國立臺灣大學基因體與系統生物學學位學程
(合辦單位:中央研究院)
博士論文
Genome and Systems Biology Degree Program College of Life Science
National Taiwan University and Academia Sinica Doctoral Dissertation
黑腹果蠅長非編碼 RNA 特性研究 Characterizing Long Non-coding RNAs in
Drosophila melanogaster
陳玫如 Mei-Ju Chen
指導教授: 陳倩瑜 博士 李文雄 博士 Advisor: Chien-Yu Chen, Ph.D.
Wen-Hsiung Li, Ph.D.
中華民國 105 年 7 月
July, 2016
國立臺灣大學(碩)博士學位論文
口試委員會審定書
黑腹果蠅長非編碼 RNA 特性研究 Characterizing Long Non-coding RNAs in
Drosophila melanogaster
本論文係陳玫如君(D99B48004)在國立臺灣大學基因體 與系統生物學學位學程之博士學位論文,於民國 105 年 7 月 20 日承下列考試委員審查通過及口試及格,特此證明
口試委員:
(簽名)
(指導教授)
系主任、所長
(簽名)
ACKNOWLEDGMENTS
“The first step in wisdom is to know the things themselves.” Carolus Linnaeus (1735)
基因體與系統生物學為一個新穎學門,其所涉範疇廣泛,在我的博士生涯裡 亦時逢茫然無據的時刻,感謝陳倩瑜教授與李文雄教授總能一語點領關鍵,引亮 學生在龐雜的基因體與系統生物學網路裡該行的道路。
「重要的科學通常概念簡單,而簡單的事情通常困難」,這一語約莫道盡這篇
博論成果背後曾有的汗水。而基因體與系統生物學學門之特殊,使得這個領域的 研究往往無法僅是一人之功,也時常需要透過不同領域實驗室的合作,感謝吳君 泰老師實驗室在黑腹果蠅的專業,讓我們在探討這個課題時能迅速尋得脈絡,也 透過在君泰老師實驗室的生物實驗訓練,讓我能在這篇論文中做出關鍵的生物實 驗證明支持。而在論文發表的過程中,多次與審查者間的論文修訂往來與數據的 補充,也衷心感銘郭建言博士於倩瑜老師實驗室的博士後研究期間不吝對論文英 文撰寫的多加指教,亦感謝倩瑜老師實驗室的學弟們所給予的強力後盾支持:昱 行、祐榆、東祈、柏均、秉翰、翊安、張平,而終成這簡單而困難的研究,期望 奠定黑腹果蠅 lncRNA 研究的堅實基礎。
從進入生物資訊領域而後從事基因體與系統生物學研究,該從大三進入倩瑜老 師實驗室進行專題研究算起,迄今已歷十年,倩瑜老師亦師亦友的指導與栽培,
以及其對於研究的熱情與態度,再再影響我許多,我想這些影響也將遠及一生,
衷心感謝能遇良師。而亦也在老師的實驗室裡遇得一生伴侶祐榆,一路支持我 專心埋首研究,也因專業互補而與我相互磨鍊拋光整個研究成果,感激這樣的幸 運,能得一這樣無論研究與生活皆相知的伴侶。
博士生涯終將階段性結束,然研究是一生志業,望尋得適宜舞台,承先啟後,
貢獻所學,不負一路栽培。
中文摘要
次世代定序技術(Next-generation sequencing; NGS)開啟 RNA 領域研究的新紀元。
過往認為只是轉錄訊號擾動的長非編碼 RNA (long non-coding RNA; lncRNA),已 由許多研究證實其在許多重要生理機制中扮演要角。然而,現今文獻對於重要模 式生物黑腹果蠅(Drosophila melanogaster)的 lncRNA 瞭解仍相當有限;究其原因,
乃黑腹果蠅 lncRNA 的基礎資訊之稀缺所致。因此,本論文追根溯源,由四個面向 對黑腹果蠅 lncRNA 進行系統性探究(1) 收集與發現:本論文開發一生物資訊方 法,自我們產生的組織特異性 RNA-seq 資料鑑定出為數不少的新 lncRNAs,並與 公開資訊可收集之已知 lncRNAs 整合,呈現迄今最新之黑腹果蠅 lncRNA 資料集;
(2) 特性註解:本論文採用大量的 RNA-seq 與 ChIP-seq 資料集(總計 93 組)增進現 有 lncRNA 的註解資訊如轉錄方向與染色質特徵之品質,並進而觀察摘要出黑腹果 蠅 lncRNA 的一般特性;(3) 基因表現:本論文以 RT-qPCR 實驗驗證了挑選之 lncRNA 的基因表現,並彰顯 RNA-seq 技術平台用於發現 lncRNA 的結果具有相當 的可信度;(4) 轉錄調控:本論文提出一結合序列特徵探勘之生物資訊方法,系統 性分析轉錄因子結合位(Transcription factor binding site; TFBS)於 lncRNA 啟動子出 現與否,以及其與 lncRNA 基因轉錄調控的關聯性。結果顯示,當使用核小體佔據 與跨物種保留性資訊,於共表現之編碼基因集進行序列探勘,其所得的序列特徵(或 稱順式因子;cis-element),多數與已知的 TFBS 相似;此外,這些順式因子可在共 表現之編碼基因與 lncRNA 基因的啟動子區域同時觀察得見(較常見於第三期幼蟲 至雄蟲階段共表現群集),顯示出共表現之編碼基因與 lncRNA 基因具有被共同調 控的可能性。簡言之,本論文彰顯系統性整合研究的優點,透過基因體與轉錄體 資料的整合,大幅加速鑑別 lncRNA 的特性;而所得之觀察結果可作為黑腹果蠅 lncRNA 功能研究的堅實基礎。
關鍵詞:整合性研究、黑腹果蠅、長非編碼 RNA、RNA 定序技術、染色體免疫沉 澱定序技術
ABSTRACT
Recent advances in sequencing technology have opened a new era in RNA studies.
Novel types of RNAs such as long non-coding RNAs (lncRNAs) have been found to play essential roles in biological processes. However, only limited information is available for lncRNAs in Drosophila melanogaster, an important model organism. Thus, this thesis aims at chracterizing fruit fly lncRNAs from four aspects: (1) collection and discovery; (2) annotation; (3) expression; and (4) regulation. I developed a computational approach to discover novel lncRNAs from the newly generated tissue-specific RNA-seq data, and then I combined the discovered lncRNAs with previously published lncRNAs into a curated dataset. Next, numerous RNA-seq and ChIP-seq datasets (93 sets) were used to improve the lncRNA annotation such as transcriptional direction and presence of conventional chromatin signatures. With these efforts, I summerized general characteristics of fruit fly lncRNAs in the thesis. In addition, I used RT-qPCR experiments to validate the expression of some randomly selected lncRNAs and demonstrated that RNA-seq is a reliable platform to discover lncRNAs. Moreover, I proposed a method to incorporate de novo motif discoveries to systemically investigate the presence of TFBSs in lncRNA promoters and how it is related to the regulation of lncRNA expression. The result revealed that most of the motifs (cis-elements) discovered from the co-expressed coding gene promoters are similar to the annotated TFBSs, where the motif dicscovery procedure considerd the information of nucleosome occupancy and evolutionary conservation. I also found that common cis-elements were usually observed in the promoters of the co-expressed coding and lncRNA genes in the development stages from L3 to male adlut. In conclusion, this thesis demostrated that integration of genomic and transcriptomic data can largely facilitate lncRNA discovery and characterization, and provided a solid foundation for studying the functions of lncRNAs in D. melanogaster.
