國立交通大學
應用化學研究所
博士論文
有機發光二極體用高分子奈米複合材料之光電
性質研究
Study on the optical and electrical properties of
polymer nanocomposites for organic light emitting
diodes
研究生:李智文 Chih-Wen Lee
指導教授:許千樹 博士 Dr. Chain-Shu Hsu
阮善法 博士 Dr. Thien-Phap Nguyen
有機發光二極體用高分子奈米複合材料之光電性質研究
Study on the optical and electrical properties of polymer
nanocomposites for organic light emitting diodes
學生:李智文 Student:Chih-Wen Lee 指導教授:許千樹 博士 Advisers : Dr. Chain-Shu Hsu 阮善法 博士 Dr. Thien-Phap Nguyen
國立交通大學
應用化學研究所
博士論文
A Thesis Submitted to Insitute of Applied Chemistry National Chiao Tung University in Partial Fulfillment
of the Requirements for the Degree of Ph.D. in
Applied Chemistry July 2008
Hsinchu, Taiwan, Republic of China 中華民國九十七年七月
有機發光二極體用高分子奈米複合材料之光電
性質研究
學生 : 李智文 指導教授 : 許千樹 博士 阮善法 博士 國立交通大學 應用化學研究所 博士班 摘要 本研究內容主要利用雙苯環取代的 PPV 聚合物(DP-PPV)衍生物 和 MEH-PPV 高分子摻混 CdSe/ZnS 量子點光電性質研究和元件陷域 能階的探討。此外我們也合成新的磷光 Pt 金屬錯合物,並探討其光 電性質與陷域能階的研究。 本研究的第一部分,我們利用兩種不同雙苯環取代的PPV聚合物 (DP-PPV)的衍生物,分別是一個苯環取代,另一個為fluorene取代基 團取代的高分子,將兩種不同比例共聚的高分子(分別為P1 和P2)和 CdSe/ZnS量子點摻混,其元件的結果可以獲得大幅的提升。在高分子 P1 中,元件的最大亮度為 697 cd/m2在,最大效率則為0.23 cd/A,在 掺混CdSe/ZnS量子點後,元件的亮度可以提升到 2794 cd/m2,效率也 提升為 0.9 cd/A。而在P2 的系統中,亮度從 3949 cd/m2提升到 8192cd/m2,效率則從0.23 cd/A提高至 1.27 cd/A。從hole-only和electron-only 元件可以發現,CdSe/ZnS量子點在元件中扮演電洞阻擋的功能,藉由 平衡電子和電洞的速率,進而增加再結合的機率,以提高元件的效能。 在本研究的第二部分,利用MEH-PPV高分子摻混CdSe/ZnS量子 點的有機發光二極體元件中,元件的提昇現象也是可以被觀察到。最 大亮度可以從65 cd/m2增加到 155 cd/m2,最大效率則從 0.005 cd/A提 高至 0.03 cd/A。利用電荷深層能階瞬間光譜儀(Q-DLTS)儀器來研究 MEH-PPV高分子摻混CdSe/ZnS量子點元件中的陷域能階和參數。從 Q-DLTS圖譜的結果中發現,CdSe/ZnS量子點的添加不但可以產生一 個新的陷域,也可以讓原本MEH-PPV高分子元件中的陷域密度降 低,進而降低電子電洞在元件中被陷域補抓的機會而增加再結合的機 率,提高元件的表現。 最後我們合成含Pt 金屬的磷光錯合物,將此材料製作成有機發 光元件,元件的驅動電壓為15 V,最大亮度為 607 cd/m2,最高效率 則為 0.28 cd/A。從Q-DLTS圖譜得知在PVK+PBD的系統中,主要有 三個不同的陷域型態,活化能分別為ET1 = 0.33 eV、ET2 = 0.4 eV和ET3
= 0.51 eV。而在掺混Pt錯合物之後,有兩個陷域依然存在(Peak I and Peak III),另一個陷域則消失了(Peak II),另外還產生了新的兩個陷域 (Peak IV and Peak V),活化能分別是ET4 = 0.25 eV和ET5 = 0.18 eV。我
們認為消失的陷域可能扮演載子的淬滅中心,而新生成的陷域可能有 利於電子和電洞的平衡,進一步提升元件的表現。
Study on the optical and electrical properties of
polymer nanocomposites for organic light emitting
diodes
Student:Chih-Wen Lee Advisers : Dr. Chain-Shu Hsu Dr. Thien-Phap Nguyen
Department (Institute) of Applied Chemistry National Chiao Tung Univeristy
Abstract
This study mainly focus on the optical and electrical properties of nanocomposites formed by blending diphenyl substituted DP-PPV derivatives and MEH-PPV polymers with CdSe/ZnS quantum dots. Furthermore, we synthesize a new Pt-contained phosphorescence material. It’s electro-optical properties and trap behavior are also investigated.
In the first part of this study, we reported the investigation of two different poly(2,3-diphenyl- 1,4-phenylenevinylene) (DP-PPV) derivatives polymers (named P1 and P2) with one phenyl and one fluorenyl substituents are used. The performance of organic light emitting devices can be improved by incorporating CdSe/ZnS quantum dots into the polymer films. The device brightness of P1 polymer can be increased from 697 cd/m2 to 2794 cd/m2. The efficiency can also be improved from
0.23 cd/A to 0.9 cd/A at 10V when CdSe/ZnS quantum dots are included. From the results of hole-only and electron-only devices, it seems that CdSe/ZnS quantum dots can act as a hole blocking layer. The balance of hole and electron mobilities results in increasing the exciton recombination.
In the second part of this study, organic light emitting diodes (OLEDs) were fabricated by using a nano-composite made by mixing MEH-PPV with CdSe/ZnS quantum-dots (QDs). The luminance and efficiency of the nano-composite devices were found to be improved. Incorporation of QDs to the polymer enhances the device luminance from 65 cd/m2 to 180 cd/m2 and the yield from 0.005 cd/A to 0.03 cd/A. The charge-based Deep Level Transient Spectroscopy (Q-DLTS) technique were used to investigate the effect of QDs on the electrical transport by determining the trap states. The trap measurements revealed that a new trap level is detected for the nano-composite devices, the result is attributed to defects of the nanoparticles. Furthermore, the density of the existing traps of the polymer decreased. We suggest that the dispersed QDs can inhibit the trapping centers, which are responsible for the quenching of luminescence.
Finally, we synthesize a new Pt phosphorescent complex. The devices based on Pt phosphorescent complex has the turn-on voltage of 15V and emit an orange light with a brightness of 607 cd/m and an external quantum efficiency of 0.28 cd/A.
2
We also use the charge based deep level transient spectroscopy (Q-DLTS) to study the trap properties and to compare the different behavior of traps with and without Pt complex. The Q-DLTS spectra reveals three different trap states in the device without Pt complex. The activation energy for each traps are E =
0.33 eV、ET2 = 0.4 eV and ET3 = 0.51 eV. Incorporating Pt complex to the
device, there are two new peaks arising (peak IV and V) and two peaks (peak I and III) remain existing . Futhermore, one peak (peak II) is disappear. We suggest that vanish trap (peak II) maybe acts as a quenching center and new peaks (peak IV and V) could balance the mobility of hole and electron to improve the device performance.
謝 誌
轉眼,博士的求學即將告一個段落。回想這段歲月,有歡笑、有 淚水、有挫折、有喜悅,我會把這些難得的感受,帶往我下個人生旅 程。 博士就讀期間,首先要感謝我的指導教授許千樹老師,許老師 對於研究上的要求與執著,確實讓我獲益良多。更感謝老師在我博四 那年,讓我到法國南特大學進行兩年的研究機會,這兩年的時間,確 實讓我獲得許多寶貴的人生經驗。同時感謝阮善法教授在我停留法國 期間,對我研究和生活上的幫忙與照顧,讓我能夠順利的完成在法國 的學業。也感謝口試委員孟心飛教授、陳信龍教授、陳文章教授和 S. Lefrant 教授對我論文的提問和指正。 實驗室就像個大家庭,實驗能夠順利的完成,實驗室的每個成員 都給了最大的幫忙。首先感謝勝雄、小百、永鑫和晉彥學長經驗的傳 承與指導,再來感謝一起打拼奮鬥的軍浩、robby、大楠、興銓、阿 慶、永明,大家彼此間的鼓勵與支持,讓這一路的苦悶的求學過程, 有了最溫暖的相伴。最後感謝學弟妹建宏、敏碩、小施、小毛、弘益、 建凱、月泙、建呈、杰修、信嵐、大pay、小 pay、又加、韋伯、迪 迪、小P、彧群、林芳、鈺婷,實驗室有了你們的加入,變得更有活 力與朝氣。還有感謝實驗室助理小燕和欣怡的幫忙,讓許多瑣碎的行政工作得以順利。
最後感謝我親愛的家人,有了你們默默的支持與鼓勵,得以讓我 在無後顧之憂的情況下順利的完成我的博士學業。
目 錄
中文摘要 ……… I 英文摘要 ……… IV 謝誌 ……… VII 目錄 ……… IX 表目錄 ……… XI 圖目錄 ……… XII 第一章 緒論……… 1.1 有機電激發光簡介……… 1 1.2 有機電激發光機制……… 5 1.3 聚對苯乙烯於發光元件上之應用……… 9 1.4 奈米材料之簡介……… 11 1.5 量子侷限效應……… 12 1.6 奈米材料在有機發光二極體元件上的應用………… 14 1.7 有機半導體缺陷簡介……… 16 1.8 有機半導體缺陷的研究……… 17 1.9 磷光元件之發展……… 19 1.10 研究動機……… 22 第二章 實驗部份……… 2.1 測試儀器……… 24 2.2 聚合物和磷光材料的合成……… 28 2.3 奈米顆粒CdSe/ZnS 合成……… 28 第三章 結果與討論……… 3.1 高分子P1 和 P2 與 CdSe/ZnS 量子點的特性分析…… 32 3.1.1 GPC 量測……… 32 3.1.2 紫外可見光譜與螢光光譜之分析……… 323.1.3 循環伏安計量分析……… 34 3.2 高分子 P1 和 P2 摻混 CdSe/ZnS 量子點發光二極體元 件製作與光電性質分析……… 37 3.2.1 發光元件的結構與製作……… 37 3.2.2 元件光電性質之量測……… 38
