行政院國家科學委員會專題研究計畫 成果報告
類固醇對 capsaicin 受體的調控作用:結構與活性間之關係
及痛覺行為之測試(2/2)
計畫類別: 個別型計畫
計畫編號: NSC91-2320-B-006-042-
執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立成功大學生理學科(所)
計畫主持人: 吳豐森
計畫參與人員: 陳淑貞、賴維斌、呂雨青
報告類型: 完整報告
處理方式: 本計畫可公開查詢
中 華 民 國 92 年 10 月 22 日
摘要
本研究計畫的主要目的在探討類固醇對 capsaicin 受體調控作用的分子機 制。我們實驗室先前的研究結果顯示,神經性類固醇還原雄性素(DHEA)及硫酸 孕烯醇酮(PS)對 capsaicin 受體具有負向調節的作用。在本研究中,我們採用快速 分離出來的成年大白鼠背根神經節細胞為材料,利用全細胞膜電位固定的電生理 記錄方法,進一步探討 DHEA 及相關類固醇對 capsaicin 受體所媒介電流反應的 調控作用及其作用機制。結果顯示 DHEA 快速且可逆地抑制 capsaicin 所誘發的 全細胞內向電流,且其抑制作用呈現劑量依賴效應,最大抑制作用為 100%,而
達到最大抑制作用一半所需的 DHEA 濃度(EC50)為 6.7 µM。DHEA 會增加
capsaicin 之 EC50,但對 capsaicin 之最大反應卻無影響作用,表示 DHEA 對 capsaicin 受體所媒介反應的抑制作用是屬於競爭型。Capsaicin 所誘發的電流及 DHEA 抑制 capsaicin 所誘發電流的能力,並不會因為細胞內液添加飽和濃度 DHEA 而有所降低,表示 DHEA 對 capsaicin 所誘發電流的抑制作用,是經由作 用到 capsaicin 受體的細胞外表面位置來達成的。此外, DHEA 並不是透過活化 去磷酸酶 1、2A 或 2B 來抑制 capsaicin 所誘發的電流。DHEA 的立體異構物 3α-DHEA 對 capsaicin 所誘發的反應不但沒有抑制作用,反而有增強的作用,顯 示類固醇對 capsaicin 受體的調控作用具立體專一性。另外,我們也利用皮內注 射的給藥方式,將 capsaicin 及其他藥物打入大白鼠的腳掌,測定 PS 對 capsaicin 所誘發的疼痛行為反應是否有抑制作用,並且進一步探討其作用機制。結果顯示 PS 可抑制 capsaicin 所誘發的疼痛反應,且其抑制作用呈現劑量依賴效應。PS 對 capsaicin 所誘發疼痛反應的抑制作用,並不會受到廣效型 opioid 受體抑制劑 (naloxone)或 cannabinoid 受體抑制劑(AM251 與 AM630)的影響而有所降低,表示 DHEA 並不是透過活化 opioid 系統或 cannabinoid 系統,來抑制 capsaicin 所誘發 的疼痛反應。此外,我們也發現 capsaicin 的注射並不會引起內生性 opioids 及 cannabinoids 的釋放。上述結果顯示,PS 可能藉由直接抑制 capsaicin 受體的活 性,來達到對 capsaicin 所誘發疼痛反應的抑制作用。
關鍵詞:Capsaicin 受體、神經性類固醇、還原雄性素(DHEA)、硫酸孕烯醇酮(PS)、
背根神經節細胞、全細胞膜電位固定記錄法、競爭型抑制作用、立體專一性、痛 覺
ABSTRACT
The major goal of this proposal is to explore the molecular mechanisms whereby steroids modulate the capsaicin receptor. Previous studies from our laboratory have shown that neurosteroids such as dehydroepiandrosterone (5-androsten-3β-ol-17-one; DHEA) and pregnenolone sulfate (PS) negatively modulate the capsaicin receptor. In the present study, we further characterized the effects of DHEA and related steroids on the capsaicin-induced current in acutely dissociated rat dorsal root ganglion neurons using the whole-cell voltage-clamp recording technique. DHEA rapidly and reversibly inhibited the capsaicin-induced current in a dose-dependent manner, with an EC50 of 6.7 µM and a maximal inhibition of 100%. DHEA increased the capsaicin EC50 with little effect on the capsaicin
maximal response, indicating that the blocking action of DHEA is competitive.
Neither the capsaicin response nor inhibition of the capsaicin response by
extracellularly applied DHEA was significantly affected by inclusion of a saturating concentration of DHEA in the electrode buffer, arguing that DHEA acted at the extracellular surface of the membrane. Moreover, DHEA did not act through protein phosphatases to inhibit the capsaicin-induced current. Furthermore, the stereoisomer of DHEA, 5-androsten-3α-ol-17-one (3α-DHEA), failed to inhibit the
capsaicin-induced current, producing instead a potentiating effect on the capsaicin response, demonstrating that the interaction of steroids with the capsaicin receptor is stereospecific. In addition, we investigated the effect of intradermal administration of PS on the nociceptive response evoked by intradermal injection of capsaicin into the rat hindpaw. Results revealed that PS dose-dependently inhibited the
capsaicin-induced nociceptive response. PS inhibition of the capsaicin-induced nociception was not significantly reduced by the non-selective opioid receptor antagonist (naloxone) or by cannabinoid receptor antagonists (AM251 and AM630), indicating that neither opioid- nor cannabinoid-dependent mechanisms mediated the effect of PS. Moreover, intradermal injection of PS did not cause the release of endogenous opioids and cannabinoids. Our results suggested that PS depressed the capsaicin-induced nociception via direct action on capsaicin receptors or associated proteins.
Keywords: Capsaicin receptors, Neurosteroids, Dehydroepiandrosterone (DHEA), Pregnenolone sulfate (PS), Dorsal root ganglion neurons, Whole-cell voltage-clamp recordings, Competitive inhibition, Stereospecific, Nociception
目錄
中文摘要
--- I
英文摘要
--- II
前言
--- 1
方法
--- 3
結果
--- 6
討論
---
11
參考文獻
--- 15
附表
---
19
附圖
---
20
自評
---
31
INTRODUCTION
Capsaicin, the main pungent ingredient in hot chili peppers (Green and Shaffer, 1993), activates a distinct subpopulation of primary sensory neurons, with somata in dorsal root, trigeminal as well as nodose ganglia (Holzer, 1991; Szallasi and
Blumberg, 1999). Capsaicin exerts its effects by binding to distinct cell-surface
receptors. Electrophysiological studies from dorsal root ganglion neurons demonstrate that capsaicin generates an inward current that results in the depolarization of the neuron (Heyman and Rand, 1985). This inward current results from the opening of a nonselective cationic channel that is largely permeable to Na+, K+ and Ca2+ (Oh et al., 1996). Capsaicin receptors have attracted particular attention by virtue of their
proposed role in nociception (Szolcsanyi, 1990) and inflammatory thermal hyperalgesia (Caterina et al., 2000; Davis et al., 2000).
Steroid hormones are known to influence profoundly the neuronal excitability (Majewska, 1987). Although the effects of steroids have been thought to be mediated by genomic steroid response elements (McEwen, 1991), recent evidence indicates that many steroids influence the neuronal excitability via direct effects on excitatory and inhibitory amino acid receptors (Majewska et al., 1986; Wu et al., 1990; Wu et al., 1991; Park-Chung et al., 1997). Certain reduced metabolites of progesterone and deoxycorticosterone, such as 5α-pregnan-3α-ol-20-one and
5α-pregnane-3α,21-diol-20-one, have been shown to be potent positive modulators of the GABAA receptor, but have no effect on the glycine response (Majewska et al., 1986; Wu et al., 1990). On the other hand, the neurosteroid pregnenolone sulfate (PS) interacts with GABAA and glycine receptors in an antagonistic fashion (Majewska et al., 1988; Wu et al., 1990; Wu et al., 1997). Surprisingly, PS also specifically
potentiates the N-methyl-D-aspartate (NMDA) glutamate receptor-mediated response while inhibiting slightly non-NMDA glutamate receptor-mediated responses (Wu et al., 1991; Wu and Chen, 1997). Interestingly, the neurosteroid
dehydroepiandrosterone (5-androsten-3β-ol-17-one; DHEA) which has been claimed to be a “super hormone” modulates both N-methyl-D-aspartate (NMDA) receptor- and γ-aminobutyric acid type A (GABAA) receptor-mediated responses in brain (Baulieu and Robel, 1996). These results are consistent with the hypothesis that steroids regulate the balance between excitation and inhibition on neurons.
