國 立 交 通 大 學
生 物 科 技 學 院
生 物 科 技 學 系
博士論文
血管收縮素對心臟細胞的第二型血管收縮素轉
化酶表現調節與第二型基質金屬蛋白酶
表現之影響
The effects of angiotensin peptides on
angiotensin converting enzyme II regulation and
matrix metalloproteinase-2 expression in cardiac cells
研 究 生: 關棠青
指導教授: 林志生 博士
血管收縮素對心臟細胞的第二型血管收縮素轉化酶表現調節
與第二型基質金屬蛋白酶表現之影響
The effects of angiotensin peptides on angiotensin converting enzyme II
regulation and matrix metalloproteinase-2 expression in cardiac cells
研 究 生:關棠青
Student:Tang-Ching Kuan
指導教授:林志生
Advisor:Chih-Sheng Lin Ph.D.
國 立 交 通 大 學
生 物 科 技 學 院
生 物 科 技 學 系
博 士 論 文
A ThesisSubmitted to Department of Biological Science and Technology National Chiao Tung University
In partial Fulfillment of the Requirements For the Degree of Ph.D.
In
Biological Science and Technology January 2013
Hsinchu, Taiwan, Republic of China
i
Acknowledgement
時光向來都在不知不覺中,飄然流逝,六年半的時間,轉眼間就這麼過去了。在博 士學位口試結束的那一刻,宣告了我博士班學程的結束,得到的,是本厚重的論文以及 一紙畢業證書,留在心底的,是許多令人值得去細細品味的經歷與回憶。 學位口試的完成,要感謝陳銘仁醫師、呂衍達醫師、張淑真老師以及曲在雯老師於 百忙之中抽空為我的博士論文進行指導,並給予寶貴的建議。而博士學位的取得,則要 感謝在我攻讀博士學位時,盡心指導我的指導教授 林志生老師。林志生老師是一位嚴 以律己且肯為學生著想的老師,他以嚴格的言教與身教指導實驗室的學生並規劃實驗室 的發展。為了讓學生畢業後在社會上具有相當的競爭力,老師對於指導學生也是相當的 嚴格,雖然在老師的指導下相當辛苦,但紮實的基本功,有效率且具邏輯性的處事方式 皆是在老師的指導下所賦予的。另外,老師也是一位很注重教學的老師,無論是課堂上 的教學或是 Lab 內的指導,他都相當認真看待,為了不讓我在攻讀博士學位時所學與碩 士期間重覆,老師領我進入了分子生物學的領域。在進林老師的 Lab 以前,我對分子生 物學可謂是一無所知,為此老師特別送我至動物科技研究所跟著師母 孫玉苓女士,學 習分子生物學的技術與知識。感謝師母在工作之餘,還抽空指導我分生相關的實驗技術 與觀念,讓我後來在跟老師討論相關研究時,不至於有太大的隔閡。 在攻讀博士學位的期間,Lab 中一直都有一群肝膽相照的戰友一起分擔老師的嚴格 指導。感謝活潑開朗的建龍學長、成熟穩建的俊旭學長以及自信且大器的思豪學長,總 是身先士卒地守護 Lab 中的學弟妹們,並適時的給予研究上的經驗與建議,即便學長們 畢業後,仍不時關心學弟妹的畢業進度並分享職場上的經驗談。感謝心臟組紹全、証皓、 子慧、首成、謝文郁醫師、鄭崑山醫師、睦元、燕秋、意涵、葛麗、怡萱、竣瑋、孟融、 勻慈、莞之、俊昇、佩衡與明慧,有了聰明酷帥的紹全、四處都有好人緣的証皓以及具 有天使臉蛋又帶點小惡魔個性的子慧其完整的論文為基礎,再加上默默做事不需人操心 的睦元、迷糊可愛的燕秋、意涵、佩衡三人組以及辦事認真負責的葛麗全力的支持,我 才能如此順利的完成此份論文,取得博士學位。ii 感謝藻類組的聖壹、千雅、筱晶、明達、佳蓉、俞任、子庭、冠華、戴樂、采郁及 郁彬,在騙死人不償命的聖壹與千雅的教導下,帶給 Lab 許多的歡笑與經典事蹟,無論 是聖壹與明達的雙人相聲、文學造詣”奇佳”的佳蓉與帥氣嬌羞的子庭其日常對話或是戴 樂因毒舌慘遭眾學妹圍勦的畫面,皆讓平時枯燥的研究生活活躍了起來。尤其感謝聖壹、 千雅以及戴樂在電腦相關問題、Lab 帳務問題以及 Lab 雜務上的協助,省去了我許多不 必要的煩惱。感謝 Sensor 組曜禎、宜貞、榕均、庭妤、瀞韓、唯婷、品萱、芳沅、碧珊、 逸柔、修兆、欣儒、怡儒、孟哲與一華的一路相伴,讓我在研究討論的過程中學習了許 多 sensor 方面的知識與技術,尤其感謝個性直率的曜禎、迷迷糊糊的唯婷以及美麗大方 的芳沅在 MMPs 方面的交流討論,令我在切入 MMPs 相關研究時能快速上手。在 Lab 中特別要感謝爽朗大方的郡誼及其玉樹臨風的男友彥谷,兩人陪我熬過了攻讀博士學位 期間實驗最低潮的那一段時期,讓我有繼續拼鬥下去的動力。 感謝 ISBL 中極富教學熱忱的牛正基老師、帥氣有型郭士民老師、高雅美麗的張淑 真老師以及樂天開朗的陳博洲老師,老師們的支持與鼓勵給予我進入交大攻讀博士學位 的機會,也給予我努力的方向與目標。感謝靖容學姊、仁材學長、家琪學姊、家宏、浩 凱、敘安、建璋、懷恩、佩樺、榆臻、淑穎、偉仲、慈緣、婷儀、佳哲、佳昕、云廷、 文泰、欣怡、丁偉與敏愉等好友的陪同與鼓勵。靖容學姊與仁材學長幽默風趣的談吐以 及其博士班歷程的經驗分享總能讓我以正面的態度面對接踵而來的各項挑戰。家琪學姊、 家宏、浩凱、敘安、建璋、懷恩、佩樺、榆臻、淑穎、偉仲、慈緣、婷儀、佳哲、佳昕、 云廷、文泰、欣怡、丁偉與敏愉等好友的日常關心讓我明白在研究的道路上我並不孤單。 最後要感謝的,是一直守護我、支持我的家人。感謝剛正不阿的父親生活上的資助 及教誨,讓我成為一位堅強、守規範且不逾矩的人。感謝母親從小到大在各方面的教誨 與用心,令我存有知性與教養。感謝弟弟支持我取得博士學位的心意,剛出社會不久的 他願意金援我至取得學位,即便是出國攻讀學位他也願意,這令我相當窩心。感謝兩位 貓女兒的陪伴,讓我這獨自在外念書的遊子每天都有回家的感覺。感謝我生命中的所遇 到的各項人、事、物,豐富我的靈魂,替我的人生增添了許多色彩,也造就了此本論文
iii 的產生。然而,取得博士學位僅僅只是人生中的一項過程,也是未來旅程的一個開端, 期盼能善用本身的所知所學,探索這美麗寬廣的世界,於名為未來的畫作中,繪出屬於 我自己璀璨的一面。 關棠青 謹誌 國立交通大學 生物科技學系 博士班 中華民國 一 百 零 二 年 一 月
iv
血管收縮素對心臟細胞的第二型血管收縮素轉化酶表現調節
與第二型基質金屬蛋白酶表現之影響
研究生:關棠青 指導教授:林志生 博士 國 立 交 通 大 學 生 物 科 技 學 院 生 物 科 技 學 系 博 士 班中文摘要
腎素-血管收縮素系統(renin-angiotensin system, RAS)為人體最重要的調節系統之 一,不正常的 RAS 與心血管疾病的病程機制有很大的關聯性。血管收縮素 II(Angiotensin II, Ang II)、血管收縮素 1-7(angiotensin 1-7, Ang 1-7)、血管收縮素轉化酶(angiotensin converting enzyme),以及第二型血管收縮素轉化酶(angiotensin converting enzyme II, ACE2)為 RAS 中的主要因子。Ang II 會刺激發炎反應、纖維母細胞生長、心肌細胞凋 零、細胞分化以及纖維化,進而導致組織修復與重塑,而 ACE2 會水解 Ang II 形成 Ang 1-7,以抑制 Ang II 所造成之不良反應。多數的文獻顯示 ACE2 於 RAS 與心臟疾病中扮 演重要角色,但其於心臟細胞中之表現調節功能仍尚未被釐清。
本研究主要目的為探討心臟細胞中 ACE2 與 ace2 基因之表現調節,以及 ACE2 的 表現調節與第二型金屬基質蛋白酶(matrix metalloproteinases-2, MMP-2)的關聯性。我 們以初代人類心纖維細胞(human cardiac fibroblasts, HCFs)來探討 Ang II 和 Ang 1-7 對 於 ACE2 的表現調節,以冷光報導分析法來探討 ace2 啟動子上與 ACE2 表現調控有關 的序列。此外,我們也用慢病毒(lentivirus)方法促使細胞高量表現 ACE2 或抑制 ACE2 生成,探討 ACE2 表現受到調控時,其對 MMP-2 表現之影響性。
本研究的重要結果歸納如下:(1) Ang II 與 Ang 1-7 分別會經由 AT1R 及 Mas 活化 ERK-MAPK 訊息傳遞途徑以刺激 ACE2 表現; (2) ace2 基因啟動子的-516/-481 序列上之 5’-ATTTGGA-3’為特定轉錄因子的結合位置並可藉此調控 ACE2 表現; (3) Ang II 會經由
v
AT1R 與其下游 ERK-MAPK 訊息傳遞鏈刺激 ace2 基因啟動子的-516/+20 序列藉此調控 ACE2 表現; (4) TGF-β1 和 TNF-α 處理 HCFs 對於其 ace2 基因啟動子活性並無顯著影響; (5) ACE2 高表現會造成 MMP-2 活性提昇,且 Ang II-AT1R-ERK1/2 傳遞途徑可降低 MMP-2 活性表現; (6) 相對於 Ang II,Ang 1-7 會經由 Mas 抑制 ERK1/2 的活化,但對於 MMP-2 的活性表現並無明顯之影響性; (7) Ang II-AT1R-ERK1/2 及 Ang 1-7-Mas-ERK1/2 這兩條傳遞途徑也可調控腫瘤壞死因子-α 轉化酶(tumor necrosis factor-α-converting enzyme; TACE or ADAM17)的表現以改變細胞膜上 ACE2 的脫落效應(shedding ACE2, shed ACE2); (8) 我們重新建立了 ACE2 基因剔除鼠(ACE2 knockout (KO) mice)群, 包括 ACE2+/-、ACE2-/-及 ACE2-/y基因型; (9) 相較於野生型小鼠,在 ACE2 KO 小鼠的心
臟組織中,MMP-2 活性表現量會顯著提昇。
本研究顯示 ACE2 會受到 Ang II 及 Ang 1-7 之表現調節,Ang II 及 Ang 1-7 會提昇 HCFs 之 ACE2 表現,且 Ang II 會經由 ace2 啟動子中的-516/+20 序列區段調控 ace2 基 因表現。於 HCFs,ACE2 的高表現會提昇 MMP-2 之活性表現,而 Ang II 會降低 HCFs/ACE2 的 ADAM17 mRNA 表現,也使 MMP-2 與 shed ACE2 的活性表現下降。這 些結果皆暗示 ACE2 於心臟重塑病程中擔任保護的角色,Ang II-AT1R-ERK1/2 及 Ang 1-7-Mas-ERK1/2 這兩條傳遞途徑可調控 ADAM17 的 mRNA 表現,以及 ACE2 與 shed ACE2 之活性表現,且 Ang II-AT1R-ERK1/2 可調控 MMP-2 之活性表現。
關鍵詞:血管收縮素轉化酶 II、血管收縮素 1-7、血管收縮素 II、心臟細胞、ACE2 基因 剔除鼠
vi
The effects of angiotensin peptides on angiotensin converting enzyme II
regulation and matrix metalloproteinase-2 expression in cardiac cells
Graduate student: Tang-Ching Kuan Advisor: Chih-Sheng Lin Ph.D.