3.3 Hole only 與electron only 元件的製作及結構…… 43
3.3.1 Hole only 與electron only 元件之結果與討論……… 43
3.4 MEH-PPV 高分子材料和 CdSe/ZnS 量子點………… 47 3.5 MEH-PPV 高分子摻混 CdSe/ZnS 量子點元件的製作… 47 3.6 測試儀器……… 47 3.7 高分子 MEH-PPV 摻混 CdSe/ZnS 量子點光學性質和 元件特性鑑定……… 48 3.7.1 CdSe/ZnS 量子點紫外可見光吸收與螢光發光光譜… 48 3.7.2 MEH-PPV 高分子摻混 CdSe/ZnS 量子點紫外可見光吸 收與螢光發光光譜……… 48 3.7.3 MEH-PPV 高分子摻混 CdSe/ZnS 量子點電子顯微鏡 50 3.7.4 元件光電性質的量測……… 53 3.8 電荷深層能階瞬間光譜儀(Q-DLTS)研究……… 57 3.9 磷光材料Pt 錯合物的結構鑑定與光學性質研究…… 68 3.9.1 Pt 磷光錯合物的結構鑑定……… 68 3.9.2 Pt 磷光錯合物的紫外可見光光譜與螢光光譜之分析 68 3.10 有機發光二極體光電性質與缺陷研究……… 70 3.10.1 元件的結構與製作……… 70 3.10.2 元件光電性質之量測……… 70 3.10.3 電荷深層能階瞬間光譜儀(Q-DLTS)研究……… 72 第四章 結論……… 82 第五章 參考文獻……… 84 學術著作……… 89
List of Tables
Table 2-1. Glass-cleaning process……….. 26 Table 3-1. Molecular weights and molecular weight distributions
of polymers P1 and P2………... 32 Table 3-2. Optical properties of P1 and P2………. 34 Table 3-3. Band gap and energy level values of P1 and P2
polymers……… 37 Table 3-4. Device performances using pristine polymers and their
nanocomposites as the active layer……… 42 Table 3-5. The devices performances for MEH-PPV and MEH-PPV
+CdSe/ZnS composites ………. 56 Table 3-6. Summary of the trap parameters in ITO/PEDOT/ MEH-
PPV+ (CdSe/ZnS) / Al devices.……… 67 Table 3-7. Summary of the trap parameters in ITO/PEDOT/ PVK+
PBD/Al devices .………... 77 Table 3-8. Summary of the trap parameters in ITO/PEDOT /PVK +
List of Figures
Fig. 1-1. Small molecular OEL device prepared by Tang et al.….. 2
Fig. 1-2. Structures of some common small molecules…………... 3
Fig. 1-3. PPV 元件結構………... 4
Fig. 1-4. Structures of some common polymer materials..……….. 5
Fig. 1-5. Structure of a single layer OLED device.……….. 6
Fig. 1-6. 激發態電子的緩解機制………... 8
Fig. 1-7. 多層元件結構………... 9
Fig. 1-8. The synthetic route of precursor approach in PPV.…… 10
Fig. 1-9. DP-PPV 和其衍生物.……….……….. 11 Fig. 1-10. 表面原子數與內部原子數的比值關係圖………... 12 Fig. 1-11. 金屬與半導體之塊材及奈米材料的能帶結構圖……... 13 Fig. 1-12. 奈米材料量子井示意圖………... 14 Fig. 1-13. 奈米材料的尺寸與發光波長變化圖。由左而右分別代 表InAs、InP、CdSe 奈米晶粒,粒徑尺寸愈小發光波 長愈短………... 15 Fig. 1-14. PPV 能階圖和陷域分布情形……….. 18 Fig. 1-15 螢 光 與 磷 光 的 放 光 機 制 OLED 元 件 放 光 機 制………... 19 Fig. 1-16. PtOEP 化學結構……….. 20
Fig. 1-17. The structure of Iridium complex materials……….. 20
Fig. 1-18. 不 同 重 金 屬 磷 光 材 料 之 能 量 轉 移………... 21
Fig. 2-1. Priciple of Q-DLTS instrument………. 28
Fig. 2-2. Chemical structure of polymer P1 and P2………. 30
Fig. 2-4. Synthesis of Pt-Fluorene comple……… 31
Fig. 3-1. UV-vis absorption and PL spectra of P1 in solution and thin film state……... 33
Fig. 3-2. UV-vis absorption and PL spectra of P2 in solution and thin film state………. 33
Fig. 3-3. UV-vis absorption and PL spectra of CdSe/ZnS quantum dot……….. 34
Fig. 3-4. Cyclic voltammograms of P1……… 36
Fig. 3-5. Cyclic voltammograms of P2……… 36
Fig. 3-6. PEDOT/PSS structure……… 37
Fig. 3-7. L-V curves for the devices of P1 and P1+CdSe/ZnS…… 39
Fig. 3-8. Y-V curves for the devices of P1 and P1+CdSe/ZnS…… 40
Fig. 3-9. EL spectra for the devices of P1 and P1+CdSe/ZnS……. 40
Fig. 3-10. L-V curves for the devices of P2 and P2+CdSe/ZnS…… 41
Fig. 3-11. Y-V curves for the devices of P2 and P2+CdSe/ZnS…… 41
Fig. 3-12. EL spectra for the devices of P2 and P2+CdSe/ZnS……. 42
Fig. 3-13. Current density versus voltage for P1 and P1+QDs…….. 45
Fig. 3-14. Current density versus voltage for P2 and P2+QDs…….. 45
Fig. 3-15. log μ versus electric field for P1 and P1+QDs………….. 46
Fig. 3-16. log μ versus electric field for P2 and P2+QDs………….. 46
Fig. 3-17. Energy diagram of P1 、 P2 and QD……….. 47
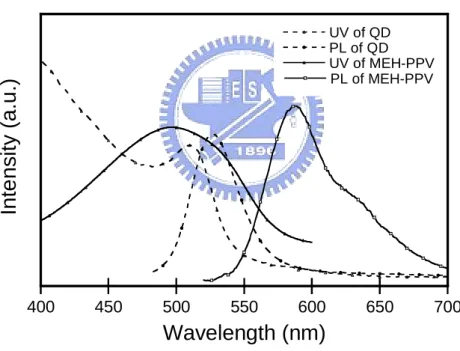
Fig. 3-18. UV-Vis absorption and PL emission spectra of CdSe/ZnS………... 48 Fig. 3-19. UV-Vis absorption and PL emission spectra of MEH-
PPV and CdSe /ZnS……….. 49
Fig. 3-20. UV-Vis absorption and PL emission spectra of MEH - PPV + CdSe / ZnS composites in film state………..
Fig. 3-21. SEM images of (a) CdSe/ZnS………... 51
Fig. 3-21. SEM images of (b) MEH-PPV+ CdSe /ZnS………. 51
Fig. 3-21. TEM images of (c) CdSe/ZnS………... 52
Fig. 3-21. TEM images of (d) MEH-PPV+ CdSe /ZnS………. 52
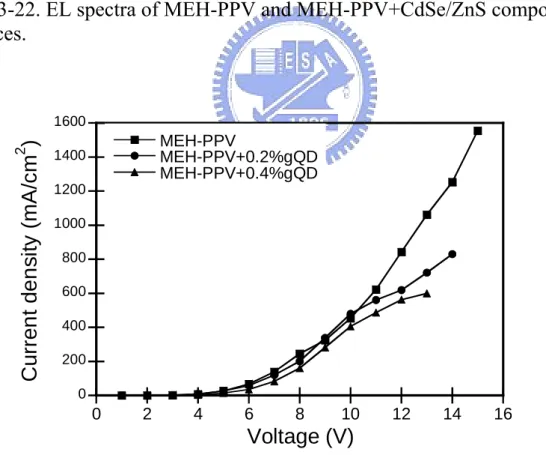
Fig. 3-22. EL spectra of MEH-PPV and MEH-PPV+CdSe/ZnS composites devices……… 54
Fig. 3-23. Current density-voltage curves of MEH-PPV and MEH- PPV+ CdSe /ZnS composites devices………... 54
Fig. 3-24. Luminance-voltage curves of MEH-PPV and MEH- PPV+CdSe/ZnS composites devices………. 55
Fig. 3-25. Yield-voltage curves of MEH-PPV and MEH-PPV + CdSe/ZnS composites devices……….. 55
Fig. 3-26. Energy level diagram of device structure ITO/PEDOT/ MEH-PPV + (CdSe/ZnS)/Al……….. 56
Fig. 3-27. Q-DLTS spectra measured on a ITO/ PEDOT:PSS/MEH-PPV/ Al device using an offset V0 = 0V, a charging voltage ΔV = + 1V for different charging time from 1 ms to 1 s at T=300 K………. 58
Fig. 3-28. Decomposition of the Q-DLTS spectra recorded in MEH-PPV diode at 300 K obtained with a charging time tc = 1 s and a charging voltage ΔV = + 1 V…………... 59
Fig. 3-29. Q-DLTS spectra measured on a ITO/ PEDOT:PSS/MEH-PPV/Al device using a charging voltage ΔV = + 1V and charge time time tc = 1 s for different temperature from T=270-320 K……….. 59 Fig. 3-30. Peak I obtained from the Q-DLTS spectra recorded in a
ITO/ PEDOT:PSS/MEH-PPV/Al diode for different temperatures in the range 270 - 320 K with a charging
time tc = 1 s, a charging voltage: ΔV = + 1 V………… 60 Fig. 3-31. Peak II obtained from the Q-DLTS spectra recorded in a
ITO /PEDOT:PSS/MEH-PPV/Al diode for different temperatures in the range 270 - 320 K with a charging time tc = 1 s, a charging voltage: ΔV = + 1 V………… 60 Fig. 3-32. Peak III obtained from the Q-DLTS spectra recorded in a
ITO /PEDOT:PSS/MEH-PPV/Al diode for different temperatures in the range 270 - 320 K with a charging time tc = 1 s, a charging voltage: ΔV = + 1 V………… 61 Fig. 3-33. Arrhenius curve for Peak I of the
ITO/PEDOT:PSS/MEH-PPV/Al device……… 61 Fig. 3-34. Arrhenius curve for Peak II of the
ITO/PEDOT:PSS/MEH-PPV/Al device……… 62 Fig. 3-35. Arrhenius curve for Peak III of the
ITO/PEDOT:PSS/MEH-PPV/Al device……… 62 Fig. 3-36. Q-DLTS spectra measured on a ITO/