Despite the important role of capsaicin receptors in nociceptive transmission and profound actions of steroids on neuronal excitability, very little is known about interactions of steroids with the capsaicin receptor. Nevertheless, recent
electrophysiological studies from our laboratory demonstrate that DHEA and PS inhibit the capsaicin receptor-mediated current in acutely dissociated rat dorsal root ganglion neurons (Chang and Wu, 2002), but the mechanisms of actions of steroids on the capsaicin receptor are less clear. In the present study, we characterized
electrophysiologically the effects of DHEA and related steroids on the capsaicin
receptor-mediated current in acutely dissociated rat dorsal root ganglion neurons using the whole-cell voltage-clamp recording technique (Hamill et al., 1981). In addition, we examined the effect of PS administered intradermally on the nociceptive response evoked by intradermal injection of capsaicin into the rat left hindpaw. We report that DHEA acts extracellularly to inhibit the capsaicin-induced current in a competitive and dose-dependent manner. Furthermore, DHEA and related steroids interact stereospecifically with the capsaicin receptor. Moreover, intraplantar injection of PS dose-dependently inhibits capsaicin-induced nociception via an opioid- and cannabinoid-independent mechanism.
METHODS
2.1. Preparation of acutely dissociated dorsal root ganglion neurons
Animal care was consistent with the guidelines set by the Laboratory Animal Center of National Cheng Kung University. All experimental procedures were approved by the Animal Research Committee of Medical College of the National Cheng Kung University. Dorsal root ganglion neurons were acutely dissociated from Sprague-Dawley male rats (200-300 g). In some experiments, dorsal root ganglion neurons obtained from female rats of similar body weight were also used. The
procedures used have been described previously (Wiley et al., 1993) with some minor modifications. Briefly, rats were anesthetized with urethane. Thoracic and lumbar dorsal root ganglia were rapidly removed, treated with 1 mg/ml Sigma type IA collagenase in Ca2+/Mg2+ free Hank’s balanced solution (GIBCO BRL, Grand Island, NY) supplemented with 4.2 mM NaHCO3, 1 mM sodium pyruvate, 20 mM HEPES, and 3 mg/ml bovine serum albumin for 45 min at 37oC, and pelleted by centrifugation (900 rpm, 3 min). The resulting pellet was suspended in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO BRL, Grand Island, NY) supplemented with 26.2 mM NaHCO3, 1 mM sodium pyruvate , 20 mM HEPES and 5% fetal bovine serum (GIBCO BRL, Grand Island, NY), triturated gently to disperse the cells, and then centrifuged. Cell pellet was resuspended in DMEM supplemented with 26.2 mM NaHCO3, 1 mM sodium pyruvate , 20 mM HEPES, 100 units/ml penicillin, 100 µg/ml streptomycin, 1.8 mM CaCl2, and 10% fetal bovine serum. Cell suspension (0.5 ml/3 ganglia) was plated on Corning 35-mm polystyrene tissue culture dishes and incubated at 37oC in an atmosphere of 5% CO2/95% air. An additional 1-ml of medium was added after 30 min. Neurons were used in experiments within 1-10 hr after plating.
2.2. Electrophysiological recordings
Electrophysiological experiments were carried out at room temperature (23-25oC) in 35-mm tissue culture dishes on the stage of an inverted phase-contrast microscope. Whole-cell currents were recorded by the whole-cell variant of the patch clamp technique (Hamill et al., 1981). Electrode resistance was 2-4 MΩ when filled with the intracellular solution containing (in mM): 140 KCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, and 4 MgATP (pH adjusted to 7.2 with KOH). Sucrose was added to make osmolarity 320 mosM. In experiments in which high concentrations of capsaicin were used, the intracellular solution was replaced with a Cs+-containing
pipet solution (in mM, 140 CsCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, and 4 MgATP, pH adjusted to 7.2 with CsOH and osmolarity to 320 mosM with sucrose).
The bath solution contained (in mM): 150 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 D-glucose (pH adjusted to 7.4 with NaOH and osmolarity to 340 mosM with sucrose). All solutions passed through 0.2 µm Millipore filters before use.
Recordings were made using an Axopatch-1D patch clamp amplifier (Axon Instruments, Foster City, CA). Cells with series resistance greater than 10 MΩ were rejected. Only cells with resting membrane potential more negative than –40 mV and input resistance in excess of 150 MΩ were used. Unless otherwise specified, all recordings were made with the cell membrane potential clamped at –50 mV. Currents were filtered at 1 kHz using an 8-pole Bessel filter and digitized (4 ms/point) using an on-line data acquisition system (pClamp 6.0, Axon Instruments, Foster City, CA).
Drug solutions were applied to single neurons by pressure ejection (15 psi) from 7-barrel pipets (Choi and Fischbach, 1981; Chan and Farb, 1985). All steroids were purchased from Steraloids (Wilton, NH), with the exception of progesterone (Sigma, St. Louis, MO). Capsaicin, okadaic acid, and cyclosporin A were purchased from Tocris Cookson (Bristol, UK). Cyclophilin A was purchased from Sigma (St.
Louis, MO). Stock solutions of capsaicin, cyclosporin A, and steroids were prepared in dimethyl sulfoxide (final concentration, 0.5%, v/v). To obviate the possible effect of dimethyl sulfoxide on the capsaicin-induced current, all the other drug solutions, including external buffer (in the pressure pipets), also contained 0.5% dimethyl sulfoxide. In all experiments, neurons received a 10-s prepulse of either external buffer or steroid solution, followed by a 30-s application of either capsaicin or
capsaicin plus steroid, and further by a 20-s pulse of external buffer solution. A period of 4 min was allowed between successive applications of capsaicin.
2.3. Capsaicin test
Male Sprague-Dawley rats (250-350 g) were housed at a temperature of 22 ± 1oC with a 12 h light-dark cycle. Food and water were available ad libitum. All experiments were performed during light cycle. Animals were acclimated to the laboratory for at least 1 hr before testing and were used once for behavioral experiments.
The procedure used for the capsaicin test was similar to that described
previously (Sakurada et al., 1992) with some minor modifications. The animals were
placed individually in a 30 cm x 30 cm x 30 cm Plexiglas observation box 10 min before the injection of capsaicin to allow adaptation to the new environment.
Following the adaptation period, 50 µl of capsaicin was injected intradermally (i.d.) under the dorsal surface of the right hindpaw using a microsyringe with a 26-gauge needle. Each rat was then returned to the observation box. The amount of time that animals spent licking the injected paw was measured with a stopwatch and was considered as an indicator of the nociceptive response. The animal was observed individually for 15 min, immediately after the injection of capsaicin in the time
course experiment. To examine the effect of PS on capsaicin-induecd licking behavior, animals were injected i.d. with capsaicin or capsaicin plus PS. To investigate the role of endogenous opioids or cannabinoids in PS-induced anti-nociception, separate groups of animals received i.d. co-administration of capsaicin with PS, PS plus an opioid antagonist, or with PS plus a cannabinoid antagonist.
The following drugs were used in the behavioral study: capsaicin, PS, AM251, AM630, WIN55,212-2 mesylate, naloxone hydrochloride and endomorphin-1. All drugs were obtained from Torcris, with the exception of PS (Steraloids). Capsaicin, PS, AM251, AM630 and WIN55,212-2 mesylate were dissolved in dimethyl sulfoxide (5%, v/v) in phosphate-buffered saline (PBS). Naloxone hydrochloride and
endomorphin-1 were dissolved in PBS. PBS contained (in mM): 137 NaCl, 2.7 KCl, 1.5 KH2PO4, and 8.1 NaH2PO4 (pH 7.4).
