行政院國家科學委員會專題研究計畫 成果報告
以超臨界流體萃取及逆流層析技術分離親水性物質(3/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2113-M-009-020- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立交通大學應用化學系(所) 計畫主持人: 余艇 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 94 年 10 月 30 日
i 中文摘要 關鍵詞:超臨界流體,冷媒,鈾,核廢料,萃取,逆流層析,蛋白質,親水性物質, 介面活性劑,反微胞 本三年期研究計畫之計劃題目為:以超臨界流體及逆流層析技術分離親水性物質,可分 為兩個主題,因此結案報告也將分兩個主題來報告,亦即超臨界流體之萃取,和逆流層析之 分離研究。以下各章都分為(甲)(乙)兩部分書寫。 (甲)於核能發電工業中,廢燃料棒中鈾及鈽金屬之回收再利用,是核能工業上主要研 究項目之一。處理核燃料棒一般需要大量之有機溶劑以及硝酸,因而會產生大量之廢液。目 前,已有研究利用超臨界二氧化碳(SF-CO2)來取代傳統有機溶劑來進行萃取,萃取完成後, 二氧化碳洩壓即可氣化而與萃取物分離,達到濃縮效果而且無需處理有機廢溶劑;文獻指出 為了減少大量濃硝酸使用以及避免硝酸直接接觸到燃料棒樣品,於前處理時先將螯合劑 TBP (Tributylphosphate)與硝酸結合形成錯合物,並溶解於 SF-CO2直接進行固相基質鈾金屬萃 取。 本實驗嘗試以液態冷媒 1,1,1,2-tetrafluoroethane(R134a)來取代 SF-CO2,其所需壓力比 使用 SF-CO2降低約 100 個大氣壓以上,且其有類似 SF-CO2經壓縮之後具有溶解度之性質, 於操作安全考量上優於 SF-CO2。前處理時亦先可將螯合劑 TBP 與硝酸結合形成錯合物,並 溶解於 R134a 直接進行固相基質鈾金屬萃取,藉由此法亦可避免直接使用濃硝酸進行鈾金屬 萃取,安全性也可以大幅提升,能夠達到經濟、安全且高效率的回收。 (乙)逆流層析(countercurrent chromatography,CCC)是一種無固體靜相支持物的液相-液相層析技術,因此不會產生樣品被固體吸附,或是靜相被雜質阻塞的問題,適合生物分子 及天然物等複雜物質的分離。以反微胞相萃取並濃縮蛋白質分子的過程,包括了正向以及反 向萃取兩個步驟,我們在逆流層析中執行這兩個步驟,可以使得萃取率增加,並且達到同時 濃縮的目的。在實驗中,進行大體積樣品之濃縮,將 10.8 mg/540mL 的 cytochrome c,濃縮 為 10.1 mg/60mL,回收率為 93.2%,濃縮為原來體積的九分之一,使反微胞結合逆流層析的 技術,可能有實際應用的價值。 除了萃取之外,我們於逆流層析儀中使用反微胞相作為靜相,逆流層析管柱容積 28 mL, 使用含有 100 mM sodium bis(2-ethylhexyl) sulfosuccinate (AOT)的正己烷作為靜相,動相是酸 鹼值以及離子強度梯度變化的緩衝溶液,在動相流速 1mL/min 以及層析儀 800 rpm 轉速下, 靜相滯留於管柱中的量為 12 mL,分離 1 mL 含有肌紅蛋白、細胞色素 C、溶菌酶各 2 mg 的 蛋白質混合溶液,以不同的 pH 值以及鹽類濃度進行梯度冲提,得到逆流層析圖並每 5mL 收 集一次,經過 HPLC、MS 分析各收集管中的蛋白質含量,可得到蛋白質相對濃度 85 %的細 胞色素 C 蛋白質溶液,有效回收率約為 64 %,在製備分離蛋白質方面,提供一新的方法。
ii
英文摘要
Key Words: supercritical fluid, refrigerant, uranium, nuclear waste, extraction, countercurrent chromatography, protein, hydrophilic components, surfactant, reverse micelle.
(A) In nuclear power industry, the recycling of uranium and plutonium metal in the form of used nuclear fuel rod is an important research topic. Nuclear fuel rods are usually dissolved in concentrated nitric acid and extracted using chelate - containing organic solvent. Uranium and plutonium can be recovered efficiently in this process. However, a large amount of waste organic solvent must be reprocessed. Supercritical fluid carbon dioxide (SF-CO2) has been introduced recently to replace the organic solvent. SF-CO2evaporates after the extraction process and separates from the extracts. Thus, it can be used to save the endeavor to reprocess the extraction solvent. Researchers also tried to synthesize a complex using TBP with nitric acid first. The complex-containing SF-CO2 was found to be able to extraction uranium metal from solid waste samples. This technique can prevent the overuse of nitric acid and avoid direct contact between nitric acid and the fuel samples.
In this Study, we attempted to replace SF-CO2with liquid refrigerant 1,1,1,2-tetrafluoroethane (R134a) as the extraction liquid due to its much lower operational pressure, and the similar salvation power compared with SF-CO2. We followed the similar idea to synthesize the complex of TBP and nitric acid. This complex-containing R134a showed great capability in extracting uranium. In addition, we did extractions for a number of other metals that would be produced during the nuclear fission process. Except for La(III)、Gd(III) and Lu(III), recoveries of other metals were negligible. Therefore, using of R134a may provide a more economical and safer way to recover uranium from used nuclear fuel rods.
(B) Countercurrent chromatography (CCC) is a technique of liquid-liquid chromatography. This technique is suitable for preparative separations of biomolecules and natural products. It usually includes two steps, forward and backward extractions, to extract and concentrate proteins. We performed these two steps in CCC in order to improve extraction rate and to achieve concentration in the same time. We accomplished to concentrate a 10.8 mg in 540 mL cytochrome c aqueous solution to a 10.1 mg in 60 mL aqueous solution. The recovery was 93.2% with a volume reduction of one ninth to the original volume.
In addition to extraction of proteins, we used a two-phase solvent system, i.e. the aqueous (mobile) and reverse micelle (stationary) phases to separate protein mixtures in CCC. The stationary phase is a 100 mM sodium bis(2-ethylhexyl) sulfosuccinate (AOT) –containing n-hexane solution. At 800 rpm of a high-speed CCC, the volume of the stationary phase retained in the separation coil (total volume 28 mL) is 12 mL. The mobile phase was eluted at a flow rate of 1 mL/min using pH and ionic strength gradients. Sample solutions are composed of myoglobin, cytochrome c and lysozyme. The effluent was collected every 5 mL and analyzed by HPLC and Mass spectrometer. The result in the individual protein collector showed that the recovered purity for cytochrome c was 85% while the recovery yield reached 64 %. We expect that applications using CCC with reverse micelle phase may provide a useful method for protein separations and purifications.
