行政院國家科學委員會專題研究計畫 成果報告
建立醣水解酵素之生物資訊分析平台-以 LPHase,MTSase,
及 MTHase 為應用模型--(子計畫三)結合生物資訊與結構模
擬研究第 64 家族 beta-glucanase 之活性區與重要胺基酸
殘基(3/3)
研究成果報告(完整版)
計 畫 類 別 : 整合型 計 畫 編 號 : NSC 99-2627-B-009-007- 執 行 期 間 : 99 年 08 月 01 日至 100 年 07 月 31 日 執 行 單 位 : 國立交通大學應用化學系(所) 計 畫 主 持 人 : 李耀坤 計畫參與人員: 此計畫無其他參與人員 處 理 方 式 : 本計畫可公開查詢中 華 民 國 100 年 10 月 18 日
行政院國家科學委員會補助專題研究計畫
█成果報告
□期中進度報告
結合生物資訊與結構模擬研究第
64 家族 beta-glucanase 之活
性區與重要胺基酸殘基
計畫類別:□個別型計畫 █整合型計畫
計畫編號:
NSC 99-2627-B-009-007
執行期間: 97 年 8 月 1 日至 100 年 7 月 31 日
執行機構及系所:國立交通大學 應化系
計畫主持人:李耀坤
共同主持人:
計畫參與人員:柳勝文,吳岳進等八員
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整報告
本計畫除繳交成果報告外,另須繳交以下出國心得報告:
□赴國外出差或研習心得報告
□赴大陸地區出差或研習心得報告
□出席國際學術會議心得報告
□國際合作研究計畫國外研究報告
處理方式:
除列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
中 華 民 國 100 年 10 月 15 日
附件一本 研 究 旨 在 探 討 自 Streptomyces matensis DIC-108 菌 株 之 β-1,3- 葡 聚 五 糖 生 產 水 解 糖 苷 酵 素 (Laminaripentaose-producing β-1,3-glucanase, LPHase)屬於醣苷水解酵素第 64 家族的水解酵素(EC
3.2.1.39),藉以單取代(single-displacement)反轉機制催化水解 β-1,3 糖苷鍵結,並專一性地生產由五個
葡萄糖所構成的laminaripentaose 寡糖產物。將著重於該酵素的性質、催化功能、反應機構與催化重要
殘基的鑑定,並進一步對此酵素的蛋白質晶體結構作解析與探討。有關蛋白質晶體結構之研究內容已
發表,為節省篇幅該內容便不再贅述.僅對其重要胺基酸之鑑定描述其研究內容。
當LPHase 酵素在各不同酸鹼值緩衝溶液中,其反應活性之 pH-profile 呈現一對稱鐘型曲線(bell-shaped
curve),由曲線得知調控 LPHase 酵素所扮演的兩個重要催化殘基之解離值 pKa1和 pKa2分別為 6.0 及
10.1。隨後,將糖苷水解酵素家族 GH-64 中,所有子家族來源菌的胺基酸序列進行多重序列比對分析,
發現了五個完全高保留度位置的胺基酸,分別為Asp143、Glu154、Asp170、Asp376及Asp377。結合酵素之
定點突變學,觀測各突變株酵素與受質卡德蘭膠水解的相對催化活性,並量測對合成之糖苷受質
p-NLPG 的反應初始速率變化;發現有兩突變株 E154G 和 D170G 酵素的反應速率 kcat/Km值,降低約為
野生株的1,700~2,000 倍。接著以利用加入親核性陰離子試劑對兩突變株 E154G 和 D170G 酵素進行化
學活性復活技術,結果得知突變株D170G 酵素的催化反應活性,可被加入的疊氮陰離子所復活並於液
相質譜與氫核磁共振磁譜分析 α-laminaripentaosyl_azide 產物的產生。鑑定出 LPHase 酵素在催化反應
過程中,扮演一般鹼催化基團(親核性基)為 Asp170和一般酸催化基團為 Glu154。並配合LPHase 酵素突
變學與蛋白晶體結構,得知胺基酸Arg115,為影響此酵素在高pH 值溶液的催化作用;和胺基酸 Tyr232,
輔助對扮演一般鹼催化基團附近水分子之氫鍵鍵結作用力有相當重要性。
關鍵字: β-1,3-葡聚五糖生產水解糖苷酵素、反應機制、重要催化基團、定點突變、化學活性復活
Abstract
Laminaripentaose-producing β-1,3-glucanase (LPHase) from Streptomyces matensis DIC-108 uniquely catalyzes the hydrolysis of β-1,3-glucan to release laminaripentaose as the predominant product. For study this novel enzyme, the gene of LPHase was reconstructed with PCR and overexpressed in E. coli. The recombinant wild-type enzyme and various mutants were further purified to >90 % homogeneity on an ion-exchange chromatograph. The catalysis of the recombinant LPHase is confirmed to follow a one-step single-displacement mechanism with 1H-NMR spectrometry. To determine the amino-acid residues essential for the catalysis, more than ten residues, including five highly conserved residues -- Asp143, Glu154, Asp170, Asp376 and Asp377, were mutated. Among the mutants, E154Q, E154G, D174N and D174G significantly lost
catalytic activity. Further investigation with chemical rescue using sodium azide on E154G and D174G confirmed that Glu154 functions as the general acid whereas Asp170 serves as the general base in a catalytic turnover. This work is the first report that provides a direct information for the identification of the essential residues of GH-64 through kinetic examination.
Key words: GH-64, Laminaripentaose-producing β-1,3-glucanase, catalytic mechanism, essential residue,
Introduction
β-Glucans are polymers composed of glucose with polymerization to various degrees, branching and diverse glycosidic linkages, such as β-1,3-, β-1,4-, β-1,6-, β-1,3/1,4- and β-1,3/1,6-linkages (Martin et. al., 2007). They can be obtained from diverse sources, including fungi, yeasts, bacteria and plants. They serve as components of the cell wall of these organisms but might also be found in extracellular secretions of microbial origin. β-1,3-Glucanases, generally termed laminarinases, including endo-β-1,3-glucanase (EC 3.2.1.39) and exo-β-1,3-glucanase (EC 3.2.1.58) are found simultaneously in several distinct families of glycoside hydrolases such as GH-5, GH-16, GH-17, GH-55, GH-64 and GH-81. These enzymes cleave the β-1,3-bond found in various β-1,3-glucans and pachyman (Blättel et. al., 2011; McGrath and Wilson, 2006) to release various oligosaccharides as products. The laminaripentaose-producing β-1,3-glucanase (LPHase), acting on the β-1,3-glycosidic bond of curdlan to release laminaripentaose from β-1,3-glucan as the predominant product, was first purified from Streptomyces matensis DIC-108 and the corresponding gene was further cloned (accession no. BAA34349 in GenBank) (Nakabayashi et. al.,1998). The enzyme, composed of 401 amino acids with the first 35 amino acids as its signal peptide, was classified as a member of GH-64. Up to now, the information of the catalytic essential residues of GH-64 has not yet been confirmed.
