國立交通大學
分子科學研究所
碩士論文
具推拉電子基之新穎紫質分子在溶液中與奈米
薄膜上的飛秒螢光動力學研究
Femtosecond Fluorescence Dynamics of Novel
Push-Pull Zinc Porphyrins in
Solutions and on Nanocrystalline Thin Films
研究生 : 徐凡軒
指導教授 : 刁維光 教授
具推拉電子基之新穎紫質分子在溶液中與奈米薄膜上的飛秒螢光動力學研究 學生 : 徐凡軒 指導教授 : 刁維光 博士 國立交通大學分子科學所碩士班 摘要 本論文研究具推拉電子基的紫質分子分別在溶液與固相奈米薄膜上的緩解 動力學。我們利用飛秒螢光上轉移技術對 ZnP1 與 ZnP2 的緩解過程作時間的解 析,其中ZnP2 具有二苯胺作為推電子基,ZnP1 則為對照組,二者的去激化過程 有十分明顯的差異。在溶液中,ZnP1 的緩解過程與 Zewail 所發表的 ZnTPP 十分 類似,但是具有二苯胺作為推電子基的ZnP2 則出現了分子內電荷轉移的過程。 固相奈米薄膜實驗中,我們比較二者在二氧化鈦與氧化鋁上的衰減速率,藉 此計算出二者電子注入的量子產率,並與製成元件後的轉換效率比較,希望能夠 有所聯結。同時我們還加入共吸附劑CDCA,希望能夠控制分子間的堆疊,以降 低分子間能量轉移的速率,而結果顯示 CDCA 確實能夠抑制堆疊的產生,並且 增加電子注入的量子產率。
Femtosecond Fluorescence Dynamics of Novel Push-Pull Zinc Porphyrins in Solutions and on Nanocrystalline Thin Films
Student : Fan Hsuan Hsu Advisor : Dr. Eric Wei-Guang Diau
Institute of Molecular Science National Chiao Tung University
Abstract
The excited-state relaxation dynamics of zinc porphyrins with electron
withdrawing and donating group were studied in this thesis. With the application of fluorescence up-conversion technique, the relaxation processes of ZnP1 and ZnP2 upon S2 excitaiton were reported. In ZnP1, the dynamics is similar to the results reported by Zewail et. al. In ZnP2, the result suggests that an additional
intramolecular charge transfer occurs between porphyrin moiety and acid group, which causes the significant change of transient
We calculate electron injection yield by comparing excited-state lifetime of each zinc-porphyrins on both TiO2 and Al2O3 solid nanocrystalline films, the relationship
between injection yield and efficiency is discussed. Mixing coadsorbate CDCA, which lowering the rate of intermolecular energy transfer, also controls aggregation of porphyrins on solid nanocrystalline films. The results show higher electron injection yield with higher concentration of CDCA.
謝誌
在我研究的過程中受到許多人的幫忙,寫謝誌的時候一一浮現於腦海中,真 的非常感謝。感謝刁老師對我碩士論文的指導及實驗方向的諸多指點;感謝光譜 組的各位同仁,一起做實驗與討論的過程非常愉快,也十分懷念與各位一起 Meeting 的時光;感謝元件組的同仁,提供我實驗所需的材料及效率的測量,我 知道他們真的很辛苦,感激不盡。最後特別感謝人在美國的志煒學長,跟著他做 實驗的那段日子十分充實使我獲益良多;感謝葉鎮宇及洪政雄兩位幫我口試的老 師,很仔細地閱讀我的論文並且給予寶貴的意見。很榮幸能夠在刁老師實驗室完 成我的碩士學位,謝謝大家。目錄 第一章 序論………1 1-1 太陽能電池的發展起源………1 1-2 染料敏化太陽能電池的研究………3 1-2-1 染料敏化太陽能電池的發展回顧………3 1-2-2 染料敏化太陽能電池的結構原理………4 1-3 敏化染料的研究………9 1-3-1 釕錯合物在染料敏化太陽能電池上的應用………9 1-3-2 質在染料敏化太陽能電池上的應用 ………11 1-3-2-1 紫質分子的光譜原理 ………12 1-3-2-2 紫質分子的相關研究 ………15 1-3-2-3 紫質分子的聚集(aggregation)………18 1-4 本論文的研究 ………20 1-5 第一章的參考文獻 ………21 第二章 實驗測量與儀器原理 ………23 2-1 紫外-可見光吸收光譜儀 ………23 2-2 螢光光譜儀 ………23 2-2-1 CCD(Charge-Coupled Device)光譜儀 ………24 2-3 時 間 相 關 單 光 子 計 數 系 統 (Time-Correlated Single PhotonCounting ,TCSPC) ………25 2-3-1 時間相關單光子計數系統的裝置圖及工作原理 ………25 2-3-2 時間相關單光子計數系統的組成元件 ………28 2-4 飛秒時間解析螢光上轉移技術(femtosecond fluorescence up-conversion) ………32 2-4-1 飛秒雷射系統 ………32
2-4-1-1 摻鈦藍寶石雷射光譜 ………32 2-4-1-2 超短脈衝雷射震盪器………33 2-4-1-3 脈衝選擇系統………38 2-4-1-4 自動校正儀………39 2-4-2 螢光上轉移技術………41 2-4-3 螢光光柵系統………43 2-5 數據分析………45 2-6 第二章的參考文獻………49 第三章 鋅紫質衍生物在溶液中的光譜與動力學研究………50 3-1 相關研究回顧………50 3-2 鋅紫質衍生物的名稱與結構………52 3-3 鋅紫質衍生物在溶液中的穩態光譜………53 3-3-1 ZnP1與ZnP2在甲苯溶液中的穩態吸收及螢光光譜………54 3-3-2 ZnP2在多種溶劑中的穩態吸收及螢光光譜………55 3-4 ZnP1與ZnP2在溶液中的緩解動力學………58 3-4-1 ZnP1在甲苯溶液中的激發態生命期測量………58 3-4-2-1 ZnP2在甲苯溶液中的激發態生命期測量………62 3-4-2-2 ZnP2在乙腈溶液中的激發態生命期測量………68 3-5 ZnP2在溶液中的緩解動力學討論………73 3-6 第三章的參考文獻………77 第四章 鋅紫質衍生物在固相薄膜上的光譜與動力學研究………78 4-1 ZnP1 在固態薄膜上的緩解過程研究 ……… 79 4-1-1 ZnP1 在二氧化鈦固態薄膜上的吸收與螢光穩態光譜 ……… 81 4-1-2 ZnP1 在二氧化鈦固態薄膜上的瞬態光譜 ……… 83 4-1-2-1 第二激發態的緩解動力學………83
4-1-3 ZnP1 在氧化鋁固態薄膜上的吸收與螢光穩態光譜 ……… 86 4-1-4 ZnP1 在氧化鋁固態薄膜上的瞬態光譜 ……… 87 4-1-4-1 第二激發態的緩解動力學………87 4-1-4-2 第一激發態的緩解動力學………88 4-1-5 ZnP1 在二氧化鈦固態薄膜上的電子注入速率常數與產率計算 … 90 4-2 ZnP2 在固態薄膜上的緩解過程研究 ……… 93 4-2-1 ZnP2 在二氧化鈦固態薄膜上的吸收與螢光穩態光譜 ……… 93 4-2-2 ZnP2 在二氧化鈦固態薄膜上的瞬態光譜 ……… 95 4-2-3 ZnP2 在氧化鋁固態薄膜上的吸收與螢光穩態光譜 ……… 97 4-2-4 ZnP2 在氧化鋁固態薄膜上的瞬態光譜……… 100 4-2-5 ZnP2 在二氧化鈦固態薄膜上的電子注入速率常數與產率計算… 102 4-3 鋅紫質衍生物在二氧化鈦固相薄膜上的介面電子轉移機制…… 107 4-4 第四章的參考文獻 ………109 第五章 結論……… 110 附錄 A ……… 112