2.4. Data analysis
The degree of modulation of the capsaicin-induced current by steroid, the percent change, was defined as (I’/I – 1) X 100%, where I and I’ were, respectively, the capsaicin-induced current in the absence and presence of steroid. I was the average of control responses obtained before and after the capsaicin response in the presence of steroid. In most cases, complete or nearly complete reversal of the steroid effect was obtained after washout. Electrophysiological and behavioral results were expressed as the mean ± SEM. Data were compared statistically by Student’s t test.
P-values less than 0.05 (P < 0.05) were considered as indicative of significance.
RESULTS
3.1. DHEA inhibits the capsaicin receptor-mediated current
At a holding potential of –50 mV, capsaicin activated inward currents (> 100 pA) in 68% (268/392) of neurons with diameters of 26-43 µm, which reversed at or near 0 mV in standard recording solutions, as expected for Na+, K+ and Ca2+-mediated currents (data not shown). The currents induced by 100 nM capsaicin were
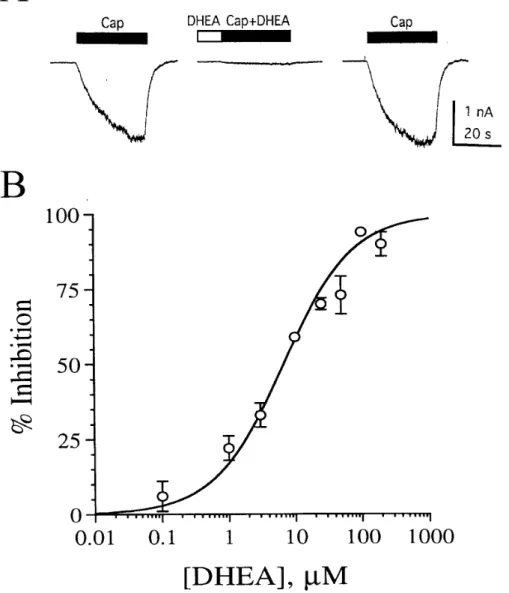
completely blocked by 10 µM capsazepine (n = 4), consistent with activation of capsaicin receptors. In agreement with the capsaicin binding site being located at the intracellular surface of the receptor (Jung et al., 1999), the current induced by 100 nM capsaicin reached slowly to its full amplitude. The effect of DHEA on the current induced by 100 nM capsaicin is illustrated in Fig. 1. Pressure application of 100 µM DHEA rapidly and reversibly inhibited the capsaicin response (Fig. 1A). DHEA alone produced little or no direct response. The magnitude of reduction of the capsaicin response (93 ± 0.7%, n = 6) by DHEA in female rat neurons was similar to that (94 ± 1.3%, n = 6) observed in male rat neurons, indicating that the effect of DHEA is sex-independent.
To quantitatively evaluate the potency and efficacy of DHEA for capsaicin receptors, pooled data were used to construct the dose-response curve for inhibition of the 100 nM capsaicin response by DHEA. DHEA inhibited the capsaicin-induced current in a dose-dependent manner, with an EC50 of 6.7 µM, a maximal inhibition of 100%, and a Hill coefficient of 0.83 (Fig. 1B). The threshold concentration for an effect of DHEA on the capsaicin-induced current was 0.1-1 µM. The maximal effect on the capsaicin response was achieved at approximately 100 µM.
3.2. Inhibition of the capsaicin response by DHEA is competitive
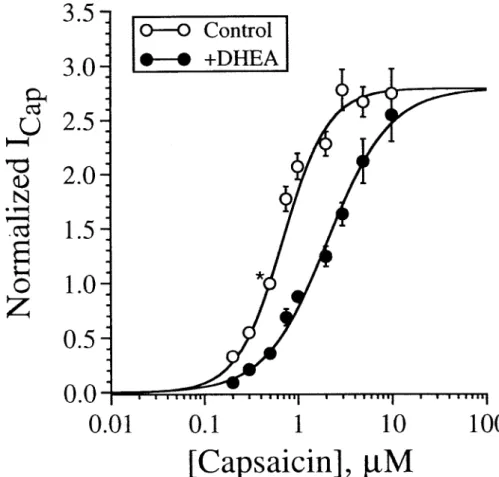
To investigate whether inhibition of the capsaicin response by DHEA is competitive, pooled data were used to construct dose-response curves for capsaicin in the presence and absence of 50 µM DHEA. In this experiment, KCl was replaced with CsCl in the pipet solution and the cell was held at –30 mV to reduce the amplitude of the capsaicin-induced current. In addition, all responses were normalized to the peak current induced by 0.5 µM capsaicin to obviate cell-to-cell variability with respect to the maximal current induced by capsaicin. As illustrated in Fig. 2, DHEA increased the capsaicin EC50 with little effect on the capsaicin maximal response, suggesting that antagonism of the capsaicin response by DHEA is competitive in nature.
3.3. DHEA does not act through protein phosphatase activation
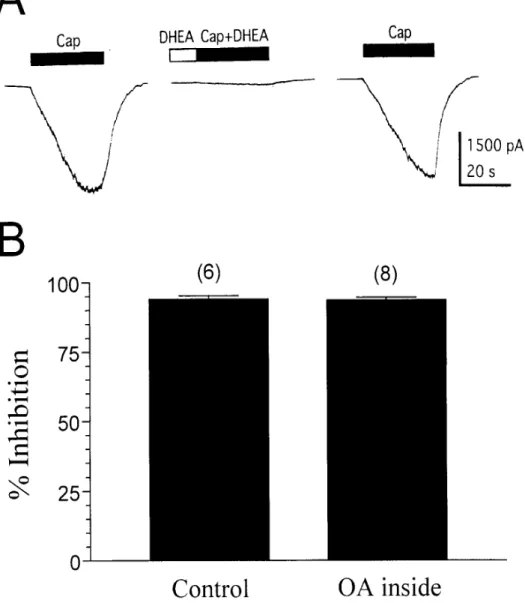
Dephosphorylation of the capsaicin receptor by protein phosphatases (PPs) has been demonstrated to reduce the capsaicin response in rat sensory neurons (Docherty et al., 1996; Koplas et al., 1997). To examine whether DHEA inhibits the capsaicin response via PP activation, we first assayed the effect of DHEA (100 µM) on the capsaicin (100 nM) response in the presence of intracellular okadaic acid (1 µM), a selective PP1/2A inhibitor. We found that addition of okadaic acid in the pipet solution stabilized the capsaicin-induced current (data not shown) and did not
significantly influence the inhibitory effect of DHEA on the capsaicin-induced current (Fig. 3A). Fifteen min after cell dialysis, the average inhibition produced by
extracellular DHEA with okadaic acid inside was 94 ± 1.0% (n = 8), which did not significantly differ from that (94 ± 1.3%, n = 6) measured without intracellular okadaic acid (P > 0.05, unpaired t-test) (Fig. 3B). Similarly, inclusion of the selective PP2B inhibitor cyclosporin A (50 nM) plus cyclophilin A (20 nM) in the electrode buffer did not significantly affect
inhibition by extracellularly applied DHEA (100 µM) of the capsaicin (100
nM)-induced current (91 ± 2.1%, n = 7 with inhibitor versus 94 ± 1.3%, n = 6 without inhibitor). These results suggest that PP1/2A and PP2B are not involved in DHEA inhibition of the capsaicin response.
3.4. DHEA acts through the extracellularly directed site
To determine whether DHEA acts intracellularly to inhibit the capsaicin response, we tested the effect of a saturating concentration (200 µM) of intracellular DHEA on inhibition of the capsaicin response by extracellularly applied DHEA (100 µM). In this experiment, 1 µM okadaic acid was added in the pipet solution to prevent possible tachyphylaxis of the capsaicin-induced current. Under this condition,
inclusion of DHEA in the electrode buffer had no significant effect on the
capsaicin-induced current at 16 min after the cell was ruptured and did not block the effect of extracellular DHEA (Fig. 4A). In six cells, average currents induced by 100 nM capsaicin at 4, 8, and 16 min after cell rupture were, respectively 1554 ±416.5, 1524 ± 336.0, and 1595 ±352.0 pA, which did not differ significantly from that (1607
± 451.3 pA) immediately (0 min) after cell rupture (P > 0.05, paired t-test). In addition, the average inhibition produced by extracellular DHEA with DHEA inside was 93 ± 2.9% (n = 6), which was not significantly different from that (94 ± 1.0%, n = 8) measured without intracellular DHEA (P > 0.05, unpaired t-test) (Fig. 4B). This
result indicates that DHEA acts at the extracellular surface of the membrane.