PEDOT:PSS/MEH-PPV + 0.2 % QD/Al device using a charging voltage ΔV = + 1V and charge time time tc = 1 s for different temperature from T=270-320 K……... 63 Fig. 3-37. Decomposition of the Q-DLTS spectra recorded in
MEH-PPV+0.2% QD diode at 300 K obtained with a charging time tc = 1 s and a charging voltage ΔV = + 1V……….. 63 Fig. 3-38. Peak V obtained from the Q-DLTS spectra recorded in a
ITO / PEDOT:PSS/MEH-PPV+0.2%QD/Al diode for different temperatures in the range 270 - 320 K with a
charging time tc = 1 s, a charging voltage: ΔV = + 1V…. 64 Fig. 3-39. Arrhenius curve for Peak V of the
ITO/PEDOT:PSS/MEH-PPV + 0.2 %QD/Al device…… 64
Fig. 3-40. Q-DLTS spectra measured on a ITO/ PEDOT:PSS/MEH-PPV + 0.4 % QD/Al device using a charging voltage ΔV = + 1V and charge time time tc = 1 s for different temperature from T=270-320 K……….. 65 Fig. 3-41. Decomposition of the Q-DLTS spectra recorded in
MEH-PPV+0.4% QD diode at 300 K obtained with a charging time tc = 1 s and a charging voltage ΔV = + 1V……….. 65 Fig. 3-42. Peak V obtained from the Q-DLTS spectra recorded in a
ITO/ PEDOT:PSS/MEH-PPV+0.4%QD/Al diode for different temperatures in the range 270 - 320 K with a charging time tc = 1 s, a charging voltage: ΔV = + 1V. 66 Fig. 3-43. Arrhenius curve for Peak V of the
ITO/PEDOT:PSS/MEH-PPV + 0.4 %QD/Al device…… 66 Fig. 3-44. Comparision of Q-DLTS spectra recorded in pure
MEH-PPV and adding 0.2% and 0.4% QD diodes at 300 K with a charging time tc = 1 s, a charging voltage: ΔV = + 1V……… 67 Fig. 3-45. Molecular structure of Pt complex……… 68 Fig. 3-46. Molecular packing diagram of π stacking for Pt complex. 69 Fig. 3-47. UV-vis absorption and PL spectra of Pt complex in
toluene………... 69 Fig. 3-48. Luminance-yield-voltage curves for the devices of Pt
Fig. 3-49. Current-voltage curve for the devices of Pt-Fluorene complex………. 71 Fig. 3-50. EL spectrum for the devices of Pt-Fluorene complex…... 71 Fig. 3-51. Q-DLTS spectra measured on a ITO/
PEDOT:PSS/PVK+PBD/Al device using a charging voltage ΔV = + 6V for different charging time from 1 ms to 1 s at T=300 K………... 73 Fig. 3-52. Resolved Q-DLTS spectra measured on a
ITO/PEDOT:PSS/ PVK+PBD/Al device using a charging voltage ΔV = + 6V for charging time 1 s at T=300 K……… 74 Fig. 3-53. Q-DLTS spectra measured on a ITO/
PEDOT:PSS/PVK+PBD/Al device in using a charging time τc = 1s and a charging voltage ΔV = + 6V for
different temperatures in the range 270-330 K…………. 74 Fig. 3-54. Peak I obtained from the Q-DLTS spectra recorded in a
ITO/ PEDOT:PSS/PVK+PBD/Al diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s, a charging voltage: ΔV = + 6 V…………
75
Fig. 3-55. Peak II obtained from the Q-DLTS spectra recorded in a ITO/ PEDOT:PSS/PVK+PBD/Al diode for different temperatures in the range 270 - 330 K with a charging
time tc = 1 s, a charging voltage: ΔV = + 6 V………… 75 Fig. 3-56. Peak III obtained from the Q-DLTS spectra recorded in a
ITO/ PEDOT:PSS/PVK+PBD/Al diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s, a charging voltage: ΔV = + 6 V………… 76 Fig. 3-57. Arrhenius plots for Peak I to III obtained from the
Q-DLTS spectra recorded in a ITO/ PEDOT:PSS/PVK+PBD/Al diode for different temperatures in the range 270 - 330 K with a charging voltage: ΔV = + 6 V……….. 76 Fig. 3-58. Q-DLTS spectra measured on a ITO/
PEDOT:PSS/PVK+PBD+Pt complex/Al device using a charging voltage ΔV = + 6V for different charging time from 1 ms to 1 s at T=300 K………. 77 Fig. 3-59. Resolved Q-DLTS spectra measured on a
ITO/PEDOT:PSS/ PVK +PBD+Pt complex/Al device using a charging voltage ΔV = + 6V for charging time 1 s at T=300 K……….. 78 Fig. 3-60. Q-DLTS spectra measured on a ITO/
PEDOT:PSS/PVK+PBD+Pt-complex/Al device in using a charging time τc = 1s and a charging voltage ΔV = +
6V for different temperatures in the range 270-330 K….. 78 Fig. 3-61. Peak I obtained from the Q-DLTS spectra recorded in a
Pt-Fluorene complex diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s, a charging voltage: ΔV = + 6 V……… 79 Fig. 3-62. Peak III obtained from the Q-DLTS spectra recorded in a
Pt-Fluorene complex diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s, a charging voltage: ΔV = + 6 V……… 79 Fig. 3-63. Peak IV obtained from the Q-DLTS spectra recorded in
a Pt-Fluorene complex diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s,
a charging voltage: ΔV = + 6 V………. 80 Fig. 3-64. Peak V obtained from the Q-DLTS spectra recorded in a
Pt-Fluorene complex diode for different temperatures in the range 270 - 330 K with a charging time tc = 1 s, a charging voltage: ΔV = + 6 V……… 80 Fig. 3-65. Arrhenius plots for Peaks obtained from the Q-DLTS
spectra recorded in a ITO/PEDOT:PSS/PVK+PBD+Pt complex/Al diode for different temperatures in the range 270 - 330 K with a charging voltage: ΔV = + 6 V……… 81
第一章 緒論
1.1 有機電激發光簡介
有機電激發光(Organic Electroluminescence, OEL)的研究肇始 於1950 年早期,1953 年Bernanose等人對分佈於高分子中的有機染料 (Acridine或Quinacrin)施以交流電壓觀察到了發光現象 [1],當時他 們解釋此發光原理應類似於傳統III-V或II-VI族元素所組合的薄膜式 電激發光板(Thin film electroluminescence panel),例如硫化鋅 (ZnS)。其後於 1963 年Pope等人於Anthracene晶體兩端跨接 400 伏 特以上之高電壓,觀察到發光現象,不過其發光強度仍低 [2]。至 1965 年 ,Helfrich 和 Schneider 利 用 含 有 AlCl3-Anthracene ( 陰 極 ) 和
Na-Anthracene(陽極)的電解質溶液成功的製備出高亮度的發光元件 [3-4],並成為第一篇有機電激發光的專利文獻。但受限於當時的設備 與製作方法,製備出來的單晶層厚度較厚,一般仍需要超過100 伏特 的電壓才能將其驅動發光,無法成為優良的EL元件,所以其後十數 年間雖然有人陸續利用Anthracene單晶製作元件,並繼續從事相關發 光機制、電荷轉移、注入電流及量子效率(Quantum Efficiency)的研 究,但距離實用階段仍有很大的差距。直到 1979 年Roberts等人以 Langmuir- Blodgett技術製造Anthracene衍生物的元件 [5],利用重複 多次的單層分子成膜,大幅降低有機發光層的厚度,連帶地使驅動電 壓下降,才使得有機電激發光研究開始步入實用化的階段。
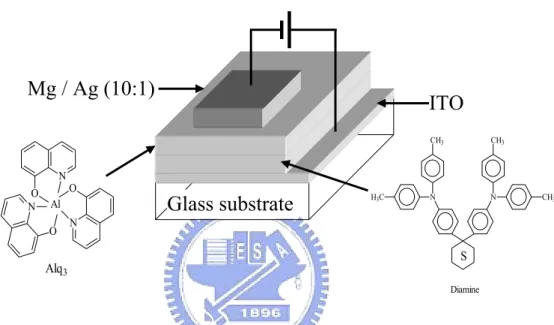
1987 年美國Eastman Kodak公司的C. W. Tang和S. A. Vanslyke利 用真空蒸鍍非晶系(Amorphous)有機薄膜的技術以及創新的異質介 面(Hetero-junction)多層有機薄膜之元件結構 [6] (如Fig. 1-1 所 示),以氧化銦錫(Indium-tin oxide, ITO)及鎂銀合金(Mg-Ag alloy) 分別當作陽極及陰極,芳香烷胺類(Aromatic diamine)作為電洞傳 輸層(Hole transport layer),8-Hydroxyquinoline aluminum(Alq3)
當作發光層(Emission layer),發表了第一個高亮度的有機薄膜發光 元件。他們大幅地改善了有機電激發光元件的特性(操作電壓<10 Volt,量子效率>1%)及穩定性,成為第一個接近實用的小分子真空 蒸鍍OEL元件,也因此激起了世人對OEL的興趣,並廣泛積極地投入 研究。 Glass substrate Mg / Ag (10:1) N O N O N O Al Alq3 N N H3C C3 CH3 CH3 H Diamine S ITO
Fig. 1-1. Small molecular OEL device prepared by Tang et al.
