當閘極偏壓高於起始電壓時才能驅動電晶體,與載子注入 能力有關;電荷遷移率的定義是在每單位電場中電荷平均飄移速度, 可由公式搭配元件之通道寬度、通道長度、介電層的電容常數以及起 始電壓即可算出;電流開關比是在 OTFT 中,閘極在最大偏壓與無外 加偏壓下,源極與汲極間的電流比值,電流開關比越大表示 OTFT 有. 4.
(25) 快速的反應且只需要較少的伏特單位即可驅動元件中源極與汲極間 的電流。 從材料導電能力的考量,由於有機分子本質的因素,其導電能力 遠低於無機半導體,但由數據中顯示,若利用分子結構設計來改善有 機分子間的堆疊方式,使分子之間能夠以有次序性(well-ordered)的方 式進行單晶相或多晶相排列,是有機會大幅改善有機分子電荷遷移率 (表 1.1)。一般而言,可藉由結構設計改善分子間 π 軌域的重疊方式, 以增加電荷在分子間的跳躍機會,例如在多環芳香烴導入適當的拉電 子基或具極性的官能基,可以平衡分子間的電荷,使分子更容易進行 π 軌域的重疊。 從材料元件製程的考量來看,由於有機分子須扮演傳遞電荷的角 色,因此需要良好的 π 共軛平面堆疊能力以利電荷的傳遞,而經常使 用具有高平面性及剛硬性的稠環分子(fuse ring),然而此類稠環結構 影響了它在一般有機溶劑中的溶解度,若要以溶液製程進行元件製 作,半導體分子需要有好的加工性,有機分子的溶解度則須納入考 量。除了溶解度問題外,有些有機分子在高溫下容易進行裂解,或在 光照下容易產生氧化反應等,此類分子做成有機半導體元件後,分子 的不穩定性將會影響元件性質及壽命,因此,一個良好的有機半導體 材料其分子必須有好的化學穩定性。. 5.
(26) 表 1.1 有機及無機半導體電荷遷移率比較. 6. 5.
(27) 第二章、文獻回顧 在文獻回顧中,本研究將對於多環芳香烴及其衍生物的發展與半 導體電性、以及如何藉由分子設計改善有機分子的排列方式和溶解度 等問題進行討論及研究。. 2.1 多環芳香烴:五環素及其衍生物 在 1970 年代,Gundlach 等人首先將五環素(pentacene)應用於有 機場效電晶體材料,當時測得電荷遷移率為 0.7 cm2 /Vs、電流開關比 為 108,進一步發現五環素晶體是典型的魚骨狀堆疊(如圖 2.1),由於 分子本身足夠的 π 共軛系統,電荷在五環素分子間能夠有效的被傳 遞,因此具有優良的電荷傳遞速率值,另外,也由於被延伸的 π 共軛 系統,使得其 HOMO 能階與電洞注入的能階相當匹配。後來在 2003 年 , Kelly 等 人 利 用 多 晶 相 的 五 環 素 作 為 有 機 半 導 體 , 搭 配 以 poly(α-methylstyrene)作為閘極的介電層,測得電荷遷移率為 5 cm2 /Vs、電流開關比為 106,為目前為止最佳的記錄 6。. 圖 2.1 五環素分子結構示意圖及其分子堆疊. 7. 7.
(28) 雖然五環素半導體電性表現優異,且如今已市售化,材料取得容 易,然而由於五環素屬於多苯環的結構,其分子剛硬性嚴重限制了它 在一般有機溶劑中的溶解度,進而影響元件製程上的應用;此外,五 環素在一般環境下並不穩定,容易有其他副反應產生 7,例如在氧氣 存在下,五環素被氧化形成 pentacenequinone (如圖 2.2);Bendikov 等 人藉由理論計算指出五環素無論在 singlet oxygen 或 triplet oxygen 存 在下,都會反應而形成 endoperoxide 的結構(如圖 2.2)8,endoperoxide 則會快速的轉變成 pentacenequinone;而 Scott 和 Chronister 等人的研 究發現 9,五環素在無氧的環境中,加熱至 125℃並且在光照的條件 下形成蝶型雙聚物(λ>400 nm) (如圖 2.3)。以上這些副反應的發生會 使得電晶體效能降低甚至失效,我們將在以下篇幅討論如何對五環素 進行化學修飾來提升其分子的溶解度及穩定度。. 圖 2.2 五環素的氧化過程. 8.
(29) 圖 2.3 五環素副產物 由上一段的討論可發現,五環素的六號、十三位置最容易產生氧 化反應或自由基反應,因此若要進行結構設計可自該位置下手。在 2001 年,Anthony 等人為了改善五環素的缺點,他們在五環素的六號 與十三號碳上導入立體結構較大的三烷基矽乙炔分子,除了成功的提 升分子的溶解度之外,並發現烷基的鏈段長度不同可誘導出分子不同 的排列方式 10,11,12(如圖 2.4):當取代基的直徑遠小於五環素長度(14 Å ) 的一半,分子堆疊主要受到共軛平面間 face-to-face 交互作用影響, 而以一維方向進行錯位堆疊(slipped π-stacking)(如圖 2.4 的 TESPen); 當取代基的直徑約等於五環素長度的一半,此時分子則從魚骨狀堆疊 轉變成以 face-to-face 的二維磚牆式排列(如圖 2.4 的 TIPSPen);當取 代基的直徑大於五環素長度的一半,此時分子排列又會從二維磚牆式 堆疊 回到一維的錯 位堆疊與魚 骨狀排列 (如 圖 2.4 的 TPSPen 與 TMSMSPen)。分子的堆疊方式與其電性表現有很大的關聯,在上述 四個分子中,經過熱昇華的 TESPen 其電荷遷移率為 0.001cm2/Vs, 但 TIPSPen 其電荷遷移率卻可達 0.4 cm2/Vs,兩者間最大的差異就在 於不同的分子排列方式,二維磚牆式堆疊使得分子間 π-共軛重疊的區 域增加,而有利於電荷在分子間的傳遞,因此有較好的電性表現。 9.
(30) 圖 2.4 分子單晶結構圖(a) TESPen; (b) TIPSPen; (c) TPSPen; (d) TMSMSPen 除了直接導入可溶性的官能基來提升五環素的溶解度外,另一種 的方法則是設計易裂解的五環素的前驅物,該前驅物再以熱或光處理 後轉換回五環素。在 1996 年,Müllen 等人利用 Diels-Alder 反應,將 四氯化苯加成在五環素的六號、十三號位置可得五環素前驅物 13, 14(如 圖 2.5),由於四氯化苯的導入,破壞了原先五環素的平面性結構,前 10.