Enzymatic hydrolysis of glycosidic bonds occurs with two possible stereochemical outcomes -- inversion or retention of the anomeric configuration at the site of cleavage (Sinnott, 1990; McCarter and Withers, 1994). Both mechanisms normally require amino acids containing side-chain carboxylic residues as essential groups. For the retaining glycosidases, enzymes catalyze the hydrolysis via a two-step double-displacement mechanism involving a covalent glycosyl-enzyme intermediate with one of the two essential amino-acid residues functioning as a nucleophile and the other as a general acid/base. In contrast, the inverting glycosidases follow a one-step single displacement mechanism with the assistance of a general acid and a general base. The general base polarizes a water molecule to develop a stronger nucleophile to attack the anomeric carbon, whereas the general acid protonates the glycosidic oxygen to accelerate the reaction (Zechel and Withers, 2000; Davies and Henrissat, 1995). Kinetic and stereochemical tests of the enzymatic reaction provide information that enables conclusions about the catalytic mechanism (Koshland, 1953; Nishimura et.
al., 2001). The molecular mechanism is conserved within the sequence-based families of glycoside hydrolases
(Henrissat, 1991; Henrissat and Bairoch, 1993; Henrissat and Bairoch, 1996, Cantarel et. al., 2009). For the six families containing β-1,3-glucanases, the GH-5 is classified as the retaining enzyme (O'Connell et. al., 2011; Baker et. al., 2005). GH-16 (Kawai et. al., 2006) and GH-17 (Chen et. al., 1995) containing various glycoside hydrolases, such as endo-β-1,3-D-glucanase, licheninase, agarase, xyloglucanase, were also found to be the retaining enzyme. Protein three-dimensional structures were available for β-1,3-glucanases from GH-5 (Dias et. al., 2004), GH-16 (Vasur et. al., 2006) and GH-17 (Receveur-Brechot et. al., 2006). Detailed enzymology and catalytic investigations, including binding model experiments of various oligosaccharide, inhibition, kinetic analysis, and essential group(s) identification were reported for the enzymes of families GH-5, GH-16 and GH-17 (O'Connell et. al., 2011; Baker et. al., 2005; Kawai et. al., 2006; Chen et. al., 1995); Varghese et. al., 1994; Armand et. al., 1994; Johansson et. al., 2004; Hrmova and Fincher, 2001). Family GH-55, GH-64 and GH-81 are classified as the inverting GH for β-1,3-glucanases. Among these families, the proteins from GH-55 were extensively studied including crystal structure and essential group identification (Ishida et. al., 2009), whereas studies on GH-64 and GH-81 were mainly focused on the subjects of protein purification (Bara et. al., 2003; Palumbo et. al., 2003), gene cloning (Martin et. al., 2006; Shen et. al., 1991)
and limited studies on catalytic features of GH-64 (Nishimura et. al., 2001; Ferrer, 2006) and GH-81 (McGrath et. al., 2009; Fliegmann et. al., 2005). LPHase, classified as a member of GH-64, was first
confirmed to be an inverting enzyme by Nishimura et. al. (Nishimura et. al., 2001). Recently, we resolved its crystal structure (3G0 in PDB) to 1.62 Å and performed computer simulations to model the enzyme-laminarihexaose structure (Wu et. al., 2009) (Figure 1). The structure provides valuable information to explore the catalytic mechanism and the candidate of the essential residues in the active site and sugar-binding site of LPHase. The present work is continuing the structure study to confirm the function of the conserved residues in the active site by site-directed mutagenesis, chemical rescue, and kinetic analysis.
EXPERIMENTS Materials
Curdlan (a laminarin β-1,3-glucan, Wako Chemical, Osaka, Japan), various polysaccharide substrates (TCI, Tokyo Kasei Kogyo Co., Ltd), oligonucleotides (synthesized by Integrated DNA Technologies, Mission Biotechnology, Taipei, Taiwan), Vent polymerase (New England Biolabs, Ipswich, MA, USA) used in PCR reactions, restriction endonucleases and T4 DNA ligase (Roche Applied Science, Basel, Switzerland), buffers and chemicals for synthesis (Sigma-Aldrich, St. Louis, MO, USA or E. Merck Co., Gibbstown, NJ, USA) and columns and gels for protein purification and protein marker for electrophoresis (GE Healthcare, Piscataway, NJ, USA) were obtained from the indicated sources. Synthetic substrates were characterized with 1H-NMR spectra (Bruker Avance spectrometer 300 MHz) and mass spectra (Q-TOF, Micromass, in the positive ESI mode).
Gene construction, protein expression and purification
PCR was employed to reconstruct the full-length gene of LPHase (accession number BAA34349) using 24 primers (sequence not shown) including 12 of sense and 12 of anti-sense primers. The PCR-amplified LPHase gene was inserted in the yT&A cloning vector. The DNA sequence was fully confirmed with cycle sequencing (ABI 3100 DNA sequencer, Perkin-Elmer Applied Biosystems). For the construction of the expression vector, the LPHase gene was inserted at the NdeI and EcoRI sites of pRSET_A.
The purifications of recombinant LPHase and mutants were identical. Complete procedures involve three steps of ion-exchange chromatographic separation. All purification steps were performed near 25 °C. The detailed procedures are described previously (Wu et. al. 2009). Protein concentration was determined with BCA (bicinchoninic acid) method, as described in the manufacturer’s protocol (Sigma; BCA-1 kit), followed by measurement of chromophore absorption at 280 nm. The molecular weight of the purified enzyme was estimated by SDS/PAGE according to Laemmli (Laemmli, 1970) and by mass spectrometry.
CD Spectra of Recombinant LPHase
Spectra were recorded at 25 °C on a spectropolarimeter (JASCO J-815; cell length 1 mm). The concentration of proteins used for CD measurement was 1 mg/mL. All spectra, with correction for the buffer background, were recorded from 200 to 255 nm. Ten spectra were recorded and averaged.
Site-directed mutagenesis in vitro
Site-directed mutagenesis for mutations was performed according to the QuikChange® method (Stratagene). The basic procedure involved PCR amplification with pRSET_A-lphase plasmid DNA as the template and two synthetic oligonucleotides containing the desired mutation as the primer. The primers used to generate the mutations are listed in Table 1. E. coli JM109 strain cells were transformed with mutation plasmid DNA. The desired mutations were confirmed with DNA sequencing. Plasmids were further transformed into E. coli BL21 (DE3) for protein over-expression. For those mutants with significant activity loss, the entire genes were sequenced to confirm that only the intended mutations had occurred.
Protein analysis with a mass spectrometer
The purity and molecular weight of the enzyme were tested with SDS-PAGE. The mass spectrometer (Q-TOF; Micromass, Manchester, U.K.) was set to scan with a ratio of mass to charge in a range m/z = 100–2500 u, with a scan 2 s/step and an interscan duration 0.1 s/step. In all ESI-MS experiments, the quadrupole scan mode was used under a capillary needle at 3 kV, a source block temperature 80 °C, and a desolvation temperature 150 °C. Purified protein (100 μL, about 25~30 μg) was precipitated with trifluoroacetic acid (25 %) at 4 °C for 30 min. After centrifugation, the precipitate was washed twice with acetone. The precipitant was redissolved in 0.1% formic acid solution for mass analysis.