Keywords: Integrative research, Drosophila melanogaster, Long non-coding RNA,
RNA-seq, ChIP-seqTABLE OF CONTENTS
博士學位論文口試委員會審定書 ... i
ACKNOWLEDGMENTS ... ii
中文摘要 ... iii
ABSTRACT ... iv
TABLE OF CONTENTS ... v
LIST OF FIGURES ... viii
LIST OF TABLES ... xi
CHAPTER 1 Introduction ... 1
1.1 Challenges of lncRNA studies in D. melanogaster ... 3
Limited numbers of known lncRNAs in D. melanogaster ... 3
Incomplete annotation of lncRNAs in D. melanogaster ... 4
Reliability of lncRNA expression detected from RNA-seq data ... 5
Transcriptional regulation of lncRNA expression ... 5
1.2 Integrative approach for characterizing lncRNAs by utilizing genomics and trnascriptomics data ... 7
1.3 Thesis structure ... 10
CHAPTER 2 Related Works ... 12
2.1 Brief history of long non-coding RNAs studies ... 12
2.2 Integrative and systemic studies on lncRNAs ... 13
2.2.1 Related works for characterizing lncRNAs in in Drosophila
melanogaster ... 13
2.2.2 Related works for transcriptional regulation of lncRNA expression 14 CHAPTER 3 Collection and Discovery of lncRNAs in Drosophila melanogaster ... 17
3.1 Known lncRNAs collected from databases and literatures ... 17
3.2 Novel lncRNAs identified from brain samples ... 18
3.3 Up-to-date list of long non-coding RNAs in D. melanogaster ... 19
3.4 Methods for collection and discovery of fruit fly lncRNAs ... 19
3.4.1 Collection of published lncRNAs ... 19
3.4.2 RNA-seq data of the fly brain ... 22
3.4.3 Novel lncRNA discovery ... 23
CHAPTER 4 Annotation of the curated lncRNAs ... 26
4.1 Improving the annotation of the lncRNAs from Young et al. ... 26
4.2 Utilizing additional RNA-seq datasets to improve the annotation of the 4,599 curated lncRNA transcripts ... 28
4.3 General characteristics of the fruit fly lncRNAs ... 31
4.3.1 Location distribution of lncRNAs in Genome ... 31
4.3.2 Length and structure of lncRNAs... 34
4.3.3 Evolutionary conservation of lncRNAs ... 37
4.3.4 Supporting evidences for lncRNA expression in the developmental stages… ... 39
4.4 Methods for annotation of the curated lncRNAs ... 46
4.4.1 Improving the annotation of curated lncRNAs ... 46
4.4.2 Genomic and transcriptomic data for supporting lncRNA expression in the developmental stages ... 49
CHAPTER 5 Reliability of lncRNA expression ... 52
5.1 Reliability of the lncRNAs newly discovered identified from brain samples… ... 52
5.2 Experimental validation of a selected set from the curated lncRNAs by RT-qPCR ... 55
5.3 Details of the RT-qPCR experiments ... 58
CHAPTER 6 Regulation of lncRNA Expression ... 60
6.1 Hierarchical clustering of co-expressed coding and long non-coding genes………....61
6.2
De novo motif discovery on the promoters of co-expressed coding genes .. 68
6.2.1 Promoter regions of genes in D. melanogaster ... 68
6.2.2 Parameter tuning for the weights of nucleosome occupancy and evolutionary conservation while conducting de novo motif discovery ... 70
6.2.3 Evaluation of the discovered motifs ... 72
6.3 Co-occurrence of TF binding motifs in the promoter regions of co-expressed coding and non-coding genes ... 74
6.4 Materials and methods for the proposed workflow ... 76
6.4.2 Hierarchical clustering ... 76
6.4.3
De novo motif discovery of cis-elements from co-expressed coding
gene promoters ... 776.4.4 Identification of shared cis-element in the co-expressed lncRNA promoters ... 78
CHAPTER 7 Limitations of this work ... 80
CHAPTER 8 Conclusions and Future Directions ... 82
REFERENCE: ... 84
APPENDIX ... 90
List of Publications ... 90
Appendix Figures ... 94
Appendix Tables ... 95
LIST OF FIGURES
Figure 1. Four challenges for characterizing lncRNAs in D. melanogaster ... 2 Figure 2. Characterizing lncRNAs in D. melanogaster ... 11 Figure 3. Procedures for discovering novel lncRNAs from RNA-seq data of
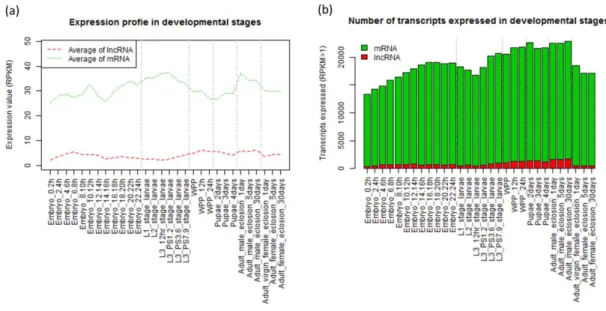
the present study. The sequencing read datasets of mRNA and total RNA were respectively mapped to the reference genome sequence using TopHat and Cufflinks. Putative lncRNAs were then discovered by Cuffcompare. Sequencing reads were again mapped to the set of putative lncRNAs to construct the final set of novel lncRNAs. ... 24 Figure 4. Distribution of lncRNA types in euchromatin. ... 33 Figure 5. Distribution of exon numbers in lncRNA and mRNA genes. ... 35 Figure 6. Expression profiles at different developmental stages of fruit fly. (a)
Averaged RPKM values at different developmental stages for lncRNAs and mRNAs. (b) Numbers of expressed transcripts (RPKM > 1) at different developmental stages for lncRNAs and mRNAs, respectively. ... 40 Figure 7. Expression profiles of the 2,926 lncRNAs which varied between at
least two developmental stages ... 41 Figure 8. Histogram of members in a co-expressed lncRNA cluster among
the 2,926 lncRNAs which varied between at least two developmental stages ... 43 Figure 9. Expression profiles of the lncRNAs which are co-expressed with at
least 9 other lncRNAs (Namely, a co-expressed lncRNA cluster is selected for this figure while it has at least 10 members). ... 43 Figure 10. Analysis of chromatin signatures (Pol II, H3K36me3 and
H3K4me3) in the curated lncRNA genes. ... 44 Figure 11. Rules for classifying lncRNAs. Black arrows (transcripts)
represent coding genes and colored transcripts are lncRNAs. (a) lncRNAs with intronic overlaps. This group includes lncRNAs (dark green and light green transcripts) located in intronic regions of coding genes (black transcripts). (b) Intergenic lncRNAs. This group includes lncRNAs (red transcripts) located in regions between two coding genes (black transcripts). (c) lncRNAs with exonic
overlaps. This group includes lncRNAs (dark blue and light blue transcripts) overlapping exonic regions of coding genes (the black transcript). ... 47 Figure 12. Occupied regions for each chromatin signature ... 51 Figure 13. RT-qPCR experiments for a selected set of lncRNAs in brains. (a)
22 novel lncRNAs discovered in the present study were selected for validation.
RpL 32 (a coding gene) and ROX1 (a non-coding gene) were included as positive controls. The horizontal line indicated delta Ct
1. The rectangle indicated the
five lncRNAs with considerably low expression, and was tested again by the second RT-qPCR experiment shown in (b). (b) The five lncRNAs from the rectangle of (a) were tested again by RT-qPCR with twofold amount of template cDNA. Ten FlyBase lncRNAs were included for comparison. The three FlyBase lncRNAs highlighted by the orange stars were selected because their RPKM values in our brain RNA-seq data was 0. ... 54 Figure 14. RT-qPCR experiments of a selected set of lncRNAs in male adults.G1: high expression with chromatin signatures (11 lncRNAs); G2: low expression with chromatin signatures (11 lncRNAs); G3: high expression without chromatin signatures (10 lncRNAs); and G4: low expression without chromatin signatures (10 lncRNAs). Three negative controls (un-transcribed region 1, 2, and 3) were all around zero. Stars were used to highlight the lncRNAs that were not from the databases (Orange stars: the selected lncRNAs from Young et al. [5]. Blue stars:
