國立交通大學
應用化學研究所
博士論文
(1) Synthesis of Analogues of N-acetyl Neuraminic Acid
Using Pd(0)-catalyzed Approach
(2) Preparation of α-Glycosyl Chloride via TCT/DMF
(3) DMF-mediated α-Stereoselective Glycosylation
研究生:張智為 撰
指導教授:蒙國光教授、Jean-Marie Beau 教授與
Stéphanie Norsikian 博士
(1) Synthesis of Analogues of N-acetyl Neuraminic Acid
Using Pd(0)-catalyzed Approach
(2) Preparation of α-Glycosyl Chloride via TCT/DMF
(3) DMF-mediated α-Stereoselective Glycosylation
研究生:張智為 撰
Graduate Student: Chih-Wei CHANG
指導教授:蒙國光教授、Jean-Marie Beau 教授與 Stéphanie Norsikian 博士
Thesis advisor: Assist. Prof. Kowk-Kong Tony Mong, Prof. Jean-Marie Beau with
Dr. Stéphanie Norsikian
國立交通大學
應用化學研究所
博士論文
A thesisSubmitted to Department of Applied Chemistry College of Science
National Chiao Tung University, Taiwan in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Science in Applied Chemistry January 2010
Hsinchu, Taiwan, Republic of China
ABSTRACTS
This dissertation is divided into four parts:
Part I. A highly effective regio- and stereoselective palladium-catalyzed allylic substitution of 2,3-unsaturated derivatives of N-acetyl neuraminic acid (Neu5Ac2en) has been developed. We show that the efficiency of the allylation reaction depends on suitable protecting groups on the starting material and that, with sodium malonate anion as a nucleophile, regioselectivity can be fine tuned by the nature of the ligands associated with the palladium complex. The reaction was also applied to other nucleophiles for constructing C-C, C-N and C-O bonds and led to the major formation of the C-4 regioisomers. The selective transformation of some of the substitution products provided easy entry to a variety of modified sialic acid derivatives.
Part II. Preparation of α-glycosyl chloride derived from glycosyl hemiacetals (1-hydroxy sugars) can be performed by treatment of Vilsmeier-Haack complex generated from stoichimetric 2,4,6-trichloro-1,3,5-triazine (TCT) and N,N-dimethylformamide (DMF) to afford the corresponding chloride in excellent yields. This mild condition has been approved being compatible with acid-labile functionalities. In addition, this protocol can be applied to the synthesis of N-acetylneuraminic acid glycal in one-pot manner.
Part III. DMF-mediated α−selective glycosylation has been systematically investigated in aspects of mechanistic understanding, the applicable scope and experimental manipulation. We found the several distinguished features:1. Only stoichiometric DMF is required; 2. Better α−selectivity is usually obtained at the low temperature; 3. Pre-activation of glycosyl chlorides with DMF is for the first time proved feasible. 4. Plausible IAD mechanism may be
contributed to stereoselectivity. This Applications by using DMF as an additive could be practical in glycol-assembly.
.
Part IV. A tetrasaccharide containing the core structure of a glycoglycerolipid isolated from Meiothermus taiwanensis ATCC BAA-400 has been efficiently synthesized by using the convergent [2+2] glycoyslation strategy. DMF-mediated α−selective and low-concentration 1,2-trans β−selective glycosylation strategy are for the first time applied to construct the specific α− or β−linkage in oligosaccharide synthesis.
RÉSUMÉ
La première partie: Nous avons mis au point une méthodologie sur l'acide sialique en utilisant la réaction de Tsuji-Trost pour obtenir des produits de couplage avec de bons rendements et de bonnes sélectivités. De plus, selon la nature de la phosphine utilisée, nous avons pu constater différentes régiosélectivité en position 2 ou 4 avec diméthylmalonate. Nous avons aussi étendu la réaction à d’autres nucléophiles et nous avons pu construire diverses liaisons. Certaines modifications des dérivés en C-2 et C-4 ont été effectuées.
La deuxième partie: Après je suis revenu à Taïwan et nous avons développer une méthode par le réactif TCT/DMF pour la préparation de α-chlorure de glycosides. Cette condition est compatibile avec diverses fonctions sensibles aux acides. L'application en chimie des sucres par une stratégie ‘one-pot’ a été examiné.
La troisième partie: Dans le projet TCT/DMF, nous avons constaté que le DMF est très important pour la sélectivité α de la glycosylation. Des études mécanistiques ont montré la formation intermédiaire d'anion glycosyl iminium.
La dernière partie: Nous avons aussi appliqué cette méthode de glycosylation α -sélective à la synthèse d'oligosaccharides.
謝誌
此論文得以順利完成,絕非僅靠個人的努力達成目標,在其背後,有
太多需要感謝的人,這些人除了在實務上與精神上的支持,更是幫助我闖
過層層困難良師益友,因為有這一群人,此論文才得以產生。
首先要感謝的是在台灣的指導教授蒙國光教授。在交通大學修習博士
期間在學術上給我許多珍貴的專業指導,同時也給我機會到法國去做學術
的交流與學習,長達兩年的光景,讓我在學術領域及生活領域有了不同的
體驗。法國的兩年修習期間我要感謝法國的指導教授 Jean-Marie Beau,在
研究工作上給我很大的空間及機會學習,讓我看到不同的科學家思維,也
再次感謝 Jean-Marie Beau 教授於百忙之中,不遠千里前來參加我的博士
論文口試。
感謝我的口試委員們:鄭彥如教授、鄭建鴻教授、李靜琪博士及來自
法國巴黎第十一大學的 Jean-Claude Fiaud 教授,感謝你們撥冗指導,讓
學生的博士口試得以順利圓滿,並且獲得許多寶貴的意見。
感謝在我投入學術研究後,在學業上與實務上總是給我靈感啟發,並
且不時傳承我寶貴經驗的何子樂教授,太景生物科技公司的 Hossein 博士、
國衛院的夏克山博士、中研院吳宗益博士與林俊宏博士,感謝你們在我的
研習過程中,擔任著良師益友的角色,讓我用更寬廣的眼界,去專注於我
的研究工作。
感謝蒙國光教授實驗室的同學們,趙晉陞、林士哲、李振偉、張世聖、
賴崑章,你們不僅是我最好的工作夥伴,也是讓求學生涯裡增添樂趣的好
朋友。
鄧桔程,謝謝你們在工作閒暇之餘,陪我在球場奔馳、吆喝、紓壓,和你
們一起揮汗打球真的很棒很過癮,也是我每個星期最期待的時光。同時也
謝謝實驗室專題生曾郁惠、洪瑋晟、張詩偉,和你們一起共同研究的期間
裡,互相學習的過程,是很寶貴的體驗過程。
感謝交通大學所有協助過我的教職人員,特別感謝許千樹教授、秘書
小燕;感謝系辦楊淑鈖小姐及林淑梅小姐在文書作業上給予我的協助、張
秋景小姐在光譜測定工作上的幫忙。
另外我要感謝的是在我職場生涯中,在太景生技公司遇到的好同事
們,特別感謝鼓勵我繼續求學做研究的林助強博士;還要感謝職場夥伴李
廣元、劉振富、葉家成、彭國樹、詹景智、羅彬等好朋友們,因為有過和
你們共事的愉快經驗,讓我回憶不已,而博士的求學生涯裡,你們也未曾
停止過對我的關心與問候。
感謝曾經給我協助與經驗交流的學長及好朋友們,中研院陳榮傑博
士、龔亮仁博士、謝昇穎、李秀停,特別感謝梁富森博士,在我前往美國
開會時提供的協助與經驗交流。
最後要感謝一群我的摯友,從年少時代一路相挺的死黨們林志仁、蕭
東昇等;還有在文獻收集的工作上助我良多的張原豪博士;在剛回台灣時
給予我們生活上協助的邱顯吉;特別感謝遠在法國巴黎的李倫和韋彤,在
我們初到法國時無條件的給我們關懷與協助,在我們經濟上最困頓的時
候,向我們伸出援手,這份情誼與恩惠我們永遠不會忘記;同時感謝法國
里昂的海武及潔芝的友情相挺,因為有你們幾位好朋友的相伴,讓我們在
孤獨艱困的法國求學生活期間,可以一次又一次的度過難關。
最後我要感謝我的家人,為了支持成全我的理想無怨無悔的付出犧
牲,尤其是一路走來始終陪伴著我、照顧我的人-我的老婆,慧穎。也希
望我們及即將出生的寶寶未來一切順利。
此論文雖然稱不上甚麼豐功偉業,但是背後因為有這麼多人的協助與
付出,讓我更珍惜這一份成果,這篇謝誌裡,要感謝的人實在太多太多,
若是還有遺漏或不足之處,請各界師長與好朋友們見諒。
TABLE OF CONTENTS
ABSTRACTS ...I RÉSUMÉ... III 謝誌... IV TABLE OF CONTENTS ...VII LIST OF FIGURES... XIII LIST OF TABLES ... XV LIST OF SCHEMES...XVII LIST OF ABBREVIATIONS ...XXII
Chapter 1
... 1Regio- and stereoselective Pd(0)-catalyzed allylic substitution of sialic acid derivatives .. 1
1. Introduction ... 1
1.1. General information of influenza virus and the related therapies... 1
1.2. Chemical synthesis of neuraminidase inhibitors from Neu5Ac2en... 6
1.2.1. The first synthesis of 4-azido derivatives using Mitsunobu Reaction... 6
1.2.2. The first synthesis for 4-epi-4-azido-Neu5Ac2en using Pd(0)... 7
1.2.3. Optimized condition for 4-azidation of Neu5Ac2en ... 7
1.2.4. C4 – Analogues synthesis from 4-chloro Neu5Ac2en... 8
1.2.5. Methylenation of ketone to the disubstituted analogues at C-4... 9
1.2.6. Derivatives synthesis of 4-azido Neu5Ac2en derivatives ... 10 1.2.7. Modification of Neu5Ac2en at C-4 for the inhibition evaluation of hPIV-1
... 10
1.2.8. Analogues derived from other positions... 11
1.2.9. Polymeric sialoside... 11
1.3. Our initial approach ... 12
1.3.1. Earlier methods: Pd(0)-catalyzed C- and O-glycosylation on sugars... 14
1.3.2. Paradox in regioselectivity - Pd(0)-catalyzed substitution in Neu5Ac2en.. 16
1.3.3. General catalytic cycle in Pd(0)-catalyzed allylic substitution (Tsuji-Trost reaction) ... 17
2. Results and discussion... 19
2.1. Substrate synthesis ... 19
2.2. Preliminary test using sodium dimethylmalonate as a nucleophile ... 20
2.3 Characterization of isomers ... 21
2.3.1. Possible isomers ... 21
2.3.2 Characterization of 13 and 16... 22
Characterization of regioisomers (C-2 or C-4)... 22
Configuration characterization of 16 via coupling constant (JH-H) and NOESY .. 23
2.3.3. Reaction procedure ... 27
2.4. Allylic substitution of 12 using sodium dimethylmalonate as a nucleophile... 28
2.4.1. Ligand effect... 28
2.4.2. Effect of the ratio of Pd/Ligand on the substrate 12... 34
2.5. Conformation highlight... 39
2.6. Solvent effect ... 40
2.7. Allylic substitution of 12 with various nucleophiles... 42
2.7.2. Type II: Unymmetric malonate... 44
2.7.3. Type III: Amine derivative as nucleophile... 46
2.8. Fuctionalization of synthesized derivatives ... 54
2.8.1. Dimethylmalonate derivative at C-2... 54
2.8.2. Piperazine derivative at C-4 ... 55
2.8.3. An attempt to synthesize the multivalent derivatives ... 55
2.8.4. Global deprotection of these synthesized derivatives... 57
3. Conclusion... 58
4. Experimental ... 59
5. References ... 79
Chapter 2
