國 立 交 通 大 學
應用化學系研究所
碩 士 論 文
利用光電流與光電壓衰減方法
研究不同管長奈米管染料敏化太陽能電池之電荷轉移動力學
Studies of Charge-Transport Kinetics in TiO
2Nanotube-based
Dye-Sensitizer Solar Cells with Different Tube Lengths
using Photocurrent and Photovoltage Decay Methods
研究生:林佳蓉
指導教授:刁維光 博士
利用光電流與光電壓衰減方法研究不同管長奈米管染料敏化太陽能電池之電荷轉移動 力學
Studies of Charge-Transport Kinetics in TiO2 Nanotube-based Dye-Sensitizer Solar Cells with
Different Tube Lengths using Photocurrent and Photovoltage Decay Methods
研究生:林佳蓉 Student:Chia-Jung Lin
指導教授:刁維光博士 Advisor:Dr. Eric Wei-Guang Diau
國立交通大學
應用化學系研究所
碩士論文
A ThesisSubmitted to
The Department of Applied Chemistry College of Science
National Chiao Tung University for the Degree of
Master of Science In
Applied Chemistry July 2010
Hsinchu, Taiwan, Republic of China
I 利用光電流與光電壓衰減方法研究不同管長奈米管染料敏化太陽能電池之電荷轉移動 力學 研究生:林佳蓉 指導教授:刁維光博士 國立交通大學應用化學系研究所
摘要
本論文主要在了解二氧化鈦奈米管的一維結構,是否有助於提升電子在半導體層的 傳遞速度。因此我們選擇不同厚度的二氧化鈦奈米管薄膜(ATO)進行實驗,來探討此問 題,並研究 ATO 的製程、染料吸附量和電解液厚度這些變因對於電子擴散速率的影響。 首先,在 ATO 製程間誤差的實驗中,我們發現製程條件相同、厚度相似、染料吸 附量相似的 ATO,其組成的 DSSC 在效率、擴散係數與電子生命期的結果皆相似。其次, 在不同染料吸附量的實驗中,我們發現染料吸附量的增加造成 DSSC 開路電壓的上升, 以及 ATO 表面覆蓋率的提高而使得電子生命期變長,但對於電子的擴散速率則無明顯 影響。再者,在不同電解液厚度的實驗中,我們發現電解液厚度會改變 ATO 上的能階 分布(DOS)分布與最低能態的位置,使的在電解液厚度的增加下,造成開路電壓的提升。 同時電解液厚度會使 DSSC 在 450 nm 以前的 IPCE 值下降,導致短路電流有些許下降。 最後,在不同 ATO 的長度的研究中,我們發現當 ATO 長度越長時,ATO 的表面態分佈 曲線越除,同時 ATO 長度增加代表染料吸附量也隨之增加,造成有較多光電子注入二 氧化鈦,使得最高表面態隨著 ATO 長度的增加而越靠近導帶。如此,使得電子在 ATO 上的去局限過程變得更為容易,也因此使擴散速率變得比較快。 我們認為 ATO 的微結構可能在成長過程中受到電場的作用力下進行有規則的排列, 使得其表面能態分布造成改變,減少深層的局限能態,造成光電子去局限至導帶之間的 能隙變小,也就是電子的擴散速率變快。因此二氧化鈦奈米管具備高長度的潛力,因為 它在高長度時,亦具有高的電荷集合效率。II
Studies of Charge-Transport Kinetics in TiO2 Nanotube-based Dye-Sensitizer Solar Cells with
Different Tube Lengths using Photocurrent and Photovoltage Decay Methods
Student:Chia-Jung Lin Advisor:Dr. Wei-Guang Diau The Department of Applied Chemistry, National Chiao Tung University
Abstract
To understand how the anodic titanium oxide’s (ATO) microstructure affecting the charge transport processes on TiO2 surface, we modified various thickness of ATO and
measured the photovoltaic properties with IV, IPCE, and charfe-transport kinetics with transient photocurrent and photovoltage methods. In addition, we also studied the effects of dye loading amount and thickness of the electrolyte layer on the performance of ATO-DSSC.
The diffusion coefficients and electron lifetimes were found to be almost the same (within the standard deviation is ~10%) at similar length of the ATO film made by the same method. The dye loading amount of N719 on ATO surface influences the open-circuit voltage (VOC) and electron lifetime (n), but it does not influence the electron diffusion coefficient
(Dn). When we modified the thickness of the electrolyte layer, it alters the distribution of
density of trapped states (DOS) and the potential of band edge of TiO2. However, the
thickness of electrolyte does not affect the Dn of the ATO. It is important to understand the
mechanism for the dependance of Dn on length of ATO when thicker ATO arrays generate
higher photo-current densities. We found that Dn becomes faster on longer ATO under the
same JSC. It means that the charge collection efficiency would become larger if the device is
III
謝誌
這本碩士論文能夠順利完成,碩士班生涯能順利結束,首先要先感謝刁維光教授收 我當專題生和研究生,以及給予我充足的發揮空間和即時的指點。此外,還要感謝口試 委員林敬堯教授以及葉鎮宇教授,在口試過程中給予我許多寶貴的意見,使論文內容的 小細節更加完整。 此外,還要特別感謝駱立揚學長從儀器設置、數據分析到論文撰寫等眾多方面的的 大力協助,包含花了一整天聽我那說得七零八落的口試預講。另外,也要感謝實驗室的 所有學長同學學弟在我實驗過程出現麻煩時的協助。 我是個不善表達的人,因此在這也就只能說一聲「謝謝大家」,以示感謝。IV
目錄
摘要 --- I Abstract --- II 謝誌 --- III 目錄 --- IV 表目錄 --- VI 圖目錄 --- VII 第一章 緒論 --- 1 1.1. 文獻回顧 --- 1 1.1.1. 太陽能電池的發展--- 1 1.1.2. 染料敏化太陽能電池的原理 --- 5 1.1.3. 染料敏化太陽能電池的材料 --- 8 1.1.4. 電子在二氧化鈦上傳遞的相關研究 --- 20 1.1.4.1. 電子在二氧化鈦上的傳遞原理 --- 20 1.1.4.2. 電子在二氧化鈦上傳遞速率的測量以及分析方法 --- 24 1.1.4.3. 二氧化鈦上化學電容的研究 --- 26 1.2. 研究動機 --- 29 第二章 實驗 --- 32 2.1. 實驗儀器設備 --- 32 2.2. 陽極處理製備二氧化鈦奈米管 --- 34 2.2.1. 鈦基材前處理 --- 34 2.2.2. 以陽極處理法制被二氧化鈦奈米管陣列 --- 34 2.2.3. 二氧化鈦奈米管之後處理 --- 37 2.3. 鉑電極製備 --- 37 2.4. 電解液製備 --- 38V
2.5. 光敏染料吸附量之檢測 --- 38
2.6. 測量 --- 39
2.6.1. 電流-電壓特性曲線(I-V curve) --- 39
2.6.2. 光電轉換效率(Incident photon-to-current conversion efficiency, IPCE) --- 39
2.6.3. 瞬態光電流與光電壓測量(Photocurrent and photovoltage transient) --- 40