iii
目錄
第一章前言
……….…….1
第二章研究目的
……….….…….2
第三章文獻探討
………..….3
第四章研究方法
………....5
第五章結果與討論
………..10
第六章文獻………19
第七章計畫自評
………..21
(一)前言 (甲)應用超臨界流體於分離科學已有三十多年的歷史,主要是利用此種流體類似液體 之溶解能力,和類似氣體之擴散速率的兩種特性,比一般液體更適用於萃取。由於二氧 化碳活性很低、無毒、臨界溫度較低、容易取得、不破壞環境等優點,因此在分離方面 的應用,大部分都是使用二氧化碳,雖然現在也可以看到使用水為超臨界流體之研究。 二氧化碳不具極性,對於極性物質之溶解度不高,因而限制了它的用途,一般為了提高 其對極性之溶解度,都可以加入極性之有機溶劑,作為修飾劑﹔然而對於蛋白質分子, 或是金屬離子,還是無法溶解。科學家試著在二氧化碳中加入介面活性劑,以形成反微 胞的方式溶解蛋白質,或是將金屬形成螯合物而溶解。然而無論介面活性劑或是螯合劑, 都必須是含氟的分子,才具有適當的溶解度。但是其操作壓力通常都很高,一般都在 200 大氣壓上下,因此工業上使用,成本也頗高。本研究因而轉向使用冷媒 R134a 作為萃取 液,取代超臨界二氧化碳,主要的研究在於核燃料棒中,鈾金屬金屬之萃取回收。 (乙)一般層析分離可分為兩種,其一是分析型,目的是分析樣品中之成分,並定量之﹔ 另一則是製備型的分離,目的是分離樣品中的主成分,並收集之,以備其他下游的研究。 製備型或半製備之分離,樣品的量比分析型的量要大很多,但分離效率不及前者﹔一般 製備型的分離,是使用大口徑的分離管柱,內填充大量矽膠,其靜相則是附著於矽膠之 上,以此種方法分離,有一潛在的問題,是矽膠本身往往還具有相當程度高極性的氫氧 基,對於高極性的物質,有時會產生永久吸附,而導致分離樣品的損失,或者會造成整 支分離管柱使用之壽命降低。 逆流層析之特色在於,動靜相都是純粹的液體,靜相是藉由離心力存在於分離管柱 中,不需要固態的填充劑作為靜相,因此不會產生上述吸附現象而造成樣品損失,或是 管柱壽命減短的情形,適用於生物分子、天然物之分離;由於靜相在管柱中通常都會達 到 40-80%,分離的樣品容量很高,很適合製備型之分離。除此之外,逆流層析的特點包 括:容易更換動靜相溶劑系統,即使無法被動相沖提出管柱之成分,也可以在分離結束 時從靜相中回收,近年來,開始吸引天然物、中藥研究者的注意。我們希望開發一些逆 流層析的新應用,尤其是在蛋白質的萃取和純化。
(二)研究目的 (甲)處理核廢料之標準流程稱之為Purex,Purex一詞為Plutonium-Uranium-Recovery by EXtraction之縮寫。此方法為利用磷酸三丁酯(tributylphosphate, TBP)於核廢料中萃取回收 鈾及鈽的程序,將使用過的核燃料棒溶解於濃硝酸,再利用含TBP螯合劑之有機溶劑,將其 中之鈾及鈽從硝酸溶液中萃取出來,得到鈾及鈽之TBP錯合物,再進行化學分離來回收及純 化鈾和鈽。此Purex程序雖然有高效率之回收,但以大量濃硝酸來處理高輻射性物質,並不是 一個相當安全的方式,還會有大量有機廢溶劑處理之問題。故發展一項新技術,能達到高回 收效率及解決Purex程序中溶劑造成之問題,為一個重要課題。 以超臨界流體(Supercritical Fluid,SF),來取代核廢料回收Purex程序中之有機溶劑,近 年來有快速的發展。超臨界流體萃取之特點在於萃取完成之後,超臨界流體一經洩壓即氣化 與萃取物分離,可將大體積中之特定物質濃縮,還可以免去處理有機溶劑問題。但是超臨界 流體(例如二氧化碳) 要具備高萃取能力,其壓力通常必須至少超過100 bar,有時甚至達到 250 bar或更高,於能源消耗與維護成本方面都會大幅增加,因此我們嘗試以R134a來取代超臨 界二氧化碳,檢視使用相類似的螯合劑,是否可以萃取鈾金屬。 (乙)生物分子中,蛋白質分子扮演了非常重要的角色,蛋白質的純化一直是生物化學研究 中一項重要的工作,過去多年來,以反微胞相來萃取蛋白質吸引了不少研究者,基本上這些 研究都是針對單一蛋白質,探討其萃取速率以及萃取回收率,而並未針對大體積樣品,進行 萃取及濃縮之實驗。如前所述逆流層析儀,很適合於生物分子之分離,因此我們將反微胞系 統應用於逆流層析儀中。研究之一是,以反微胞相萃取蛋白質,以逆流層析儀的高萃取效率 來純化並濃縮蛋白質。研究之二是,以反微胞溶液作為固定相來分離蛋白質。一方面擴大逆 流層析的應用領域,一方面使得反微胞萃取的研究,可以應用於蛋白質的濃縮和分離,而不 只是停留在,對單一蛋白質的萃取。
(三)文獻探討
(甲)處理核廢料之標準流程為Purex程序[1],其Purex一詞為Plutonium-Uranium-Recovery by EXtraction (另一個說法為Plutonium Uranium Reduction EXtraction )之縮寫。其為利用磷酸三丁 酯(TBP)於核廢料中萃取回收鈾及鈽之一種化工過程。20 世紀 50 年代中期,該流程首先 被用來提取核廢料中核武器所需要之鈽,後來被用來處理輻照核燃料,將使用過的核燃料棒 溶解於濃硝酸,再利用有機溶劑含TBP螯合劑,將其中之鈾及鈽從硝酸溶液中萃取出來,得 到鈾及鈽之TBP錯合物,再進行化學分離來回收及純化鈾和鈽[1,2]。此Purex程序雖然有高效 率之回收,但以大量濃硝酸來處理高輻射性物質,並不是一個相當安全的方式,還會有大量 有機廢溶劑處理之問題[3,4]。故發展一項新技術,能達到高回收效率及解決Purex程序中溶劑 造成之問題,為一個重要課題。 目前,有相關文獻[5]提出利用超臨界流體(Supercritical Fluid,簡稱SF),來取代核廢 料 回 收 Purex 程 序中 之有 機 溶 劑 。近 十 餘年來 , 超 臨 界流 體 萃取法 ( Supercritical Fluid Extraction,SFE)廣泛地發展及應用[6-8],且有相當多文獻提出利用螯合劑結合超臨界流體 之技術[9],可成功地萃取金屬,包括鹼金族、鹼土族、過渡金屬、重金屬、鑭系金屬以及錒 系金屬[10,11]。超臨界流體萃取之特點在於萃取完成之後,超臨界流體一經洩壓即氣化與萃 取物分離[12],並將大體積中之特定物質有效濃縮,免去處理有機溶劑問題,此為SFE相對於 傳統有機溶劑萃取最大優勢所在。目前,最普遍的超臨界流體為二氧化碳[13],由於二氧化 碳之臨界條件(Tc= 31.1 ℃,Pc = 73.76 bar)相較於其他超臨界流體而言較不嚴苛,且具有 環保、無毒、製備成本低、化學惰性高等特點,容易被使用者所接受。因此,一系列有關超 臨界二氧化碳取代Purex程序中有機溶劑使用之文獻陸續出現,但其中有一點值得關注:
SF-CO2 要具備高萃取能力,其壓力通常必須超過100 bar,有時甚至達到250 bar或更高,表示
必須使用能承受高壓之儀器裝置,於能源消耗與維護成本方面都會大幅增加,而且要考慮工 業安全性。 另外,於螯合劑選用上,傳統Purex程序所使用的螯合劑為有機磷試劑( Organophosphorus Reagents ),例如 TBP,於SF-CO2之文獻[7,8,11]中亦都利用此螯合劑。另有文獻[5,7,8]使用其 他類螯合劑,其中ß-二酮類( ß-diketones )為另一種可用於萃取鈾金屬之螯合劑。若選用 ß-diketones螯合劑,其本身必須含氟原子[14],或是於SF-CO2中添加修飾劑(Modifier)[15,16] 才能提升回收效率。物質氟化必須透過特殊合成步驟,價格上必然會高出許多,然而,修飾 劑通常為有機醇類或烷類,此會造成萃取後溶劑分離上的問題。綜合以上所述,本實驗之研 究目標為使用液態冷媒R134a以取代SF-CO2萃取。 (乙)反微胞(Reverse micelle) 當疏水型的界面活性劑處於有機溶劑之中,也能產生類似水中微胞的聚集結構[17],因 為這個環境之中,界面活性劑的兩端與水中微胞的排列方向相反,疏水端碳鏈朝外溶於有機 溶劑,親水端朝內,這種聚集結構稱為反微胞(reverse micelle)(圖 2-4,圖 2-5)。反微胞中心是 一個極性高的親水環境叫做水核(water pool),因為水核存在於有機溶劑之中,一般稱之為 water in oil。 反微胞萃取親水性物質的機制,主要包括: (1) 靜電作用力(electrostatic interactions)。離子型界面活性劑帶有正(負)電荷,利用界面活性 劑聚集而成的反微胞萃取親水性物質,離子型界面活性劑和水溶液中的帶電物質之間的靜電 力是重要的關鍵所在[18,19]。譬如從反微胞有機相萃取水溶液中的帶電染料(表 2-3),對於帶 負電荷的染料分子,需採用陽離子型的界面活性劑,相反地,萃取帶正電的染料分子需要用 到陰離子型的界面活性劑[20]。藉由正負電荷相互吸引的力量達萃取效果,此萃取過程中靜 電力是最主要的作用力。(2)疏水性作用力(hydrophobic interactions),而對非離子型的界面活 性劑而言,改變 pH 值對其萃取的效果影響不大,亦即上述所提到的靜電作用力對非離子型 的界面活性劑並非影響其萃取的主要因素,因此有些學者認為水溶性分子的疏水基與界面活