3.5. Effects of other chemically related steroids on the capsaicin-induced current
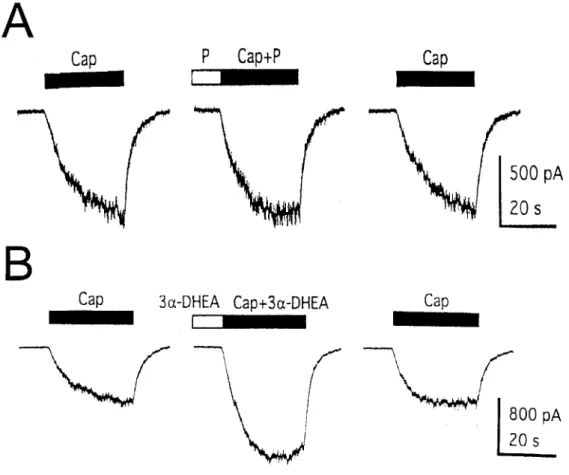
We also examined the effects of other chemically related steroids, including progesterone and some reduced metabolites of DHEA (Table 1). Not all steroids inhibited the capsaicin response. Progesterone (100 µM) did not exert any significant effect on the capsaicin response (Fig. 5A and Table 1), suggesting that inhibition of the capsaicin-induced current by DHEA is a specific effect.
5α-Androstan-3β-ol-17-one and 5β-androstan-3β-ol-17-one, reduced derivatives of DHEA, had activity similar to that of DHEA, whereas DHEA sulfate (DHEAS) and pregnenolone sulfate (PS) produced lesser effect. Thus, a double bond at C-5 is not required, and stereochemistry at C-5 is not critical for inhibition, whereas addition of a sulfate group at C-3β results in weaker activity. Interestingly, the stereoisomer of DHEA, 5-androsten-3α-ol-17-one (3α-DHEA), failed to inhibit the capsaicin-induced current, producing instead a potentiating effect on the capsaicin response (Fig. 5B and Table 1). These findings indicate that the interaction of steroids with the capsaicin receptor is stereospecific.
3.6. PS dose-dependently inhibits the capsaicin-induced nociceptive response in rats
Recently, Sakurada et al. reported that the intradermal (i.d.) injection of capsaicin into the dorsal surface of the mouse hindpaw produced nociception of rapid onset (5 min), characterized by licking of the injected hindpaw (Sakurada et al., 1992).
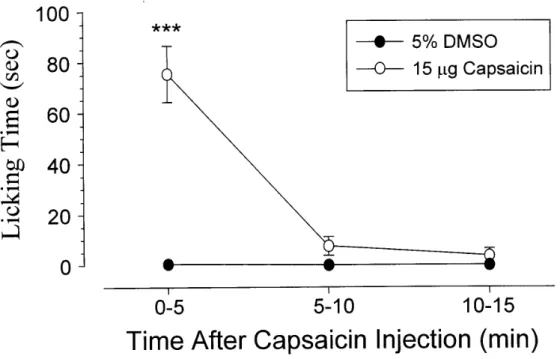
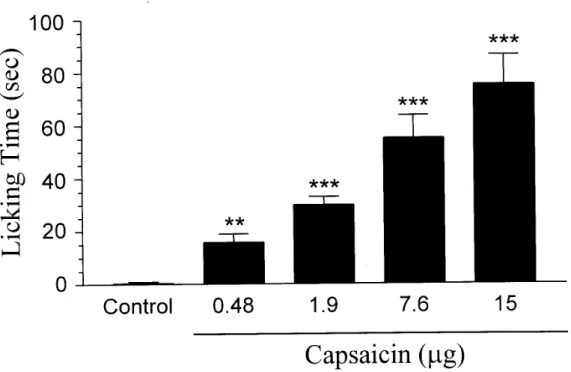
To investigate whether capsaicin-induced nociceptive response in rats has similar temporal characteristics as those in mice, we determined the time course of capsaicin-induced licking in rats. Similar to that elicited in mice, the nociceptive response evoked by capsaicin (15 µg) in rats appeared immediately, peaked at 0-5 min and decayed at 5 min after capsaicin (Fig. 6). Control injection of 5% DMSO had little effect upon the rats. During the first 5 min, capsaicin (0.48-15 µg) elicited a dose-dependent increase in paw licking (Fig. 7). The minimal dose for capsaicin to induce a nociceptive response was 0.48 µg. The duration of paw licking was 75 ± 11.1 sec/5 min for a maximal effective dose of capsaicin (15 µg). Unless otherwise indicated, in all following experiments, 15 µg of capsaicin was therefore used, in combination with various drugs.
To determine whether PS inhibits capsaicin-induced nociception in rats in vivo,
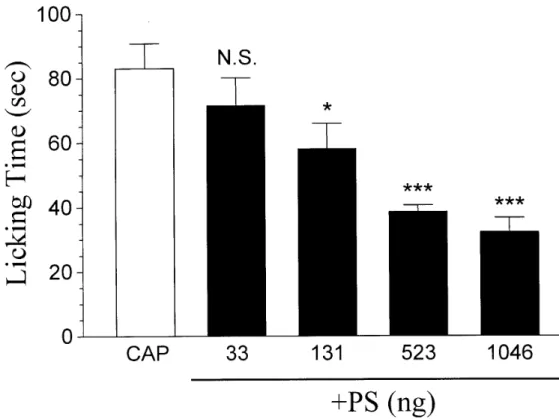
we explored the effect of the i.d. injection of PS on capsaicin-induced licking behavior. If PS produces similar effects on capsaicin receptors located at neuronal somata and peripheral nerve terminals, it is anticipated that PS would depress the capsaicin-induced licking response. Indeed, this is the case. As illustrated in Fig. 8, i.d.
injection of PS (33-1046 ng) dose-dependently inhibited the nociceptive response elicited by 15 µg capsaicin. The threshold dose for an effect of PS on the
capsaicin-induced nociceptive response was 131 ng. The maximal effect on the capsaicin response was achieved at approximately 523 ng.
3.7. PS inhibits the capsaicin-induced nociceptive response through a mechanism distinct from the opioid system
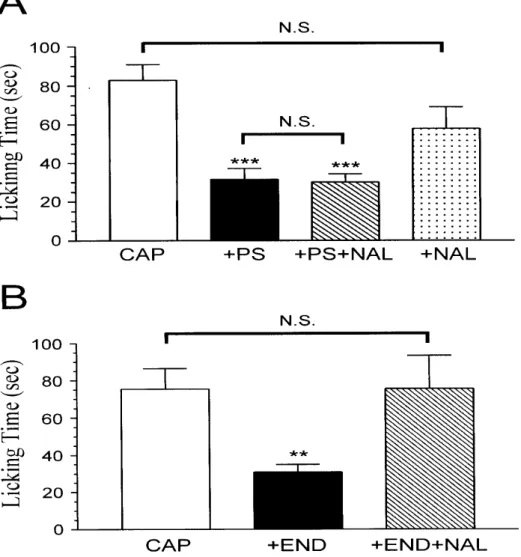
To investigate whether PS acts through the opioid mechanism to inhibit the capsaicin-induced nociceptive response, the non-selective opioid receptor antagonist naloxone was co-injected i.d. with capsaicin and PS. We found that naloxone failed to block the inhibition by PS of capsaicin-induced licking behavior (Fig. 9A). The mean duration of paw licking during the first 5 min was 30 ± 4.3 sec (n = 6), which did not differ significantly from that in capsaicin plus PS-injected rats (32 ± 5.6 sec, n = 7) (P
> 0.05, unpaired t test). This result suggests that PS does not act through opioid receptors located on peripheral endings of pain afferent fibers to inhibit
capsaicin-induced nociception. In addition, block of opioid receptors with naloxone produced no potentiating effect on the capsaicin-induced nociceptive response (Fig.
9A), arguing against the possibility that endogenous opioids acting at opioid receptors participate in attenuating capsaicin-evoked nociception.