2.6.4. 化學電容測量 (Chemical Capacirance)--- 43 第三章 結果與討論 --- 45 3.1. ATO 製程誤差的影響 --- 46 3.2. 染料吸附量的影響 --- 51 3.3. 電解液厚度的影響 --- 57 3.4. ATO 厚度的影響 --- 66 第四章 結論 --- 77 第五章 參考文獻 --- 79 第六章 附錄 --- 86
VI
表目錄
第二章
表 2.1二氧化鈦奈米管染料敏化太陽能電池效率測量之電解液配方 F’ ... 38
第三章 表 3.1對於不同次 ATO 製程的 ATO-DSSC 的實驗結果,N719 的吸附密度與其相關 I-V 特性係數。 ... 48
表 3.2對於改變染料吸附量的 ATO-DSSC 的實驗結果,N719 的吸附密度與其相關 I-V 特性係數。... 54
表 3.3對於改變電解液厚度的 ATO-DSSC 的實驗結果,N719 的吸附密度與其相關 I-V 特性係數。... 59
表 3.4電解液厚度與 2 MM樣品槽的光徑長度比較表。 ... 61
VII
圖目錄
第一章
圖 1.1染料敏化太陽能電池工作原理示意圖。[15] ... 6
圖 1.2AM-1.5G 照射下的太陽光譜圖[19]。 ... 9
圖 1.3 以 RU金屬為中心的撮合物染料結構,(A)為 N3,(B)為 N719,(C)為BLACK DYE。TBA= TERT-BUTYLAMMONIUM。 [20] ... 10
圖 1.4應用於染料敏化太陽能電池的有機染料結構:(A) INDOLINE ( =9%)[25]、(B) PORPHYRIN (7.1%)[26]、(C) PHTHALOCYANINE (3.5%)[27]、(D) SQUARINE (4.5%)[28]、(E) COUMARIN (6.5%)[29]與(F) HEMICYANINE (5.2%)[30]。 ... 11 圖 1.5各種製作對電極觸媒層材料的比較[35]。... 12 圖 1.6開路電壓的修正策略[42]。 ... 14 圖 1.7MEOTAD結構圖[45]。 ... 15 圖 1.8DSSC 陽極半導體材料之能階圖[46]。 ... 16 圖 1.9典型 NP-DSSC 示意圖[50]。 ... 16
圖 1.10 一維 TIO2 奈米結構; (A) WIRE[51]、(B) TUBE[50]、(C) ROD[56]與(D) FIBER[56]。 ... 17
圖 1.11 陽極處理 TNT 之 SEM 顯微結構:(A)1MH2SO4/0.15 WT.%HF 水溶液[68];(B)0.5 WT.%NH4F/GRYCEROL 電解液[69]。 ... 18 圖 1.12 背照式 NT-DSSC 元件示意圖[70]。 ... 19 圖 1.13 陽極處理法製備 TNT 陣列之電化學過程競爭反應示意圖。 ... 20 圖 1.14DSSC 中電子在傳遞過程中重要的參數與路徑示意圖[75]。 ... 22 圖 1.15 波長 514NM探測雷射照射到電池後電池電流的提升情形。 ... 24 圖 1.16 以脈衝雷射微擾所得到的瞬帶電流變化圖,虛線是以擴散方成是擬合的結果,所得的擴散係數分 別是(A)10、(B)3.2、(C)4.0×10-5 CM2S-1。[84] ... 25
VIII 圖 1.17 使用輸出電壓可調式雷射所得到的(A)瞬態光電流變化圖,(B) 瞬態光電壓變化圖。... 26 圖 1.18 奈米結構二氧化鈦電極行為的電子能量示意圖[91]。 ... 27 圖 1.19 化學電容實驗與擬合結果[91]。 ... 29 圖 1.20 不同奈米粒與奈米線比例的 DSSC 短路電流及效率與薄膜厚度的關係圖。 ... 30 圖 1.21 不同奈米粒與奈米線比例的 DSSC 擴散係數與薄膜厚度的關係圖。 ... 30 圖 1.22ATO-DSSC 短路電流及效率與薄膜厚度的關係圖。 ... 31 第二章 圖 2.1陽極處理製備 ATO 的實驗裝置示意圖。... 35 圖 2.2陽極處理定電壓法之電壓參數控制示意圖。 ... 35 圖 2.3陽極處理混合法之電壓與電流密度參數控制示意圖。 ... 36 圖 2.4以定電壓法進行陽極處理之陽極處理時間與 ATO 厚度的關係圖。 ... 36 圖 2.5二氧化鈦奈米管燒結升溫條件 ... 37 圖 2.6瞬態光電流與光電壓測量光路圖(一)... 41 圖 2.7 瞬態光電流與光電壓測量光路圖(二) ... 42 圖 2.8瞬態光電流與光電壓對時間的關係圖。(A)與(B)分別為使用脈衝雷射得到的瞬態光電流與光電壓對 時間的關係圖,(C)與(D)分別為使用連續波雷射得到的瞬態光電流與光電壓對時間的關係圖。 ... 43 第三章 圖 3.1不同次奈米管製程的奈米管的實際厚度圖。 ... 46 圖 3.2以 0.1MNAOH(AQ)脫附不同次奈米管製程的 ATO 的吸收光譜圖。 ... 47 圖 3.3 不同次奈米管製程的 DSSC 的 I-V 特性曲線圖。 ... 47 圖 3.4不同次奈米管製程的的 DSSC 在不同波長下的 IPCE 圖。 ... 48 圖 3.5不同次奈米管製程的的 DSSC 在 532 NM下的電子流量與光通量的關係圖。 ... 49 圖 3.6不同次奈米管製程的的 DSSC 在不同強度的 532 NM雷射下的擴散係數與短路電流關係圖。 ... 50 圖 3.7不同次奈米管製程的的 DSSC 在不同強度的 532 NM雷射下的擴散係數與光通量關係圖。 ... 50 圖 3.8不同次奈米管製程的的 DSSC 在不同強度的 532 NM雷射下的電子生命期與開路電壓關係圖。 .... 51
IX 圖 3.9不同染料吸附量的奈米管的實際厚度圖。 ... 52 圖 3.10 以 0.1MNAOH(AQ)脫附不同染料吸附量的 ATO 的吸收光譜圖。 ... 52 圖 3.11 不同染料吸附量的 DSSC 的 I-V 特性曲線圖 ... 53 圖 3.12 不同染料吸附量的 DSSC 的開路電壓、短路電流、填充率與效率對染料吸附量的關係圖 ... 53 圖 3.13 不同染料吸附量的 DSSC 在不同波長下的光電轉換效率(IPCE)圖。 ... 55 圖 3.14 不同染料吸附量的 DSSC 在 532 NM下的電子流量與光通量的關係圖。 ... 55 圖 3.15 不同染料吸附量的 DSSC 在不同強度的 532 NM雷射下的擴散係數與短路電流關係圖。 ... 56 圖 3.16 不同染料吸附量的 DSSC 在開路電路時,在不同強度的 532 NM雷射下的電子生命期與開路電壓關 係圖。 ... 57 圖 3.17 不同電解液厚度的 DSSC 奈米管的實際厚度圖 ... 58 圖 3.18 以 0.1MNAOH(AQ)脫附不同電解液厚度的 DSSC 的奈米管的吸收光譜圖。 ... 58 圖 3.19 不同電解液厚度的 DSSC 的 I-V 特性曲線圖 ... 59 圖 3.20 不同電解液厚度的 DSSC 的開路電壓、短路電流、填充率與效率對電解液厚度的關係圖 ... 60 圖 3.21 模擬不同厚度電解液對光的吸收情形。 ... 61 圖 3.22 不同電解液厚度的 DSSC 在不同波長下的光電轉換效率(IPCE)圖 ... 62 圖 3.23 不同電解液厚度的 DSSC 在 532 NM下的電子流量與光通量的關係圖。 ... 63 圖 3.24 不同電解液厚度的 DSSC 在不同強度的 532 NM雷射下的擴散係數與短路電流關係圖。 ... 63 圖 3.25 不同電解液厚度的 DSSC 電容與開路電壓的關係圖。 ... 64 圖 3.26 不同電解液厚度的 DSSC 在不同強度的 532 NM雷射下的電子生命期與開路電壓關係圖。 ... 65 圖 3.27 不同電解液厚度的 DSSC 電子生命期與電容的關係圖。 ... 66 圖 3.28 不同奈米管管長的 DSSC 奈米管的實際厚度圖。 ... 67 圖 3.29 以 0.1MNAOH(AQ)脫附不同厚度的 ATO 的吸收光譜圖。... 67 圖 3.30 不同 ATO 厚度的 DSSC 的 I-V 特性曲線圖 ... 68 圖 3.31 不同 ATO 厚度的 DSSC 的染料吸附量、開路電壓、短路電流密度,填充率以及效率對奈米管長度 的關係圖。 ... 69
X 圖 3.32 不同 ATO 厚度的 DSSC 在 532 NM下的電子流量與光通量的關係圖。 ... 70 圖 3.33 不同 ATO 厚度的 DSSC 在不同強度的 532 NM雷射下的擴散係數與短路電流關係圖 ... 70 圖 3.34 不同 ATO 厚度的 DSSC 在不同強度的 532 NM雷射下的擴散係數與短路電流的相關參數 與 ATO 厚 度的關係圖。 ... 71 圖 3.35 不同 ATO 厚度的 DSSC 電容與開路電壓的關係圖。 ... 72 圖 3.36 借用阿瑞尼斯公式表示電子從表面態釋放到導帶而傳到外電路的示意圖。 ... 72 圖 3.37 不同 ATO 厚度的 DSSC 在不同強度的 532 NM雷射下的擴散係數與開路電壓關係圖。 ... 74 圖 3.38 不同 ATO 厚度的 DSSC 去局限能量障礙 VA與 ATO 厚度係圖。 ... 74 圖 3.39 不同 ATO 厚度的 DSSC 在不同強度的 532 NM雷射下電荷收集效率與光通量的關係圖。 ... 75 圖 3.40 電子在長管與短管奈米管上傳遞的示意圖。 ... 76
1
第一章 緒論
1.1. 文獻回顧