該元件的特徵是採用具有孤電子對(Lone electron pair)的芳香 烷胺類做為電洞傳輸層,增加電洞的傳輸效果,另採用能吸引電子的 鋁錯合物Alq3做為電子傳輸層(Electron transport layer)兼發光層,
將載子封閉在有機層中,使再結合(Recombination)效率大幅提昇, 而且有機層膜厚只有1000 Å,才得以實現以 10 Volt低電壓驅動,得 到1000 cd/m2以上的劃時代亮度。從Pope等人至柯達公司所用的材料 以有機小分子為主(見Fig. 1-2)。 在有機高分子 OEL 元件的發展方面,1982 年 Patridge 等人已利 用旋轉塗佈方式(Spin-coating)製作出第一個高分子的 OEL 元件 [7],當時所利用的材料是 Poly(N-vinylcarbazole)(PVK)。但真正讓
有機高分子用於OEL 受到重視的則是 1990 年英國劍橋大學卡文迪西 實驗室(Calvendish Lab.)所發表,利用 Poly(p-phenylene vinylene) (PPV)所製造的 OEL 元件[8],結構如 Fig. 1-3 所示。
N N
Hole Transport Materials
TPD
N N
NPB
Electron Transport / Emitting Materials
N O N O N O Al N O Be O N Alq3 BAlq BeQ2 DPVBi N N N N N N N HTM-1 MTDATA N O N O Al O
Fig. 1-3. PPV 元件結構 他們利用前驅物法(Precursor route)將高分子前驅物塗佈於導電 玻璃表面,再經過熱脫去反應製成了共軛聚合物,並鍍上鋁金屬做為 陰極,製作出第一個以共軛高分子為發光材料的 OEL 元件,引發了 第二波的OEL 元件的研究熱潮。該元件之驅動電壓為 14 V,量子效 率 為 0.05% 。 劍 橋 大 學 更 於 1992 年 成 立 Cambridge Display Technology(CDT)公司,致力於發展高分子發光材料與元件技術。 自此Kodak 與 CDT 各自成為發展小分子與高分子 OEL 研究的兩大巨 擘。常見的OEL 高分子材料結構如 Fig. 1-4 所示。
S R N CH2 CH R R PPP PPV PT PVK PF n n n n n
Fig. 1-4. Structures of some common polymer materials.
1.2 有機電激發光機制
OEL 元件本質上為一固態半導體元件,其所使用的材料是有機 半導體而非傳統的無機半導體。傳統的發光二極體是利用無機材料 III-V A 族或 II-VI A 族化合物半導體所製成,以原子為基本單位;而 大部分的有機半導體則是由所謂的“共軛分子"或“共軛高分子" 所構成,由於分子間的交互作用遠較原子間之交互作用為弱,因此有 機半導體通常帶著強烈的分子本身的色彩。共軛高分子在聚合物主鏈 上含有大量的π電子,在主鏈中形成不定形位移(Delocalization), 此不定形位移被認為是影響共軛高分子能階差(Band-gap)之主要因 素,也進而影響其光電性質。在共軛高分子中又以 PPV(見 Fig. 1-4) 系統最常做為有機發光二極體的發光材料,將 PPV 薄膜夾於金屬陰 極和陽極之間(見下頁Fig. 1-5),整個元件製作在透明基板如玻璃 或 PET(塑膠基板)上 [9],透明陽極通常是由 ITO(摻雜錫之氧化 銦)所構成,陰極則選用功函數(Work function)較小的金屬如鈣或 鋁,當一順向偏壓加諸於陽極和陰極之間時,電洞和電子分別自陽極和陰極注入有機半導體之最高填滿分子軌域(Highest occupied molecular orbital , HOMO ) 與 最 低 未 填 滿 分 子 軌 域 ( Lowest un-occupied molecular orbital,LUMO),二者在有機薄膜中傳導而相 遇,經由再結合形成高能態的激子(Excition)。當激子由激發態降 回基態時,伴隨輻射性衰變(Radiative decay)的方式而產生光子 (Photon),透過透明基板而發光。
Fig. 1-5. Structure of a single layer OLED device.
激子緩解(Relaxation)的過程如 Fig. 1-6 所示,大概可分成下列 幾種過程 :
(1) 振動鬆弛(vibrational energy relaxation):
分子被激發至電子激發能(S1)的某一振動激發態上,緊接著由於 分子間或與溶劑分子的相互碰撞而以熱的方式耗散其部份能量,從振 動激發態鬆弛到S1的最低振動態上,此一過程即為振動鬆弛。振動鬆 弛發生的時間大約在 10-14~10-12s的量級,小於螢光輻射躍遷的壽命 (10-8),所以螢光輻射躍遷的始態幾乎都是S1 的最低振動態。 (2) 內轉換(internal conversion,IC) 激發態分子經過非輻射躍遷耗散能量而落回相同自旋度低能態
程。內轉換發生的速率為 10-12s左右,由於它的存在,我們很難觀察 到由S2以上的激發單重態至基態的螢光輻射躍遷,絕大多數的螢光躍 遷是S1ÆS0。此外,螢光和內轉換是相互競爭的,一個分子的螢光性 能好壞,不但決定於螢光發射速率常數(kf),還受到內轉換速率常數 (kic)所控制。 (3) 外轉換(external conversion) 指激發分子會因與未激發分子或溶劑分子等其它分子間相互作 用而進行能量轉移,此時能量轉移以熱或其他型式釋出。 (4) 系統間跨越(intersystem crossing,ISC) 若S1與T1能階交疊或有很好的耦合,電子則可能改變其自旋方向 進入T1,此過程即為系統間跨越。於T1的電子以輻射方式回到基態的 過程即為磷光(phosphorescence)。如果兩能階的耦合較小,則大部份 激發分子仍將在S1態內鬆弛,最終以螢光(fluorescence)或內轉換的方 式回至基態。 單重態激子經輻射性衰變回到基態而放出螢光,三重態激子則會 以磷光或是非輻射方式衰退,不會放出螢光。然而形成單線態激子的 機率為四分之一,形成三重態激子的機率為四分之三,因此理論上 OEL 的內部量子效率(Internal quantum efficiency)最大值為 25%。
Fig. 1-6. 激發態電子的緩解機制 OEL 材料其電子和電洞的遷移率(mobility)並不相同,當電子和 電洞注入後,電子和電洞再結合的區域會比較靠近某一個電極,當在 結合區離電極太近,電荷容易被金屬面驟熄(quenching),因而大幅降 低發光效率,故元件的設計上會採用多層膜的設計使再結合區遠離電 極。這種多層結構的優點是能夠藉由其他傳輸層來平衡電子和電洞的 遷移率,以修正再結合的區域。此外,由於傳輸層之間的介面能障 (energy barrier),在適當的電場下,電子和電洞會停留在這個介面之 間,使得再結合機會增加,而提高EL 效率。
為使電子和電洞躍遷時所跨越之能障最小,製作元件時會考量介 面之間能階的匹配,而進行多層結構的蒸鍍,如Fig. 1-7 所示。 Fig. 1-7. 多層元件結構
1.3 聚對苯乙烯(poly(p-phenvylene vinylene))於發光元
件上之應用
近十年來,高分子有機發光二極體在材料的研發上有相當多的研 究,其中PPV由於成膜性佳及具有高螢光的特性,因此目前有相當多 以PPV作為發光層的文獻報導。PPV 能階差( band gap )大約是2.5 eV,最大放射波長為551 nm,是屬於一個黃綠光的螢光材料。由於 其為一不可溶之材料,故一般製備方法為使用Wessling method前驅 物法[10]。其方式為先將可溶性的前驅物高分子(precursor polymer) 旋轉塗佈於基材上,再進行加熱脫去反應而形成所需的共軛高分 子,如Fig. 1.8所示。由於前驅物轉換成PPV的熱處理過程中,若有 氧氣的存在,則會產生化學缺陷,使PPV的發光效率變差[11]。為 增加其應用性,美國加州大學的Heeger等人合成雙邊烷氧基團取代 之MEH-PPV[12],此類聚合物可溶於一般有機溶劑,大量的簡化其製程步驟。其電激發光最長波長為 610 nm,為橘紅光,能隙為2.1eV, 也因柔軟碳鏈的導入,使得玻璃轉移溫度(Tg)較PPV低,量子效率可 達2%。原因是因為MEH-PPV 側鏈烷氧基上的氧原子上孤對電子具 推電子效應,可以使其主鏈上共軛性增加,導致PPV 高分子共軛主 鏈上的電子密度增加,即可增加PPV高分子主鏈上共軛系統之電子共 軛性,而降低PPV之最高階填滿分子軌域(HOMO)與最低階未填滿分 子軌域(LUMO)之間能隙,使發光波長紅位移至橘紅光到紅光波長範 圍[13]。 ClH2C CH2Cl Cl Cl S S n a b, c, d e Cl S 1 o a) tetrahydrothiophene , MeOH, 65
b) NaOH, MeOH/H2O or Bu4NOH, NaOH, 0 C
c) neutralization(HCl) d) dialysis(water) e) 180 - 300 C , vacuum, 12 Hr o C o
Fig. 1-8. The synthetic route of precursor approach in PPV
另外,以芳香族取代基為側鏈導入PPV,不僅可降低共軛長度, 具可溶性,同樣也避免主鏈間堆疊而降低發光效率。Hsieh於1998年 發表2,3-diphenyl為側取代的PPV衍生物(如Fig.1-9),此高分子在製成 薄膜後的光學量子效率高達65% [14]。這顯示芳香族的立體效應可避 免PPV主鏈的堆疊,也降低因堆疊而產生的self-quenching。
Fig. 1-9. DP-PPV 和其衍生物
1.4 奈米材料之簡介
所謂的奈米(Nanometer),即表示億分之一米,為介於原子、分子 與巨觀尺度之間,一般定義為直徑小於100 nm的粒子。而所謂的奈米 科技,就是將各式元件之基本構造縮小至奈米的程度。奈米有多小, 舉例來說,人髮的直徑大約是八萬奈米。由於奈米粒子體積非常小, 造成表面積與體積的比例大增,即粒子越小,裡面的原子數越少,暴 露在表面上的原子所占的比率就越高。由Fig. 1-10. 所示,隨奈米晶 粒變小其暴露於表面原子數與粒子總原子數的比率隨之增大[15],改 變了晶體自由能與其熱力學上特性,如相變能量的提升、熔點下降、 純矽半導體的能隙為1.12電子伏特,但矽奈米晶體能隙卻大於1.12電 子伏特等。 因此,當顆粒的直徑減少時,會引起他的表面原子數、表面積和 表面活性的大幅增加。同時,表面原子具有高的表面能,且不穩定, 很容易與外來的原子吸附鍵結,形成穩定的結構。因此,表面原子與 內部原子相比,具有更大的化學活性和提供大面積的表面活性原子。 對外界環境如溫度、光、濕度、氣體等十分敏感,對於環境些微的改 變,能迅速引起材料表面離子的價電子態和電子傳輸明顯的變化。例 如光的吸收明顯增加使得金奈米粒子呈現黑色,二氧化鈦奈米粒子 (光觸媒)的表面光化學反應分解有機物質等[16]。Fig. 1-10. 表面原子數與內部原子數的比值關係圖