(31) 趨物的溶解度因此大幅提升而可溶解於二氯甲烷、甲苯等溶劑中,進 一步將前驅物分子加熱至 180℃時,這些前驅物逐漸轉變成五環素及 四氯化苯,因此他們五環素前驅物以溶液製程(旋轉塗佈法)得到薄 膜,再將該薄膜置於 200℃的環境加熱五秒鐘後製成半導體元件並測 量其電性,可得電荷遷移率 0.2 cm2/Vs、電流開關比 106。. 圖 2.5 含四氯化苯之五環素前驅物結構示意圖 在 2002 年,Afzali 等人則是用步驟較少的合成方式,在五環素 的六號、十三號位置導入 N-磺醯胺基得到另個五環素前驅物 15 (如圖 2.6-a),此前驅物的溶解度亦有明顯的提升,以溶液製程得到半導體 元件後,進行熱處理後可得 0.89 cm2/Vs 的電荷遷移率,Afzali 等人 隨後對 N-磺醯胺基五環素進行改良,得到具感光特性的五環素前驅 物 (如 圖 2.6 b), 因 此 可 藉 由 微 影 製 程 將 有 機 薄 膜 直 接 圖 案 化 (photopattern)於電晶體元件上,再經由紫外光照射使五環素前驅物裂 解,如此一來可避免熱處理對五環素造成傷害 16。藉由合成五環素前 驅物的方式,可以大幅改善五環素的溶解度,使得五環素的元件製程 可由溶液製程取代真空蒸鍍,然而,部分前驅物的殘留影響了五環素 的規則排列,造成元件電性下降。. 11.
(32) 圖 2.6 (a) N-磺醯胺基五環素;(b)具感光性 N-磺醯胺基五環素前驅物 在 2005 年,Nuckoll 等人將醌基(quinone)導入多環芳香烴中 17, 使得分子內電荷分布不均,誘使較缺電子(electron-poor)的醌基與較多 電 子 (electron-rich)的 苯 環 以 face-to-face 交 錯 方 式進 行堆 疊 (如 圖 2.7),因此分子能夠以較緊密的方式以頭尾相疊 face-to-face 的方式排 列 (分 子 間 的 距 離 3.25Å ) , 成 功 改 善 原 先 五 環 素 為 人 詬 病 的 edge-to-face 魚骨狀排列。. 圖 2.7 醌基型五環素結構示意圖及其分子排列 從上述的討論可以發現,五環素天生擁有很好的導電特性,可惜 因為分子本身的結構特性影響其溶解度與分子穩定性,使其在電晶體 方面的應用受到限制,雖然有許多材料學家提出各種分子修飾的辦法. 12.
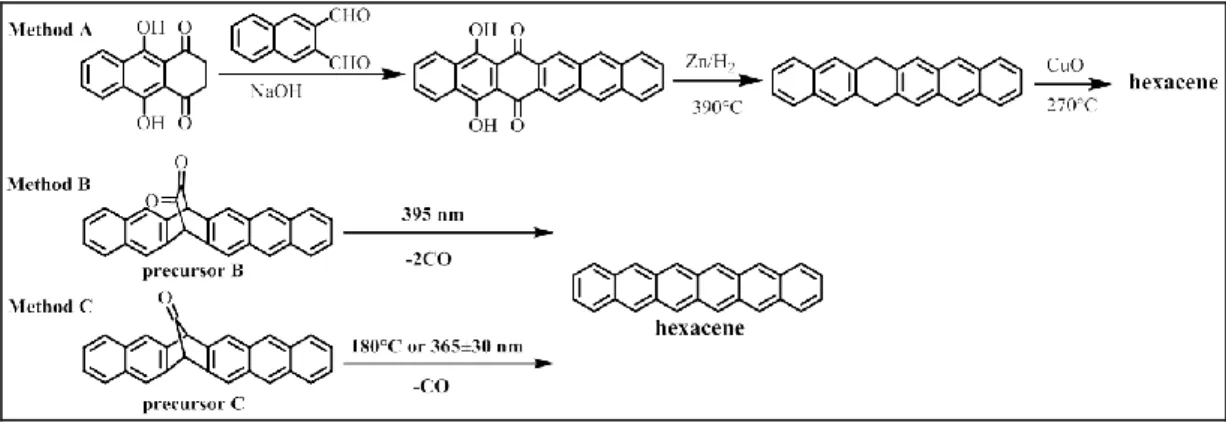
(33) 來解決五環素的問題,部分方法成功的改善分子排列方式以及分子溶 解度問題,但這些做法並未提升電荷遷移率。. 2.2 多環芳香烴:六環素 理論上,隨著稠環(fuse ring)數量增加(五環素→六環素),分子共 軛長度隨之增加,分子間 π-π 堆疊區域也可增加,對於電荷在分子間 跳躍有正面的影響,然而有別於合成容易且已商業化的五環素,六環 素(hexacene)的合成困難且煩瑣,因此六環素在 OFET 的發展較緩慢。 早期,Stacey 等人將 9,10-dihydroxyanthracene-l,4-dione 與萘-2,3 二甲醛進行縮合反應後,再以鋅/氫氣在 390℃進行還原反應,以氧化 銅在 270℃、二氧化碳環境(0.15 Torr)下進行脫氫反應得到六環素 18, 這種合成方法的條件較嚴苛,須在高溫減壓環境下進行反應,反應產 率也不高(如圖 2.8,method A);後來在 2007 年,Shah 與 Neckers 等 人以較溫和的方式合成出六環素前驅物 19 (如圖 2.8 的 precursor B), 經由照射 395 nm 的 UV 光後進行光致脫羰反應(photodecarbonylation) 而得六環素,可惜這個方式得到的六環素其穩定性極差,六環素分子 在低濃度下(<10-4 M)就會產生二聚體,在氧氣環境中會迅速與氧反 應產生過氧化物,因此,以此方式無法真正得到六環素晶體(如圖 2.8,method B)。. 13.
(34) 圖 2.8 六環素的合成演進 在 2012 年,有別於 Shah 與 Neckers 等人的方法進行光催化一氧 化碳脫去的步驟時會生成具有兩個自由基的中間物進而得到雙聚體 結構 19,Chow 等人以合成五環素前驅物的方法合成六環素前驅物 20, 再以加熱或光照脫除一分子的一氧化碳後得到六環素. 21. (如圖 2.8,. method C),並且順利以物理氣相沉積法得到六環素單晶分子,從 XRD 的分析中得知六環素與五環素的排列方式幾乎相同,皆以魚骨狀的方 式進行 edge-to-face 排列(如圖 2.9),這樣的排列方式較不易形成六環 素二聚體 22,因此六環素分子可在不受光照下的一般環境放置 30 天 以上而不變質,這對六環素而言是一個重要的突破!Chow 等人將六 環素單晶分子製成電晶體材料後,與五環素、四環素、三環素以及萘 做電性比較(如圖 2.9 中的表),由於六環素重組能量(reorganization energy, λ+)最小、電子耦合常數最大(t +),因此在這些多環芳香烴中擁 有最大的電荷遷移率。. 14.