the lncRNAs from the present study). The horizontal line indicated the cutoff (delta Ct 2) used to define a validated lncRNA. Green stars: the transcripts that are now annotated as other types of transcripts by FlyBase, and thus were removed from the list of the curated lncRNAs in the present study. ... 56 Figure 15. Workflow for identifying shared cis-elements of co-expressed
coding and long non-coding genes ... 60 Figure 16. Frequency distribution of clusters along with different member
numbers in a cluster ... 62 Figure 17. Expression profiles for the lncRNAs of the 27 co-expressed
clusters……….. ... 65 Figure 18. Second-phase hierarchical clustering for the 27 co-expressed
clusters with at least 50 transcripts. The rainbow color bar indicates the groups categorized by the second-phase hierarchical clustering. The gray color bar
represents the number of lncRNAs within a cluster. Details of group and cluster information can be found in Table 13. ... 66 Figure 19. Second-phase hierarchical clustering for the co-expressed clusters
with at least 30 transcripts. The rainbow color bar indicates the groups categorized by the second-phase hierarchical clustering. The gray color bar represents the number of lncRNAs within a cluster. Details of Group 1-7 can be found in Table 13, while Group X represents the clusters that contain members of 30~49 transcripts……. ... 67 Figure 20. Distribution and Conservation scores (CS) analysis of the 2,059
annotated binding sites collected from REDfly database [90] (170 TFs and 2,048 target genes included). (a) Position distribution. The averages CS of TFBSs located within (500 to 200 bp) is 0.482; (b) Frequency of TFBSs that have a CS value 0.482…. ... 69 Figure 21. Parameter tuning for the weights (b, c) which are given to
nucleosome occupancy and evolutionary conservation. Different colors denote different weights for nucleosome occupancy. The colors, (blue, red, green, orange), indicate b
(0, 1, 2, 3). Different types of lines represent different weights for
evolutionary conservation. The line types, (solid line, thick broken line, broken line), indicate c (1, 2, 3). ... 71Appendix Figure 1. Parameter tuning for different pattern supports (ratio of pattern-hit promoters/all promoters in the positive set) and different weights used for pattern ranking. Precision is calculated by the ratio of (True Positives/Predicted instances), and presented as percentage. ... 94
LIST OF TABLES
Table 1. Numbers of fly lncRNAs from different data sources ... 4
Table 2. Statistics of the public data used for studying transcriptional regulation in yeast, fruit fly and human ... 6
Table 3. Summary statistics of datasets used in thesis. ... 8
Table 4. Statistics of exon numbers in lncRNA and mRNA genes from different sources. ... 27
Table 5. Statistics of transcriptional direction in the lncRNA genes from different sources. ... 28
Table 6. Types of lncRNA transcripts. ... 29
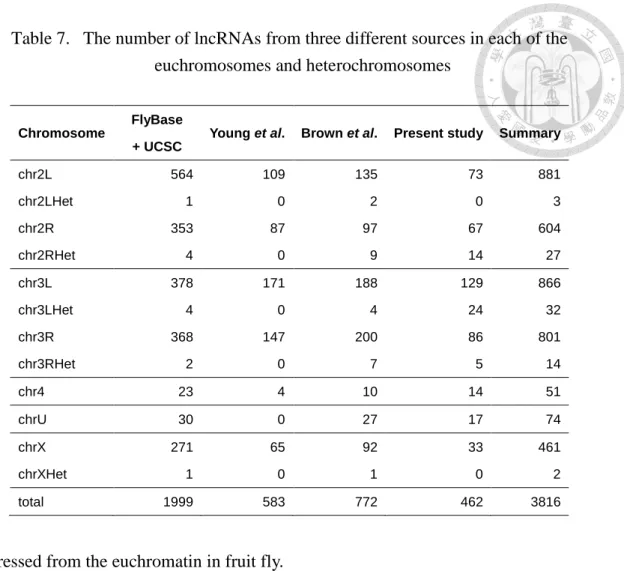
Table 7. The number of lncRNAs from three different sources in each of the euchromosomes and heterochromosomes ... 32
Table 8. Length of lncRNA transcripts ... 34
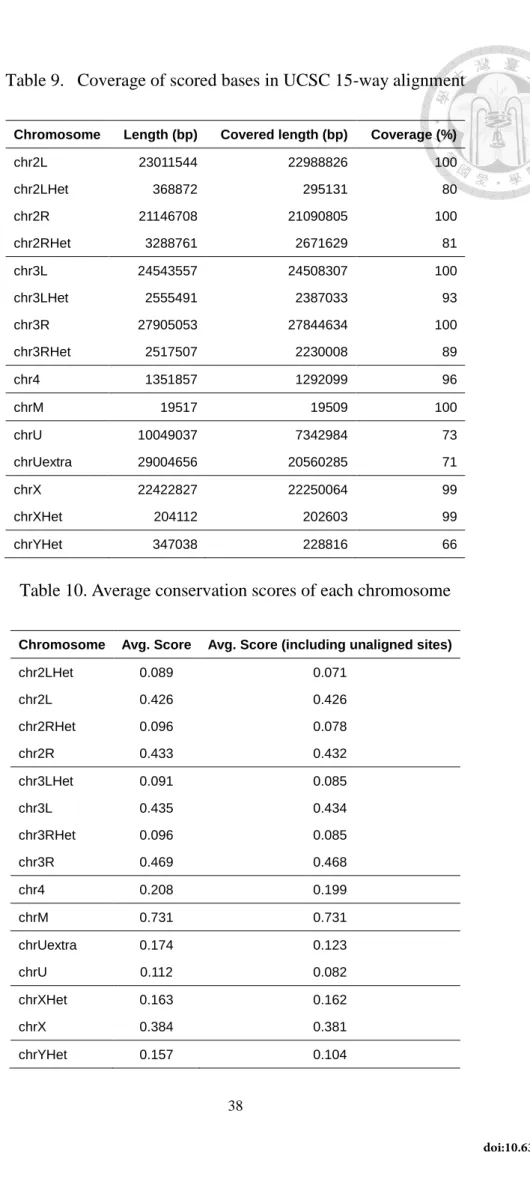
Table 9. Coverage of scored bases in UCSC 15-way alignment ... 38
Table 10. Average conservation scores of each chromosome ... 38
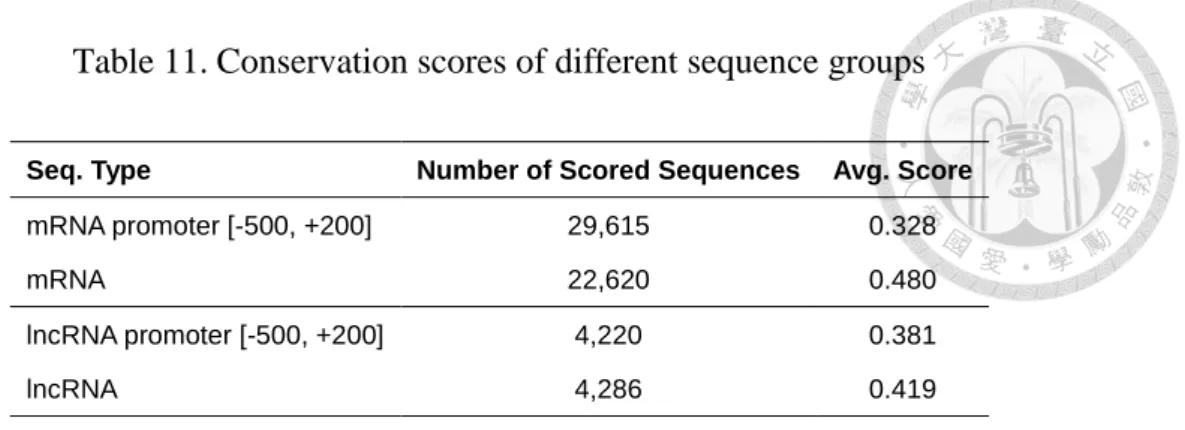
Table 11. Conservation scores of different sequence groups ... 39
Table 12. Statistics of clusters with different cutoff of correlation ... 61
Table 13. Summary of the selected clusters which contain at least 50 transcripts…… ... 64
Table 14. Summary of de novo motif discovery results ... 73
Table 15. Investigation of similarity between lncRNA and mRNA promoters…… ... 73
Table 16. Summary of cis-elements shared by co-expressed coding and long non-coding genes ... 75
Appendix Table 1. Primer list of the selected lncRNAs for RT-qPCR experiments……… ... 95
Appendix Table 2. Raw Ct values of RT-qPCR experiments for un-transcribed regions and the selected lncRNAs. ... 97
CHAPTER 1 Introduction
Recent advances in sequencing technology, such as RNA-seq, have opened a new era in RNA studies. Novel types of RNAs such as long non-coding RNAs (lncRNAs) have been discovered by transcriptomic sequencing and some lncRNAs have been found to play essential roles in biological processes such as development and diseases [1, 2].
More and more studies have discovered and investigated lncRNAs in many organisms such as human and mouse. However, only limited information is available for lncRNAs in Drosophila melanogaster (fruit fly), an important model organism.
Through considerable literature survey, most of lncRNA studies were found to be conducted in human or mouse, while only a few in D. melanogaster. For example, some lncRNAs have been observed to regulate developmental processes in D. melanogaster.
Two genes, roX1 and roX2 recruit the MSL (male specific lethal) chromatin remodelling complex to genes on the male X chromosome, but not the autosomes or the female X chromosomes, to increase the acetylation of histone H4K16 [3]. This regulation can coordinate the dosage compensation required for male development.
While the functionality of some lncRNAs in fruit fly was known, most of the lncRNAs have not yet been functionally characterized. The reason behind is probably owing to the fact that some of fundamental knowledge is currently scarce for fly lncRNAs.
Therefore, characterization of lncRNAs in D. melanogaster is an important area of research.
To characterize fly lncRNAs, four essential questions would need to be clarified (Figure 1). First, it remains unclear whether the current set of fly lncRNAs is comprehensive. This question could be answered by collecting know lncRNAs to assess the current state. Nevertheless, discovery of novel lncRNAs from newly generated RNA-seq data also helps to infer whether additional lncRNAs could be found. Second, properties of fly lncRNAs are not well characterized because of incomplete annotation.
Integrating multiple data sources from fly genomics and transcriptomics could improve annotations of lncRNAs. Third, the reliability of novel lncRNAs discovered from RNA-seq need to be assessed. Quantitative reverse transcription polymerase chain reaction (RT-qPCR) is the gold standard to validate the expression of the discovered
Figure 1. Four challenges for characterizing lncRNAs in D. melanogaster
lncRNAs. Finally, it remains challenging to infer transcriptional regulation of lncRNA expression, because experimentally validated TF binding sites (TFBS; usually represented as sequence motifs) are currently scarce. In this regard, in silico predictions are needed for characterizing this issue in a large scale. In this thesis, we will discuss these four problems in detail and give an integrative approach to solve these problems by adopting multiple data sources from fly genomics and transcriptomics.