... 84An Easy Access to α-Glycosyl Chloride via TCT/DMF... 84
1. Introduction ... 84
1.1. Previous methods in preparations of -glycosyl chlorides ... 84
2. Results and discussion... 86
2.1. Preliminary results and condition optimizations... 86
2.2. Mechanistic aspect ... 88
2.3. Applications in carbohydrate chemistry... 89
2.3.1. Preparation of α-glycosyl chlorides from glycosyl hemiacetals ... 89
2.3.2. Preparation of α-glycosyl chlorides from glycosyl orthoesters... 91
2.3.3. Chemoselective chlorination and formylation... 92
2.3.4. Synthesis of N-acetylneuraminic acid glycal... 92
3. Conclusion... 94
4. Experimental ... 96
5. References ... 110
Chapter 3
... 113DMF functions as a “brake” molecule in highly α-stereoselective glycosylation... 113
1. Introduction of α-stereoselective O-glycosylation... 113
1.1. Earlier strategies for α-selective O-glycosylation ... 115
1.1.1. Lemieux’s in situ anomerization strategy... 115
1.1.2. Solvent influrence in α-selective glycosylations... 116
1.1.3. α-Selectivitive glycosylations enhanced by promoters... 118
1.1.4. α-Selective glycosylation with special anomeric leaving groups ... 119
1.1.5. α-Selective glycosylation by special protecting group... 120
1.1.6. α-Selective glycosylations by neighboring group participation... 122
1.1.7. α-Selective glycosylation by additives... 127
1.1.8. α-Selective O-glycosylation mediated by DMF-type molecules ... 131
2. Our strategy using DMF as an additive in α-selective glycosylation ... 135
3. Results and discussion... 137
3.1. DMF as an additive: Effect in stereoselectivity of glycosylations... 139
3.2. Screening of additives... 141
3.3. Examination of solvent effect ... 142
3.4. Mechanistic investigation ... 143
3.4.1. The correlation of α/β ratio and amount of DMF ... 143
3.4.3. Identification of the 6-O-formyl derivative from glycosylation results .... 150
3.4.4. Summarize the plausible reaction pathways... 153
3.5. Standardizing the reaction procedure... 154
3.6. Extend the scope to other donors and acceptors ... 156
3.6.1. Glycosylation of α-galactosyl chlorides with primary alcohol acceptors . 157 3.6.2. Glycosylation of α-glucosyl chlorides with primary alcohol acceptors.... 161
3.6.3. The secondary alcohol acceptor... 167
4. Conclusion... 171
5. Experimental ... 173
6. References ... 192
Chapter 4
... 197Synthesis of oligosaccharides - ... 197
Glycolipids derivative of Meiothermus taiwanensis ATCC BAA-400... 197
1. Introduction ... 197
2. Retrosynthesis of glycolipid derivatives – convergent (2+2) synthesis... 199
3. Results and discussion... 200
3.1. Synthesis of building blocks ... 200
3.1.1. Preparation of disaccharide 99 ... 200
3.1.2. Preparation of Gal-N3 donor 130... 201
3.1.3. Preparation of glucosyl acceptor 134 ... 201
3.1.4. Preparation of disaccharide E - 136... 202
3.1.5. Convergent [2+2] approach for synthesis of tetrasaccharide 140 ... 204
5. Experimental ... 206
6. References ... 217
APPENDIX ... 219
Spectra list of the synthesized compounds... 219
LIST OF FIGURES
Chapter 1
Figure 1 - Possible pathways for preventing the infection of influenza viruses ... 2
Figure 2 - Intervention of neuraminidase inhibitors and process of anchoring to host cell through HA... 3
Figure 3 - Nomenclatures of different subtypes of influenza virus... 3
Figure 4 - Design of neuraminidase inhibitors mimicking the cleaved intermediate 3... 4
Figure 5 - The binding of inhibitors with oseltamivir-resistant influenza virus neuraminidase 5 Figure 6 - Modifications of Neu5Ac2en at C4, 5, 6, 7, 8, 9... 11
Figure 7 - Pd(0)-catalyzed allylic substitution on Neu5Ac2en ... 13
Figure 8 - Pd(0)-catalyzed O-glycosylation and it’s application in oligosaccharide synthesis15 Figure 9 - Pd(0)-catalyzed regioselective allylic substitution ... 16
Figure 10 - Paradox in regioselectivity - Pd(0)-catalyzed substitution in Neu5Ac2en... 17
Figure 11 - General catalytic cycle in Pd(0)-catalyzed allylic substitution... 18
Figure 12 - Possible routes for the formation of isomers ... 21
Figure 13 - NOE correlation of 16 and 3D diagram... 23
Figure 14 - NOE correlation of 13 and 3D diagram... 25
Figure 15 - TBS protection of 13 and X-ray of 15 ... 25
Figure 16 - Regioselectivity predicted by chemical shift of 13C-NMR... 29
Figure 17 - Tolman's cone angle... 30
Figure 18 - Conversion between η3 and η1 η2 bonding mode... 31
Figure 20 - Plausible conformation due to ligand property... 33
Figure 21 - Trans effect induce regioselective substitution... 37
Figure 22 - J value between peracetylated Neu5Ac2en and 12 ... 39
Figure 23 - Symmetric malonate type ... 43
Figure 24 - Plausible epimerization... 43
Figure 25 - Phosphonate derivatives ... 44
Figure 26 - Acetoacetate derivatives ... 46
Figure 27 - Morpholine derivatives... 47
Figure 28 - Aniline and benzylamine derivative ... 50
Chapter 2
Figure 1 - Common chlorinating reagent for preparation of glycosyl chlorides... 84Chapter 3
Figure 1 - Biological functions of glycoconjugates ... 113Figure 2 - Examples of bioactive α-glycosides... 114
Figure 3 - Ethereal solvents induce α-selective glycosylation... 117
Figure 4 - Proposed mechanism of ClO4- playing a role in α-selective glycosylation... 119
LIST OF TABLES
Chapter 1
Table 1 - Conditions for the regioselective allylic substitution of 12... 29
Table 2 - Regioselectivity in relation to bite angle ... 31
Table 3 - Regioselectivity in relation to bite angle ... 32
Table 4 - Dppb as a ligand and examination of Pd/P ratio in regioselectivity of 12 ... 34
Table 5 - Dppe as a ligand and study of Pd/P ratio in regioselectivity of 12... 35
Table 6 - PPh3 as ligand and study of Pd/P ratio in regioselectivity of 12 ... 35
Table 7 - PBu3 as a ligand and study of Pd/P ratio in regioselectivity of 12 ... 36
Table 8 - Solvent effect ... 40
Chapter 2
Table 1 - Lactosyl hemiacetal 56 as a model substrate for condition optimizations ... 87Table 2 - Preparation of α-glycosyl chlorides via TCT/DMF ... 90
Chapter 3
Table 1 - The effect of promoter for activation of thioglycosides ... 118Table 2 - Crich’s study: The effect of diaryl sulfoxides for α-sialylation using 2-propanol as an acceptor ... 128
Table 3 - Comparative study of DAMA as an additive in α-O-sialylation... 131
Table 4 - DMA as an additive in α-O-glucosylation ... 133
Table 6 - DMF as an additive: Optimization of conditions in α-selective glycosylations using
70b and 82... 140
Table 7 - Screening of the additive in α-selective glycosylations using 70b and 82 ... 142
Table 8 - Examination of solvent effect using 70b and 82 ... 142
Table 9 - The correlation diagram regarding to α/β ratio and amount of DMF ... 144
Table 10 - Glycosylation with the putative glycosyl iminium II... 147
Table 11 - Glycosylation of 70b with the simple primary alcohol acceptors ... 157
Table 12 - Orthogonal glycosylation of 70b with the primary glycosyl acceptors... 158
Table 13 - Glycosylation of 69b with the primary alcohol acceptors... 159
Table 14 - Glycosylation of 103 with the primary alcohol acceptor 82 ... 160
Table 15 - Glycosylation of 105 with the primary glycosyl acceptors ... 161
Table 16 - Glycosylation of 110 with the primary glycosyl acceptor 82... 162
Table 17 - Glycosylation of 110 with the primary alcohol acceptors ... 165
Table 18 - Glycosylation of 117 with the primary alcohol acceptor 82... 166
Table 19 - Glycosylation with the secondary alcohol acceptor 119 ... 167
LIST OF SCHEMES
Chapter 1
Scheme 1 - The first chemical synthesis for the azido derivatives of Neu5Ac2en... 6
Scheme 2 - The first synthesis of 4-epi-4-azido-Neu5Ac2en using Pd(0) ... 7
Scheme 3 - Optimized condition for azidation of Neu5Ac2en... 8
Scheme 4 - Analogues synthesis from 4-chloro Neu5Ac2en ... 8
Scheme 5 - Methylenation of ketone to the disubstituted analogues at C-4... 9
Scheme 6 - The derivatives synthesis using Click chemistry ... 10
Scheme 7 - C4 – Modification of Neu5Ac2en at C4 ... 10
Scheme 8 - Pd(0)-catalyzed C-glycosylation at anomeric carbon ... 14
Scheme 9 - Pd(0)-catalyzed O-glycosylation ... 14
Scheme 10 - Synthetic route of substrates ... 19
Scheme 11 - Allylic substitution of Neu5Ac2en derivative 11... 20
Scheme 12 - Allylic substitution of Neu5Ac2en derivative 12 ... 21
Scheme 13 - Characterization of regioisomers ... 23
Scheme 14 - Formation of bicyclic product... 26
Scheme 15 - Preparation of the palladium complexes and their reaction... 38
Scheme 16 - Standard reaction ... 42