(35) 圖 2.9 六環素分子單晶結構圖與多環芳香烴電荷傳遞速率的比較. 2.3 多環芳香烴:四環素 在 2004 年,Bao 等人在四環素中導入鹵素原子,藉此破壞四環 素原先 edge-to-face 的作用力(如圖 2.10-a),使分子堆疊自魚骨狀轉變 為 π 軌域的平面堆疊 23,其中當四環素鍵結一個鹵素時(如圖 2.10-b), 分子兩兩之間以 face-to-face 的方式堆疊,而分子對之間仍以魚骨狀 堆疊;當四環素鍵結兩個鹵素時(如圖 2.10-c),分子的排列則轉變成 以 face-to-face 的方式進行一維柱狀堆疊,導入鹵素後兩者排列方式 的差異直接反應在電性表現上,後者的電荷遷移率為 1.6 cm2/Vs,幾 乎是前者的 1000 倍!. 15.
(36) 圖 2.10 四環素及其衍生物之分子堆疊示意圖. 16. 在 2006 年,Swager 等人在四環素的末端苯環同時導入四個氟原 子,在另一苯環之不同位置導入烷基或烷氧基 24 (如圖 2.11),從分子 的單晶結構發現,由於氟原子與苯環的 π 軌域的作用力之下,使得這 些分子幾乎以頭尾相疊的方式堆疊,其中,長烷鏈導入的位置對於分 子共軛平面間的距離及分子共軛平面間的平移距離皆有影響,但整體 而言可幫助分子的排列,誘導分子以柱狀方向進行堆疊,如此排列方 式的改善成功提升了電荷遷移率。. 圖 2.11 四環素衍生物分子示意圖 16.
(37) 2.4 多環芳香烴:三環素 在多環芳香烴中,並苯的數量越多(如五環素、六環素),分子共 軛長度越長,有助於分子間 π-π 堆疊,然而當苯環數量增加時,分子 的穩定性和溶解度皆越差,增加了分子合成及元件製作上的困難度; 小分子並苯(如萘、蒽) 的共軛長度較短,使得其電性效果不佳,但 是其分子的穩定性及溶解度皆較五環素佳,也較易進行分子加工,因 此,可藉由此特性在分子上以寡聚物的方式延伸分子的共軛系統,來 改善小分子並苯電性不佳的問題。 以蒽而言,其寡聚物之間的連接位置可分成:peri-position 與 end-position25(如圖 2.12-a)。其中,若以苯環做為取代基導入在蒽的 peri-position,此苯環和蒽之間會存在一個扭曲角,反之,當苯環導入 在蒽的 end-position, 則苯環 與蒽之間會以平 面的方式延伸 (如圖 2.12-b)。. 圖 2.12 蒽的 peri-與 end-position 示意圖. 17.
(38) 在 2003 年,Suzuki 等人利用在蒽的二號、六號位進行延伸,發 表了雙聚及三聚蒽寡聚物 26 (如圖 2.13),利用寡聚物的方式得到線性 分子,除了可以延伸分子的 π 共軛長度外,在此也降低了分子的氧化 電位而增加了電荷注入電極的機會。在 OFET 特性方面,增加蒽分子 的數量以及在分子末端導入長烷鏈皆增加電荷遷移率(如圖 2.13)。可 惜的是,這類分子存在溶解度問題,2A 及 DH-2A 僅溶解在二氯苯熱 溶液中,3A 及 DH-3A 則無法溶解在一般有機溶劑,因此,以寡聚物 的方式在蒽的二號及六號位進行延伸可解決蒽共軛長度不足的問 題,但當寡聚物數量增加時,在分子設計上仍須考量溶解度問題。. 圖 2.13 寡聚蒽衍生物結構示意圖與其電荷遷移率比較. 18.
(39) 在 2005 年,Meng 等人在蒽的二號、六號位導入噻吩(thiophene) 及己烷噻吩(hexyl thiophene)27 (如圖 2.14),這兩個結構皆可溶於熱甲 苯中。從分子單晶結構發現,導入的噻吩與蒽幾乎是在同一平面上, 如此平面且對稱的結構有利於分子緊密堆積,可惜的是,分子間仍以 魚骨狀的方式堆疊。. 圖 2.14 DTAnt 與 DHTAnt 分子單晶結構與半導體電性比較 在 2006 年,Meng 等人. 28. 進一步發表了 DPPVAnt(如圖 2.15),. DPPVAnt 分子在溶解度方面與 DTAnt 和 DHTAnt 相似,可溶於熱甲 苯中。有趣的是,雖然 DPPVAnt 分子間仍以魚骨狀的方式堆疊,但 其分子與分子間的距離較五環素短,如圖 2.15 所示,a(五環素)與 a’(DPPVAnt)層與層的距離相差了 0.55Å,而 b(五環素)與 b’(DPPVAnt) 層與層的距離相差了 0.42Å ,如此對於電荷在分子間跳躍是有利的。 我們可由 DTAnt、DHTAnt 與 DPPVAnt 這三個例子得知,在蒽的二. 19.
(40) 號及六號位置導入單一簡單的芳香環對於分子間堆疊方式並無太大 影響,仍然是以 edge-to-face 之魚骨狀方式進行排列。. 圖 2.15 五環素(左圖)與 DPPVAnt(右圖)之分子排列方式比較 在 2005 年,Anthony 等人將三異丙基矽乙炔基導入在蒽的九號、 十 號 位 (TIPSAnt)29(如 圖 2.16), 有 別 於該 團隊 在 2001 年 發 表 的 TIPSPen 結構 10,由於三異丙基矽乙炔取代基的直徑遠大於蒽的分子 長度,反而撐開了原先未修飾蒽分子之間的距離,可從其單晶結構證 實此分子為魚骨狀堆疊,因此當時是被應用在有機發光二極體(OLED) 中,若要應用在 OFET 領域則需進一步做修飾。 於是在 2007 年,Shim 等人在 TIPSAnt 的二號、六號位導入己烷 噻吩及苯乙烯,發表了 TIPSAntHT 與 TIPSAntPV 兩個結構 30(如圖 2.17),藉由己烷噻吩及苯乙烯除了可延伸共振長度之外,並藉由這兩 20.