1.1 Challenges of lncRNA studies in D. melanogaster
Limited numbers of known lncRNAs in D. melanogaster
To assess the current state of lncRNA in the fruit fly, this thesis collected fruit fly lncRNAs from databases and literature and found that the number of known lncRNA genes in fruit fly (Table 1) was much smaller than those reported in human (~102,000) and mouse (~87,000) [4]. We suspect that the set of known lncRNAs in fruit fly is far from exhaustive. In this thesis, we first collect known lncRNA loci from databases and literature to establish an extensive list of annotated lncRNAs. Second, we produce two tissue-specific RNA-seq datasets from brain samples, respectively using the poly(A)-enriched and the ribo-zero method, and develop a computational pipeline to identify new lncRNAs from the two RNA-seq datasets.
Table 1. Numbers of fly lncRNAs from different data sources
Type Source Number of fly lncRNA genes Database FlyBase (Release 6.06) 2,460
Database UCSC genome browser 980 Literature Young et al. (2012) 1,119 Literature Brown et al. (2014) 1,875
Incomplete annotation of lncRNAs in D. melanogaster
The annotations of the collected lncRNAs are found to be incomplete. For example, Young et al. [5] reported 1,119 lincRNAs for D. melanogaster in 2012, but provided no detailed information because the RNA-sequencing reads were not generated with a strand-specific library construction [6]. In particular, transcriptional directions and exon regions are scarce for some of the previous published lncRNAs. Transcriptional direction is an important characteristic in lncRNAs. The transcripts of lncRNAs are able to disrupt the transcription of coding genes, a phenomenon known as convergent transcription in which the transcriptional direction of the lncRNA and the mRNA are head-to-head against each other [7, 8]. Conversely, for divergent transcription, the lncRNA/mRNA gene pair exhibit coordinated changes in transcription [9]. In this regard, the direction of lncRNA transcription is an important feature to be annotated.
Another essential characteristic is the exon regions, which is important for most of subsequent biological experiments such as quantitative reverse transcription polymerase chain reaction (RT-qPCR). This thesis improves lncRNA annotation by integrating a
large number of sequencing datasets (93 sets in total) from multiple sources (lncRNAs, RNA-seq and ChIP-seq). With these efforts, four general characteristics of lncRNAs are summarized in this thesis, including (1) genomic location distribution of lncRNAs, (2) length and structure of lncRNAs, (3) evolutionary conservation of lncRNAs, and (4) supporting evidences for lncRNA expression in the developmental stages.
Reliability of lncRNA expression detected from RNA-seq data
RNA-seq as a kind of high-throughput technology remains a possibility of certain bias and errors; for example, false lncRNAs detection might be caused by contaminated genomic DNA or unprocessed pre-mRNA during library construction. Recent studies have also revealed that the quantification results might be estimated differently by using different types of reads [10] or different bioinformatics/statistics methods [11, 12] . Therefore, it remains uncertain whether a lncRNA discovered from RNA-seq data is truly expressed. In this thesis, the reliability of lncRNA expression is assessed by adopting additional supporting evidences from genomics (ChIP-seq) or transcriptomics (RNA-seq) data, other data sources (such as coding potential predictors, and Conserved Domains database), and RT-qPCR validation.
Transcriptional regulation of lncRNA expression
While many studies have focused on annotating the function of lncRNAs, the
knowledge about how the expression of lncRNAs is regulated is considerably limited.
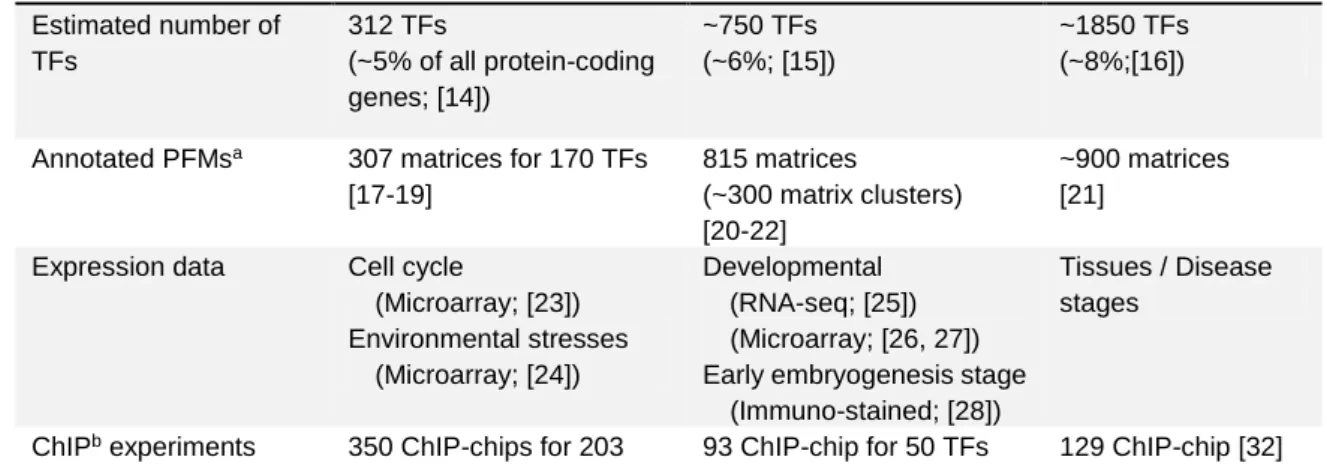
Only a few studies went upstream to ask how lncRNAs are regulated [13]. In fact, it is quite challenging to study this issue in a genome-wide level owing to the fact that transcription factor binding sites (TFBSs) with experimental validation are currently scarce (Table 2). In this regard, in silico predictions of TFBSs may be needed to investigate the regulation of lncRNA expression. This thesis incorporates de novo motif discovery to systemically investigate the presence of cis-elements shared by the promoters of coding and long non-coding (C-LNC) genes.
Table 2. Statistics of the public data used for studying transcriptional regulation in yeast, fruit fly and human
Data types Yeast Fruit fly Human
Estimated number of TFs
312 TFs
(~5% of all protein-coding genes; [14])
~750 TFs (~6%; [15])
~1850 TFs (~8%;[16])
Annotated PFMsa 307 matrices for 170 TFs [17-19]
815 matrices
(~300 matrix clusters) [20-22]
~900 matrices [21]
Expression data Cell cycle
(Microarray; [23]) Environmental stresses
(Microarray; [24])
Developmental (RNA-seq; [25]) (Microarray; [26, 27]) Early embryogenesis stage (Immuno-stained; [28])
Tissues / Disease stages
ChIPb experiments 350 ChIP-chips for 203 TFs [29]
93 ChIP-chip for 50 TFs [30, 31]
6 ChIP-seq for 2 TFs [30]
129 ChIP-chip [32]
16 ChIP-seq [33]
a. PFM: position frequency matrix, which is utilized for representing the frequency of nucleotides (A, T, C and G) in a TF binding motif.
b. ChIP: Chromatin Immunoprecipitation
1.2 Integrative approach for characterizing lncRNAs by utilizing genomics and trnascriptomics data
To assess the current state of lncRNAs and their annotation in D. melanogaster, we collected known fly lncRNAs from databases and the literature, and then used strand-specific RNA-seq datasets (Table 3) to add to the characterization of the annotations. The collected lncRNAs contained approximately 3,300 genes. To investigate whether many more lncRNAs could be discovered, we obtained additional RNA-seq datasets from the brain (Table 3). We selected the brain, instead of the whole body, because many lncRNAs were tissue-specific according to lncRNA studies in mammals [34]. Also, the brain is important for studying neuron-related diseases. Since some lncRNAs may not contain poly(A) tails, both poly(A)-enriched and ribo-zero libraries were constructed in this thesis. For the purpose of discovering novel lncRNAs, we developed a reference-based assembly approach to identify potential lncRNA transcripts.
The next question addressed in this thesis is whether RNA-seq is a reliable platform for the discovery of novel lncRNAs. A previous study used chromatin immunoprecipitation sequencing (ChIP-seq) data of chromatin signatures to detect transcription of lncRNAs [35]. A lncRNA locus, similar to that of a protein coding gene, contains the promoter and gene body and associates with the active chromatin
Table 3. Summary statistics of datasets used in thesis.