1.5 量子侷限效應(Quantum confinement effect)
對單一原子而言,最高電子佔據軌域(HOMO)和最低電子未佔據 軌域(LUMO)之間的能量差即為原子的能隙。對塊材而言,能隙則是 指價帶與傳帶之間的能量差。就混成軌域的觀念來看,當原子的數目 由一個逐漸增多時,由於能階的數目也隨之增加,故能帶與價帶之間 的能量差會逐漸減小,也就是其能隙會不斷地改變,直到原子數目增 加到某一數目之後,晶體的能階就不會再改變。而奈米晶體能隙隨著 晶體縮小而產生藍位移(blue shift)的現象,就稱做量子局限效應,如 Fig. 1-11.所示[17]。
Fig. 1-11. 金屬與半導體之塊材及奈米材料的能帶結構圖 由於電子本身具有波粒二重性,當電子的波長和奈米粒子的尺寸 可比較時,就會有量子效應產生,此時電子可視為一波動,我們可將 奈米粒子假想成一個量子井(如Fig. 1-12.),而電子被限制在其中無法 脫離,按照電子之波動方程式及恰當之邊界條件,我們可解出在此量 子井中的電子能階公式: En= n2h2/8mL2 (1) ΔE = En+1-En = (2n+1)h2/8mL2 (2) ΔE 為電子能階能量差(在半導體中當n=1 時則視為能隙),其中L 則 可視為粒徑大小。當粒徑變小時,預期能階能量差將會變大(在半導 體中可視為能隙增大),故就半導體而言,隨粒子粒徑變小,其吸收 與放光譜帶會漸往短波長偏移,而此即限量化效應在光學上所造成的 影響[18,19]。
Fig. 1-12.奈米材料量子井示意圖
.6 奈米材料在有機發光二極體元件上的應用
料。有些
e 混入 PPV 中,在不同的電壓驅動下,
化之TiO2及SiO2混入MEH-PPV
中,而提升了MEH-PPV元件的發光強度和效率[30],元件效能的提升
1
量子點(quantum dots)為在光學上具有特殊性質的奈米材 半導體量子點只要改變其粒徑大小就可以改變其發光光色[20,21],發 光的光色幾乎可以涵蓋整個可見光光譜(如 Fig. 1-13 所示),量子點的 粒徑變化從 2 nm 到 6 nm,發光光色可以從藍光變化到紅光。也因為 此種特性,使的量子點在發光二極體中有很大的應用性,其中又以 II–VI 核殼(core-shell)結構的 CdSe/ZnS 最受到矚目,此材料的發光效 率可達到 50%以上[22,23]。 Colvin 於 1994 年將 CdS 可以分別得到 PPV 或者 CdSe 的發光[24],然而此元件的效率非常低。 2002 年,Coe 等人利用旋轉塗佈摻混 CdSe 和 TPD 有機材料而得到 效率大於 1 %的量子點發光元件[25,26],類似的元件結構也被用來製 作白光的發光二極體元件[27-29]。 在 1997 年,Carter等人利用奈米奈米粒子在有機發光二極體元件中所扮演之角色及功能,至目前 為止並無較明確之結論。有些研究發現奈米粒子對於元件的電荷傳遞 並無影響[33],有些研究則觀察到奈米粒子對於元件中電荷的傳遞會 有顯著的改變[25,30,32],亦有文獻認為奈米粒子能減緩發光分子之 光氧化反應[34],進而增進其效率。因此,將奈米粒子和有機發光二 極體成結合成為具潛力之研究方向。 Fig. 1-13. 奈米材料的尺寸與發光波長變化圖。由左而右分別代表 InAs、InP、CdSe 奈米晶粒,粒徑尺寸愈小發光波長愈短。
1.7 有機半導體缺陷簡介
幾十年來共軛高分子在有機發光二極體上的應用一直被廣泛地 。電荷的移動率和電荷的補抓在共軛高分子的元件電性上扮演重 及電子電洞再結合的區域對發光元 ,主要原因是因為材料結構發生其他反應,如: 氧化、還原、 之,位於價帶(valence band)上方的 荷的時間會大於傳輸時間( tt )。因此,當 研究 要的角色。電子和電洞的移動率以 件的結果有很大的影響。其中,陷域(trap)的存在會導致電荷移動率 的降低或者扮演電子電洞再結合(recombination)或焠滅(quenching)的 中心[35]。因此對於陷域的研究與了解,在發光二極體元件有莫大的 幫助。 陷域(trap)可以定義為自由電荷的補獲中心,陷域的產生跟材料 的缺陷(defect)有關。缺陷的形成可能有兩個原因,第一 :材料的化學 結構改變 分子鏈斷裂...。第二 :在有機材料的合成或有機薄膜成膜時化學不純 物(impurities)的導入或者因為分子間彼此的反應所產生。如 :堆疊 (aggregate)或激發雙體(excimer),對於這樣的情況,缺陷的形成是很 難控制,但卻也不能夠被忽略。 陷域往往會形成一個新的能階介於半導體能隙(energy gap)之間 或者形成於金屬和有機層的介面。如果新的能階位於導帶(conduction band)下方,則可以補抓電子。反 則是補抓電洞。由於在有機半導體材料中能帶的寬度非常的窄,因此 我 們 常 用 最 低 未 填 滿 分 子 軌 域 (LUMO ) 和 最 高 填 滿 分 子 軌 域 ( HOMO )來分別取代導帶和價帶。特別在非晶態的有機薄膜中,軌 域的分布類似高斯曲線。 存在有機半導體能帶中有兩類的陷域:一為淺層陷域( shollow traps ),其主要抓住電荷的時間會小於傳輸時間( tt ),二為深層陷域 ( deep traps ),其主要抓住電 電荷被陷域在深層陷域時,則電荷將無法對傳輸的過程有所貢獻。(a) 光學研究 發光光譜是一種典型用來鑑定陷域存在有機材料中的儀器。控制 不同的條件下合成相同的材料,透過光譜的比較,進而觀察陷域的存在 述。唯有透過電性的研究才能將陷域的參數,例如 : 活化 區域, 陷域密度等。陷域的型態(電子或電洞陷域)也可 oerer 人利用熱促進電流(thermally stimulated currents, TSC)研究不同元
Campbell等人在2000年則利用 / 機 el [36,37]。 (b)電性研究 光學的研究可以提供陷域存在的證據,但是對於陷域的特性卻無 法清楚的描 能, 俘獲交錯 以進一步被鑑定出來。一般常用的儀器如下 : 如電流-電壓特性 (current-voltage characteristics) [38] 、 阻 抗 光 譜 儀 (impedance spectroscopy) [39] 、 飛 行 時 間 式 電 荷 傳 輸 特 性 量 測 (time of flight measurement) [40]、熱促進電流(thermally stimulated currents, TSC) [41] 以及深層能階瞬間光譜儀(deep level transient spectroscopy, DLTS) [42]。這些儀器的基本原理為量測被陷域捕獲而釋放的電荷,利用施 加正、負電壓,更可以進一步定義為主要陷域或者次要陷域。
1.8 有機半導體缺陷的研究
電荷的補獲在元件中對於電荷的傳遞具有重要的影響。Schw 等 件結構中高分子PPV的陷域分布[43]。深層能階瞬間光譜儀(deep level transient spectroscopy, DLTS)研究 PPV高分子中的陷域[44]。同年,R. H. Friend團隊研究元件結構為Si MEH-PPV/Au的I-V特性而定義出MEH-PPV高分子中具有五種不同 的陷域[45]。至此,許多文獻發表了利用TSC和DLTS儀器來研究有 分子的陷域情形[46-48]。
如 利 用 電 荷 - 深 層 能 階 瞬 間 光 譜 儀 (charge-based deep lev transient spectroscopy, Q-DLTS)研究PPV材料的陷域性質 [49],可以
量測到PPV材料的活化能(activation energies)、俘獲交錯區域(capture cross section)和陷域濃度(traps concentrations)。在某些情況下,可以被 分 辨 出 介 於 多 數 和 少 數 兩 者 間 的 電 荷 陷 域 。 主 要 元 件 結 構 為 ITO/PPV/MgAg 的發光二極體,有兩類的電荷陷域可以被發現。元 件為ITO/PPV/MgAg的能階圖如Fig. 1-14 所示,第一類的活化能( EI ) 在 0.49-0.53 eV範圍以及俘獲交錯區域在 10-16-10-18 cm2級數間,此能 階可以被視為接受型的多數電荷陷域(如:電洞)。第二類的活化能( EII ) 在 0.40-0.42 eV範圍以及俘獲交錯區域在 10-19級數間,此能階可以被 視為少數電荷陷域(如:電子)。因此,第二類的陷域形式在正向電壓 下會限制了少數電荷(指電子)注入PPV發光層,而降低電激發光效率。 Fig. 1-14. PPV 能階圖和陷域分布情形
當電子、電洞在有機分子中再結合時,會因電子自旋對稱方式不 ,產生兩種不同的激發狀態,一種是非自旋對稱(anti-symmetry)的 方式回到基態。另外一種則是自旋對稱 (spin auli exclu 同 單重激發態,是以螢光的 -symmetry)的三重激發態,是以磷光的形式放光回到基態。 電子由單重激發態回到基態的過程是被允許的,所以一般較容易 觀察到分子產生螢光。但從三重激發態回到基態的過程,會在基態產 生一對自旋方向相同的電子,因此違反了鮑利不相容原理(p sion priciple),所以在常溫下是很難觀察到磷光,螢光和磷光分 別的緩解過程如 Fig. 1-15 所示。 Fig. 1-15. 螢光與磷光的放光機制 OLED 元件放光機制 在 ature」 0]所發表的成果指出,將高效率的磷光材料(PtOEP) (如Fig. 1-16 所 示)摻雜在主體材料中,能量從主體傳遞到磷光材料中發光,可以將 外部 1998 年,美國普林斯頓大學(Princeton) Forrest小組於「N [5 量子效率提高到 4%,從此開啟了磷光發光材料的熱潮。具有高 效率的磷光摻雜物(dopant)多是含重原子的材料,這是由於重原子可
使得電子自旋-軌道偶合(spin-orbital coupling)作用增強,有效地混合 單重態與三重態,增加S1ÆT1系統間穿越的機率,此現象即為”重原
子效應(heavy atom effect)。除此之外,重原子的加入也可以減低三重 態的放光生命期。
Fig. 1-16. PtOEP 化學結構
1994 年Forrest and Thompson[51]發表利用Ir(ppy)3做出綠色的磷
光材料。並且利用選則不同之芽基(ligand),可發射出紅、綠、藍三 原色光[52,53],如Fig. 1-17.中所示。 O O N Ir
Fig. 1-17. The structure of Iridium complex materials 2 O F F O N Ir 2 O O N Ir S 2 Ir 2 O O N Ir S (ppy)2(acac) (green)