(41) 個取代基使分子與分子錯開,在此,由於蒽分子的共軛延伸使得加入 矽烷炔基能夠幫助分子間的排列,從單晶結構可以發現,TIPSAntHT 與 TIPSAntPV 不再以魚骨狀的方式排列,反而因為中心蒽分子與兩 旁的芳香環在同一平面上(圖 2.17-b,d),使得分子間能更緊密的以 face-to-face 方式堆疊,兩者皆以二維共軛平面堆疊而成。另外,相較 於先前 Meng 等人發表的 DHTAnt只能溶解在熱甲苯中 27,TIPSAntHT 與 TIPSAntPV 由於矽烷炔基的導入提升了分子的溶解度,兩者皆可 溶在一般有機溶劑中。雖然這兩個分子之電性表現並不突出,但在蒽 的九號、十號位導入烷炔結構確實能增加分子溶解度,再利用延長分 子共振長度誘使分子從魚骨狀排列轉變為其他排列方式。. 圖 2.16 TIPSAnt 分子單晶結構圖. 21.
(42) 圖 2.17 (a)(b)TIPSAntHT 與(c)(d)TIPSAntPV 分子單晶結構圖 在 2010 年,Shim 等人延續了他們在 2007 年的研究,將三異丙 基矽乙炔基(TIPS)蒽的二號、六號位的加入噻吩(TIPSAntT)、苯並噻 吩 (benzothiophene, TIPSAntBeT) 以 及 苯 基 噻 吩 (phenyl thiophene, TIPSAntPT)作為取代基,探討這些取代基對於電性的影響. 31. (如圖. 2.18),他們發現導入越剛硬(rigid)的取代基,分子的溶解度就越差, 因此 TIPSAntBeT 僅能溶解在二氯苯熱溶液中。有別於無取代基的 TIPSAnt 分子以魚骨狀的進行堆疊,觀察其單晶結構可以發現這些分 子皆以 face-to-face 的方式進行堆疊,其中 TIPSAntT 與 TIPSAntBeT 分子間在堆疊時,中心的共軛結構與兩個鄰近分子的三異丙基矽乙炔 基形成錯位堆疊(slipped stacking)的方式(如圖 2.18-a,b),而 TIPSAntPT. 22.
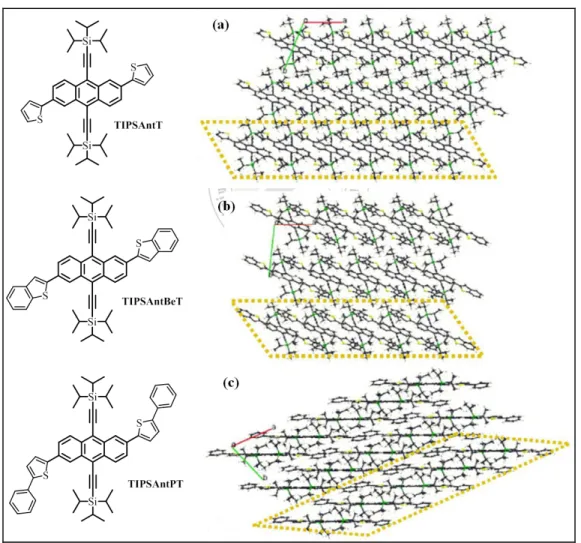
(43) 則是相反,苯環取代基避免了中心共軛結構與鄰近分子的 TIPS 相 鄰,使得中心共軛結構能與鄰近分子的共軛結構進行堆疊,而形成了 磚牆式排列,在半導體電性方面,由於 TIPSAntBeT 擁有較剛硬的結 構,分子的結晶能力較佳,但其高結晶性卻也增加了分子薄膜與介電 層之間缺陷(hole trapping sites)的數量,造成驅動元件的不易,須用較 大的閘極電壓(-28.7 V)才能啟動元件(相較於 TIPSAntPT 只需-4.0 V)。. 圖 2.18 TIPSAntT、TIPSAntBeT 與 TIPSAntPT 分子排列方式示意圖. 23.
(44) 在 2006 年,Geng 等人發表了一系列蒽的寡聚物分子 32,他們在 蒽的九號、十號位上加入辛炔(octylnyl)以增加分子的溶解度,其單體 和雙體對一般有機溶劑的溶解度很好,然而當蒽的重複數量增加三時 (即三體蒽分子),溶解度的問題再度浮現,有趣的是,他們用癸炔 (decynyl)取代辛炔導入在蒽上(如圖 2.19),竟能大幅提升溶解度,使 得 OA-5b 分子足以溶解在氯仿中!另外,從 OA-1 的單晶結構中可以 發現導入的長烷鏈與蒽幾乎是在同一平面上,如此一來有利於分子間 緊密堆疊。. 圖 2.19 OA-5a 與 OA-5b 分子示意圖及 OA-1 與其單晶結構圖. 24.
(45) 第三章、研究動機 3.1 本計畫選擇蒽分子的理由 目前在以多環芳香烴做為有機半導體材料的領域中,以五環素的 電性表現最佳,然而五環素存在著下列幾個問題:(一)五環素的氧化 電位較低,使其在空氣中及光照下容易分別生成醌基五環素及五環素 雙聚物 (二)五環素剛硬性的稠環結構使其溶解度不佳,而不易進行 分子加工 (三)分子間以 edge-to face 魚骨狀方式進行 π 共軛堆疊,此 方式對於電荷傳遞的效率較差(如圖 3.1)。為了避免碰到五環素所遇 到的問題,本研究選擇蒽做為分子的主體,這是因為蒽有較高的氧化 電位可避免在空氣中被氧氣氧化,穩定性較佳,而且分子剛硬性較 差,因此溶解度較好,可惜蒽的 π 共軛長度較短,分子間重疊的區域 較小,因此電荷遷移能力不佳,因此本研究將在蒽上藉由分子修飾調 整其分子間的排列方式,從魚骨狀排列轉變為其他對於電荷遷移較有 效率的方式(如:磚牆式堆疊),以改善原先不佳的電性表現。. 圖 3.1 多環芳香烴之稠環數量與其特性比較. 25.
(46) 本研究將從兩個部分著手改變蒽的排列模式: (一)延伸分子的共軛長度。從文獻回顧中的討論可以發現,若將 蒽以並苯的方式連接(即得六環素,如圖 3.2),可以順利延伸共振長 度,然而六環素雖有高的電荷傳遞速率,但分子合成不易且仍有穩定 性的疑慮 21;若將蒽以寡聚物的方式連接亦可延伸共振長度 26,文獻 指出若在其二號、六號位進行芳香環取代,則能以線性、平面的方式 延伸,雖然共軛長度延伸範圍不及六環素,但分子的穩定性與溶解度 卻可大幅提升(圖 3.2),因此本研究將使用後者的方式延伸蒽的共軛 長度,做成蒽的雙聚物(dimer)及三聚物(trimer)。. 圖 3.2 延伸蒽的構想 (二)如何藉由分子設計將魚骨狀排列轉變為磚牆式排列。在許多 文獻中都證實藉由分子設計可大幅調整分子間排列的方式 12, 28, 33,除. 26.