Failure of naloxone to block the inhibition by PS of the capsaicin-induced nociceptive response or to potentiate the capsaicin response may be due to an
ineffective dose of naloxone (50 µg) used in these experiments. To test this possibility, we determined whether the anti-nociceptive effect of the endogenous opioid
endomorphin-1 could be blocked by the same dose of naloxone. As shown in Fig. 9B, capsaicin-evoked pain behavior was blocked when endomorphin-1 was injected into the paw together with capsaicin. Moreover, the anti-nociceptive effect of
endomorphin-1 was completely reversed by this dose of naloxone. These results strongly suggest that 50 µg is an effective dose of naloxone for inhibiting opioid receptors and that endomorphin-1 acts through opioid receptors to inhibit the capsaicin-induced nociceptive response.
3.8. PS inhibits the capsaicin-induced nociceptive response via a mechanism different
from the cannabinoid system
To determine whether PS acts through the cannabinoid mechanism to inhibit the capsaicin-induced nociceptive response, the selective CB1 receptor antagonist AM251 was injected into the paw together with capsaicin and PS. As illustrated in Fig.
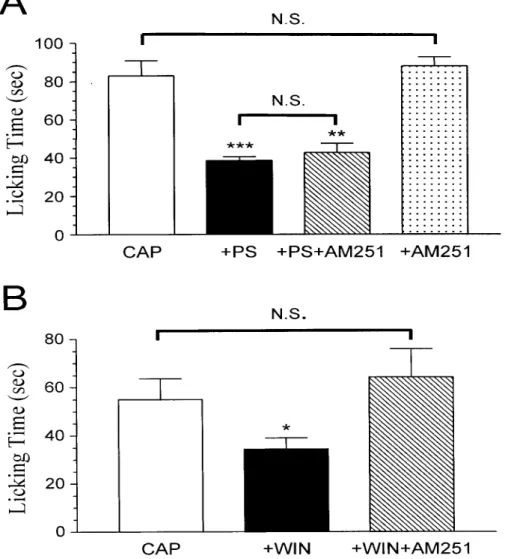
10A, inhibtion by PS of the capsaicin-induced pain response was not blocked by AM251. The mean duration of paw licking during the first 5 min was 43 ± 4.9 sec (n
= 6), which was not significantly different from that in rats without AM251 injection (39 ± 2.1 sec, n = 6) (P > 0.05, unpaired t test). This result indicates that PS does not act through CB1 receptors located on peripheral endings of pain afferent fibers to inhibit capsaicin-induced nociception. Moreover, block of CB1 receptors with AM251 did not significantly enhance the capsaicin-induced nociceptive response (Fig. 10A), excluding the possibility that endogenous cannabinoid agonists acting at CB1
receptors participate in reducing capsaicin-evoked nociception.
Failure of AM251 to block the inhibition by PS of the capsaicin-induced nociceptive response or to potentiate the capsaicin response may be caused by an ineffective dose of AM251 (1 µg) used in these experiments. To examine this possibility, we determined whether the anti-nociceptive effect of the synthetic CB1
receptor agonist WIN55,212-2 could be blocked by the same dose of AM251. As shown in Fig. 10B, co-injection of WIN55,212-2 with capsaicin blocked the
capsaicin-induced nociceptive response. Furthermore, the anti-nociceptive effect of WIN55,212-2 was completely reversed by the same dose of AM251 used in Fig. 10A.
These results argues that 1 µg is an effective dose of AM251 for inhibiting CB1
receptors and that WIN55,212-2 acts through CB1 receptors to inhibit the capsaicin-induced nociceptive response.
Similar to AM251, co-injection of the selective CB2 receptor antagonist AM630 together with capsaicin and PS did not block PS inhibition of the
capsaicin-induced nociceptive response (Fig. 11). The mean duration of paw licking during the first 5 min was 57 ± 8.4 sec (n = 7), which did not differ significantly from that in rats without AM630 injection (39 ± 2.1 sec, n = 6) (P > 0.05, unpaired t test).
This result strongly suggests that PS does not act through CB2 receptors located on peripheral endings of pain afferent fibers to inhibit capsaicin-induced nociception.
Furthermore, the capsaicin-induced nociceptive response was not significantly
enhanced by the blockade of CB2 receptors with AM630 (Fig. 11), arguing against the idea that endogenous cannabinoid receptor agonists acting at CB2 receptors participate in attenuating capsaicin-evoked nociception.
DISCUSSION
The present study can be separated into two parts. In the first part of our study, we demonstrates that the neurosteroid DHEA acts competitively and extracellularly to inhibit the capsaicin-induced current in a dose-dependent manner. In addition to the capsaicin receptor, the GABAA receptor is also negatively modulated by DHEA (Demirgoren et al., 1991; Baulieu and Robel, 1996; Kroboth et al., 1999; Park-Chung et al., 1999). Inhibition by DHEA of the GABA-induced current is dose-dependent, with an EC50 of 35 µM and maximal inhibition of 68% (Demirgoren et al., 1991), indicating that DHEA is less potent and efficacious at the GABAA receptor than at the capsaicin receptor (EC50 = 6.7 µM; maximal inhibition = 100%; Fig. 1B). Like DHEA, its sulfated derivative, DHEAS, also antagonizes the GABA response, but with greater potency and efficacy (Demirgoren et al., 1991). This contrasts with our observation that DHEAS has lesser activity at the capsaicin receptor than DHEA (Table 1). These results, together with the findings that progesterone potentiates the GABA response (Wu, et al., 1990) but has little effect on the capsaicin response (Fig. 5A and Table 1), suggest that structure-activity requirements for steroid interaction with the capsaicin receptor and the GABAA receptor are different.
The high lipophilicity of the steroids raises the possibility that the effect of DHEA is due to the nonspecific mechanism of action, such as perturbation of the membrane lipids surrounding the capsaicin receptor protein. However, we have shown that progesterone does not produce any significant effect on the capsaicin response (Fig. 5A and Table 1). This finding indicates that inhibition of the
capsaicin-induced current by DHEA is a specific effect. Moreover, in contrast to the inhibitory effect of DHEA on the capsaicin response, the stereoisomer of DHEA, 3α-DHEA, potentiates the capsaicin-induced current (Fig. 5B and Table 1), arguing that the interaction of steroids with the capsaicin receptor is stereospecific. This high degree of structural and stereochemical specificity for steroid effects suggests the existence of a specific site of interaction closely associated with the capsaicin receptor.
There are several potential sites at which DHEA could exert its blocking action including: (1) competitive inhibition at the capsaicin binding site, (2)
competitive inhibition through a site different from the capsaicin binding site, or (3) non-competitive inhibition at a distinct site. First, DHEA induces a parallel, rightward shift of the capsaicin dose-response curve (Fig. 2), demonstrating that the depressive action of DHEA is competitive. Second, intracellular DHEA affects neither the
capsaicin response nor inhibition by extracellularly applied DHEA of the capsaicin response (Fig. 4), indicating that the DHEA modulatory site is most likely to be on the external surface of the membrane. This contrasts with the binding site for capsaicin being present in the intracellular surface of the receptor (Jung et al., 1999) and suggests that DHEA competitively inhibits the capsaicin response via a site distinct from the capsaicin binding site. In addition to DHEA, ruthenium red and capsazepine also negatively modulate the capsaicin receptor (Dray et al., 1990; Amann and Maggi, 1991; Bevan et al., 1992). Inhibition of the capsaicin response by ruthenium red is non-competitive (Bevan et al., 1992), indicating that DHEA and ruthenium red do not act through a common site. Like DHEA, antagonism of the capsaicin response by capsazepine is competitive (Bevan et al., 1992). Whether DHEA acts through the capsazepine modulatory site to inhibit the capsaicin response needs further investigation.