性劑的疏水基之間的凡得瓦耳力是影響這一類界面活性劑的最主要因素[21,22]。 影響蛋白質萃取的因素,可區為:
(1)水溶液的酸鹼值(pH)[23-26]。蛋白質由許多氨基酸組成,是一種具有許多離子基團的帶電 大分子,每一種蛋白質都有它的等電點(isoelectric point, pI),在酸鹼值小於 pI 的環境中,蛋 白質表面淨電荷為正電,相反地,在酸鹼值大於 pI 值的環境中,蛋白質表面靜電荷為負電。 離子型界面活性劑的親水端帶電,於有機溶劑中形成反微胞,想要使蛋白質能被萃取進入反 微胞,則蛋白質必須和反微胞帶相反電荷,產生靜電吸引力,此一萃取過程我們稱為正向萃 取(forward extraction)。而當在另一個相反的 pH 環境之下,蛋白質表面淨電荷轉變成和反微 胞帶相同電荷,產生靜電排斥力,蛋白質離開反微胞回到水相,此一過程我們稱為反向萃取 (backward extraction)。我們可說 pH 值和萃取主要機制(靜電作用力)的關係為:pH 值直接影 響蛋白質帶正電或負電、決定萃取的排斥力或吸引力。 (2)水溶液中的離子強度(ionic strength) 水溶液中帶電粒子之間的靜電作用力,會因為溶液中的離子強度增加,造成靜電吸引或 排斥力降低,稱之 Debye screening effect,此效應可減低蛋白質和界面活性劑的淨電力。在一 個以 AOT 反微胞相萃取細胞色素蛋白(cytochrome c)的實驗中,固定水溶液的 pH 值,使細胞 色素 C 蛋白和反微胞之間電荷相反,兩者之間存在靜電吸引力,以不同的 KCl 離子濃度觀察 萃取效果,發現在 0.1 M KCl 的環境下,細胞色素 C 分子很容易地被反微胞萃取,而提高離 子強度之後,在高達 1 M KCl 情況下, 細胞色素 C 分子無法以反微胞萃取。同時又有研究 發現,若是水中的離子強度不高(濃度< 0.1 M),含有界面活性劑的有機相和水溶液兩相不容 易分層,無法有效萃取蛋白質[27], 離子強度與萃取主要作用力(靜電吸引力)的關係為:雖無法改變蛋白質電荷(也就是吸引 力和排斥力),但其濃度可調整靜電作用力大小,進而影響萃取效果。 (3)離子種類(Type of Electrolyte) 水溶液中的鹽類種類也會影響蛋白質的萃取效果,鹽類解離之後產生的離子會產生遮蔽 效應(screening effect),有文獻發現在 AOT/isooctane 系統下只改變離子種類[28],對蛋白質的 萃取效果是 Li+
>Na+>Cs+>Rb+>K+,推測是金屬的水合離子電荷的遮蔽效果所影響,鉀離子 遮蔽的效果最大導致萃取率不高。鋰離子的遮蔽效果最差,所以蛋白質的萃取效果最好。 (4)界面活性劑
界面活性劑[29]的選擇是非常重要的一部分,目前常見用在反微胞萃取的陰離子型界面 活性劑有 sodium bis(2-ethylhexyl)sulfosuccinate (AOT)以及 di-(2-ethylhexyl)phosphoric acid (DEHPA)兩種[30],AOT 是屬於磺酸型陰離子界面活性劑,DEHPA 則是磷酸型陰離子,除了 這兩種陰離子型界面活性劑之外,尚有一些研究試圖合成不同種類的界面活性劑[31],目的 就是為了達到更有效、迅速、選擇性高的蛋白質萃取。Trioctyl methyl ammonium chloride (TOMAC)和 cetyltrimethylammonium bromide (CTAB)則是常用在蛋白質萃取的陽離子界面活 性劑,它們都是四級銨鹽,在有機溶劑中形成帶正電荷的反微胞[32,33]。 此外,增加界面活性劑的濃度可增加有機相萃取蛋白質的量 21,濃度高的界面活性劑雖 然有利於正向萃取,但將會使蛋白質的反向萃取相對困難,因此如何調整界面活性劑濃度, 在正向-反向萃取率之間達到最佳化,也可能是設計萃取條件的參考之一。 (5)蛋白質種類 因為蛋白質是一種複雜的巨大分子,不同的蛋白質除了 pI 值、分子量不同之外,它們的 表面電荷分佈也不相同[35]。不同的 pI 值可造成在同樣的 pH 值環境下,一種蛋白質帶正電、 另一種則帶負電。反微胞的水核大小有限,比較容易萃取低分子量的蛋白質,然而高分子量 的蛋白質(分子量大於 100000)因為過於巨大造成萃取效果不佳[36],所以需要用到較高的界面 活性劑濃度,造成有機-水兩相不容易分層。
(6)共界面活性劑(cosurfactant)和共溶劑(cosolvent) 因為反向萃取的速率緩慢,於是在反微胞系統中,在主要的界面活性劑加入非離子型界 面活性劑[37]或是共溶劑[38],嘗試改變反微胞的電荷分佈或是反微胞形狀,期望減低蛋白質 和反微胞的電荷作用力,減低反萃取時間,所以加入這類添加物的目的是為了加強反萃取效 果。另外有研究使用終結反微胞電荷的方法,在使用陰離子界面活性劑的情況之下,加入陽 離子型界面活性劑會瓦解反微胞[39],產生電荷中和,蛋白質便迅速回到水相。 至於將反微胞相應用於逆流層析儀中,分離親水性物質,並未有任何文獻之記載。
(四)研究方法 (甲)以 R134a 萃取鈾金屬 (A) 硝酸與有機磷試劑形成之錯合物對鈾金屬萃取探討 (A-1) 以 HNO3-有機磷試劑於固相基質中萃取鈾金屬 1. HNO3(65.0 %)與 2 倍體積比(或重量比)之螯合劑 TBP 混合,震盪 1 hr 後,以 6000 rpm 轉速離心 20 min。靜置隔天,取上層相,此層即為 HNO3-TBP 結合之液態。以相同之實 驗步驟合成其他 HNO3-Ligand(Ligand =有機磷試劑)之錯合物。 2. 裝置如圖 4-1 所示。 3. 取液態 HNO3-Ligand 合成物 5 mL 置入 20 mL 不鏽鋼攪拌槽,加入磁石。 4. 取三氧化鈾 10 mg 與矽藻土 0.1 g 混合置入固相萃取槽中。 5. 將控溫水槽與固相萃取槽設定好溫度(Temperature, T ℃)。 6. 將定壓之 R134a 注滿 20 mL 不鏽鋼攪拌槽,關閉進出口閥門,靜態攪拌 90 min。 7. 靜態攪拌完之後,將連接於固相萃取槽之閥門打開,注滿固相萃取槽,進行靜態萃取 60 min。
8. 接續進行動態萃取流速(Flow rate, f)以 0.3~0.4 mL/min,定量收集 20 mL(Volume, V) R134a,以乙醇體積 7 mL 為收集液,收集完之後,再以乙醇沖洗管路,一同置入收集液 當中,將總體積加至 10 mL。 9. 以 ICP-MS,進行萃取之定量。 10. 改變溫度、壓力條件及 HNO3-有機磷試劑合成物,重複上述之實驗。 11. 各條件實驗均操作四次,取平均值。 圖 4-1 萃取裝置圖 (A-2) 分別進行兩種鈾氧化物 UO2及 UO3萃取 操作步驟如同上。以液態 HNO3-TBP 合成物 5 mL,於壓力 60 bar、溫度 60 ℃下進行萃
取,以 UO2及 UO3(各 10 mg)分別進行動態萃取流速以 0.3~0.4 mL/min,以每 5 min、10 min、 15 min、20 min、30 min、40 min、50 min、60 min 共八管,以乙醇體積 7 mL 為收集液來進 行收集。
(A-3) 以不同濃度的硝酸與 TBP 合成後進行萃取
液態 HNO3-TBP 合成物 5 mL,於此操作條件為壓力 60 bar、溫度 60 ℃,分別為 1 N、3
(A-4) 硝酸與 TBP 依不同體積比例錯合後進行萃取
以液態 HNO3-TBP 合成物 5 mL,於此為壓力 60 bar、溫度 60 ℃之下,改變 HNO3與 TBP 體積比例(1:2、1:1 及 2:1)進行錯合。
(B) 以不同螯合劑相結合對鈾金屬萃取之探討