Scheme 17 - The formation of byproduct via 3,3-sigmatropic rearrangement... 45
Scheme 18 - Allylic azidation of 12... 46
Scheme 20 - Diphenyl methanamine as a nucleophile ... 50
Scheme 22 - Phenol as a nucleophile... 52
Scheme 23 - Zinc methoxide as a nucleophile ... 52
Scheme 24 - Plausible mechanism via zinc-alkoxide mediation ... 53
Scheme 25 - Functionalization of C-2 dimethylmalonate derivative ... 54
Scheme 26 - Functionalization of piperazine derivative at C-4... 55
Scheme 27 - Retrosynthesis of multivalent NA inhibitors ... 56
Scheme 28 - TBS deprotection of 34... 57
Scheme 29 - Functionalization of piperazine derivative 36 ... 57
Scheme 30 - Global deprotection of the morpholine derivative 35 ... 57
Chapter 2
Scheme 1 - Conversion from thioglycoside to glycosyl chloride via chlorosulfonium chloride ... 85Scheme 2 - Preparation of glycosyl chloride via TCT/DMF... 86
Scheme 3 - Plausible mechanism for TCT/DMF chlorination at C-1 ... 88
Scheme 4 - Preparation of α-glycosyl chlorides from glycosyl orthoesters... 91
Scheme 5 - Chemoselective chlorination and formylation of 77a and 78a... 92
Scheme 6 - One-pot synthesis of NANA glycal 81 ... 93
Scheme 7 - Sequential chlorination–glycosylation... 94
Chapter 3
Scheme 1 - α-selective glycosylation via in situ anomerization ... 115Scheme 2 - Gervay's synthesis of KRN7000 ... 116
Scheme 4 - Torsional effects in donor activation and examples... 121 Scheme 5 - DTBS directed α-selective galactosylation\ ... 121 Scheme 6 - An isotopic labeling probe to investigate the effect of ester participating groups
... 123 Scheme 7 - Neighboring group participation by a S auxiliary at C-2 leading to
1,2-cis-glycosides... 123 Scheme 8 - Oxathiane glycosyl donors direct α-selective glycosylation ... 124 Scheme 9 - Gin’s proposed mechanism of α-sialylation via the amide auxiliary ... 125 Scheme 10 - Nguyen’s proposed mechanism of α-selective glycosylation via nickel-mediation
... 125 Scheme 11 - Demchenko’s α-selective glycosylation via the metal-coordinated glycosyl donor
... 126 Scheme 12 - Bogusiak’s α-selective glycosylation enhanced by the polar additives... 127 Scheme 13 - Crich’s proposed mechanism: Sulfoxide derivatives involving in α-selective
sialylation ... 129 Scheme 14 - Boon’s method: PhSEt as an additives involved in α-selective glycosylation. 130 Scheme 15 - The influence of additives for supramer structures via IR spectroscopy... 130 Scheme 16 - Xylosylation of terpenols in DMF ... 132 Scheme 17 - Nishida’s dehydrative glycosylation method using Appel agents in DMF... 132 Scheme 18 - Koto’s plausible mechanism using DMA as an additive in α-O-glucosylation 134 Scheme 19 - DMF-mediated α-selective glycosylation via glycosyl iminium intertemediates
... 136 Scheme 20 - The formation of per-O-acetylated glucopyranosyl uronium triflate... 141 Scheme 21 - Control experiment without addition of DMF ... 145
Scheme 22 - Trap of iminium using DMF as a “brake” molecule... 146 Scheme 23 - 1H-NMR study to investigate the existence of glycosyl iminium salts ... 149 Scheme 24 - Dehydrative α-mannosylation mediated by DMF... 149 Scheme 25 - Identification of 6-O-formyl derivative 89 ... 151 Scheme 26 - Plausible mechanism for the formation of 6-O-formyl derivative 89 ... 151 Scheme 27 - Plausible mechanism for α-selective glycosylation through IAD... 152 Scheme 28 - IAD approaches in the literature ... 152 Scheme 29 - The plausible reaction pathways... 153 Scheme 30 - Lin’s proposed mechanism for α-selective galactosylation via 4- and 6-acyl
remote participation... 160 Scheme 31 - Proposed mechanism for α-selective glucosylation effected by a bicyclic
protecting group ... 164 Scheme 32 - Orthogonal glycosylation of 110 with the primary glycosyl acceptor 114 ... 165 Scheme 33 - Previous synthesis of Gb3 derivative in our lab... 169
Chapter 4
Scheme 1 - The first synthesis of the precursor of glycolipid isolated from Meiothermus taiwanensis ATCC BAA-400 ... 198 Scheme 2 - Retrosynthesis of tetrasaccharide derivatives ... 199 Scheme 3 - Preparation of disaccharide 99... 200 Scheme 4 - Synthetic route for preparation of Gal-N3 donor 130 ... 201
Scheme 5 - Preparation of glucosyl acceptor 134... 202 Scheme 6 - Preparation of disaccharide 136... 202
Scheme 7 - Preparation of disaccharide 139... 203 Scheme 8 - Convergent [2+2] synthesis of tetrasaccharide 140 ... 204
LIST OF ABBREVIATIONS
Ac acetyl
Ac2O acetic anhydride
AgOTf silve trifluoromethanesulfonate
All allyl Boc tert-butyloxycarbonyl Bn benzyl bs broad singlet Bz benzoyl Bu butyl CH3CN acetonitrile c concentration cat. catalytic
CIP contact ion-pair
Cq quarternary carbon atom
CSA camphor sulfonic acid
δ chemical shift d doublet dba dibenzylideneacetone DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DCE dichloroethane DCM dichloromethane
DCMME α-dichloromethyl methyl ether
dd doublet of doublets
DMA N,N-dimethyl acetamide
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
DAMA N-acetyl acetamide
DMSO dimethylsulfoxide DN donacity dppb bis(diphenylphosphino)butane dppe bis(diphenylphosphino)ethane DPS diphenylsulfoxide DTBMP 2,6-di-tert-butyl-4-methylpyridine equiv. equivalent
EDCI 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
ESI electrospray ionization
Et ethyl EtCN propionitrile
EtOAc ethyl acetate
Et3N triethylamine Fmoc 9H-fluoren-9-ylmethoxycarbonyl h hour Hex hexane HMPA hexamethylphosphoramide HMPT hexamethylphosphoric triamide
HOBT hydroxybenzotriazole HRMS high resolution mass spectrometry
Hz Hertz
IAD intramolecular aglycon delivery
IDCP iodonium di-collidine perchlorate
IR infrared spectroscopy J coupling constant KHMDS potassium hexamethyldisilazide LG leaving Group m multiplet M molar min minute mp melting point Me methyl MS molecular sieves n normal
NANA N-acetylneuraminic acid
NBS N-bromosuccinimide
Neu5Ac2en 5-acetamido-2,6-anhydro-3,5-dideoxy-D-glycero-D-galacto-non-2 -enoic acid
NIS N-iodosuccinimide
NMM N-methylmorpholine
NMR nuclear magnetic resonance
PG protecting group Ph phenyl
PMB p-methoxybenzyl
ppm parts per million
PTSA p-toluenesulfonic acid
Py pyridine
Rf retardation factor
RT room temperature
s singlet
SSIP solvent-separated ion pair
t tertiary t triplet
TBAB tetra-n-butylammonium bromide
TBAF tetra-n-butylammonium fluoride
TBS tert-butyldimethylsilyl
TBSOTf tert-butyldimethylsilyl methanesulfonate
TCT 2,4,6-trichloro-1,3,5-triazine
tert tertiary
TFA trifluoroacetic acid
Tf2O trifluoromethanesulfonic anhydride
TfOH trifluoromethanesulfonic acid
THF tetrahydrofuran
TLC thin layer chromatography
TMSOTf trimethylsilyl methanesulfonate
TMU 1,1,3,3 tetramethylurea
t.o.f turn over frequency
Chapter 1
Regio- and stereoselective Pd(0)-catalyzed allylic substitution of
sialic acid derivatives
1. Introduction
1.1. General information of influenza virus and the related therapies
Very recently, the new influenza A (H1N1) reassorted from the swine viruses first emerged in North America in February 2009. The virus has readily spread in other regions and become the worldwide pandemic. Generally, its higher mortality than seasonal influenza may be explained because this new strain virus is capable of affecting the lower respiratory tract and cause potential severe syndromes in some serious cases.1 In the human history, three influenza pandemics occurred in the 20th century and killed tens of millions of people, with each of these pandemics being caused by the appearance of a new strain of the virus in humans. Often, these new strains result from the spread of an existing flu virus to humans from other animal species. Moreover, a deadly avian strain of H5N1 has posed the greatest risk for a new influenza pandemic since it first killed humans in Asia in the 1990s.2
Although vaccination is the primary strategy for the prevention of influenza, an antigenic drift in the virus may occur after the seasonal formulation of vaccine has been used, rendering the vaccine less protective. Furthermore, vaccine production by current methods cannot be produced with the speed required to halt the progress of a new strain of influenza virus; therefore, it is likely that vaccine would not be available for the first wave of spread of virus.
agents were developed against spreading of viruses in different stages (Figure 1). These antiviral drugs are able to affect the virus itself and may be used for either prevention or treatment.
