國 立 交 通 大 學
機械工程學系
碩 士 論 文
利用分子動力模擬
探討石墨烯奈米複合材料機械性質
Investigating
Mechanical Properties of Graphene
Nanocomposites
by Using Molecular Dynamics Simulation
研 究 生 :許嵩群
指導教授 :蔡佳霖 博士
利用分子動力模擬探討石墨烯奈米複合材料機械性質
Investigating Mechanical Properties of Graphene Nanocomposites by
Using Molecular Dynamics Simulation
研 究 生:許嵩群 Student:Sung-Chiun Shiu 指導教授:蔡佳霖 Advisor:Jia-Lin Tsai 國 立 交 通 大 學 機 械 工 程 學 系 碩士論文 A Thesis
Submitted to Department of Mechanical Engineering College of Engineering
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Mechanical Engineering August 2012
Hsinchu, Taiwan, Republic of China
i
利用分子動力模擬探討石墨烯奈米複合材料機械性質
學生:許嵩群 指導教授:蔡佳霖 博士
國立交通大學機械工程學系碩士班
摘
要
本研究主要目的藉由分子動力模擬石墨烯奈米複合材料機械性質,探 討聚酰亞胺與樹脂兩種高分子基材中添加三種不同形式之石墨烯,形成群 聚、分散與分散表面改質石墨烯奈米複合材料,並研究不同形式下石墨烯 奈米複合材料的機械性質,其中機械性質包含了楊氏係數、玻璃轉換溫度 以及熱膨脹係數。除了機械性質之外,石墨烯對高分子基材的密度分布及 分子結構排列方式的影響亦為本研究重點。結果顯示,當高分子靠近石墨 烯周圍時密度較高,遠離石墨烯時密度下降至高分子平均密度,亦觀察到 當高分子鏈靠近石墨烯介面時形成緊密壓縮且貼附於石墨烯之情形。此外, 在機械與熱性質中,分散石墨烯奈米複合材料相對於群聚石墨烯奈米複合 材料呈現較高的楊氏係數、玻璃轉換溫度以及較低的熱膨脹係數。主要原 因由分散石墨烯使奈米複合材料中高分子呈現高度的秩序,進而提高了奈ii
米複合材料的綜合性質。此外在三種石墨烯奈米複合材料中,分散表面改 質石墨烯為最佳的補強材料,藉著互動能量的計算了解表面改質石墨烯可 有效的提高其互動能量,而互動能量的增強可能改善表面改質石墨烯奈米 複合材料的機械性質。
iii
Investigating Mechanical Properties of Graphene
Nanocomposites by Using Molecular Dynamics Simulation
Student : Sung-Chiun Shiu Advisor : Dr. Jia-Lin Tsai
Department of Mechanical Engineering National Chiao Tung University
Abstract
The research aims to investigate the mechanical properties of graphene nanocomposites using molecular dynamics (MD) simulation. Three different formats of graphene, i.e., graphene flakes, intercalated graphene and intercalated graphene oxide, were incorporated respectively in polymer matrix to form the graphene nanocomposites. Both polymer systems, i.e. polyimide and epoxy, were considered respectively as matrix in the nanocomposites. The mechanical properties of the nanocomposites including Young’s modulus, glass transition temperature (Tg) and coefficient of thermal expansion (CTE), in terms of the
different formats of graphene were characterized in this study. In addition to the mechanical properties, the influences of graphene on the morphology, density and order parameter of the polymers were also examined. Results illustrated that the local density in the vicinity of the graphene is relatively high and then decreases to the bulk value as the region is away from the interface. Furthermore, it was found that the polymer chains near the graphene are densely compacted and flattened down parallel to the graphene interface. On the other
iv
hand, for the mechanical and thermal properties, the nanocomposites with dispersed graphene exhibit higher Young’s modulus, higher glass transition temperature and lower thermal expansion coefficient than those with graphene flakes. This is because the dispersed graphene leads to high degree of ordered polymer in the nanocomposite and thus enhances the overall properties of the nanocomposite. In addition, the interacted graphene oxide provides the best reinforcement among the three cases of nanocomposites. Based on the calculation of interaction energy, it was validated that the oxide modification on graphene surface can effectively enhance the interaction energy, and such enhancement in interaction energy may be responsible for the improvement of mechanical properties of graphene oxide nanocomposites.