不含氟之-乙酮類 1-Benzoylacetone 1.25 g 與有機磷試劑 TBP 3.75 mL 混合,震盪 1 hr 後, 以 6000rpm 轉速離心 20min,即為 1-Benzoylacetone-TBP 合成物。液態 1-Benzoylacetone-TBP 合成物 5 mL 於此操作條件為 60 bar 60 ℃及 15bar 30℃。 (C) 其他金屬於固相基質之萃取 1. 裝置如圖 4-1 所示。 2. 取液態 HNO3-TBP 合成物 5 mL 置入 20 mL 不鏽鋼攪拌槽,加入磁石。 3. 取三氧化鈾 1 mg 與矽藻土 0.2 g 混合置入固相萃取槽中。 4. 將控溫水槽與固相萃取槽設定好溫度 60 ℃。
5. 將壓力 60 bar 之 R134a 注滿 20mL 不鏽鋼攪拌槽,關閉進出口閥門,靜態攪拌 90 min。
6. 靜態攪拌完之後,將連接於固相萃取槽之閥門打開,注滿固相萃取槽,進行靜態萃取 30 min。 7. 接續進行動態萃取流速以 0.3~0.4 mL/min,定量收集 20 mL R134a,以乙醇體積 7 mL 為 收集液,收集完之後,再以乙醇沖洗管路,一同置入收集液當中,將總體積加至 10 mL。 8. 以 ICP-MS,進行萃取之定量。 9. 更換其他金屬之氧化物或硝酸物,重複上述之實驗。 10. 各條件實驗均操作三次,取平均值。 (乙)以逆流層析之分離研究 (A) 萃取部分 (1)使用往覆式幫浦以 5mL/min 將有機靜相填滿整個管柱。見儀器圖 4-2. (2)起動高速逆流層析儀,使其轉速達到 800rpm。 (3)當轉速穩定後,以 2mL/min 之流速將 pH = 7 之動相水溶液打入管柱中。 (4)待靜相不再流出,表示動靜相達到平衡,將動相水溶液換成蛋白質樣品溶液,以 2mL/min 之流速打入管柱中。 (5)打開 UV-Vis 偵測器並開啟色層分析儀數據處理系統,完成參數的設定。 (6)當樣品完全注入後,改打入 pH = 11 之動相水溶液,將流速改為 1mL/min,同時轉動六向 閥,使流析出來的分析物進入 UV-Vis 偵測器中來做線上偵測。 (7)等到訊號出現後,即以每十毫升為一管收集被萃取並濃縮的回收液,共收集六管。 (8)完成實驗後,慢慢將轉速降到零,再關掉溫控箱、UV-Vis 偵測器及色層分析儀數據處理系 統。 (9)使用氮氣將管柱內的溶液推出以獲得靜相滯留量。 (10)先以甲醇以 3mL/min 之流速清洗管柱,再以 5mL/min 之流速打入去離子水,最後再以氮 氣推出清洗的溶劑,並以較低的氮氣流速吹乾管柱(over night) 。 定量的方法: 使用標準添加法來做定量。在此方法中,所使用的標準溶液為已知濃度為 100ppm 的蛋白質 水溶液,其步驟為先取四個 5mL 之定量瓶,每一瓶中分別加入 1mL 之回收液,再分別加入 0、 1、2、3mL 之標準溶液,再以 pH = 7 的動相水溶液稀釋到刻度線。接著以生物分子定量用 UV-Vis 偵測器偵測四瓶溶液在 408nm 的個別吸收度,並根據此四個吸收值及所分別添加的標 準溶液體積為軸做圖,再將圖形趨勢線的斜率和截距,代入公式中,重覆上述步驟五次,即 可得到每一管回收液的濃度 ; 再進而得求得樣品的回收率及濃縮倍率。
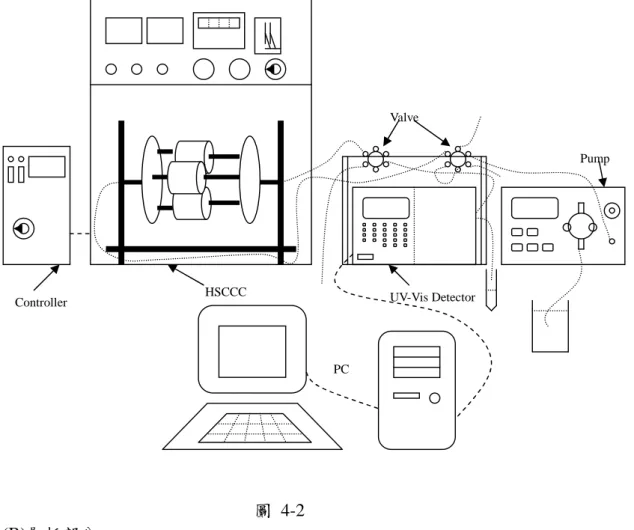
圖 4-2 (B)層析部分 實驗裝置如圖 4-3 所示 (1)使用往復式幫浦以流速 3mL/min 將有機靜填滿整個管柱。 (2)啟動高速逆流層析儀器,使其轉速達到 800 rpm。 (3)當轉速穩定後,梯度幫浦將動相 A 以流速 1 mL/min 打入高速逆流層析儀中。 (4)當有機相不再流出,表示動靜相達到平衡,打開 UV 偵測器(280 nm),等待訊號穩定。 (5)注入蛋白質混合溶液 1 mL,並利用動相 A + B 在不同條件下的梯度比例。並同時紀錄層析 訊號。 (6)每 5 分鐘收集一管。即每管中含有 5 mL 收集液。紀錄下各收集管的 pH 值。 (7)完成實驗後,將高速逆流層析停止轉動,以氮氣將管柱內的液體吹出並收集,記錄有機相 和水相的體積。上層有機相的體積就是靜相滯留量。 (8)以丙酮與甲醇(1:2)混合物 60 mL 沖洗管柱,再以 60 mL 去離子水清洗。最後再用氮氣吹乾 管柱。 (9)收集液以 HPLC 分析之 高速逆流層析動相溶液-緩衝溶液 實驗一: 動相 A1:含有 50 mM Tris-HCl、0.1 M KCl、pH 7 緩衝溶液 動相 B1:含有 50 mM K2HPO4、0.1 M KCl、pH 12 緩衝溶液 實驗二; 動相 A2:含有 50mM Tris-HCl、0.2M KCl、pH 7 緩衝溶液 動相 B2:含有 50mM K2HPO4、0.2M KCl、pH 12 緩衝溶液 實驗三 動相 A3:含有 50mM Tris-HCl、0.2M KCl、pH 7 緩衝溶液 動相 B3:含有 50mM K2HPO4、0.6M KCl、pH 12 緩衝溶液 Pump UV-Vis Detector HSCCC PC Valve Controller
(五)結果與討論 (甲) (A)不同溫度對固相基質萃取之影響 本實驗探討溫度之改變,對於 HNO3-TBP 於固相基質中,萃取鈾金屬之效率。實驗結果 由表 5-1 得知,增高溫度(30℃-50℃)對萃取鈾金屬無顯著之影響,但當 R134a 於定壓 60 bar 之下,當溫度升高至 60 ℃時,於固相基質當中,以 R134a 收集 20 mL 的萃取回收率明顯 地提高約 10 %,文獻中,在超臨界二氧化碳中,所使用之萃取溫度亦為此溫度,因此,接下 來的實驗條件溫度以 60℃為操作條件作為比較。 表 5-1 不同溫度之固相基質萃取
Complex Pressure(bar) Temperature(℃) Extractionefficiency(E%) RSD(%)
30 56.2 16.3 40 59.7 7.06 50 59.6 8.02 HNO3-TBP 60 60 70.8 6.97
Sample: UO310.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a= 20 mL; RSD 值為重複實驗三次所得
(B)壓力對固相基質萃取之影響
本實驗探討壓力之改變,對於 HNO3-TBP 於固相基質中,萃取鈾金屬之效率。實驗結果
由表 5-2 得知,於不同壓力所得鈾金屬之萃取回收率相差不大。 表 5-2 不同壓力之固相基質萃取
Complex Temperature(℃) Pressure(bar) Extractionefficiency(E%) RSD(%)
15 67.2 5.67 30 68.1 2.35 45 67.0 2.71 HNO3-TBP 60 60 70.8 6.97
Sample: UO310.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a= 20 mL; RSD 值為重複實驗三次所得 由文獻得之,利用 SF-CO2於固相基質中,萃取鈾金屬之萃取率,二氧化鈾於 125 bar 所 得萃取效率僅有 52.5%,二氧化鈾與三氧化鈾之混合物,於 125 bar 所得萃取率僅有 61.9%左 右,而本實驗結果,由圖 5-1 所示,R134a 於 15 bar 時,鈾之萃取效率可達 67.2%,當壓力 提高為 60 bar 時,則鈾之萃取率可達 70.8%,雖然兩者實驗條件不完全一致,但利用此法可 於較 SF-CO2低壓之下,達到對鈾金屬有七成左右之萃取效率。
10 20 30 40 50 60 70 0 20 40 60 80 100 E x t r a c t i o n e f f i c i e n c y ( % ) Pressure(bar) 圖 5-1 利用 R134a 於不同壓力下進行固相基質萃取鈾金屬之萃取率
Sample: UO310 mg, 矽藻土 0.1 g;fR134a= 0.3~0.4 mL/min;VR134a=20 mL;T=60℃ (C)二氧化鈾及三氧化鈾之固相基質萃取
於文獻提到,利用含有 HNO3-TBP 錯合物之超臨界二氧化碳,於固相基質中可直接萃取
UO2與 UO3。UO2之萃取效率較差,是由於將 HNO3與 TBP 先結合,HNO3本身之濃度含量
較少,因此,當 U(IV)要氧化成 U(VI)所需時間較長,但不管為 UO2或 UO3於超臨界二
氧化碳當中,其 HNO3仍保有其氧化能力,HNO3-TBP 亦具有螯合鈾金屬之能力。
本實驗之在類似條件下,使用 R134a 於固相基質中萃取鈾金屬。實驗結果如圖 5-2 所示, 於 R134a 中,不管鈾金屬是以正四價或正六價形式存在,鈾金屬皆能夠被萃取出來,且 U(IV)
所需時間亦同樣需要較長之時間,由本實驗得知,含 HNO3-TBP 之 R134a 於固相基質中,萃