圖目錄 圖 1-1 染料敏化太陽能電池結構示意圖………5 圖 1-2 染料敏化太陽能電池電子轉移過程示意圖,其中 S* 為染料的激發 態,S 為染料的基態,S+ 為染料的氧化態。CB 和 VB 分別為 TiO2的 導帶與價帶………7 圖 1-3 三種釕-多吡啶錯合物的分子結構……… 10 圖 1-4 紫質環分子的基本結構 ……… 11 圖 1-5 (a)原血紅素 (b)葉綠素的分子結構……… 12 圖 1-6 (a)H2TPP (b)ZnTPP 的分子結構及在苯溶劑中的吸收、靜態螢光光 譜……… 13 圖 1-7 四軌域模型中紫質的 LUMOs(上列)及 HOMOs(下列),藍色與紅色分 別表示不同相之π軌域……… 15 圖 1-8 紫質中電子在不同 (a)軌域 (b)能態 間的躍遷示意圖………… 15 圖 1-9 Zewail 等人提出的(a) H2TPP (b)ZnTPP 在苯溶劑中的去激化動力學 模型……… 17 圖 1-10 聚集體偶極矩與分子中心連線間的夾角θ ……… 18 圖 1-11 激子模型中各種排列方式的雙分子聚集體能階圖 ……… 19 圖 1-12 ZnP1 與 ZnP2 鋅紫質衍伸物的結構……… 20 圖 2-1 CCD 光譜儀內部結構示意圖………24 圖 2-2 時間相關單光子計數系統工作原理 ……… 26 圖 2-3 Fluo Time 200 儀器配置圖………27 圖 2-4 時間相關單光子計數系統模組訊號計時運作流程 ……… 28 圖 2-5 時間鑑別器根據門檻電壓判別訊號類別 ……… 29 圖 2-6 TAC 偵測單一光子的計時機制………30
圖 2-8 超快雷射脈衝震盪器 Mira900D 裝置圖……… 34 圖 2-9 克爾透鏡鎖模示意圖 ……… 36 圖 2-10 群速分散補償裝置示意圖 ……… 37 圖 2-11 脈衝選擇器結構示意圖 ……… 39 圖 2-12 自動校正儀裝置圖 ……… 40 圖 2-13 非線性光學晶體上的混頻現象 ……… 42 圖 2-14 螢光上轉移的原理示意圖 ……… 43 圖 2-15 FOG100 螢光光柵系統結構 ……… 44 圖 3-1 T. Peltola 等人提出的 ZnTPPS 4-- MV2+ 錯何物電子轉移動力學模型, 其中 kic為內轉換過程的速率,k2、k1分別為 S2與 S1電子轉移速率, kr為電子回傳速率 ……… 51 圖3-2 ZnTPP-PI 與 ZnTPP-β-PhPI 的分子結構………52 圖 3-3 Osuka 等人發表之電子轉移速率與能距在四種溶劑中的關係趨勢 52 圖3-4 ZnP1與ZnP2紫質衍生物分子的名稱與結構……… 53 圖3-5 (a)ZnP1與(b)ZnP2 在甲苯中的穩態吸收及螢光光譜,溶液濃度為1 ×10-5 M,激發波長為435 nm ……… 54 圖3-6 ZnP2在各種溶劑中的吸收及螢光光譜,藍色線條為吸收光譜,紅色 線條為螢光光譜,溶液濃度為1×10-5 M,螢光測量激發波長為435 nm……… 57 圖3-7 ZnP2在不同溶劑中的史托克位移對該溶劑Δf的變化關係圖……57 圖3-8 ZnP1在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,觀測波長 為480nm,紅色圓圈為原始數據,黑色實線為擬合結果,綠色實線 為原始數據與擬合結果之差值 ………59 圖3-9-1 ZnP1在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值 ………… 60
圖3-9-2 ZnP1在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值 ………… 61 圖3-10 ZnP2在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,觀測波 長為480 nm,紅色圓圈為原始數據,黑色實線為擬合結果,黑色虛 線為空白溶劑的測量結果,綠色實線為原始數據與擬合結果之差 值……… 63 圖3-11-1 ZnP2在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值………… 65 圖3-11-2 ZnP2在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值………… 66 圖3-11-3 ZnP2在甲苯溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值………… 67 圖3-12 ZnP2在乙腈溶液中的螢光瞬態光譜。激發波長為430 nm,觀測波長 為480 nm,紅色圓圈為原始數據,黑色實線為擬合結果,綠色實線 為原始數據與擬合結果之差值……… 69 圖3-13-1 ZnP2在乙腈溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值………… 70 圖3-13-2 ZnP2在乙腈溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態
圖3-13-3 ZnP2在乙腈溶液中的螢光瞬態光譜。激發波長為430 nm,紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為各能態 的模擬結果,綠色實線為原始數據與擬合結果之差值………… 72 圖3-14 ZnP1在苯溶液中的緩解動過程示意圖,激發波長為430 nm………73 圖3-15 ZnP2在(a)甲苯 (b)乙腈 溶液中的緩解動力學模型示意圖。其中τ IC、τVR、τISC、τCT1及τCT2分別為內轉換、振動緩解、系統間交叉、 S1能態及S2能態的電子轉移過程時間常數………76 圖 4-1 二氧化鈦、氧化鋁與 ZnP1、ZnP2 的絕對能階圖……… 79 圖 4-2 (a)二氧化鈦(b)氧化鋁 固相薄膜的 SEM 照片……… 80 圖 4-3 ZnP1 在二氧化鈦薄膜上的穩態吸收光譜及螢光光譜,實線為吸收光 譜 虛 線 為 螢 光 光 譜 , 與 CDCA 的 莫 耳 濃 度 比 (a)1:0(b)1:2(c)1:10………82 圖 4-4 ZnP1 吸附在二氧化鈦薄膜上的螢光瞬態光譜。激發波長為 430 nm,觀測波長為 480 nm,紅色圓圈為原始數據,黑色實線為擬合 結果,綠色實線為原始數據與擬合結果之差值……… 84 圖 4-5 ZnP1 在二氧化鈦薄膜上的螢光瞬態光譜。激發波長為 430 nm,紅 色圓圈為原始數據,黑色實線為擬合結果,藍色、粉紅色及橘色為 各組成的模擬結果,綠色實線為原始數據與擬合結果之差值… 85 圖 4-6 ZnP1 在氧化鋁薄膜上的穩態吸收光譜及螢光光譜,實線為吸收光譜 虛線為螢光光譜,與 CDCA 的莫耳濃度比(a)1:0(b)1:2(c)1:10…86 圖 4-7 ZnP1 吸附在氧化薄膜上的螢光瞬態光譜。激發波長為 430 nm,觀 測波長為 480 nm,藍色、紅色、綠色分別表示 ZnP1 與 CDCA 的濃 度比為 1:0、1:2 與 1:10,圖形為原始數據,實線為擬合結果 ……88 圖 4-8 ZnP1 在氧化鋁薄膜上的螢光瞬態光譜。激發波長為 430nm,紅色圓 圈為原始數據,黑色實線為擬合結果,紫色、藍色、粉紅色及橘色
為各組成的模擬結果,綠色實線為原始數據與擬合結果之差值…89 圖 4-9 ZnP1 吸附在氧化薄膜上的螢光瞬態光譜。激發波長為 430 nm,分 別比較 S2的衰減與 S1的上升過程,藍色、紅色、綠色分別表示 ZnP1 與 CDCA 的濃度比為 1:0、1:2 與 1:10,圖形為原始數據,實線為 擬合結果……… 90 圖 4-10 ZnP2 在二氧化鈦薄膜上的穩態吸收光譜及螢光光譜,實線為吸收光 譜 虛 線 為 螢 光 光 譜 , 與 CDCA 的 莫 耳 濃 度 比 (a)1:0(b)1:2(c)1:10(d)1:30 ……… 94 圖 4-11 ZnP2 陽 離 子 在 二 氧 化 鈦 薄 膜 上 於 (a) 黑 暗 中 (b) 照 光 (c) 經 由 Ferrocene 還原的吸收特徵峰表現 ……… 95 圖 4-12 ZnP2 在二氧化鈦薄膜上的螢光瞬態光譜。激發波長為 430 nm,紅 色圓圈為原始數據,黑色實線為擬合結果,藍色、粉紅色為各組成 的模擬結果,綠色實線為原始數據與擬合結果之差值………96 圖 4-13 ZnP2 在氧化鋁薄膜上的穩態吸收光譜及螢光光譜,實線為吸收光譜 虛 線 為 螢 光 光 譜 , 與 CDCA 的 莫 耳 濃 度 比 (a)1:0(b)1:2(c)1:10(d)1:30 ……… 97 圖 4-14 ZnP2 在(a)二氧化鈦(b)氧化鋁 固態薄膜上的穩態螢光強度比 較………100 圖 4-15 ZnP2 在氧化鋁薄膜上的螢光瞬態光譜。激發波長為 430 nm,紅色 圓圈為原始數據,黑色實線為擬合結果,紫色、藍色、粉紅色及橘 色為各組成的模擬結果,綠色實線為原始數據與擬合結果之差 值………101 圖 4-16 ZnP2 在(a)二氧化鈦(b)氧化鋁 薄膜上不同觀測波長的生命期比 較………103
圖 4-17-1 ZnP2 在二氧化鈦薄膜上不同觀測波長的螢光瞬態光譜。激發波長 為 430 nm,紅色圓圈為原始數據,黑色實線為擬合結果,藍色、 粉紅色及橘色為各組成的模擬結果,綠色實線為原始數據與擬合結 果之差值………104 圖 4-17-2 ZnP2 在氧化鋁薄膜上不同觀測波長的螢光瞬態光譜。激發波長為 430 nm,紅色圓圈為原始數據,黑色實線為擬合結果,紫色、藍色、 粉紅色及橘色為各組成的模擬結果,綠色實線為原始數據與擬合結 果之差值………105 圖 4-18 鋅紫質衍伸物以 430 nm 激發光源激發至 S2能態後的緩解機制示意 圖………107
表目錄 表 2-1 標準偏差的衍伸計算 ……… 48 表3-1 各種溶劑的Δf值與ZnP2在其中的吸收螢光位置及史托克位移… 57 表3-2 ZnP1在甲苯溶液中的螢光上轉移瞬態光譜擬合結果………62 表3-3 ZnP2在甲苯溶液中的螢光上轉移瞬態光譜擬合結果………68 表3-4 ZnP2在乙腈溶液中的螢光上轉移瞬態光譜擬合結果………73 表 4-1 ZnP1 的電子注入速率常數與量子產率………92 表 4-2 ZnP2 在二氧化鈦薄膜上的穩態吸收光譜與螢光光譜特徵峰位置…94 表 4-3 ZnP2 在氧化鋁薄膜上的穩態吸收光譜與螢光光譜特徵峰位置……98 表 4-4 ZnP2 在二氧化鈦薄膜上電子注入過程的量子產率………99 表 4-5 ZnP2 的電子注入速率常數與量子產率 ……… 102 表 4-6 ZnP2 在不同觀測波長的電子注入速率常數與量子產率………… 106