Substrate preparation and enzymatic assays
Curdlan (5 g) was washed with aqueous ethanol (20 % by volume) and then suspended in phosphate buffer (100 mL, 50 mM, pH 10.5) with stirring near 25 0C for 30 min. The reaction solution was neutralized with HCl (0.1 M) to obtain gel-like curdlan. After centrifugation, the precipitant was washed with ddH2O to remove salt and re-suspended in ddH2O at 70~75 °C for 2 h. The supernatant was obtained after centrifugation to remove the insoluble portion and evacuated to dryness. The resulting powder was redissolved (final concentration 5 % w/v) in phosphate buffer (50 mM, pH 7.5) for enzymatic assay and application. The activity of LPHase was analyzed on estimating the amount of the reducing ends of sugars using the dinitrosalicylic acid (DNS) method (Miller, 1959). The standard assay mixture (0.5 mL) contained enzyme solution properly diluted, curdlan substrate (2 % w/v) in phosphate buffer (50 mM, pH 7.5). The reactions were performed for 2h at 37 °C; DNS reagent (0.5 mL) was then added, and the resulting mixture was boiled for 15 min, chilled and centrifuged to isolate the insoluble curdlan. The resulting adducts of reducing sugars were analyzed and measured spectrophotometrically at 540 nm. The absorption coefficient of the resulting adducts was determined to be 788 M-1.cm-1 when D-glucose served as control sample. One unit of LPHase activity is defined as the amount of enzyme required to release 1 µmol of detectable reducing sugars at 37 °C in 1 min. The products of enzymatic reaction were analyzed with a mass spectrometer (triple quadrupole , Quattro Micro; Micromass).
The optimum pH for LPHase activity was determined on incubating the purified enzyme with substrate curdlan (2 %), in buffers with pH in a range 2.0~11.5. Buffers used were glycine (pH 1.8-3.5 and 9.0-10.0), sodium acetate (pH 4.0-5.5), morpholinoethanesulfonic acid (pH 5.5-6.5), phosphate (pH 6.5-7.5), Tris (pH 7.5-8.5), Caps (10.5-11.5). The optimum temperature was determined on assaying the enzyme activity (at pH 7.0) at temperatures from 4 °C ~ 95 °C. All activities were measured following a standard protocol as described above. For tests of pH stability, LPHase was pre-incubated in buffers (pH 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0) at 25 °C and was further assayed at varied intervals using a standard protocol. For tests of thermal stability, LPHase was pre-incubated at pH 7.0 and temperatures 25, 40, 50, 60, 65, 75 and 85 °C, and the residual activity of the enzyme was measured after various periods.
The effects of various metal ions and reagents (Cu2+, Ni2+, Ba2+, Cd2+, Zn2+, Co2+, Mn2+, Ca2+, Mg2+, Fe2+, Hg2+, EDTA and DTT) on LPHase activity were investigated by incorporating 1-10 mM ion (or reagent) to the standard assay system.
Preparation of laminaripentaose, synthesis of p-NPLP and kinetic assay
To prepare laminaripentaose, we poured the reaction mixture containing curdlan (100 mL, 5 % w/v), prepared in Caps buffer (50 mM, pH 10.5) and purified LPHase (5 mL, 10 mg/mL) into tubular regenerated-cellulose membrane (molecular weight cut-off 3000), which was kept in ddH2O (250 mL), 45 oC for 12 h. The enzymatic reaction was performed in the dialysis system and ddH2O (250 mL) was changed every 4 h. The reaction was stopped on adding ethanol to final of 85 %. The resulting solution was kept at -20 °C for 4 h. The precipitated protein was removed via centrifugation and the supernatant ethanol solution containing laminaripentaose was concentrated. The product was analyzed with a mass spectrometer with electrospray ionization. p-Nitrophenyl-β-1,3-D-laminaripentaoside (p-NPLP) was synthesized according to the literature (Smits et. al., 1996).
Enzyme activity and kinetic parameters were determined at pH 7.0 on monitoring the hydrolysis of
p-NPLP to release p-nitrophenol (or p-nitrophenolate); the absorption coefficient (△ε) was 7280 M-1.cm-1
(400 nm, pH 7.0). A spectrophotometer (Agilent 8453) equipped with a circulating water bath at 40 0C was used. All kinetic data were calculated as the average of at least three experiments.
Chemical rescue and 1H -NMR spectral analysis
The rescue of the activities of E154G and D170G was performed on adding sodium azide or sodium formate (0.01-2.0 M) to the assay reagent that contained enzyme (1.6 mg/mL) and curdlan (2 %) in phosphate buffer (50 mM, pH 7.5). The reaction was performed at 37 °C for 4 h. The activity was estimated from the amount of the reducing-end sugar. The products derived from the catalysis were determined from 1H-NMR spectra measured at 27 °C, as described previously (Nishimura et. al., 2001). In preparation for a temporal 1H-NMR experiment, the wild-type and mutated LPHases and curdlan were deuterated on repeated lyophilisation and solvent exchange with 2H2O (99.8 %). The final condition of the reaction mixture contained wild-type LPHase (1.6 mg/mL for D170G) and substrate (2 %) in 2H2O (99.96 %). The spectra were recorded at various intervals after the addition of enzyme. The conditions for the temporal 1H-NMR spectral tests of chemical rescue are identical to the above description except D170G (1.6 mg/mL) and azide (1.5 M) were
employed.
Chemical modification of D170C
The labeling reactions were performed at 25 °C for 12 h in Tris buffer (50 mM, pH 8.0) containing D170C (1 mM) and labeling reagents (5 mM, iodoethanoic, dithiodiglycolic acid). The excess labeling reagent was removed with ultra-filtration. The efficiency of the labeling reaction was evaluated with a mass spectrometer (ESI-MS). The catalytic activities of the resulting mutants were determined with curdlan (2 %, w/v) in phosphate buffer (50 mM, pH 7.5). p-NPLP served as the substrate for determination of the kinetic parameters.
RESULTS AND DISCUSSION
Protein expression, purification and characterization
β-1,3-D-glucanase promotes the hydrolysis of the β-1,3-glycosidic linkage of a linear biopolymer of β-1,3-linked polysaccharides. Among β-1,3-D-glucanases, LPHase, releasing laminaripentaose as the predominant product (Nakabayashi et. al., 1998), is of particular interest. The gene of LPHase was reconstructed with primers and PCR. The mature LPHase gene (without the first 35-aa signal peptide) fused with a methionine at the N-terminus of the mature protein was inserted into pRSET_A. This new clone, pRSET_A-lphase, was transformed into E. coli BL21 (DE3) for expression. The recombinant LPHase was expressed as a soluble form. After three consecutive steps of purification, the purity of LPHase was enhanced 26-fold; the recovery yield was 34 %. The purity (> 95 % homogeneity) and the Mr of the purified LPHase were confirmed with SDS-PAGE and mass spectrometry (Mr 39424±3 (calculated Mr 39421). The apparent Mr of LPHase was estimated to be about 38,000 with a size-exclusion chromatograph, indicating LPHase to have a monomeric form in solution and is consistent with the observation in crystal form.
The catalytic activity of recombinant LPHase was evaluated on several polysaccharides containing the β-1,3-glucosidic bond such as curdlan, laminarin, lentinan, schizophyllan and pachyman. The reactions were carried out with 2% (w/v) substrate in phosphate buffer (50 mM, pH 7.5) for 2h at 37 °C. Of those, only curdlan and laminarin (~60 % activity of that with curdlan) were effective substrates. The specific activity of LPHase with curdlan was determined to be 7.1 µmol/ min/mg. Other soluble β-glucans (highly branched β-1,3/1,6 glucans) were digested mildly by LPHase (<10 % activity of that with curdlan). No significant activity was detected when polysaccharides containing the α or β-1,4-glucosidic bond, such as lichenan, xylan, cellulose, starch, chitin and chitosan were employed as the substrate for LPHase. The LPHase specifically catalyzes the hydrolysis of the β-1,3-glucosidic bond.