Platforms Types
Total number of datasets
Experimental condition
Number of datasets Public RNA-seq
(59 datasets in total)
Paired-end without strand-specific
30 Time course / whole body 30
Paired-end with strand-specific
29
Tissue / head 9
Tissue / ovary 2 Tissue / accessory glands 1 Tissue / testis 1 Tissue / carcass 4 Tissue / digestive system 4
Tissue / CNS 2
Tissue / fat body 3 Tissue / imaginal discs 1 Tissue / salivary glands 2 In-house RNA-seq
(2 in total)
Paired-end with poly(A)-enriched
1 Tissue / brain 1
Paired-end with ribo-zero
1 Tissue / brain 1
ChIP-seq (32 in total)
H3K36me3 3
Embryos 1
Larvae 1
Mixed Adult 1
H3K4me3 14
Embryos 7
Larvae 3
Pupae 1
Adult Female 1
Adult Male 1
Mixed Adult 1
RNA polymerase II 15
Embryos 8
Larvae 5
Pupae 1
Mixed Adult 1
Detailed information of these datasets can be seen in Additional File 3: Table S2 of the published work [36] and Appendix Table 1 in this thesis.
signatures such as H3K4me3 and H3K36me3 [37-39]. It is also known that lncRNA expression also requires specific binding of transcription factors to promote RNA polymerase II (Pol II)-mediated transcription [40-42]. In combination with the information of expression profiles and these three chromatin signatures which are believed to be present in the actively transcribed regions, an lncRNA with these three chromatin signatures would be considered to be transcribed with higher confidence. As for a lncRNA discovered from a specific tissue sample, three more analyses could be conducted to investigate the reliability. For one of this kind of lncRNAs, it could be examined whether (1) it was observed to be expressed in the RNA-seq datasets from developmental stages; (2) it was predicted with a low coding probability by two or more predictors; and (3) it was not predicted to contain any conserved domains of proteins.
Last but not least, RT-qPCR validation is the gold standard to assess a lncRNA transcribed or not.
While we integrated multiple sets of RNA-seq and ChIP-seq data (Table 3) to investigate transcription of lncRNAs during the development of D. melanogaster, we observed that a large proportion of genomic regions for lncRNAs expressed in RNA-seq were not occupied by chromatin signatures (H3K4me3, H3K36me3 and Pol II) that are usually associated with active transcription. However, no studies have discussed which feature (chromatin signatures or expression intensities) is better for inferring the
existence of lncRNAs. To answer this question, we designed RT-qPCR experiments to evaluate the confidence level of lncRNAs discovered from RNA-seq.
Additionally, to investigate transcriptional regulation of lncRNA expression, this thesis incorporated de novo motif discovery to systemically investigate the presence of cis-elements shared by the promoters of coding and long non-coding (C-LNC) genes.
For this purpose, the time-course RNA-seq data set of 30 developmental stages of D.
melanogaster (Table 3) was adopted. Co-expressed C-LNC gene clusters were constructed by applying hierarchical clustering on the expression profiles of fly mRNAs and the compiled lncRNAs in this thesis. To identify potential regulatory elements, de
novo motif discovery was conducted on the promoters of coding genes in a cluster. Then,
the discovered motifs were examined to see whether they are also present in the promoters of LNC genes in the same cluster. The discovered motifs were also used to identify potential common regulators of these C-LNC genes.
In summary, this thesis aims to demonstrate that ambitious integration of sequencing data followed by computational procedures can largely facilitate novel lncRNA discovery as well as enhance lncRNA annotation and characterization.
1.3 Thesis structure
This thesis address the above challenges from the four corresponding aspects as showed in Figure 2: (1) collection and discovery; (2) annotation; (3) expression; and (4)
regulation. CHAPTER 2 provides literature reviews for the related works about lncRNA studies and the current status in Drosophila melanogaster. CHAPTER 3 presents the collection and discovery of fruit fly lncRNAs. A computational approach is developed for identifying novel lncRNAs from the generated RNA-seq data with two types of library constructions. CHAPTER 4 is then focused on improving the annotations of the published and the newly discovered fly lncRNAs. Several general properties are characterized for the curated fly lncRNAs. Next, the reliability of the lncRNA expression was investigated and validated by RT-qPCR in CHAPTER 5. Then, CHAPTER 6 moves to the upstream of the lncRNA expression. A novel method incorporating motif discovery is proposed for systematically investigating the potential
cis-elements and how it affects lncRNA expression. CHAPTER 7 discusses the
limitations of this work. The conclusion and future work are given in CHAPTER 8.
CHAPTER 2 Related Works
2.1 Brief history of long non-coding RNAs studies
Okazaki et al. (2002) investigated the mouse transcriptome by using 60,770 cDNAs, and found that around two third of mouse transcriptome was consisted by non-coding RNAs (ncRNAs) [43]. At the time, ncRNAs was comprehended as transcriptional noises. The fact of that ncRNAs is the major component of the transcriptome brought the attention of researchers to these geek transcripts. In 2004, Cawley et al. found that a great proportion of ncRNAs have transcription factor binding sites (TFBSs) in their promoters by an unbiased mapping of human TFBSs on chromosome 21 and 22 [44].
This study revealed the potential for ncRNAs to be transcriptionally regulated. This idea was relayed by Ravasi et al. In 2006, they provided experimental validation for the expression of several ncRNAs in mouse, and demonstrated that transcription of ncRNAs is the real event [45]. Thus, the view of transcriptional noises on ncRNAs was completely overthrown. In the next ten years, ncRNAs, including short ncRNAs (such as miRNAs) and long ncRNAs (lncRNAs), became the hot spots in RNA research. A RNA sequence is classified as a lncRNA if it lacks coding potential and has a length
>200 base pairs (bp) [46]. The functional roles of lncRNAs have been investigated in several studies [47-50]. A review paper reported that lncRNAs serve as regulators of
diverse cellular functions such as epigenetic silencing or transcriptional regulation [48].
Moreover, the advance of sequencing technology has facilitated the accumulation of a large amount of data. Thus, developing systematic approaches for integrating and interpreting these data is essential for the current academia research.
2.2 Integrative and systemic studies on lncRNAs
In the state-of-art of lncRNAs studies, several integrative and systemic studies have been conducted for the investigation of lncRNAs. These studies could be roughly categorized into four types: (1) LncRNA identification [51, 52]; (2) RNA-protein interactions [53, 54]; (3) LncRNA function identification [55]; and (4) Transcriptional regulation of lncRNA expression [13]. However, most lncRNAs studies were for mammalian species such as human and mouse. The accumulated information about
Drosophila melanogaster lncRNAs is lacking when compared with mammalian
organisms. Besides, over the past years, most studies have focused on investigating lncRNA functions [47-50], but few studies went upstream to ask how lncRNAs are regulated [13].
2.2.1 Related works for characterizing lncRNAs in in Drosophila
melanogaster
Many studies have developed bioinformatics methods to systematically identify and
related works are only a few. Young et al. (2012) [5] was the first work which systematically identified a large amounts of lncRNAs from RNA-seq data in fruit fly.
But due to the RNA-seq datasets that were not constructed by strand-specific library, only limited annotations and characteristics of fly lncRNAs could be provided in their study. In 2014, Brown et al. [56] incorporated RNA-seq data from 10 types of tissues to study all types of transcripts in fly transcriptome, which also included lncRNAs. This study provided some interesting insights of lncRNAs, but failed to comprehensively discuss characteristics of fly lncRNAs. In this thesis, we integrated the information provided by the above two studies, compensated the scarce information of them, and thus presented the most up-to-date list of fly lncRNAs with comprehensive annotations. .
2.2.2 Related works for transcriptional regulation of lncRNA expression
Studies on mouse and human have reported that lncRNA genes are similar to protein coding genes in that they contain promoters and transcribed regions [44]. Upon transcription, these regions will have active chromatin signatures such as the tri-methylation of histone H3 lysine 4 (H3K4me3) and the tri-methylation of histone H3 lysine 36 (H3K36me3) [38, 39, 57]. It has also been revealed that lncRNA expression may require specific binding of transcription factors to drive RNA polymerase II (Pol II)-mediated transcription [40-42]. Wu et al. (2010) found that the expression of
lncRNAs was regulated though EzH2-mediated H3K27 methylation on embryonic stem cells, which is known as a similar way to the regulation of protein coding genes [58]. In plant, it has been demonstrated that the expression of the lncRNA, COOLAIR, was inhibited by covered COOLAIR promoter with AtNDX aim to form R-loop in
Arabidopsis [59]. Moreover, Yang et al. (2013) showed that histone
acetylation-mediated modulation of the promoter region could suppress lncRNA, and cause low expression in tumor (lncRNA-LET) [60]. The above-mentioned studies have provided a firm support to that lncRNA expression is associated with the molecular modification of its promoted region.
To fully understand the function of lncRNA, the key driver of lncRNA expression may be also essential but less study systematically investigated this issue. For example, Yang et al. [13] constructed the ChIPBase database providing a user-friendly interface for users to browse transcription factor (TF) binding sites from ChIP-seq experiments in the regulatory region of a lncRNA. Though, the information provided by the ChIPBase included all of the peaks across different cell lines or tissues without telling from which experimental condition a TFBS is. Therefore, users cannot obtain specific TFBS information in a specific experimental condition. This inspired Jiang et al. [61]
developed a web-based tool, TF2LncRNA, to enables users to obtain the specific information of TFs, TFBSs, and the experimental conditions. However, both of the two
studies highly replied on ChIP experiments, where only limited number of ChIP datasets for TFs is available in D. melanogaster. To be more specific, only ~100 ChIP experiments for ~50 TFs are available currently, the number of which is far less than the estimated number of TFs (as showed in Table 2). An alternative approach is to adopt de
novo motif discovery on the promoters of co-expressed genes for investigate
transcriptional regulation. This approach may be easily frustrated by the fact that the number of co-expressed lncRNAs is usually limited. In this regards, this thesis proposed a procedure that performing de no motif discovery only on coding gene promoters in a co-expressed gene cluster, and then used the discovered motifs to identify regulatory elements in the co-expressed lncRNA promoters.