v
致謝
感謝指導教授 蔡佳霖博士兩年來細心的教誨,並且在研究的過程中 不斷的給予建議以及指導,使得論文能夠順利完成,在此致上由衷的感謝。 同時感謝清華大學動機系葉孟考教授以及工業研究院邱博士撥冗擔任學生 口試委員,並且給予寶貴的建議,使得此篇論文更加完善。 感謝學長曾世華、王泰元、黃健洋、徐正文、洪健峰、黃奕嘉及學姊 高菁穗這兩年來研究的建議與協助,使我收穫良多。此外感謝劉少淇、賴 彥錕、林子晨、李佳旻、聶奕心,陪伴著實驗室的生活,度過充滿歡笑與 辛勞的兩年。 在漫長的求學生涯中,感謝我親愛的家人在背後的支持與鼓勵,讓我 順利完成碩士學位,並感謝一路支持我的湯雅,使我倍感衝勁。vi
目錄
中文摘要 ... i 英文摘要 ... iii 致謝 ... v 目錄 ... vi 圖表目錄 ... ix 第一章 緒論 ... 1 1.1 研究背景與文獻回顧 ... 1 1.2 研究目標及方法 ... 7 第二章 分子動力學模擬 ... 9 2.1 運動方程式與積分演算法 ... 9 2.2 勢能函數與截斷半徑 ... 10 2.3 週期性邊界條件 ... 16 2.4 系綜(Ensemble)之選擇 ... 17 第三章 石墨烯奈米複材原子模型 ... 19 3.1 石墨烯/聚酰亞胺奈米複合材料模型建構與模擬 ... 19 3.1.1 模型 I:不同分散程度石墨烯奈米複合材料模型 ... 19 3.1.2 模型 II:表面改質石墨烯抽出模型 ... 22vii 3.1.3 分析方法與計算 ... 23 3.2 石墨烯/樹脂奈米複合材料模型建構與模擬 ... 31 3.2.1 建立樹脂分子模型 ... 32 3.2.2 模型 I:不同分散程度石墨烯奈米複合材料模型 ... 33 3.2.3 模型 II:表面改質石墨烯抽出模型 ... 34 3.2.4 分析方法與計算 ... 35 第四章 結果與討論 ... 38 4.1 石墨烯/聚酰亞胺複合材料機械/物理性質的討論 ... 38 4.1.1 勢能函數正確性之驗證 ... 38 4.1.2 石墨烯的添加對聚酰亞胺高分子幾何和結構的討論 ... 39 4.1.3 石墨烯的分散性與表面改質對複材之機械/物理性質的討論 ... 40 4.1.4 表面改質石墨烯對聚酰亞胺複合材料介面強度之影響 ... 42 4.2 石墨烯/樹脂複合材料機械/物理性質的討論 ... 43 4.2.1 勢能函數正確性之驗證 ... 43 4.2.2 石墨烯的添加對樹脂幾何和結構的影響 ... 44 4.2.3 石墨烯的分散性與表面改質對複材之機械/物理性質的討論 ... 45 4.2.4 表面改質石墨烯對樹脂複合材料介面強度之影響 ... 47 第五章結論與未來展望 ... 48 5.1 結論 ... 48
viii
5.1 未來展望 ... 49
參考文獻 ... 50
附 表 ... 56
ix
圖表目錄
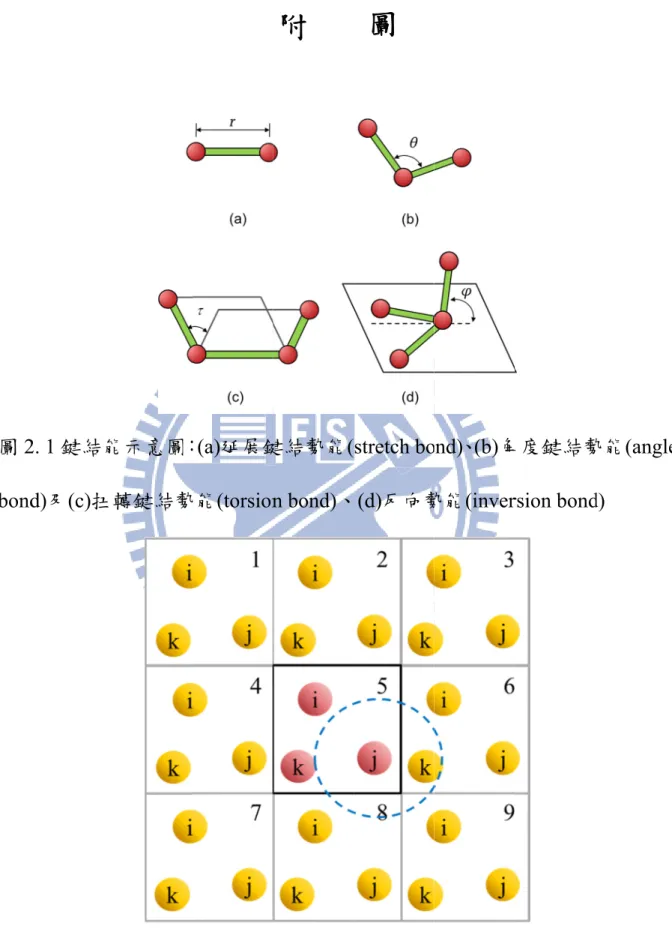
表 1 原子代號與原子質量[36] ... 56 表 2 凡得瓦勢能參數表[36] ... 57 表 3 延展鍵結勢能參數表[36] ... 58 表 4 角度勢能參數表[36] ... 58 表 5 扭轉勢能參數表[36] ... 59 表 6 聚酰亞胺與聚酰亞胺奈米複合材料之機械與熱性質 ... 59 表 7 聚酰亞胺高分子與石墨烯之互動能量與相對剪切應力 ... 60 表 8 樹脂與樹脂奈米複合材料之機械與熱性質 ... 60 表 9 樹脂與石墨烯之互動能量與相對剪切應力 ... 61 圖 2. 1 鍵結能示意圖:(a)延展鍵結勢能(stretch bond)、(b)角度鍵結勢能 (angle bond)及(c)扭轉鍵結勢能(torsion bond)、(d)反向勢能(inversion bond) ... 62 圖 2. 2 主胞室與映像胞室示意圖 ... 62 圖 3. 1 聚酰亞胺單體 ... 63 圖3.2 建構石墨烯示意圖 ((a)石墨烯上視圖 (b)群聚石墨烯前視圖 (c)分散 石墨烯前視圖) ... 63 圖 3.3 (a)氫氧基(-OH) (b)環氧基(-O-) (c)氫氧基與環氧基以 10%的比例植x 入石墨烯中之原子結構模型 ... 64 圖 3. 4 模型 I:石墨烯/聚酰亞胺奈米複合材料平衡結構分子模型 ((a)群聚 石墨烯 (b)分散石墨烯 (c)表面改質分散石墨烯) ... 65 圖 3. 5 模型 II:石墨烯抽出奈米複合材料平衡結構分子模型 ... 66 圖 3. 6 聚酰亞胺沿著 Z 方向切割成數個小區塊 ... 66 圖 3. 7 聚酰亞胺單體上碳原子與氮原子所連結之代表向量示意圖 ... 67 圖 3. 8 參考向量 Z 軸與 C-N 代表向量所夾角度之示意圖 ... 67 圖 3. 9 平衡結構施予模擬室單方向應變示意圖 ... 68 圖 3. 10 溫度與密度關係圖 ... 68 圖 3. 11 石墨烯抽出分子模型圖 ... 69 圖 3. 12 (a)單獨石墨烯分子模型圖 (b)高分子基材模型圖 ... 69