取鈾金屬具有螯合鈾金屬之能力,且 HNO3亦保有其氧化能力,使 U(IV)氧化成 U(VI)。
0 10 20 30 40 50 60 70 0 2 4 6 8 10 W e i g h t o f U ( V I ) ( m g ) Time(min) 圖 5-2 UO3與 UO2於固相基質之萃取結果
(▲)UO3(●)UO2;Sample: UO3、UO2各 10.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a = 0.3~0.4 mL/min;VR134a=20 mL;T=60℃;P=60 bar
(D)利用不同硝酸濃度結合 TBP 進行固相基質萃取 本實驗針對不同濃度之硝酸與有機磷試劑 TBP 錯合後,再與 R134a 混合進行固相基質萃 取,主要目的為探討先將不同濃度之硝酸與 TBP 錯合後,於固相基質萃取鈾金屬之影響。所 得結果如表 5-3 所示,硝酸濃度的增高,於固相基質之 U(VI)萃取率無明顯的增加,因此, 先將不同濃度之硝酸與 TBP 結合,再進行固相基質萃取。實驗結果顯示硝酸濃度之不同,對 在固相基質萃取 U(VI)並無太大之影響,反而於濕式反應中,硝酸溶液濃度的改變,對鈾 之萃取率影響較大。 表 5-3 不同硝酸濃度結合 TBP 之固相基質萃取 HNO3濃度(N)
Pressure(bar) Temperature(℃) Extractionefficiency(E%) RSD(%)
1 53.5 16.0 3 68.4 6.09 6 70.8 6.91 9 71.1 3.57 14.4 60 60 67.4 3.77
Sample: UO310.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a=20 mL; RSD 值為重複實驗三次所得 (E)不同 HNO3與 TBP 體積比例合成之錯合物對固相基質之萃取 本實驗先將 HNO3與 TBP 以不同體積比例(1:2、1:1 及 2:1)結合後,再與 R134a 混合後,一同進入固相基質中萃取鈾金屬,探討 HNO3與 TBP 混合比例不同,對於進行固相 基質萃取鈾金屬之影響。實驗結果由表 5-4 所示,將 HNO3與 TBP 以不同體積比例結合後, 進行固相基質萃取鈾金屬,所得結果並無顯著之差異,因此,先將 HNO3與 TBP 依不同比例 結合,利用冷媒 R134a 進行固相基質萃取鈾金屬,並無顯著之差異。 表 5-4 不同 HNO3與 TBP 結合比例之錯合物於固相基質中萃取鈾金屬之效率 HNO3-TBP 體積比例 Pressure(bar) Temperature(℃) Extraction efficiency(E%) RSD(%) 1:2 70.4 2.74 1:1 71.8 2.46 2:1 60 60 72.7 1.32
Sample: UO310.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a=20 mL; RSD 值為重複實驗三次所得 (F)-二酮類 1-Benzoylacetone 與有機磷試劑 TBP 結合之固相基質萃取 本實驗選用一不含氟之-二酮類 1-Benzoylacetone 與有機磷試劑 TBP 相結合後,於固相 基 質 中 萃 取 鈾 金 屬 。 實 驗 結 果 由 表 5-5 所 得 之 結 果 看 來 , 使 用 不 含 氟 之-二 酮 類 1-Benzoylacetone 與有機磷試劑 TBP 相結合,再與 R134a 混合後,於固相基質中萃取鈾金屬 之效率相當差,其中有很多因素是需要被考慮,在此提出幾個可能的原因,可能鈾不易形成 Uranyl-TBP 之形式被萃取出來,且在本系統當中不會產生協同效應來改善螯合劑對鈾的萃取 效率,因此,1-Benzoylacetone-TBP 錯合物在固相基質中,對鈾沒有萃取能力。
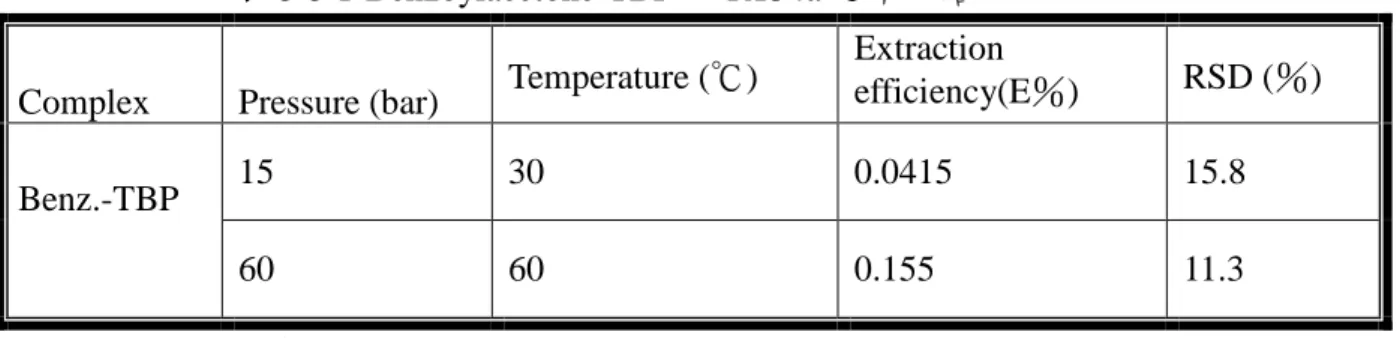
表 5-5 1-Benzoylacetone-TBP 於 R134a 之萃取結果 Complex Pressure (bar) Temperature (℃)
Extraction efficiency(E%) RSD (%) 15 30 0.0415 15.8 Benz.-TBP 60 60 0.155 11.3 Benz. = 1-Benzoylacetone
Sample: UO310.0 mg, 矽藻土 0.1 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a=20 mL; RSD 值為重複實驗三次所得 (G)其他金屬於固相基質之萃取 本實驗主要目的為證明含有 HNO3-TBP 之 R134a,於固相基質進行萃取,此法是否對鈾 金屬有萃取專一性。由表 5-6 可得知,利用含有 HNO3-TBP 之 R134a,於固相基質中去萃取 各類金屬,除了錒系與鑭系金屬之外,其他金屬幾乎無法被萃取。於一般螯合劑中,U(VI) 之金屬離子基本上只接受 O 之配位,於特殊條件下才會與 N、S 或 P 配位,於此所用的螯合 劑 TBP 含有 O 之原子,而其他金屬例如:Ni(II)之離子僅接受 N 及 S 之配位,其他詳細 之配位情形,可參考 2.2.1 螯合劑簡介,因此,Ni(II)很難被含有 HNO3-TBP 之錯合物,於固 相基質中被萃取出來。然而,La 和 Gd 金屬也很容易被萃取出來,對於專一性是比較不利 的。 表 5-6 含 HNO3-TBP 之 R134a 於不同金屬之萃取結果
Metal U(VI) U(IV) La(III) Gd(III) Lu(III) Sr(II)
Extraction
efficiency(E%) 79.5 19.6 27.2 47.6 23.0
-RSD(%) 15.9 13.2 10.4 11.5 24.6
-Metal Ba(II) Zr(IV) Mo(VI) Ni(II) Cr(III)
Extraction efficiency(E%)
<0.1
- - <0.02
-RSD(%) - - - -
-Sample: Metal 1.0 mg, 矽藻土 0.2 g;Complex 5 mL;fR134a= 0.3~0.4 ml/min;VR134a=20 mL; T = 60 ℃;P = 60bar;RSD 值為重複實驗三次所得;(-)表示 ICP-MS 測不到此金屬之含量 結論:以超臨界流體二氧化碳來取代傳統有機溶劑,從核廢料中萃取回收鈾金屬,很可能會 被核能工業採用,本研究企圖以冷媒 R134a 來取代超臨界流體。以實驗數據比對判斷,是有 可能的,而且萃取效率或許比超臨界萃取更好,至於可否實際應用,還需要更多的評估。 (乙) (A)萃取部分 (A-1) 離子濃度對回收率的影響 離子濃度是影響萃取率的因素之一,一般而言,離子濃度越高,蛋白質分子就越難進入 反微胞中,這是由於隨著離子濃度的提高,反微胞的粒徑會隨之縮小,而這也使得調整離子 濃度可以作為反向萃取的控制條件。在實驗中,將離子濃度作為變異的因素,探討它對回收 率的影響,其得到其結果如表 5-7。
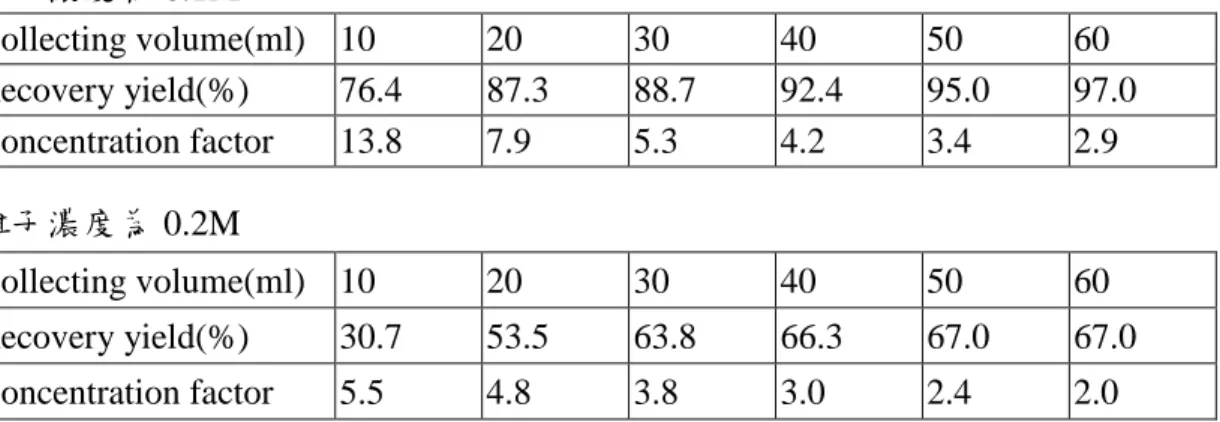