Figure 1 - Possible pathways for preventing the infection of influenza viruses
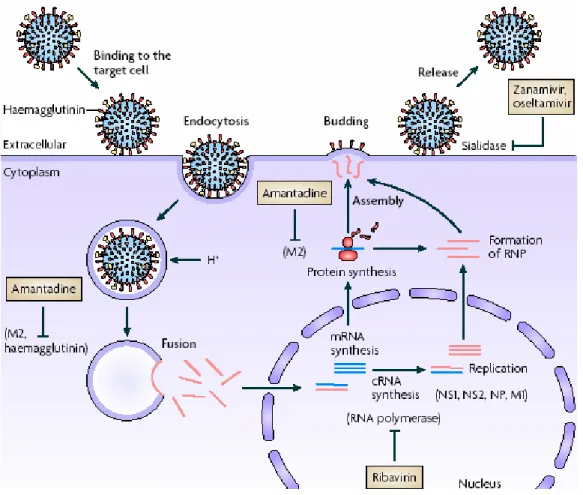
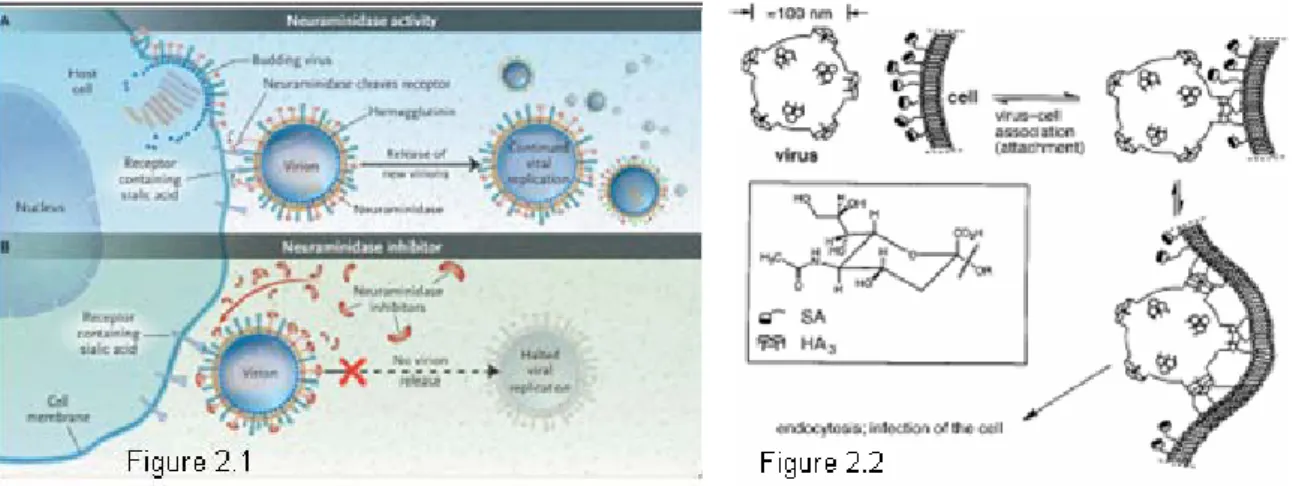
Neuraminidase (NA) is a glycohydrolase enzyme (EC 3.2.1.18) which catalyzes the cleavage of terminal sialic acid α-ketosidically linked to antigenic glycoproteins on the surface of the influenza virus. This action can prevent aggregation of viruses and promote the release of progeny viruses from infected cells. Administration of chemical inhibitors of neuraminidase is a treatment that limits the severity and spread of viral infections(Figiure 2.1).3
Hemagglutinin (HA) is a homotrimeric glycoprotein found on the surface of the influenza viruses which possesses the three binding sites. It is responsible for binding the virus to the
terminal sialylated glycoprotein on the membrane surface of the host cell followed by embedding into the cell (Figure 2.2).4
Figure 2 - Intervention of neuraminidase inhibitors and process of anchoring to host cell through HA
The subtypes of influenzas are characterized and named according to the type of nuclear materials, the origin of area, and different groups of hemagglutinin (HA) and neuraminidase (NA), such as the stereotype of influenza A viruses, found in Fujian, China in 2002, hence named as A/Fujian/411/2002/H3N2 (Figure 3).
Figure 3 – Nomenclatures of different subtypes of influenza virus
particularly effective and convenient. As a mater of fact, 2,3-unsaturated N-acetylneuraminic acid 2 (5-acetamido-2,6-anhydro-3,5-dideoxy-D-glycero-D-galacto-non-2-enoic acid, Neu5Ac
-2en) was first discovered by Meindl and Tuppy in 1969 as a potent neuraminidase inhibitor (Figure 4).5-7 According to the enzyme-based rational design mimicking the cleaved intermediate 3, inhibitors towards neuraminidase of viruses were discovered and developed as the two anti-influenza drugs currently used to treat infected patients, Zanamivir 1 (Relenza®) and Oseltamivir phosphate 4 (Tamiflu®).8-10
1- Zanamivir (Relenza) O HN AcHN NH H2N O OH OH OH HO O COOH HO O glycoprotein Neuraminidase (Sialydase) R=NHAc Sialymimetic structures 4- Oseltamivir phosphate (Tamiflu) O OH OH HN O H2N NH HN Peramivir 1 2 3 4 5 6 7 8 9 O COOH HO AcHN OH OH HO OH OH HO AcHN O COOH HO AcHN OH OH HO + 3- Neu5Ac2en COOEt NH2
.
H3PO4 AcHN O 3- cleaved intermediateFigure 4 - Design of neuraminidase inhibitors mimicking the cleaved intermediate 3
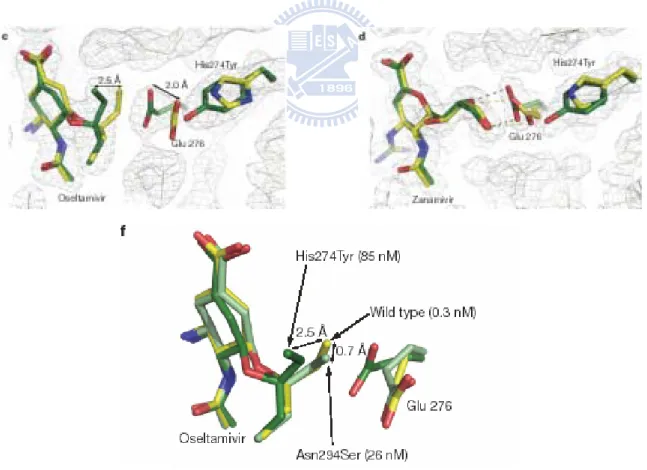
Even though the anti-influenza drugs are available for the treatment, the emergence of drug resistance to oseltamivir11,12 still occurred recently and caused the humans health risk.
Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants have been reported by Collins et al. in UK.13 They demonstrated that the hydrophobic pentyloxy group of oseltamivir is an influential group. To bind with pentyloxy group of oseltamivir, the carboxyl group of Glu 276 needs a conformational change (Figure 5-c). In contrast, the polar glycerol group of Zanamivir has the tight affinity with Glu276 through hydrogen bonding (Figure 5-d). However, the mutated NA bearing His294Try residue affects the binding between the side chain of oseltamivir and Glu276 (Figure 5-f). That is a key factor which resulted in the drug resistance of oseltamivir. Therefore, the development of next-generation chemotherapeutic agents derived from Zanamivir would be advantageous instead of oseltamivir analogues.
1.2. Chemical synthesis of neuraminidase inhibitors from Neu5Ac2en
In 1993, von Itzstein et al. developed the anti-influenza drug, Zanamivir 1 (Relenza®) based on the structure-basis rationale involving the probable sialosyl cation transition-state intermediate.14 Afterwards, a large number of sialymimetic analogues were synthesized and evaluated for probing the biological binding activity and indeed encouraged the current drug design.15,16 In this dissertation, we will focus on the synthetic strategies derived from Neu5Ac2en.
1.2.1. The first synthesis of 4-azido derivatives using Mitsunobu Reaction
The first chemical synthesis for the azido derivatives of Neu5Ac2en has been reported by Schauer et al. in 1991.17 The azido product suffered from 3,3 sigmatropic rearrangement and lost its chirality at C-4 (Scheme 1).
O OH HN H AcO OAc OAc O O
OMe PPh3, DEAD, NaN3
Mitsunobu Reaction O N3 HN H AcO OAc OAc O O OMe O N3 HN H AcO OAc OAc O O OMe Toluene (67%) = 1 : 3 THF (34%) = 3 : 2 3,3 rearrangement O N HN H AcO OAc OAc O O OMe N+N -O HN H AcO OAc OAc O O OMe N -N+ N O+ HN H AcO OAc OAc O O OMe -N3 O N HN H AcO OAc OAc O O OMe N+ N
1.2.2. The first synthesis for 4-epi-4-azido-Neu5Ac2en using Pd(0)
The pioneering attempt to use the organometallic catalyst in synthesizing the analogue of Neu5Ac2en was conducted by von Itzstein et al. in 1997. However, only epi-Neu5Ac2en allowed to be accessed by this method.18 They demonstrated that a pseudo-axially disposed acetate better facilitate the initial oxidation addition of the allyl acetate to Pd(0) (Scheme 2).