第一章 序論
在這一章節裡面,我們回顧了染料敏化太陽能電池的發展歷史並介紹其結構 原理,最後說明紫質(porphyrin)在其中所扮演的角色和功能。 1-1 太陽能電池的發展起源 近百年來,人類的經濟活動蓬勃發展。然而在現代化的過程中,伴隨著各種 風險,能源危機便是其中之一。從20 世紀直至今日,由於石化燃料的逐漸短缺 加上全球氣候的異變,人們開始注意到能源的問題,因此綠色能源的議題開始在 各國之間發酵。 太陽能是綠色能源中最具有潛力的項目之一,普遍、長久、零污染是其無可 取代的優勢。太陽能分怖廣闊,取得方便,儘管會有地理以及氣象方面的差異使 得各地能夠利用的太陽能資源有所不同,但它不需要開採、運輸,能夠就地利用。 此外,太陽能的利用不會產生任何廢料、廢水,亦沒有任何噪音,對於環境沒有 傷害,於地球的永續發展有極大的意義。地球接受來自太陽的能量換算成電力有 1.22x1017瓦1,這樣的能量比全世界的電力需求量還多了三萬倍,因此太陽能具 有非常高的發展價值。 太陽能的利用主要分為三種,光電轉換,光熱轉換以及光化學轉換。三者之 中尤以光電轉換的部分最受矚目,因此如何提高光電的轉換效率被廣泛地研究。 太陽能電池是太陽能光伏發電的基礎和核心,是一種能把光能轉變成電能的器件 (Photovoltaic device)。依照材料能夠分為矽太陽能電池、無機化合物半導體太陽 電池(Inorganic Compound Semiconductor Solar Cell)、有機化合物太陽電池 (Organic Compound Solar Cell)、塑膠太陽電池以及敏化柰米晶太陽電池 (Sensitized Nanocrystal Solar Cell),本論文所研究的染料敏化太陽能電池(Dye Sensitized Solar Cell,DSSC)即屬於此類太陽電池。稱為光伏效應(Photovoltaic effect)。光伏效應可追溯到西元 1839 年,法國的物理 學家Becqurel 等人發現用光照射電解質後,在電解質中會產生電壓,進而產生 電流2。1876 年,在硒的全固態系統中也發現類似的現象。1882 年就製成了硒的 光伏電池。1928 年又製成了銅-氧化銅(Cu-Cu2O)的光伏電池。1929 年建立了固 體能帶理論,首次論證了能夠利用太陽電池將太陽能轉化為電能。1939 年,製 成矽結(Silicon junction)型電池。雖然在 1941 年就存在著有關矽電池的報導,但 是一直到1954 年 D. M. Chapin 等人在美國貝爾實驗室首次製成了光電轉換效率 6%的單晶矽太陽電池3,光伏發電技術才算真正進入實用階段,並有了矽電池的 第一代產品,它的出現象徵著太陽電池研發進度的重大突破。1955 年建立了太 陽電池的理論,此後太陽電池在理論上以及製成方面都有非常快速的發展。1956 年人們開始把太陽應用到地面的航標燈、閃光燈和通訊機上,隨後更運用到了太 空船以及人造衛星上,此時的矽太陽電池光電轉換效率已經有了顯著的突破。隨 著人類探索宇宙的腳步加快,同時也促進了太陽電池的發展。儘管當時太陽電池 在航太領域有了十分不錯的成果,但是成本昂貴,生活在地面上的人們對於太陽 電池的利用依然十分有限,直到西元1973 年爆發了石油危機,人類的能源結構 產生重大變革,才漸漸地將太陽電池利用於地面日常生活當中。1974 年,Haynos 等人利用矽之非等方性(anisotropic)的蝕刻(etching)特性,慢慢地將太陽電池表面 的矽(111)結晶面刻出許多類似金字塔的特殊幾何形狀,能夠有地改善太陽光在電 池表面反射的情況,當時的電池轉換效率已達17%。1976 年後,整個產界將重 心放在降低成本、提高轉換效率以及提高電池穩定性等目標。 在1990 年之前,太陽電池的材料是以矽基的單晶矽、多晶矽以及非晶矽為 主,這是因為矽基材料的製造以及取得比較容易,價格相對上也比較低廉。然而 商業化的矽基太陽電池在效率上始終難以突破13-16%的限制,因此人們開始嘗 試以其他材料製作太陽電池的可能性。以III-V 族所形成的半導體材料也能夠應 用在太陽電池上,如GaAs,GaInP 等。由於這類半導體材料擁有直接能隙(direct
長。然而此類太陽電池的價格高出矽晶太陽電池的數十倍,因此並沒有辦法普及 化。II-VI 族的化合物半導體同樣也具有直接能隙,所以同樣能運用於太陽電池 上,如CdTe 太陽電池。由於 CdTe 的能隙值為 1.45 eV,正好位於理想太陽電池 的能隙範圍之間,因此算是很好的電池材料。然而在環保意識高漲的今日,鎘污 染的問題成為了這類電池發展上的隱憂。此外還有Cu(InGa)Se2、CuInSe24等多 元化合物在大面積、可撓式等應用領域都有不錯的表現,但是同樣受製成複雜、 材料昂貴以及毒性問題的限制。 除了前述幾種半導體太陽電池以外,在過去幾十年間利用光電化學反應所製 造的太陽電池也被廣泛地研究著。這類電池的組成分為幾個部分,光導電極 (photoelectrode)、氧化還原電解質(redox electrolyte)以及催化用的輔助電極 (counter electrode)等。Si、GaAs 等半導體雖然也能夠做為光導電極的材料,但是 在光線照射之下,這類電極在電解質中容易發現腐蝕現象,造成電池穩定性不良 而限制了發展。寬帶隙半導體,如TiO2、SnO2等在電解質溶液中有著高穩定性, 但是其吸收範圍只有在紫外光,因此捕獲太陽光的能力非常差,無法直接用太陽 能的轉換。科學家們在研究中發現,將與這些寬帶隙半導體之導帶價帶相配的有 機染料附著於其上,能夠藉由這些染料對於太陽光的吸收讓整個系統的吸收範圍 擴展到可見光區,此現象稱作半導體的染料敏化作用,而以這樣的原理製作而成 的太陽電池就叫做染料敏化太陽能電池。 1-2 染料敏化太陽能電池的研究 1-2-1 染料敏化太陽能電池的發展回顧 半導體染料敏化技術的形成可以追溯到照相術發展的初期。1949 年, Putzeiko 和 Trenin 首次報導了有機光敏染料對半導體的敏化作用5。隨後在70 到 90 年代 R. Memming 和 H. Gerischer 等人大量地研究各種有機敏化劑與半導體薄 膜間的光敏作用6-9。早期這方面的研究主要是集中在平板電極上,然而這類的 電極僅能在表面吸附一層染料分子,由於相對的表面積較小,表面上的單分子層
染料對於光的捕獲能力較差,能量效率大概都在0.1%以下。人們試圖利用多層 吸附以增加光的捕獲效率,然而內層的染料對外層染料的電子轉移過程會造成阻 礙作用而降低光電轉化量子效率。這樣的問題是20 世紀 90 年代以前限制染料敏 化太陽電池發展的主要因素,光電轉換效率始終在1%以下,並不實用。 1985 年,瑞士洛桑高等工業學校的 M. Grätzel 利用奈米晶二氧化鈦半導體電 極提高了吸附表面積,而克服了平板電極因為吸附表面積小造成光補獲效率差的 問題10。奈米晶半導體薄膜的多孔性結構使其表面積遠遠大於幾何面積,以10 mm 厚的二氧化鈦薄膜為例,其總表面積可以增加約兩千倍。1991 年 M. Grätzel 等人首次將金屬釕有機錯合物作為染料吸附在二氧化鈦的奈米晶多孔膜上製成 電池,其光電轉換效率高達7.1%11,此後這種製作簡單、成本低廉的太陽電池因 此吸引了眾多研究者的目光。 1-2-2 染料敏化太陽能電池的結構原理 染料敏化奈米薄膜太陽電池具有穩定、廉價以及製程簡單的優點,其中主要 的半導體薄膜材料是奈米二氧化鈦,由於其具有含量豐富、成本低廉、穩定、無 毒和抗腐蝕的優勢,加上電池製作過程採用大面積網印技術和簡單的浸泡方法, 所以適用於工業化大面積的生產。在一般的p-n 結光伏電池中,半導體同時具有 捕獲入射光以及傳導光生載流子的兩種作用,但是在染料敏化太陽電池中,這兩 種作用的進行是分開的。如圖1-1 所示,染料敏化太陽能電池是由一吸附著染料 的二氧化鈦多孔薄膜作為陽極,與電解質、金屬電極形成了”三明治”的結構。 二氧化鈦擁有三種晶體結構,板鈦礦、金紅石以及銳鈦礦,其共同的組成基本單 元為[TiO6]8-八面體。板鈦礦是由氧密堆積而成,鈦原子處於八面體中心位置; 紅金石結構也是建立在氧的密堆積上,儘管它的晶體結構不是一種密堆積方式, 和板鈦礦一樣相對於理想八面體有些許的變形,並且是以共頂點的方式相連接; 銳鈦礦是由八面體共邊結構所組成,實際上可視為一四面體結構,而鈦-氧之間
圖1-1 染料敏化太陽能電池結構示意圖。 石,由於無法合成且在自然界含量有限,所以工業上的應用價值不高。紅金石則 是最穩定的結晶型態,在自然界中為數不多,多為人工製造。銳鈦礦在常溫下是 穩定的,但在高溫下會轉化為紅金石,由於穩定性不如金紅石,在導帶(Ti 3d)中 的軌域重疊比較多,能夠更有效地接受價帶(O 2p)上端所激發出來的電子而擁有 更好的光催化活性,因此一般的染料敏化太陽能電池都是使用銳太礦相的二氧化 鈦。經由半導體的光吸收值(λg)與能隙寬度(Eg)的關係公式 λg (nm) = 1240 / Eg (eV) (1-1)
可以得知其吸收主要是在紫外光區,因此入射光的捕獲必須由吸附於半導體薄膜 上的染料敏化劑所完成。在受到光的激發以後,染料中的電子從基態躍遷到激發 態,接著注入到半導體的導帶中,注入到導帶中的電子可以很快地移動到薄膜與 導電玻璃的接觸面而流到外電路中,到達另一端的金屬製陰極,最後透過電解質 的傳輸,完成整個迴路。 電解質在染料敏化太陽能電池中有著傳輸電子與還原染料的作用。此外一些 研究也指出電解質還具有改變二氧化鈦氧化還原能階的能力12,因此對於光電壓 的影響很大。目前的電解質主要由碘和碘的化合物(如 LiL、KI 等)溶解在有機溶 劑中組成。I-/I3-氧化還原對具有很好的穩定性、可逆性與高的擴散常數,更重要 的是在可見光的範圍吸收可忽略,因此備受青睞。 圖 1-2 描述了染料敏化太陽能電池系統的電子轉移過程,藉由這些過程完成 圖 1-2 染料敏化太陽能電池電子轉移過程示意圖,其中 S*為染料的激發態,S 為染料的基態, S+為染料的氧化態。CB 和 VB 分別為 TiO 2的導帶與價帶。