表 5-7 不同離子濃度對回收率的影響 離子濃度為 0.1M Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 76.4 87.3 88.7 92.4 95.0 97.0 Concentration factor 13.8 7.9 5.3 4.2 3.4 2.9 離子濃度為 0.2M Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 30.7 53.5 63.8 66.3 67.0 67.0 Concentration factor 5.5 4.8 3.8 3.0 2.4 2.0 由表 5-7 的實驗結果可以得知,當離子濃度由 0.1M 增為 0.2M 時,其回收率會大幅的降低, 推測其原因,主要應該和靜相滯留量有關。根據之前本實驗室已畢業的李心怡學姐之論文 36 指出:當所打入樣品的體積與靜相滯留量的比值越高,其回收率也會隨之提高,靜相滯留量與 回收率的關係如表 5-8。 表 5-8 靜相滯留量與回收率的關係 VR 2 5 10 Recovery yield(%) 67 78 82 在表 5-8 中,VR代表打入樣品的體積與靜相滯留量的比值,而當我們使用 0.1M 之離子濃度 時,其靜相滯留量約為 28mL,打入的樣品體積為 180mL,則 VR約為 6.4 ; 而使用 0.2M 之離 子濃度時,其靜相滯留量約為 60mL,則 VR約為 3。與表 4-7 相比較下,當我們使用 0.1M 之 離子濃度時,其 VR約為 6.4,但其回收率已達 97%,遠超過表 4-7 中,VR值為 10 的回收率 82%,而表 4-7 中所使用之離子濃度亦為 0.2M,因此我們可以推測,在這裡影響回收率的因 素除了 VR值外,應該還有其它的因素存在,而根據我們所得到的數據來看,離子強度很可能 就是其中之一。 (A-2) pH 值對回收率的影響 改變 pH 值在本實驗中乃是進行反向萃取的因素,理論上當 pH 值高於 pI 值,反向萃取 就會發生,但是事實上並非如此,根據李心怡學姐之論文,雖然 cytochrome c 的 pI 值為 9.4, 應該 pH 值為 10 時,就有反向萃取的發生,然而由表 5-9 中可看出,必需要在 pH =12.8 的情 況下,其回收率才能達到 67%。 表 5-9 pH 值與回收率的關係 pH 10.0 11.0 12.0 12.8 Recovery yield(%) 9 24 49 67 但是上述的實驗結果都是在以 5mL 的高 pH 值鹼液來做反相萃取的情況下,在此方法中, 除了 5mL 的高 pH 值鹼液外,用來做收集的動相仍然是低 pH 值的,因此在反向萃取的過程 中,pH 值將劇烈的變動,而導致必需使用極高 pH 值之鹼液,才可以在 pH 值改變後,pH 值 還在 pI 值以上,而有反向萃取的效果。 因此,在本實驗中,使用 pH 值為 7 及 11 之緩衝溶液,分別作為正向萃取及反向萃取的 動相,其實驗結果如表 5-10。 表 5-10 使用 pH 值為 11 之緩衝溶液做反向萃取,所得到的結果 Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 35.0 50.6 58.2 63.1 66.5 68.5 Concentration factor 6.3 4.5 3.5 2.8 2.4 2.1
由表 5-11 之結果,可以證實當使用 pH 值為 11 之緩衝溶液做反向萃取,其最後的收集液其 pH 值亦在 11 上下,且其回收率可以達到 68.5%,與原先使用一小段 pH 為 12.8 之鹼液做反 向萃取,所得到的回收率差不多。 而當我們將緩衝溶液之 pH 值調整到 12,其回收率與 pH 值為 11 之緩衝溶液相比差異不 大,有時甚至還會得到較低的回收率如表 4-10 ,推測其原因應是其 VR值皆約為 2,且尚未 達到 AOT 結構被完全破壞的 pH 值,故改變 pH 值在本實驗中對回收率並沒有幫助。 表 5-11 使用 pH 值為 12 之緩衝溶液做反向萃取,所得到的結果 Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 39.4 46.8 51.8 56.4 59.7 62.1 Concentration factor 7.1 4.2 3.1 2.5 2.1 1.9 (A-3) EA 含量對回收率的影響 由於我們在樣品瓶中的實驗可以證實添加 EA 確實可以在相同的時間內,在反相萃取的 過程中,提高樣品的回收率;因此我們將此一方法應用在逆流層析上,觀察它對回收率的影 響。在實驗中分別配製 EA 所占比例為 0%、5%及 10%的有機靜相,觀察其對回收率的影響, 其結果列於表 5-12。 表 5-12 EA 含量對回收率的影響 EA 含量為 0% Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 64.4 80.6 80.6 80.6 80.6 80.6 Concentration factor 11.6 7.3 4.8 3.6 2.9 2.4 EA 含量為 5% Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 72.5 83.2 83.2 83.2 83.2 83.2 Concentration factor 13.1 7.5 5.0 3.7 3.0 2.5 EA 含量為 10% Collecting volume(ml) 10 20 30 40 50 60 Recovery yield(%) 76.4 87.3 88.7 92.4 95.0 97.0 Concentration factor 13.8 7.9 5.3 4.2 3.4 2.9
*表 5-12 之實驗條件:靜相為 50mM AOT 溶於含有 0%,5%,10%EA 之 n-hexane 中 ; 動相為 pH=6.99,0.1M NaCl 的緩衝溶液 ; 反相萃取之動相為 pH=11,0.1M NaCl 的緩衝溶液 ; 樣品為 20mg/L 之 cytochrome c (B) 層析部分 (B-1)實驗一 使用兩動相:50 mM Tris-HCl 、0.1 M KCl、 pH 7 緩衝溶液,以及 50 mM K2HPO4、0.1 M KCl 、pH 12 緩衝溶液,其餘實驗條件不改變,進行梯度沖提,管柱總體積 28 mL,得到靜 相滯留量大約是 10 mL,逆流層析訊號如圖 5-3 所示。
m v minutes 0 20 40 60 80 0 500 1000 圖 5-3 實驗一的逆流層析訊號。動相流速 1 mL/min,UV 偵測器:280 nm 由層析圖中可見在實驗流析的前 40 分鐘,整個系統相當不穩定,因為溶液混濁而造成偵 測到巨大且雜亂的訊號,注入蛋白質和動相梯度變化之後,從管柱流出的液體呈現不穩定的 混濁狀態,表示逆流層析內部的相分佈遭到擾動,蛋白質的分離效果也非常不理想。我們認 為是因為離子強度不夠,使得有機-水相界面不容易迅速分層而產生混濁。 (B-2)實驗二 由於實驗一造成有機-水相界面混濁的原因,可能是因為離子強度不夠,為了解決此項缺 點,我們將 KCl 濃度提高到 0.2 M,希望能解決混濁以及靜相流失的問題。實驗二的逆流層 析圖(圖 5-4)較為穩定許多, m v minutes 0 20 40 60 80 0 500 1000 圖 5-4 實驗二的逆流層析訊號。動相流速 1 mL/min,UV 偵測器:280 nm 添加離子濃度可幫助有機相-水相在逆流層析中的分層,因為有機靜相能有效地滯留在管柱中 而非迅速流失,增加了對於蛋白質樣品的分離能力,隨著動相 pH 值梯度改變,可明顯分辨 訊號波峰,但是圖中之層析訊號圖並不能完全表示純凈的蛋白質,流析的動相分別收集 17 管,從各收集管的 pH 值,可知動相的 pH 值從 7 到 12 線性攀升。
實驗(二)分管收集以 HPLC 分析,各管中蛋白質之濃度,以及各蛋白質之相對濃度比 (因為各蛋白質無法完全分離)之結果顯示於圖 5-5。 1 3 5 7 9 11 13 15 17 c y to c h ro m e c 0.0 40.0 80.0 120.0 160.0 200.0 ppm Tube number ( a ) cytochrome c Lysozyme myoglobin 1 3 5 7 9 11 13 15 17 cytochrome c 0.0% 20.0% 40.0% 60.0% 80.0% 100.0% Tube number (b)
cytochrome c
lysozyme
myoglobin