Considerable evidence suggests that pro-inflammatory prostaglandins, such as PGE2, sensitize capsaicin responses in rat sensory neurons through an activation of cAMP-dependent protein kinase (PKA) to phosphorylate the capsaicin receptor (Pitchford and Levine, 1991; Lopshire and Nicol, 1998; Bhave et al., 2002). In contrast, dephosphorylation of the capsaicin receptor either by PP2B or by ATP-free pipet solutions reduces the capsaicin response in rat dorsal root ganglion neurons (Docherty et al., 1996; Koplas et al., 1997; Piper et al., 1999). Therefore, it is possible that DHEA inhibition of the capsaicin-induced current might be mediated by PP activation to dephosphorylate the capsaicin receptor. However, inclusion of PP1/2A (Fig. 3) or PP2B inhibitors in the pipet buffer do not significantly affect inhibition of the capsaicin response by DHEA, demonstrating that PP1/2A and PP2B are not involved in the inhibitory action of DHEA.
Free DHEA and DHEAS are metabolically interconvertible by sulfotransferase for conjugation and sulfatase for hydrolysis in many tissues (Baulieu, 1996; Kroboth et al., 1999). DHEA and DHEAS, which are collectively designated DHEA(S), are secreted in high amounts in humans and a few other primates. The highest reported plasma concentration of DHEA(S) in humans is about 10 µM (Parker and Odell, 1980;
Guillemette et al., 1996; Hornsby, 1997), which is close to our EC50 value (6.7 µM ) for DHEA effect on the capsaicin response (Fig. 1B). In sharp contrast, plasma concentrations of DHEA(S) in rats are < 0.1 µM (Robel and Baulieu, 1995;
Guillemette et al., 1996), below the concentration range at which neuromodulation of the capsaicin receptor is observed. However, because neurosteroids such as DHEA(S) can be synthesized locally in the nervous system of rats from cholesterol (Robel and
Baulieu, 1995; Kroboth et al., 1999), high local DHEA(S) concentrations could occur.
Moreover, there is evidence that neurosteroid levels can vary in response to
environmental changes. For example, increases in brain DHEA(S) levels have been observed after exposure of male rats to females (Robel and Baulieu, 1995) and after stress (Corpechot et al., 1981). Therefore, it is likely that DHEA under physiological conditions may modulate the capsaicin receptor in the mammalian nervous system.
This work establishes DHEA as an endogenous negative modulator for the capsaicin receptor. Given the prominent role for the capsaicin receptor in nociception (Szolcsanyi, 1990) and inflammation-induced heat hyperalgesia (Caterina et al., 2000;
Davis et al., 2000), DHEA may reduce the capsaicin receptor-mediated pain sensation and contribute to prevent the formation of inflammatory thermal hyperalgesia.
In the second part of our study, we investigated the effect of PS on
capsaicin-induced nociception in rats in vivo. We show that intraplantar injection of PS dose-dependently inhibits capsaicin-induced nociceptive response (Fig. 8). To our best knowledge, this is the first time to demonstrate that capsaicin-induced
nociception can be reduced by the neurosteroid, suggesting that PS may potentially act as a pain-relieving agent.
Activation of peripheral opioid receptors has been shown to alleviate
capsaicin-induced thermal nociception in primates (Ko et al., 1998; Ko et al., 1999).
In animal models of inflammatory hyperalgesia, peripheral opioid receptors attenuate pain behavior and there is evidence for tonic activation of opioid receptors by
endogenous opioids, which may functionally synergise to regulate pain initiation (Stein et al., 1989; Stein, 1995). It is possible that PS inhibition of the
capsaicin-induced nociception might be mediated by opioid-dependent mechanism.
However, non-selective opioid receptor antagonist naloxone fails to block the inhibition by PS of capsaicin-induced pain behavior, demonstrating that PS does not act through opioid receptors located on peripheral endings of pain afferent fibers to inhibit capsaicin-induced nociception. In addition, block of opioid receptors with naloxone produced no potentiating effect on the capsaicin-induced nociception (Fig.
9A), excluding the possibility that endogenous opioids acting at opioid receptors participate in attenuating capsaicin-induced pain behavior.
Cannabinoids have been shown to modulate nociceptive processing in models of acute, inflammatory and neuropathic pain (Martin and Lichtman, 1998; Pertwee, 2000). In animal models of inflammation-induced hyperalgesia, both CB1-like and
CB2-like receptors inhibit pain behavior and there is also evidence for
inflammation-induced release of endocannabinoids , which may function to reduce inflammatory hyperalgesia (Calignano et al., 1998; Jaggar et al., 1998). In a model of neuropathic hyperalgesia, a CB1 receptor agonist reduces, and an antagonist enhances hyperalgesia (Herzberg et al., 1997). This raises a possibility that inhibition by PS of the capsaicin-induced nociception might be mediated by cannabinoid-dependent mechanism. However, Neither the selective CB1 receptor antagonist AM251 nor the selective CB2 receptor antagonist AM630 blocks inhibition by PS of
capsaicin-induced pain behavior (Fig. 10A and Fig. 11), indicating that PS does not act through CB1 or CB2 receptors to inhibit capsaicin-induced nociception.
Furthermore, the capsaicin-induced nociceptive response was not significantly enhanced by the blockade of CB1 or CB2 receptors (Fig. 10A and Fig. 11), arguing against the possibility that endcannabinoids acting at CB1 or CB2 receptors participate in attenuating capsaicin-induced nociception.
In summary, PS inhibits capsaicin-induced nociception via an opioid- and cannabinoid-independent mechanism. These findings, together with the observation that PS rapidly and reversibly reduces the capsaicin-induced current in acutely dissociated rat dorsal root ganglion neurons (Table 1), strongly suggest that PS
depresses the capsaicin-induced nociception via direct action on capsaicin receptors or associated proteins. Whether other chemically related steroids also have
anti-nociceptive effect remains to be determined.
REFERENCES
Amann, R., and Maggi, C.A. (1991). Ruthenium red as a capsaicin antagonist. Life Sci. 49, 849-856.
Baulieu, E.-E. (1996). Dehydroepiandrosterone (DHEA): a fountain of youth? J. Clin.
Endocrinol. Metab. 81, 3147-3151.
Baulieu, E.-E., and Robel, P. (1996). Dehydroepiandrosterone and
dehydroepiandrosterone sulfate as neuroactive neurosteroids. J. Endocrinol. 150, S221-S239.
Bevan, S., Hothi, S., Hughes, G., James, I.F., Rang, H.P., Shah, K., Walpole, C.S.J., and Yeats, J.C. (1992). Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br. J. Pharmacol. 107, 544-552.
Bhave, G., Zhu, W., Wang, H., Brasier, D.J., Oxfford, G.S., and Gereau IV, R.W.
(2002). cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721-731.
Calignano, A., La Rana, G., Giuffrida, A., and Piomelli, D. (1998). Control of pain initiation by endogenous cannabinoids. Nature 394, 277-281.
Caterina, M.J., Leffler, A., Malmberg, A.B., Martin, W.J., Trafton, J., Petersen-Zeitz, K.R., Koltzenburg, M., Basbaum, A.I., and Julius, D. (2000). Impaired
nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306-313.
Chan, C.Y., and Farb, D.H. (1985). Modulation of neurotransmitter action: control of the γ-aminobutyric acid response through the benzodiazepine receptor. J. Neurosci.
9, 2365-2373.
Chang, T.-J., and Wu, F.-S. (2002) Effects of steroids on the capsaicin
receptor-mediated current in acutely dissociated rat dorsal root ganglion neurons.
The Seventeenth Joint Annual Conference of Biomedical Sciences, Taipei, Taiwan. P266. (Abstract)
Choi, D.H., and Fischbach, D. (1981). GABA conductance of chick spinal cord and dorsal root ganglion neurons in cell culture. J. Neurophysiol. 45, 605-620.
Corpechot, C., Robel, P., Axelson, M., Sjovall, J., and Baulieu, E.-E. (1981).
Characterization and measurement of dehydroepiandrosterone sulfate in rat brain.
Proc. Natl. Acad. Sci. USA 78, 4704-4707.
Davis, J.B., Gray, J., Gunthorpe, M.J., Hatcher, J.P., Davey, P.T., Overend, P., Harries, M.H., Latcham, J., Clapham, C., Atkinson, K., Hughes, S.A., Rance, K., Grau, E., Harper, A.J., Pugh, P.L., Rogers, D.C., Bingham, S., Randall, A., and Sheardown, S.A. (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183-187.