圖 3. 13 (a)硬化劑 TETA (b)樹脂 EPON862化學式 ... 70
圖 3. 14 EPON862以及TETA構成交叉鍵結樹脂(Cross-linked epoxy)代表 結構 ... 70 圖 3. 15 模型 I:石墨烯/樹脂奈米複合材料平衡結構分子模型 ((a)群聚石 墨烯 (b)分散石墨烯 (c)表面改質分散石墨烯) ... 71 圖 3. 16 交叉鍵結樹脂上碳原子與碳原子所連結之代表向量示意圖 ... 72 圖 4. 1 聚酰亞胺高分子溫度與密度之關係圖 ... 73 圖 4. 2 聚酰亞胺高分子在群聚石墨烯複合材料中密度分布 ... 73
xi 圖 4. 3 聚酰亞胺高分子在分散石墨烯複合材料中密度分布 ... 74 圖 4. 4 聚酰亞胺高分子在分散表面改質石墨烯複合材料中密度分布 ... 74 圖 4. 5 聚酰亞胺高分子在群聚石墨烯複合材料中秩序分布 ... 75 圖 4. 6 聚酰亞胺高分子在分散石墨烯複合材料中秩序分布 ... 75 圖 4. 7 聚酰亞胺高分子在分散表面改質石墨烯複合材料中秩序分布 ... 76 圖 4. 8 聚酰亞胺複材溫度與密度之關係圖((a)群聚石墨烯 (b)分散石墨烯 (c)分散表面改質石墨烯) ... 77 圖 4. 9 不同抽出距離石墨烯與聚酰亞胺高分子間的互動能量 ... 78 圖 4. 10 樹脂高分子溫度與密度之關係圖 ... 79 圖 4. 11 樹脂在群聚石墨烯複合材料中密度分布 ... 79 圖 4. 12 樹脂在分散石墨烯複合材料中密度分布 ... 80 圖 4. 13 樹脂在分散表面改質石墨烯複合材料中密度分布 ... 80 圖 4. 14 樹脂在群聚石墨烯複合材料中秩序分布 ... 81 圖 4. 15 樹脂在分散石墨烯複合材料中秩序分布 ... 81 圖 4. 16 樹脂在分散表面改質石墨烯複合材料中秩序分布 ... 82 圖 4. 17 樹脂複材溫度與密度之關係圖((a)群聚石墨烯(b)分散石墨烯 (c)分 散表面改質) ... 83 圖 4. 18 不同抽出距離石墨烯與樹脂間的互動能量 ... 84
1
第一章 緒論
1.1 研究背景與文獻回顧
複合材料(Composites)具有高度剛性、高強度、耐腐蝕、質量輕等特性, 已廣泛地運用於不同工程領域上如航太工業、汽車工業及運動用品等。傳 統複合材料加強材均以纖維為主,例如碳纖維及玻璃纖維,一般的纖維尺 度多在微米範圍。近年來隨著奈米科技的發展,藉由分散技術的開發,已 將奈米加強材料添加於複材中。因為奈米加強材具有高比表面積 (Specific surface area)的特性,所以與基材有較多的接觸面積;此外,奈米加強材亦 具有高剛度及剛強度的特性,所以添加奈米加強材,可明顯改善整體複合 材料的機械性質[1-3]。一般以奈米材料為加強材之複合材料,稱為奈米複 合材料(Nanocomposites)。常見的奈米加強材料有許多種類,例如管狀的碳 管、球狀的奈米粉體及板狀的石墨等。 近年來 Novoselov 等人[4, 5]運用膠布反覆剝開的方法,成功製備出奈 米石墨烯(Graphene sheet)。石墨烯[6, 7]係由碳原子以六邊型排列所形成的 二維結構,厚度約為3.4Å,經實驗測量其楊氏係數與拉伸強度分別為 1TPa 與130GPa[8],熱傳導係數約為 5000W/(m‧K) [9]。由於石墨烯是二維的結 構,具高比表面積(Specific surface area),添加於高分子基材中,可增加與2
基材的接觸面積,因此,石墨烯的特性及其奈米複合材料已成為複合材料 領域學者研究的重點。
Ramanathan 等人[10]將改質石墨烯(Functionalized graphene sheet)分別 添加於聚丙烯腈 PAN (Polyacrylonitrile)基材與聚甲基丙烯酸甲酯 PMMA (Polymethyl methacrylate)基材中,製成奈米複合材料。實驗結果顯示,當添 加 1 wt%的改質石墨烯於聚丙烯腈中,玻璃轉換溫度(Glass transition temperature, Tg)增加了 40℃。此外,將 1wt %的改質石墨烯加入於聚甲基丙 烯酸甲酯基材中,發現其楊氏係數和破壞強度分別提升 80%與 20%。Jang 等人[11]亦將石墨烯添加於聚甲基丙烯酸甲酯基材中,結果發現當添加 0.8wt%的石墨烯,其玻璃轉換溫度提升 10.4°C。Xu 等人[12]探討添加石墨 烯於聚乙烯醇(Polyvinyl alcohol, PVA)基材對楊氏係數與破壞強度的影響。 他們發現該複材僅添加 3wt%的石墨烯,其楊氏係數提升 129%,破壞強度 則提升72%。Das 等人[13]利用奈米壓痕的技術,探討石墨烯添對聚乙烯醇 奈米複合材料,楊氏係數與材料硬度的影響。結果顯示,當添加 0.6wt%的 石墨至聚乙烯醇基材中,楊氏係數約提升35%;此外,材料硬度提升 47%。 當石墨添加的比例增加時,聚乙烯醇的結晶度亦提升。Liang 等人[14]亦研 究添加石墨於聚乙烯醇基材對機械性質的影響。結果發現該奈米複合材料 添加0.7wt%的石墨,楊氏係數隨著提升 62%,而破壞強度則上升 72%,玻 璃轉換溫度亦提升3.3℃。