Department of Biological Science and Technology
National Chiao Tung University
Abstract
Renin-angiotensin system (RAS) is an important regulation system in the human circulatory system. The abnormal RAS is associated with the pathogenesis of cardiovascular diseases. Angiotensin II (Ang II), angiotensin 1-7 (Ang 1-7), angiotensin converting enzyme (ACE) and angiotensin converting enzyme II (ACE2) are the mainly members in RAS. Ang II stimulates inflammation, fibroblasts growth, cardiac myocyte apoptosis, fibrogenesis, and differentiation to cause tissue repair/remodeling, and it has been hydrolyzed by ACE2 to obtain the Ang 1-7 to against the disadvantageous effect of Ang II. ACE2 plays a significant role in RAS and heart diseases, but the regulation mechanisms of ACE2 in the heart and cardiac cells are unclear.
This study demonstrates the regulation mechanism of ACE2 and ace2 gene expression in cardiac cells, and the association between ACE2 and matrix metalloproteinases-2 (MMP-2) in the cells. The ACE2 expression in human cardiac fibroblasts (HCFs) treated with angiotensin peptides, Ang II and Ang 1-7, and analyzed the promoter activity of human ace2 using luciferase report assay to identify regulatory elements of ace2 gene. In addition, we also revealed the relationship with ACE2 and MMP-2 in HCFs utilized the technology of lentivirus to investigate the role of ACE2 regulation in cardiac cells.
The major results of this study are: (1) Ang II-AT1R and Ang 1-7-Mas axes were via ERK-MAPK signal pathway to regulate ACE2 expression, respectively; (2) the sequences of
vii
5’-ATTTGGA-3’ within -516/-481 domain of the ace2 promoter could regulate ACE2 expression and this sequences was the binding site of the transcription factor; (3) Ang II regulated ACE2 expression through AT1R to activate ERK-MAPK signal pathway to
stimulate the -516/+20 domain within the ace2 promoter; (4) the ace2 promoter activity was no significant response when HCFs treated with TGF-β1 and TNF-α; (5) ACE2
overexpression enhanced MMP-2 activity and Ang II-AT1R-ERK1/2 axis decreased MMP-2 activity; (6) compared to Ang II, Ang 1-7 through Mas receptor inhibited the activation of ERK1/2, but no significant effect of MMP-2 activity; (7) the tumor necrosis
factor-α-converting enzyme (TACE or ADAM17) expression could be regulated through Ang II-AT1R-ERK1/2 and Ang 1-7-Mas-ERK1/2 axes to alter ACE2 shedding; (8) the ACE2 knockout (KO) mice were re-established in our laboratory, including the mice with ACE2+/-, ACE2-/- and ACE2-/y genotypes; (9) ACE2 deficiency enhances MMP-2 activity in heart tissue of ACE2 KO mice compared to WT mice.
This study reveals the ACE2 regulation by angiotensin peptides. Ang II and Ang 1-7 could enhance the ACE2 expression in HCFs and indicated that Ang II through the -516/+20 sequence domain within ace2 promoter to regulate ace2 gene expression. ACE2
overexpression enhances MMP-2 activity in HCFs, Ang II decrease the mRNA expression of ADAM17, MMP-2 activity and shed ACE2 activity in HCFs/ACE2. These results show ACE2 plays a protect role against the mechanism of heart remodeling by Ang II induction. Two mainly axes, Ang II-AT1R-ERK1/2 and Ang 1-7-Mas-ERK1/2, regulate the mRNA expression of ADAM17 and the activity of ACE2 and shed ACE2, and MMP-2 activity also could be regulated through Ang II-AT1R-ERK1/2 pathway.
Keywords: angiotensin-converting enzyme II, angiotensin II, angiotensin 1-7, cardiac cells, ACE2 knockout mice
viii
Contents
Acknowledgement i Abstract in Chinese iv Abstract in English vi Contents viii List of Tables x List of Figures xi1. Research background and significance 1
1-1. Heart remodeling and cardiac fibrosis 1
1-2. Renin angiotensin system 4
1-3. Angiotensin converting enzyme II 6
1-4. ACE2 with heart diseases and heart remodeling 9
1-5. Matrix metalloproteinases 11
1-6. ACE2 and gelatinase (MMP-2 and MMP-9) 16
2. Research Approaches 18
3. Materials and Methods 20
3-1. Chemicals and reagents 20
3-2. Cell culture and treatment 21
3-3. Human ace2 constructs 21
3-4. Transient transfection 24
3-5. Lentivirus infection 24
3-6. ACE2 knockout mice 25
3-7. Protein extraction 26
3-8. Luciferase reporter assay 27
3-9. Nuclear extraction 27
3-10. Electrophoretic mobility shift assay (EMSA) 28
3-11. RNA isolation and quantification 28
3-12. Reverse transcription-polymerase chain reaction (RT-PCR) and Real time polymerase chain reaction 29
3-13. Immunocytofluorescence assay 32
3-14. Western blotting 32
3-15. ACE2 activity assay 33
3-16. Gelatin zymography 33
3-17. Statistics 34
4. Results 35
4-1. Ang II up-regulates ACE2 expression in HCF cells via the AT1R and the ERK-MAPK pathway 35
ix
4-2. Ang 1-7 up-regulates ACE2 expression via the Mas receptor 39
4-3. Expression levels of deletion constructs in the ace2 promoter 43
4-4. Identification of the regulatory domain within the ace2 promoter 44
4-5. Identification of the regulatory element for ace2 45
4-6. Effect of Ang II on the transcriptional activation of ace2 49
4-7. Effect of pro-inflammatory factors on the transcriptional activation of ace2 52
4-8. The ACE2 activity of HCFs treated with Ang II and Ang 1-7 54
4-9. The ACE2 and MMP-2 activity of HCFs infected with ACE2 lentivirus 55
4-10. The ACE2 and MMP-2 activity of HCFs/ACE2 infected with ACE2 shRNA 56
4-11. The MMP-2 activities of HCFs/ACE2 treated with Ang II and Ang 1-7 58
4-12. The ERK1/2 expression of HCFs/ACE2 treated with Ang II and Ang 1-7 60
4-13. The shed ACE2 activity of HCFs/ACE2 treated with Ang II and Ang 1-7 62
4-14. The mRNA expression of MAPK1 and ADAM17 in HCFs/ACE2 treated with Ang II and Ang 1-7 64
4-15. ACE2 knockout mice 66
5. Discussion 71
5-1. Regulation of angiotensin converting enzyme II by angiotensin peptides 71
5-2. Identifying the regulatory element for human angiotensin-converting enzyme 2 (ACE2) expression 75
5-3. The association between ACE2, MMP-2 and angiotensin peptides 77
6. Conclusions 83
7. References 85
8. Appendix 107
8-1. The study of ACE2 and heart diseases 107
8-2. ACE2 and gelatinase (MMP-2 and MMP-9) 126
8-3. The study of ACE2 overexpression 128
x
List of Tables
Table 1-1. Types of different matrix metalloproteinases and their substrate specificity 13
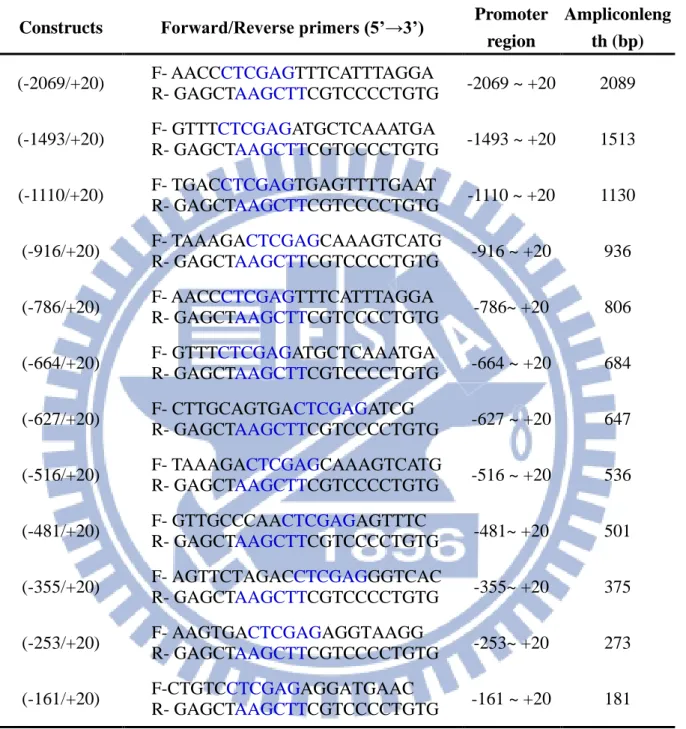
Table 3-1. Sequences of the primer pairs used for human ace2 promoter constructs 23
Table 3-2. Information of the PCR primers and condition performed in this study 31 Table 4-1. Sequences of the primer pairs used for generating the mutant constructs of
xi