光電轉換。其中的電子轉移過程分別為: (1) 於可見光照射之下,被以化學吸附於 TiO2半導體薄膜上的染料吸收,其電子 由基態躍遷至激發態: S+hν →S∗ (2)由於電子在激發態並不穩定,因此會透過染料分子與TiO2半導體薄膜表面間 的化學鍵結或者隔空的穿遂13注入到能量較低的TiO2導帶中,而染料本身因 為失去電子而被氧化: 2 ( ) S∗ →e TiO− +S+ 選擇能階匹配的染料與半導體薄膜能有助於此電子注入的過程,更利於迴路 的完成。 (3)一旦電子進入TiO2導帶中將很快地通過TiO2層進入收集電極流向外電路: 2 ( ) e TiO− →e− (4)電解質 I3-被來自TiO2導帶通過外電路而到達陰極的電子還原成I-: 3 2 ( ) 3 I −+ e Cathode− → I− (5)氧化態的染料分子從電解質 I-得到電子而恢復為還原態:
3 3 S++ I− → +S I − 至此為一完整的迴路。 除了迴路中的過程以外,同時會有其他的過程降低電池的光電流。 (6)導帶中的電子與氧化態的染料分子複合: 2 ( ) S++e TiO− →S (7)少量被氧化的電解質不經過外電路而直接從導帶上得到電子還原,形成暗電 流,因此應該避免電解質與TiO2的直接接觸: 3 2 ( 2) 3 I −+ e TiO− → I− 通常在染料分子中,電子處於激發態的時間越長,越有利於電子的注入;反 之,若是電子在激發態的時間很短,就越可能經由其他非輻射的過程緩解回到基 態而來不及注入到TiO2的導帶中。過程(2)和(6)分別是電子注入的順與逆反應, 二者的速率常數比反映了注入過程的效率好壞。過程(7)是造成電流損失的主要 原因之一,因此若是電子在TiO2層中的移動速度越快,和電解質的複合過程產 生的機會就越小,也就能夠降低電流的損失。由於奈米晶半導體薄膜之中並沒有 空間電荷層,因此電荷的有效分離仰賴各反應的速度,也就是說電子注入、染料 還原等正向反應的速率應遠大於逆向的複合過程。 電池的開路電壓(Voc)如圖 1-2 所示,由二氧化鈦的導帶與電解質之氧化還原 電位的電位差所決定:
其中 2 , CB TiO E 為TiO2導帶電位,ER R/ −為電解質的氧化還原電位,q 為完成氧化還 原過程所需要的電子數。 1-3 敏化染料的研究 敏化染料能夠藉由可見光的吸收,將被激發的電子傳送到 TiO2電極的導 帶,並同時接受來自電解質的電子而還原。理想的敏化染料須具備下列幾種條件: (1) 在可見光範圍有良好的吸收度。 (2) 在 TiO2表面能夠有效吸附而不易脫落,因此染料分子需要具有易與TiO2奈
米半導體薄膜表面結合的官能基,如-COOH、-SO3H 和-PO3H2等。以-COOH
為例,能夠加強TiO2的3d 軌與染料分子π軌域間之電子偶合,使電子轉移 更容易。 (3) 染料的氧化態(S+)和激發態(S*)需具有穩定性與高活性。 (4) 激發態的氧化還原電位要夠高,以確保激發態的電子能有效地轉移至 TiO2。 (5) 激發態的壽命長,不易有其他緩解路徑與電子轉移相競爭。 (6) 染料分子激發態和 TiO2導帶的能階需匹配以增進電子轉移的過程。 1-3-1 釕錯合物在染料敏化太陽能電池上的應用 目前應用在染料敏化太陽能電池的各種有機染料之中,以 Grätzel 團隊所發 展出來的釕-多吡啶錯合物使用最為普遍14,15。這類的有機染料在可見光範圍有 很廣的吸收,又因為它的氧化態穩定耐久度高,優良的光電化學性質,在製成太 陽能電池以後能有 10%以上的轉換效率。圖 1-3 是三種常見的釕-多吡啶錯合物 分子結構:
N N N N N N Ru COOH COOH COOH HOOC HOOC COOH N N N N NCS NCS Ru COOH HOOC HOOC COOH
Ru(dcbpy)3 N3 dye : Ru(dcbpy)2(NCS)2
N N N NCS NCS NCS Ru HOOC HOOC HOOC Black dye : Ru(tctpy)(NCS)3 圖1-3 三種釕-多吡啶錯合物的分子結構。 其中N3 與 Black dye 在可見光的吸收範圍都很廣,光電轉換效率也非常好,尤 其是N3 染料,其相關的研究亦十分蓬勃16-18。這類染料上的羧基能夠與TiO2薄 膜表面形成穩定的化學鍵結,NCS 官能基也有助於可見光的吸收。雖然目前還 沒有發展出比釕錯何物更好的染料,但是在地球上釕元素的含量十分稀少,加上 N3 染料在合成使用上有專利權的考量,因此我們希望能夠開發出轉換效率接近
N3 且成本較低的染料,讓染料敏化太陽能電池於應用上更為普及也更符合經濟 效益。 1-3-2 紫質在染料敏化太陽能電池上的應用 紫質(Porphyrin)來自於希臘文 porphura,意思是紫色,其基本結構如圖 1-4 所示,由四個吡咯(pyrrole)以及四個未飽和的碳橋相接而成。雖然紫質環周圍的 取代基可以有無數種組合,但是存在於自然界中的形式卻只有少數幾種而已。通 常人工的紫質環會在9~12 的位置加上取代基,而天然形式的紫質環則是在 1~ 8 的位置被取代。 N NH N HN 1 2 3 4 5 6 7 8 9 10 11 12 圖1-4 紫質環分子的基本結構。 如果在紫質環分子的中央以氮原子上的孤對電子與金屬離子嵌合形成錯合 物,我們稱作為金屬離子紫質(metal ion porphyrin),而這類反應也是紫質最顯著 的化學特性。大多數具有生理功能的紫質都是屬於金屬離子紫質,例如以二價的 鐵離子為中心的金屬離子紫質稱作原血紅素(Heme),存在於血紅素及肌紅蛋白
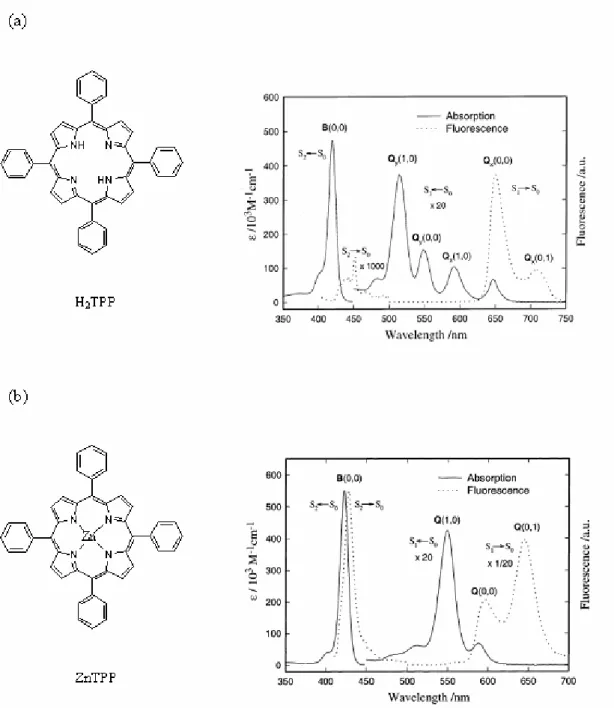
中;而若是以二價鎂離子為中心則稱為葉綠素(Chlorophyll),存在於高等植物的 葉肉細胞中,參與光合作合,如圖1-5。 (a) (b) N N N N Fe COOH COOH N N N N Mg R COOphytyl O H3COOC R=CH3 or CHO 圖1-5 (a)原血紅素 (b)葉綠素的分子結構。 由於過渡金屬通常擁有許多的氧化態,因此這類金屬所形成的金屬離子紫質在激 發態的去激化過程中可能包含了各種氧化態的緩解過程以及電子轉移,太過於複 雜而不利於非輻射緩解過程及電子轉移方面的研究,因此我們選擇了只有單一氧 化態(+2 價)的鋅金屬離子紫質及其衍生物作為研究的對象。 1-3-2-1 紫質分子的光譜原理 紫質的吸收光譜是由一個較強的 B 頻帶(或者稱作 Soret 頻帶),以及另一較 弱的Q 頻帶組成,二者的位置大約分別在 400 nm 及 550 nm 附近(依取代基而有 所不同),如圖 1-6。
圖1-6 (a)H2TPP (b)ZnTPP 的分子結構及在苯溶劑中的吸收、靜態螢光光譜。
紫質環是一含有 18 個π電子的共軛系統,其吸收躍遷與共軛環多烯(cyclic polyene)類似屬於π-π*躍遷。Gouterman 在 1961 年提出了四軌域模型(four-orbital model)來解釋紫質的吸收光譜特徵 19,所謂的四軌域指的是兩個 HOMO 及兩個
如圖所示,HOMO 的軌域對稱性有 a2u及a1u兩種,而LUMO 則均為 eg,紫質的 吸收來自於電子在這四個軌域間的躍遷,如圖1-8(a)。吸收躍遷經由線性組合後 在能量上產生分裂而得到兩個對稱性均為Eu 的能態,較高的能態其振子強度 (oscillator strength)也較強,對應的吸收峰即為 B 頻帶;另一較低能的能態振子 強度較低,吸收峰為Q 頻帶,如圖 1-8(b)。 圖1-8 紫質中電子在不同 (a)軌域 (b)能態 間的躍遷示意圖 1-3-2-2 紫質分子的相關研究
1880 年,Seyler 從血紅素中製得 hematoporphyrin,並且在 chlorophyll 中分 離得到phylloporphyrin,發現兩種錯合物在去掉中心金屬離子以後有著類似的分 子結構20。1902 年,Zelaki 首次合成二價的銅與鋅金屬離子紫質21,此後化學上 的研究進展突飛猛進,例如三明治夾心結構的稀土金屬離子紫質22、酞菁23等, 為紫質的研究應用開闢了許多不同的領域。
1987年,隨著飛秒級超快脈衝雷射的問世,動力學的研究進入了飛秒化學 (femtosecond chemistry)的新領域,皮秒(picosecond)與飛秒級的超快速光物 理化學反應漸漸能夠被人們解析出來。1997年,Gurzadyan等人利用螢光上轉換 技術(fluorescence up-conversion,見第二章)觀察ZnTPP在乙醇溶劑中的S2螢光衰 減過程發現了其時間常數為2.35 ps24,而此值與S1螢光的上升(rise)過程的時間常 數一樣,因此推測這個過程是由S2->S1的內轉換過程(internal conversion,IC)。 2000年Mataga等人同樣對於ZnTPP在乙醇溶劑中的螢光衰減過程作觀察而 得到了類似的S2->S1內轉換過程時間常數2.3 ps,同時他們還在較短的觀測波長發 現了屬於S1的高振動能態衰減過程,比起S1低振動能態的衰減過程快上許多,因 此提出了電子由S2內轉換到達S1以後,能夠有兩種緩解途徑的可能模型:一是緩 解至接近S1最低振動能態附近的能態,另一則是經由分子內電子振動重新分配 (intramolecular vibronic redistribution,IVR)來到高振動能態,接著再與其他能態 偶合緩解25。 2002年Zewail的研究團隊分別對H2TPP及ZnTPP在苯溶劑中的螢光緩解過程 作了詳細的研究26,27。首先是H2TPP的研究,他們藉由激發B、Qy以及Qx頻帶分離 出各個能態所發生的過程,而得到電子從S2能態緩解至基態的完整過程。他們提 出的動力學模型如圖1-9(a)所示,當電子被激發到S2激發態以後,經由內轉換到 達Qy能態,這個過程的時間常數小於50飛秒,接著再以小於100飛秒的速度到達 Qx能態,這時候會產生分子內電子振動重新分配,其時間常數約為100到200飛秒 之間,之後藉由溶質與溶劑間的碰撞產生振動緩解(vibrational relaxation,VR)過 程,由於他們觀察到兩種時間常數,因此推測是非彈性與彈性碰撞所致,其值分 別是1.4皮秒與10到20皮秒。在到達處於熱平衡的Qx能態以後,經由12奈秒的系 間轉換(intersystem crossing,ISC)到T1能態,最後回到基態。