圖 5-5、HPLC 分析實驗二收集管中的蛋白質含量 (a)蛋白質的濃度(ppm) (b)蛋白質的蛋白質相對濃度(%) 由於肌紅蛋白的 pI 值約為 7,在動相 pH 7 條件下,蛋白質表面淨電荷與陰離子型界面活 性劑 AOT 的電荷作用力較小,肌紅蛋白不易進入反微胞靜相。cyrochrome c 和溶菌酶兩蛋白 質在 pH 7 環境中,表面正電荷和 AOT 反微胞有強大的吸引力因而容易進入靜相,而與肌紅 蛋白分離開來。分離出肌紅蛋白之後,再提高動相 pH 值可逐漸減低蛋白質的表面正電荷, 細胞色素 C 分子從 pH 7 升高到 pH 9 的過程,表面正電荷逐漸微弱,蛋白質與反微胞的靜電 吸引力逐漸減小,細胞色素 C 離開反微胞回到動相水溶液,與溶菌酶分離開來。隨著 pH 值 逐漸增高至 11 左右,溶菌酶表面電荷和 AOT 反微胞的吸引力也逐步減弱,因而回到動相水 溶液中。 但是蛋白質溶菌酶的回收率不高,溶菌酶的等電點 pI = 11,表面正電荷和反微胞之間有 強大的電荷吸引力,即使以 pH 12 動相沖提,溶菌酶有效回收率只有 16 %,實驗中不使用更 高的酸鹼值( pH 13 or 14 )是為了避免破壞蛋白質活性,我們將嘗試在後續的實驗中更改酸鹼值之外的其他參數。 (B-3)實驗三 為了提高溶菌酶的回收率,我們除了使用 pH 動相梯度之外,再加入 KCl 增加動相的離 子強度,形成了 pH 7~12 的酸鹼梯度和 KCl 0.2~0.6M 的離子強度梯度,藉由 pH 值梯度逐步 改變蛋白質表面電荷,離子強度的增加可減低溶液中蛋白質和反微胞的電荷作用力。 實驗三的逆流層析訊號圖如圖 4-6,依序收集之後得到 20 支蛋白質收集管,量測各管中 的 pH 變化,隨著流析時間增加,沖提收集管中的 pH 值如預期地進行梯度升高。利用 HPLC 定量蛋白質含量和蛋白質相對濃度,確認可得到蛋白質相對濃度 85 %以上,回收率 60 %以 上的效果。 m v minutes 0 50 100 0 50 100 150 200 250 300 350 400 圖 5-6 實驗三 逆流層析訊號圖。動相流速 1 mL/min,UV 偵測器:280 nm 1 3 5 7 9 11 13 15 17 cytochrome c 0.0 50.0 100.0 150.0 200.0 ppm Tube number (a) cytochrome c lysozyme myoglobin
1 3 5 7 9 11 13 15 17 cytochrome c 0.0% 20.0% 40.0% 60.0% 80.0% 100.0% Tube number ( b) cytochrome c lysozyme myoglobin 圖 5-7 HPLC 分析實驗三收集管中的蛋白質含量 (a)蛋白質的濃度(ppm) (b)蛋白質的蛋白質相對濃度(%) 比較實驗二和實驗三的分析結果,兩實驗中的肌紅蛋白和細胞色素 C 的回收濃度和蛋白 質相對濃度相差不大,但溶菌酶的濃度和蛋白質相對濃度均有提升,回收率從 16 % 大幅提 升到 60 %,因為兩實驗的 pH 值梯度條件相同,能大幅提升溶菌酶蛋白質的回收率是因為提 高了 KCl 離子濃度,有效降低蛋白質和反微胞的靜電吸引力,使溶菌酶反萃取回水中。 結論:使用反微胞相於逆流層析中,對於萃取蛋白質或是蛋白質的分離,都有相當的潛力, 主要是利用逆流層析儀,有效率的濃縮能力,和層析分離能力。目前在萃取方面所遭遇的困 難是在於反向萃取速率較慢,往往會影響回收率;至於層析方面,由於不同蛋白質分子之間 的作用力之牽引,還不能得到完全純粹的分離,但是,在加入酸鹼以及電解質濃度梯度沖提, 結果已經大幅改進,相信未來可以達到完全分離。 (六)文獻:
1. S. Jarudilokkul, L. H. Poppenborg and D. C. Stuckey, “Backward extraction of reverse
micellarencapsulated proteinsusing a counterionicsurfactant.”Biotechnol. Bioeng. 62 (1999)
593-601.
2. D. G. Hayes, “Mechanism of protein extraction from the solid state by water-in-oil
microemulsions.”Biotechnol. Bioeng. 53 (1997) 583-593.
3. M.Goto,T.Ono,F.Nakashio and T.A.Hatton,“Design ofsurfactantssuitableforprotein
extraction by reversed micelles.”Biotechnol. Bioeng. 54 (1997) 26-32.
4. E. P. Melo, S. M. B. Costa, J. M. S. Cabral, P. Fojan and S. B. Petersen, “Cutinase-AOT interactions in reverse micelles : the effect of 1-hexanol.”Chemistry and Physics of Lipids 124
(2003) 37-47.
5. D.G.Hayesand C.Marchio,“Expulsion ofproteinsfrom water-in-oil microemulsions by
treatmentwith cosurfactant.”Biotechnol. Bioeng. 59 (1998) 557-566.
6. P. L. Luisi and L. J. Magid, “Solubilization of enzymes and nucleic acid in hydrocarbon
micellarsolutions.”Crit. Rev. Biochem. 20 (1986) 409-474.
7. M. Dekker, R. Hilhorst and C. Laane, “Isolating enzymes by reversed micelles.” Anal.
Biochem. 178 (1989) 217-226.
8. E.B.Leodidisand T.A.Hatton,“Amino acidsin AOT reversed micelles.1.determination of interfacial partition coefficients using the phase-transfermethod.”J. Phys. Chem. 94 (1990)
6400-6411.
9. E. B. Leodidis and T. A. Hatton,“Amino acidsin AOT reversed micelles.2.thehydrophobic effectand hydrogen bonding asdriving forcesforinterfacialsolubilization.”J. Phys. Chem. 94
(1990) 6411-6420.
10. P.L.Luisi,“Enzymeshosted in reversemicellesin hydrocarbon solution.”Angew. Chem. Int. Ed. Engl. 24 (1985) 439-450.
11. M. J. Pires, M. R. Aires-Barrosand J.M.S.Cabral,“Liquid-liquid extraction of proteins with
reversed micelles.”Biotechnol. Prog. 12 (1996) 290-301.
12. K. E. GÖklen and T. A. Hatton, “Liquid-liquid extraction of low molecular proteins by selectivesolubilization in reversed micelles.”Sep.Sci.Technol.22 (1987)831-841.
13. K.Naoe,O.Ura,M.Hattori,M.Kawagoeand M.Imai,“Protein extraction using non-ionic
reversemicellesofSpan 60.”J. Biochem. Eng. 2 (1998) 113-119.
14. T.Nishiki,K.Nakamura and D.Kato,“Forward and backward extraction ratesofamino acid
in reversed micellarextraction.”J. Biochem. Eng. 4 (2000) 189-195.
15. G.J.Lye,J.A.Asenjo and D.L.Pyle,“Protein extraction using reversemicelles-kinetics of
protein partitioning.”Chem. Eng. Sic. 49 (1994) 3195-3204.
16. M. Hentsch, P. Menoud, L. Steiner, E. Flaschel and A. Remken, “Optimization of the
surfactant(AOT)concentration in areversed micellarextraction process.” Biotechnol. Tech. 6
(1992) 359-364.