(47) 了能讓分子層與層間的堆疊更緊密外,甚至能夠明顯改善分子巨觀上 的堆疊方式,得以從 face-to-edge 的魚骨狀堆疊轉變為其它能夠更有 效的傳遞電荷之堆疊方式。因此本研究將在蒽分子中的不同位置導入 取代基(如圖 3.3),藉由取代基在分子的不同位置來影響分子的排列 特性,促使分子間以不同程度的方式交錯著,減少 edge-to-face 的排 列方式,提升分子間 face-to-face 的排列機會。 有鑑於文獻提到三體蒽的溶解度不佳 26,本研究在三體蒽分子之 中心蒽的九號及十號位置導入可提升分子溶解度的長烷鏈,以避免出 現蒽三體所遇到的問題(如圖 3.3)。. 圖 3.3 蒽分子之官能基修飾示意圖. 27.
(48) 3.2 分子設計藍圖 圖 3.4 係本研究綜合上述動機的考量,所期望合成的十個蒽衍生 物,除了蒽單體外,為了延長蒽的共軛長度,本研究在蒽的二號、六 號位進行延伸,設計出蒽雙體及蒽三體衍生物,用來比較分子共軛長 度對分子基本特性的影響。另外,藉由線性方式延長分子長度的同時 亦可延伸分子間魚骨狀的排列距離,而調整分子的排列方式。為了提 升三體蒽分子的溶解度,因此本研究參考文獻作法在三體蒽分子的中 心蒽之九號、十號位以炔基做為延伸並導入己烷基 32,其中,炔基的 導入可做為長烷鏈結構的緩衝,避免長烷鏈之立體結構直接影響中心 蒽分子的堆疊。 另外,在所有蒽分子中導入甲氧基,除了可改變分子內的電荷分 布之外,本研究另將甲氧基導入在蒽的不同位置上,以研究取代基的 位置差異對分子基本特性的影響,並期待藉由不同位置的取代基誘使 分子以不同的方式進行固態堆疊,進而能調整分子排列的型態。. 28.
(49) 圖 3.4 目標分子示意圖. 29.
(50) 第四章、結果與討論 4.1 目標分子的合成討論 為了研究寡聚蒽分子不同官能化結構位置對材料的堆疊形態以 及半導體特性的影響,本研究嘗試合成不同甲氧基位置的蒽單體(如 圖 4.1),並針對不同的蒽單體的合成進行討論:. 圖 4.1 蒽單體示意圖 流程 4.1 是二辛炔蒽<D2>的合成路徑。<D2>的合成可由市售的 2,6-二氨基蒽醌搭配溴化亞銅以及有機偶氮試劑(tert-butyl nitrite)進 行 Sandmeyer reaction30,將 2,6-二氨基蒽醌轉換成 2,6-二溴蒽醌後, 此二溴蒽醌再參考 Geng 等人的合成方法 32,以正丁基鋰在低溫下拔 去辛炔的末端氫後,在蒽醌的九號、十號位進行親核性加成反應 (nucleophilic addition),接著在酸性水溶液中以含二結晶水的氯化亞錫 進行還原反應,得到二辛炔蒽<D2>。. 30.
(51) 流程 4.1 <D2>之合成路徑 流程 4.2 是單體<B2>的合成。<B2>的合成是使用市售的 2-氨基 蒽醌進行,使用與合成<D1>相同的試劑在相同條件下進行反應,即 可成功得到<B1>,接著在<B2>的合成中,將醌基還原的反應則是參 考文獻作法 33,利用硼氫化鈉將九號、十號位的醌基還原成二羥基蒽 後,再以硫酸二甲酯進行甲基化後即可得<B2>(流程 4.2),在這個反 應中<B2>產率為 30%,另有兩個副產物生成:30%副產物經 NMR 鑑 定為 2-溴蒽(2-bromoanthracene),剩下的 30%副產物無法由 NMR 確 認為何種結構。. 流程 4.2 <B2>之合成路徑 <9,10DMA>的合成方法與<B2>類似,使用市售的 9,10-蒽醌做為 起始物,上述<B2>的合成方法 33 即可得到<9,10DMA>,產率為 30%, 過程中也是產生了兩種副產物,其一經 NMR 鑑定確認為蒽(產率為 30%),另一則是未知物(產率為 30%)。 31.
(52) 流程 4.3 <9,10DMA>之合成路徑 在<1,4DMA>的合成中,曾經嘗試過兩種方式(流程 4.4),路徑 A 是利用市售的 1,4-二羥基-9,10-蒽醌進行甲氧基化後再將醌基還原而 得,路徑 B 則是將<1,4AQ>還原成 1,4-二羥基蒽後再進行甲氧基化而 得,兩種方法皆可以得到<1,4DMA>,但<1,4AQ>直接購買的成本較 高,自行合成所 需的步驟也較多,因此選擇以路徑 A 進行量產 <1,4DMA>。 在流程 4.4 路徑 B 的研究中,我們先以萘醌的還原進行測試(流 程 4.5),嘗試過數種方法,其中包括以連二亞硫酸鈉(sodium dithionite, Na2S2O4)做為還原劑搭配水和醚類溶劑 34,35 (流程 4.5 方法 A)、以鋅 做為還原劑搭配醋酸溶液 36 (流程 4.5 方法 B)等,改變許多反應條件, 結果皆失敗,最後參考文獻的做法 37(流程 4.5 方法 C),使用含二結 晶水的氯化亞錫搭配甲醇為溶劑,可成功還原醌基,且產率可達 90%。 得到<1,4DMA>後,接著須分別在<1,4DMA>的二號與六號位置進行 溴化取代得到<A2>與<C5>單體(流程 4.6),<A2>可從<1,4DMA>以 N-溴代丁二醯亞胺(NBS)進行溴化反應而得到<A2>,然而<C5>無法 直接自<1,4DMA>以一般溴化的方式將溴原子接在六號位置,因此我 們的構想是使用四號位已有溴取代的 4-溴鄰苯二甲酸衍生物與對二 甲氧基苯進行 Fridel-Craft acylation 而得到 6-溴蒽醌衍生物,再將醌 32.
(53) 基還原成 6-溴蒽衍生物即可。在 Fridel-Crafts acylation 反應中,適用 的官能基可以是羧酸(再進一步做成醯氯)以及酸酐,因此以下將進行 <C1>與<C2>的合成討論。. 流程 4.4 <1,4DMA>之合成路徑. 流程 4.5 萘醌還原為羥基萘之測試 O. O. O. Br Br O. O <1,4DMA>. <C5>. O <A2>. O O O. O O Br O <C2>. OH OH. or Br O <C1>. 流程 4.6 <C5>與<A2>的合成差異. 33.