List of Figures
Fig. 1-1. Pathologic cardiac intercellular communication 2
Fig. 1-2. Schematic representation of the renin-angiotensin system (RAS) cascade 6
Fig. 1-3. Family of enzymes and proteins belonging to the ACE family of proteins 8
Fig. 1-4. Family of enzymes and proteins belonging to the MMPs family of proteins 15
Fig. 2-1. The flowchart of research strategies 19
Fig. 3-1. Strategy for producing targeted disruption of the ace2 gene 26
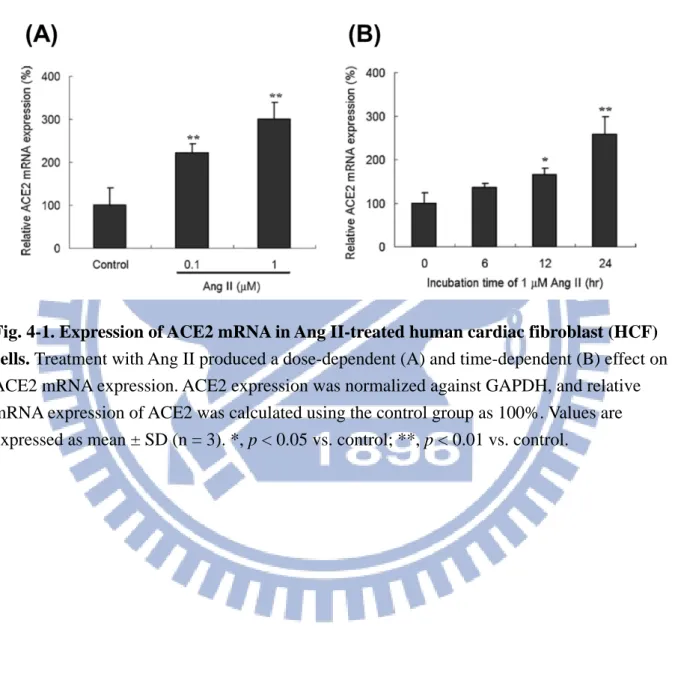
Fig. 4-1. Expression of ACE2 mRNA in Ang II-treated human cardiac fibroblast (HCF) cells 36
Fig. 4-2. Role of the ERK–MAPK signaling pathway of AT1R in Ang II-mediated ACE2 up-regulation 37
Fig. 4-3. Distribution and expression of ACE2 in Ang II-treated HCF cells 38
Fig. 4-4. Expression of ACE2 in HCF cells following Ang 1-7 treatment 40
Fig. 4-5. Distribution and expression of ACE2 in HCF cells treated with Ang 1-7 41
Fig. 4-6. Effect of blocking the AT1R-mediated signaling pathway on Ang 1-7 affected p-ERK1/2 and ACE2 protein expression in HCF cells 42
Fig. 4-7. Composition and promoter activity of the constructs on the expression of the reporter enzyme, luciferase, in HCFs 44
Fig. 4-8. Analyses of the promoter activity of the deleted and reversed domain within the upstream region of ace2 45
Fig. 4-9. Identification of the regulatory element within the –516/–481 domain 48
Fig. 4-10. Interaction of nuclear extracts from HCFs with (–516/–481) and mutant (M1 - M7) oligonucleotides by EMSA 49
Fig. 4-11. The effects of Ang II stimulation on ACE2 expression in HCFs 51
Fig. 4-12. The effect of Ang II stimulation on endogenous ACE2 expression in HCFs 52
Fig. 4-13. The promoter activity of ace2 in HCFs treated with pro-inflammatory factors 53
xii
Fig. 4-15. The ACE2 and MMP-2 activity of HCFs infected with ACE2 lentivirus 56
Fig. 4-16. The ACE2 and MMP-2 activity of HCFs/ACE2 infected with ACE2 shRNA 58
Fig. 4-17. The MMP-2 activities of HCFs/ACE2 treated with Ang II and Ang 1-7 60
Fig. 4-18. The ERK1/2 expression of HCFs/ACE2 treated with Ang II and Ang 1-7 62
Fig. 4-19. The shed ACE2 activity of HCFs/ACE2 treated with various doses of Ang II and Ang 1-7 63
Fig. 4-20. The shed ACE2 activity of HCFs/ACE2 treated with Ang II and Ang 1-7 64
Fig. 4-21. The mRNA expression of ADAM17 and MAPK1 in HCFs/ACE2 treated with Ang II and Ang 1-7 66
Fig. 4-22. PCR genotyping protocol for ACE2 knockout mouse 68
Fig. 4-23. The ACE2 expression of ACE2 knockout mice 69
Fig. 4-24. The ACE2 and MMP-2 activity of ACE2 knockout mice 70
Fig. 5-1. Schematic diagram representing the interplay of Ang II and Ang 1-7 on cardiac ACE2 regulation 74
1
1. Research background and significance
1-1. Heart remodeling and cardiac fibrosis
Organ fibrosis is a complex process that is defined as excess ECM deposited and
accumulated in the tissues including skin, heart, lung, kidney and vessels (Jinnin, 2010). Heart remodeling occurs in response to injury and an increase in wall stress plays a key role in the progressive deterioration of cardiac function that leads to heart failure (Pfeffer and Braunwald, 1990; Sharpe, 2000). Remodeling is characterized by cardiac hypertrophy and dilatation as well as conformational changes in the shape of the heart (Fig. 1-1). The remodeling process consists of a series of timed molecular events that include the inflammatory response to injury, proliferation of cardiac fibroblasts and differentiation to myofibroblasts, and formation of the fibrotic scar tissue in defected myocardium. Myocardium is comprised of a number of cell types such as cardiomyocytes, cardiofibroblasts, endothelial cells and smooth muscle cells, cardiac fibroblasts are also the highest cell population in the myocardium, accounting for about two-thirds of the cells (Camelliti et al., 2005). Cardiac fibroblasts are a critical element of myocardial repair that produce collagens, providing the tensile strength for cardiac tissue (Camelliti et al., 2005). The morphology of cardiac fibroblasts is the flat and spindle shaped cell in myocardium, it is the only cell population lack a basement membrane in the
myocardium. Furthermore, cardiac fibroblasts has more function such as homeostasis and remodeling of the cardiac ECM, electrical activity, production of growth factors and cytokines, and intercellular signaling with cardiomyocytes, endothelial or smooth muscle cells to impact cellular angiogenesis, cell proliferation, cardiomyocyte hypertrophy or apoptosis, appear that play a key role during pathological remodeling of the heart by maintaining normal cardiac structure, function, biochemical and electrical features of the heart (Fan et al., 2012).
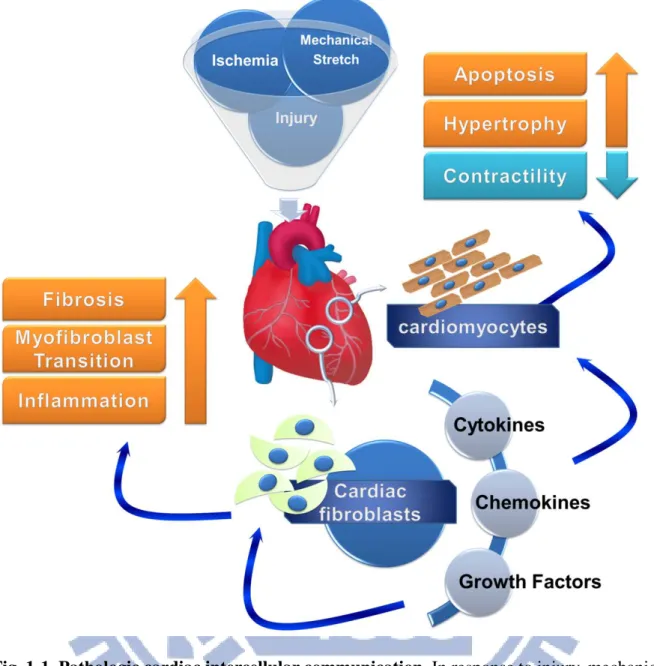
2
Fig. 1-1. Pathologic cardiac intercellular communication. In response to injury, mechanical stretch, and ischemia, the cardiac fibroblast undergoes a phenotypic transition to a
myofibroblast, releasing a variety of growth factors, chemokines and cytokines that act both in an autocrine and paracrine fashion. Stimulation of cardiac fibroblast results in a positive feedback loop to further enhance their activation, collagen deposition, and cytokine release, resulting in fibrosis and chronic inflammation. Cytokine effects on cardiomyocytes leads to pathologic effects including hypertrophy, apoptosis, and impaired contractile responses. [Martin and Blaxall, 2012]
In response to cardiac injury or stress, cardiac fibroblasts have been triggered and differentiated into myofibroblasts, which have greater synthetic ability to produce ECM
3
proteins, chemokines and cytokines such as IL-1α, IL-1β, IL-6, IL-10, TGF-β1 and TNF-α (Petrov et al., 2002; Baum and Duffy, 2011). These ECM, chemokines and cytokines mediate migration and contractile of cardiac fibroblasts and myofibroblasts at the site of injury, and maintain the inflammatory response to injury (Eghbali, 1992; Baum and Duffy, 2011). In addition, the TGF-β1 that cardiac fibroblasts and myofibroblasts released accelerate differentiation of cardiac fibroblasts into myofibroblasts and increase collagen expression (Butt et al., 1995; Walker et al., 2004), the accumulation of fibrotic depositions that can interrupts the connection between the myocardial cells and blood vessels in the myocardium leading to overall impairment of cardiac function. These results reveal that cardiac fibroblasts and myofibroblasts have been demonstrated to play a key role in reparative fibrosis in the infarcted heart (Díez et al., 2002; Calderone et al., 2006).