1.1.1. 太陽能電池的發展 自18世紀工業革命以來,人類對能源的需求日益增加。目前,能源的消耗主要來自 化石燃料,由此引發的能源危機和環境污染成為極待解決的嚴重的問題。20世紀70年代 的石油危機使這一矛盾更加突出。根據美國能源部預估,目前全球石油存量約40 年、 天然氣存量約60年、煤炭存量約200年,而全球能源需求在2050 年將達到目前的兩倍, 2100 年將達到目前的三倍,因此將可預期未來全球能源供需失調的困境。有鑑於此, 各種可在生能源包括生質能源、生物能、風能、水能、核能、以及太陽能成為解決全球 性的能源危機和環境問題的幾個重要途徑。這其中,太陽能作為一種可再生能源,具有 其他能源所不可比擬的優點;與化石燃料相比,太陽能取之不盡,用之不竭;與核能相 比,太陽能更為安全,其應用不會對環境造成任何汙染;與水能、風能相比,太陽能利 用的成本比較低,而且不受地理條件的限制。 太陽每年向地球輻射的能量大約為5.4×1024 J[1]。全世界每年需要的最終能源相當於 8×109噸的煤,也就是1.09×1050 J地能量(1噸的煤大約可產生2.93×1010J的能量)[2]。如果地 球上一小部分的太陽能被利用的話,許多能原問題都可以迎刃而解。因此利用太陽能的 研究和應用受到世界各國政府的重視。目前太陽能的轉換主要有四種形式:太陽能轉換成 熱能、太陽能轉換成熱電能、光電太陽能轉換與化學太陽能轉換。其中光電太陽能轉換 是將太陽能直接轉成電能,其為世紀各國最重視的研究課題之一。 光伏效應可追溯到166年前。1839年,Becquerel等人將可個電極放在電解液裡,光 照其中其中的一個電極,能夠檢測到光電壓的產生[3]。在此相當長的時間裡,光伏效應 僅僅是一種現象而已,沒有實用化器件的產生。五十年代以來,以矽為代表的半導體材 料得到迅速的發展。1954年,轉換效率為6%的p-n型太陽能電池在貝爾實驗室誕生,這 是一個實用化的光電轉換器件。從此,半導體矽太陽能電池得到蓬勃的發展,並且廣泛2 應用於衛星、航空、軍事以及偏遠地需的通訊和電力供應。由於這種光電轉換系統非常 穩定,開始人們很少關心他的生產成本。太陽能科技於1950 年從太空科技轉移至一般 民生商業用途,然而,由於太陽能發電的設備成本偏高,使其在民生應用上無法普及。 近年來由於奈米技術與先進材料之開發,有助於提升太陽能電池之光電轉換效率,因此 單(多)晶矽太陽能電池、非晶矽太陽能電池、薄膜太陽能電池、有機太陽能電池等技術 之改良,正受到各先進國家之研究機構、大學與工業界的重視。 目前許多研究機構發展了各類太陽能電池的轉換效率約介於 10-30 %,其發展過程 及分類概述如下: (1) 第一代晶圓(wafer)太陽能電池: 1954年貝爾實驗室[4]開發第一顆太陽電池,此第一代電池一般以摻雜少量之三 價雜質原子的 p型半導體(如硼(B))當作基板,再利用高溫熱擴散將濃度略高於 B的五價磷原子(P)摻入p型基板內,此五價之雜質原子因其中四個價電子分別與 鄰近的四個 Si原子形成共價鍵,而多出一個自由電子並取代 Si原子的位置,因 此形成一 p-n介面。當太陽光照射到 Si內部的 p-n介面後,光子在 Si內將生電 子−電洞對,其中一部分的電子-電洞對由 p-n接合內部電場的作用而分離而產生 電位差,再由外部迴路產生電流通過。晶圓型太陽電池依基材不同可區分為: 單晶矽太陽能電池、多晶矽太陽能電池及三五族(砷化鎵,GaAs)太陽能電池。
A. 單晶矽(single crystal silicon)太陽能電池(15-24 %)
單晶矽太陽能電池因具有較高的轉換效率,是目前太陽能電池市場之主要佔有 者,目前單晶矽太陽能電池的最高光電轉換效率為 24.4 %,由 UNSW (University of New South Wales)於 1998年製作[5]。雖然單晶矽太陽能電池的操 作穩定性極佳,但單晶矽晶圓材料之昂貴,使得太陽能電池的製作成本增加。
B. 多晶矽(polycrystalline silicon)太陽能電池(10-20 %)
多晶矽太陽能電池的光電轉換效率相對於單晶矽電池雖相對較低,但因製作步 驟簡單,成本較低,因此許多低功率需求的電力系統皆選用此電池。2004年由
3 法蘭克博士(Frank Dimroth)領軍的太陽能電池團隊(FhG-ISE)製作的多晶矽太 陽能電池[6],其光電轉換效率已可達 20.3 %。 C. Ⅲ-Ⅴ族太陽能電池(19-40 %) Ⅲ-Ⅴ族元素如砷化鎵(GaAs)及磷化銦(InP),高效率聚光型多接面太陽能電池 為利用透鏡與反射鏡將陽光聚焦於太陽能電池晶片上,並採用三層半導體材料: 鎵銦磷化合物、鎵銦砷化合物和鍺;各層材料被調適為各捕捉太陽光譜中一部 分,其聚光系統可將陽光強度增至 240倍。1995年 GaAs太陽能電池的轉換效 率可達 32 %,而 2005年美國再生能源實驗室(NREL)團隊與美國波音光譜實 驗室(Spectrolab)聯合研發成功首個超越 40 % 效率障礙的太陽能電池[7]。砷化 鎵電池雖擁有如此出眾的表現,但材料及製造成本非常昂貴,因此應用途徑較 為特殊,如人造衛星之發電系統及軍事用途。 (2) 第二代薄膜(thin-film)太陽能電池: 薄膜太陽能電池顧名思義是以薄膜型為主要基材的電池,其材料製作為薄膜成 形的方法。薄膜電池技術開發較晚,轉換效率較單晶矽太陽能電池低,由於使 用材料的節省、製造成本降低、製成簡單、重量輕穎,所以用途寬廣,廣受各 研究單位歡迎。薄膜太陽能電池依所用材料種類及其結晶可分為非晶矽、多晶 矽、微晶矽、碲化鎘、銅銦鎵硒等。
A. 非晶矽(amorphous silicon, a-Si)薄膜太陽能電池(8-13%)
非晶矽太陽能電池之基本結構主要是由 p-i-n或 n-i-p的三層式結構所構成,其 中 p與 n代表高濃度 p型及 n型半導體薄膜層。其功能在於產生內建高電場, 而 i薄膜層為不含雜質的本質性半導體薄膜層,用於產生電子−電洞對,此電子 −電洞對並可因其高電場而快速的被分離。非晶矽薄膜一般成長於塑膠或玻璃 基板上,其光學吸收係數高,因此元件厚度可控制到很薄。但此晶態因有史坦 柏−勞斯基效應(Steabler-Wronski Effect),照光時的熱會使部分未飽和狀態的矽 晶格毀損而有漏電流的產生,進而影響光電轉換效率,故其穩定度有待改善。
4 目前穩定狀況下之非晶矽薄膜太陽能電池為 2003年 U. Neuchatel團隊所研發, 其研究結果為 9.5 %[8]。 B. 碲化鎘(CdTe)薄膜太陽能電池(10-15 %) Ⅱ-Ⅵ族太陽能電池製程技術中主要以硫化鎘(CdS)及 CdTe薄膜技術為主,在 n型的 CdS以及 p型的 CdTe接面處之活性層會發生載子產生及收集作用,因 而產生光電流。美國第一太陽能 ( FSLR)發展之 CdTe薄膜太陽能電池,目前 轉換效率約 9-10 %,最高可達 16 % [9],是此類電池中最早實現量產和低成 本的成果。目前 CdTe薄膜太陽能電池在實驗室的成果可達 16.5 %,由美國 NREL完成[10]。但畢竟 Te元素在地球上相當貧乏,且 CdTe太陽能電池材料 佔總成本 53 %的高成本,CdTe如果在太陽能電池方面發展太快速,將會面臨 材料短缺或 Cd元素毒性的疑慮,這些問題都會成為未來發展的限制因素。 C. 銅銦硒(CuInSe2)/銅銦鎵硒化物(CuInGaSe2)薄膜太陽能電池(10-20 %) Wagner博士首先使用銅銦硒(CIS)單晶材料,並於其表面沉積 n型 CdS而製作 出單晶型 CIS化合物的太陽能電池,其元件之光電轉換效率為 12.0 % [11]。p 型的 CIGS與 n型的 CdS緩衝層/窗層為此型電池主要產生電能的部分,元件在 不照光的情況下,由於空間中濃度分佈不均,電子會從 CdS緩衝層擴散至CIGS 吸收層,導致部分 n型緩衝層帶正電,另一部分 p型吸收層帶負電,而在介面 處形成空乏區及內建電位。目前美國 NREL所發展的「三階段製程(three-stage process)」技術多晶 CIGS太陽能電池的光電轉換效率已可達 19.5 % [12]。 (3) 第三代有機(organic)太陽能電池: 由於第一代與第二代太陽能電池製程必需在無塵室與真空設備的操作下完成, 所以設備成本投資大,因此使得第三代太陽能電池的低成本製程被業界看好。 第三代太陽能電池因其製程簡單、設備成本低廉、容易製成大面積化與可撓曲 電池等優點,所以在近幾年來發展快速。但其光電轉換效率目前處於瓶頸,因 此在元件的改善方面也是本研究的主要方向。
5 A. 有機半導體太陽能電池(3-6 %) 又稱有機薄膜太陽能電池,其利用有機分子層吸收光後激發放出電子並傳遞至 寬能隙的無機奈米層而產生電壓。電荷分離發生在中間的混合層,並透過電子 施體及受體的有機半導體層傳遞至對電極。南韓光州科學技術學院 (GIST)的 研究人員在 2009年 4月 26日的學術雜誌《Nature Photonics》[13]上宣佈,已 將有機薄膜太陽能電池的單元轉換效率提高到了 6.1 %。其具體測量值如下: 轉換效率為 6.1 %、開路電壓 (Voc)為 0.88 V、短路電流密度 (Jsc)為 10.6 mA cm-2、填充因子(FF)為 0.66。以上是在 AM (air mass)為 1.5 G,光線入射強度 為 100 mW cm-2的條件下測量的結果。
B. 染料敏化太陽能電池(Dye-Sensitized Solar Cells, DSSC) (7-11 %)