O OAc HN H AcO OAc OAc O O OMe Pd(PPh 3)4, NaN3 THF/H2O, 50oC, 84% O N3 HN H AcO OAc OAc O O OMe O OAc HN H AcO OAc OAc O O OMe Pd(PPh3)4, NaN3 THF/H2O, 50oC O N3 HN H AcO OAc OAc O O OMe X O COOMe OAc Pd(L)4 L=PPh3 O COOMe Pd L L Nu OAc O COOMe Nu Plausible mechanism
Scheme 2 - The first synthesis of 4-epi-4-azido-Neu5Ac2en using Pd(0)
1.2.3. Optimized condition for 4-azidation of Neu5Ac2en and activity evaluation of the 4-guanidino Neu5Ac2en derivatives
inhibition. None of them showed a better inhibitory activity than Zanamivir. It is worth of note that the optimized procedure for synthesizing the 4-azido Neu5Ac2en has been scaled up to a large quantity (600 g) (Scheme 3).19
O O N H AcO OAc OAc O OMe
TMSN3(1.5 eq.), tert-butyl alcohol
76% O N3 HN H AcO OAc OAc O O OMe O HN HN H AcO OAc OAc O O OMe NH NH2 O HN HN H AcO OAc OAc O O OMe NH NHR
R= nitro, ethoxycarbonyl, methyl, amino, hydroxy Optimized Conditon
reflux, 10 h,
Scale: Starting material (600 g)
Scheme 3 - Optimized condition for azidation of Neu5Ac2en
1.2.4. C4 – Analogues synthesis from 4-chloro Neu5Ac2en at C-4 by Stille coupling
O Cl HN H AcO OAc OAc O O OMe Bis(dibenzylideneacetone) -palladium(0), PPh3,R-SnBu3 THF, 50oC,33-49% O R HN H AcO OAc OAc O O OMe O Cl HN H AcO OAc OAc O O OMeBis(dibenzylideneacetone) -palladium(0), PPh3,Vinyl-SnBu3 THF, 50oC,18% O HN H AcO OAc OAc O O OMe O O N H AcO OAc OAc O OMe + H3 H4 major minor R= COOMe SiMe3 Ph
Kok and von Itzstein developed an organometallic-mediated approach (Stille coupling) for the stereoselective C-C bond formation at C-4. However, they emphasized that the instability of the chlorosubstrate would be a problematic issue. Moreover, the excessive use of toxic stannyl reagents limits the scope of this method (Scheme 4).20, 21
1.2.5. Methylenation of ketone to the disubstituted analogues at C-4
The serial studies for synthesizing C-4 disubstituted derivatives of Neu5Ac2en from the olefinated moiety have been also reported by von Itzstein’s group (Scheme 5).22, 23
O OH HN H O O OH O O OMe X= OAc, OMe, N3, Cl, CN MeO O O HN H O O OH O O OMe MeO O HN H O O OH O O OMe MeO O HN H O O OH O O OMe MeO O O N H O O OH O O OMe X O AcHN H O O OH OH O OMe X PCC Cp2ZrCl2 Zn, CH2I2 O AcHN H O O OH OH O OMe X MeO Basic Acidic MCPBA
Highly prone to enolisation and not amenable to Wittig or Peterson Reaction
+ X
1.2.6. Derivatives synthesis of 4-azido Neu5Ac2en derivatives– using Click chemistry Several Zanamivir analogues with different substituted triazoles derived from 4-azido NeuAc2en using click chemistry has been described by Zuo et al. and also evaluated in anti-avian influenza virus (H5N1) activities, which showed the comparable inhibition with Zanamivir 1 (Scheme 6).24 O N3 HN H HO OH OH O O OH O N AcHN H HO OH OH O OH N N R Scheme 6 - The derivatives synthesis using Click chemistry
1.2.7. Modification of Neu5Ac2en at C-4 for the inhibition evaluation of hPIV-1
Human parainfluenza virus type 1 (hPIV-1) is an important pathogen causing upper and lower respiratory disease in infants and young children. Referred to the reported information of enzyme interaction, a serial of C-4 modified inhibitors of Neu5Ac2en showed the superior inhibitory activity than Zanamivir (Scheme 7). This finding implied that these analogues showed the different binding affinity with neuraminidases from the different strain of virus.25
1) 80% AcOH 2) 0.1 M KOH-MeOH R = Me, Et, n-Pr, CH2CH CH2, CH2C CH 59 - 99% Ag2O, TBAI or NaH 0 - 69% DMF, O OH HN H O O OH O O OH O OR HN H O O OH O O OH O OR HN H HO OH OH O O OH
1.2.8. Analogues derived from other positions
Modifications of Neu5Ac2en at C-4, 5, 6, 7, 8, 9 - The truncated 6-glycerol substituted series related to Zanamivir also contributed the significant binding information with neuraminidase.13 Variations at other positions, such as C-5, C-7, C-8, C-9 were also investigated for the inhibitory activity against neuraminidase (Figure 6).
O NH2 HN H N R1 O O OH R2 R3 O HN HN H N R1 O O OH R2 R3 R4 O HN RHN H HO OH OH O OH NH NH2 O HN AcHN H Z Y X O OH NH NH2 O HN AcHN H O O OH NH NH2 HO HO
Modification at C4, C5 with the truncated glycerol chain Modification at C5
Modification at C7, C8, C9
Figure 6 - Modifications of Neu5Ac2en at C4, 5, 6, 7, 8, 9
1.2.9. Polymeric sialosides
Multivalent compounds derived from C-4 - Polymeric 4-N-linked sialoside - Polymeric sialosides have been synthesized and acted as multivalent inhibitors of hemagglutinin.26
Multivalent compounds derived from C-7 - Polymeric 7-O-linked sialoside - All polymeric sialosides displayed less potent activity for the inhibition of influenza A sialidase. However, a much greater in vivo efficacy against influenza A in the animal mice model by intranasal
administration than Zanamivir was reported.27, 28
Multivalent compounds derived from C-7 - Polymeric , tri- or tetrameric 7-O-linked sialoside - The X-ray crystal structure of Zanamivir bound in the influenza NA active sites shows that 7-OH group is not involved in any significant interactions. Some polymeric sialosides designed by Krippner et al. and derived from C-7 showed the prolonged efficacy due to the slow lung clearance of high molecular weight (500 kD) compounds.29, 30
After a survey of the recent reports regarding the synthesis of neuraminidase inhibitors, many versatile molecules were built up to address either in-vivo or in-vitro potency against the influenza viruses. However, the new synthetic routes to prepare the novel constructs and more economic direct-access approach derived from each position of sialic acid are still challenging and of the great interest for the organic chemists.
1.3. Our initial approach
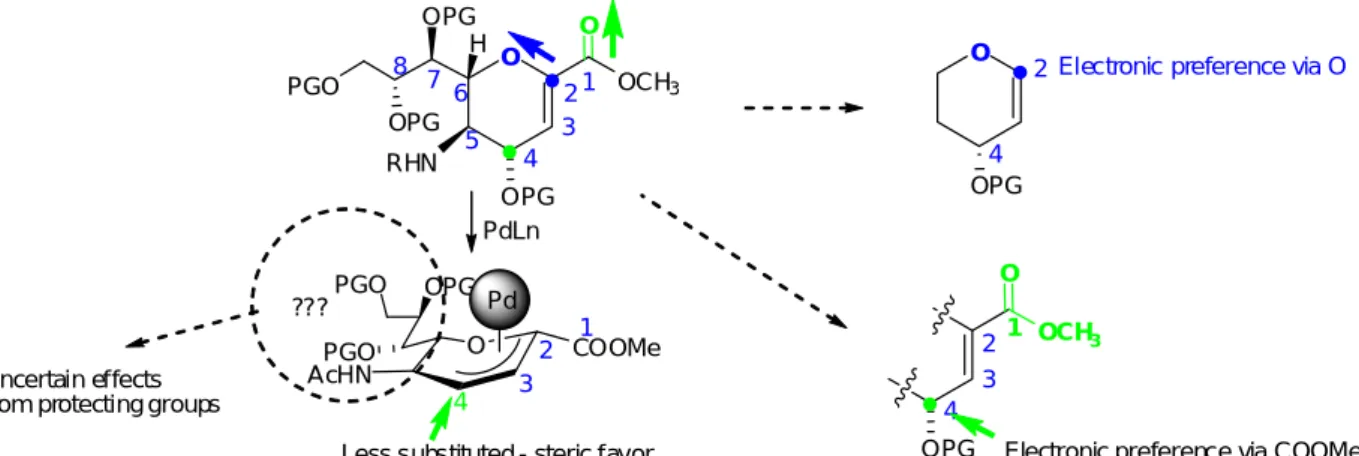
Pd(0)-catalyzed allylic substitution is a powerful tool for the different types of bond formation, such as C-C, C-N, C-S, C-O, C-P, and widely applied in asymmetric synthesis in the past decades.31-33 Although certain synthetic methodologies using organo- metallic catalysts, such as the Stille coupling mentioned above, have been reported on Neu5Ac2en, we envisaged that the starting substrate with the different protecting or functional groups probably give us certain advantages in terms of reactivity and selectivity. Our initial idea was to synthesize the protected substrate and test its feasibility in Pd(0)-catalyzed substitution, also named “Tsuji-Trost” reaction. The successive formation of Pd π-allyl complex would be a key intermediate for the later nucleophilic substitution (Figure 7).
Pd(0) PG = Protecting Group O COOMe OPG PGO GPO AcHN PdL2 O COOMe OPG PGO PGO AcHN Nu O COOMe Nu OPG PGO PGO AcHN Nu
-- OPG attack at C-2 attack at C-4Pd(0)-catalyzed allylic substitution
2 4 O COOMe OPG OPG PGO PGO AcHN C-4 adduct C-2 adduct
Regio-select iv it y ? Ster eo-selectivity ?
Figure 7 - Pd(0)-catalyzed allylic substitution on Neu5Ac2en
Before proving this concept, the outcome in reactivity, regioselectivity and stereoselectivity is almost unpredictable due to the functional complexity for this type of sugar substrate. However, similar strategies using palladium catalyst on the pyranose systems have been developed for C- and O-glycosylation on the relatively simple structures. Those results will be summarized in the later section and expected as an useful information to guide us for the following investigation.
1.3.1. Earlier methods: Pd(0)-catalyzed C- and O-glycosylation on sugars
Pd(0)-catalyzed C-glycosylation
RajanBabu et al. reported a general method for Pd(0)-catalyzed C-glycosylation on glycols.23 Several C-glycosides derived from C-1 were easily accessed in excellent regio- and stereoselectivity due to the anomeric stabilization in the pyranose ring (regioselectivity) and the preferential substitution from the opposite face of π-allyl Pd complex (stereoselectivity) (Scheme 8).
O
X O
O
X = OCOCH3, OCOCF3, OP(O)(OCH2CH3)2 +K-CH(CO 2CH3)2 Pd(dba)2, DIPHOS THF, rt O O O CO2CH3 CO2CH3
Scheme 8 - Pd(0)-catalyzed C-glycosylation at anomeric carbon
Pd(0)-catalyzed O-glycosylation
Lee et al. developed a new method for O-glycosylation of glycal with the aid of zinc(II) alkoxide under Pd(0)-mediation.34 The variation of α/β stereoselectivity is dependant of the different ligand used (Scheme 9). The mechanistic explanations for the excellent selectivity have not been mentioned in this context.