在ZnTPP 中,金屬離子能夠改變紫質的能階及分子對稱性,因此其緩解的 過程與H2TPP 並不相同。如圖 1-9(b)所示,因為 x 軸與 y 軸簡併所以 Q 能態並 沒有分裂。他們利用不同的激發波長397 nm、550 nm 分別激發 B 和 Q 頻帶,觀 察電子在S2及S1能態的緩解過程。在S2能態他們發現一個1.45 皮秒的衰減過 程,而在S1能態所看到的上升過程比1.45 來得快,因此他們推測有更高的能態 S2’存在,於是利用 550 nm 雷射光源所產生的雙光子(two photon)吸收嘗試將電子 激發到更高的激發態,在同樣的觀測波長下他們看到了不同於1.45 皮秒的雙指 數衰減(bi-exponential)過程,其時間常數分別是 200 飛秒及 1 皮秒,故而更確定 了S2’存在的可能性。S2’及 S2中的電子經過內轉換以後分別到達S1中不同的振 動能態,因此在S1的觀測中可以看到兩種振動緩解的過程,時間常數分別是5 皮秒與12 皮秒,隨後藉 2 奈秒的系間轉換過程到 T1能態。 1-3-2-3 紫質分子的聚集(aggregation) 當紫質分子互相靠近時,會因為π-π間的作用力形成聚集體。根據 Kasha 所提出的理論28,當兩個分子的躍遷偶極矩以平行的方式排列時,二分子中心的 連線會與偶極矩間會形成一個夾角θ,如圖1-10 所示。 圖1-10 聚集體偶極矩與分子中心連線間的夾角θ。
當θ>54.7°時,分子是屬於面對面(face-to-face)的排列,稱作 H 聚集體,此時的 吸收光譜相較於單體分子會有藍位移(blue shifted)的現象;若是θ<54.7°,屬於 頭對尾(head-to-tail)的排列,稱為 J 聚集體,而其吸收光譜則是會有紅位移 (red-shifted)產生。無論是藍位移或者紅位移,其位移的程度均取決於分子間的 π-π作用力,我們能用激子模型(exciton model)來描述這樣的現象29。所謂的激 子指的就是聚集體的激發態,圖1-11 為激子模型中各種排列方式的雙分子聚集 體能階及躍遷示意圖: 圖1-11 激子模型中各種排列方式的雙分子聚集體能階圖。 當雙分子的偶極矩是以平行(parallel)的方式排列時,偶極矩方向相反的組合在能 量上較為穩定,而方向相同的組合能量則較高,但由於前者的躍遷屬於禁制 (forbidden)躍遷,而後者為允許(allowed)躍遷,因此吸收光譜會有藍位移產生; 而在頭對尾的排列中,方向相同的組合能量較低與平行排列相反,因此會有紅位 移;若是雙分子排列介於二者之間時,二種排列均為允許躍遷,因此其吸收光譜
會出現兩個吸收峰。 1-4 本論文的研究 本論文研究鋅紫質衍生物 ZnP1 與 ZnP2 在不同環境下的緩解動力學,研究 的目的是希望了解其在二氧化鈦固相薄膜上的介面電子轉移機制,其中ZnP1 為 ZnP2 的對照組,二者的結構如圖 1-12 所示, N N N N Zn COOH N N N N Zn COOH N ZnP1 ZnP2 圖1-12 ZnP1 與 ZnP2 鋅紫質衍伸物的結構。 在溶液的實驗中,我們藉由二者的光譜比較與改變不同溶劑,希望探討 ZnP2 是否有分子內電荷轉移的現象;在固相薄膜的實驗部分,我們主要針對電子注入 與分子間的能量轉移過程進行相關控制實驗,以瞭解二者對緩解過程所造成的影 響。
1-5 第一章的參考文獻
1.林明獻,全華圖書股份有限公司,2008,1,4.
2.E. Becqurel; C. R. Acad, Sci. Paris 1839,9,561-567.
3.D. M. Chapin; C. S. Fuller; G. L. Pearson,J. Appl. Phys.,1954,51,676. 4. H. W. Schock1 ; R. Noufi, Prog. Photovolt. Res. Appl.,2000,8,151-160.
5. E. K. Putseiko; A. N. Terenin;Photosensitization of the internal photoeffect in zinc oxide and other semiconductors by adsorbed dyes, Zhur. Fiz. Khim., 1949, 23,676. 6. W. Kautek; H. Gerischer; H.Tributsch, J. Electrochem. Soc.,1980,127,2472. 7. W. Kautek; H. Gerischer, Electrochim. Acta,1981,26,1771.
8. C. Sinn;D. Meissner; R. Memming, J. Electrochem. Soc.,1990,137,168. 9. K. Tubbesing; D. Meissner; R. Memming; B. Kastening, J. Electroanal.
Chem.,1986, 214,685.
10.G. Rothenberger; M. Grätzel; J. Moser;N. Serpone; D. K. Sharma, J. Am. Chem. Soc.,1985,107,8054.
11. B. O’Regan, M. Grätzel; Nature,1999,353,737. 12. 孟慶波;林原;戴松元,物理,2004,33,177.
13. L. Luo; C. W. Chang; C.Y. Lin; W. G. Diau, Chem. Phy. Lett.,2006,432,452-456. 14. M. K. Nazeeruddin; A. Kay, I. Rodicio; R. Humphry-Baker; E. Mueller; P. Liska,
N. Vlachopoulos; M. Grätzel , J. Am. Chem. Soc.,1993,115,6382.
15. M. K. Nazeeruddin; S. M. Nazeeruddin; J. J. Lagref; P. Liska; P. Comte; C. Barolo; G. Viscardi; G. Viscardi; K. Schenk; M. Grätzel, Coor. Chem. Rev.,2003,248,1317 . 16.Y. Lu, D. Choi, J. Nelson, O. B. Yang, B. A. Parkinsona; Journal of the
Electrochemical Society,2006,153,E131.
17. A. Fillinger, B. A. Parkinson; Journal of the Electrochemical Society, 1999, 146,4559.
A,2004,163,331.
19. M. J. Gouterman; Mol. Spectroscopy,1961,6,138.
20. G. Y. Kennedy; Annals of the New York Academy of Sciences,1975,244,662. 21. J. Zelaski, Z.Physiol.; Chem.,1902,37,54.
22. J. W. Buchler, et al.; Z.Naturforsch.,1983,B38,1339.
23. J. He, G. Benko, F. Korodi, T. Polyvka, R. Lomoth, B. Akermark, L. Sun, A. Hagfeldt, V. Sundstrom;J. Am. Chem. Soc,2002,124,4922.
24. G. G. Gurzadyan, T. H. Tran-Thi, T. Gustavsson; J. Chem. Phys.,1998,108 (2),385.
25. N. Mataga, Y. Shibata, H. Chosrowjan,N. Yoshida, A. Osuka; J. Phys. Chem.,2000,104(17),4001.
26. J. S. Baskin, H. Z. Yu, A. H. Zewail; J. Phys. Chem. A,2002,106,9837-9844. 27. J. S. Baskin, H. Z. Yu, A. H. Zewail; J. Phys. Chem. A,2002,106, 9845-9854. 28. E. G. Mdrae, M. Kasha;Physical processes in radiation biology New York :
Academic Press,1964.