DSSC主要利用有機光敏染料吸收光能後,電子從染料的基態躍升至激發態, 經由作為電子傳輸層的工作電極傳至基材及外電路到達對電極,此時失去電子 的染料將可被電解質中的氧化反應還原至基態,而傳到對電極的電子則將氧化 態的電解質還原,如此形成 DSSC的電子傳輸迴路。經過十餘年的發展,目前 染料敏化太陽能電池的最高轉換效率已達 11 % [14]。 綜合以上所述,DSSC因具有製程設備及成本低廉、製作簡便、可透光、可製成可 撓曲電池,以及可藉由吸附不同染料得到色彩繽紛的元件等優點。 1.1.2. 染料敏化太陽能電池的原理 染料敏化太陽能電池早期的工作發現:由一個低功函金屬、一個有機層和一個高功 函金屬(或導電玻璃)組成夾心式電池,便會觀察到光伏效應。電池器件通常的結構為: 玻璃/ITO(三氧化銦)/染料/金屬電極。太陽光首先穿過透明電極(導電玻璃)照射到有機薄 膜上;有機薄膜吸收光之後,產生許多電子/電洞對(即正負電荷分離),正電荷向負極移 動,負電荷向正極移動,這種已經分離的電荷統稱電荷載流子,電荷載流子到達電極提 供外電路時,表現為電流。
6
圖 1. 1 染料敏化太陽能電池工作原理示意圖。[15]
目前染料敏化太陽能電池的幾本結構包含:提供電子的光敏染料(photosensitizer)、 由透明導電氧化物(transparent conducting oxide, TCO)與多孔性二氧化鈦奈米薄膜組成用 來提供傳遞電子的工作電極(working electrode)、轉移電洞的電解質(electrolyte)和鉑對電 極(Pt counter electrode)。染料敏化太陽能電池的基本結構與電子在各個介面的傳遞途徑 如圖 1. 1 所示,圖中○1 ~○5 (以實線表示)為 DSSC 的工作機制,○6 ~○8 (以虛線表示)為損 耗機制,箭頭表示電子傳遞方向。 電子在 DSSC 中的傳遞過程與工作機制反應時間[16, 17]如下: ○1 光激反應(Photoexcitation): (1. 1) 染料分子吸附在多孔性無機半導體薄膜(二氧化鈦)上,當染料分子吸收光能後,在小 於 10-15秒的時間內被激發成電子-電洞對,電子從基態躍遷到激發態。 ○2 電子注入(Electron injection): (1. 2) 染料上位於激發態的電子迅速注入二氧化鈦的導帶(conduction band, CB),使染料形成
7 正離子。電子注入的速度取決於染料與多孔性半導體薄膜能階的匹配、鍵結強度或聚 集程度,此過程發生的時間一般約為 10-15 ~10-12秒。 ○3 電子擴散(Electron diffusion): 電子注入二氧化鈦導帶後,以擴散的方式將電子傳遞到透明導電玻璃上,再經由外電 路將電子傳回到 DSSC 的對電極。早期研究發現使用二氧化鈦奈米粒做為陽極的 DSSC,在電荷收集的過程需要耗時 10-4~10-3秒才能完成,可能原因是奈米粒的排列 方式散亂無序,使得電子可能擴散向任何方向。因此,為了改善電子擴散速度,在半 導體層加入了如奈米棒(nanorod)、奈米線(nanowire)或奈米管(nanotube)等一維結構, 提供電子明確的傳遞方向。 ○4 染料再生(Dye regeneration): (1. 3) 當染料於 1 形成電子-電洞對時,一方面電子注入半導體的導帶,另一方面電動則從 染料分子轉移至電解質中,使電解液產生氧化反應 (1. 4) 而使氧化態的染料正離子被還原至基態。 (1. 5) 此染料再生反應的反應時間約為 10-9 ~10-6秒。
○5 電解質再生(Redox couple regeneration):
(1. 6) 電子經由外電路回到對電極表面,並與擴散到電解質-電極介面的氧化鈦電解質 進行 還原反應,完成 DSSC 的循環。由於 在導電玻璃上的還原能力較差,而在此介面會 造成太大的過電位[18]。為了減少電子在此介面的損失,所以在導電玻璃上多增加一 層鉑的觸媒層,加速 的還原反應。陰極上電子藉由鉑的催化將 還原成 耗時約 12 ns。 DSSC 除了上述五項工作機制提供電子順利形成循環的傳遞路徑之外,還有數個會
8 使電子產生損耗而影響整體效率的傳遞路徑: ○6 電子-電動再結合(non-radiative relaxation): (1. 7) 染料分子激發態的電子藉由釋放光或熱的過程回到基態,而使電子無法順利注入半導 體導帶,因而早成損失,此過程約耗時 10-9 ~10-7秒 ○7 電荷重組(charge recombination): (1. 8) 注入半導體導帶的電子與染料正離子再結合而造成電子損失,此步驟所需時間約為 10-6~10-3秒。 ○8 電子攔截(interception): (1. 9) 注入半導體導帶的電子被電解質中的氧化鈦攔截而造成電子無法傳至外電路,此步驟 所需時間約為 10-3秒。 染料敏化太陽能電池的材料設計上必頇要能對工作機制○1 ~○5 作最佳化的改善與選 擇,另外也必頇要降低損耗機制○6 ~○8 的影響,以提升 DSSC 的光電轉換效率。我們可 以發現○2 和○7 與○4 和○6 分別是一對競爭機制,從上述個機制的反應時間看來,損耗機制 的反應時間遠長於工作機制的反應時間,因此對 DSSC 的影響很小。但是○3 和○8 這對競 爭機制的反應時間差異就沒有其他競爭機制對明顯,因此若電子在半導體導帶內的擴散 時間太長,電子就有很高的機會被電解質攔截而進行還原反應,形成反向電流,此稱為 暗電流(drak current)。因此陽極材料的設計必頇要能使電子的擴散速率越快越好,也就 是材料的擴散係數要儘可能提高。 1.1.3. 染料敏化太陽能電池的材料 染料敏化太陽能電池的基本結構如 1.1.2 所述,主要包含四個部分:光敏染料、對 電極、電解質和半導體層。
9 1.1.3.1. 光敏染料 圖 1. 2 AM-1.5G 照射下的太陽光譜圖[19]。 染料敏化太陽能電池,顧名思義,即若 DSSC 想要產生電流,則染料的激發態電子 必頇能夠順利的傳到半導體層。因此,染料激發態的能階必頇要非常靠近且高於半導體 導帶的邊緣,才能夠有效的傳導電子。既然染料在 DSSC 中的角色是吸收太陽光後丟出 電子,因此染料的吸光範圍越寬則可以越有效率利用太陽光。但從太陽光譜如圖 1. 2 可 發現,太陽提供能量的波長從 300 nm~2500 nm,而常見的染料的吸光波長都只有在可 見光,少數染料可以吸收近紅外光,同時強吸收的波長範圍窄。如此一來,往長波長方 向的光區就很少被利用到。 光敏染料的吸收光譜,會明顯的影響 DSSC 的效能,尤其對 DSSC 電流密度有決定 性的影響,因此染料的設計上大至有幾項指標: 吸收係數(absorption coefficient, ): 此為染料在特定波長下對光子的吸收能力。目前常使用的染料的吸光區間(400 nm~900 nm)的太陽光能量,就占太陽光譜總能量的 1/3~1/2,因此若能提高此區間 內的吸光能力,則有助於提升 DSSC 的效率。
10 吸收光譜(absorption spectrum) : 或稱吸收帶;不同染料對於不同波長的光有不同的吸收能力,若染料的吸收帶寬度 能延伸到近紅外光區,則可以增加對太陽光的利用,因此陸續有各種不同顏測的光 敏染料的發展,如有機染料。但染料的吸收帶越靠近紅外光區,能隙(band gap)就越 小,因此也就越頇注意到染料基態與激發態的能階位置,這是因為染料激發態的電 子要能傳入半導體層,因此激發態的能階必頇高於半導體的導帶能階,同時電子會 從電解質中將電子傳回到染料基態,因此染料基態的能階必頇低於電解質的能階。 因此,若保持使用相同的半導體層以及電解質,則染料的能隙的縮小就有一定的極 限,超過了極限,電子就無法流動,這樣的染料對染料敏化太陽能電池就沒有意義。 光電轉換效率光譜: 針對不同波長下染料對於入射光子-光電流的轉換效率(Incident Photon-to-current Conversion Efficiency, IPCE),此量測式觀察染料特性的一項重要指標,因為即使染 料在某個波段對光有吸收能力,但卻無法傳出電子的話,對染料敏化太陽能電池就 沒有意義。
(a) (b) (c)
圖 1. 3 以 Ru 金屬為中心的撮合物染料結構,(a)為 N3,(b)為 N719,(c)為 black dye。TBA= tert-butylammonium。[20]
11
在目前的研究中,效率大於10 %的染料敏化太陽能電池所使用的染料多是以釕金屬 錯合物(ruthenium polypyridyl complexes)為主[21-23],並吸附於具結晶性、多孔性的高比 表面積的二氧化鈦光做電極。最著名的Ru-polypyridyl 染料分子為N3、N719與black dye, 其化學結構如圖1. 3所示。從Robertson [20]及Argazzi [24]的相關研究顯示,這些 Ru-polypyridyl 染料與半導體層間具有很強的鍵結,並且由理論計算證明其電荷重組機 會最小。因此近年來這些Ru-polypyridyl 染料被廣泛的使用及討論。 (a) (b) (c) (d) (e) (f) 圖 1. 4 應用於染料敏化太陽能電池的有機染料結構:(a) indoline (=9 %)[25]、
(b) porphyrin (7.1 %)[26]、(c) phthalocyanine (3.5 %)[27]、(d) squarine (4.5 %)[28]、(e) coumarin (6.5 %)[29]與(f) hemicyanine (5.2 %)[30]。
12 的吸附量,以增加對光的吸收量,例如提高半導體層的總表面積,或是加入散色層以提 高對光的使用率並擴展DSSC的IPCE光譜。但這些改善的效果遠遠不及直接提升染料的 吸收係數或是加寬染料的吸收光譜,同時因為Ru為貴重金屬,在成本的考量下不利於染 料敏化太陽能電池的發展。因此科學家式圖調整硫氰化物(-SCN)配位基[32]、改變聯吡 啶配位基[23]、將Ru-polypyridyl 中心金屬替換為鋨金屬[31]等改善,期望將其吸收範圍 往長波長延伸。另外,學者們亦積極尋找高效能且具有高吸收係數與吸收帶的替代染料, 如indoline、coumarin、porphyrin 及phthalocyanine 系統等(圖1. 4)。 1.1.3.2. 對電極 在染了敏化太陽能電池的工作原理中,染料分子吸收入射光的光子後,電子從激發 態迅速注入半導體之導帶並由外部電路傳回對電極;其次,染料的再生由 完成;最後 離子游離至對電極表面,被來自外部電路的電子還原。一般來說,有機溶劑中的 離 子游離至 ITO 或 FTO 進行還原的過程,是非常緩慢的[33, 34]。為了減少此反應中的過 電位,因此必頇在對電極表面塗佈具有催化作用的材料。 圖 1. 5 各種製作對電極觸媒層材料的比較[35]。