Besides of ECM production, cardiac injury causes chronic cardiac fibroblasts and myofibroblasts activation to produce matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), leading to imbalanced collagen/MMP secretion. A number of growth factors, cytokines, and chemokines have been identified that can regulate production of MMPs and TIMPs to maintain ECM homeostasis by cardiac fibroblasts (Moore et al., 2012). In various MMPs that cardiac fibroblasts released, MMP-2 and MMP-9 have been shown to release the ECM-bound latent TGF-β1, thereby inducing collagen synthesis and further contribute to the adverse remodeling (Yu and Stamenkovic, 2000). In MI and unstable angina patients, MMP-2, MMP-9 and TIMP-1 in the serum were significantly elevated compared with healthy controls, suggesting that these MMPs and TIMP-1 and
proinflammatory cytokines could play an important role in the pathophysiology of acute coronary syndrome (Tziakas et al., 2004). In addition, overexpression of MMP-2 led to severe myocardial fibrosis (Bergman et al., 2007) and MMP2-deficient mice showed the reduced myocardial hypertrophy and fibrosis (Matsusaka et al., 2006), while MMP-9 deficiency
4
partially improved myocardial hypertrophy and fibrosis following pressure overload (Heymans et al., 2005).
1-2. Renin angiotensin system
The renin-angiotensin system (RAS) is a classically hormonal system consists of endocrine, paracrine and intracrine system (Fyhrquist and Saijonmaa, 2008). The manly function of RAS involved the balance of salt and water, blood pressure and natriuresis, it also plays an important local role to regulate regional blood flow and nutrition in several target organs such as heart (Giani et al., 2012; Guimarães et al., 2012), blood vessels (Khakoo et al., 2008), and lungs (Imai et al., 2008; Shrikrishna et al., 2012; Wong et al., 2012). Furthermore, abnormal activation of the RAS is associated with the pathogenesis of cardiovascular and renal diseases such as hypertension (Jan Danser, 2012; Lo et al., 2012), myocardial infarction (Connelly et al., 2011; Burchil et al., 2012) and heart failure (Agarwal et al., 2012; Birner et al., 2012).
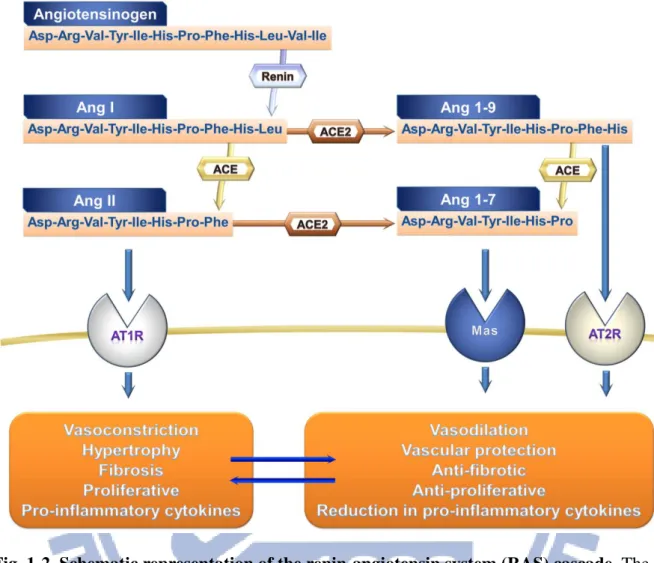
In a classical RAS, the glycoprotein angiotensinogen (AGT;
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile) released from the liver is degraded by the enzyme renin that originates in the kidney, generating the inactive angiotensin I (Ang I; Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) (Ferrario and Strawn, 2006). Subsequently, the dipeptide carboxypeptidase, angiotensin-converting enzyme (ACE) hydrolyzes the C-terminal dipeptide His-Leu of decapeptide Ang I to generate octapeptide angiotensin II (Ang II;
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe) (Kokubu et al, 1979), and the C-terminal peptide Phe of Ang II is metabolised by the carboxypeptidase, ACE2 to produce the vasodilator, angiotensin (1-7) (Ang 1-7; Asp-Arg-Val-Tyr-Ile-His-Pro) (Donoghue et al., 2000; Turner and Hoope, 2002; Rice et al., 2004). Finally, the C-terminal dipeptide His-Pro of Ang 1-7 was been
5
hydrolyzed by ACE to obtain inactive peptide angiotensin (1-5) (Ang 1-5; Asp-Arg-Val-Tyr-Ile) (Ferreira et al., 2012).
Ang II is the main regulator of the RAS, has been revealed that stimulate inflammation, cell growth, apoptosis, fibrogenesis, and differentiation to cause tissue repair/remodeling (Ruiz-Ortega and Ortiz, 2005; Mehta and Griendling, 2007). It has a very short half-life and is quickly degraded to Ang III and Ang 1-7, a similar function peptide and an oppose function peptide, respectively (Sun, 2010). Ang II stimulates a wide variety of biological functions in the heart (Zheng et al., 2012), blood vessels (Wehlage et al., 2012), kidneys (Pinheiro et al., 2012), adipose tissue (Kalupahana and Moustaid-Moussa, 2012), pancreas (Lau and Leung, 2011; Chan and Leung, 2011) and brain (Chrissobolis et al., 2012; Vargas et al., 2012) mediated the specific receptors Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R) (Skeggs et al., 1980; Corvol et al., 1995; Komatsu et al., 2009). The majority
physiological and pathophysiological effects of Ang II are mediated by the AT1R. Compared with those effects through AT1R, Ang II binding to the AT2R generally causes opposite effects such as stimulated bradykinin and nitric oxide to induce a counterregulatory vasodilatation (Horiuchi et al., 1997; Touyz etal., 1999; Sun, 2010).
Ang 1-7 is an important regulator in the RAS activity acts as an endogenous inhibitor of Ang II. Ang 1-7 binding to the Mas receptor and triggering signal pathways to release of bradykinin (Isa et al., 2011; Gembardt et al., 2012), prostaglandins (Yousif et al., 2012; Costa et al., 2012), and endothelial nitric oxide (Ferrario et al., 2005; Shah et al., 2012) and induce opposite effects to those elicited by Ang II such as apoptosis (Santos et al., 2003; Wang et al., 2012), vasodilation (Savergnini et al., 2010; Pringle et al., 2011), anti-fibrosis (Grobe et al., 2007; Nadu et al., 2008; Ferreira et al., 2010), anti-hypertrophic (Santos et al., 2004; Mercure et al., 2008; Santiago et al., 2010) and anti-proliferative (McCollum et al., 2012; Ni et al., 2012). The schematic representation was present in Fig. 1-2.
6
Fig. 1-2. Schematic representation of the renin-angiotensin system (RAS) cascade. The significant counterregulatory axes of the RAS are composed by ACE-Ang II-AT1R and ACE2-Ang 1-7-Mas. ACE, angiotensin-converting enzyme; Ang II, angiotensin II; AT1R, Ang II type 1 receptor; AT2R, Ang II type 2 receptor; ACE2, angiotensin-converting enzyme 2; Ang 1-7, angiotensin 1-7; Mas, Ang 1-7 receptor. [Wang et al., 2012]
1-3. Angiotensin converting enzyme II
ACE2 was cloned as a first homolog of human ACE and mapped to the X chromosome by two independent research groups in 2000 (Donoghue et al., 2000; Tipnis et al., 2000). The ACE2 is an 805 amino acid zinc-metallopeptidase and type I integral membrane glycoprotein encoded from 18 exons with a molecular weight of approximately 120 kDa (Turner and Hooper, 2002), it is predominantly observed in the heart, kidneys and testes (Tipnis et al.,
7
2000) such as cardiomyocytes (Gallagher et al., 2008), luminal surface of tubular epithelial cells (Donoghue et al., 2000; Tipnis et al., 2000) and adult Leydig cells (Douglas et al., 2004). In addition, ACE2 also had been confined at a lower level in a wide variety of tissues
including the brain (Xia and Lazartigues, 2008; Xu et al., 2011), liver (Lambert et al., 2008; Pereira et al., 2009) and lung (Kuba et al., 2006; Imai et al., 2008).
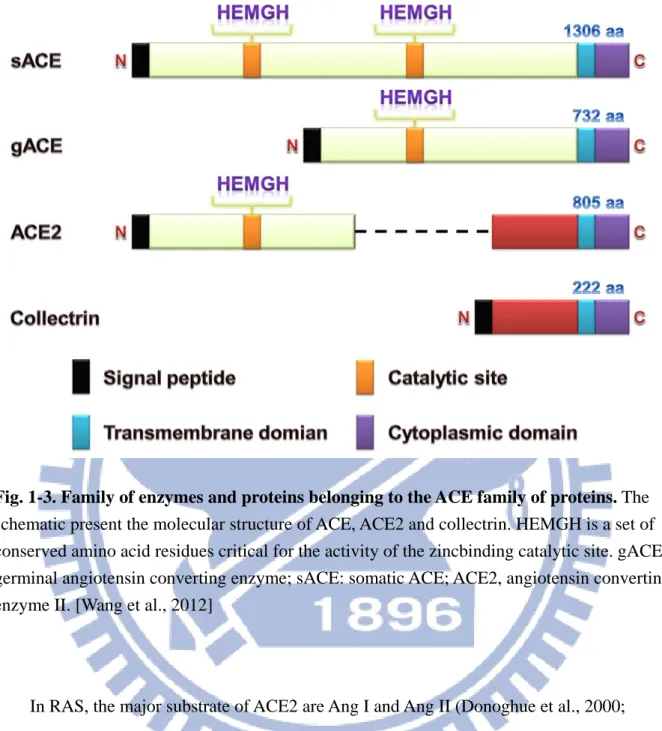
In molecular structure, the human ace2 gene comprise 18 exons, the first 12 exons of ace2 is similar to the first 11 exons of the ace gene. Moreover, the zinc-binding motif
(HEMGH) of ACE2 is located within exon 9, compared to exon 8 of the ace gene (Donoghue et al., 2000; Tipnis et al., 2000). As like ACE, ACE2 has 2 domains of the amino-terminal catalytic domain and the carboxy-terminal domain, shares 42% sequence identity and 61% sequence similarity with the catalytic domain of ACE (Donoghue et al., 2000; Tipnis et al., 2000; Douglas et al., 2004). Unlike somatic ACE, ACE2 only contains a single catalytic site with the prototypical zinc-binding HEMGH motif, and functions as a carboxymonopeptidase removing a single C-terminal residue from peptide substrates whereas ACE acts as a
carboxy-dipeptidase (peptidyldipeptidase), removing a C-terminal dipeptide (Clarke and Turner, 2012). In addition, the carboxy-terminal domain of ACE2 shows 48% sequence identity with collectrin, which was a non-catalytic protein that has a critical role in amino acid absorption in the kidney (Danilczyk et al., 2006; Malakauskas et al., 2007), pancreatic
beta-cell proliferation (Akpinar et al., 2005) and insulin exocytosis (Fukui et al., 2005). The molecular structure of ACE, ACE2 and collectrin was present in Fig. 1-3.