CHAPTER 3 Collection and Discovery of lncRNAs in
Drosophila melanogaster
In this thesis, we compiled an the most update list of fruit fly lncRNAs from databases and literature and found that the number of known lncRNA genes in fruit fly (~3,300) was much smaller than those reported in human (~102,000) and mouse (~87,000) [4].
We suspected that the set of known lncRNAs in fruit fly was far from exhaustive.
Indeed, 462 novel lncRNA genes were discovered when two brain-specific RNA-seq datasets were produced in the present study. Thus, more lncRNA genes will likely be found when more RNA-seq studies of fruit fly are conducted in the future. The final set of curated fly lncRNAs, including known and novel lncRNAs, contains 3,816 lncRNA genes (4,599 lncRNA transcripts).
3.1 Known lncRNAs collected from databases and literatures
A non-redundant set of 1,999 lncRNA genes (2,347 transcripts) from FlyBase (r5.57) [62] and the UCSC genome browser [63] was first constructed. Next, the long intergenic non-coding RNAs (lincRNAs) reported in the study by Young et al. [5] and Brown et al. [56] were collected to expand the list. Among the 1,119 lincRNAs reported by Young et al. and the 3,088 lncRNAs by Brown et al., some potentially redundant
lincRNAs or lncRNAs were excluded by a selection procedure (see the section of 3.4.1).
In the end, 583 lincRNA genes (583 transcripts) from Young et al. and 772 lncRNA genes (1,207 transcripts) form Brown et al. were added to the non-redundant set reported in the present study.
3.2 Novel lncRNAs identified from brain samples
We developed an approach to discover lncRNAs from the brain-specific RNA-seq datasets of fruit fly produced in this thesis (SRP051132), which were obtained using two types of library construction, the poly(A)-enriched and ribo-zero protocols. This approach can be applied to future studies for the same purpose. The proposed pipeline consists of several steps, including reference-based assembly (using an earlier version of gene annotations downloaded from UCSC genome browser on March 13th, 2013), coding potential estimation, ribosomal RNA exclusion, and read remapping (see the section of 3.4.3). The results consisted of 754 intergenic transcripts that have not been previously annotated. After excluding transcripts with lengths less than 200 bp, 725 transcripts remained as putative lncRNAs. Then, we retained 591 putative lncRNA genes which showed a low potential to encode proteins. After excluding ribosomal RNA contamination, 587 putative lncRNA transcripts remained. We further excluded 57 transcripts that had no sufficient read support during the follow-up read remapping.
Before finalizing the list, we compared the discovered lncRNAs with the most updated
gene annotations from UCSC genome browser (Sep. 21st, 2015), and removed 68 transcripts that overlapped some newly reported coding genes in the sense direction.
Finally, we obtained 462 novel lncRNA transcripts that have not been reported previously. RT-qPCR experiments were conducted for validation. The results showed that all of the selected novel lncRNAs were validated, which revealed the high reliability of the discovered novel lncRNA genes (details in CHAPTER 5).
3.3 Up-to-date list of long non-coding RNAs in D. melanogaster
In total, a set of 3,816 curated lncRNA genes (4,599 transcripts) in D. melanogaster was constructed in this thesis (Additional File 1 and Additional File 2 of the published work [36]). The final set of curated fly lncRNAs is larger than the 2,460 lncRNA genes in FlyBase (Release 6.06 [62]), and the 2,446 lncRNA transcripts recently reported by Matthews et al. [64]. Our final list is also larger than the latest version (version 4) of a well-known lncRNA database, NonCode (961 lncRNA genes) [65].
3.4 Methods for collection and discovery of fruit fly lncRNAs
3.4.1 Collection of published lncRNAs
The lncRNAs were collected from FlyBase [62], the UCSC genome browser [63], Young et al. [5], and Brown et al. [56]. A set of lncRNAs was obtained using the keyword term “non_protein_coding_genes” when querying FlyBase D. melanogaster
(r5.57). LncRNA transcripts shorter than 200 bp were filtered out. First, the lncRNA transcripts from FlyBase were chosen as the primary set of lncRNA sequences. Second, BLASTn [66] was used to align the lncRNA transcripts collected from the UCSC genome browser against the primary set. Afterwards, by checking the alignments with E-value < 10-10 in the BLASTn results, redundant lncRNA transcripts were removed when either of the following two conditions was satisfied: (1) a lncRNA has the same loci with another lncRNA, or (2) a lncRNA overlaps another lncRNA with an overlapping region covering 50% of the transcript length. With the specified criteria, 972 redundant sequences were excluded. Third, 1,119 lincRNAs were collected from the study by Young et al. [5], where 415 sequences were excluded because they contained overlapping regions with the non-redundant set of lncRNA transcripts from FlyBase and the UCSC genome browser. Additionally, 3,088 lncRNA transcripts were collected from Supplementary Data 2 of the study of Brown et al. [56]. We removed 49 lncRNA transcripts with a length
200 bp and 19 transcripts that were annotated as
coding genes in the file provided by Brown et al. The remaining 3,020 lncRNA transcripts were next aligned to the above non-redundant set of lncRNA transcripts from FlyBase, UCSC, and Young et al. by using BLASTn. The alignments with E-value <10-10 in the BLASTn results were further examined by the following selection procedure.
We removed lncRNA transcripts that were annotated with an already included FlyBase
lncRNA ID. LncRNA transcripts containing overlapping regions with the curated FlyBase/UCSC lncRNA transcripts (covering
50% of the either transcript length)
were removed unless the new lncRNA transcripts contain multiple exons and the number of exons differs from that of FlyBase/UCSC lncRNA transcripts. Afterwards, lncRNA transcripts aligned to lncRNA transcripts of Young et al. were removed only if they have the same loci or have an overlapping region covering 90% of transcript length.As a result, 1,635 redundant lncRNA transcripts were removed. All lncRNA transcripts were then aligned to 156 ribosomal RNAs collected from FlyBase r6.07 (2 sequences) and the NCBI database (154 sequences) using BLASTn. Sequences (10 sequences) with E-value < 10-10 and identity
99% were removed to exclude ribosomal RNA
contamination.To ensure that the lncRNAs curated in this thesis did not contain newly reported coding genes present in the most updated FlyBase annotations, we retrieved ‘Feature Type’ and ‘Gene Model Status’ for the curated lncRNA transcripts from FlyBase by submitting transcript IDs to the batch download tool of FlyBase r6.07. Additionally, we utilized ‘Coordinates Converter’ provided by FlyBase to see whether a transcript location is no longer present in the release 6 genome (R6). Moreover, for the lncRNA transcripts from Young et al., FlyBase recently incorporated these lncRNA transcripts and provided update annotations based on a manual review (FBrf0220965). By taking
the above-mentioned information from FlyBase into account, we removed 673 transcripts that were annotated as protein coding genes, pseudogenes, rRNA genes, snRNA, snoRNA, scaRNA, out-of-date IDs, or located within TE regions or the sequences dropped by the BDGP in the R6 genome. In the end, this thesis constructed a set of lncRNAs from FlyBase, the UCSC genome browser, and the studies by Young et
al. [5] and Brown et al. [56], consisting of 3,354 lncRNA genes, corresponding to 4,137
lncRNA transcripts.
3.4.2 RNA-seq data of the fly brain
Brain samples were collected from four-day post-eclosion Canton S male adults. At a time, 20 to 30 adults were gassed with carbon dioxide and dissected. The collected brains were preserved in refrigerator until 100 brains were collected. Afterwards, total RNA was purified from the 100 brains, using the NucleoSpin® RNA II Purification Kit.
RNA-seq was performed using the strand-specific library with poly(A)-enriched protocol or Ribo-Zero™ Gold Kit to generate paired-end 90-bp reads on the Illumina Hi-seq 2000 platform. In total, ~25 million and ~50 million pair-end reads of 90-bp in length were obtained from the poly(A)-enriched library and the total RNA (with Ribo-Zero™ Gold Kit) library, respectively. The raw reads have been submitted to NCBI Sequence Read Archive database (SRP051132).