13
近年來對於 DSSC 對電極的研究,Pt 材料為最常用的催化材料[34, 36-38]。而其製 作方法的選擇,通常取決於催化效果及成本的考量。其製作方法如濺鍍法或
Papageorgiou 等人[33]發展利用熱分解法 (Thermal Cluster Platinum catalyst, TCP),其將 5 mM 氯鉑酸/無水異丙醇溶液旋轉塗佈於 FTO 玻璃上,接著於空氣陰乾 3 分鐘後,置 入空氣爐熱處理 385℃10 分鐘而完成對電極觸媒層的製作。這個方法所塗佈的 Pt 層具 有較低的 Pt 量、較高的催化效果與物理穩定性。為了減少對電極的製造費用,Suzuki 等人[35]將奈米碳管取代昂貴的 Pt 應用於對電極上;Saito 等[39]使用高分子聚合物 (3,4-ethylenedioxythiophene)材料塗佈於導電玻璃做為對電極。這些新材料的單位成本雖 比 Pt 低,但這些材料需塗佈更大厚度的量於對電極基材上,才能達到有效的催化效果, 因而提高製造成本;再者,高厚度的觸媒層將降低對電極的光穿透度,此將不利於背照 式 DSSC 元件的測試,如本論文所使用的奈米管 DSSC。Suzuki 等人[35]亦對其他對電 極觸媒層材料如單層奈米管 (SWCNT)、碳絲 (carbon filament)、奈米角 (nanohorn)以及 Pt 做效率比較,如圖 1. 5 所示。由圖中可得知,無塗佈觸媒層對電極之電池效率最差, 而以 Pt 觸媒層的效果最好。 對電極的研究重點主要在於製程簡單、降低成本以及高催化效率。另外,製程若可 於室溫、不需昂貴設備,則可更進一步降低製作成本。對電極觸媒層修飾的好壞雖然對 於 DSSC 元件效率無太大的影響,但一個具有高催化效果且物理穩定性佳的對電極,與 其他最佳化元件組合後將有助於提升光電轉換效率。綜合以上對電極的介紹,本論文中 的 DSSC 元件量測,將使用 TCP 方法製備 Pt 觸媒層/ ITO 作為元件的對電極。 1.1.3.3. 電解質 電解液中氧化還原對的主要作用有:將氧化態的染料還原,完成染料再生;另一方 面, 接受由對電極傳回的電子後被還原,完成電化學迴路。對於高效率 DSSC 而言, 氧化還原對的作用過程必頇符合以下限制:(1) 它必頇在染料抓取陽極上的電子而再結 合之前將染料正離子還原;(2) 不允許從光陽極上攔截電子而還原。因此,設計電解質
14 需考慮兩個條件:快速的染料再生動作與緩慢的攔截作用。目前文獻上報導轉換效率在 4 %以上的 DSSC,皆使用 / 氧化還原對做為電解質。而 / 在元件中具有高效 率的染料再生能力,以及對於 TiO2上電子的緩慢攔截作用[40]。另外,半導體層導帶上 的電子被氧化態的電解質攔截,因而無法傳至外電路回到對電極。此步驟發生時間(約 10-3秒)遠慢於電子注入半導體層的速度(10-15 -10-12秒),因此對元件效率的影響較小。
電解液中氧化還原對的能司特電位(Nernstian potential)與 TiO2半導體層的費米能
階(Fermi level, EF)之間的電位差,可定義為元件所能產生的最大電壓值 (開路電壓, VOC)[41],如圖 1. 6 所示。以最高效率 TiO2/ Dye/( / )之 DSSC 為例,其 VOC最佳 化值為 0.8 V [22]。Hupp 研究團隊[42]指出, EF尚有 200 mV 的上修空間, I-/ I3-的氧 化還原電位亦有約 550 mV 的下修空間。因此,要提升開路電壓,可將 EF往負電位方 向修正,或是將氧化還原對的氧化還原電位往正電位方向修正。 圖 1. 6 開路電壓的修正策略[42]。 此外,在 I -/ I3-電解液的改良上,Grätzel 團隊[43]在 2003 年發表的結果中,以含氟 的聚合物 PVDF-HFP (poly(vinylidenefluoride-co-hexafluoropropylene))與 / 電解液混 合後,加熱使電解液中的溶劑揮發, / 與聚合物結合而形成膠態的 / 電解質。其 大幅改善了液態電解質在化學及熱穩定性的缺點,延長元件的使用壽命,其元件效率可
15
達 ~6 %;同年, Grätzel [44]又將鈷錯合物之氧化還原對(CoII
/ CoIII)應用於 DSSC 中,
其光電轉換效率在較低強度的光照下可達 8 %。另外,高穩定性之固態 DSSC 元件在 後續研究中,利用有機電洞傳導媒介 2,2′-7,7′-tetrakis(N,N-di-p-methoxyphenylamine) 9,9′-spirobifluorene (spiro-MeOTAD)作為固態電解質,其結構如圖 1. 7 所示。MeOTAD
搭配有機染料 indoline,可以成功地將電解質的電位往正的方向修正至比 / 高出 450 mV [45]。 圖 1. 7 MeOTAD 結構圖[45]。 欲發展高速電荷轉移的 DSSC 元件,應考慮更多各種不同的氧化還原對或添加劑。 至今成功改良的電解液研究中,皆降低了電解質的氧化還原能力。電解質於未來進一步 的發展,則以高穩定性的固態電洞傳輸層最引人注目[43]。 1.1.3.4. 半導體層 染料敏化太陽能電池陽極的半導體層,主要的功能為接收染料激發態電子、將電子 傳至外部電路。為了使這個電子的傳遞過程更為順利,必頇考慮到半導體的導帶能階與 光敏染料的激發態能階之間的搭配。另外,為了減少電子於半導體層中發生電荷重組的 機會,此材料應具有較寬的能隙。由於二氧化鈦(TiO2)具有較寬能隙(3.2 eV),且可與 Ru-polypyridyl 染料的能階匹配,因此被廣泛地使用作為 染料敏化太陽能電池的陽極材 料。各種陽極半導體能階如圖 1. 8 所示,圖中氧化鋅 (ZnO)與 TiO2相同具有寬能帶; 雖然 ZnO 也可作為陽極材料,但其化學穩定性較差(例如:不耐酸性溶液),因此,大部 分的陽極材料還是以 TiO2為主。
16 圖 1. 8 DSSC 陽極半導體材料之能階圖[46]。 TiO2材料的形貌上,最早是以奈米粒 (nanoparticles, NPs)薄膜作為電子傳輸層[36]。 二氧化鈦奈米粒為最早發展也是至今研究最為廣泛的 TiO2型態,具有高孔隙率以及較 高的比表面積,可增加光敏染料的吸附量。目前以 二氧化鈦奈米粒為主要陽極材料的 染料敏化太陽能電池發展至今,DSSC 的光電轉換效率(η)最高已達 11 %[21, 22, 47-49]。 然而,在 NP-DSSC 中電子在工作電極的傳遞及擴散速度已幾乎達到極限,使得其元件 之效率至今始終無法進一步的突破。 圖 1. 9 典型 NP-DSSC 示意圖[50]。
17 科學家們認為電池效率之所以無法突破,主要的原因之一在於 TiO2 NP 為零維結構, 如圖 1. 9 所示,電子在此半導體層粒子間的擴散為散亂無方向性 (random work),因而 降低了電子在此材料中的傳遞速度[42, 51-54],一般預估此一電荷收集的步驟需要 10-4~10-3秒才能完成,因此,在陽極半導體層的材料設計上,必頇提供電子一個直接且 方向明確的傳輸途徑。 為了增加工作電極的電荷收集效率,以提升電子在半導體的傳遞速度,進而減少染 料電荷重組以及電子在傳輸過程中被電解質攔截的機會。因此,針對電子在工作電極的 傳遞方面,研究者陸續於 DSSC 的工作電極導入一維 TiO2奈米結構,如奈米線(wire)[51]、
奈米管(tube)[50, 52-54]、奈米棒(rod)[55, 56]及奈米纖維 (fiber)[56]等結構,如圖 1. 10 所示,即是提供電子一個單一方向性的傳輸路徑。
圖 1. 10 一維 TiO2 奈米結構; (a) wire[51]、(b) tube[50]、(c) rod[56]與(d) fiber[56]。 文獻上製備一維 TNT 陣列有許多方式,如最早發展的模版製造(template replica)[57]、溶膠−凝膠(sol-gel)[58]、水熱法(hydrothermal processes)[59]與陽極處理
18 (anodization)[53, 54, 60-65]等方法。其中以低設備成本及簡易製程的陽極處理法最普遍, 所以具有極高的發展潛力[62-65]。 隨著陽極處理法技術的提昇使 TNT 薄膜的開發日趨成熟,這也促使 NT-DSSC 的研 究在學界受到普遍的重視。文獻上以陽極處理法製備垂直有序的 TNT 陣列有以下數: (1) 從 2001 年 Grimes 等人[66]開始,以 0.5 wt.%氫氟酸(HF)水溶液為陽極處理電解液, 在反應溫度 18 ℃下施加 20 V 工作電壓,反應 20 min 後,可於純鈦板上得到 250 nm 長的 TNT 陣列;(2) Schmuki 團隊在 2003 年[67]於 1 cm2作用面積的 Ti 片上,利用 1 M 硫酸(H2SO4)水溶液加入幾滴 HF 以及 20 V、24 h,得到 580 nm 的 TNT 陣列。以 上所製備之 TNT 管長皆小於 1 μm,這是因為 TNT 管口處的 pH 值會有很大的變化(氟 離子聚集),而增加了 TNT 上端溶解速率[68];因此(3) Schmuki 於 2005 年的 Angew.