第二章 實驗測量與儀器原理
在本章節裡我們對於實驗中所使用的各種儀器及其原理加以說明,從紫外-可見光的吸收光譜及穩定態(steady state)螢光光譜,到皮秒與飛秒的時間解析測 量系統,最後是樣品的處理及數據的分析方法。 2-1 紫外-可見光吸收光譜儀 在本實驗當中我們使用 Varian 公司的產品型號為 Cary50 的紫外-可見光光譜 儀來測量吸收光譜。儀器中所使用的光源為氙燈,其光區範圍是190~1100 nm, 我們所使用的掃描速度為每秒1200 nm,其解析度為 0.25 nm。 本實驗的樣品可以分為液態溶液及固態薄膜兩種。液態的溶液部分,為了避 免在測量上出現吸收飽和的情況,我們視情形使用1 mm 及 1 cm 的石英比色管, 而溶液濃度均控制在 10-5 M 左右。測量前我們使用與該溶液相同的溶劑吸收光 譜作為基底線(baseline),因此所測得的吸收光譜會將其視為背景值而扣除。 在固態的薄膜測量上,由於兩種奈米級的固態薄膜(TiO2、ZrO2)均有很好的 透光性,因此同樣使用穿透式的方法測量。相同地我們使用空白的半導體薄膜作 為基底線,在測得的吸收光譜中將其扣除。 2-2 螢光光譜儀 本實驗的螢光光譜測量使用脈衝式的二極體雷射作為激發光源,在打入樣品 後於90°方向上收光,並且加上必要的 long pass filter 以除去激發光源本身的干 擾。激發後產生的螢光經過透鏡收集進入光纖傳遞到CCD 光譜儀(USB2000-FLG, Ocean Optics)加以分光及偵測。偵測的波長範圍是 378~1050 nm,入射孔徑為 200 μm,解析度為 10 nm,偵測器為 2048-圖素線性 CCD 陣列(2048-element linear CCD array)。2-2-1 CCD(Charge-Coupled Device)光譜儀
本實驗所使用的 CCD 光譜儀是由 Ocean Optics 所生產的 USB2000-FLG, 圖2-1 為該光譜儀內部結構示意圖1: 圖2-1 CCD 光譜儀內部結構示意圖。 其中各個元件的的功能介紹如下: (1) SMA905 連接器(connector),用以連接 CCD 和光纖,讓螢光能夠精確地進入 CCD 中。 (2) 固定的狹縫(slit),控制光通量。 (3) 吸收濾鏡(absorbance filter),其功能為過濾掉二級或者三級的入射光,及色彩 平衡。 (4) 準直鏡(collimating mirror),將原本發散的入射光經由此鏡反射後成為準直的 光束進入光柵。 (5) 光柵(grating),能夠將入射光按照波長區分開。
(6) 聚焦鏡(focus mirror),將第一級反射的入射光聚焦後再反射至偵測器 (detector)。
(7) L2 偵測器收集透鏡(L2 detector collection lens),能夠增加光的收集效率。 (8) OFLV 濾鏡,能夠避免二級或者三級反射光進入偵測器。
(9) UV2 升級偵測器(UV2 detector upgrade),增加光譜儀的偵測範圍。 (10) 偵測器,由一線性的 CCD 陣列(array)所組成。
入射光會依(1)~(10)的路徑傳送到偵測器之中,最後轉換成數位訊號送入電腦中 得到我們要的螢光光譜。
2-3 時間相關單光子計數系統(Time-Correlated Single PhotonCounting , TCSPC) 在本實驗中對於時間解析的螢光衰減過程我們利用兩套系統來測量,時間相 關單光子計數系統是其中之一,從50 皮秒到奈米尺度的過程都能夠用這套系統 重現出來。 2-3-1 時間相關單光子計數系統的裝置圖及工作原理 我們所使用的時間相關單光子計數系統是由 PicoQuant 公司所生產的 Fluo Time 200,其工作原理如圖 2-2 所示2。藉由週期性的脈衝雷射將樣品激發並對 單一的光子加以收集,透過與雷射同步觸發的訊號(synchronization,SYNC trigger) 間的比較得到一相對時間,接著將每個光子到達的相對時間加以累計就能夠重現 出一螢光對著時間衰減的波形圖案。由於偵測器在每個週期都只會偵測到最快到 達的光子,因此如果在一個週期內出現兩個以上的光子會造成波形的改變而失 真,此現象稱作pileup effect3。所以實驗的條件必須是”每個週期中所偵測到的光 子數目遠小於1”才能夠準確地重現出樣品螢光隨著時間衰減的過程。
圖2-2 時間相關單光子計數系統工作原理
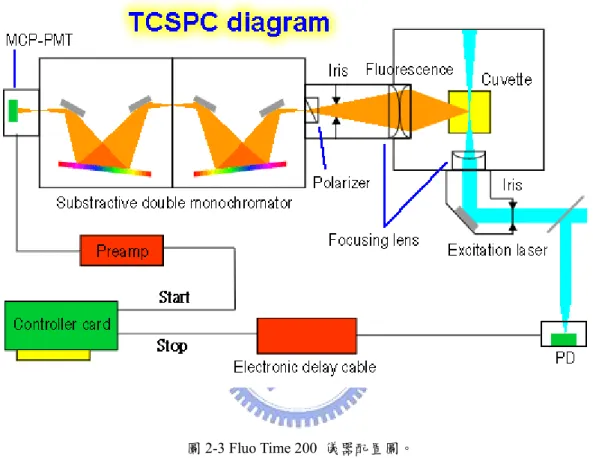
時間相關單光子計數系統的儀器配置圖如圖2-3 所示,我們所使用的激發光 源為脈衝式的二極體雷射(pulsed diode laser),每個脈衝波的半高寬(full width half maximun,FWHM)約是 50 皮秒。在雷射輸出的同時,光源本身的控制器(driver ; PDL 800-B,PicoQuant)會送出一同步觸發訊號,經過分數式時間鑑別器(constant fractional discriminator,CFD)判別訊號的有效與否,接著由可調節式延遲器 (variable delay line ; 425A,ORTEC)調整進入時間相關單光子計數系模組
(SPC630 ,PicoQuant)的延遲時間。雷射激發源經過一光圈(iris)進入系統,藉由調 整光圈的大小能夠控制雷射光的通量,接著通過一面反射鏡與聚焦透鏡(focusing lens)將光源聚焦進入樣品槽(sample chamber),而在反射鏡面上有兩個旋鈕用以 控制雷射光的入射方向。樣品的放光利用兩面透鏡加以收集後,通過一光圈調節
(perpendicular)、水平(horizontal)及魔術角度(magic angle)等,最後經過相減式的 雙光柵光譜儀(substractive double monochromator ; 9030DS,Sciencetech)後進入偵 測器。
圖2-3 Fluo Time 200 儀器配置圖。
偵測器為微頻道光電倍增管(micro-chanel plate photon-multiplier tube,MCP-PMT ; R3809U-57,Hamamatsu),其輸出的電流經由一前置放大器(pre-amplifer)將訊號 轉換成正電壓值並放大達到時間相關單光子計數系統模組可以接受的範圍,接著 進入時間相關單光子計數系統模組進行訊號的計時工作。
圖 2-4 為時間相關單光子計數系統模組進行訊號計時的運作流程。利用兩個 分數式鑑別器分別判別螢光以及訊號有效與否並決定其到達的時間,再以標準訊 號(NIM)送入時間-振幅轉換器(time-to-amplitude converter,TAC)中。在 TAC 收 到螢光訊號以後,內部的電容便開始充電,直到下一個觸發訊號到達時才將充電 停止,並產生電壓輸出,所輸出的電壓正比於兩訊號輸入 TAC 的相對時間差。
最後以類比數位轉換器(analog-to-digital converter,ADC)將電壓振幅轉換為個別 的時間頻道(channel),送入多頻道分析儀(multi-channel analyzer,MCA)進行個別 時間頻道的累計,完成一次單一光子的偵測。重複上述的步驟將單一光子持續地 計時與累積,便可將螢光隨著時間改變的過程完整重現。 圖2-4 時間相關單光子計數系統模組訊號計時運作流程。 2-3-2 時間相關單光子計數系統的組成元件
1. 激發光源-皮秒二極體雷射(picosecond diode laser)
我們進行時間相關單光子計數系統相關實驗時所使用的激發光源為
Picoquant 公司所生產的脈衝式二極體雷射 LDH-P-C 435,激發波長在 435 nm, 脈衝波的半高寬為59 皮秒。可由控制器選擇脈衝的重複率(2.5 MHz~40 MHz)及 調整雷射輸出強度。
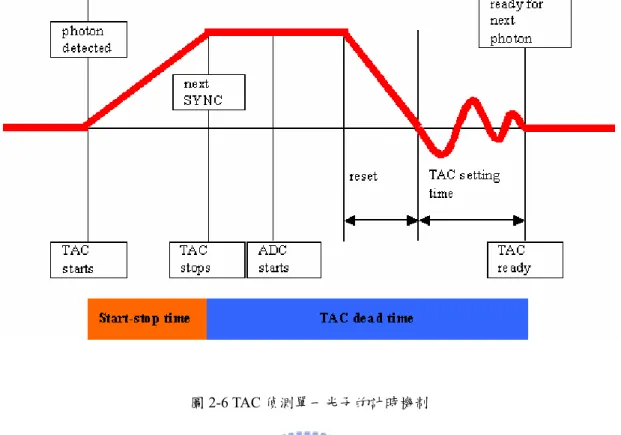
2. 分數式時間鑑別器 時間鑑別器的作用在於辨別訊號是否有效,並且判定其到達的時間。一般的 時間鑑別器會設定一個門檻電壓(threshold)作為判別的標準,當外來的訊號強度 高過所設定的電壓時,便可被處發而被視為有效訊號;反之,如果輸入的訊號小 於該門檻電壓時,則會被視為電路中的雜訊而忽略之,如圖2-5 所示。 圖2-5 時間鑑別器根據門檻電壓判別訊號類別。 而分數式時間鑑別器判讀訊號到達時間的方式如下所述:將輸入的脈衝訊號分成 兩部分,其中之一的電壓振幅以固定比例縮小;而另一個電壓則以訊號反轉 (invert)的方式並且延遲 Td時間,此時前者的振幅出現在後者前緣相同振幅的位 置上,接著再將處理後的訊號加總,將電壓值為零的時間作為原始脈衝的到達時 間。 3. 時間-振幅轉換器 TAC 是一種電容裝置,功能類似精確的碼表,能夠精確地得到樣品被脈衝 雷射激發後,產生的單一光子到達時間。其作用機制如圖2-6 所示,在接受到「開 始訊號」時開始充電,直到接受到「停止訊號」時停止,並產生一類比輸出電壓, 此電壓振幅正比於兩訊號的輸出時間差。