3 Kai 等人[15]探討以石墨烯為加強材之高性能奈米複合材料,當添加 5wt%的石墨,可使聚己內酯(Polycaprolactone, PCL)楊氏係數約提升 108%, 而破壞強度提升 28.2%。此外結晶溫度亦提升約 7°C。Mack 等人[16]利用 鉀離子將石墨拆層成石墨奈米板(Graphite nanoplatelet),並依照不同比例添 加於聚丙烯腈基材。結果發現,楊氏系數隨著加強材料含量的增加而上升, 當加強材料的含量為4wt%時,其楊氏係數基本上高出基材一倍以上。此外 在熱性質方面,藉由熱重分析實驗(Thermal gravimetric analysis, TGA),當 添加了 2 wt% 與 4wt % 的石墨奈米板於聚丙烯腈基材,熱穩定溫度 (Thermal stability)分別提升 10℃與 25℃。Rafiee 等人[17]將石墨奈米板、單 壁奈米碳管與多壁奈米碳管三種加強材分別添加於環氧樹脂(Epoxy)中,比 較添加不同加強材料之奈米複合材料機械性質,結果發現,當添加相同比 例之加強材料時,以石墨奈米板複合材料之楊氏係數與破壞強度具有較好 表現。Wang 等人[18]比較添加不同加強材料之奈米複合材料熱傳導效率, 結果顯示,當添加 1wt% 石墨於環氧樹脂(Epoxy)中,熱傳導效率優越於以 單壁奈米碳管為加強材料之環氧樹脂奈米複材。研究亦指出,添加石墨烯 於環氧樹脂,可降低材料之熱膨脹係數(Coefficient of Thermal Expansion, CTE),並使玻璃轉換溫度有所提升。 Kim 與 Macosko[19, 20]研究石墨烯對 聚酯(Polyethylene, PEN)與聚碳酸酯(Polycarbonate, PC)奈米複合材料機械性 質與熱性質影響,結果發現添加 4.0wt%的石墨烯,可使聚酯楊氏係數提升
4 57%,材料熱膨脹係數降低約 4.1%。石墨烯亦使聚碳酸酯楊氏係數有所提 升並降低熱膨脹係數。Steurer 等人[21]探討添加石墨烯於聚碳酸酯與聚丙 烯(Polypropylene, iPP)對機械性質與導電性質影響,發現添加 5wt %的石墨 烯,使聚碳酸酯與聚丙烯楊氏係數分別提升 52%與 43%,實驗結果亦指出 石墨烯可改善材料導電性質。Fang 等人[22]發展以石墨為加強材的高性能 奈米複合材料,藉著均勻分散技術,他們發現僅添加0.9 wt%的石墨,可將 聚苯乙烯玻璃轉換溫度提高 15℃,拉伸強度與楊氏係數則分別提升 70%和 57%。Ansari 與 Giannelis[23]探討添加改質石墨烯於聚偏氟乙烯對楊氏係數 的影響,實驗結果顯示添加4wt%的石墨烯可使複材楊氏係數提升 92%。 根據上述眾多學者實驗觀察,我們歸納,將石墨烯添加於高分子基材 中,可大幅提升奈米複合材料的楊氏係數、破壞強度及玻璃轉換溫度,並 降低熱膨脹係數以及增加熱傳導係數等機械性質;此外為了瞭解加強材料 與基材間負載傳遞與補強的效果,目前眾多學者則是藉由原子力顯微鏡 (Atomic force microscopy, AFM)、拉曼波峰偏移法(Raman peak shift)以及奈 米抽出試驗等方式了解加強材與基材間之介面強度。
Barber 等人[24]使用原子力顯微鏡對多壁奈米碳管進行抽離基材實驗, 結果發現,多壁碳管與基材間剪切應力約為10MPa 至 90MPa。此外,作者 亦說明添加奈米碳管可強化碳管附近高分子基材之機械性質,使其周圍基 材能承受較高的應力。Cooper 等人[25]發展以掃描探針顯微鏡技術,了解多
5
壁奈米碳管與環氧樹脂間介面剪切強度,結果顯示介面剪切的強度隨著碳 管的長度與斷面面積有所差異,其剪切應力的範圍35MPa 至 380MPa 間。 Roy 等人[26]使用拉曼波峰偏移法(Raman peak shift)來量測試片介面剪切應 力,探討單壁奈米碳管與聚乙烯醇間介面強度,經由實驗觀察其相對剪切 強度約160MPa,並說明改質碳管附載傳遞的能力優越於未改質碳管。Tsuda 等人[27]則是以奈米等級抽出試驗測量不同環境下多壁奈米碳管與聚醚醚 酮(Polyether ether ketone)間相對剪切應力,他們發現在常溫下剪切應力落於 3.5-7MPa 之間,若是將試片升溫至 573K 並施予 1MPa 的壓力放置一小時, 其介面剪切應力提升至 6-14MPa,此結果指出不同環境造成其介面強度之 改變。 