Ir(btp)2(acac) (red)
Ir(thp)2(acac) (green) Ir(Fppy)
2(acac) (blue) 2 Irppy3 (green) N N Ir O N N Ir F F O 2 Firpic
並非重金屬原子都可以帶來高的元件效益,如果激子產生在 ligand上(如PtOEP結構),自旋-軌道偶合(spin-orbital coupling)效應便 不顯著而造成磷光激子半生期高達100 μs,因此在高電流密度下,造 成三重態-三重態的驟熄(T-T annihilation)以及熱形式衰退的能量損失 都會降低元件的效益。反之在Ir(ppy)3材料中,激子產生在重金屬和 ligand上,自旋-軌道偶合(spin-orbital coupling)效應明顯而使的半生期 縮短至1 μs,大幅減少因非光形式所造成的能量損失,因此可提高元 件的外部量子效率,如Fig. 1-18 所示。 Fig. 1-18. 不同重金屬磷光材料之能量轉移 磷光有機電激發光元件的結構包括電洞傳輸層、摻雜磷光材料 的 發 光 層 、 抑 止 激 發 子 擴 散 的 電 洞 阻 擋 層 和 電 子 傳 輸 層 (HTL/EML/HBL/ETL)。由於磷光的三重激發態生命期很長,三重態 激子具有較長的生命期,所以在元件中的擴散範圍較單重態電子大上 許多,可達1000Å,相較於單重態激子在元件中的擴散距離約為數十
到 100 Å。因此為了提高元件效率,必須將三重態激子限制在發光層 中,如此才能有效地將能量從主體傳遞到客體,因此需要有電洞阻擋 層(hole blocking layer,HBL)的存在[54]。作為 HBL 的材料具有極大 的游離能,可以阻擋激子的擴散,尤其當主體與客體軌道重疊弱時, 此層的功用更是重要。 磷光材料結構大多由過渡金屬及配位基組成,此類過渡金屬錯合 物常見的有三價銥錯合物和二價鉑錯合物。之所以選用此類過渡金屬 錯合物是因為這些金屬擁有d6電子組態、具較強的鍵結、long-lived 激發態和較高發光效率等特性[55],得以加強發光強度和減少非輻射 衰退。對於原子序Z=75~77 的重金屬離子而言,因為擁有較強的自旋 軌域耦合(spin-orbit coupling),增加系統間跨越能力,使得單重態激 發態跳躍至三重態激發態的機率增高,進而增加磷光放光效率。
1.10 研究動機
有機發光二極體近年來廣為學術界、工業界所廣泛研究。在共軛 高分子有機材料的部分主要以聚對苯乙烯 poly(p-phenvylene vinylene) 系列衍生物為主。而半導體奈米粒子本身的尺寸效應或光電特殊特 性,其導入共軛發光高分子之中來達到奈米粒子本身發光或者改善原 本主體高分子的發光特性已被廣泛的研究,然而奈米粒子本身扮演的 角色及功能未被清楚的解釋。 因此,本研究利用新合成的雙苯環取代之聚(1,4-仲苯基乙烯) (DP-PPV)衍生物以及 MEH-PPV 高分子摻混 CdSe/ZnS 量子點,來 探討量子點在提昇元件表現上的貢獻。並且利用不同的元件結構, 來觀察量子點對不同電荷的影響。為了更進一步探討量子點在元件 中所扮演的角色和功能,將 MEH-PPV 高分子摻混 不 同 濃 度 的 CdSe/ZnS 量子點,接著利用電荷-深層能階瞬間光譜儀(charge-based deep level transient spectroscopy, Q-DLTS)來比較摻混 CdSe/ZnS 量子點前後的光譜差異,藉以清楚的分析 CdSe/ZnS 量子點在元件中的影 響和行為。 高效能的無機磷光錯合物應用於有機電激發光元件(OLED)在近 幾年來引起廣泛的討論及研究。相對於螢光系統來說,磷光系統可同 時利用單重態及三重態激子,理論上可達到內部 100 %的量子效率。 所以本研究的另一部份為合成新的磷光材料鉑金屬(Pt)錯合物, 並將其製作成有機發光元件來探討其元件特性,接著同樣利用電荷-深層能階瞬間光譜儀( charge-based deep level transient spectroscopy, Q-DLTS )來研究此元件材料的陷域行為,以期對磷光材料的陷域情形 做更深入的了解。
第二章 實驗部份
2.1 測試儀器
為了鑑定或測試所得的聚合物及相關材料的特性,採用下列測試儀器:
2.1.1 凝膠滲透層析儀( Gel Permeation Chromatography,GPC )
使用 Series III Pump 型高效能管柱層析幫浦。偵測器為 Viscotek T50A Differential Viscometer 和 Viscotek LR125 Laser Refractometer , 管 柱 為 American Polymer Standards Corporation 所生產。樣品濃度取 2.0 mg/mL, 並使用 polystyrene ( PS ) 標準樣品製作分子量校正曲線沖提液為 THF,流 速為 1.0 mL/min,並保持於 35℃的恆溫槽中。樣品溶液以 0.2 μm 的 Nylon filter 過濾後使用。
2.1.2 紫外線與可見光譜儀 ( UV-Vis Spectrophotometer )
分別使用 HP 8453 型和 CARY 5G UV-Visible 光譜儀。用以偵測樣品 之吸收光譜,量測時樣品以溶劑溶解後置於石英盒內,或直接旋轉塗佈成 膜於石英玻璃表面量測。2.1.3 螢光光譜儀 ( Photoluminescence Spectrophotometer)
分別使用 ARC SpectraPro-150 和 Fluorolog HORIBA 型螢光光譜儀。 用以偵測樣品之放射光譜,儀器使用之激發光源為450 W 之 Xenon 燈,量 測時激發波長根據個別樣品之吸收光譜而有所不同,所得數據為光激發光 (potoluminescence,PL)光譜。
2.1.4 循環伏安計( Cyclic voltammetry,CV)
使用Autolab的ADC 164 型電位儀來記錄氧化-還原電位,將高分子溶 液塗佈於Pt上當作工作電極,以飽和甘汞電極(standard calomel electrode, SCE)當作參考電極(reference electrode),鉑(Pt)當作工作電極及對應電極 (counter electrode),以 0.1 M的 (n-Bu)4NBF4 / acetonitrile為電解質液,以 50
mV/sec的速度進行掃描。
2.1.5 穿透式電子顯微鏡 ( Transimisson Electron Microscopy )
使用 Hitachi H-9000-NAR 電子顯微鏡 ,操作電壓為 300 kV.
2.1.6 LED 元件性質的量測
在元件加以通電壓使其發光後,其放射光使用 Photo Research PR-650 Spectra Scan 分光儀收集並記錄各項光電性質。
2.1.7 ITO 玻璃 pattern 的製作
本實驗所使用的玻璃基板為Merck Display Tecnology公司之阻值為 20 Ω/ square的indium-tin oxide ( ITO )玻璃,使用時並切割為 30 ×30 mm2之正 方形。由於我們欲將所製作之元件圖形化( pattern ),故必須有以下之步 驟: (1)上光阻:本研究所使用之光阻為長春人造樹酯股份有限公司 AF5040 乾 式光阻。 (2) 曝 光:依照所需 pattern 在 300 ~ 400 nm 波長紫外光曝光 55 秒。 (3) 顯 影:以 1% ~ 2% 重量百分率濃度之碳酸鈉水溶液顯影。 (4) 蝕 刻:再將顯影過後的 ITO 玻璃基板浸入 50℃的濃鹽酸水溶液蝕刻約 30 秒。 (5) 去光阻:以 1 % ~ 3 % 重量百分率濃度之氫氧化鈉水溶液剝除光阻。 圖形化後的 ITO 玻璃,再經過 Table 2-1 的清洗步驟後,即可用來作 為發光元件的基材。
Table 2-1. Glass-cleaning process.
Cleaning step Time Detergent
10 min
H2O 10 min
NaOH(aq) 10 min D.I water 10 min
Acetone 10 min
IPA 10 min
Oven 150oC 12 hr
2.1.8 電荷深層能階瞬間光譜儀 ( Charge-based Deep Level
Transient Spectroscopy Q-DLTS )
本儀器測量配合由 InOmTech 公司所提供的 AS--MEC-02 自動化系統 來進行量測,其量測環境在真空下。
為了計算出在元件中的電荷陷域,我們利用此技術來量測。這項技術 是相似傳統的DLTS,但是利用量測釋放電荷變化(released charge variation ) 來取代量測電容(capacitance)。其原理可經由Fig. 2-1 簡單的表現,主要為在 一個很短的時間內( charging time, tc ),提供一個充電電壓( charging voltage,
△V ),目的是要去充滿材料中的陷域。然後,將偏壓設定為零,則陷域捕 獲的電荷會因熱力學因素而慢慢釋放。瞬間釋放電荷在兩個連續的時間內 ( t1, t2 )被記錄下來,則可以定義此視窗速率 τ ( rate window )為: τ = (t2-t1)/ln(t2/t1) (1)
Q-DLTS圖譜則是以電荷差( △Q = Q(t1)-Q(t2) )和視窗速率( τ )的函數 繪圖。假設瞬間電流是時間和所提供t1/t2比例定值為指數函數,則在所測 量的溫度T下電荷變化值( △Q )會在釋放時間( τm )出現最大值,從不連續 的陷域能階上因熱力釋放電荷, 沒有再陷域效應發生( re-trapping) ,可以被寫 成 : Q(t) = Q0[1-exp(en(p)t)] (2) 這裡Q0是在充滿過程的起始陷域電荷,en(p) 是電子( 電洞 )放光速率等 於 1/ τ,也可被寫成: en(p) = σΓn(p)T2exp(-EA/kT) (3)
σ是俘獲交錯區域( capture cross section ),EA是活化能( activation energy ),
和Γn(p)是載子的有效質量為一個定值。 當視窗速率( τ-1 )達到陷域放光( trap emittion )時,△Q(τ)圖譜會出現最 大值,因此,從公式(3)可以得到以溫度的函數圖形Q-DLTS圖譜為溫度的 函數圖形,並且可以計算得到σ和EA 值。再則,陷域密度NT 也可以從Q-DLTS訊號的最大值△QM被計算得到。 利用此測量技術,測量在不同溫度下的Q-DLTS圖,可以得到一系列 的釋放時間( τm )。EA和σ也可以從-ln (τT2) v.s. 1/KT圖譜的斜率和截距得 知 ,而陷域密度也可以從Q-DLTS圖譜中的最大值得到。
Fig. 2-1. Priciple of Q-DLTS instrument.