Chem. Int. Ed. [69]發表中,選擇了黏度較高的甘油(Grycerol)作為溶劑並加入 0.5 wt.%
NH4F、20 V 工作電壓以及 1 Vs-1的升壓速率,可得到較長且外觀帄滑的 TNT 陣列(7 μm), 如圖 1. 11 所示。 圖 1. 11 陽極處理 TNT 之 SEM 顯微結構:(a) 1M H2SO4/ 0.15 wt.% HF 水溶液 [68];(b) 0.5 wt.% NH4F/ Grycerol 電解液[69]。 此外,Grimes 率先於 2006 年[70]成功將 6 μm 長之 TNT 陣列應用於 DSSC 陽極 材料,其元件結構如圖 1. 12 所示。其光陽極 作用面積為 0.4 cm2,吸附 N719 染料後 之背照式 NT-DSSC 元件效率為 4.24 %。而此 TNT 使用之陽極處理電解液使用 0.1 M
19 鈉(NaOH)將電解液調至 pH = 5,施加 25 V 電壓,成長時間為 17 h。 圖 1. 12 背照式 NT-DSSC 元件示意圖[70]。 其他應用於 DSSC 的 TNT 陣列有:(1) Frank 的研究團隊[50]利用 0.5 wt.% NH4F/ glycerol 為陽極處理電解液,施加工作電壓 20 V,經過 70 h 得到 5.7 μm 管長之 TNT, 其光陽極(1 cm2 )經吸附 N719 染料後之元件效率為 3 %;(2) Grimes 及其研究團隊[62] 使用 8 wt.% 四丁基氟化銨(tetra-butyl ammonium fluoride, TBAF)/ 甲醯胺(formamide) 為電解液,經過 20 V 工作電壓、反應時間 24 h 而得到長度為 20 μm 之 TNT,經表面 粗糙化 TNT 陽極(1 cm2 )吸附 N719 染料後之元件效率為 6.89 %;(3) 本實驗室[63]利用 0.5 wt.% NH4F / EG 電解液、 60 V 工作電壓,可於 3 h 得到 19 μm 之 TNT,其 0.28 cm2 作用面積之光陽極經過表面粗糙化以及吸附 N719 染料,其 NT-DSSC 元件可得到 7 % 的之元件效率。 我們參考已發表的文獻得知,陽極處理製備 TNT 陣列之表面型態,會因為各種陽 極處理條件參數而有所不同[60]。例如,TNT 的形貌會因不同電解液而改變,目前陽極 處理製備 TNT 電解液的使用,最常見的以含氟離子為基底的電解液為主;TNT 管內徑 隨著陽極處理電壓的增加而越大。其他如陽極處理環境溫度、電解液含水量或電解液 pH 值等,皆會造成不同 TNT 的成長結果;另外, TNT 管長通常會隨著電解液離子濃度 或陽極處理時間的增加而加長,但亦可能因電解液離子濃度太高而使 TNT 管長不一定 有正向的結果。那是因為 TNT 反應過程中,由於水的電解在鈦陽極產生氧氣,並於鈦
20
陽極表面進行氧化反應而生成緻密的 TiO2氧化膜,同時電解液中的氟離子會對此氧化
膜進行蝕刻。因此,陽極處理過程中包括 Ti 金屬/ TiO2 (metal/ oxide, MO)界面為電化學
成長反應(electrochemical growth process)、TiO2/電解液(oxide/ electrolyte, OE)界面為化學
溶解反應(chemical discussion process)同時進行,如圖 1. 13 所示。當成長反應的速度高 於溶解反應,則 TNT 管長可隨反應時間而增加,反之則無法成功製備有序之 TNT。因 此氧化膜的形成與電解液選擇、環境溫度、電壓、電流、pH 值和成長時間等因素有密 切的關係。 圖 1. 13 陽極處理法製備 TNT 陣列之電化學過程競爭反應示意圖。 1.1.4. 電子在二氧化鈦上傳遞的相關研究 從章節 1.1.2 中我們可以發現,在電子正向傳遞的過程中,最慢的過程為電子在二 氧化鈦上的擴散,因此影響電池效能的最大因素便在於電子在半導體層上的傳遞速度 (傳遞時間等級是 10-4~10-3秒)。此外,DSSC 陽極材料的多孔性使得材料的比表面積極 大,擴散於孔洞間的電解液與陽極的接觸面積極大,使得電解液自半導體層上攔截電子 的速率也影響整體電池效率。 1.1.4.1. 電子在二氧化鈦上的傳遞原理 電子傳遞過半導體層可以描述成連續性方程式(continuity equation)[71] (1. 10)
21
其中 n 是光照下的電子密度,J 是半導體薄膜上的電流密度,G 是電子的產生速率,而
R 是電子的再結合速率。在均勻吸附染料的多孔性二氧化鈦薄膜上而言,產生速路可以
被表示成 G=ainj I0exp(-ax),其中 I0是光通量,a 是染料敏化薄膜上波長相關的吸收係
數,inj是電子從激發態的染料注入二氧化鈦的效率。再結合速率我們假設與電子濃度 成一次相關,因此表示成 R=(n-n0)/t0,其中 n0是在非照光下的電子密度,t0是與位置無 關電子生命期。電子在半導體層上的漂移與擴散都對電流密度有所貢獻, (1. 11) 其中n是電子遷移率,是薄膜上的電場,而 D 是電子的擴散係數。因為奈米級的粒子 尺寸太小以致於無法形成明顯的電場,所以電子在奈米級粒子構成的半導體層上的傳遞 主要來自於電子的化學位能的梯度(擴散),而非電場梯度(漂移)。從能量學的觀點而言, 電子傳輸主要是電子的準費米能階梯度造成的[72]。如此一來。時間相關的電子傳遞方 程式可以從式(1. 11)帶入式(1. 10)得到。在忽略了電子的漂移後,連續性方程式變成為 (1. 12) 左邊項代表電子濃度變化與時間的關係,右邊第一項代表電子流量,第二項代表再結合 速率,第三項代表從光激過程得到的電子注入的速率。式(1. 12)以變數分離(separation of variables)與傅立葉轉換(Fourier transformation)分析後[72],我們可以發現穩定態的光電 流與光強度成正比,與實驗結果相同[73],但另一方面也表現出時間常數光強無關,這 與實驗結果不合[72],因此表示式(1. 12)無法充分的描述電子在半導體層的瞬態表現。 此外,多孔性二氧化鈦薄膜上電子的擴散係數與在單晶二氧化鈦導帶上並不相符。根據 電子的移動速率 1cm2 V-1 s-1,從愛因斯坦方程式(Einstein equation)Dn=nkBT/q 得到在室 溫下的電子擴散速率級數[74]約在 10-2 cm2s-1,遠大於實驗結果。
22 圖 1. 14 DSSC 中電子在傳遞過程中重要的參數與路徑示意圖[75]。 我們已知在非晶相(amorphous)與無序(disordered)導體中,電子被局限(trap)置局限態 會導致傳遞速路的降低[76, 77]。這些材料具有高密度局限態的特徵,導致電子傳遞主要 受局限態的性質主導。局限態分布在很寬的能量範圍,因此電子的移動速率會根據局限 態的填充情況(費米能階),也就是電子密度改變,如圖 1. 14。因此式(1. 12)可以改寫成 (1. 13) 其中 ncb(x)是在位置 x 時,在導帶的電子密度,Dn是自由電子(未被陷住)擴散係數。右 邊第三項表示電子被局限的速率,右邊第四項是電子經由熱發射從局限態回到導帶,電 子與電解液的再結合速率即為這兩項的差值。 在短路電路時,在位置 x 穩定態自由電子濃度可從式(1. 13)得到: = (1. 14) 其中邊界條件為在二氧化鈦與電解液的界面沒有電子進入或離開(Jn(d)=0)。在無照光 (EF0)與照光(EF)下的費米能階與電子密度的關係表示成 (1. 15)
23 成一次指數型式: (1. 16) 其中 Nss0是在 EF0時的表面態密度,而 mc代表表面態分布曲線的斜率,也代表帄均局限 態的深度。因為再結合的速率遠慢於局限和去局限(detrap)[78],因此電子在局限態的準 費米能階和 EF相同。在局限態的電子密度 nt(x)可以從自從 EF0到 EF積分式(1. 16)得到 (1. 17) 其中 k 為波茲曼常數(Boltzmann constant, 1.38×10-23 J K-1,T 為絕對溫度。事實上,ncb(x) >> ncb(0),表示 ncb(x)與 nt(x)間以 ncb(x)nt(x)mc/kT的關係呈現正相關。因為被局限的電子 密度遠大於自由電子的電子密度[78, 81],電子濃度 n (x)= ncb(x)+ nt(x)~nt(x),因此 ncb(x) 與 n(x)之間的關係可表示為 (1. 18) 因為 n (x)~nt(x),所以薄膜上的電子束 Q~Qt可以從式(1. 17)積分得到。將式(1. 15)帶入 式(1. 17)同時維持在短路電路時 ncb(x) >> ncb(0)的假設,我們可以得到 (1. 19) 從式(1. 19)我們可以發現 (1. 20) 以及短路電流密度(Jsc)正比於光通量,我們可以得到 (1. 21) 式(1. 21)表示由於表面態與能量的一次指數關係,使得短路電流與薄膜上的電子數間有 power-law 的關係。同時因為 Q=JSC,而為將在侷限態內的所有電子釋放出來所需的時
24 間,因此我們可以得到 (1. 22) 其中 (1. 23) 1.1.4.2. 電子在二氧化鈦上傳遞速率的測量以及分析方法
常見的測量方法有電化學阻抗分析圖譜(Electrochemical Impedance Spectroscopy, EIS)、強度調製光電流/光電壓分析圖譜(Intensity Modulated Photocurrent Spectroscopy , IMPS、Intensity Modulated Photovoltage Spectroscopy , IMVS)與瞬態光電流/光電壓測量 (Transient Photocurrent/Photovoltage Measure)。本篇論文中使用的方法為接為瞬態光電流 /光電壓測量,因此在此介紹文獻中常用的儀器設計。
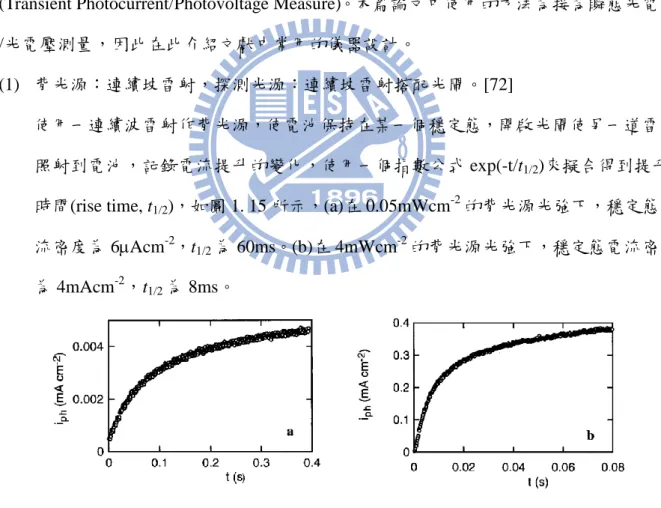
(1) 背光源:連續坡雷射,探測光源:連續坡雷射搭配光閘。[72]
使用一連續波雷射作背光源,使電池保持在某一個穩定態,開啟光閘使另一道雷射
照射到電池,記錄電流提升的變化,使用一個指數公式 exp(-t/t1/2)來擬合得到提升