(54) <C1>與<C2>雖已市售化,但單位價格過高,因此我們選擇購買 4-溴鄰二甲苯自行合成<C1>與<C2>。作法是將 4-溴鄰二甲苯以過錳 酸鉀氧化成 4-溴鄰苯二甲酸<C1>,接著在醋酸酐的催化下脫去一個 水分子,即可得 4-溴鄰苯二甲酸酐<C2> (流程 4.7)38。將甲基氧化成 羧酸的反應中,本研究嘗試了不同的鹼性試劑(流程 4.7 中的表),測 試結果發現以吡啶為鹼可達較高的產率 39,由於吡啶可與 4-溴鄰二甲 苯互溶,亦可與水互溶,並且同時扮演鹼與溶劑的角色,將原先不互 溶的水層和有機層充分混合,使兩者之間能夠充分反應。不過起初吡 啶的用量大(61 當量),經過調整及追蹤後,吡啶的用量可降至 26 當 量,過錳酸鉀也可從 18 當量降至 10 當量。. 流程 4.7 <C1>合成之條件測試及<C2>之合成方法 我們在<C5>的合成上花費了許多時間進行許多反應條件的測 試,主要以 Fridel-Crafts acylation 進行流程 4.6 中的構想。起初參考 本實驗室的方法,將<C1>醯氯化後,進行 Fridel-Crafts acylation 應可. 34.
(55) 得到<C4>(流程 4.8 方法 A),然而首先碰到的是<C1>溶解度的問題, 使得<C1>在醯氯化的步驟就受到限制,於是開始搜尋能夠溶解<C1> 的溶劑與反應方法,參考文獻 40 的方法(流程 4.8 方法 B),使用三氟 醋酸與三氟醋酸酐進行反應,在反應途中確實能使<C1>溶解在溶劑 中,但卻得到無法預期的產物;有些文獻. 41,42,43,44. 則是在三氟醋酸與. 三氟醋酸酐中加入 85% 磷酸以形成更好的離去基,嘗試過數個條件 仍無法順利得到<C4>(流程 4.7 方法 C);在流程 4.8 方法 D 即是合成 <2,3DMA>和<E5>的方法(流程 4.10),該方法對於合成<2,3DMA>與 <E5>有很高的反應性,但當反應物換成對-苯二甲醚卻無效;最後在 數篇文獻的整理中. 45,46. ,發現以 Fridel-Crafts acylation 要進行一步驟. 的蒽醌合成時,須要在較嚴苛的反應條件下才會進行反映,因此參考 文獻做法,改以三氯化鋁與氯化鈉於高溫下(150~170℃)所形成的融 熔態進行(無使用溶劑)(流程 4.8 方法 E),實際參考該方法後果然成 功得到<C3>!有趣的是,在合成<C3>的反應中,無論使用對-苯二甲 醚還是對-苯二酚皆得到<C3>,將<C3>進行甲基化後,再將醌基還原 即 可 得 <C5>, 流 程 4.8 方 法 E 的 產 率 相 較 於 以下 將 討 論的 以 Diels-Alder reaction 的合成方法(流程 4.9),除了實驗步驟較少之外, 產率可明顯提升。. 35.
(56) 流程 4.8 以 Fridel-Crafts acylation 合成<C5>之反應測試與流程 除了以 Fridel-Crafts acylation 的方法外可合成<C5>,有些文獻則 是使用 Diels-Alder reaction 得到 6-溴-1,4 蒽醌<C10>(流程 4.9)47,反 應所需的二烯(diene)前驅物<C9>可自<C1>在酸性環境下酯化後,使 用氫化鋁鋰將羰基還原成羥基<C8>48,再以三甲基氯矽烷與溴化鋰將 羥基換成溴後 reaction 後. 49,50. ,在碘化鉀的置換下與對苯醌進行 Diels-Alder. 47. ,以氯化錫和甲醇將醌基還原為羥基後再進行羥基的甲. 基化而得<C5>。在此方法中,合成 <C1>、<C7>、<C8>和<C9>每個 步驟的產率都在 90%左右,然而<C9>到<C10>的步驟產率卻驟降, 僅 10~15%,原因為此反應中產生了許多溶解度極差的副產物,在本 36.
(57) 實驗室先前的研究中,發現以對苯醌進行 Diels-Alder reaction 時,由 於對苯醌是個好的親雙烯體(dienophile),其兩側皆可和二烯(diene)進 行反應,反應性佳且不易控制只接單邊,因此雖然流程 4.9 可得 <C5>,相較於流程 4.8 的方法 E 而言產率太低,所以本研究主要仍以 流程 4.8 方法 E 進行<C5>的合成及量產。. 流程 4.9 以 Diels-Alder reaction 合成<C5>之流程 <2,3DMA>與<E5>的合成則是參考文獻做法(流程 4.10)51,鄰苯 二甲酸酐在路易士酸三氯化鋁的作用下與鄰二甲醚進行 Fridel-Crafts acylation,而得<E1>,接著<E1>在濃硫酸的催化下脫水、環化成蒽醌 <E2>。其中,由於<C2>為非對稱結構,因此在反應進行時會形成<E3> 與<E3'>混合物,將混合物進行環化後皆可得<E4>,並不影響反應; 另外,鄰苯二甲醚在反應中為過量試劑,反應完後會有殘留,由於鄰 苯二甲醚在常溫常壓下之沸點高達 206℃,利用再沉澱的方式得到的 <E1>、<E3>與<E3’>固體產物易有鄰苯二甲醚的殘留,此問題可藉由 溶解度的差異來純化之,使用二氯甲烷稍微潤洗產物(產物對二氯甲 烷的溶解度不佳),而帶走多數的鄰苯二甲醚,得到較純的產物。. 37.
(58) 流程 4.10 <2,3DMA>與<E5>之合成 在環化的部分(流程 4.10 中,<E1>至<E2>與<E3>至<E4>),本研 究發現反應時間和硫酸的用量對環化反應影響很大:當反應時間為三 十分鐘以上,反應結束後會生成深墨綠黑色黏稠狀液體,造成後續處 理的不易,且產率略為下降,此乃反應過度造成,若反應時間控制在 三十分鐘內,在後續處理上容易許多,且產率較高;硫酸在此同時扮 演溶劑以及酸催化試劑的角色,其用量必須能夠讓反應物充分分散, 但用量太多時,濃硫酸的強氧化力特性會將有機物氧化成焦黑的碳, 經過數次實驗的追蹤及比較,1 毫升的硫酸與 3.5 毫莫耳的反應物為 最佳比例。得到<E2>與<E4>後,再將醌基還原,此步驟與流程 4.4 中<A1>反應至<1,4DMA>的方法相同,產率也都在 70%以上! 合成出含溴的單體蒽衍生物後,接著為硼酯衍生物的合成(流程 4.11),係以鈀金屬錯合物做為催化劑、搭配聯硼酸頻那醇酯做為硼酯 的來源、使用醋酸鉀為鹼的條件下,即可合成出含硼酯的蒽衍生物 <D3>、<A3>、<B3>、<C6>以及<E6>,產率在 40%~66%。. 38.