2.2 聚合物和磷光材料的合成
本 次 實 驗 所 使 用 的 聚 合 物 P1 、 P2 和 poly[2-methoxy-5(2’-ethyl hexyloxy)-1,4-phenylenevinylene] (MEH-PPV)的合成步驟如本實驗室之前的 論文所述[56]。其結構分別如 Fig. 2-2 和 Fig. 2-3 所示。 磷光材料Pt錯合物的合成步驟如Fig. 2-4 所示。以CuI為催化劑將化合 物 1 和化合物 2 混合在CH2Cl2 溶劑中,並以triethylamine (C2H5)3N為acid quencher,即可成功合成出Pt 錯合物。2.3 奈米顆粒 CdSe/ZnS 合成
硒化鎘/硫化鋅量子點製備以0.0386 g 氧化鎘(CdO)與 5.88 g HDA,5.88 g TOPO,在氣體置換後於氬氣下加熱2-3小時至320 oC以形成無色CdO-HDA錯合物,而後將反應液降溫並維持在270 oC 時,藉由快速打入0.25 g Se (in 5 ml TBP),隨反應時間不同而得到不同大小的CdSe。 等溫度自然降溫至180 oC 後,將0.316 g Zinc stearate / 0.016 g S/ TBP 前驅液(5 ml) 緩慢注入,反應1小時,最後將反應溫度降回室溫後,加入適 量的氯仿並離心後,將不溶解的固體移除。CdSe/ZnS 奈米晶體最後由甲醇 將其沈澱出來,經過離心之後可得到CdSe/ZnS奈米晶體。
M1 C6H13 C6H13 CH2Cl ClH2C H3CO O CH2Br BrH2C + M2 t-BuOK THF C6H13 C6H13 * H3CO O * * X Y P1, X=50, Y=50 P2, X=90, Y=10
H3CO
O
*
*
n
Fig. 2-3. Chemical structure of MEH-PPV polymer
N N Pt N N Pt Cl
+
H CuI ,(C2H5)3N CH2Cl2 1 2 3第三章 結果與討論
3.1 高分子 P1、P2 與 CdSe/ZnS 量子點的特性分析
3.1.1 GPC 量測
P1 和 P2 是以單體 M1 為主體,配合不同比例的 M2 做共聚合反應。 Table 3-1 列出其 GPC 的數據,從 PDI 值可以看出半透膜透析可以達到好 的高分子純化的效果。因為 M1 的分子立體阻礙比 M2 大,而 P1 所含 M1 的比例較少,因此 P1 的分子量略大於 P2。
Table 3-1. Molecular weights and molecular weight distributions of polymers P1 and P2 a b Polymer Mw Mn PDI (Mw Mn/ ) P1 762900 713000 1.07 P2 709200 611020 1.18 a:重量平均分子量 b:數目平均分子量 PDI(polydispersity):Mw/Mn,表示分子量分佈大小
3.1.2 紫外可見光譜與螢光光譜之分析
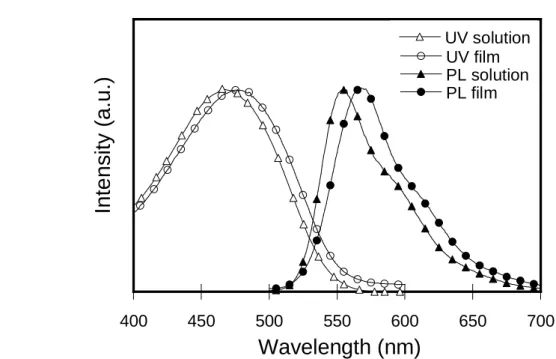
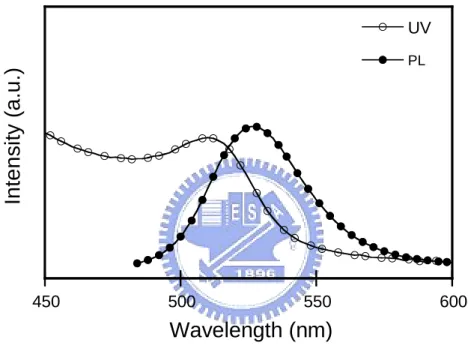
Fig. 3-1 和 3-2 分別為 P1 和 P2 的 UV 吸收和 PL 放射光譜圖。隨著 M2 的導入的比率增加,P1 最大放射有紅位移的情況也增加不少,明顯的可以 看出,因為 M2 的烷氧基之推電子效應以及 M2 的聚合產物本身是橘紅光 的材料,因此我們所得的 P1 最大放射峰都會較 P2 往長波長區域移動。 薄膜的吸收或放射位置和溶液態相比皆有紅位移的現象,是因為當分 子在薄膜狀態時,其分子與分子之間的距離較溶液態來的靠近,其堆疊 (aggregation)造成高分子鏈的能階形成簡併態(degenerency),因此薄膜時最 大放射光譜會有紅位移的現象。 Fig. 3-3 顯示 CdSe/ZnS 量子點的 UV 吸收和 PL 放射光譜圖,本次實 驗所選用的量子點為一個發橘光的材料。400 450 500 550 600 650 700 UV film PL solution PL film UV solution Inte nsity (a.u.) Wavelength (nm)
Fig. 3-1. UV-vis absorption and PL spectra of P1 in solution and thin film state.
400 450 500 550 600 650 UV solution UV film PL solution PL film Inte n s ity ( a .u.) Wavelength (nm)
450 500 550 600 650 700 Absorption Photoluminescence Wavelength (nm) Inte nsi ty (a.u.)
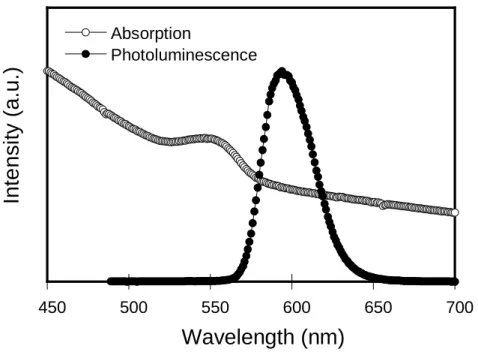
Fig. 3-3. UV-vis absorption and PL spectra of CdSe/ZnS quantum dot.
Table 3-2. Optical properties of P1 and P2.
UV-Vis (nm) P L (nm)
Polymer Film Solution Film Solution P1 477 468 567 553 P2 454 449 526 519
3.1.3 循環伏安計量(Cyclic voltammetry)分析
為了瞭解發光材料於光激發光或電激發光過程中HOMO及LUMO等能 帶的高低關係,我們對高分子P1 和P2 進行了循環伏安計量分析。發光材 料之能帶結構會直接影響其PL及EL的光色,故這一方面的數據亦是評估發光材料的一項重要指標。對於發光材料之電子游離能( Ionization potential, IP )與電子親和力( Electronic affinity,EA )數據的取得,最簡單的方法就是 以CV數據配合UV-visible光譜之吸收波長數據來計算。一般發光材料IP、 EA及能隙( Energy gap,Eg )的標準表示法如下:
HOMO = 4.4 + Eox,onset LUMO = 4.4 + Ered,onset 由於高分子材料在量測CV所表現出的還原曲線會因為有水或其他物質 的出現而變得非常雜亂,導致材料本身的還原曲線並不明顯,因而無法直 接得到EA值,故對於高分子能隙的取得一般藉由 UV-visible光譜中的最長 波長吸收峰的起始波長(λonset )來計算: Eg = 1240 /λonset
其中λonset單位為nm,而所得Eg的單位為eV, 再利用Eg = HOMO – LUMO
關係式解得LUMO能帶值。Fig. 3-4 和 3-5 表示高分子P1 和P2 在氧化方面 的CV圖,而Table 3-3 則列出計算之後的結果。
-0,001 0 0,001 0,002 0,003 0,004 0,005 -0,5 0 0,5 1 1,5 2 2,5 3 C u rren t (mA ) Potential (V)
Fig. 3-4. Cyclic voltammograms of P1.