The thermal (25 °C – 85 °C) and acid (pH 2 – 11) stability of LPHase was investigated with curdlan as substrate. In general, the recombinant LPHase was stable up to 60 °C with incubation for 4 h at pH 7.0 and rapidly lost its activity at temperature > 65 °C; the optimum temperature for LPHase is approximately 55 °C. The recombinant protein was stable in pH range 5.0 – 9.0 (at 25 °C for at least 4 h) and maintained ~80% activity for pH between 9.5 – 11.0. The enzyme dramatically lost its catalytic activity in solution with pH ≥
11.5 or < 3.0. Tests of the pH-dependent activity showed that the recombinant enzyme had optimum activity in pH range 7.5 – 8.5.
To investigate the effect of metal ion, we added various divalent metal cations (Cu2+, Ni2+, Ba2+, Cd2+, Zn2+, Co2+, Mn2+, Ca2+, Mg2+, Fe2+, Hg2+) and dithiothreitol (DTT) up to 10 mM in enzymatic assay. No significant loss on activity was observed except for Hg2+ (>20 μM) and DTT (>100 μM), for which 90 – 95% inhibition were observed.
β-1,3-D-laminaripentaose on a large scale was prepared from curdlan by enzymatic catalysis using recombinant LPHase. Laminaripentaose was further used to synthesize p-nitrophenyl-β-1,3-D- laminaripentaoside (p-NPLP), which was employed for kinetic tests on LPHase.
Mutagenic study of LPHase
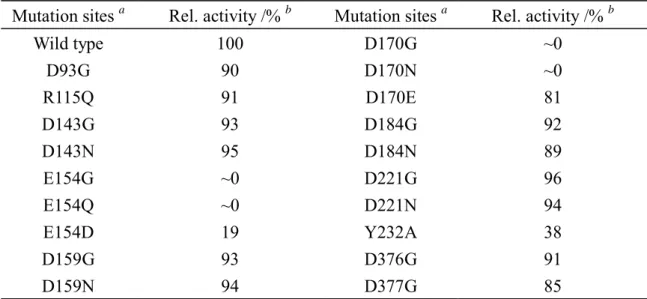
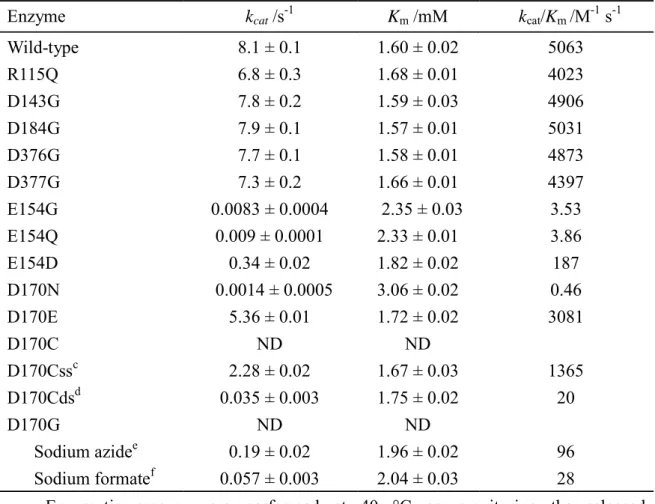
As the molecular mechanism is believed to be conserved within the same family of glycosyl hydrolases (Henrissat, 1991; Henrissat and Bairoch, 1993; Henrissat and Bairoch, 1996, Cantarel et. al., 2009), a successful investigation of one particular enzyme is valuable to characterize general features of the family. The residues in the catalytic domain are involved in the physical or chemical steps to orient the enzyme appropriately and to break or to form chemical bonds. Insight into the catalytic residues is derived from the x-ray structure, but to confirm the function of the essential residues demands extensive kinetic tests coupling with various strategies, including mechanism-based inhibition, the combination of amino-acid sequence analysis and mutagenesis, and chemical rescue. For most retaining enzymes, the formation of a covalent enzyme intermediate is expected. In such a case, mechanism-based or active-site-directed irreversible inhibition is a common strategy to assist the identification of key residues; 2-fluoroglycosides were employed to trap the glycosyl-enzyme intermediate and to facilitate the identification of the nucleophile through peptide sequencing with LC/MS (Ferrer et. al., 2005; Li et. al., 2002; Hart et. al., 2000). As LPHase is an inverting enzyme, its molecular mechanism involves no formation of a covalent glycosyl-enzyme intermediate. Although many GH-64 β-1,3-D-glucanases were cloned, the extensive test for functional characterization and the essential residues involved in catalysis are less experimentally identified. Most glycohydrolases require two carboxylic acids for their catalytic reactions. The conserved glutamate and aspartate of GH-64 glycohydrolases, derived from the multi-alignment of 14 sequences of β-1,3-D-glucanase, were selected as potential candidates (Figure 2). Five (glutamate and aspartate) residues -- Asp143, Glu154, Asp170, Asp366 and Asp367 -- were highly conserved. An extensive mutagenesis test on all possible candidates was performed. Other residues (partially conserved) -- Asp93, Asp159, Asp184 and Asp221 -- were also chosen for testing. We varied the putative catalytic residues, glutamate and aspartate to glutamine, asparagines or glycine by site-directed mutagenesis. All mutants were overexpressed in E. coli. The procedures of protein purification were similar to that of wild-type LPHase, as described in Experiments. The relative activities of hydrolyzing curdlan and kinetic parameters of p-NPLP substrate for mutants are summarized in Tables 2 and 3. The activity assay showed that D93G, D143G/N, D159G/N, D184G/N, D221G/N, D376G and D377G retained activity (> 85 %) relative to that of the wild-type enzyme. In contrast, E154Q, E154G, D170N, D170C and D170G lost catalytic activity (<0.1%). The measurement of kinetic parameters using p-NPLP as substrate revealed that mutation at Glu154 and Asp170 caused a large effect on activity perturbation. For instance, the activities (kcat/Km) of D170N and E154Q (or E154G) decreased at least 10000-fold and 1300-fold, respectively.
The Km values of mutants were slightly perturbed with a range 1.6 – 3.06 mM. The structural alteration of E154G and D170G seemed to be minor as their CD spectra were nearly identical to that of wild-type or active mutants (Figure 3). The kinetic outcome indicates that Glu154 and Asp170 play important roles for the catalytic activity of LPHase.
Chemical rescue
Activity rescue with exogenous nucleophiles, such as azide, formate and other anions, enhances the catalytic activity of enzymes mutated at residues that are essential for catalysis of glycoside hydrolases (Zechel and Withers, 2001; Bravman et. al., 2001; Cobucci-Ponzano et. al., 2005; Shallom et. al., 2005). For retaining glycosidases, an exogenous nucleophile such as azide ion might rescue the activity of mutant with mutation at the nucleophile residue (or the acid-base residue) to yield the glycosyl azide with inverting configuration (or retaining configuration) as product. For inverting glycosidases, the catalytic hydrolysis proceeds via a single-displacement mechanism in which the general acid residue promotes the catalysis via protonation of the glycosidic oxygen, and the general base is employed to polarize a water molecule in the active site to attack the anomeric carbon of the substrate. The addition of exogenous nucleophile might activate a mutant with dysfunction on general-base residue but not on general-acid residue (MacLeod et. al., 1994; Zechel et. al. 2003).