(59) 流程 4.11 硼酯衍生物的合成及產率整理 本研究以 Suzuki-Miyaura 交叉耦合反應進行二聚體及三聚體的 合成,在許多文獻上都有以蒽為主體進行交叉耦合反應的研究. 28,26,. 32. ,幾乎都以 Pd(PPh3)4 同時作為鈀金屬催化劑及 ligand,再搭配碳酸. 鈉水溶液為鹼性試劑在甲苯的迴流溫度下進行反應(流程 4.12 方法 A),<A2>與<A3>在此條件下反應可順利進行,可得接近六成的產 率,然而由於 Pd(PPh3)4 對於水氣及氧氣較為敏感,易在空氣中被氧 化而降低反應性,且保存上也較不易,因此反應產率容易因為藥品的 問題而有波動(0%~58%),因此調整實驗方法及條件,參考實驗室學 長陳柏志之實驗方法,改以 Pd(OAc)2 作為鈀金屬催化劑、使用 Sphos. 39.
(60) 作為 ligand、搭配磷酸鉀水溶液為鹼性試劑在四氫呋喃的迴流溫度下 進行反應(流程 4.12 方法 B),相較於 Pd(PPh3)4,Pd(OAc)2 對於水氣和 氧氣的容忍度較大,雖然反應性及反應產率不及 Pd(PPh3)4,但產率 較穩定,因此其餘的雙體結構和所有的三體分子皆以方法 B 進行合 成(流程 4.13)。. 流程 4.12 二聚體的合成及產率整理. 40.
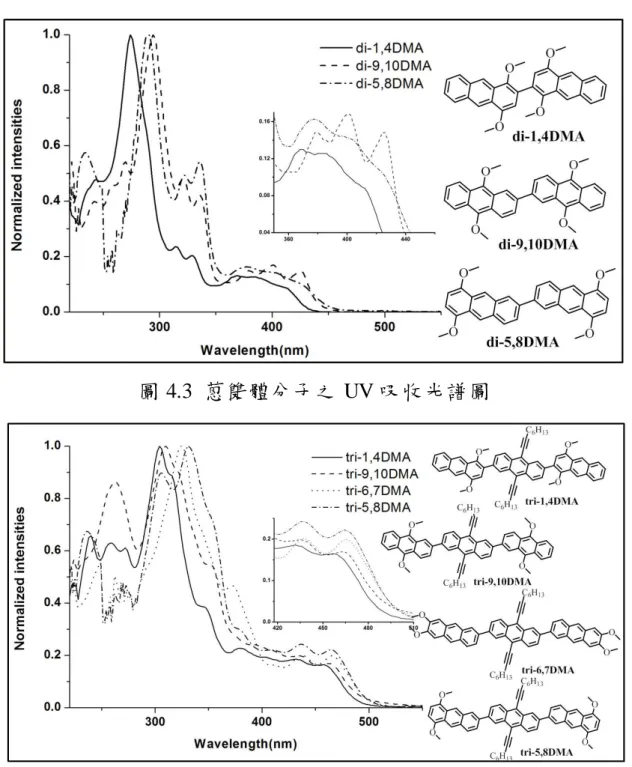
(61) 流程 4.13 三聚體的合成及產率整理. 4.2 分子溶解度比較 本研究合成出一系列取代基位置不同的蒽衍生物,藉由在蒽的二 號及六號位置進行分子共軛長度的延長,以單體分子進行交叉耦合反 應後得到雙體分子及三體分子,本研究合成出的蒽衍生物皆可溶在二 氯甲烷中,相較於文獻中 26 所提到的 3A 對於一般溶劑的溶解度不佳 已有明顯改善。 單體分子對於一般溶劑(二氯甲烷、丙酮、乙酸乙酯、甲苯等)都 有 很好 的溶解 度; 雙體 分子的 溶解 度就開 始稍 微有 所限制 , <di-1,4DMA>仍然有好的溶解度,然而<di-5,8DMA>與<di-9,10DMA>. 41.
(62) 的溶解度開始下降,它們溶於重氫氯仿的量可進行 1H-NMR 的鑑定, 但已不足以進行 13C-NMR 的測量。 而 三體 分子的溶 解度依 序以 <tri-1,4DMA> 、 <tri-9,10DMA> 、 <tri-5,8DMA>、<tri-6,7DMA>遞減,其中<tri-1,4DMA>依舊溶解度最 佳 , 一 般 溶 劑 皆 可 溶 解 之 , 而 <tri-9,10DMA> 溶 解 度 雖 不 如 <tri-1,4DMA> , 但 尚 可進 行. 13. C-NMR 的 測 量 ; <tri-5,8DMA> 及. <tri-6,7DMA>仍可溶解在二氯甲烷中,其溶解度卻與<di-5,8DMA>與 <di-9,10DMA>相近,對於雙體分子而言,再增加一個具剛硬性的蒽 而成為三體分子勢必降低分子的溶解度,然而三體分子卻與雙體分子 的溶解度相似、甚至更好(<tri-9,10DMA>優於<di-9,10DMA>),證明 在三體分子的中心蒽導入長烷鏈的結構可成功改善分子的溶解度。 另外,取代基在一號及四號位置的分子,無論是單體、雙體還是 三體溶解度都明顯優於其他取代位置;相反的,取代基在五號及八號 位置雙體及三體分子溶解度最差。. 4.3 光學性質分析 本研究利用紫外光/可見光吸收光譜(UV-Vis absorption spectra)及 螢光放射光譜(Fluorescence emission spectra),以了解材料取代基位置 對蒽單體及其寡聚物分子的光物理性質影響。 在吸收光譜的結果中,取代基的位置對於最大吸收波長的位移有 明顯的關聯性:單體分子中(如圖 4.2),分子的最大吸收波長的順序. 42.