8
Fig. 1-3. Family of enzymes and proteins belonging to the ACE family of proteins. The schematic present the molecular structure of ACE, ACE2 and collectrin. HEMGH is a set of conserved amino acid residues critical for the activity of the zincbinding catalytic site. gACE, germinal angiotensin converting enzyme; sACE: somatic ACE; ACE2, angiotensin converting enzyme II. [Wang et al., 2012]
In RAS, the major substrate of ACE2 are Ang I and Ang II (Donoghue et al., 2000; Turner and Hooper, 2002; Rice et al., 2004), ACE2 efficiently cleaves a single residue phenylalanine from Ang II to generate Ang 1-7, with about 400-fold higher catalytic efficiency than the conversion of Ang I to Ang 1-9 by removing the C-terminal leucine residue (Vickers et al., 2002). Furthermore, ACE2 is also a multifunctional enzyme as a monocarboxypeptidase to degrade other biological substrates such as vasoactive bradykinin (1–8) (Donoghue et al., 2000), [des-Arg9]-bradykinin (Vickers et al., 2002; Warner et al., 2004), Apelin-13 (Kalea and Batlle, 2010), Apelin-17 (Vickers et al., 2002; Oudit and
9
Penninger, 2011) and Apelin-36 (Kuba et al., 2007). ACE2 hydrolyzed apelin-13 and apelin-36 peptides with high catalytic efficiency (Vickers et al., 2002), and that apelin peptides were mediated APJ receptors to activate the G-protein coupled
seven-transmembrane-domain receptor (GPCR) family predominantly expressed to regulate cardiovascular function and fluid homeostasis in the heart and lungs (Lee et al., 2000; Kleinz and Davenport, 2005; Pitkin et al., 2010).
Ang II-ACE2-Ang 1-7 and apelin-APJ are two important peptide systems with various and fundamental cardiovascular effects, and ACE2 may prevent or supress a variety of vascular and cardiac disorders (Kalea and Batlle, 2010; Oudit and Penninger, 2011; Wang et al., 2012). The major function of ACE2 is to counter-regulate ACE activity by reducing Ang II bioavailability and increasing the vasoprotective/antiproliferative peptide, Ang 1-7 formation. As a result, ACE2 plays a crucial role in maintaining the balance between the two axes ACE2-Ang 1-7-Mas and ACE-Ang II-AT1R of the RAS, chronic and sustained imbalance may lead to pathophysiology of the cardiovascular, renal, pulmonary and central nervous systems. ACE2 is effectively control fibrosis and structural remodeling in heart (Huentelman et al., 2005; Dong et al., 2012), lung (Shenoy et al., 2010; Rey-Parra et al., 2012) and liver (Paizis et al., 2005; Osterreicher et al., 2009), and extremely beneficial for pulmonary hypertension (Ferreira et al., 2009; Li et al., 2012).
1-4. ACE2 with heart diseases and heart remodeling
In the heart damage such as hypertension, myocardial infarction (MI) and chronic heart failure (CHF) (Cohn et al., 2000), cardiac myocytes were die and been replaced by fibroblasts and collagen to form fibrous tissue, these changes are referred to as “heart remodeling” (Opie et al., 2006). Ang II is a mainly factor of the RAS, activate cardiac fibroblast functions via
10
AT1R to increase the amount of ECM in the heart (Villarreal et al., 1993; Kim et al., 1995) to induce cardiomyocyte hypertrophy, cardiac remodeling and left ventricular dysfunction (Iwata et al., 2005; De et al., 2006; Whaley-Connell et al., 2007). In the failing heart, the local Ang II concentration is increased and related to the pathological signs of heart failure (Serneri et al., 2001). ACE2 is a significant regulator in RAS to degrade Ang II to suppress the heart
dysfunction that Ang II stimulated (Bikkavilli et al., 2006).
In the heart, ACE2 had been certified a dramatic decrease with aging (Xie et al., 2006) and expressed in coronary microcirculation (Donoghue et al., 2000), macrophages (Burrell et al., 2005), myofibroblasts (Guy et al., 2008), cardiofibroblasts (Zhong et al., 2010), and cardiomyocytes (Gallagher et al., 2008). ACE2 polymorphism was also been reported that four single nucleotide polymorphisms were associated with higher left ventricular mass index, higher septal wall thickness and increased odds ratio for left ventricular hypertrophy (Lieb et al., 2006). In addition, ACE and ACE2 immunoreactivity were higher in cardiac tissue of patients with ischemic heart failure compared to normal subjects (Burrell et al., 2005), the ACE2 activity was also increased in failing human heart ventricles obtained from patients with either idiopathic dilated cardio-myopathy or primary pulmonary hypertension (Zisman et al., 2003). These results appear that ACE2 plays a significant role in cardiac diseases.
In MI rats, cardiac ACE2 mRNA expression and activity were decreased after 2-4 weeks ligation of the left coronary artery (Karram et al., 2005), increased at 4 weeks (Burrell et al., 2005) and down-regulation at 8 weeks post-MI (Ocaranza et al., 2006), these results suggest that regulation of cardiac ACE2 expression and activity varies depending on disease state and time point at which measurements are obtained.
ACE2 overexpression protects the heart from Ang II-induced hypertrophy (ez-Freire et al., 2006), MI (Der et al., 2008), fibrosis (Huentelman et al., 2005), and also improved left ventricular remodeling after experimental MI (D´ıez-Freire et al., 2006). In MI rat,
11
overexpression of ACE2 inhibited the development of early atherosclerotic lesions by suppressing the growth of vascular smooth muscle cells (Rentzsch et al., 2008) and ameliorated left ventricular remodeling and dysfunction (Zhao et al., 2010). In addition, cardiac fibroblasts infected with ACE2 lentivirus decreased the collagen production that acute hypoxic exposure induced (Grobe et al., 2007).
Loss of ACE2 worsened the pathological remodeling and progressive reduction in LV contractile function to cause systolic dysfunction and heart failure (Crackower et al., 2002; Yamamoto et al., 2006; Bodiga et al., 2011). In ACE2 null mice, transverse aortic constriction or exacerbated pressure overload stimulated cardiac Ang II and AT1R activation increased. This result reduced cardiac contractility and induced cardiac dysfunction and heart
remodeling (Gurley et al., 2006; Yamamoto et al., 2006). Furthermore, the response of Ang II stimulated via hypoxia-activation was greater in cardiomyocytes isolated from ACE2−/y mice than isolated from WT mice (Keidar et al., 2007). In the absence of ACE2, p47phox NADPH oxidase subunit plays a critical role to activate myocardial NAPDH oxidase system to
increase superoxide and activate MMPs leading to the severe adverse myocardial remodeling and dysfunction in ACE2 KO mice (Bodiga et al., 2011). Almost reports appear that ACE2 plays a protector in the heart and loss of ACE2 severely impaired cardiac function was probably related to the Ang II accumulation. The significant references were listed in Appendix 8-1.
1-5. Matrix metalloproteinases
The tissue fibrosis is caused by excessive accumulation of extracellular matrix (ECM) components, especially types I and III collagen, in various pathological manifestation diseases (LeRoy et al., 1974; Uitto et al., 1979). The balance of ECM components is maintained by
12
matrix metalloproteinases (MMPs) and MMPs inhibitors, tissue inhibitors of
metalloproteinases (TIMPs) (Clutterbuck et al., 2009). MMPs are essential components for various normal biological processes such as embryonic development, morphogenesis,
reproduction tissue resorption and remodeling (Szarvas et al., 2011), they also implicated in a number of key pathologic processes including inflammation, fibrosis, arthritis, pulmonary diseases and cancer (Amălinei et al., 2010), because of the abnormally ECM deposition that imbalance MMPs and the TIMPs caused.
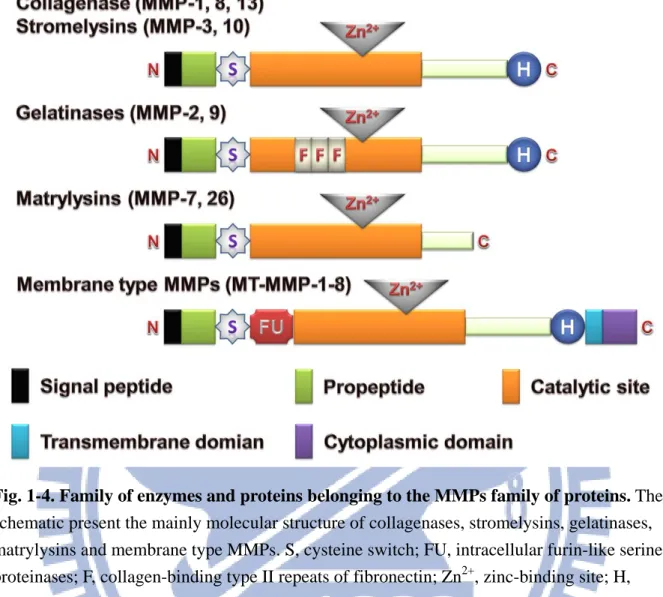
MMPs had been discovered by Gross and Lapiere in 1962, are a group of Zn2+ and calcium dependent endopeptidases of common significant peptide chain sections, however glycosylated in different amount and different locations (Sternlicht and Werb 2001). MMPs comprise a large family of protease and share several similarities in terms of their structure, regulation and function (Nagase and Woessner, 1999; Bode and Maskos, 2001). Up to now, 28 types of MMPs have been identified, and they are further divided into six major subfamilies based on structure and substrate specificity, including collagenases, gelatinases, stromelysins, matrilysins, membrane-type MMPs and other MMPs (Table 1-1; Vargová et al., 2012).