Demirgoren, S., Majewska, M.D., Spivak, C.E., and London, E.D. (1991). Receptor binding and electrophysiological effects of dehydroepiandrosterone sulfate, an antagonist of the GABAA receptor. Neuroscience 45, 27-135.
Docherty, R.J., Yeats, J.C., Bevan, S., and Boddeke, H.W. (1996). Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflügers Arch. 431, 828-837.
Dray, A., Forbes, C.A., and Burgess, G.M. (1990). Ruthenium red blocks the capsaicin-induced increase in intracellular calcium and activation of membrane currents in sensory neurones as well as the activation of peripheral nociceptors in vitro. Neurosci. Lett. 110, 52-59.
Green, B.G., and Shaffer, G.S. (1993). The sensory response to capsaicin during repeated topical exposures: differential effects of itching and pungency. Pain 53, 323-334.
Guillemette, C., Hum, D.W., and Belanger, A. (1996). Levels of plasma C19 steroids and 5α-reduced C19 steroid glucuronides in primates, rodents, and domestic animals. Am. J. Physiol. 271, E348-E353.
Hamill, O.P., Marty, A., Neher, E., Sakmann, B., and Sigworth, F.J. (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches, Pflügers Arch. 391, 85-100.
Herzberg, U., Eliav, E., Bennett, G.J., Kopin, I.J. (1997). The analgesic effect of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci. Lett. 221, 157-160.
Heyman, I., and Rand, H.P. (1985). Depolarizing responses to capsaicin in a subpopulation of rat dorsal root ganglion cells. Neurosci. Lett. 56, 69-75.
Holzer, P. (1991). Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol. Rev. 43, 144-201.
Hornsby, P.J. (1997). DHEA: a biologist’s perspective. J. Am.n Geriatr. Soc. 45, 1395-1401.
Jagga, S.I., Hasnie, F.S., Sellaturay, S., Rice, A.S.C. (1998). The antihyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 76, 189-199.
Jung, J., Hwang, S.W., Kwak, J., Lee, S.-T., Kang, C.-J., Kim, W.B., Kim, D., and Oh, U. (1999). Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J. Neurosci. 19, 529-538.
Ko, M.C., Butelman, E.R., and Woods, J.H. (1998). The role of peripheral mu opioid receptors in modulation of capsaicin-induced thermal nociception in rhesus monkeys. J. Pharmacol. Exp. Ther. 286, 150-156.
Ko, M.C., Butelman, E.R., and Woods, J.H. (1999). Activation of peripheral kappa opioid receptors inhibits capsaicin-induced thermal nociception in rhesus monkeys.
J. Pkarmacol. Exp. Ther. 289, 112-118.
Koplas, P.A., Rosenberg, R.L., and Oxford, G.S. (1997). The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J.
Neurosci. 17, 3525-3537.
Kroboth, P.D., Salek, F.S., Pittenger, A.L., Fabian, T.J., and Frye, R.F. (1999). DHEA and DHEA-S: a review. J. Clin. Pharmacol. 39, 327-348.
Lean, A.P., Munson, P.J., and Rodbard, D. (1978). Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J.f Physiol. 235, E97-E102.
Lopshire, J.C., and Nicol, G.D. (1998). The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J. Neurosci. 18, 6081-6092.
Majewska, M.D. (1987). Steroids and brain activity: essential dialogue between body and mind. Biochem. Pharmacol. 36, 3781-3788.
Majewska, M.D., Harrison, N.L., Schwartz, R.D., Barker, J.L., and Paul, S.M. (1986).
Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004-1007.
Majewska, M.D., Mienville, J.-M., and Vicini, S. (1988). Neurosteroid pregnenolone sulfate antagonizes electrophysiological response to GABA in neurons. Neurosci.
Lett. 90, 279-284.
Martin, B.R., and Lichtman, A.H. (1998) Cannabinoid transmission and pain perception. Neurobiol. Disease 5, 447-461.
McEwen, B.S. (1991). Non-genomic and genomic effects of steroids on neural activity. Trends Pharmacol. Sci. 12, 141-147.
Oh, U., Hwang, S.W., and Kim, D. (1996). Capsaicin activates a nonselective cation channel in cultured neonatal rat dorsal root ganglion neurons. J. Neurosci. 16, 1659-1667.
Park-Chung, M., Wu, F.-S., Purdy, R.H., Malayev, A.A., Gibbs, T.T., and Farb, D.H.
(1997). Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol. Pharmacol. 52, 1113-1123.
Park-Chung, M., Malayev, A., Purdy, R.H., Gibbs, T.T., and Farb, D.H. (1999).
Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 830, 72-87.
Parker, L.N., and Odell, W.D. (1980). Control of adrenal androgen secretion. Endocr.
Rev. 1, 392-410.
Pertwee, R.G. (2000) Cannabinoid receptors and pain. Prog. Neurobiol. 63, 569-611.
Piper, A.S., Yeats, J.C., Bevan, S., and Docherty, R.J. (1999). A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. J. Physiol. (London) 518, 721-733.
Pitchford, S., and Levine, J.D. (1991). Prostaglandins sensitize nociceptors in cell culture. Neurosci. Lett. 132, 105-108.
Robel, P., and Baulieu, E.-E. (1995). Dehydroepiandrosterone (DHEA) is a neuroactive neurosteroid. Ann. NY Acad. Sci. 774, 82-110.
Sakurada, T., Katsumata, K., Tan-No, K., Sakurada, S., and Kisara, K. (1992). The capsaicin test in mice for evaluating tachykinin antagonists in the spinal cord.
Neuropharmacology 31, 1279-1285.
Stein, C. (1995) The control of pain in peripheral tissue by opioids. N. Engl. J. Med.
332, 1685-1690.
Stein, C., Millan, M.J., Shippenberg, T.S., Peter, K., and Herz, A. (1989). Peripheral opioid receptors mediating antinociception in inflammation. Evidence for
involvement of mu, delta and kappa receptors. J. Pharmacol. Exp. Ther. 248, 1269-1275.
Szallasi, A., and Blumberg, P.M. (1999). Vanilloid (capsaicin) receptors and mechanisms. Pharmacol. Rev. 51, 159-211.
Szolcsanyi, J. (1990). Capsaicin, irritation and desensitization. In: Green, B.G., Mason, J.R., Kare, M.R., (Eds), Chemical Senses. Dekker, New York, pp. 141-168.
Wiley, J.W., Gross, R.A., and MacDonald, R.L. (1993). Agonists for neuropeptide Y receptor subtypes NPY-1 and NPY-2 have opposite actions on rat nodose neuron calcium currents. J. Neurophysiol. 70, 324-330.
Wu, F.-S., Gibbs, T.T., and Farb, D.H. (1990). Inverse modulation of γ-aminobutyric acid- and glycine-induced currents by progesterone. Mol. Pharmacol. 37, 597-602.
Wu, F.-S., Gibbs, T.T., and Farb, D.H. (1991). Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol. Pharmacol.y 40, 333-336.
Wu, F.-S., and Chen, S.-C. (1997) Mechanism underlying the effect of pregnenolone sulfate on the kainite-induced current in cultured chick spinal cord neurons.
Neurosci. Lett. 222, 79-82.
Wu, F.-S., Chen, S.-C., and Tsai, J.-J. (1997) Competitive inhibition of the glycine-induced current by pregnenolone sulfate in cultured chick spinal cord neurons. Brain Res. 750, 318-320.
Table 1
Effects of steroids on the 100 nM capsaicin-induced current
Steroid Structure Change of response (%)
DHEA (50 µM) (100 µM)
-73 ± 6.3 (6) -94 ± 1.3 (6)
5α-Androstan-3β-ol-17-one (100 µM)
-92 ± 1.2 (4)
5β-Androstan-3β-ol-17-one (100 µM)
-94 ± 1.5 (5)
DHEAS (100 µM) -45 ± 6.4 (7)
PS (100 µM) -57 ± 5.3 (14)
3α-DHEA (50 µM) +78 ± 5.7 (5)
Progesterone (100 µM) +2 ± 7.9 (6)
Holding potential is –50 mV. Values are means ± standard errors. Number of cells is indicated in parentheses.