(63) 為<2,3DMA>> <9,10DMA>> AN><1,4DMA>,兩兩之間差異約為 10 nm, 以 取 代 基 在 二 號 與 三號 位 置 分 子 的共 軛 長 度 最長 , 而 <1,4DMA>相較於未有取代的蒽(簡稱<AN>),呈現出藍位移的現象, 表示在<AN>的一號及四號位進行取代時,對於共軛長度的延伸效果 不 佳 ; 三 體 分 子 中 (如 圖 4.4), 分 子 的 最 大 吸 收 波 長 的 順 序 為 <tri-5,8DMA> > <tri-6,7DMA> > tri-9,10DMA > <tri-1,4DMA> , 以 <tri-5,8DMA>的最大吸收波長為 330nm 最長,表示取代基在五號及 八號位置最能有效延伸三體分子的共軛長度。對於雙體分子而言,結 果就有些不同(如圖 4.3),反而是<di-9,10DMA>具有最大的吸收波長 (294 nm),但與<di-5,8DMA>最大的吸收波長差異不大(290 nm)。 在單體、雙體及三體分子的 UV 吸收光譜中,除了在短波長的部 分(240~300 nm)有來自單一芳香環的電子躍遷外(最大吸收波長),在 較長波長的部分(320~390 nm)另有三個訊號(部分分子該訊號的峰值 數據較不明顯),推測該訊號來自於分子間堆疊的 π-π transistion31,52 (如圖 4.2~4.4)。其中就單體而言,取代基在一號、四號位置其長波長 部分有最佳的延伸,暗示其分子堆疊較佳,然而本研究發現在雙體及 三體分子中,以取代基在五號、八號位置以及六號、七號位置其長波 長處有最大的延伸,綜合以上兩個現象推測當取代基導入在分子外側 時(例如五號、八號位置以及六號、七號位置),分子間的堆疊較佳, 因此共振長度越長,能量轉移的訊號也更加紅位移,進一步分子堆疊 的特性仍需以分子單晶結果予以確認。. 43.
(64) 相同取代位置的單體、雙體及三體 UV 吸收結果整理在圖 4.5 中,隨著蒽的數量增加,即具有相同取代位置的分子從單體延伸至雙 體、三體時,其最強吸收訊號之位置有明顯的紅位移(兩兩之間差異 約為 30nm),證明在蒽的二號及六號位置進行多環芳香烴的修飾,能 夠成功延長分子的共軛長度。. 圖 4.2 蒽單體分子之 UV 吸收光譜圖. 44.
(65) 圖 4.3 蒽雙體分子之 UV 吸收光譜圖. 圖 4.4 蒽三體分子之 UV 吸收光譜圖. 45.
(66) 圖 4.5 蒽分子 UV 吸收光譜圖 測量放射光譜前,先預先使用螢光光譜儀中的 3D 全波長掃描模 式進行各個波段的激發(excitation)與放光(emission),用來得知各分子 放光最強處所需使用的激發波長,結果發現大部分的分子放光最強所 需的激發光源與分子在 UV 吸收圖中最強的吸收波長相符。 在放射光譜的結果中(如圖 4.6~圖 4.9),幾個分子在激發光源的 兩倍波長處有一放光訊號,該訊號為倍頻光,不進行討論(例如圖 4.6 中<9,10DMA>於 520 nm 處之訊號)。除了<di-5,8DMA>、<2,3DMA> 和<tri-6,7DMA>的主要放光波長在 360nm 左右外,其餘分子的主要 放光波長都紅位移至 440~530nm。在單體分子中(如圖 4.6),當甲氧. 46.
(67) 基導入在分子的一號、四號位置與九號、十號位置時,相較於未修飾 的蒽皆有紅位移的現象;在所有雙體分子中(如圖 4.7),在 363nm 左 右都可見到一放射波長,從文獻中可得知中心辛炔蒽的放光波長約為 443nm 與 467nm32,本研究所設計的三體蒽分子除了<tri-6,7DMA>之 外,其他放光波長可紅位移至 505~540nm。 特別的是,<2,3DMA>與<tri-2,3DMA>兩者共振長度差異很大, 但其放光波長卻幾乎相同,在此推測<tri-2,3DMA>的結構不易形成激 發雙體(excimer),分子也不易進行有序堆疊,因此而無能量轉移的現 象,放光波長未有紅位移出現,兩者分子放光波長僅在 364nm。在 <di-5,8DMA>的結構中也有上述相似的情形,其放射波長為 362nm, 相較於<1,4DMA>的 470 nm 藍位移了約 110nm。. 圖 4.6 蒽單體分子之 FL 放射光譜圖. 47.
(68) 圖 4.7 蒽雙體分子之 FL 放射光譜圖. 圖 4.8 蒽三體分子之 FL 放射光譜圖. 48.
(69) 圖 4.9 蒽分子 FL 放射光譜圖. 49.
(70) 4.4 電化學性質分析 場效電晶體材料的氧化還原性質與材料得到電子或失去電子後 的 穩 定 性 有 關 , 且 當 這 些 蒽 分 子 應 用 在 OFET 材 料 中 , 其 HOMO/LUMO 能階需與電極能階符合,因此本研究利用循環伏安儀 (Cyclic Voltammetry) 測 量 蒽 分 子 的 氧 化 電 位 , 換 算 出 分 子 的 HOMO/LUMO 能階。 進行測量時,係以碳電極為工作電極(working electrode)、以白金 電極為輔助電極(counter electrode)、以 Ag/AgCl 電 極為參考電極 (reference. electrode) , 電 解 質 為 四 丁 基 六 氟 磷 酸 胺. (tetra-n-butylammonium hexafluorophosphate)溶於二氯甲烷的 0.1M 電 解質溶液,再以 25 mL 容量瓶配置含 0.1M 電解質溶液和 10-3M 待測 物之二氯甲烷溶液,取待測溶液(約 15 mL)放入樣品瓶中,將上述三 根電極放入溶液中,施加直流電後即可開始測量。為了以內插法計算 分子實際 HOMO 值,須另行測量二茂鐵之氧化半電位來計算分子的 HOMO 能階。 在測量蒽分子之氧化半電位的過程中,意外發現這些分子亦有還 原 能力, 因此以較 適合進 行還原 電位測量 的二甲 基甲醯 胺 (dimethylformamide)為溶劑,配置 0.1M 四丁基六氟磷酸胺電解質溶 液,再以 25 mL 容量瓶配置含 0.1M 電解質溶液和 10-3M 待測物之二 甲基甲醯胺溶液進行還原半電位之測量,並另行測量 TPBI(1,3,5-tris(N-phenylbenzimidizol-2-yl)benzene)之還原半電位當作 50.
數據
+7
Outline
相關文件
In BHJ solar cells using P3HT:PCBM, adjustment of surface energy and work function of ITO may lead to a tuneable morphology for the active layer and hole injection barrier
200kW PAFC power plant built by UTC Fuel Cells.. The Solar
Lately, the chairperson of the Business Education Club, Louise, approached Sandy and proposed the idea of starting up a short term business with Organic Farming Club during
Solar panels generate electrical power by transforming solar radiation into direct current electricity using semiconductors that displays the photo-voltaic
In order to use the solar rays more efficient and improve the conversion efficiency of solar cell, it is necessary to use antireflection layer to reduce the losses of
(2009) Relating freshwater organic matter fluorescence to organic carbon removal efficiency in drinking water treatment. (1993) Filter mechanisms in
(2009) Relating freshwater organic matter fluorescence to organic carbon removal efficiency in drinking water treatment. (1993) Filter mechanisms in
Regarding to characteristic of organic matter of filtrates from three algal species, the percentage of carbon content of hydrophobic neutral (HPON) in whole incubation periods