13
Table 1-1. Types of different matrix metalloproteinases and their substrate specificity
Subgroups MMPs Name Substrate
Collagenases
MMP-1 Collagenase-1
Collagen I, II, III, VII, VIII, X, and gelatin
MMP-8 Collagenase-2
MMP-13 Collagenase-3
Gelatinases
MMP-2 Gelatinase A
Collagen I, IV, V, VII, X, XI, XIV, and gelatin
MMP-9 Gelatinase B
Stromelysins
MMP-3 Stromelysin-1
Collagen II, IV, IX, X, and gelatin, α-casein, β-casein
MMP-10 Stromelysin-2
MMP-11 Stromelysin-3
Matrilysins
MMP-7 Matrilysin-1
Collagen I, II, III, V, IV, X and casein
MMP-26 Matrilysin-2
Membrane-type MMPs
MMP-14 MT1-MMP Gelatin, fibronectin and laminin
MMP-15 MT2-MMP Gelatin, fibronectin and laminin
MMP-16 MT3-MMP Gelatin, fibronectin and laminin
MMP-17 MT4-MMP Fibrinogen and fibrin
MMP-24 MT5-MMP Gelatin, fibronectin and laminin
MMP-25 MT6-MMP Gelatin
Other MMPs
MMP-12 Metalloelastase Collagen IV, elastin and gelatin
MMP-19 RASI-1 Collagen I, IV and gelatin
MMP-20 Enamelysin Collagen I, IV, and gelatin
MMP-23 CA-MMP Gelatin
MMP-26 Matrilysin-2, endometase Collagen IV and gelatin
MMP-28 Epilysin Gelatin
14
The major structure of all MMPs consists of three domains: N-terminal hydrophobic signal sequence, a propeptide domain region and a catalytic domain (Nagase, 1997; Visse and Nagase, 2003). The N-terminal hydrophobic signal sequence decides the MMPs which been released out or maintained in the cell membrane. For example, membrane-type MMPs utilize transmembrane and cytosolic domains anchoring them to the cell membrane (Nagase and Woessner 1999; Stoker and Bode, 1995). The function of the propeptide domain is to maintain latency of the MMPs until a signal for activation is given. Catalytic domain contains two ions of zinc and at least one ion of calcium bound on various amino acid residues. The catalytic domain of all MMPs contains the consensus motif HExGHxxGxxH and three histidines that coordinate with the zinc ion in the active center. Second ion of zinc and calcium are bound in inactive part of catalytic domain with high affinity, but their role remains still unknown (van Wart and Hansen-Birkedal 1990; Nagase and Woessner 1999). The molecular structure of different type of MMPs was present in Fig. 1-4.
15
Fig. 1-4. Family of enzymes and proteins belonging to the MMPs family of proteins. The schematic present the mainly molecular structure of collagenases, stromelysins, gelatinases, matrylysins and membrane type MMPs. S, cysteine switch; FU, intracellular furin-like serine proteinases; F, collagen-binding type II repeats of fibronectin; Zn2+, zinc-binding site; H, hemopexin domain. [Vargová et al., 2012]
The ECM decreased by MMPs is mainly impressed by TIMPs, MMPs and TIMPs play a critical role in maintaining the balance between ECM deposition and degradation in
physiological processes (Hulboy et al., 1997; Vu and Werb, 2000). Four TIMPs, TIMP-1, -2, -3, and -4, have been identified (Cruz-Munoz and Khokha, 2008), these TIMPs are secreted by a variety of cell lines such as smooth muscle cells and macrophages. TIMPs also involved in the process of inflammation and fibrosis, their activity is increased by PDGF and TGF-β and either increased or decreased by different ILs (Jones et al., 2003). In addition, evidences suggest that fibrotic livers have high expression of the TIMP-1 and TIMP-2, and thus the
16
combination of low expression of MMPs and high TIMPs may prevent the degradation of the fibrillar collagens.
1-6. ACE2 and gelatinase (MMP-2 and MMP-9)
ACE2 is a newly identified component of RAS and plays a negative regulator of Ang II in the RAS. Most of published papers reveal that Ang II could break the balance of MMPs expression in heart and induce heart remodeling (Brassard et al., 2005; Yaghooti et al., 2011), but the relative between ACE2 and MMPs are still unknown. In 2009, Kassiri’s group utilized the left anterior descending artery ligation and ACE2 KO mice to investigate the role of ACE2 in MI (Kassiri et al., 2009). In wild-type mice, ACE2 was persistent increased in the infarct zone of heart, ACE2-deficient was increased interferon-γ, interleukin-6, phosphorylation of ERK1/2 and JNK1/2 signaling pathways and MMP-2 and MMP-9 levels in response to MI. Loss of ACE2 also associated with the increased expression and phosphorylation of p47phox, Ang II levels, NADPH oxidase activity, and superoxide generation, which could lead to enhanced MMP-mediated degradation of the extracellular matrix in ACE2-deficient
myocardium and eccentric remodeling, increased pathological hypertrophy, and worsening of systolic performance (Bodiga et al., 2011; Patel et al., 2012).
Interesting, ACE2 overexpression inhibited cell growth, MMP-2 and MMP-9 expression, VEGFa production, and ACE and AT1R expression in human lung cancer xenografts and A549 cells in vitro (Feng et al., 2011). These evidences reveal that Ang II mediated AT1R to induce NADPH oxidase and MMP activation, AT1R blocker and Ang 1-7 supplementation inhibited NADPH oxidase and MMP activation (Kassiri et al., 2009; Bodiga et al., 2011). Furthermore, these results suggest that ACE2 serves as a protective mechanism and associated
17
with MMPs expression, especially MMP-2 and MMP-9, but the detail signal pathway still not clear. The significant references were listed in Appendix 8-2.
18
2. Research Approaches
Heart remodeling is causing of the process of heart repair and making heart structure change, which heart structure change forms heart remodeling and leads to most heart diseases such as cardiac hypertrophy, heart failure and atrial fibrillation. ACE2 is a novel element in RAS and plays a significant role against the harmful effect of Ang II induced. The Ang II has been degraded by ACE2 and form Ang 1-7 to suppress the tissue remodeling that Ang II induced. Although ACE2 inhibit Ang II induced disadvantageous effect is well known, the regulation of ACE2 in the process of heart fibrosis is unclear.
The aim of this study is investigating the regulatory mechanism of ACE2 and ace2 gene expression in cardiac cell, and the association between ACE2 and gelatinase (MMP-2 and MMP-9) in the process of heart remodeling. First, the condition and ACE2 expression in HCFs treated with angiotensin peptides, Ang II and Ang 1-7, have been detected. The signal pathway regulated ACE2 expression was also estimated. Second, the serial fragments of ace2 promoter have constructed into pGL3-basic vector to drive luciferase expression. The
luciferase expression has been detected and represents the ace2 promoter activity. These constructs are the powerful tool to identify the regulatory element within ace2 promoter and confirm the signal pathway that regulated ACE2 expression. Third, HCFs infect with the ACE2 lentivirus, TLC-ACE2, and shRNA to create ACE2 overexpression and knockout HCFs. The association between gelatinase, ACE2 overexpression and knockout has been indicated. The signal pathway and shed ACE2 activity that ACE2 overexpression and knockout HCFs treated with angiotensin peptides also estimate. The flowchart of research strategies was present in Fig. 2-1.
19
Fig. 2-1. The flowchart of research strategies. The purpose of this study is to identify the regulatory elements of ACE2 and the molecular mechanism of ACE2 regulation on heart remodeling using by angiotensin peptides, lentivirus and shRNA. First, we want to identify the regulation and the signal pathway of ACE2 expression by angiotensin peptides. Second, we using the constructs included serial fragments of ace2 promoter to identify the
transcription factors which affect ACE2 promoter activity. Finally, we using lentivirus and shRNA to establish the ACE2 overexpression and knockout HCFs and estimate the
20
3. Materials and Methods
3-1. Chemicals and reagents
The goat polyclonal IgG, glyceraldehyde-3-phosphate-dehydrogenase antibody (V-18; #sc20357), horseradish peroxidase (HRP) labeled secondary antibodies (donkey anti-goat IgG and goat anti-rabbit IgG; #sc2020 and #sc2004), and the rabbit polyclonal IgG, Ikaros
antibody (H-100; #sc13039), were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-phospho-MEK1/2 (#9121) and anti-phospho-ERK1/2 (#4370)
antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-ACE2 (#ab59351) and anti-AT1R (#ab9391) antibodies were purchased from Abcam (Cambridge, MA, USA). Alexa FluorTM 488-conjugated secondary antibody was obtained from
Invitrogen (#A11034; Eugene, OR, USA). Ang II (#H1705), Ang 1-7 (#H1715) and Ang 1-7 Mas receptor blocker A779 (#H2888) were purchased from Bachem (Merseyside, United Kingdom). The Ang II type-1 receptor (AT1R) antagonist, valsartan (Val; #1708762), was obtained from U.S. Pharmacopeia (Rockville, MD, USA), and the mitogen-activated protein kinase kinase (MEK) inhibitor (PD98059; #P215), and poly-L-lysine (0.01% solution; #P4832) were obtained from Sigma-Aldrich (St. Louis, MO, USA). A commercial medium (#2301) was obtained from (ScienCell Research Laboratories, SanDiego, CA, USA) for cell culture. The luciferase reporter vectors, pGL3-Control Vector (#E1741) and pGL3-Basic Vector (which lacks a promoter; #E1751), and the Luciferase Assay System (#E1500) were purchased from Promega (Madison, WI, USA). The ACE2 overexpression lentivirus, TLC-hACE2, and ACE2 shRNA, TRCN-46697, were purchased from Vectorite Biomedica Inc. (Vectorite Biomedica, Taipei, Taiwan) and National RNAi Core Facility Platform
(Institute of Molecular Biology/Genomic Research Center, Academia Sinica, Taipei, Taiwan), respectively. The ACE2 inhibitor, DX600 and ACE2 fluorescence substrate, Mca-APK(Dnp), were purchased from Ana Spec (Fremont, CA, USA). All other reagents were obtained from
21 Sigma-Aldrich.
3-2. Cell culture and treatments
Primary human cardiac fibroblasts (HCFs; #6300; ScienCell Research Laboratories, San Diego, CA, USA) were cultured according to our published protocol (Lin et al., 2010). In brief, the HCFs were seeded in 100-mm Petri dishes (2 x 106 cells/dish) or 12-well plates (1 x 105 cells/well) that had been pre-coated with 0.01% poly-L-lysine (Sigma), and were cultured in Fibroblast Medium (#2301; ScienCell Research Laboratories), which included 2% fetal bovine serum (#0010; ScienCell Research Laboratories). The cells were incubated at 37°C in a humidified 5% CO2 atmosphere and the culture medium as exchanged with fresh medium
every 2 days. The cells at passages 3 or 4 were used in all experiments and were placed in serum-free medium for 24 h prior to their use in further experiments.