We employed chemical rescue of the activities of E154G and D170G to elucidate the functions for both residues. Replacing the essential group with a glycine residue presumably results in obtaining sufficient space in the active site to accommodate a small nucleophile (or base) in the catalytic cavity. The activities of such mutated enzymes might be partially recovered if the small ion functions as a nucleophile or a general base. We employed sodium azide and formate to reactivate E154G and D170G. With azide or formate at various concentrations (0.05-2 M), the catalytic activity of D170G mutant was enhanced. The specific activity of the D170G mutant increased 52- and 31-fold with the addition of sodium azide (2 M) and sodium formate (1 M), respectively, whereas the catalysis of E154G remained unaltered (Figure 4 and Table 3). In summary, the kinetic outcome derived from site-directed mutagensis and chemical rescue indicates that Glu154 functions as the general acid and Asp170 serves as the general base in a catalytic turnover.
Two possible products are obtainable from the chemical rescue; one product is the α-anomeric azide-sugar derived from the direct nucleophilic attack of azide at the anomeric carbon (shown in Scheme 1a), and the other is the hydrolytic product released from the substrate through the attack of a water molecule that may be polarized with azide (shown in Scheme 1b). The products derived from the azide rescue of D170G catalysis were laminaripentaose and laminaripentaosyl azide, which were analyzed with a mass spectrometer (ESI/MS), m/z= 851.1 (M+Na+) and 876.2 (M+Na+), respectively (Figure 5). An enzymatic temporal 1H-NMR test was performed with curdlan as substrate to examine both anomeric preference and production of azide-sugar in the chemical rescue of D170G. Weak signals assignable to a free hemiacetal H-1 were observed at 4.67 ppm (β-anomer) and 5.24 ppm (α-anomer) consistent with the polymeric structure of the substrate (Nishimura et. al., 2001). The temporal course of the reaction is shown in (Figure 6). The spectrum corresponding to 20 min of hydrolysis (Figure 6b) showed a new signal centred at 5.24 ppm (J1, 2 = 3.6 Hz) assigned to C1-H of a reducing oligosaccharide in an α-configuration, indicating that LPHase performs the catalysis with the inverting mechanism. For the reaction at 85 min (Figure 6c), a new signal appeared at
4.67 ppm (J1, 2 = 8.1 Hz), assigned to C1-H of the reducing-end sugar in β-configuration. The β-form sugar was derived from the spontaneous mutarotation of the α-anomer. When the reaction approached 140 min, another line appeared, centered at 5.52 ppm (J1,2 = 4.3 Hz), corresponding to the reducing–end C1-H of the pentaosylazide in the α-configuration (Viladot et. al., 1998) (Figure 6d). This outcome indicates that azide acts as a base to polarize a water molecule to promote the inverting catalysis and to function as a nucleophile to attack the anomeric center to give α-pentaosylazide. The test of azide rescue on D170G catalysis confirmed that Asp170 function as the general base and, thus, Glu154 serves as the general acid of LPHase.
Catalytic feature of E154 and D170 mutants
Although the laminaritetraose-complex protein structure was resolved (Wu et. al., 2009), the catalytic role of Glu154 and Asp170 is not confirmed. For an improved understanding of the catalytic mechanism and the substrate binding feature, a computer simulation to model an enzyme-laminarihexaose structure was performed. The LPhase-laminaritetraose complex shows that the glucose moieties from the reducing end of tetraose bound to the +5 to +2 subsites of the substrate binding site. The best model of the LPHase-laminarihexaose simulation was obtained by placing the first three glucose moieties from the reducing end of hexaose at the +5 to +3 subsites in the LPhase-laminaritetraose complex (Wu et. al., 2009). The result shows that Glu154 is within hydrogen-bonding distance of the glycosidic oxygen (~1.9 Å), indicating that it may function as the proton donor. Asp170 is ~6 Å from the anomeric carbon; in this interval a water molecule can be accommodated between Asp170 and the sugar. Asp170 is predicted to serve as the general base for the catalysis. Of mutational tests on nine conserved carboxylic acid residues, only E154 and D170 mutants showed a large loss of activity without significant structural alteration. When these mutants were converted into the one with the other carboxylic residue, i.e. E154D and D170E, the activity was partially recovered. For example, with curdlan as substrate, E154D retains 19 % and D170E retains 81 % of the activity of the wild-type enzyme. When p-NPLP served as substrate, the catalytic activity (kcat/Km) of
E154D and D170E was recovered to 4 % and 61 %, respectively (Table 3). The weak activity of E154D is likely due to the insufficient distance of the general acid to protonate effectively the glycosidic oxygen for rate acceleration. In D170E mutant, the carboxyl group of Glu170 can provide a suitable orientation to enhance the reaction. The temporal 1H-NMR tests (data not shown) confirmed that D170E also catalyzed the hydrolysis of curdlan with inversion.
An inspection of the protein structure from various GH families revealed that the distance between the two essential carboxylates is approximately 5.5 Å for retaining GH (Sinnott, 1990; McCarter and Withers, 1994) and ~10 Å for the inverting GH (Sinnott, 1990; Davies and Henrissat, 1995). To extend the carboxylic side chain at the general base might result in converting LPHase into a retaining enzyme. For this approach, the D170C mutant was prepared; the mutant was inactive. Two labeling reagents -- iodoethanoic acid and dithiodiglycolic acid -- were used to modify the residue of Cys of D170C mutant, separately. Only a stoichiometric label was observed on D170C. Yet, no labeling reaction occurred with the wild type LPHase, D170G and E154G mutants. Although Cys306 and Cys326 are present in LPHase, the labeling reaction indicated that both residues seem to be inaccessible for chemical modification. The resulting proteins derived from the labeling reaction of D170C were analyzed with a mass spectrometer. The relative molar mass (Mr) of the modified D170C mutants increased by 57 and 89, corresponding to the addition of
residue –CH2COO- (designated as D170Css) and –SCH2COO- (designated as D170Cds), respectively (Figure
7). D170Css retained 27 % of the activity (kcat/Km) relative to wild-type LPHase. Laminaripentaose is the predominant catalytic product. The catalytic activity of D170Cds was recovered only 0.4 % (Table 3) and a mixture of laminaribi-, tri-, tetra-, and pentaose was produced. The temporal 1H-NMR experiments demonstrated both D170Css and D170Cds still catalyzed the hydrolysis of curdlan with the inversion mechanism (data not shown). Although the attempt was unsuccessful in converting LPHase into a retaining enzyme by genetically or chemically re-placing a carboxylic side-chain back to the position corresponding to the Asp170, the restore of the enzymatic activity of the mutants (D170Css and D170Cds) suggested that Asp170 of LPHase indeed function as the general base in the enzymatic reaction.
Conclusion
Our recent study (Wu et al. 2009) described the X-ray structure of LPHase from Streptomyces matensis belonging to family GH64 and strongly suggested that Glu154 and Asp170 function as the general acid and the general base, respectively, of this inverting enzyme. In this study, by using a combination of site-directed mutagenesis, kinetic analysis, chemical rescue, and product analysis, the catalytic role of Glu154 and Asp170 was experimentally confirmed. This study appears to be the first direct identification of these essential residues in GH64.
References
Armand, S., Tomita, H., Heyraud, A., Gey, C., Watanabe, T., and Henrissat, B. (1994) FEBS Lett., 343, 177-180.