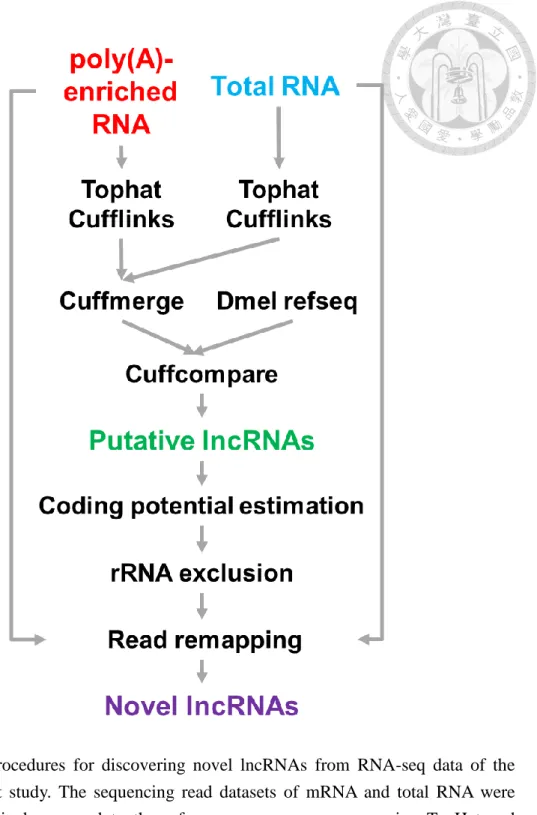
3.4.3 Novel lncRNA discovery
To discover novel lncRNAs from the two new datasets described above, we first mapped all short reads onto the unmasked D. melanogaster genome sequences (BDGP R5/dm3; from the UCSC genome browser), using TopHat [67]. Cufflinks [67] was then used to assemble the mapped reads and the assembled transcripts were compared to the reference annotation (Dmel refseq) from the UCSC genome browser (downloaded on March 13th, 2013) using Cuffcompare, a utility included in Cufflinks. The two sets of assembled transcripts, from poly(A)-enriched RNA and total RNA, respectively, were compared to the reference annotation at the same time to get a union set of intergenic transcripts. We set a length of 200 bp as the cutoff to exclude shorter non-coding RNAs.
We then calculated the coding potential of all putative lncRNA loci using the Coding Potential Calculator (CPC) [68]. The putative lncRNA transcripts were then aligned
against a set of ribosomal RNAs (the same set described in the “Collection of published lncRNAs” section) to exclude ribosomal RNA contamination. Afterwards, we remapped
both poly(A)-enriched RNA and total RNA sequencing reads to the putative lncRNA transcripts, using Cufflinks. After remapping, we excluded transcripts with no read support as reported by Cufflinks. The developed computational pipeline is shown in Figure 3. Then, we compared the identified lncRNAs with the most updated R5 genome annotations downloaded from the UCSC genome browser (Sep. 21st, 2015), and
Figure 3. Procedures for discovering novel lncRNAs from RNA-seq data of the present study. The sequencing read datasets of mRNA and total RNA were respectively mapped to the reference genome sequence using TopHat and Cufflinks. Putative lncRNAs were then discovered by Cuffcompare.
Sequencing reads were again mapped to the set of putative lncRNAs to construct the final set of novel lncRNAs.
removed lncRNA transcripts that overlapped with some newly reported coding genes in a sense direction. The resulting set of putative lncRNA transcripts were then compared
to the set of non-redundant lncRNA transcripts collected from FlyBase, the UCSC genome browser, and the studies by Young et al. [5] and Brown et al. [56] to remove redundant sequences.
CHAPTER 4 Annotation of the curated lncRNAs
This thesis showed that integrating multiple public datasets can largely facilitate the annotations and characterization of fly lncRNAs. A great amount of sequencing datasets, including 59 RNA-seq datasets and 32 ChIP-seq datasets collected from the modENCODE database, were used for improving the annotation. Next, according to the improved annotations, we observed four general characteristics of fruit fly lncRNAs and discussed these characteristics in this chapter.
4.1 Improving the annotation of the lncRNAs from Young et al.
Young et al. [5] reported 1,119 lincRNAs for D. melanogaster in 2012, but provided no detailed information because the RNA-sequencing reads were not generated with a strand-specific library construction [6]. In this thesis, we collected the original 30 RNA-seq datasets [6] used by Young et al. (Table 3and modENCODE IDs: 4433-4462 as shown in Additional File 3: Table S2 of the published work [36]) and adopted 29 additional stranded poly(A)-enriched RNA-seq datasets at different developmental stages (Table 3 and modENCODE IDs: 4291-4319 as shown in Additional File 3: Table S2 of the published work [36]) to determine the exon regions and transcriptional directions for the lincRNAs reported in Young et al.’s study. After excluding redundant lincRNAs against the annotated lncRNAs from the databases and removed transcripts
which are no longer lincRNAs in the current FlyBase annotations (FBrf0220965), 583 lincRNA genes remained. To identify the exon regions of these 583 lincRNA genes, we remapped the 30 RNA-seq datasets to the lincRNA sequences using Cufflinks [67]. We found that most of lincRNA genes from Young et al. consisted of only one or very few exons (Table 4 and Additional File 4 of the published work [36]). As for transcriptional
Table 4. Statistics of exon numbers in lncRNA and mRNA genes from different sources.
Exon num. FlyBase + UCSC Young et al. Brown et al. Present study mRNA
1 1167 444 465 422 2751
2 495 93 163 33 4739
3 196 32 60 6 4109
4 68 12 35 1 3659
5 36 1 15 0 2863
6 17 0 8 0 2268
7 8 0 7 0 2003
8 2 1 7 0 1586
9 3 0 2 0 1281
10 1 0 2 0 995
11 1 0 5 0 781
12 3 0 0 0 612
13 0 0 0 0 471
14 0 0 0 0 391
15 0 0 1 0 331
16 0 0 0 0 240
17 0 0 0 0 200
18 1 0 0 0 145
>=19 1 0 2 0 837
Total 1999 583 772 462 30262
Table 5. Statistics of transcriptional direction in the lncRNA genes from different sources.
Transcriptional direction FlyBase + UCSC Young et al. Brown et al. Present study mRNA
Positive (+) 1011 200 392 268 14,941
Negative (-) 988 192 380 194 15,321
Unknown (*) 0 191 0 0 0
Total 1999 583 772 462 30262
direction, similar procedures were conducted. We annotated the direction of transcription in about 67% of the 583 lincRNA genes from the study by Young et al.
(Table 5). To be more specific, 200 lincRNA genes were identified on the positive strand and 192 on the negative strand of the fruit fly genome (Table 5 and Additional File 2 of the published work [36]).
4.2 Utilizing additional RNA-seq datasets to improve the annotation of the 4,599 curated lncRNA transcripts
We utilized the RNA-seq datasets from multiple sources as well as those generated in this thesis to improve the annotation of the curated lncRNAs. Three properties were emphasized here: (1) the classification of a lncRNA in terms of its genome location and transcriptional direction; (2) whether the lncRNA is expressed in the brain or not; and (3) whether the lncRNA has a poly(A) tail or not.
The lncRNAs collected in the present study were classified into several groups according to their genome locations with respect to the closest adjacent coding gene.
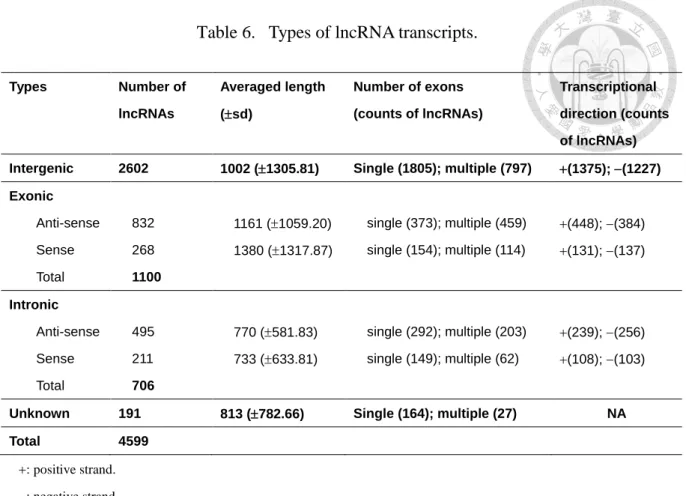
Table 6. Types of lncRNA transcripts.
Types Number of
lncRNAs
Averaged length (sd)
Number of exons (counts of lncRNAs)
Transcriptional direction (counts of lncRNAs) Intergenic 2602 1002 (1305.81) Single (1805); multiple (797) (1375); (1227) Exonic
Anti-sense 832 1161 (1059.20) single (373); multiple (459) (448); (384) Sense
Total
268 1100
1380 (1317.87) single (154); multiple (114) (131); (137)
Intronic
Anti-sense 495 770 (581.83) single (292); multiple (203) (239); (256) Sense
Total
211 706
733 (633.81) single (149); multiple (62) (108); (103)
Unknown 191 813 (782.66) Single (164); multiple (27) NA
Total 4599
: positive strand.
: negative strand.
NA: not available.