O X BnO BnO X= OAc, OBoc 0.5 eq. Et2Zn 10% Pd(OAc)2 Ligand, THF, rt O O O OR
Pd(0)-catalyzed O-glycosylation and it’s application in oligosaccharide synthesis
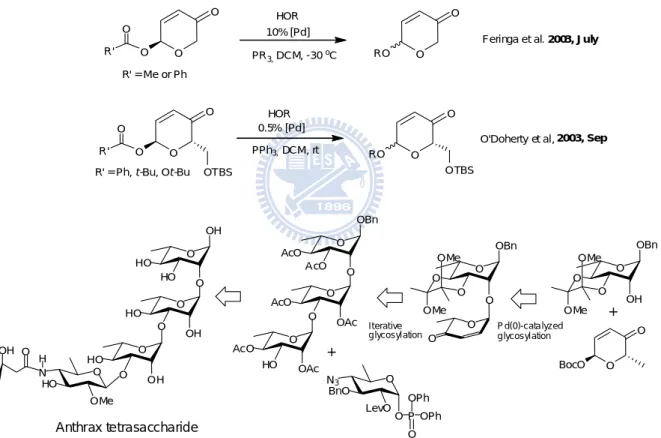
Feringa et al. also described a Pd(0)-catalyzed approach for O-glycosylation in enone-pyranose system. The property of enone enables the following O-glycosylations.35
Interestingly, at almost the same time, O’Doherty et al. published similar results using a lower Pd catalyst loading and further applied this method for the total synthesis of anthrax tetrasaccharide (Figure 8).36 O O O R' O R' = Me or Ph O O O R' O
R' = Ph, t-Bu, Ot-Bu OTBS
10% [Pd] PR3,DCM, -30oC 0.5% [Pd] PPh3,DCM, rt HOR O O RO O O RO OTBS HOR
Feringa et al. 2003, July
O'Doherty et al, 2003, Sep
O N HO OMe O O HO OH O HO HO O OH O HO O OH H O OH Anthrax tetrasaccharide HO O AcO OAc O AcO AcO O OBn O AcO O OAc O N3 BnO LevO O P OPh O OPh O O O OBn OMe OMe O O BocO + OH O O O OBn OMe OMe O O O Iterative g lycosyl ation P d(0)-cata lyzed g lycosyl ation +
Figure 8 - Pd(0)-catalyzed O-glycosylation and it’s application in oligosaccharide synthesis
Pd(0)-catalyzed regioselective allylic substitution induced by a carbonyl group
In the enone system, the excellent regioselectivity at the γ carbon with C-, N-, and O-nucleophile using Pd(0)-catalyzed allylic substitution approach is attributed to the electron-withdrawing property of carbonyl group. The other stereoisomer run through
π−α−π rearrangement can be converted to the same reactive species in line with the geometry of intermediate complex, thus leading to the same product(Figure 9).37
R1 O OR* OP R1 O OR* OP R1 O OR* Nu PdLn, Nu (via π−α−π rearrangement) PdLn, Nu (with retention)
Figure 9 - Pd(0)-catalyzed regioselective allylic substitution
1.3.2. Paradox in regioselectivity - Pd(0)-catalyzed substitution in Neu5Ac2en
The unsaturated Neu5Ac2en bearing carboxylate moiety at C-1, an acetamido group at C-5, and a glycerol chain at C-6 provides a unique electronic environment, usually make synthetic endeavors less productive, such as the low yield in O-sialylation.38 In general, the electron-withdrawing property of carbonyl at carboxylate group reduces electron density at C-4 and further induce the regioselectivity at C-4 position. In contrast, more electronegative oxygen in pyranose ring could stabilize the anomeric cation to facilitate the attack of the incoming nucleophile (Figure 10).39 Apparently, two types of forces from oxygen and carboxylate seem opposite to each other and possibly cause regio-discrimination and even diminish the reactivity as well. Besides, unexpected factors may be arising from other functionalities, such as the acetamide at C-5 and substitutents at the glycerol chain, could make the entire process more complicated.
O OPG RHN H PGO OPG OPG O OCH3 1 2 3 4 5 6 7 8 O OPG O OCH3 1 2 3 4 OPG 2 4
Electronic preference via O
Electronic preference via COOMe
O COOMe OPG PGO PGO AcHN 1 2 3 4
Less substituted - steric favor Pd
???
PdLn
Uncertain effects from protecting groups
Figure 10 - Paradox in regioselectivity - Pd(0)-catalyzed substitution in Neu5Ac2en
1.3.3. General catalytic cycle in Pd(0)-catalyzed allylic substitution (Tsuji-Trost reaction)
Palladium-catalyzed allylic substitution is a versatile process. η3-allylpalladium complexes were first isolated and identified over 30 years. In 1965, Tsuji first disclosed the bond forming involved π-allylpalladium complex with certain nucleophiles.40,41 Afterwards,
numerous mechanistic studies and applications regarding to this approach were profoundly investigated in regioselectivity, enantioselectivity and a wide range of substrates and their nucleophilic partners. From Pd(0) catalylic cycle (Figure 11), substrates bearing the different leaving groups in combination with the diverse Pd(0) sources, ligand (mono/bidentate/chiral/ hetero), types of nucleophiles (measured by pKa or steric dimension) and counter ions under different concentrations, solvent and temperature usually influence the distinct scenario in regioselectivity and enantioselectivity.42-45 Among these parameters, the derivatives of
highly-functionalized Neu5Ac2en possess an unique electronic and steric environment probably inducing more difficulties in organometallic-mediated reaction. For examples, interconversion of intermediate complex, uncertain interactions between substrates, metals
and ligands may cause problems during the reaction course. Pd L L R X R' ionization R X R' Pd L L R R' Pd L L R R' Pd -X -R' R Nu Pd L L * R R' Nu Pd L L * R' R Nu * R R' Nu * + or nucleophilic addition complextion decomplextion interconversion PdL2 L L
Oxidative add ition ( right side)
2. Results and discussion
2.1. Substrate synthesis O COOMe AcO OAc AcO AcO AcHN OAc O COOH HO OH OH HO AcHN OH O COOMe HO OH OH HO AcHN OH O COOMe OAc OAc AcO AcO AcHN a b c d e f g 5 6 7 8 9 10 11 12 O COOMe OH OH HO HO AcHN O COOMe OH O O HO AcHN O COOMe OBz O O HO AcHN O COOMe OBz O O TBSO AcHNReagent and conditions : a) Dowex 50 H+, MeOH, rt, 24 h, 99%; b) Ac
2O, 0oC to rt, 24 h, 99%; c) TMSOTf (2.5 equiv.)
AcOEt, 0oC to rt, 4 h, 89%; d) NaOMe, MeOH, rt, 30 min; e) CSA, 2,2-dimethoxypropane, acetone, reflux, 1-2 h;
f) BzCl, pyridine, DCM, 0oC, 2 h , 72% for the 3 steps; g) TBSOTf, DCM, lutidine, rt, 18 h, 88%. Scheme 10 - Synthetic route of substrates
For the purpose of this study, the substrate synthesis started from sialic acid 5 to proceed in esterification with methanol followed by acetylation in the presence of pyridine and acetic anhydride.19 The resulting per-acetylated methyl ester 7 was treated carefully with trimethylsilyl triflate (TMSOTf) in ethyl acetate to induce β-elimination to obtain the unsaturated derivative 8 in an excellent 89% yield. Zemplén de-O-acetylation followed by 8,9-isopropylidenation furnished a diol 10,46 which was subsequently selectively protected with benzoyl group to furnish the benzoate 11 in 72% yield over 3 steps.47 In order to prevent
the influence of the 7-OH group, 11 was further silylated with tert-butyldimethylsilyl triflate (TBSOTf) to give the fully-protected derivative 12.48
2.2. Preliminary test using sodium dimethylmalonate as a nucleophile
In a first trial, the treatment with per-acetylated Neu5Ac2en 8 under Pd(0)-catalyzed allylic substitution conditions using freshly-prepared sodium dimethylmalonate as a nucleophile failed to give any desired product, and only starting materials were recovered quantitatively. This result agrees with the previous works reported by von Itzstein,18 who undertook azidation under Pd(0)-mediation in use of Neu5Ac2en 7 (Section 1.2.2). Thus, we turned our attention to other substrates. In contrast, allylic benzoate 11 readily reacted with sodium dimethylmalonate in the presence of Pd(OAc)2 (20 mol%) in combination with PPh3
(40 mol%). The reaction, carried out at 50 oC for 18 h, provided regioselectively alkylated product 13 at C-2 on the α face of the sugar (C-2/C-4 ratio of 81:19, 66% yield; Scheme 11). Different palladium sources {[Pd2(dba)3]•CHCl3, [(allylPdCl)2], etc.} associated with different
ligands (PPh3, PBu3, dppb, etc.) did not significantly improve this transformation.
11 O COOMe OBz O O HO AcHN O COOMe O O HO AcHN COOMe COOMe 2 13 (C-2 adduct) CO2Me CO2Me Na MeOOC COOMe 4 14 (C-4 adduct) O COOMe O O HO AcHN + Pd(OAc)2(20 mol%) PPh3(40 mol%) THF, 50oC, 18 h 66%, 13:14 = 81:19a a The ratio of C-2/C-4 was determined by 1H-NMR.
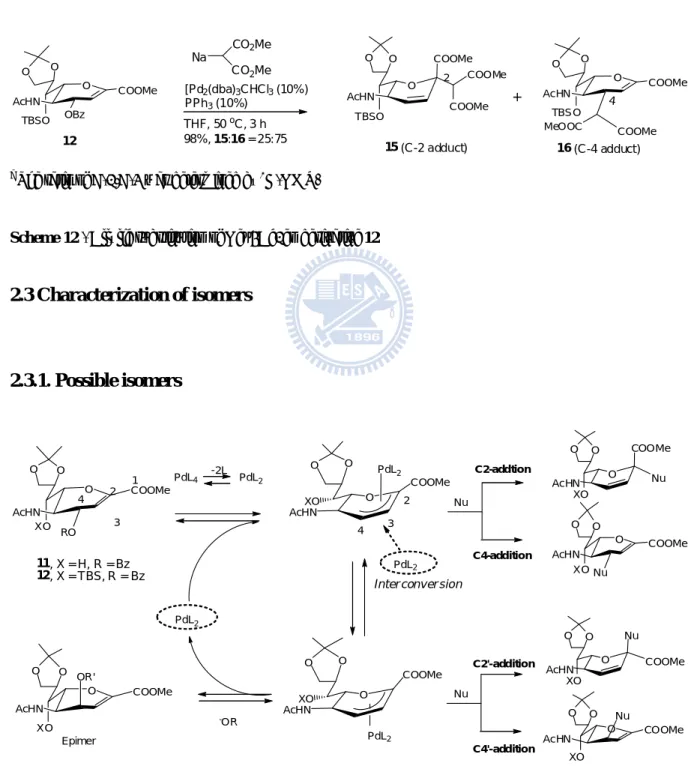
To our surprise, silylated allylic benzoate 12 provided the major alkylated product 16 at C-4 in an excellent 98% yield (C-2/C-4 ratio of 25:75, Scheme 12) under similar conditions. Before investigating these inverse regioselectivity between C-2 and C-4, exact configurations and conformations of these isomers 13, 14, 15 and 16 need to be characterized.