-0,001 0 0,001 0,002 0,003 0,004 0,005 0,006 0,007 -0,5 0 0,5 1 1,5 2 2,5 3 Curren t (mA) Potential (V)
Table 3-3. Band gap and energy level values of P1 and P2 polymers band gap HOMO LUMO
polymer (eV) (eV) (eV) P1 2.42 5.42 3.00 P2 2.22 5.61 3.39
3.2 高分子 P1 和 P2 摻混 CdSe/ZnS 量子點發光二極體元
件製作與光電性質的量測
3.2.1 發光元件的結構與製作
為探討本研究所合成出高分子 P1 和 P2 摻混 CdSe/ZnS 量子點的電激 發光性質,因此我們製作了高分子發光二極體元件,在元件方面,其結構 為 ITO/PEDOT/polymer+QD/Ca/Al,作元件時多加一層 poly(3,4-ethylene dioxythiophene) (PEDOT)作為電洞傳輸層,PEDOT 具有高導電度及很好的 熱穩定性,其結構如 Fig. 3-6 所示。由於其為水溶性,因此不會有與有機 發光層互溶的問題,藉由此層之加入,能增加元件的發光效率。在元件的製作上,首先是ITO玻璃的選擇,我們採用 20 Ω/square的 ITO玻璃,經過裁切為 3 cm x 3 cm大小,再經適當的清洗程序 (如Table 3-4 所示)後使用。高分子溶液的配置則是將高分子P1 和P2 溶解在toluene中, 以超音波震盪溶解後,與溶解在toluene的量子點依比例混合。PEDOT與發 光層均是經由旋轉塗佈成膜,在PEDOT的旋轉塗佈方面,我們以 6500 rpm 旋轉 40 秒,塗佈完成後於 150oC下烘烤 1 小時,發光層則是 1500 rpm旋轉 40 秒,塗佈完成後於 80oC下烘烤 1 小時。 至於陰極則是使用功函數較低的鈣,使電子更容易地注入發光材料的 LUMO軌域。但因鈣陰極氧化的程度會嚴重影響元件的效率,為避免在元 件封裝時陰極的鈣金屬接觸到氧氣,於是我們另外蒸鍍了一層鋁金屬作為 鈣陰極的保護層。於真空度 9 × 10-7 torr下,分別蒸鍍上 35/100 nm的Ca/Al 金屬。
3.2.2 元件光電性質之量測
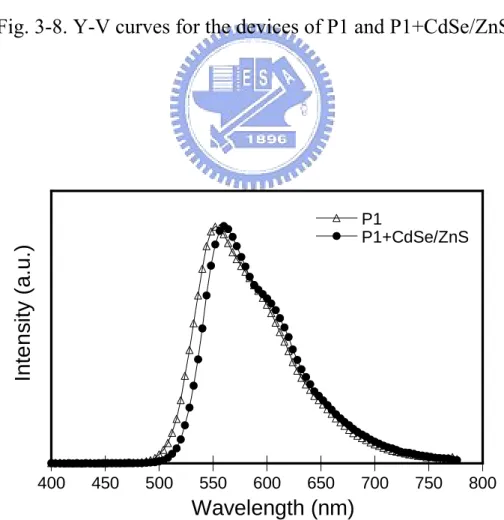
Fig. 3-7 和 3-8 分別為高分子P1 和P1 掺混CdSe/ZnS之亮度對電壓圖與 效率對電壓圖。在純高分子P1 中,最大亮度在電壓 10V時,其值為 697 cd/m2,最大效率則為 0.23 cd/A,而高分子P1 掺混CdSe/ZnS之元件中,亮 度可以提升到在電壓 10V時的 2794 cd/m2,最大效率也提升為 0.9 cd/A。 Fig. 3-9 為P1 和P1 掺混CdSe/ZnS的電激發光光譜圖,兩個圖譜的形狀大致 相同,皆屬於高分子P1 的放射,並未觀察到量子點CdSe/ZnS的放射,因此 量子點的放射在本實驗中應該是可以被忽略的。同樣的,在高分子P2 的系 統中,Fig. 3-10 和 3-11 分別為高分子P2 和P2 掺混CdSe/ZnS之亮度對電壓 圖與效率對電壓圖。元件的提升效果一樣可以被觀察到,亮度從P2 的 3949 cd/m2提升到 8192 cd/m2,效率則從 0.23 cd/A提高至 1.27 cd/A,Fig. 3-13 為P2 和P2 掺混CdSe/ZnS的電激發光光譜圖元件的所有結果整理並列 於Table 3-4。高分子掺混 CdSe/ZnS 在元件表現上的提升往往伴隨著電流密度的降 低,這意味著 CdSe/ZnS 在元件中對電荷的傳輸有著直接的影響,為了更 進一步研究,我們製作了 hole-only 和 electron- only 元件來比較電洞和電子 在元件中的傳輸情形。 0 2 4 6 8 10 12 0 500 1000 1500 2000 2500 3000 L u m inance ( cd /m 2 ) Voltage (V) P1 P1+CdSe/ZnS
0 0.2 0.4 0.6 0.8 1 1.2 0 2 4 6 8 10 12 P1 P1+CdSe/ZnS Voltage (V) Yield (cd/A)
Fig. 3-8. Y-V curves for the devices of P1 and P1+CdSe/ZnS.
400 450 500 550 600 650 700 750 800 P1 P1+CdSe/ZnS Inte n s ity ( a .u.) Wavelength (nm)
0 2 4 6 8 10 0 1000 2000 3000 4000 5000 6000 7000 8000 9000 Lum ina nc e ( cd/ m 2 ) Voltage (V) P1 P1+CdSe/ZnS
Fig. 3-10. L-V curves for the devices of P2 and P2+CdSe/ZnS.
0 0.2 0.4 0.6 0.8 1 1.2 1.4 0 2 4 6 8 10 12 P2 P2+ CdSe/ZnS Yield (Cd /A ) Voltage (V)
400 450 500 550 600 650 700 750 800 P2 P2+CdSe/ZnS Inte n s ity ( a .u.) Wavelength (nm)
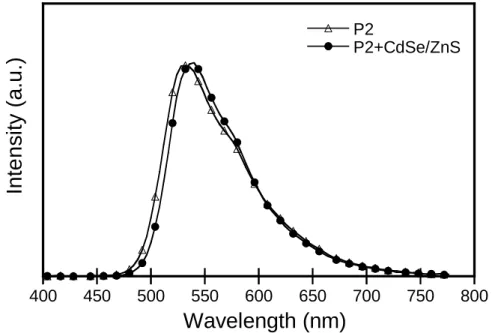
Fig. 3-12. EL spectra for the devices of P2 and P2+CdSe/ZnS.
Table 3-4. Device performances using pristine polymers and their nanocomposites as the active layer.
EL Turn-on Max. current Max.
emission voltage efficiency brightness active layer λmax (nm)a (V) (cd/A) (cd/m2)
P1 552 5 0.23 697 P1+QDs 560 5 0.9 2794
P2 531 4 0.27 3949 P2+QDs 538 3 1.27 8192
3.3 Hole only 及 electon only 元件的製作及結構
通常 hole-only 元件為 ITO/PEDOT/Polymer/Au,此元件中電洞為主要 的傳輸電荷,另一方面,electron-only 元件為 ITO/Al/Polymer/Ca/Al,因為 電洞會被阻擋在 ITO 和 Al 電極之間,所以元件中電子為主要傳輸電荷。 Hole only 元件之製作原理為利用陰極及陽極皆為高工作函數之材料, 藉由其工作函數較接近,達到只傳電洞之目的,而electon only 元件之製作 原理則是藉由選擇陰極及陽極為低工函數之材料,藉此探討發光層本身是 屬於易傳電洞或易傳電子之材料。在本實驗中此 hole only 元件之結構,陽 極為 ITO,並塗佈一層 PEDOT 來作為電洞注入層及修飾層。發光層之製 作上,直接以旋轉塗佈之方式製作而成。陰極之選擇則為工作函數較高之 Au 金屬。而 electron only 元件之結構,則是於 ITO 上先蒸鍍上一層 Al 金 屬,再塗佈上發光層。最後再蒸鍍上Ca 金屬作為陰極。3.3.1 Hole only 及 electon only 元件之結果與討論
Fig. 3-13 表示 P1 和 P1 摻混 CdSe/ZnS 量子點的電流密度-電壓圖。在 相同電壓下,其電洞的電流密度遠大於電子,表示在 P1 材料中的電洞傳 輸速率較快。Fig. 3-14 則為 P2 和 P2 摻混 CdSe/ZnS 量子點的電流密度-電 壓圖。一樣的結果可被觀察到,電洞的電流密度大於電子。因此,P1 和 P2 材料均屬於偏向電洞傳輸的材料,另一方面,由於 P1 的電洞和電子傳 輸速率相差較大,所以導致元件的表現效果不如P2。 在摻混 CdSe/ZnS 量子點之後,可以發現電洞的電流有下降的趨勢, 對於電子電流的影響似乎不大,接著我們利用空間電荷限制電流( space charge limited current )公式,如下所示 :
J = 9εε0 μV2/8L3
μ : 電荷移動速率 L : 高分子層厚度
V : 外加電壓 ε0 : 高分子電容值 透過公式,將電洞和電子的電流-電壓經過計算轉換成電洞和電子遷移率-電場圖,分別顯示於 Fig. 3-15 和 3-16。從圖中可以更清楚的發現,在高分 子 P1 中,電洞和電子的遷移速率相差較大。反之,P2 的電洞和電子的遷 移速率較為接近,因此 P2 比 P1 有較佳的元件表現。而在摻混 CdSe/ZnS 量子點之後,電洞的遷移率皆有下降的情形,電子的遷移率則沒有較大的 變化,由此可知 CdSe/ZnS 量子點在高分子中對於電洞的傳輸有較顯著的 限制作用。 Fig. 3-17 列出了元件結構為 ITO/PEDOT/Polymer/QDs/Ca/Al 的能階 圖,從能階圖中可以發現,CdSe/ZnS 量子點的 HOMO 能階遠低於 P1 或 P2 的 HOMO 能階,因此我們認為,在高分子摻混量子點的系統中,部份 的量子點會聚集並且和高分子層產生相分離,因為量子點的 HOMO 值相 對於高分子來說較低,因此有較多的電洞被阻擋在高分子層和量子點小區 域之間,使得電洞和電子的遷移率較為平衡,進而增加電洞和電子再結合 的機會,提高元件的亮度與效率表現。這個現象和文獻上的結果有很大的 不同,許多文獻發現,摻混奈米粒子往往可以增加電荷的傳遞效果,進而 增加元件的電流[30,32,57]。另ㄧ方面,這些相分離現象也可能修飾了有機 層和金屬層的接觸介面,避免了兩層直接的接觸而造成激子的淬滅。
0 2 4 6 8 10 0 50 100 150 200 250 Current De nsity (mA/c m 2 ) Voltage (V) P1 (hole-only) P1/QDs (hole-only) P1 (electron-only) P1/QDs (electron-only)
Fig. 3-13. Current density versus voltage for P1 and P1+QDs.
0 2 4 6 8 10 0 200 400 600 800 1000 Current De nsity (mA/c m 2 ) Voltage (V) P2 (hole-only) P2/QDs (hole-only) P2 (electron-only) P2/QDs (electron-only)
4000 6000 8000 10000 12000 14000 -13,5 -13,0 -12,5 -12,0 -11,5 -11,0 -10,5 -10,0 -9,5 -9,0 -8,5 -8,0 log u (m 2 /V. S ) E1/2 (V/m)1/2 P1-hole P1+QD P1-electron P1+QD-electron
Fig. 3-15. log μ versus electric field for P1 and P1+QDs.
2000 4000 6000 8000 10000 12000 14000 -12,0 -11,5 -11,0 -10,5 -10,0 -9,5 -9,0 -8,5 -8,0 lo g u (m 2 /V .S ) E1/2 (V/m)1/2 P2-hole P2+QD-hole P2-electron P2+QD-electron