時間(rise time, t1/2),如圖 1. 15 所示,(a)在 0.05mWcm-2的背光源光強下,穩定態電
流密度為 6Acm-2,t1/2為 60ms。(b)在 4mWcm-2的背光源光強下,穩定態電流密度 為 4mAcm-2,t 1/2為 8ms。 圖 1. 15 波長 514nm 探測雷射照射到電池後電池電流的提升情形。 (2) 背光源:連續坡雷射,探測光源:脈衝式雷射。[82] 使用一連續坡雷射作背光源,使電池保持在某一個穩定態,在以脈衝雷射對電池進
25
行微擾。在文獻中背光源波長選擇在染料的強吸光區與弱吸光區皆有,選擇弱吸光 區是想避免染料吸光能力太強而使得光無法穿透整個陽極材料,導致靠近背光測的 染料無法進行激發,因此選擇強吸光區則要注意雷射光源強度是否足夠。由於脈衝 雷射的脈衝寬度只有 5~10ns,遠快於 DSSC 電流的傳遞,因此我們可以在瞬態電流 變化圖看到完整的擴散曲線,並使用由 Fick’s second law 推導得到的擴散方程式[83] (1. 24)來擬合,如圖 1. 16: (1. 24) 其中 q 為電子單位電量,d 為二氧化鈦薄膜厚度,N 為受激發的電子數。 圖 1. 16 以脈衝雷射微擾所得到的瞬帶電流變化圖,虛線是 以擴散方成是擬合的結果,所得的擴散係數分別是(a)10、 (b)3.2、(c)4.0×10-5 cm2s-1。[84] 瞬態電壓變化使用一個指數公式 exp(-t/)來擬合,得到電子生命期(lifetime, )。 (3) 光源:輸出電壓可調式雷射。[85] 此方法所使用的雷射可輕微調整供應給雷射的電壓。維持相同電壓一段時間等到電 池達到穩定態,輕微減少供應的電壓,使雷射的強度有稍許的下降,造成電流/電壓 的變動。
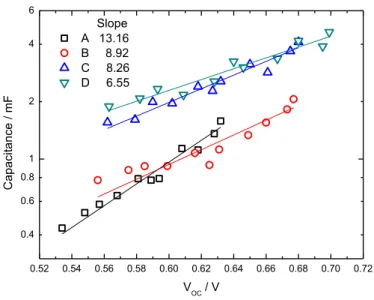
26 圖 1. 17(a),再以 (1. 25) 換算得到擴散係數。 瞬態電壓變化的分析方法與上一個實驗方式修同,如圖 1. 17(b)。 圖 1. 17 使用輸出電壓可調式雷射所得到的(a)瞬態光電流變化圖,(b) 瞬態 光電壓變化圖。 1.1.4.3. 二氧化鈦上化學電容的研究 DSSC 的電容通常有數種方式決定:EIS[75, 86, 87]、循環伏安法(cyclic voltammetry)[88]、積分在不同電壓下的電流[89],以提供能量狀態密度(Density of States, DOS)和電子密度。 通常在教科書中有關電容的概念為靜電電容,也就是兩個有等量且性質相反電荷的 帄行金屬板中的電場。但是這個敘述只有在當電位差引起的過多的電荷被限制在帄行板 表面中非常小的區域的時候有效,特別是在介觀(mesoscopic)系統中的情形就與一般觀 念相去甚遠。一般而言,電子儲存槽的介觀帄板電化學位能變化,導致過量電荷造成的 電場的調視和帄板上的導帶的費米能階位置的變化。因此,電化學電容被表示成與 DOS 相關 (1. 26)
27
其中 dN/dE 是薄膜的所有 DOS[90]。當 dN/dE 非常大時,就可視為一個金屬膜電容而忽
略掉式(1. 26)。將 DSSC 的組態以此方式表現如圖。當電子的費米能階在i,電化學位 能的變化為 dV,並假設導帶能階(Ec)與電解液氧化還原能階保持固定時,奈米結構二氧 化鈦電極行為的示意圖如圖 1. 18。TCO/二氧化鈦介面因吸光而位能變化。二氧化鈦內 的奈米結構中,費米能階的表現傾向於導帶。電子的化學位能變化 dn導致導帶 dnc和 被局限電子的定域能階(localized level) dnL的能階填充改變。 圖 1. 18 奈米結構二氧化鈦電極行為的電子能量示意圖[91]。 此外,從一般熱力學條件獲得的化學電容被視為理想電容元件。若體積單元儲存化 學能是由於熱力學位移,則單位體積下的化學電容為 (1. 27) 這其實是式(1. 26)的另一種寫法,但這代表了此電化學電容考慮了在電子密度 Ni下,因 化學位能i的改變造成的電容接受或釋出外加電子的能力。因為在第 i 個部份的化學位 能是 (1. 28) 的理想狀態,其中 kB 是波茲曼常數而 T 是溫度,因此從式(1. 27)我們可以得到
28 (1. 29) 這表示當熱能改變時,能量與可逆的方式儲存在化學電容內。 因此,將導帶的電子密度以波茲曼分布的形式敘述 (1. 30) 其中 Nc 是導帶態的有效密度,可以將式(1. 27)轉變成 (1. 31) 圖中的總化學電容為將式(1. 31)對整個薄膜厚度積分。 考慮到圖中有自由電子與定域電子的改變,我們將此 DSSC 中的化學電容改寫成 (1. 32) 其中 (1. 33) 在此 g(E)是當能量為 E 時,在能隙定域態的密度,並已 Tc取代 T,其中(tailing parameter)=T/Tc,我們可得到 (1. 34) 從將式(1. 34)代入式(1. 33)後可得到化學電容表式為 (1. 35) 結果,式(1. 31)與式(1. 35)顯示了奈米結構二氧化鈦中的化學電容與背光電壓間呈現一 次指數關係。但從式(1. 31)得到的(dlnC/dV) -1的理論斜率(e/kBT)-1 =0.026 V decade-1無法 從實驗中得到[75, 88]。實驗結果為(dlnC/dV) -1 =0.100 V decade-1,表示被局限的電子的 電容表現公式式(1. 34)在實驗結果中佔有較公式(1. 31)更多比例。
29 圖 1. 19 化學電容實驗與擬合結果[91]。 從圖 1. 19 中我們可以發現在較高位能的區域,實驗數據與擬合結果相符合,但在 較低位能區則實驗數據與擬合結果差異較大,顯示二氧化鈦的真實電容與位能關係,在 費米能階較靠近導帶時,會如理論所式呈現指數關係,但費米能階遠離導帶時,電容與 位能關係曲線會有扭曲的現象。
1.2. 研究動機
從文獻中發現[92], DSSC 陽極材料若使用傳統的二氧化鈦奈米粒或奈米線時,起 初 DSSC 的短路電流與效率會隨著薄膜厚度的增加而提高,但超過某個臨界值時,隨著 薄膜厚度的持續增加,DSSC 的短路電流與效率呈現大幅度的下降,如圖 1. 20 所示。經 由進一步實驗發現,當薄膜厚度超過這些臨界值時,隨然染料吸附量隨著薄膜厚度的增 加而提升,但電子在二氧化態上的擴散速率大幅下降,如圖 1. 21 所示,導致 DSSC 表 現出來的短路電流與效率大幅下降。30 圖 1. 20 不同奈米粒與奈米線比例的 DSSC 短路電流及效率與薄膜厚度的關係圖。 圖 1. 21 不同奈米粒與奈米線比例的 DSSC 擴散係數與薄膜厚度的關係圖。 而我們實驗室以二氧化鈦奈米管做為陽極材料的 DSSC,當薄膜厚度增加至接近 60 m 時,短路電流與效率依然隨著薄膜厚度的增加而上升,如圖 1. 22 所示。在我們的實 驗條件範圍內並沒有發現與奈米粒與奈米線類似的臨界值,因此我們想要了解當我們使
31 用二氧化鈦奈米管做為陽極材料時,除了染料的吸附量隨著薄膜厚度的增加而上升之外, 電子在二氧化態上的擴散速率是否也能對短路電流與效率的提升有所貢獻。 100 200 300 8 10 12 14 10 20 30 40 50 60 4.5 5.0 5.5 6.0 D ye lo a d in g / n mo l cm -2 JSC / mA cm -2 / % ATO Length / m 圖 1. 22 ATO-DSSC 短路電流及效率與薄膜厚度的關係圖。
32
第二章 實驗
2.1. 實驗儀器設備
(1) 電源供應器 本研究使用必穎科技之可程式控制電源供應器進行陽極處理反應,其型號為 BPW-20006/ 3002,輸出電壓為 200 V DC、脈衝±30 V DC。(2) 恆溫水槽 (Temperature controlled container)
恆溫水槽的溫度範圍可控制在−10 ℃至50 ℃,廠商為健升儀器有限公司,型號: JS-210H,電源電壓為110 V/ 14 A。
(3) 高溫爐 (Muffle Furnaces)
研究中使用之高溫爐為三杰電機所組裝,其型號為MF-20,加熱空間為20×20×20 cm3,
溫度極限達1200 ℃。
(4) 掃描式電子顯微鏡 (Scanning Electron Microscope, SEM)
顯微結構的觀察使用本校貴儀中心提供的日本捷東(JEOL)熱場發掃描式電子顯微 鏡,型號JSM-6500F,其中燈絲為HITACHI-S2500 熱離子發射式燈絲。 (5) 旋轉塗佈機 (Spin coater) DSSC中對電極表面需塗佈一層鉑,將利用琦太企內徑26 cm 規格的旋轉塗佈機來 完成。 (6) 太陽光源模擬器 (Solar simulator) DSSC的光電流測定使用SAN-EI製造,型號為XES-502S之AM-1.5G太陽光模擬器, 並利用矽標準參考電池(VLSI standards, Oriel PN 91150V)將光源校正為一個太陽光
強度(100 mW cm−2)。
(7) 光電轉換效率光譜儀 (Incident photo-to-current conversion efficiency, IPCE)
本研究中的IPCE量測為實驗室自行架設之量測系統[1],其配備PTi 公司所製型號 A-1010之氙燈(Xe lamp, 150 W)燈源以及Dongwoo公司之DM150i單光儀
33
(Monochromator, 1200 gr mm−1 blazed at 500 nm)。
(8) 數位電源電錶 (digital source meter)
DSSC元件光電流的檢測(IV及IPCE)皆搭配程式控制之Keithley 2400 數位電源電錶, 用於量測元件之電流−電壓值,其最大功率為20 W。 (9) 紫外-可見光光譜儀 (UV-visible spectrometer) 本研究中染料吸附量的檢測使用Varian公司所製,型號Cary-50紫外−可見光光譜儀, 掃描波長範圍為190 nm至1100 nm。 (10) 連續波雷射 StockerYale 公司所生產之二極體雷射。分別 10 mW 的 532 nm 雷射與 10 mW 的 635 nm 雷射 (11) Nd:YAG 雷射 EKSPLA 公司所生產,型號為 NT342/1/UV 的波長可調式脈衝雷射。 (12) 電流放大器