Baker, J. O., McCarley, J. R., Lovett, R., Yu, C. H., Adney, W. S., Rignall, T. R., Vinzant, T. B., Decker, S. R., Sakon, J., and Himmel, M. E. (2005) Appl. Biochem. Biotechnol., 121, 129-148.
Bara, M. T., Lima, A. L., and Ulhoa, C. J. (2003) FEMS Microbiol. Lett., 219, 81-85.
Blättel, V., Larisika, M., Pfeiffer, P., Nowak, C., Eich, A., Eckelt, J., and König, H. (2011) Appl. Environ. Microbiol. 77, 983-990.
Bravman, T., Mechaly, A., Shulami, S., Belakhov, V., Baasov, T., Shoham, G., and Shoham, Y. (2001) FEBS
Lett., 495, 115-119.
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009) Nucleic
Acids Res. 37, D233-238.
Chen L., Sadek, M., Stone, B. A., Brownlee, R. T., Fincher, G. B., and Høj, P. B. (1995) Biochim. Biophys.
Acta, 1253, 112-116.
Cobucci-Ponzano, B., Mazzone, M., Rossi, M., and Moracci, M. (2005) Biochemistry, 44, 6331–6342.
Davies, G. and Henrissat, B. (1995) Structure, 3, 853-859.
Dias, F. M. V., Vincent, F., Pell, G., Prates, J. A. M., Centeno, M. S. J., Tailford, L. E., Ferreira, L. M. A., Fontes, C. M. G. A., Davies, G. J., and Gilbert, H. J. (2004) J. Biol. Chem., 279, 25517-2552.
Ferrer, P. (2006) Microbial cell Factories, 5, 1-8.
Ferrer, M., Golyshina, O. V., Plou, F. J., Timmis, K. N., and Golyshin, P. N. (2005) Biochem. J., 391, 269-276.
Fliegmann, J., Montel, E., Djulić, A., Cottaz, S., Driguez, H., and Ebel, J. (2005) FEBS Lett., 579, 6647-6652. Hart, D. O., He, S. M., Chany, C. J., Withers, S. G., Sims, P. F. G., Sinnott, M. L., and Brumer, H. (2000)
Biochemistry, 39, 9826-9836.
Henrissat, B. (1991) Biochem. J., 280, 309-316.
Henrissat, B. and Bairoch, A. (1993) Biochem. J., 293, 781-788.
Henrissat, B. and Bairoch, A. (1996) Biochem. J., 316, 695-696.
Hrmova, M. and Fincher, G. B. (2001) Plant Mol. Biol., 47, 73-91.
Ishida, T., Fushinobu, S., Kawai, R., Kitaoka, M., Igarashi, K., and Samejima, M. (2009) J. Biol. Chem., 284, 10100-10109.
Johansson, P., Brumer, H., Baumann, M. J., Kallas, A. M., Henriksson, H., Denman, S. E., Teeri, T. T., and Jones, T. A. (2004) Plant Cell, 16, 874–886.
Kawai R., Igarashi K., Yoshida M., Kitaoka M. and Samejima M. (2006) Appl. Microbiol. Biotechnol., 71,
898-906.
Koshland, D. E. (1953) Biol. Rev. Camb. Philos. Soc., 28, 416–436.
Laemmli, U. K. (1970) Nature, 227, 680–685.
Li, Y. K., Chir, J., Tanaka, S., and Chen, F. Y. (2002) Biochemistry, 41, 2751-2759.
MacLeod, A. M., T. Lindhorst, S. G. Withers, and R. A. J. Warren. (1994) Biochemistry, 33, 6371-6376.
Martin, K., McDougall, B. M., McIlroy, S., Chen, J., and Seviour, R. J. (2007) FEMS Microbiol. Rev., 31, 168-192.
Martin, K. L., McDougall, B. M., Unkles, S. E., and Seviour, R. J. (2006) Mycol. Res., 110, 66-74.
McGrath, C. E., Vuong, T. V., and Wilson, D. B. (2009) Protein Eng. Des. Sel. 22, 375-382.
McGrath, C.E. and Wilson, D. B. (2006) Biochemistry, 45, 14094-14100. McCarter, J. D. and Withers, S. G. (1994) Curr. Opin. Struct. Biol., 4, 885-892.
Miller, G. L. (1959) Anal. Chem., 31, 426–429.
Nakabayashi, M., Nishijima, T., Ehara, G., Nikaidou, N., Nishihashi, H., and Watanabe, T. (1998) J. Ferment.
Bioeng., 85, 459-464.
Nishimura, T., Bignona, C., Alloucha, J., Czjzeka, M., Darbona, H., Watanabe, T., and Henrissat, B. (2001)
FEBS Lett., 499, 187-190.
O'Connell E., Piggott C., and Tuohy M. (2011) Appl. Microbiol. Biotechnol., 89, 685-696.
Palumbo, J. D., Sullivan, R. F., and Kobayashi, D. Y. (2003) J. Bacteriol., 185, 4362-4370.
Receveur-Brechot, V., Czjzek, M., Barre, A., Roussel, A., Peumans, W. J., Van Damme, E. J. M., and Rouge, P. (2006) Proteins, 63, 235-242.
Shallom, D., Leon, M., Bravman, T., Ben-David, A., Zaide, G., Belakhov, V., Shoham, G., Schomburg, D., Baasov, T., and Shoham, Y. (2005) Biochemistry, 44, 387–97.
Shen, S. H., Chretien, P., Bastien, L., and Slilaty, N. (1991) J. Biol. Chem., 266, 1058-1063.
Sinnott, M. L. (1990) Chem. Rev., 90, 1171-1202.
Smits, E. Engberts, B. F. N., Kellogg, R. M., and Doren, H. A. (1996) J. Chem. Soc., Perkin Trans. 1, 2873-2877.
Varghese, J. N., Garrett, T. P., Colman, P. M., Chen L., Hoj, P. B., and Fincher, G. B. (1994) Proc. Natl. Acad. Sci., 91, 2785-2789.
Vasur, J., Kawai, R., Larsson, A. M., Igarashi, K., Sandgren, M., Samejima M., and Ståhlberg, J. (2006) Acta
Cryst. D, 62, 1422-1429
Wu, H. M., Liu, S. W., Hsu, M. T., Hung, C. L., Lai, C. C., Cheng, W. C., Wang, H. J., Li, Y. K., and Wang, W. C. (2009) J.Biol.Chem., 284, 26708-26715.
Zechel, D. L. and Withers, S. G. (2000) Acc. Chem. Res., 33, 11-18.
Zechel, D. L. and Withers, S. G. (2001) Current Opinion in Chemical Biology, 5, 643-649.
Zechel, D. L., Reid, S. P., Stoll, D., Nashiru, O., Warren, R. A. J., and Withers, S. G. (2003) Biochemistry, 42, 7195-7204.
Table 1. Primer sequences for various mutationsa
a: Note that the forward and reverse primers designed for different mutants at the same position were used for site-directed mutagenesis. For example, D143G(+) and D143N(-) were used as primers to produce both D143G and D143N. Other cases such as E154Q(-), E154D(+), D159G(+), D159N(-), D170N(-), D170E(+), D170C(+), D184G(+), D184N(-), D221G(+), D221N (-), Y232A(+) and Y232F(-) primers were used to perform all
mutations at the corresponding sites. DNA sequencing was employed to identify and confirm the mutations achieved.
b: Mutations are underlined.