12 O COOMe OBz O O TBSO AcHN O COOMe O O TBSO AcHN COOMe COOMe 2 15 (C-2 adduct) CO2Me CO2Me Na MeOOC COOMe 4 16 (C-4 adduct) O COOMe O O TBSO AcHN + [Pd2(dba)3CHCl3(10%) PPh3(10%) THF, 50oC, 3 h 98%, 15:16 = 25:75
a The ratio of C-2/C-4 was determined by 1H-NMR.
Scheme 12 - Allylic substitution of Neu5Ac2en derivative 12 2.3 Characterization of isomers 2.3.1. Possible isomers O COOMe RO O O XO AcHN O Nu O O XO AcHN PdL4 11, X = H, R = Bz 12, X = TBS, R = Bz O COOMe O O XO AcHN PdL2 O COOMe O O XO AcHN PdL2 O COOMe O O XO AcHN Nu O COOMe OR' O O XO AcHN -OR C2-addtion C4-addition O COOMe Nu O O XO AcHN O COOMe O O XO AcHN Nu C2'-addition C4'-addition -2L PdL2
Inter conver sion
1 2 3 4 COOMe 4 3 2 Nu PdL2 PdL2 Epimer Nu
Stereoselectivity of allylic substitution in use of metal-olefin complex is commonly modulated by the conformation of intermediate to form the “stereo-retention” product. In reality, the stability of the complex varies widely in the reversible catalytic cycle and finally leads the reaction to the state in the compromised energy level. As illustrated in Figure 11, the substrate is catalyzed by Pd(0) to form the intermediate complex which was called “complexation and ionization”, followed by either undergoing interconversion to the “stereo-inverse” complex or directly coupling with the nucleophiles to achieve the “stereo-retention” product in line with the regio-dominance regulated by the electronic and steric factors of active species.
2.3.2 Characterization of 13 and 16
Configuration of two regioisomers 13 and 16 were determined using the relevant 2D-NMR (COSY, HMQC) in combination with DEPT and 1H-NMR to assign carbons and protons.
Characterization of regioisomers (C-2 or C-4)
In terms of regioselectivity, the chemical shifts of the particular protons (13, H-3 = 6.2 ppm, H-4 = 6.0 ppm, H-5 = 4.6 ppm; 16, H-3 = 6.0 ppm, H-4 = 3.5 ppm, H-5 = 3.7 ppm ) provide the significant evidence to explain the alkylated position. It would be attributed to the allylic downshifting effect and further approved by hydrogenation of 13 with two additional methylene group (CH2). Thereupon peracetylation of 17 was made to facilitate the observation of NOE. Unfortunately, the overlapped signals between H-3(axial) and H-3(equatorial) makes
the judgment more difficult in stereoselectivty of the nucleophilic attack (Scheme 13). O COOMe OAc AcO OAc AcHN COOMe COOMe H1' 2 4 O COOMe O O OH AcHN COOMe COOMe 1' 2 4 1 O COOMe OH HO OH AcHN MeOOC COOMe H3 H3'
Reagent and conditions : (a) 5% Pd/C, H2, MeOH, rt, 24h,; (b) Ac2O, Py, rt, 80% (over 2 steps).
NOE
a b
13 17 18
Scheme 13 - Characterization of regioisomers
Configuration characterization of 16 via coupling constant (JH-H) and NOESY
To solve problem, based on the 1H-NMR analysis of 16, the coupling constant calculation of H-4, H-5 andH-6 can provide the crucial information to determine the absolute configuration
in a direct manner. Therefore, the large coupling constants (JH5,H4 = 10.0 Hz, J H6,H5 = 10.0 Hz, J
H4,H5 = 10.1 Hz) which were respectively found in 1H-NMR succinctly revealed the trans
relationship between H-4, H-5 andH-6 (Figure 13).
H3 H6 H4 H5 H H5' O O O H6 O O O HN O O O O AcHN COOMe E E 16 TBS Malonate Pyranose ring H6 H5' H4 H3 H5 Si NOE correlation
The 3D energy-minimized drawing is created by Chem3D software
In principle, two neighboring protons eclipsed with the larger dihedral angle correspond to a larger coupling constant. This relationship has been established by Karplus. Additionally, to reconfirm the assignment of configuration, conducting NOESY experiments to realize the spatial relationships of each proton can be a complementary route.
As illustrated in Figure 12, H-6of compound 16 showed the evident NOE signals with either H-4 or NH, which further indicate the relatively closer relationship and ensure the chirality of C-4. Meanwhile, we also observed the proximity between TBS group and H-5. This evidence supports that malonate group might be away from TBS group due to the steric repulsion.
Characterization of 13 via NOESY
Similarly, compound 13 could be examined with the same procedure and certain unexpected messages from NOESY confirmed our initial assumption. Firstly, we found the long-range and larger coupling constant of JH3-H5 (~2.6 Hz) than JH3-H4 (~1.7 Hz). The
preliminary assignment of H-3 and H-4 in conflict with the later NOESY correlation pattern suggested us to examine their correspondence in the related spectrums. From the singlet peak of H-1’, the corresponding NOE signal with H-3 was observed. The methyl ester of C-1 seems bent inward and contributes NOE signals with either H-3 or H-4. In addition, NH of amido group also correlated to H-6 and H-4. All of evidences pointed out that dimethylmalonate and protected glycerol side chain are prone to situate on the same face with the boat-like pyranose ring (Figure 14).
H3 H4 H1' H5 H6 N H O O O O O OH AcHN MeOOC COOMe 13 OH O Pyranose ring COOMe Acetonide Malonate H3 H4 H6 H1' O O Me
The 3D energy-minimized drawing is created by Chem3D software
Figure 14 - NOE correlation of 13 and 3D diagram
X-ray of 15
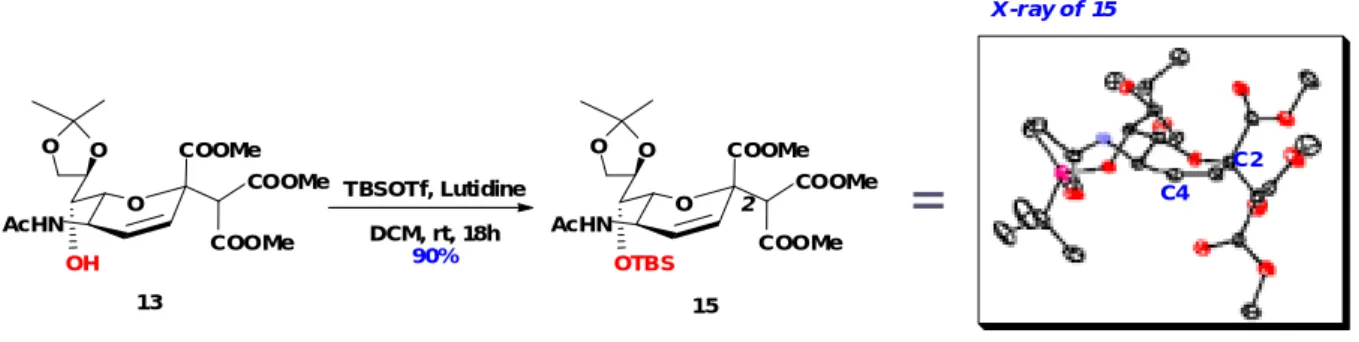
To prove our assignment, 13 was protected with TBS group at 7-OH leading to the same product with 15 (complete matching of 1H-NMR). Furthermore, the configuration of 15 can be reconfirmed by X-ray analysis (Figure 15).
13 TBSOTf, Lutidine DCM, rt, 18h 90% O COOMe O O OTBS AcHN COOMe COOMe 2 15
=
X -r ay of 15 C4 C2 O COOMe O O OH AcHN COOMe COOMeFigure 15 - TBS protection of 13 and X-ray of 15
On the other hand, bicyclic acetal derivative was formed in high yield by treating 12 with the palladium catalyst without an external nucleophile (Scheme 14). This revealed a probable
transitory π-allyl palladium complex close in geometry to the bicyclic product and therefore to conformer B (Scheme 14). This explains logically the selective formation of the C-2 adduct based on steric grounds, with the bulky side chain at C-6 screening the C-4 attack of the incoming nucleophile. 12 O COOMe O O HO AcHN Pd L2 L1 Pd L2 L1 O CO2Me O O OH AcHN O COOMe OBz O O TBSO AcHN 2.5 mol% [Pd2(dba)3].CHCl3, PPh3, DBU, DCM 50oC, 2 h, 93% O CO2Me O O O AcHN bicy clic pr oduct Conformer A Conformer B
2.3.3. Reaction procedure
During the catalytic cycle of Pd(0)-catalyzed allylic substitution, the first step is to form active Pd(0) species stabilized by the ligands, followed by adding the substrate and nucleophile successively. We intended to examine the different adding sequences in order to streamline the operational steps.
Procedure A - To the starting material was added the solution of Pd(0) followed by addition of ligand, and stirred at rt until the solution turned to be the corresponding color which implied the formation of the pre-catalyst. After addition of freshly-prepared nucleophile, the reaction mixture was stirred at rt for 10 mins, and then heated at the indicated temperature (usually adopt THF as a solvent).
Procedure B - To the starting material was added the solution of Pd(0) followed by addition of ligand and freshly-prepared nucleophile. The reaction mixture was heated at the indicated temperature.
Procedure C – To begin mixing all of the solid reactants and additives was followed by adding the liquid reactants and solvent. The reaction mixture was directly heated at the indicated temperature.