目前除了以實驗方式觀察石墨烯奈米複合材料之機械性質以及奈米碳 管複合材料介面強度外,有部分學者藉由分子模擬,來研究加強材對於高 分子基材在熱性質、機械性質與分子幾何結構方面的影響。Pan 等人[28]使 用分子動力學研究改質石墨烯添加至聚乙烯醇高分子中,對玻璃轉換溫度 的影響。結果顯示,當添加改質石墨烯於聚乙烯醇,其玻璃轉換溫度從385K 降至360K;然而,此模擬結果卻與實驗趨勢相反。Zheng 等人[29, 30]探討 不同手性(Chirality)、不同改質比例之單壁碳管與高分子基材間介面強度之 影響,藉由單壁碳管抽出模擬,分析互動能量與剪切應力,學者指出以碳 管(10,10)與高分子基材間具有較高之互動能量與相對剪切應力,此外模擬
6 結果亦指出當碳管表面改質比例提升,其剪切應力與互動能量也相對提升。 Lv 等人[31, 32]探討表面改質之石墨烯與高分子基材間之介面強度,藉由石 墨烯抽出(Pull-out)模擬,分析石墨烯與高分子基材間互動能量(Interaction energy)以及剪應力(Shear stress)。結果顯示,表面改質之石墨烯有效的增加 高分子與其接觸面積,當接觸面積增加可使介面間互動能量相對提升。此 外,並指出介面剪切強度主要受到兩點因素影響,一為當互動能量越高, 結合能量則(Bonding energy)越高,使得介面剪切應力有所提升;二為在改 質官能基植入石墨烯造成石墨烯表面有著奈米尺度的粗糙度,當抽出模擬 時官能基團與高分子間會有機械互鎖(Mechanical interlocking)的現象,進而 提升介面剪切強度,故石墨烯表面改質可有效的改善奈米複合材料中,石 墨烯與基材間負載傳遞效率(Load transfer efficiency)。然而,改質石墨烯對 奈米複合材料,楊氏係數、機械性質與熱性質方面之影響,在文獻中並無 探討。關於分子幾何結構方面,Tsai 與 Gao [33]發現在石墨板周遭,高分子 基材的分子鏈會有明顯規則排列,使得高分子局部密度提高,楊氏係數增 加。 從上述文獻回顧,我們發現尚無有系統地探討石墨烯碳板對奈米複合 材料機械性質的影響,也因為石墨烯奈米複合材料為新發展的材料系統, 所以相關的文獻非常有限。事實上,奈米複合材料的性質主要受到內部奈 米加強材的分散性及加強材本身與周遭基材介面特性的影響。分散性和介
7 面特性同時會影響周遭高分子的排列及結構(morphology),進而影響到介面 的接合能量(bonding-energy)及介面強度。因此,本研究主要探討不同分散 程度的石墨烯碳板及添加不同表面改質的石墨烯碳板,對奈米複合材料機 械性質的影響。我們考慮奈米複合材料的機械/物理性質包括,楊氏係數、 熱膨脹係數、玻璃轉換溫度及在石墨烯周遭高分子基材分子的排列及密度 分佈等。透過有系統的分子模擬,我們可以完整瞭解石墨烯在奈米複合材 料中扮演的角色。並藉由分析模擬結果來改善奈米複合材料的製程,以期 能達到最佳的機械/物理性質。
1.2 研究目標及方法
本研究主要目的藉由分子動力學模擬,探討不同分散程度之石墨烯碳 板及三種表面改質之石墨烯碳板對奈米複合材料機械/物理性質的影響。此 模擬所使用的加強材料為石墨烯,高分子基材則為聚酰亞胺(Polyimide, PI) 與樹酯。 第二章將介紹分子動力學的基本理論,對分子動力學的計算方法進行 說明,包含勢能函數、截斷半徑、運動方程式、週期性邊界條件及系綜等;, 第三章介紹聚酰亞胺/石墨烯與樹酯/石墨烯奈米複合材料模型建構的方式、 勢能函數的選用、結構平衡以及機械與熱性質分析的方法,其中包含楊氏 係數、熱膨脹係數、玻璃轉換溫度模擬方式以及奈米複合材料中聚酰亞胺8
區域密度(Local density)與區域秩序參數(Local order parameter)計算的方法, 最後說明石墨烯抽出模擬時複合材料介面之互動能量(Interaction energy)及 剪切應力的計算方式。第四章討論奈米複合材料中不同分散情況的石墨烯 以及不同分散情況的改質石墨烯對複合材料楊氏係數、熱膨脹係數、玻璃 轉換溫度的計算與以及奈米複合材料中聚酰亞胺的區域密度與區域秩序參 數的影響,最後將研究不同官能基植入石墨烯當中,並將石墨烯抽出高分 子基材的模擬,比較不同官能基下複合材料互動能量和介面剪應力之差異。 第五章將歸納出本論文模擬分析的結論以及未來展望。
9
第二章
分子動力學模擬
分 子 動 力 學(Molecular dynamics, MD)理論於 1950 年由 Irving 與 Kirkwood[34]提出,其主要概念是建立一個系統,以微觀的角度來模擬分子 間的物理現象,利用統計力學的方式計算分子間的作用力,接著藉由運動 方程式即可得到分子的位置、速度,求得相對應的物理性質及其動態的特 性。
2.1 運動方程式與積分演算法