Mutation site Primer sequenceb
D93G (+): 5’- GGATGGAGGCGCCTGGAGCTGGAGTAGG-3’ (–): 5’- CCTACTCCAGCTCCAGGCGCCTCCATCC-3’ R115Q (+): 5’- CAAGTTCTCTGGCCAGATCTACTTCTCGTAC-3’ (–): 5’- GTACGAGAAGTAGATCTGGCCAGAGAACTTG-3’ D143G (+): 5’- CAGAACCCGACTGGCCCGAACCGCGAC-3’ D143N (–): 5’- GTCGCGGTTCGGGTTAGTCGGGTTCTG-3’ E154G (+): 5’- CTTCAACTGGTCCGGCTACACGCTCAACG-3’ E154Q (–): 5’- CGTTGAGCGTGTACTGGGACCAGTTGAAG-3’ E154D (+): 5’- CTTCAACTGGTCCGACTACACGCTCAACG-3’ D159G (+): 5’- GTACACGCTCAACGGCTCCGGGCTCTG-3’ D159N (–): 5’- CAGAGCCCGCAGTTGTTGAGCGTGTAC-3’ D170G (+): 5’- CAGTACGCAGGTCGGCATGTTCTCAGCTC-3’ D170N (–): 5’- GAGCTGAGAACATGTTGACCTGCGTACTG-3’ D170E (+): 5’- CAGTACGCAGGTCGAGATGTTCTCAGCTC-3’ D170C (+): 5’- CAGTACGCAGGTCTGCATGTTCTCAGATC-3’ D184G (+): 5’- GTGCGGCGCGGCGGCGGCACCACACTGAGC-3’ D184N (–): 5’- GCTCAGTGTGGTGCCGTTGCCGCGCCGCAC-3’ D221G (+): 5’- CAGACGCGATCCGGCGGTACCGTGCTTCG-3’ D221N (–): 5’- CGAAGCACGGTACCGTTGGATCGCGTCTG-3’ Y232A (+): 5’- CGCTCTCTCCACTCGCTGGTGTCGAGACAGG-3’ Y232F (–): 5’- CCTGTCTCGACACCGAAGAGTGGAGAGAGCG-3’ D376G (+): 5’- CGGCTTCGCATTCGGCGACGTGGGACATCAC-3’ (–): 5’- GTGATGTCCCACGTCGCCGAATGCGAAGCCG-3’ D377G (+): 5’- GTGATGTCCCACGCCGTCGAATGCGAAGCCG-3’ (–): 5’- CGGCTTCGCATTCGACGGCGTGGGACATCAC-3’
Table 2. Relative activities of LPHase and mutants with curdlan as substratec
Mutation sites a Rel. activity /% b Mutation sites a Rel. activity /% b
Wild type D93G R115Q D143G D143N E154G E154Q E154D D159G D159N 100 90 91 93 95 ~0 ~0 19 93 94 D170G D170N D170E D184G D184N D221G D221N Y232A D376G D377G ~0 ~0 81 92 89 96 94 38 91 85
a. All mutational sites are conserved in family 64 except Asp93, Arg115, Asp159, Asp184 Asp221 and Tyr232. b. The assay of enzymatic activity was performed at 37 °C with curdlan substrate (2 %) in sodium
phosphate buffer (50 mM, pH 7.5).
Table 3. Michaelis-Menten parameters of LPHase and mutants with p-NPL as substratea
Enzyme kcat /s-1 Km /mM kcat/Km /M-1 s-1
Wild-type R115Q D143G D184G D376G D377G 8.1 ± 0.1 6.8 ± 0.3 7.8 ± 0.2 7.9 ± 0.1 7.7 ± 0.1 7.3 ± 0.2 1.60 ± 0.02 1.68 ± 0.01 1.59 ± 0.03 1.57 ± 0.01 1.58 ± 0.01 1.66 ± 0.01 5063 4023 4906 5031 4873 4397 E154G E154Q E154D 0.0083 ± 0.0004 0.009 ± 0.0001 0.34 ± 0.02 2.35 ± 0.03 2.33 ± 0.01 1.82 ± 0.02 3.53 3.86 187 D170N D170E D170C D170Cssc D170Cdsd 0.0014 ± 0.0005 5.36 ± 0.01 ND 2.28 ± 0.02 0.035 ± 0.003 3.06 ± 0.02 1.72 ± 0.02 ND 1.67 ± 0.03 1.75 ± 0.02 0.46 3081 1365 20 D170G Sodium azidee Sodium formatef ND 0.19 ± 0.02 0.057 ± 0.003 ND 1.96 ± 0.02 2.04 ± 0.03 96 28
a. Enzymatic assays were performed at 40 °C on monitoring the released p-nitrophenolate at 400 nm in sodium phosphate buffer (50 mM, pH 7.1).
b. ND: the activity was undetectable even with enzyme up to 0.2 mg/mL in the assay. c. resulting protein of D170C labeled by iodoethanoate.
d. resulting protein of D170C labeled by dithiodiglycolate.
e. Chemical rescue of D170G was performed at 40 °C in sodium phosphate buffer (50
mM, pH 6.8) containing sodium azide (2.5 M) with D170G.
f. Chemical rescue of D170G was performed at 40 °C in sodium acetate buffer (50
Figure 2
Figure 4 Figure 5
Figure 6 Figure 7
Legend
Scheme 1. Proposed mechanism of LPHase for inverting catalysis and activity rescue of general base mutant
by azide. (a) The inverting catalysis of LPHase. (b) Azide ion replaces the nucleophilic water molecule and attacks the anomeric carbon directly, forming the glycosylazide product with inverted anomeric configuration. (c) Azide ion functions as the general-base residue to activate the nucleophilic water molecule that subsequently attacks the anomeric carbon, forming free sugar with inverted anomeric configuration. Note that the glycosidic bond is β-1,3 linkage and the laminaripentaose is the predominant product (ROH) if curdlan is used as substrate. In this study, the p-NPL was used as substrate for azide rescue, the ROH is p-nitrophenol.
Figure 1. Docking model of LPHase·laminarihexaose complex. The docked laminarihexaose is drawn as
sticks with carbon (green), oxygen (red) and proton (gray) atoms. Two catalytic residues --- Glu154 and Asp170 -- are shown in blue and magenta, respectively in the structure shown with a surface model. Other residues at the substrate binding site such as Arg115 (red), Tyr232 (yellow), Tyr371 (yellow) and Asp377 (brown) are shown. Other highly conserved residues (Asp93, Asp143, and Asp376) and partially conserved residues (Asp159, Asp184, Asp221) are shown in figure (a). Note that Asp93 and Asp221 are located on the back side of the cleft and Asp376 is buried inside the structure. The close view of the active site with the docked laminarihexaose was shown in figure (b).
Figure 2. Partial gene multi-alignment of GH-64 β-1,3-glucanase members. Data from a multialignment
exercise, using partial sequences of family GH64 β-1,3-glucanases. Biology WorkBench 3.2 CLUSTALW (San Diego Supercomputer Center, CA, USA) software was used. All enzyme sequences were derived from published gene sequences. NCBI Genbank accession details are : Eukaryota : EAA_48717(Magnaporthe
grisea70-15), CAD_21311 and EAA_27909 (Neurospora crassa OR74A), others was belong to bacteria :
BAA_04892 (Arthrobacter sp. YCWD3 ), AAA_25502 (Cellulosimicrobium cellulans), CAJ_60367 (Frankia
alni ACN14a), AAT_77161 (Lysobacter enzymogenes C3), AAN_77504 (Lysobacter enzymogenes N4-7),
BAC_68763 and BAC_68677 (Streptomyces avermitilis MA-4680).CAB_69688, CAC_16456 and CAC_16439 (Streptomyces coelicolor A3), BAA_34349 (streptomyces matensis DIC-108). Only partial sequences are shown. The conserved Glu and Asp are marked and numbered based on the S. matensis LPHase sequence.