17. Kotlarchyk, M. Huang, J. S., Chen, S.H., “Structure of AOT Reversed micelles Determined by Small-angle Neutron Scattering.” J. Phys. Chem. (1985), 89, 4382-4386.
18. P. L. Luisi, L. J. Magid, “Solubilization of Enzymes and Nucleic Acids in Hydrocarbon Micellar solutions.”Crit. Rev. Biochem.(1986), 20, 409-474.
19. T. A. Hatton,“In Surfactant-based processes,”New York, (1989), 33, 55-90.
20. P. Panditands, Basu., ”Removal of Ionic Dyes from Water by Solvent Extraction Using
ReverseMicelles,”Environ. Sci. Technol. (2004), 38, 2435-2442
21. E. B.Leodidis, T. A. Hatton, “Amino Acids in AOT Reversed Micelles. Determination of Interfacial Partition Coefficients Using the Phase-TransferMethod.”J. Phys. Chem.(1990), 94,
6400-6411.
22. E. B. Leodidis, T. A. Hatton, “Amino Acids in AOT Reversed Micelles. The Hydrophobic Effectand Hydrogen Bonding asDriving ForcesforInterfacialSolubilization.”J. Phys. Chem.
(1990), 94, 6411-6420.
23. K.E.Goklen,T.A.Hatton,“Liquid liquid Extraction of Low Molecular Proteins by Selective Solubilization in Reversed Micelles.”Sep. Sci. Technol. (1987), 22, 831-841.
24. M.van’tRietDekker,J. W. Baltussen, , A. Bijsterbosh, B.H. Hilhorst, R.; Laane, C. ”In
Proceedings of the 4th EuropeanCongress on Biotechnology.”(1987),2,507-510.
25. G. Masahiro, H. Yukihisa, F. Taka-aki, O. Tsutomu, F. Shintaro,” Important Parameters
Affecting Efficiency ofProtein Refolding by Reversed Micelles.”Biotechnol. Prog. (2000), 16,
1079-1085
26. W.-Y. Chen, Y.-W. Lee, S.-C. Lin, C.-W. Ho.”Renaturation and Interaction of Ribonuclease A with AOT Surfactantin ReverseMicelles”.Biotechnol. Prog. (2002), 18, 1443-1446
27. K. E. Go¨klen, T. A. Hatton,“Protein extraction using reversemicelles.”Biotechnol. Prog.
(1985), 1, 69-74.
28. K. Takumi, K. Aki, “Effects of ion species in aqueous phase on protein extraction into
reversed micellarsolution,”Sep. Purif. Technol ,(2003),31,251-259
29. C. Regalado, J. A. Asenjo, D. L. Pyle,”Protein extraction by reverse micelles: Studies on the recovery ofhorseradish peroxidase.”Biotechnol Bioeng, (1994),44,674
30. Z. Hu, E. Gulari. ”Protein Extraction Using the Sodium Bis(2-ethyIhexyI) Phosphate (NaDEHP)ReverseMicellarSystem.” Biotechnol Bioeng,(1996), 50, 203-206.
31. M. Goto, T. Ono, F. Nakashio, T. Alan Hatton, ”Design ofSurfactantsSuitable for Protein Extraction by Reversed Micelles.”Biotechnol Bioeng, (1997), 54, 1
32. R. B. G. Wolbert, R. Hilhorst, G. Voskuilen, H. Nachtegaal, M. Dekker, K. van’tRiet,B. H.Bijsterbosch, “Protein transferfrom an aqueous phase into reversed micellessthe effect
33. M. J. Pires, J. M. S. Cabral,“Liquid-liquid extraction of a recombinant protein with a reverse
micellephase.”Biotechnol. Prog. (1993), 9, 647-650.
34. M. Hentsch, P. Menoud, L. Steiner, E. Flaschel, A. Renken,”Optimization ofthe surfactant
(AOT)concentration in areversemicellarextraction process.”Biotechnol. Tech. (1992), 6,
359-364.
35. M. J. M. Castro, J. J. G. Moura, J. M. S.Cabral, In Advances in Gene Technology: Protein
Engineering and Protein Production; Oxford IRL Press: Oxford, U.K., (1988); pp 136.
36. H. Ishikawa, K. Noda, T. Oka,“KineticPropertiesofEnzymesin AOT-Isooctane Reversed
Micelles.”J. Ferment. Bioeng. (1990), 6, 381-385.
37. M. Dekker, K. van’tRiet,J. W. A. Baltussen, B.H. Bijsterbosh,; R. Hilhorst, C. Laane, In
Proceedings of the 4th EuropeanCongress on Biotechnology; O. M. Neijssed, , R. R. van der
Meer, K.Ch. A. M. Luyben, , Eds.; Amsterdam, (1987); Vol. 2, pp 507-510.
38. S.-S. Lee, B.-K. Lee, J.-S. Choi, J.-P. Lee,”EffectofAlcoholAddition on Back-Extraction of BSA and Cytochrome C Using AOT Reverse Micellar System.” Bull. Korean Chem. Soc. (2001), 22, 897
39. S. Jarudilokkul, L. H. Poppenborg, D. C. Stuckey “Backward Extraction of Reverse Micellar Encapsulated Proteins Using a Counterionic Surfactant.”Biotechnol Bioeng, (1999), 62, 593-601 學生論文: 1. 翁基育“於逆流層析儀中以反微胞萃取及濃縮細胞色素分子”,國立交通大學,碩士論文, 2004年7月。 2. 傅月杏“利用HNO3-TBP錯合物之冷媒HFC-134a 於固相基質中萃取鈾金屬” 國立交通大 學,碩士論文,2005年6月。 3. 沈經緯“於逆流層析中以反微胞製備分離蛋白質”國立交通大學,碩士論文,2005年6月。
(七)計畫自評 (甲)以超臨界流體二氧化碳萃取核廢料,非常可能會成為ㄧ個新的工業技術,尤其是用“乾 式萃取法”,可避免直接使用硝酸接觸樣品,但是操作的壓力必須超過 150 bar 才有效率, 因此我們嘗試使用冷媒 R134a,原因是在相當低壓之下(15 bar)就能夠提供很高的萃取率,是 比較安全的條件,而且冷媒之回收再利用,也可以很容易的進行。本實驗相當成功的證明 R134a 可以有效率的萃取鈾金屬。但是,可能之缺點有二,其一是 R134a 或有可能在未來被 禁用,其二是此冷媒所造成之溫室效應比二氧化碳高很多。不過若是考慮其高工業安全性, 和可能節省之能源,上述二缺點或許就可以忽略。我們實驗之結果,相當程度的展顯使用 R134a 可以取代超臨界流體二氧化碳,對實際工業上而言,則必須有先導性工廠(pilot plant) 的實驗,才能確實驗證其可行性。 (乙)本實驗室使用反微胞相於逆流層析中,以萃取並濃縮蛋白質或以層析方法分離蛋白質。 以反微胞萃取蛋白質,過去文獻的研究,都是針對單一蛋白質正向以及反向萃取,作熱力學 或是動力學的研究,並未實際的來作大量樣品之萃取和濃縮,我們以逆流層析儀作為ㄧ個高 效率的濃縮器具,的確可以將大量低濃度的蛋白質水溶液,回收並濃縮至小體積中,目前比 較大的困難是回收率會受制於 pH 值的大小,若是使用特別高(例如 pH > 12)的酸鹼度,回 收率很高,但是可能會造成蛋白質變質,另外,有一些實驗條件,會讓回收速率降低,也可 能減少回收率,這些是需要克服的問題。但是,我們已經明確的彰顯出,用這個方法,可以 達到濃縮大量低濃度蛋白質的目的 至於層析方面,我們是首先將反微胞相應用於逆流層析來分離蛋白質。由於不同蛋白質分子 之間似乎有一些的作用力存在,雖然我們大體上可以將三種蛋白質分開,還不能得到完全純 粹的分離,亦即我們可以分出三大部分,每一部份都有一個蛋白質是主成分,參雜少部分另 外兩種蛋白質。在加入酸鹼以及電解質濃度梯度沖提,結果已經大幅改進。我們預期未來可 以達到完全分離,為蛋白質的分離,提供另外一種方法。 此兩部分的研究,第一部份和原計畫有一些出入,原先我們希望以超臨界流體二氧化碳來萃 取親水性物質,包括蛋白質和金屬。由於需要可溶於超臨界流體二氧化碳的介面活性劑和螯 合物,一般都含有氟原子,合成困難,而且,萃取操作壓力很高。近幾年來文獻已經大幅減 少,一般批評這種方法,理論上可行,但實際上困難度很高,也不容易工業化,因此我們改 採使用冷媒 R134a,實驗數據顯示,的確可以取代超臨界流體二氧化碳,當然還是需要工業 上實際的評估。第二部份,和原先構想一致,也達成了預期的目標。成果都正在寫成論文, 預備發表於 SCI 期刊。