分子動力學主要概念為藉由勢能函數(Potential function)主導各分子間 之作用力,並且假設所有分子動力學遵守古典力學之牛頓運動定律如下列 方程式所示
Nj j i j i i ir
r
r
r
dr
r
d
dt
r
d
m
F
2 1 2
(
)
(2.1.1) 其中Fi為第i 顆原子所受到的力,mi為第i 顆原子之質量,ψ為勢能函數, 其中勢能函數主導了原子所到的力,對於整個模擬而言是影響結果的重要 角色。ri與 rj分別為 i、j 原子之座標。藉由方程式(2.1.1)中,可得到時間 t 時原子上之受力情況,若已知時間點t 時之原子座標,即可運用跳蛙演算法 ( leap-frog algorithm)[35]。 m t F t t t v t t v( ) ( ) ( ) 2 1 2 1 (2.1.2)10 t t t v t r t t r( ) ( ) ( ) 2 1 (2.1.3)
Verlet leap-frog algorithm 藉由已知時間點 t 時的原子所受到的力 F(t),將 F(t) 除以原子質量得到時間點t 之加速度,如方程式(2.1.2),選定時間步階Δt, 從t t 2 1 時之速度推算出t t 2 1 時之速度,進而利用方程式(2.1.3),由已知 t 時的位置推算出tt時之位置。
2.2 勢能函數與截斷半徑
分子動力學模擬主要是藉由勢能函數(potential function)來計算分子間 的作用力,本研究的勢能函數分別選用 Dreiding 勢能函數[36]描述石墨烯/ 聚酰亞胺複材分子模型以及Compass 勢能函數[37, 38]描述石墨烯/交叉鍵結 樹脂複材分子模型,選用不同勢能函數描述分子結構的原因主要是在不同 勢能下,高分子在機械與物理性質上也有所不同,經由模擬的結果,由 Dreiding 勢能函數下的聚酰亞胺與 Compass 勢能函數下的交叉鍵結樹脂, 在楊氏係數以及玻璃轉化溫度與實驗十分接近,其結果在第四章將有詳細 的描述。接下來將介紹Dreiding 勢能函數與 Compass 勢能函數。 分子動力學模擬中,分子間的作用力是藉由勢能函數來描述,勢能函 數, Dreiding 勢能函數[36]可表示如下 Dreiding bond Dreiding nonbond DreidingU
U
U
(2.2.1)11
其中Unonbond-Dreiding與Ubond-Dreiding分別表示Dreiding 勢能函數中非鍵結勢能與
鍵結勢能,本篇所使用之原子代號以及質量列於表1,在非鍵結勢能選用凡 得瓦力(van der Walls force),採用 Lennard Jones-12-6 (LJ-12-6)勢能函數[39]
6 ij ij 12 ij ij ij Dreiding nonbondr
σ
r
σ
4ε
U
(2.2.2) 其 中 方 程 式(2.2.2) 中 , 分 為 前 項 排 斥 力 (repulsive force) 與 後 項 吸 引 力 (attractive force)兩部分,其中ε
ij為能量參數(Energy well)、σ
ij為平衡距離、 rij為兩原子間的距離;在L-J 勢能中的參數僅考慮同一種原子間,例如碳與 碳間的能量參數ε
cc為 0.0951 Kcal/mol,平衡距離σ
cc為 3.473Å,當需要考 慮碳對於其他原子時,如碳與氫之間之凡得瓦勢能,則必須藉由結合法則 (combination rule)[40]計算得知,計算方法分別如下方程式所示。 jj ii ij
(2.2.3)2
)
σ
(σ
σ
ij
ii
jj (2.2.4) 藉由方程式(2.2.3)、方程式(2.2.4)可推算出不同原子間的凡得瓦參數;將兩12 個不同種類原子i 與 j 各自平衡距離
ii與
jj相加取平均最為兩種分子間之 平衡距離
ij;將兩種不同原子i 與 j 各自能量參數
ii與
jj相乘並且開根號, 作為此兩種分子間的能量參數
ij。藉由結合法則可計算出不同原子間結合 能量及平衡距離,本篇所使用之凡得瓦參數列於表2 中[36]。此外,在本文 中的的凡得瓦勢能截斷半徑均為 10Å,當兩原子的距離大於截斷半徑,凡 得瓦勢能視為零。 除了非鍵結勢能之外,還有描述分子內的鍵結勢能,Dreiding 勢能函數 的鍵結勢能U
bondDreiding 表示如下 inversion torsion angle stretch Dreiding bondU
U
U
U
U
(2.2.5)其中Ustretch表示延展鍵結勢能,Uangle表示角度鍵結勢能,Utorsion表示扭轉鍵
結勢能,Uinversion表示反向鍵結勢能,分別表示如下: 1. 延展鍵結勢能(stretch bond):由兩顆原子間距離的變化所產生的能量,可 表示為 2 0 r stretch
k
(r
r
)
2
1
U