3-3. Human ace2 constructs
Human genomic DNA was used as the template to obtain the upstream of ace2 via polymerase chain reaction (PCR) and DNA cloning. A 2.1-kb DNA fragment was obtained by PCR with primers based on the sequence for human ace2 (GenBank ID: AY217547). The sequences for the forward (Hace2-proF) and reverse (Hace2-proR) primers were
5′-AACCCTCGAGTTTCATTTAGGA-3′ and 5′-GAGCTAAGCTTCGTCCCCTGTG-3′, respectively; Xho I and Hind III sites are indicated by underlined nucleic acids in the forward and reverse primers, respectively.
The DNA fragment was then cloned into the pGL3-Basic luciferase reporter vector at the Xho I and Hind III sites to generate the –2069/+20 construct. A series of deleted DNA
22
fragments of the upstream region of ace2 were obtained by PCR using the plasmid DNA of the –2069/+20 construct as template with the specific recognition primer pairs (Table 3-1). These deleted DNA fragments were also cloned into the pGL3-Basic vector at the Xho I and Hind III sites to generate a series of deletion constructs to test the promoter activity of ace2. All of the constructs generated in this study were checked by restriction-mapping and sequencing to confirm their authenticity.
23
Table 3-1. Sequences of the primer pairs used for human ace2 promoter constructs Constructs Forward/Reverse primers (5’→3’) Promoter
region
Ampliconleng th (bp) (-2069/+20) F- AACCCTCGAGTTTCATTTAGGA
R- GAGCTAAGCTTCGTCCCCTGTG -2069 ~ +20 2089 (-1493/+20) F- GTTTCTCGAGATGCTCAAATGA
R- GAGCTAAGCTTCGTCCCCTGTG -1493 ~ +20 1513 (-1110/+20) F- TGACCTCGAGTGAGTTTTGAAT
R- GAGCTAAGCTTCGTCCCCTGTG -1110 ~ +20 1130 (-916/+20) F- TAAAGACTCGAGCAAAGTCATG
R- GAGCTAAGCTTCGTCCCCTGTG -916 ~ +20 936 (-786/+20) F- AACCCTCGAGTTTCATTTAGGA
R- GAGCTAAGCTTCGTCCCCTGTG -786~ +20 806 (-664/+20) F- GTTTCTCGAGATGCTCAAATGA
R- GAGCTAAGCTTCGTCCCCTGTG -664 ~ +20 684 (-627/+20) F- CTTGCAGTGACTCGAGATCG
R- GAGCTAAGCTTCGTCCCCTGTG -627 ~ +20 647 (-516/+20) F- TAAAGACTCGAGCAAAGTCATG
R- GAGCTAAGCTTCGTCCCCTGTG -516 ~ +20 536 (-481/+20) F- GTTGCCCAACTCGAGAGTTTC
R- GAGCTAAGCTTCGTCCCCTGTG -481~ +20 501 (-355/+20) F- AGTTCTAGACCTCGAGGGTCAC
R- GAGCTAAGCTTCGTCCCCTGTG -355~ +20 375 (-253/+20) F- AAGTGACTCGAGAGGTAAGG
R- GAGCTAAGCTTCGTCCCCTGTG -253~ +20 273 (-161/+20) F-CTGTCCTCGAGAGGATGAAC
R- GAGCTAAGCTTCGTCCCCTGTG -161 ~ +20 181 The recognition sequences of restriction enzymes, CTCGAG for Xho I in the forward primers
and AAGCTT for Hind III in the reverse primers, were shown in blue letters.
The promoter region was defined according to the position relative to the transcription start site (+1) in ACE2 mRNA sequence (GenBankno. AF_291820).
24
3-4. Transient transfection
Transient transfection was carried out according to our published protocol (Sun et al., 2005) with some minor modifications. Briefly, 2 x 105 HCFs were seeded in a 6-well culture plate one day before DNA transfection, and grown to approximately 70% confluence. The cells were washed with GIBCO Dulbecco’s phosphate-buffered saline (D-PBS) (Invitrogen, Carlsbad, CA, USA) to remove the remaining medium, then 400 μl of cell growth medium containing 4 μg of plasmid DNA mixed with 6 μl of TurboFect Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) was added gently. The DNA-transfected cells were then incubated at 37°C and under 5% CO2 in an incubator. After 24 h the cells were collected
and lysed, and assayed for luciferase activity.
3-5. Lentivirus infection
The cloning of human ACE2 in lentiviral vector and production of lenti-hACE2 viral particles was according to Huentelman et al. with slight modifications (Huentelman et al., 2005). Homo sapiens angiotensin I converting enzyme (peptidyl-dipeptidase A) 2, mRNA (cDNA clone MGC:57146 IMAGE:5297380) was used as a template with primer pairs: Spe-ACE2-F, 5’ - GAACCCACTGCTTACTGGCTTATCG - 3’; and Spe-ACE2-R, 5’ - GCTGGCAACTAGAAGGCACAGTCG - 3’ to carry out PCR amplification, the PCR production was cloned into pCR II-TOPO vector (Invitrogen, Carlsbad, CA) to obtain pACE2-TOPO. The complementary DNA encoding human ACE2 in pACE2-TOPO was subcloned into VBI-TLC vector using the SpeI sites to obtain TLC-ACE2 clone and produced lenti-hACE2 viral particles by Vectorite Biomedica Inc.
A pLKO.1-shRNA plasmid encoding a short hairpin RNA (shRNA) with sequences targeting human ACE2 was introduced into HEK293T cells with lentiviral packaging vectors
25
pMD.G and pCMV 8.91 by National RNAi Core Facility, Taiwan. The RNAi Consortium Numbers (TRCNs) and sequences of this shACE2 are 5’-GCCCTTATTTACCTGGCTGAA-3’ (TRCN0000046693); 5’-GCCCAAATGTATCCACTACAA-3’ (TRCN0000046694);
5’-GCAAAGTTGATGAATGCCTAT-3’ (TRCN0000046695); 5’-GCTGGACAGAAACTGTTCAAT-3’ (TRCN0000046696) and 5’-GCCGAAGACCTGTTCTATCAA-3’ (TRCN0000046697).
The ACE2 overexpression and knockdown experiments were performed as previously described, with optimization (Lee et al., 2008; Lin et al., 2012), HCFs were infected with the collected viruses 24 h in the presence of polybrene (8 μg/ml) at different MOI. After virus infection, cells were cultured in fresh growth medium for 24 h prior to their use in further experiments.
3-6. ACE2 knockout mice
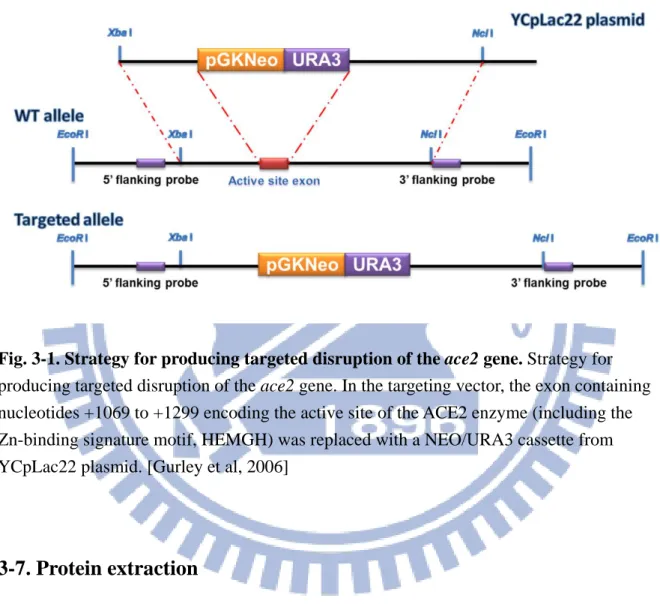
ACE2 knockout mice were established by Gurley et al. (2006). The ace2 gene consists of 18 exons, and the exon 1 was targeted by homologous recombination. The exon containing nucleotides +1069 to +1299 encoding the active site of the ACE2 enzyme (including the Zn-binding signature motif, HEMGH) was replaced with a NEO/URA3 cassette to obtain the targeting vector which disrupted ace2 gene (Fig. 3-1). The targeting construct was
electroporated into MPI1-12D ES cells that had been derived from 129/SvEvfBRTac mice and then injected into C57BL/6H blastocysts to generate chimeras.
The male chimeras were crossed with C57BL/6J female mice to obtain male hemizygous mutants and female heterozygous and homozygous females mutants. The ACE2 KO mice utilized in this study was named B6;129S5-Ace2tm1Lex/Mmcd (MMRRC:31665) and obtain from Mutant Mouse Regional Resource Centers (MMRRC). The first generation of ACE2 KO
26
mice we obtained from MMRRC was bred in National laboratory animal center (NLAC) and distinguished between hemizygous, heterozygous and homozygous mutants by DNA
genotyping.
Fig. 3-1. Strategy for producing targeted disruption of the ace2 gene. Strategy for producing targeted disruption of the ace2 gene. In the targeting vector, the exon containing nucleotides +1069 to +1299 encoding the active site of the ACE2 enzyme (including the Zn-binding signature motif, HEMGH) was replaced with a NEO/URA3 cassette from YCpLac22 plasmid. [Gurley et al, 2006]
3-7. Protein extraction
The protein extraction was performed as our previous report (Sun et al, 2008). HCFs washed twice by D-PBS and lysis by PRO-PREPTM Protein Extraction Solution (iNtRON Biotechnology, Inc., Kyungki-Do, Korea) on ice for 10 min incubation. After incubation, the cell lysis solution was transfer to 1.5 ml eppendrof tube and sonicated 3 times for 5 s with interval 10 s on ice by ultrasonic processor (UP950A, Hansor, Taichung, Taiwan). Finally the protein extraction was isolated by 13,000 rpm centrifugation at 4°C for 5 min (Biofuge primo
27
R, Sorvall, Osterode, Germany). The total amount of protein in homogeneous extract was measured by the Bradford dye binding assay (Bio-Rad Laboratories, Hercules, CA, USA) and bovine serum albumin as the standard.