圖2-6 TAC 偵測單一光子的計時機制 在單一個脈衝週期中若是出現兩個光子,TAC 在接受到第一個光子後就會 開始充電,此時的TAC 形同「blind」,因此無法接受到第二個較慢到達的光子, 致使光譜不能完整地紀錄到實際的光子分佈而變形,相較於真實的光譜,在這種 情況下所得到的會是較快的衰減過程,為了避免發生這樣的情形,必須確定每個 激發脈衝所產生的螢光光子被偵測到的機率遠小於一,這樣才能避免 TAC 在同 一個週期中接收到兩個光子。因此,時間相關單光子計數系統的實驗條件必須使 螢光光子數小於脈衝重複頻率的 1/100,可藉由控制雷射光的強度及光圈(iris)大 小來調整。
TAC 有兩種充電模式:一種為正常的開始-結束模式(normal start-stop mode),其開始訊號為同步觸發訊號,停止訊號則是螢光訊號。另一種為反轉的 開始-結束模式(reverse start-stop mode),此時則反過來以螢光訊號為開始訊號, 同步觸發訊號作為結束。為了取得真實的訊號必須降低每個週期接收到光子的機 率,如果TAC 使用正常的開始-結束模式,在大多數的週期中都無法接收到光子
以後都能夠接收到結束的訊號,減少dead time(等待系統回到可進行下一回偵測 所需的時間)及提高讀取訊號的速率。 4. 類比-數位轉換器 類比-數位轉換器的功能是將 TAC 所產生振幅正比於光子到達時間的電壓轉 換為相對應的時間頻道(數位訊號)。 5. 多頻道分析儀 MCA 能夠將每次的螢光時間偵測結果紀錄在個別的時間頻道中,藉著重複 螢光時間偵測取樣,可將樣品螢光產生的時間分佈完整再現。時間頻道數目可經 由軟體調整,分別可以選擇4096、1024、256、64 個頻道。 6. 可調節式延遲器 TAC 所輸出的電壓會經由類比-數位轉換器轉換成其相對應的時間頻道並進 入多頻道分析儀儲存累計。各個頻道代表了不同的電壓值(時間差),但由於頻道 的數目有限,所以需要利用延遲器調整同步觸發訊號進入 TAC 的時間來改變其 輸出電壓值,如此可在螢幕上呈現完整的衰減訊號。 7. 微通道型光電管 微通道型光電管包含了產生光子的陰極,毛細管平行堆疊而成的微通道平 板,以及接收訊號的陽極。在內徑為十微米(micrometer,μm)的毛細管內鍍上電 極材質,並在兩端加上高電壓,當電子進入毛細管內,則在管內進行放大,由於 放大過程只侷限於相當短的距離內,因此可得到相當好的時間解析度,在此系統 可達到30 皮秒以下。由於此單光子偵測器相當敏感,若突然間入射太多光子, 其結構相當容易被破壞,因此一般要求即使在不工作時也必須置於陰暗處,工作 時的入射光子數也被要求在每秒十萬以內。
8. 雙光柵光譜儀 如果系統所要求的時間解析度到達數十皮秒的範圍內,就必須考慮到光的路 徑所造成的影響,而此因素對於單光儀的影響尤其明顯。當一光束打在光柵不同 的位置時,其所產生的繞射光將因行經不同的光程而造成時間上的差異,為此利 用兩個光柵藉由相對的轉動以補償光行進時在時間上所產生之偏差。光柵適用的 波長範圍是 350-900 奈米,可選擇寬度為 0.5、1.0 及 2.0 毫米(millimeter,mm) 的狹縫進行實驗,提供光譜頻寬(spectral bandwidth)為 4、8 及 16 奈米。
2-4 飛秒時間解析螢光上轉移技術(femtosecond fluorescence up-conversion) 2-4-1 飛秒雷射系統
“Laser”是 Light Amplification by Stimulated Emission of Radiation 的首字母縮 略字。意即:藉由受激所引發的放射來進行光放大作用。因為雷射是一種震盪放 大 的 光 , 所 以 具 有 高 亮 度(brightness) 、 高 方 向 性 (directionality) 、 單 色 性 (monochromatity,意即頻寬非常的窄),以及高相干性(coherent)。所謂高相干性, 可以解釋成它所輸出的光相位(phase)是相同的。基於上述的特性讓雷射成為一種 相當有用的光源和偵測工具。超快雷射脈衝是指其輸出雷射的脈衝半高寬(Full Width Half Maximum,FWHM)小於一皮秒(10-12秒)。由於飛秒雷射可以具有高時 間解析度,因此被廣泛地應用在超快動力學研究領域中,包括化學、物理和材料 科學等等。
2-4-1-1 摻鈦藍寶石雷射光譜
摻鈦藍寶石雷射(Ti:sapphire laser)是一典型的固態雷射,以 Ti:Al2O3晶體
為增益介質(gain medium),由 P. F. Moulton 博士於 1982 年在美國麻省理工學院 Lincoln 實驗室首先製造出來4,5。摻鈦藍寶石屬於六方晶系,比重3.98,硬度 9, 折射率1.76(在 800 nm),在 Ti:Al2O3晶體裡以Ti3+離子取代Al3+離子,濃度為
質。摻鈦藍寶石雷射具有兩項特點:(1)輸出雷射光頻率可在 700 至 1000 nm 之 間調變;(2)它可以產生大約 100 飛秒超短脈衝(甚至更短),而且每一個脈衝都具 有高能量。前一特點使它迅速地將染料雷射取代,因為不需要使用流動的染料, 在方便性、穩定性、安全性方面都優越許多。第二個特性與該晶體的非線性本質 有關,在化學動力學研究上,可達到分子運動等級的解析度。 由於摻鈦藍寶石晶體具有很寬的增益帶,所以結合鎖模(mode-locking)技術 後能夠提供非常窄的脈衝寬度。圖 2-7 顯示摻鈦藍寶石晶體的 UV-Vis 吸收光譜 與螢光光譜。UV-Vis 吸收光譜的範圍在 400~600 nm 之間,螢光放光的範圍則是 在600~1000 nm 之間。 圖2-7 摻鈦藍寶石晶體的 UV-Vis 吸收光譜與螢光光譜6。 2-4-1-2 超短脈衝雷射震盪器 由圖 2-8 超短脈衝雷射裝置圖可以看出,超短脈衝雷射有兩個主要的部分, 分別是增益的部分以及群速分散(Group Velocity Dispersion,GVD)補償系統。產 生超短脈衝雷射具有三個關鍵因素首先要有寬頻範圍夠大的雷射增益介質;第二
是適當的鎖模機制;第三為精確的色散補償。三者兼備才能夠達到產生超短雷射 脈衝的條件。 圖2-8 超快雷射脈衝震盪器 Mira900D 裝置圖7。 (1) 寬頻雷射增益介質:由傅立葉轉換(Fourier transform)可知,脈衝時寬越短, 其光脈衝頻寬越大8。所以若要得到超短時寬的雷射脈衝,增益介質的放射光 譜(emission spectrum)必須要有夠大的頻寬,如此才能提供足夠的頻率成分來 合成超短脈衝。目前最普遍被使用的增益介質是摻鈦藍寶石(Ti:Al2O3)。在 圖2-7 中可看出,放射光譜的頻寬範圍在近紅外光波段高達 300 nm,非常適 合作為飛秒脈衝雷射的增益介質。不但如此,由於 Ti:Al2O3 具有高損壞閥 值(damage threshold=23 GW/cm2 at 200 ps),可使用高功率光原來激發不會損 壞;並且其飽和通量(saturation fluence)高達 0.9 J/cm2,能夠有效地轉換激發 光源所輸入的能量;同時又具備良好的導熱性(thermal conductivity=0.42
P:Brewster prism M:Mirror
因此摻鈦藍寶石適用於作為超短脈衝雷震盪放大器的增益介質。 (2) 要達到超短脈衝輸出,必須在共振腔內加入適當的鎖模機制,迫使原本共振 腔的穩定雷射光由一小擾動開始成長而形成超短脈衝。由頻域(frequency domain)觀點來看,藉由強迫雷射震盪器同時工作在許多頻率上,並且鎖定不 同頻率之間的相位,這類機制稱為鎖模。 目前在固態雷射,尤其是摻鈦藍寶石雷射,應用最為廣泛的技術為克爾 透鏡鎖模技術(Kerr lens mode locking,KLM)9。在雷射發明之前,材料的光 折射率n 與照光強度無關。直到雷射發明後,有了足夠的光強度才可以觀測 到材料值折射率的變化。在一級近似下,材料的光折射率n 與照光強度關係 如公式2-1: n = n0 + n2I (2-1) 其中 n0為與光強度無關的折射率,n2為折射率的非線性係數,這個數值非常 小且與材料本身的性質有關,I 為照光強度。當光強度弱時,n2I 比 n0小很多, 對折射率n 的影響可以忽略,但是對於一高強度的短脈衝而言,n2I 值很大, 由這種折射率的變化所引起的光相位延遲不能忽略。表現在時間上會出現自 相位調控(self-phase modulation),使得脈衝頻寬增加;表現在空間上,正的 n2會導致自聚焦效應而改變其空間模態。 利用這種特性我們可以設計雷射共振腔如圖 2-9,使得高強度的空間模態 具有比較大的增益(或是比較小的損失),而低強度的空間模態則有比較小的 增益(或是比較大的損失),如此強者越強、弱者越弱,脈衝時寬因此被壓縮 得更短。由於這種鎖模機制是利用克爾效應所引起的自然聚焦現象,故稱之 為克爾透鏡鎖模。在選擇適當的雷射震盪腔體參數和功率條件下,可以產生 皮秒或者飛秒等級的超短光脈衝。