(2.2.6) 其中 r 為兩顆原子間的距離,如圖 2.1(a)所示,其勁度常數(stiffness13
constant) kr與兩顆原子之平衡距離 r0如表3 所列[36]。
2. 角度鍵結勢能(angle bond):由三顆原子所構成之角度,角度變化所產生 的能量,可表示為
angle kθ(cosθ cosθ0)2 2 1 U (2.2.7) 其中 θ 為三顆原子所形成的角度,如圖 2.1(b)所示,其能量參數 kθ與 三顆原子的平衡角度θ0如表 4 所列[36]。 3. 扭轉鍵結勢能(torsion bond):由四顆相鄰原子所組成的兩個平面,兩平 面夾角變化所產生之能量,可表示為
δ)]
cos(mτ
[1
A
U
torsion
τ
(2.2.8) 其中τ為四顆原子組成兩個平面所夾之角度,如圖 2.1(c)所示,其能量 參數Aτ、參數δ 與參數 m 如表 5 所列[36]。 4. 反向鍵結勢能(inversion bond):由四顆相鄰原子所組成,其中一顆原子 與另外三顆所組成之平面所夾之角度變化而產生之能量,可表示為)]
cos(
[1
A
U
inversion
φ
(2.2.9) 其中 為一顆原子與另外三顆原子組成平面所夾之角度,如圖2.1(d)所14
示,其能量參數Aψ則參照Dreiding 力場[36]選用 40 Kcal/mol-rad2。
Compass 勢能函數[37, 38]藉由基本原理(ab initio)計算方法,計算分子 內的鍵結與分子間非鍵結參數,使用此勢能函數可以在很大的溫度、壓力 範圍內精準的預測出各種分子的結構、機械以及物理性質。Compass 勢能 函數可表示如下 Compass bond Compass nonbond Compass
U
U
U
(2.2.10)其中 Unonbond-Compass與 Ubond-Compass分別表示 Compass 勢能函數中非鍵結勢能
以及鍵結勢能,在非鍵結勢能考慮凡得瓦力(van der Walls force),採用 Lennard Jones-9-6 (LJ-9-6)勢能函數[41]
6 ij ij 9 ij ij ij ompass C nonbondr
σ
3
r
σ
2
ε
U
(2.2.11) 其中ε
ij為能量參數(Energy well)、σij為平衡距離、rij為兩原子間的距離;在 LJ-9-6 勢能若考慮不同原子間時,則藉由六階結合法則(6th order combination rule)[42]計算得知,計算方法分別如下方程式所示。15 1/6 6 jj 6 ιι ιj
2
)
σ
(σ
σ
(2.2.12)
6 jj 6 ii 3 jj 3 ii jj ii ijσ
σ
σ
σ
ε
ε
2
ε
(2.2.13) 藉由方程式(2.2.12)、方程式(2.2.13)之六階結合法則可推算出不同原子間的 結合能量及平衡距離;將兩個不同種類原子i 與 j 各自平衡距離
ii與
jj帶 入方程式(2.2.12)求得平衡距離
ij;將兩種不同原子i 與 j 各自能量參數
ii、 jj
以及平衡距離
ii 、
jj 帶入方程式(2.2.13)求得兩種原子間的能量參數 ij
。 Compass 勢能中鍵結勢能[37, 38]可表示如下
x coupling cross 2 x φ 3 2 1 θ 4 0 4 3 0 3 2 0 2 b 4 0 4 3 0 3 2 0 2 Compass bond U χ K cos3φ 1 V cos2φ 1 V cosφ 1 V ) θ (θ H ) θ (θ H ) θ (θ H ) b (b K ) b (b K ) b (b K U (2.2.14) 其中分別表示延展鍵結勢能、角度鍵結勢能、扭轉鍵結勢能、反向鍵結勢 能以及交互耦合勢能Ucross-coupling,而交互耦合勢能又可表示如下16
cosφ
)
θ
)(θ
θ
(θ
F
cos3φ
V
cos2φ
V
cosφ
V
)
θ
(θ
cos3φ
V
cos2φ
V
cosφ
V
)
b
(b
)
θ
)(θ
b
(b
F
)
θ
)(θ
θ
(θ
F
)
b
)(b'
b
(b
F
U
θ θ' φ ' 0 0 φ θθ' θ φ θφ 3 θφ 2 θφ 1 0 b φ bφ 3 bφ 2 bφ 1 0 b θ 0 0 bθ θ θ' ' 0 0 θθ' b b' ' 0 0 bb' coupling cross
(2.2.15) 其中分別表示延展鍵結與延展鍵結、角度鍵結與角度鍵結、延展鍵結與角 度鍵結、延展鍵結與扭轉鍵結、角度鍵結與扭轉鍵結、以及角度鍵結與角 度鍵結和扭轉鍵結之交互耦合勢能函數,詳細的勢能函數可參考文獻[37, 38] 所示。本研究Compass 勢能使用 Material Studio 軟體[43]來進行分子動力模 擬,由於此軟體為商用化軟體,詳細的參數已附加在軟體下尚無對外公開。2.3 週期性邊界條件