Stanford Research Systems, Inc.所生產的低雜訊電流放大器(Low-noise current preamplifier),型號為 SR570。
(13) 電壓放大器
Stanford Research Systems, Inc.所生產的低雜訊放大器(Low-noise preamplifier),型號 為 SR560。 (14) 示波器 LeCroy 公司所生產的示波器,型號為 LT372,500 MHz,4 GS/s。 (15) 粗糙度儀 Veeco 公司所生產的表面粗度儀,型號為 Dektak 150。 (16) 光強度測量儀
34
OP-2VIS。
(17) 脈衝產生器(Pulse generator)
Stanford Research Systems, Inc.所生產的四通道數位延遲/脈衝產生器(Four channel digital delay/pulse generator),型號為 DG535。
(18) 電控光閘
ThorLabsInc 所生產的電控光閘,控制器型號為 SC10。
2.2. 陽極處理製備二氧化鈦奈米管
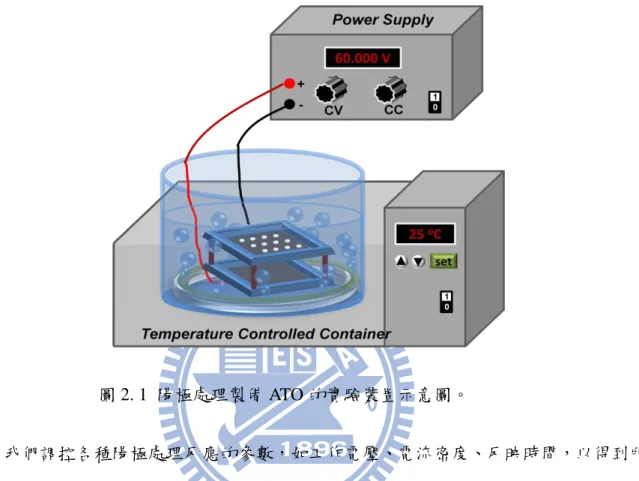
2.2.1. 鈦基材前處理
遠用厚度約 150m 的純鈦片(純度 99.9% Grade 1,Kobe steel),鈦基材前處理步驟
如下: (1) 以丙酮沖洗鈦片表面。 (2) 將鈦片以鋁箔紙包覆,盡可能隔絕空氣,置於兩片帄整表片的不銹鋼板之間,並 在上方重壓 5 公斤,於高溫爐內以 580℃,3.5 小時的條件進行退火熱處理 (Annealing)。 (3) 以 240 號(#240)砂紙均勻磨除氧化層。 (4) 使用 17% 的氫氟酸水溶液蝕刻鈦片表面。 (5) 將蝕刻後的鈦片利用超音波震盪器清洗 5 分鐘,再以丙酮沖洗鈦片表面。 2.2.2. 以陽極處理法制被二氧化鈦奈米管陣列 我們前處理完成的鈦片至於模具內並放入電解槽中進行陽極處理(Anodization)來製 備二氧化鈦陽極處理膜(Anodic titanium oxide, ATO),如選用適合的電解質及工作電壓,
可於鈦金屬表面形成二氧化鈦奈米管。在本篇論文中,我們選擇以氟化胺(NH4F)為主,
溶於乙二醇(, EG)以及 2 vol.%去離子水(DI water),做為電化學反應的電解液,其配方為
35 的鈦片,陰陽兩極的距離為 2.7 公分,置入模具及曝氣裝置放入電解液中,以恆溫水槽 控制電化學反應的環境溫度(25℃),實驗裝置如圖 2. 1 所示。鈦片經陽極處理後的 ATO 從模具中取出,並以乙醇沖洗後進行後處理。 圖 2. 1 陽極處理製備 ATO 的實驗裝置示意圖。 我們調控各種陽極處理反應的參數,如工作電壓、電流密度、反映時間,以得到所 需的 ATO 厚度。本篇論文所使用的陽極處理參數分別為兩種型式:定電壓法
(Potentiostatic Method)與混合法(Hyberid Anodic Method)。
36 (1) 定電壓法 電壓以 4V / min 的上升速度將電壓升高至 60V,並維持固定電壓數小時,其示意圖 如圖 2. 2 所示。陽極處理時間與 ATO 厚度的關係如圖 2. 4 所示。 圖 2. 3 陽極處理混合法之電壓與電流密度參數控制示意圖。 (2) 混合法 電壓以以 4V / min 的上升速度將電壓升高至 60V,並維持固定電壓 1 小時候,轉換 電流密度至 5.6mAcm-2,並維持固定電流密度數小時,其示意圖如圖 2. 3 所示。。 陽極處理時間與厚度關係圖如圖 2. 4 所示。 0 1 2 3 4 5 6 7 8 0 10 20 30 40 50 60 Slope = 10.7 m h-1 Hybrid-5.6 mA cm-2 Potentiostatic-60 V L / m t /h 圖 2. 4 以定電壓法進行陽極處理之陽極處理時間與 ATO 厚度的關係圖。
37 2.2.3. 二氧化鈦奈米管之後處理 陽極處理使鈦上成長出二氧化鈦奈米管陣列,此的二氧化鈦奈米管為非晶相,因此 需要精果高溫燒結使其具有結晶相。本實驗的熱處理溫度為 460℃,升溫條件如圖 2. 5 二 氧化鈦奈米管燒結升溫條件: 圖 2. 5 二氧化鈦奈米管燒結升溫條件 二氧化鈦奈米管陣列經過高溫燒結後,由於表面具有緻密層,因此將試片浸入乙醇 並置於超音波震盪器內以最小功率震盪 15 分鐘,將表面清除,最後以乙醇洗淨烘乾。 二氧化鈦奈米管陣列的緻密層清除程度及其表面形貌(如孔徑大小)將利用 SEM 鑑定, 奈米管陣列的厚度利用表面粗糙度儀(-step)測量。
2.3. 鉑電極製備
本研究使用 TCP 法製備染料敏化太陽能電池的鉑對電極,步驟如下: (1) 將 FTO 玻璃裁切成 1×2cm2的大小後, (2) 氯鉑酸溶液配置:2mg H2PtCl6˙6H2O/1ml isopropanol。 (3) 旋轉塗佈:將 H2PtCl6/isopropanol 溶液以 28l cm-2的量均勻滴於 FTO 導電面上, 以 2000 轉(rpm)的轉速進行 10 秒的旋轉塗佈。 (4) 鉑還原:以 360℃將塗佈氯鉑酸溶液的 FTO 玻璃燒烤 15 分鐘,使氯鉑酸溶液還原38 成白金並附著於 FTO 上。 (5) 重複步驟(3)、(4)一次,以增加 FTO 表面觸媒層的量及粗糙度。
2.4. 電解液製備
論文中使用配方編號為 F’的 I -/I3-離子液體配方,如表 2. 1。 表 2. 1 二氧化鈦奈米管染料敏化太陽能電池效率測量之電解液配方 F’藥品 LiI I2 TBP BMII GuNCS CH3CN n-C4H9CN
濃度 0.1M 0.01M 0.5M 0.6M 0.1M 85:15
2.5. 光敏染料吸附量之檢測
我們為每個條件準備兩片相同的二氧化鈦奈米管試片,同時浸泡於同一瓶染料相同 時間。取出試片後以變性酒精將電極尚未吸附的染料清除,以吹風機烘乾。其中一片封 裝程元件進行量測,另一片進行染料吸附量測量。 在染料吸附量檢測方面,選擇 0.1M NaOH 水溶液將染料自二氧化鈦奈米管上脫附, 再測量溶液得到紫外光-可見光光譜,並把 NaOH 水溶液的吸收度當作基準線(baseline)。 染料 N719 在 0.1M NaOH 水溶液中的莫耳吸收係數(),在波長 500nm 的位置約為 10690M-1cm-1。因此,若使用 1ml 0.1M NaOH 水溶液脫附面積 0.16 帄方公分的奈米管上 的染料,並使用 0.2 公分的石英槽(cuvette)來測量吸收度,則根據比爾定律(Bill’s Law) (2. 1) 其中 A 代表吸收度;代表莫耳吸收係數,單位為 M-1 cm-1;b 代表光徑,單位是 cm;c 代表溶液濃度,單位是 M。由於 N719 在水溶液中的莫耳吸收係數在 500nm 時為 10690 M-1cm-1,此時比爾定律可寫成 (2. 2) 奈米管的染料吸附量可寫成39 (2. 3)
2.6. 測量
2.6.1. 電流-電壓特性曲線(I-V curve) 首先在一個太陽光強度(100 mW cm-2 )的太陽光源模擬器(solar simulator)下得到 DSSC 的最大開路電壓後,施予 DSSC 一相等負載電壓,此時電流值為 0,逐次遞減所 施予的負載電壓直至負載電壓為 0,並記錄對應的電流值,最後可得到一完整的電流-電壓特性曲線(I-V Curve)。 在電流-電壓特性曲線中,電流為 0 時的電壓稱為開路電壓(Open-circuit voltage, VOC), 電壓為 0 時的電流稱為短路電流(Short-circuit current, ISC)。電流-電壓特性曲線中每一組 相對應的電壓與電流相乘可得到相對應的功率值,能提供最大的功率值的電壓與電流分 別表示為 Vmp與 Imp。此時 DSSC 所能提供的最大功率與理論上的輸出功率的比值,即 稱為填充率(Fill factor, FF),可表示為: (2. 4) 而電池的光電轉換效率(conversion efficiency, )可表示為: (2. 5) 其中 Pin為 DSSC 所照射到的光強度,即太陽光強度(100 mW cm-2)乘以 DSSC 的面積。2.6.2. 光電轉換效率(Incident photon-to-current conversion efficiency, IPCE)
以 氙 燈 (Xe) 為 光 源 , 經 過 單 光 儀 (Monochromator) 色 散 出 不 同 波 長 的 光 , 在 350~800nm 的範圍內每隔 10 nm 測量在該波長下光的能量以及 DSSC 的電流值,經式(2. 6)換算可得到不同波長時的光電轉換效率。
![圖 1. 1 染料敏化太陽能電池工作原理示意圖。[15]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8239397.171272/18.892.273.658.120.469/圖11染料敏化太陽能電池工作原理示意圖15.webp)
![圖 1. 3 以 Ru 金屬為中心的撮合物染料結構,(a)為 N3,(b)為 N719,(c)為 black dye。TBA= tert-butylammonium。[20]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8239397.171272/22.892.162.793.486.993/圖1金屬為中心的撮合物染料結構a為N3b為N719c為blackdyeTBA=tertbutylammonium.webp)
![圖 1. 10 一維 TiO2 奈米結構; (a) wire[51]、(b) tube[50]、(c) rod[56]與(d) fiber[56]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/8239397.171272/29.892.130.750.495.992/圖11一維TiO2奈米結構awire51btube5crod56與dfiber56.webp)