Fig. 1. DHEA inhibits the capsaicin response. (A) DHEA (100 µM) inhibits rapidly and reversibly the current induced by 100 nM capsaicin (Cap). Horizontal bar above each trace, period of drug application. (B) Dose-response curve for inhibition of the capsaicin (100 nM) response by DHEA. Data points, percentage change in peak current in the presence of DHEA (mean of four to seven experiments). Error bars, standard errors. Error bars are not indicated when smaller than the size of the circle.
DHEA dose-response curve is fitted with the logical equation (Lean et al., 1978).
(% inhibition)/(% inhibition)max = [DHEA]nH/([DHEA]nH + EC50nH)
where [DHEA] is the concentration of DHEA, and nH is the Hill coefficient. Curve-fit analysis reveals an EC50 of 6.7 µM, a maximal inhibition of 100%, and nH of 0.83.
Fig. 2. Antagonism of the capsaicin response by DHEA is competitive. Dose-response curves for capsaicin in the presence and absence of 50 µM DHEA. All responses are normalized to the peak current (*) induced by 0.5 µM capsaicin. Data points, normalized peak current (mean of four to eight experiments). Error bars, standard errors. Error bars are not indicated when smaller than the size of the circle. Each set of data points is fitted with the logistic equation (Lean et al., 1978).
I/Imax = [capsaicin]nH/([capsaicin]nH + EC50nH)
Where Imax is the maximal normalized current, [capsaicin] is the concentration of capsaicin, and nH is the Hill coefficient. In the absence of DHEA, EC50 = 0.70 µM, Imax = 2.81, and nH = 1.78. In the presence of DHEA, EC50 = 2.11 µM, Imax = 2.80, and nH = 1.30.
Fig. 3. DHEA inhibition of the capsaicin response is not mediated by activation of PP1/2A. (A) Inclusion of the selective PP1/2A inhibitor okadaic acid (1 µM) in the electrode buffer does not significantly affect inhibition by extracellularly applied DHEA (100 µM) of the capsaicin (100 nM)-induced current. (B) Average data for the effect of extracellularly applied DHEA (100 µM) on the capsaicin-induced current in the absence (Control) and presence (OA inside) of intracellular okadaic acid (1 µM).
Number of cells is indicated in parentheses.
Fig. 4. DHEA acts extracellularly to inhibit the capsaicin response. (A) Dialyzing the inside of the cell with a saturating concentration (200 µM) of DHEA influences neither the capsaicin (100 nM)-induced current nor inhibition by extracellularly applied DHEA (100 µM) of the capsaicin-induced current. Time after cell rupture is shown above each tracing. (B) Average data for the effect of extracellularly applied DHEA (100 µM) on the capsaicin-induced current in the absence (Control) and presence (DHEA inside) of intracellular DHEA (200 µM). Number of cells is indicated in parentheses.
Fig. 5. Effects of progesterone and 3α-DHEA on the capsaicin response. (A) Application of 100 µM progesterone (P) has no significant effect on the 100 nM capsaicin-induced current. (B) 3α-DHEA (50 µM) enhances rapidly and reversibly the current induced by 100 nM capsaicin.
Fig. 6. Time course of paw licking of the injected paw after the intradermal injection of capsaicin into the dorsal surface of the rat left hindpaw. Control rats receive an intradermal injection of vehicle (5% DMSO). Each value represents the mean ± SEM of 6 rats during 5 min interval as indicated. *** P < 0.001 when compared to vehicle-injected controls.
Fig. 7. Capsaicin dose-dependently induces the nociceptive response . Each bar represents the amount of time (sec) the animals spent licking the injected paw during a 5 min observation period (mean ± SEM of 6-7 rats in each group). *** P < 0.001 and ** P < 0.01 when compared to vehicle (5% DMSO)-injected controls.
Fig. 8. PS inhibits the capsaicin-induced nociceptive response in a dose-dependent manner. Each bar represents the amount of time (sec) the rats spent licking the injected paw during a 5 min observation period (mean ± SEM of 6-7 rats in each group). N.S. = not significant. *** P < 0.001 and * P < 0.05 when compared to 15 µg capsaicin-injected rats (CAP).
Fig. 9. PS does not act via the opioid mechanism to inhibit capsaicin-induced nociceptive response. (A) Naloxone does not attenuate the inhibition of the capsaicin-induced nociceptive response by PS. The labels +PS, +PS+NAL, and +NAL represent the effects of co-administration of capsaicin (15 µg) with PS (523 ng), with PS (523 ng) plus the non-selective opioid receptor antagonist naloxone (50 µg), and with naloxone (50 µg), respectively. (B) Endomorphin-1 acts through the opioid receptor to inhibit the capsaicin-induced nociceptive response. The labels +END and +END+NAL represent the effects of co-injection of capsaicin (15 µg) with endomorphin-1 (1 µg) and with endomorphin-1 (1 µg) plus naloxone (50 µg), respectively. In both (A) and (B), each bar represents the amount of time (sec) the animals spent licking the injected paw during a 5 min observation period (mean ± SEM of 6-7 rats in each group). N.S. = not significant. *** P < 0.001 and ** P < 0.01 when compared to capsaicin-injected rats.
Fig. 10. PS does not act via the CB1 mechanism to inhibit capsaicin-induced nociceptive response. (A) AM251 has no effect on the inhibition of the capsaicin-induced nociceptive response by PS. The labels +PS, +PS+AM251, and +AM251 represent the effects of co-administration of capsaicin (15 µg) with PS (523 ng), with PS (523 ng) plus the selective CB1 receptor antagonist AM251 (1 µg), and with AM251 (1 µg), respectively. (B) WIN55,212-2 acts through the CB1 receptor to inhibit the capsaicin-induced nociceptive response. The labels +WIN and +WIN+AM251 represent the effects of co-injection of capsaicin (7.6 µg) with WIN55,212-2 (3 µg) and with WIN55,212-2 (3 µg) plus AM251 (1 µg), respectively.
In both (A) and (B), each bar represents the amount of time (sec) the animals spent licking the injected paw during a 5 min observation period (mean ± SEM of 6-7 rats in each group). N.S. = not significant. *** P < 0.001, ** P < 0.01 and * P < 0.05 when compared to capsaicin-injected rats.
Fig. 11. PS does not act via the CB2 mechanism to inhibit capsaicin-induced nociceptive response. The labels +PS, +PS+AM630, and +AM630 represent the effects of co-administration of capsaicin (15 µg) with PS (523 ng), with PS (523 ng) plus the selective CB2 receptor antagonist AM630 (5 µg), and with AM630 (5 µg), respectively. AM630 does not significantly decrease the inhibition of the capsaicin-induced nociceptive response by PS. Each bar represents the amount of time (sec) the rats spent licking the injected paw during a 5 min observation period (mean
± SEM of 6-8 rats in each group). N.S. = not significant. *** P < 0.001 and * P < 0.05 when compared to capsaicin-injected rats.
SELF-EVALUATION
This study demonstrates that the neurosteroid DHEA acts extracellularly to inhibit the capsaicin receptor-mediated current in a dose-dependent and competitive manner. In addition, structure-activity studies indicate that interaction of steroids with the capsaicin receptor is stereospecific. Moreover, PS injected intraplantarly
dose-dependently inhibits capsaicin-induced nociceptive response in rats in vivo through an opioid- and cannabinoid-independent mechanism. Capsaicin receptor activation has been proposed to play an important role in nociception and
inflammatory thermal hyperalgesia. This work should shed some light on the mechanism of actions of steroids on the capsaicin receptor and this knowledge may lead to new strategies for reducing the pain and inflammation-induced hyperalgesia.
Our work produces two manuscripts. One manuscript concerning the
mechanism of DHEA action on the capsaicin-induced current in acutely dissociated rat dorsal root ganglion neurons has been submitted for publication. The other
manuscript regarding inhibition by PS of capsaicin-induced nociception in rats in vivo is in the preparation and will be submitted for publication in near future. In addition, we will perform further experiments to address the following questions: a) Is
inhibition by PS of the capsaicin-induced current competitive? b) Is the effect of PS on the capsaicin-induced current voltage- and agonist-dependent? c) Does PS act intracellularly to inhibit the capsaicin-induced current? d) What is the effect of PS on kinetic properties of single-channel activity induced by capsaicin? This will produce at least one more manuscript.