3-8. Luciferase reporter assay
The luciferase assay was performed according to the manufacturer’s instructions of Luciferase Assay System (Promega). The DNA-transfected HCFs were rinsed twice with D-PBS (Invitrogen) and lysed with luciferase cell culture lysis reagent included in the kit (CCLR; Promega). Cell lysates were centrifuged at 4°C for 2 min, and the supernatants were removed and mixed with the luciferase assay reagent (Promega). Luciferase activity was measured using a single tube luminometer (Lumat LB9507, Brethold Technologies, Bad Wildbad, Germany).
3-9. Nuclear extraction
Nuclear protein was extracted using a Nuclear Extraction kit (P/N 13938; Panomics, Redwood City, CA, USA) according to the manufacturer’s protocol. HCFs (1 x 107 cells) were collected and washed twice with D-PBS, then centrifuged at 500 x g for 5 min. The cells were resuspended in 1 ml of Working Reagent and the tubes were shaken at 200 rpm on ice for 10 min. The sample was centrifuged at 14,000 x g for 3 min at 4°C and the supernatant, consisting of cytoplasmic extract, was then removed. Forty μl of Buffer B Working Reagent was added to each pellet and then the sample was vortexed for 10 s. The mixture was incubated on ice for 60 min with gentle agitation by hand every 20 min. The nuclear extract was obtained as supernatant after centrifugation at 14,000 x g for 5 min at 4°C.
28
3-10. Electrophoretic mobility shift assay (EMSA)
EMSA was performed using an EMSA Gel Shift kit (P/N 13009; Panomics). The double-stranded oligonucleotides comprising the sequence –516/–481 of ace2 were labeled with biotin. Nuclear extracts of HCFs were incubated in the Reaction Buffer for 5 min, before adding the biotin-labeled DNA probe. After incubating for 30 min at 15°C, the mixture was separated by electrophoresis in a 6% polyacrylamide gel operating at 120 V, with 0.5 x TBE as the running buffer, for 1 h. In competition assays, 66-fold molar excess of unlabeled double-stranded oligonucleotide was added to the binding reaction 5 min before the labeled oligonucleotides. After electrophoresis, the DNA-protein complexes were transferred to positively charged nylon membranes (BrightStar®-Plus; Ambion, Austin, TX, USA) by semi-dry electroblotting (HoeferTM; Amersham Biosciences, Uppsala, Sweden) and immobilized using a Spectroline Spectrolinker UV Crosslinker (Spectronics Corporation, New York, NY, USA). The membrane was blocked in 1 x Blocking Buffer, incubated with streptavidin-horseradish peroxidase for 15 min and incubated in 1 x Detection Buffer for 5 min. Working Substrate Solution (200 μl Solution I, 200 μl Solution II, and 1.6 ml Solution III) was added to develop the results (All of the aforementioned solutions were included in the Panomics Gel Shift kit). The developed bands were visualized by exposing the membrane to X-Ray film (Super Rx Medical X-Ray Film; Fujifilm, Kanagawa, Japan).
3-11. RNA isolation and quantification
Total cellular RNA was extracted using TRIzol Plus RNA Purification System
(Invitrogen) following the manufacturer’s recommendations and procedures reported by Pan et al. (Pan et al., 2008). Briefly, 1 ml of TRIzol reagent was added to 5 x 106 cells. The
29
mixture was vigorously agitated for 30 s and incubated at room temperature for 5 min. Next, 200 μl chloroform was added and the solution was centrifuged at 12,000 x g for 15 min. The aqueous phase was transferred to a clean tube, precipitated with 500 μl of isopropyl alcohol, and centrifuged at 12,000 x g for 15 min. The resulting RNA pellet was washed with 1 ml of 75% cold ethanol (−20°C) and centrifuged at 12,000 x g at 4°C for 5 min. The pellet was dried at room temperature, resuspended in 25 μl of diethylpyrocarbonate (DEPC)-treated water, and stored at −80°C. RNA was quantified by measuring the absorbance at 260 nm and 280 nm, and was electrophoresed on a denaturing 1% agarose gel. The integrity and relative amounts of RNA were evaluated using ultraviolet visualization of ethidium bromide-stained RNA.
3-12. Reverse transcription-polymerase chain reaction (RT-PCR) and Real
time polymerase chain reaction
The cDNA was synthesized using ReverTra Ace Set (Toyobo, Osaka, Japan). For cDNA synthesis, 3 μg of RNA was reverse transcribed in a total reaction volume of 20 μl with 1 x reverse transcription buffer, 0.5 mM of dNTPs, 2.5 μM of oligo-dT (TOYOBO, Osaka, Japan), 1U/μl of RNase inhibitor (TOYOBO), and 5 U/μl of ReverTra AceTM reverse transcriptase (TOYOBO). After incubation for 60 min at 42°C, the mixture was incubated for 5 min at 95 °C to denature the products. PCR primers for RT-PCR analysis were shown in Table 3-2. The PCR reactions contained 2 μl of cDNA, 2 μl of each primer (10 μM), 5 μl of 10 x PCR buffer, 2 μl of 10 mM of dNTPs, 1 μl of 5 U/μl Taq polymerase (Promega, Madison, WI, USA), and 36 μl distilled water in a total volume of 50 μl. Thermal cycler (MiniCyclerTM; MJ Research, Waltham, MA, USA) conditions were 5 min at 94°C followed by 18-36 cycles of denaturation (94°C for 30 s), annealing (55°C for 30 s), and elongation (72°C for 45 s). The resulting PCR products were visualized on 2% agarose gels stained with ethidium bromide. The stained
30
image was recorded by an image analyzer (Kodak DC290 Digital Camera SystemTM; Eastman Kodak, Rochester, NY, USA). Band intensity was quantified using densitometric analysis by ImageJTM. The relative mRNA expression of the determined gene was normalized as a ratio to GAPDH expression.
Semi-quantitative real-time (RT) PCR was performed using SYBR Green Realtime PCR Master Mix Plus (Toyobo) with 20 pM of each primer and 5 μl cDNA, in a total volume of 25 μl and monitored using Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s recommendations. Specificity of the
real-time PCR was confirmed by routine agarose gel electrophoresis and melting-curve analysis, according to a published method (Livak and Schmittgen, 2001). Expression of the GAPDH (GenBank ID: NM_002046.3) gene was used as an internal standard. The primers for ACE2 (GenBank ID: AF291820 and NM002046.3), ADAM17 (GenBank ID:
NM_003183.4) and MAPK1 (GenBank ID: NM_002745.4) were: ACE2 forward, hACE2-F, 5′-CATTGGAGCAAGTGTTGGATCTT-3′, and, ACE2 reverse, hACE2-R,
5′-GAGCTAATGCATGCCATTCTCA-3′; ADAM17 forward, hADAM17-F,
5′-CTTTCAGCATTCTTGTCCATTGTGTG-3′, and, ADAM17 reverse, hADAM17-R, 5′-GCTCAGCATTTCGACGTTACTGGG-3′; MAPK1 forward, hMAPK1-F,
5′-CAAGTCCATTGATATTTGGTCTGTAGGC-3′, and, MAPK1 reverse, hMAPK1-R, 5′-CAAGAATACCCAAAATGAGGTTCAGC-3′; GAPDH forward, hGAPDH-F, 5′-ACAGTCAGCCGCATCTTCTT-3′, and, GAPDH reverse, hGAPDH-R, 5′-GTTAAAAGCAGCCCTGGTGA-3′.
31 Table 3-2. Information of the PCR primers and condition performed in this study
Gene GenBank accession no.
Sequence of forward primer (5’→3’)
Sequence of reverse primer (5’→3’) PCR condition (cycle number)
Size of PCR product (bp) ACE2 NM021804 ACGACAATGAAATGTACCTGTTCCG TCCGATCTCTGATCCCAGTGAAG 94°C, 30 s → 55°C, 30 s → 72°C, 45 s (36 cycle) 399 AT1R NM004835 CCAAAAGCCAAATCCCACTCAAACC TCTGACATTGTTCTTCGAGCAGCC 94°C, 30 s → 55°C, 30 s → 72°C, 45 s (26 cycle) 362 GAPDH AF261085 GGTGATGCTGGTGCTGAGTA
TTCAGCTCTGGGATGACCTT 94°C, 30 s → 55°C, 30 s → 72°C, 45 s (18 cycle) 413 ACE2, angiotensin converting enzyme II; AT1R, angiotensin II type 1 receptor;
32
3-13. Immunocytofluorescence assay
HCF cells were grown overnight on 0.01% poly-L-lysine-coated coverslips (1.2 mm
diameter) and treated with Ang II or Ang 1-7 for 24 h. The cells were washed in PBS, fixed with 4% formaldehyde for 15 min, and permeabilized with 0.5% saponin (Sigma-Aldrich) for 15 min. Non-specific binding sites were blocked with 1% BSA for 30 min. The cells were then incubated with anti-ACE2 (1:100 dilu-tion) at 37°C for 1 h, followed by Alexa FluorTM 488-conjugated secondary antibody (1:200 dilution) at 37°C for 1 h in a humidified chamber. The cells were also counterstained with DAPI (1:10,000 dilution) at 37°C for 5 min in a humidified chamber. After washing in PBS, the coverslips were mounted in DakoCytomation Fluorescent Mounting Medium (DakoCytomation, Denmark A/S, Denmark), and fluorescent signals were observed using the FluoViewTM FV500 Confocal Microscope (Olympus, Tokyo, Japan).
3-14. Western blotting
The western blot for ACE2, ERK1/2 and GAPDH was carried out as our previous report (Kuan et al., 2011). Aliquots containing 30 μg protein were electrophoresed on 8%
SDS-PAGE gels and then transferred electrophoretically to polyvinylidene fluoride
membranes (Immobilon-PTM; Millipore, Bedford, MA, USA) by semi-dry electro-blotting (HoeferTM). Briefly, nonspecific binding sites were blocked by incubating the membranes in 5% non-fat milk in Tris-buffered saline. Primary antibodies against proteins were diluted 1:1,000 for ACE2, ERK1/2 and for GAPDH. The secondary antibodies were applied using a dilution of 1:2,000. Substrates were visualized using enhanced chemiluminescence detection (Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate; PerkinElmer, Boston, MA, USA) and exposing the membranes to X-ray film (Fujifilm). The bands on the film were