At beginning, we used 12 as a model substrate to carry out the Pd(0)-catalyzed allylic substitution and added sodium dimethyl malonate (either solution or solid form) as a nucleophile. After examining these different procedures A, B and C, the similar reaction time and product distribution were found respectively. The explanation towards the results is given that the following formation of π-allylpalladium complex derived from 12 or the rate-determining step of decomplexation might occur at the higher temperature (50 oC). Generally, we adopted the procedure C as our standard protocol unless specified in some
unusual conditions.
2.4. Allylic substitution of 12 using sodium dimethylmalonate as a nucleophile
2.4.1. Ligand effect
As described in the first trial of 12 (4-OBz-7OTBS), the isomer from C-4 addition coupling with dimethyl malonate was first obtained. To realize the role of ligand in the reaction, a serial of conditions have been examined by a panel of ligands, such as dppe, dppb, dppf, P(OPh)3. Surprisingly, the inverse regioselectivity on the substrate 12 using the bidendate
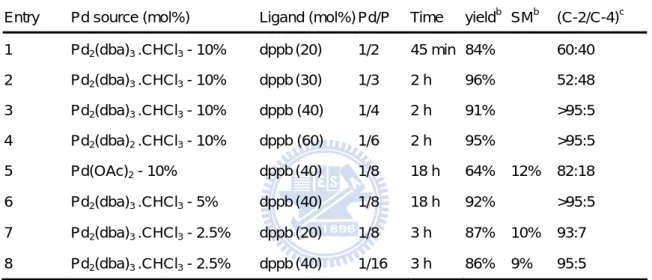
ligands was achieved (entry 2-8 of Table 1). The preliminary results are summarized in Table 1, categorized by each individual ligand, the ratio of Pd/ligand. Using 10% of Pd2(dba)3 .CHCl3 as
Pd source in the absence of the external ligands as a control reaction proceed the poor regioselectivity with ratio of 47/53 (C-2/C-4) in a moderate yield (66%, entry 1 of Table 1). While adding the bidentate ligands (dppe, dppb and dppf), C-2 adducts have been preferably obtained within 2 h with excellent yields (84-96%) (entry 2-8). On the other hand, the similar regioselectivity was performed with more basic triphenylphophite, which possess the stronger π-acceptor property and caused the larger electronic difference between C-2 and C-4 (entry 9, Table 1). An elegant series of 13C NMR experiments support this assumption (Figure 16).49, 50 In general, the more stable position for the positive charge character is the more substituted terminus, and therefore this centre becomes more reactive to an incoming nucleophile. To realize the inverse regioselectivity mediated by bidentate ligands, we set out the more detailed investigation in aspects of functions of ligands.
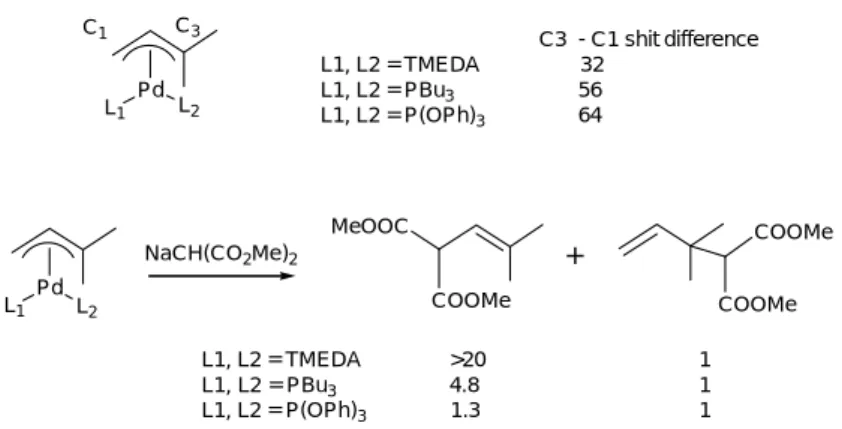
MeOOC COOMe NaCH(CO2Me)2 L1, L2 = TMEDA >20 1 L1, L2 = PBu3 4.8 1 L1, L2 = P(OPh)3 1.3 1 Pd L1 L2 COOMe COOMe + Pd L1 L2 C1 C3 C3 - C1 shit difference L1, L2 = TMEDA 32 L1, L2 = PBu3 56 L1, L2 = P(OPh)3 64
Figure 16 - Regioselectivity predicted by chemical shift of 13C-NMR
Table 1 - Conditions for the regioselective allylic substitution of 12
Entry Pd source (mol%) Ligand (mol%) Pd/P e Time Yieldf
15:16 (C-2/C-4)f 1 Pd2(dba)3 .CHCl3 - 10% None 3 h 66% 47:53 2 Allylpalladium(II) chloride dimmer a - 10% dppe b (40) 1/4 3 h 96% 93:7 3 Bis(benzonitrile)-palladium (II) Chloridea -10% dppeb (40) 1/4 3 h 86% >95:5 4 Pd2(dba)3 .CHCl3 - 10% dppeb (40) 1/4 3 h 91% >95:5 5 Pd2(dba)3 .CHCl3 - 10% dppb c (40) 1/4 2 h 91% >95:5 6 Pd2(dba)3 .CHCl3 - 5% dppb c (20) 1/4 2 h 84% 89:11 7 Pd2(dba)2 .CHCl3 - 2,5% dppbc(20) 1/8 3 h 87% 93:7 8 Pd2(dba)3 .CHCl3 - 10% dppf d (40) 1/4 19 h 94% >95:5 9 Pd2(dba)3 .CHCl3 - 10% P(OPh)3 (40) 1/2 19 h 87% 87:13 10 Pd2(dba)3 .CHCl3 - 10% PBu3 (40) 1/2 2,5 h 95% 5:95 11 Pd2(dba)3 .CHCl3 - 10% PCy3 (40) 1/2 2,5 h 91% 9:91 12 Pd2(dba)3 .CHCl3 - 10% PPh3 (20) 1/1 20 h 98% 5:95
a Allylpalladium(II) chloride dimer contains two Pds; Bis(benzonitrile)-palladium(II) Chloride contains one Pd,
b dppe = 1,2-Bis(diphenylphosphino)ethane ; c dppf = 1,1'-bis(diphenylphosphino)ferrocene; d dppb = 1,4-bis(diphenylphosphino)butane e bidentate ligand contains two phosphors
In terms of the property of mono ligand, “cone angle” was introduced by Tolman to describe the size of ligand which further influences the reactivity in the catalytic process (Figure 17).51 However; the differences in cone angle can not address the question concering about the dramatic change in regioselectivity so far.
P
M
θ (cone angle)
Figure 17 - Tolman's cone angle
To answer it, the resulting bite angle for each intermediate complex with bidentate ligand needs to be taken into account. Thus, in 2001, van Leeuwen et al. demonstrated that the distorted coordination of a substituted allyl moiety to palladium can induce more electronic preference on more substituted electrophilic carbon which simply means “geometry induce electronic effect”.52 This relevant studies on the simple substrate possibly render a plausible explanation.
In principle, coordination geometry of π-allylpalladium is symmetrically square planar. The bidentate ligand bonding to palladium will disturb the symmetry due to the nature of the fixed linker. Thus, this distortion of an η3 toward an η1,η2 bonding mode will result in a lower value of the overlap integral on the η2 site and a higher value of this integral on the η1 site. On the η2 site, both the donating interaction (ligand to metal) and the back-donating interaction (metal to ligand) will be decreased. At the same time, the Pd-C1 distance is decreased and the C1-C2 distance is increased so that the unsymmetrically π-bonded allyl moiety is formed accordingly. This assumption has been approved by the crystal structure of π-allylpalladium
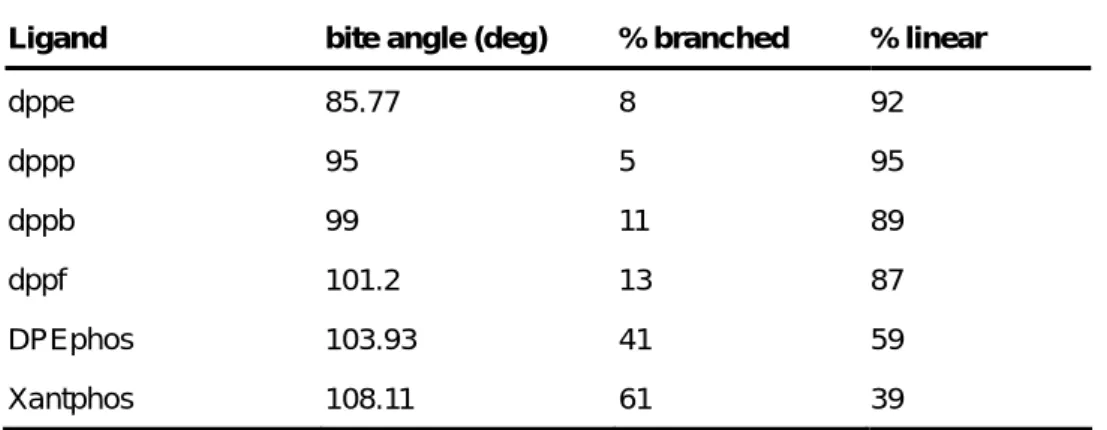
complex with the bidentate ligand. It implies that an increase in the bite angles results in a decrease of the back-donation. As a consequence, the allylic LUMO orbital, which are the sites of reaction in the nucleophilic attack, remain less occupied. Therefore, a larger bite angle results in an increase of the reactivity of the allyl moiety toward nucleophilic attack. This has indeed been observed in this case of trans-but-2-enyl acetate (Figure 18 and Table 2).53, 54
η3- bonding mode Pd L L X COOMe MeOOC Na MeOOC COOMe COOMe COOMe + branched lineared + 1 3 Pd L L 1 3 Pd L L longer shorter Nu
Lar ger bite angle η1η2- bonding mode
Figure 18 - Conversion between η3 and η1 η2 bonding mode
Table 2 - Regioselectivity in relation to bite angle
In earlier time, the steric interference in regioselectity has been also investigated by van Leeuwen and co-workers. They conducted the mechanistic study in the relatively simple
Ligand bite angle (deg) % branched % linear
dppe 85.77 8 92 dppp 95 5 95 dppb 99 11 89 dppf 101.2 13 87 DPEphos 103.93 41 59 Xantphos 108.11 61 39