圖2-9 克爾透鏡鎖模示意圖。 (3) 前面提到,脈衝時寬越短,其光脈衝頻寬越大。換句話說,一個超短脈衝內 含有許多不同波長的光子。如果這脈衝通過一個不吸收這些波長的介質,這 個介質的折射率 n 與波長λ有關,即折射率為 n(λ)。脈衝在介質中的速度 v(λ)能以公式 2-2 表示之: ( ) ( ) c v n λ λ = (2-2) 其中 c 為光速。也就是說,由於介質材料的折射率對應於不同波長的光有不 同的值,因而造成不同波長的光在介質內的傳播速度不同,而使得超短雷射 脈衝時寬變得比進入該介質前還要寬,這種現象稱為群速分散(GVD)。在一 般的光學材料,長波長的光折射率要比短波長光還要小,使得長波長光以較 快的速度通過該介質,造成脈衝的前端為長波長的光子,後面則是短波長的 光子,稱為正的群速分散。反之,如果長波長光通過該介質較慢,則是負的
通過長度為5 mm 的 Ti:Al2O3晶體後,其時寬將延長為40fs,這表示在共振 腔內材料色散若沒有得到精確的補償,超短雷射脈衝將無法穩定存在。 目前最簡單的補償方式是利用稜鏡對(prism pair)來引入負值的色散,其 原理如圖 2-10 所示,由於低頻成分通過稜鏡對的有光程(optical path)較高頻 成分長,在適當的稜鏡材料與稜鏡距離下,將可補償增益介質所引入的材料 色散而將脈衝壓縮回原本的時寬。目前已經很少人使用四塊稜鏡的方式,取 而代之的是如Mira900 裝置圖(圖 2-8)中,使用兩塊稜鏡加上一面反射鏡。也 可以使用光柵取代稜鏡達到將群速分散壓縮的目的。 圖2-10 群速分散補償裝置示意圖。 在我們的實驗中所使用的超快雷射是屬於商業化的產品,由Coherent 公司所 生產的 Mira900D 型,使用摻鈦藍寶石作為增益介質的鎖模雷射。其調控輸 出波長的範圍約是700~1000 nm 之間,使用克爾透鏡鎖模技術達到鎖模的目 的。該雷射的腔體設計能夠讓摻鈦藍寶石晶體裡產生光學克爾效應而達到自 聚焦的結果。自聚焦在鎖模操作中可造成更高的來回增益(round trip gain)。 在Mira900D 雷射中,光抽運(pumping)來源是由 Coherent 公司所生產的二極 體雷射(型號為 Verdi V10),使用時正常輸出功率為 10 瓦。Mira900D 雷射輸 出的脈衝半高寬約在100~200 fs 之間。在波峰為 800 nm 平均輸出功率是 1.6
瓦。重複速率接近76 MHz。在雷射腔體的輸出端,包含一個可以微調的狹縫 (slit)置於 output coupler 之前。藉由調整該狹縫的寬度,可以讓雷射鎖模的操 作更為容易,鎖模後也更穩定。Mira900D 使用雙折射濾光片(birefringent filter) 作為波長選擇裝置,當光束進入雙折射晶體後,會分離成兩個具有不同相位 的部分,分別是ordinary 和 extraordinary。當這兩部分重新結合在一起後,會 造成極化方向的改變。如果將雙折射晶體的厚度梢作改變,只有兩個相位差 符合整數的波長光束能夠通過該晶體,而該光束的極化方向則沒有改變,在 雷射腔體中,雙折射濾光片設置符合 Brewster’s angle。藉由這些技術, Mira900D 摻鈦藍寶石雷射可以調整輸出光的波長介於 700 到 1000 nm 之間。 2-4-1-3 脈衝選擇系統
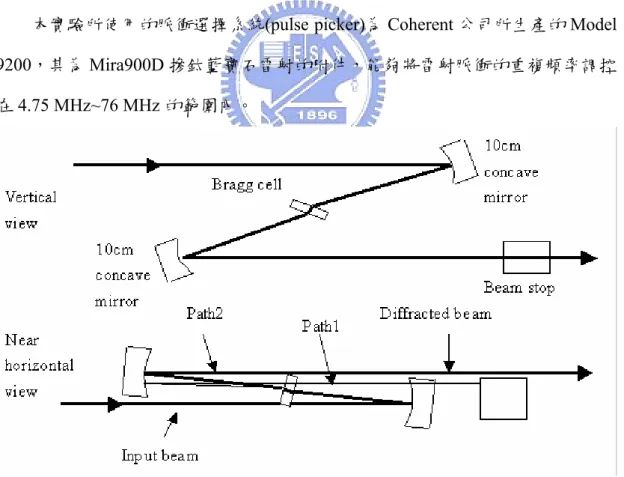
本實驗所使用的脈衝選擇系統(pulse picker)為 Coherent 公司所生產的 Model 9200,其為 Mira900D 摻鈦藍寶石雷射的附件,能夠將雷射脈衝的重複頻率調控 在4.75 MHz~76 MHz 的範圍內。
圖 2-11 為脈衝選擇器內部構造的示意圖,其中 Bragg cell 是一聲光調變器 (acousto-optic modulator) , 由 一 壓 電 聲 光 轉 換 器 (piezoelectric acousto-optic transducer)及 TeO2晶體所組成,能夠週期性地從脈衝序列中擷取出單一的脈衝,
以降低雷射重複頻率 9。當壓電聲光轉換器未接受到外來的 RF 脈衝時,雷射光 的行進路線如path1 所示,此時雷射光會受到 beam stop 的阻擋而無法通過脈衝 選擇器;若是控制提供一短時寬RF 脈衝作用在壓電聲光轉換器,壓電聲光轉換 器便能將其轉換為一短時寬聲波進入 TeO2晶體,並在晶體上形成駐波,當聲波
擠壓介質時,會改變介質的密度,進而使折射率改變,此時的晶體猶如一光學相 位光柵(optical phase grating),使通過的雷射光受到光柵的繞射而改變其行進方 向,如path2 所示,因此能夠通過脈衝選擇器。經由上述的過程,我們知道能夠 藉由控制RF 脈衝的頻率來決定雷射光的重複頻率。本系統的繞射效率(diffraction efficiency)大於 60%,而適用的波長範圍是 700~1000 nm。由於在時間相關單光 子計數系統中我們所測量的是50 皮秒到奈秒尺度的衰減過程,若是使用重覆頻 率為76 MHz 的 Mira900D 作為激發光源,會造成在一個衰減過程中出現多個激 發脈衝的情形,因此能夠利用脈衝選擇器適度地降低光源重複頻率,配合在入射 光徑上所架設的光二極體(photodiode)發送同步觸發訊號,將 Mira900D 的光源使 用在單光子計數系統的實驗測量上,相較於皮秒二極體雷射能夠有更強的強度及 更短的脈衝時寬。 2-4-1-4 自動校正儀 自動校正儀(auto-correlator)能夠測量超快雷射輸出脈衝寬度,是當前最常被 使用的技術之一。這個技術由Maier 等人於 1966 年所提出10,11。圖2-12 為自動 校正儀的基本架構,類似 Michelson 干涉儀,使用非線性光學的倍頻(Second Harmonic Generation,SHG)原理來測量脈衝寬度。用一塊分光鏡(Beam splitter) 將入射的雷射光分成兩束強度各半的光束,其中一束雷射光經過不同的延遲距 離,再將兩束雷射光聚焦到一塊二階非線性光學晶體產生二倍頻。藉由改變不同
的延遲距離能夠造成脈衝波在二倍頻晶體上的時間分佈產生產生差異,因此會得 到不同強度的訊號,再將光程差換算成光行進的時間差(Δt),如此可以描繪出訊 號強度對時間的關係圖12。
圖2-12 自動校正儀裝置圖。
在這裡,我們可以假設兩個相同的脈衝在相差時間τ上所呈現的強度分別是 I(t-τ)和 I(t)函數,則 I(t)的自動校正函數如公式 2-3 所示:
( ) ( ) ( ) ( ) ( ) ac A τ I t I t ∞ I t I t τ dt −∞ = ⊗ =
∫
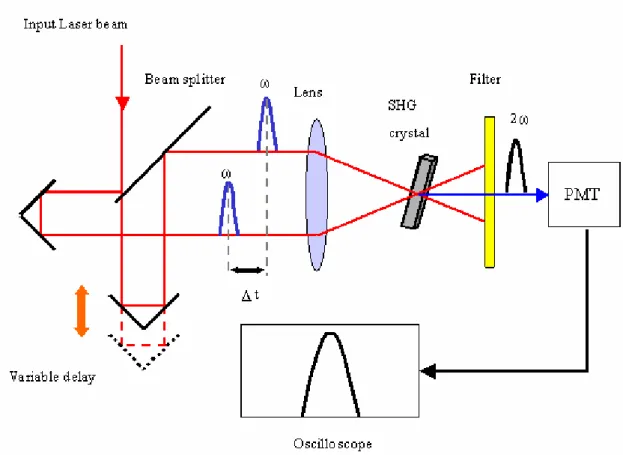
− (2-3) 因為訊號經由自動校正的處理後必然是左右對稱的函數,所以其缺點在於有關脈衝形狀的資訊相當有限,無法確定原始的脈衝形狀是否對稱。為了決定脈衝的寬 度,最普遍的做法是假設脈衝形狀為一高斯函數(Gaussian function,G(t)),經由 擬合(fitting),找出自動校正函數的半高寬(FWHM),進而反推得知雷射脈衝的真 實半高寬。但由於自動校正儀僅能測到脈衝雷射剛從系統中射出時的半高寬(和 我們架設的位置有關),再經過許多光學元件後,其 GVD 效應會讓雷射的半高寬 變寬,因此在實驗中我們亦會使用螢光上轉移技術測量激發光的三倍頻訊號以求 得真實的半高寬。 2-4-2 螢光上轉移技術13 在 2-3 節中我們所介紹的時間相關單光子計數系統擁有 50 皮秒左右的時間 解析度,這是目前電子裝置的極限14,15,因此若欲觀測更快速的反應過程,就必 須得利用其他的方法代替傳統的電子裝置。 螢光上轉移技術(fluorescence upconversion)配合飛秒雷射脈衝,利用非線性 的光學原理,能夠達到數十飛秒的時間解析度。此一技術與2-4-1-4 中所述的自 動校正儀原理類似,讓螢光與另一束閘門雷射脈衝(gating pulse)於時間和空間上 都重疊在一塊非線性光學晶體上產生混頻(sum frequency)如圖 2-13,藉著偵測混 頻出來的光束與調整閘門脈衝的到達時間來紀錄螢光隨著時間的衰減過程。 根據能量守恆原理,我們能夠利用公式 2-4 與 2-5 來計算混頻光束的頻率:
sum fluorescence gate
ω =ω +ω (2-4)
1 1 1
sum fluorescence gate