For lncRNAs located in regions that overlap with coding genes, the transcriptional direction was also considered to be an essential aspect for classification. In this regard, lncRNAs are classified into anti-sense exonic, sense exonic, anti-sense intronic and sense intronic lncRNAs, according to the transcriptional direction with respect to the overlapping coding gene. Among the curated 4,599 lncRNA transcripts, 2,602 were classified as intergenic lncRNA transcripts, 1,100 as exonic lncRNA transcripts (Table 6 and Additional File 2 of the published work [36]) and 706 as intronic lncRNA transcripts. There were 191 lncRNA transcripts for which the transcriptional direction could not be determined and were classified as ‘unknown’.
Additionally, this thesis provided two sets of sequencing reads of RNA samples from the brain (Table 3). With the two datasets, we could infer which lncRNAs were expressed in the brain. If the criterion ‘RPKM > 1’ was used, the data revealed that about one third of lncRNAs (1,464 transcripts, Additional File 2 of the published work [36]) were expressed in the brain. In Figure 13(b) we showed the RT-qPCR experiments of seven lncRNA genes with RPKM > 1 and three lncRNA genes with RPKM = 0. The
RT-qPCR results showed that the
delta Ct values of the seven lncRNA genes with
‘RPKM > 1’ were distinguishable from the three lncRNA genes with ‘RPKM = 0’. In
this regard, ‘RPKM > 1’ is considered as a safe criterion to infer the expression of lncRNAs in the brain. In summary, we found that 33% of the 3,816 lncRNA genes were expressed in the brain, when the criterion ‘RPKM > 1’ was used (Additional File 2 of the published work [36]). This number is considerably higher than that observed in other tissues reported by Brown et al. [56]. The study of Brown et al. incorporated RNA-seq data from 10 types of tissues and the testis tissue showed the highest number of expressed lncRNA genes (~30% of the 1,875 lncRNA genes).
We further examined whether a lncRNA contains the poly(A) tail. Both poly(A)-enriched and ribo-zero library constructions were used in the present study because some lncRNAs were previously found to contain no poly(A) tails in mammals [69-71]. Among the 1,464 lncRNA transcripts observed in the brain RNA-seq data,
there were 190 lncRNA transcripts with a high probability of not containing poly(A) tails when expressed in the brain (Additional File 2 of the published work [36]).
4.3 General characteristics of the fruit fly lncRNAs
To understand the general characteristics of lncRNAs, we further processed the improved annotation in the previous sections, and characterized lncRNAs from four aspects, including (1) Location distribution of lncRNAs in Genome; (2) Length and structure of lncRNAs; (3) Evolutionary conservation of lncRNAs; and (4) Supporting
evidences for lncRNA expression in the developmental stages.
4.3.1 Location distribution of lncRNAs in Genome
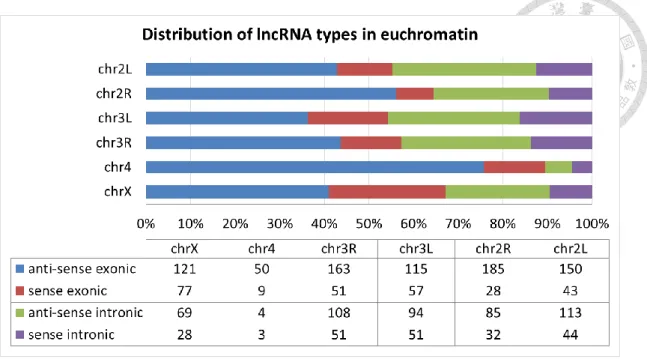
The numbers of lncRNAs from the three different sources are shown in Table 7 which indicated that lncRNAs are everywhere in the genome. In general, the euchromosome acquired more lncRNAs than the heterochromosome. Among the curated 4,599 lncRNA transcripts, 2,602 were classified as intergenic lncRNA transcripts, 1,100 as exonic lncRNA transcripts (Table 6 and Additional File 2 of the published work [36]) and 706 as intronic lncRNA transcripts. Table 6 shows that the number of lncRNAs for the four groups decreased as follows: anti-sense exonic lncRNAs > anti-sense intronic lncRNAs
> sense exonic lncRNAs > sense intronic lncRNAs. The lncRNA numbers of the four groups in the different euchromatin regions were also provided (Figure 4). Here, we only considered lncRNAs located in euchromatin because most lncRNAs were
Table 7. The number of lncRNAs from three different sources in each of the euchromosomes and heterochromosomes
Chromosome
FlyBase + UCSC
Young et al. Brown et al. Present study Summary
chr2L 564 109 135 73 881
chr2LHet 1 0 2 0 3
chr2R 353 87 97 67 604
chr2RHet 4 0 9 14 27
chr3L 378 171 188 129 866
chr3LHet 4 0 4 24 32
chr3R 368 147 200 86 801
chr3RHet 2 0 7 5 14
chr4 23 4 10 14 51
chrU 30 0 27 17 74
chrX 271 65 92 33 461
chrXHet 1 0 1 0 2
total 1999 583 772 462 3816
expressed from the euchromatin in fruit fly.
However, in the curated list, we observed that there are some lncRNA transcripts from different sources partially sharing common genomic regions. These lncRNA transcripts might be in fact the same lncRNA, might be different splicing forms of a single lncRNA gene, or might be actually independent lncRNA genes. We realized that it remained difficult to learn the fact and determine the exact boundaries for these putative lncRNAs based on the limited information collected so far. Before a mature methodology can be developed, manual examination on RNA-seq data in a genome
Figure 4. Distribution of lncRNA types in euchromatin.
browser is highly recommended. We highlighted the overlap information in Additional File 2 of the published work [36] to remind the readers that more investigations on such lncRNAs are needed. In addition, we also observed that the types of lncRNA transcripts (exonic, intronic, or intergenic lncRNAs) would potentially be changed once the annotation of protein-coding genes is updated. As the loci and boundaries of protein-coding genes continue to be refined, noncoding RNAs originally classified as intergenic may be found to be exonic, intronic or even become a new splicing form of a coding gene. In addition, Some of the Young et al. lincRNAs have been found by a follow-up FlyBase analysis (FBrf0220965) to overlap UTRs and are probably not lncRNAs. Therefore, the readers should be aware that the number of exonic sense lncRNAs in the curated list might be inflated by these lncRNAs.
4.3.2 Length and structure of lncRNAs
Transcriptional length of lncRNAs
The average length of the curated lncRNA transcripts is 1,008 bp with a diverse range and which is shorter than the average length of mRNAs (2,869 bp). More than 97% of the lncRNA transcripts have lengths from 200 bp to 4,000 bp (Table 8) which are consistent to the numbers reported by Novikova et al. [72].
Transcriptional direction of lncRNAs
When comparing lncRNAs with fruit fly mRNAs, we found that about half of the curated lncRNA genes were transcribed in the positive strands and half in the negative strands (Table 5). For each specific group of the lncRNA transcripts in Table 6 (the classification of a lncRNA in terms of its genome location and transcriptional direction), the lncRNA transcripts were equally derived from both strands. Moreover, 988 lncRNA genes (25.89% among the 3,816 lncRNA genes) were found to be transcribed in a
Table 8. Length of lncRNA transcripts
Range 200~500 500~1000 1000~2000 2000~4000 4000~up Total
FlyBase + UCSC 707 997 463 131 49 2347
Young et al. 189 179 130 60 25 583
Brown et al. 390 443 240 104 30 1207
Present study 130 214 93 23 2 462
Total 1416 1833 926 318 106 4599
direction antisense to protein coding genes. This number is larger than that (15%) reported in human [73].
Exons of lncRNAs
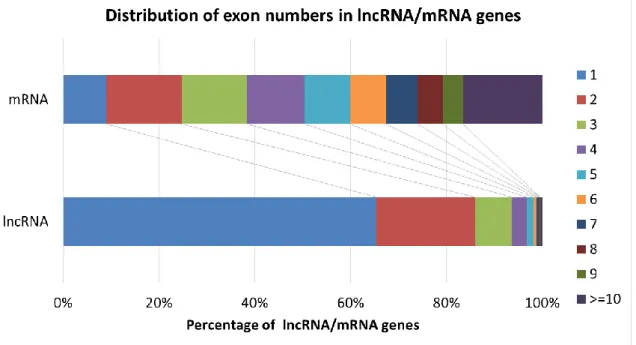
As for the number of exons in lncRNAs, fruit fly lncRNAs tend to have fewer exons than mRNAs (Table 4), which is consistent with the observation in rat by Wang et al.
[74]. Figure 5 showed that ~60% of mRNAs contain no more than five exons. The percentage of mRNAs with different exon numbers were roughly equally distributed (9% for one exon, 16% for two exons, 14% for three exons, 12% for four exons and 9%
for five exons). In contrast, ~94% of lncRNAs contain one to three exons, and more than half of the lncRNAs contain only single exon. The exon numbers of lncRNAs were
Figure 5. Distribution of exon numbers in lncRNA and mRNA genes.