Figure 3. CD spectra of recombinant LPHase and mutants. E154G (○) and D170G (+) exhibit CD spectra
similar to those of wild-type LPHase (solid line) and other mutants with significant activity, such as D184G (●).
Figure 4. Activity enhancement of E154G and D170G mutant by exogenous nucleophiles. The activity
of E154G (○) and D170G (●) in the presence of sodium azide at various concentrations; E154G (□) and D170G (■) with sodium formate. The assays were performed with curdlan (2%, w/v) as substrate at 37 °C for 4 h in phosphate buffer (50 mM, pH 7.5).
%, w/v) in sodium phosphate (50 mM) with sodium azide (2.5 M), pH 7.5, at 37 0C for 4 h. Centrifugation, ESI/MS analyses of supernatant product. The laminaripentaose and laminaripentaose azide of the hydrolysis product have m/z = 851.12 and 876.21, respectively.
Figure 6. 1H-NMR spectra during hydrolysis of curdlan catalyzed by D170G in the presence of sodium azide. (a) spectrum of substrate containing sodium azide (1.5 M) without D170G; (b~d): Spectra of a sample
after addition of D170G for 20, 85 and 140 min, respectively. The signals corresponding to the C1-H of α- anomer, β-anomer of laminaripentaose and α-laminaripentaosylazide are indicated.
國科會補助專題研究計畫成果報告自評表
請就研究內容與原計畫相符程度、達成預期目標情況、研究成果之學術或應用價
值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)
、是否適
合在學術期刊發表或申請專利、主要發現或其他有關價值等,作一綜合評估。
1. 請就研究內容與原計畫相符程度、達成預期目標情況作一綜合評估
■達成目標
□ 未達成目標(請說明,以 100 字為限)
□ 實驗失敗
□ 因故實驗中斷
□ 其他原因
說明:
2. 研究成果在學術期刊發表或申請專利等情形:
論文:■已發表 □未發表之文稿 □撰寫中 □無
專利:□已獲得 □申請中 □無
技轉:□已技轉 □洽談中 □無
其他:(以 100 字為限)
3. 請依學術成就、技術創新、社會影響等方面,評估研究成果之學術或應用價
值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)(以
500 字為限)
本案發表二篇論文,分別解析 GH64 家族水解酵素中 LPHase 之結構,並深入探
討此酵素之催化機制,利用定點突變和酵素動力學技術,我們亦證實 LPHase
催化反應中之重要胺基酸殘基,學術價值備受肯定,成果已分別發表於著名之
國際生化期刊 (JBC 和 PEDS).此外,我們亦建立利用此酵素發展靈芝寡醣之
生物技術製程,對開啟靈芝寡醣之生理活性研究與應用將有重要價值.
附件二國科會補助計畫衍生研發成果推廣資料表
日期: 100 年 10 月 15 日國科會補助計畫
計畫名稱:結合生物資訊與結構模擬研究第64 家族 beta-glucanase 之活性區與重要胺基酸殘基 計畫主持人:李耀坤 計畫編號:NSC 99-2627-B-009-007 領域:生物化學研發成果名稱
(中文)無此項成果資料 (英文)成果歸屬機構
發明人
(創作人)
技術說明
(中文) 無此項成果資料 (200-500 字) (英文)產業別
技術/產品應用範圍
技術移轉可行性及預期
效益
註:本項研發成果若尚未申請專利,請勿揭露可申請專利之主要內容。國科會補助專題研究計畫項下出席國際學術會議心得報告
日期: 100 年 10 月 15 日一、參加會議經過---- 本案未補助出席國際會議
二、與會心得
三、考察參觀活動(無是項活動者略)
四、建議
五、攜回資料名稱及內容
六、其他
計畫編號
NSC 99-2627-B-009-007
計畫名稱
結合生物資訊與結構模擬研究第64 家族 beta-glucanase 之活性區與重要胺基酸殘 基出國人員
姓名
服務機構
及職稱
會議時間
年 月 日至
年 月 日
會議地點
會議名稱
(中文)
(英文)
發表論文
題目
(中文)
(英文)
附件四國科會補助專題研究計畫項下赴國外(或大陸地區)出差或研習心得報告
日期: 100 年 10 月 15 日一、國外(大陸)研究過程----本案未補助國外(大陸)研究
二、研究成果
三、建議
四、其他
計畫編號
NSC 99-2627-B-009-007
計畫名稱
結合生物資訊與結構模擬研究第64 家族 beta-glucanase 之活性區與重要胺基酸殘 基出國人員
姓名
服務機構
及職稱
出國時間
年 月 日至
年 月 日
出國地點
附件五國科會補助專題研究計畫項下國際合作研究計畫國外研究報告
日期: 100 年 10 月 15 日一、國際合作研究過程----本案無國際合作研究
二、研究成果
三、建議
四、其他
計畫編號
NSC 99-2627-B-009-007
計畫名稱
結合生物資訊與結構模擬研究第64 家族 beta-glucanase 之活性區與重要胺基酸殘 基出國人員
姓名
服務機構
及職稱
合作國家
合作機構
出國時間
年 月 日至
年 月 日
出國地點
附件六國科會補助計畫衍生研發成果推廣資料表
日期:2011/08/02國科會補助計畫
計畫名稱: (子計畫三)結合生物資訊與結構模擬研究第64家族beta-glucanase之活性區 與重要胺基酸殘基(3/3) 計畫主持人: 李耀坤 計畫編號: 99-2627-B-009-007- 學門領域: 生物資訊跨領域研究無研發成果推廣資料
99 年度專題研究計畫研究成果彙整表
計畫主持人:李耀坤 計畫編號:99-2627-B-009-007- 計畫名稱:建立醣水解酵素之生物資訊分析平台-以 LPHase,MTSase,及 MTHase 為應用模型--(子計 畫三)結合生物資訊與結構模擬研究第 64 家族 beta-glucanase 之活性區與重要胺基酸殘基(3/3) 量化 成果項目 實際已達成 數(被接受 或已發表) 預期總達成 數(含實際已 達成數) 本計畫實 際貢獻百 分比 單位 備 註 ( 質 化 說 明:如 數 個 計 畫 共 同 成 果、成 果 列 為 該 期 刊 之 封 面 故 事 ... 等) 期刊論文 0 0 100% 研究報告/技術報告 0 0 100% 研討會論文 0 0 100% 篇 論文著作 專書 0 0 100% 申請中件數 0 0 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 6 6 100% 博士生 2 2 100% 博士後研究員 0 0 100% 國內 參與計畫人力 (本國籍) 專任助理 0 0 100% 人次 期刊論文 2 2 100% J. Biol. Chem. 和 PEDS 各一篇 研究報告/技術報告 0 0 100% 研討會論文 0 0 100% 篇 論文著作 專書 0 0 100% 章/本 申請中件數 0 0 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 0 0 100% 博士生 0 0 100% 博士後研究員 0 0 100% 國外 參與計畫人力 (外國籍) 專任助理 0 0 100% 人次其他成果