由於分子動力學模擬是探討原子尺度下各種微觀的物理現象,若要模 擬系統的真實尺寸,其所包含的原子數量是相當可觀的,如欲模擬真實尺 寸的所有原子,對於現今的電腦來說是十分困難的。因此在分子動力學模 擬中通常僅選取具代表性的單位晶胞(unit cell)來進行模擬,藉由單位晶胞17
周期性的重複排列,達到與真實系統相近的結果,此方法稱為周期性邊界 條件(periodic boundary condition)[44]。
分子動力學模擬中,真實模擬的單位晶胞稱為主胞室(primary cell),在 二維的情形下,周圍包含了八個映像胞室(image cell),圖 2.2 中編號為 5 的 即為主胞室,其餘八個則是映像胞室;而在三維的情形下則包含了二十六 個映像胞室。當考慮周期性邊界條件時,在計算原子間的作用力時,除了 計算主胞室內原子間的作用力外,主胞室與映像胞室內的原子間的作用力 必須一併計算,為了避免在計算作用力時產生錯誤,必須找到主胞室與映 像胞室內原子之間的最小作用距離進行計算,此即最小映像法則(minimum image criterion) [39]。以圖 2.2 中的系統來說,主胞室內的 j 原子與 k 原子之 間的距離已大於截斷半徑,依據截斷半徑的原則,兩者間的作用力應不需 計算,但由於周期性邊界條件,主胞室內的j 原子與其中一個映像胞室內的 k 原子距離小於截斷半徑,因此兩者間的作用力必須納入計算。
2.4 系綜(Ensemble)之選擇
分子動力學模擬中,為維持模擬系統之穩定,在微觀系統中會引入熱 力學系統的統計規律性概念,藉由特定的約束條件進行處理。依據控制條 件的不同,可分為三種系綜,分別為NVE、NVT 和 NPT[44]三種。在 NVE 系綜中,系統內的原子數目、體積為定值,藉由能量守恆調節系統總能量,18 使能量維持定值。NVT 為固定系統內的原子數目、體積並進行溫度的調節, 使系統溫度達到給定的初始值。NPT 為固定系統內的原子數目,並進行壓 力及溫度的調節,使其三方向之平均壓力及溫度接近設定的初始值。不同 的系綜其代表的意義亦不同,在NPT 系綜中,可施予壓力與目標溫度可模 擬現實環境;而NVT 系綜中,固定模擬室體積,可觀察在指定目標溫度下 分子間的互動。
19
第三章
石墨烯奈米複材原子模型
3.1 石墨烯/聚酰亞胺奈米複合材料模型建構與模擬
本研究探討不同分散程度之石墨烯以及表面改質石墨烯對奈米複合材 料機械性質的影響。首先說明兩組模型建構與模擬的方式,模型I 研究不同 分散程度之石墨烯與表面改質石墨烯造成奈米複合材料中楊氏係數、熱膨 脹係數、玻璃轉換溫度以及在石墨烯周遭高分子鏈的排列與密度分佈的改 變;此外為了探討表面改質對奈米複材介面性質的影響,我們建置模型 II 進行表面改質石墨烯抽出之模擬,分析表面改質石墨烯與高分子基材間之 互動能量及相對剪切應力。3.1.1 模型 I:不同分散程度石墨烯奈米複合材料模型
為了比較基材中添加石墨烯對機械/物理性質的影響,首先建立單純聚 酰亞胺(Pure PI)分子模型做為對照模型,其中聚酰亞胺分子模型為 12 條高 分子鏈所組成,每條鏈長均為20 個重複單體,聚酰亞胺單體如圖 3.1 所示。 接著為了瞭解不同分散程度之石墨烯以及不同分散程度之表面改質石墨烯 對奈米複合材料機械/物理性質的影響。我們藉由 Materials Studio 軟體[43] 建構石墨烯複材模型,石墨烯 x 方向(Armchair)與 y 方向(Zigzag)之長度分 別為60.50Å與56.08Å,其中碳與碳之間距離為1.42Å、鍵結角度為 120 度, 根據Schniepp 等人[45]經實驗觀察石墨烯之邊界接有氫原子,由於本篇選用20 有限長度的石墨烯,為了使模擬更貼近實驗結果,我們在石墨烯邊界接上 氫原子,圖3.2(a)為石墨烯上視圖。接著,將上述建構之石墨烯以群聚與分 散兩種不同方式放置,群聚石墨烯放置方式如圖3.2(b)所示,將八層石墨烯 以A-B-A 的方式群聚堆疊成石墨板,相鄰層與層間距為 3.4Å;分散石墨烯 放置方式如圖3.2(c)所示,將三層石墨烯均勻放置於模擬室中,彼此的間距 為 18Å,群聚與分散石墨烯所占重量百分比為 18%,且三方向均為週期性 邊界。最後分別將 48 條以及 18 條聚酰亞胺分子鏈填滿模擬空間,以建構 出初始群聚石墨烯與分散石墨烯奈米複材模型,其中每條分子鏈長均為 20 個重複單體。 此外為了瞭解表面改質石墨烯對奈米複合材料機械性質的影響,在此 將建構分散改質石墨烯奈米複材分子模型,經由文獻[21, 45, 46]實驗發現, 常見的表面改質石墨烯常接上羥基(Hydroxy group)與環氧基(Epoxy group), 其官能基結構分別如圖 3.3(a)、圖 3.3(b)所示,文獻[46]並指出表面改質石 墨烯化學式可能為 C2O0.77H0.29,藉此文獻所提供碳氫氧比例,估算羥基與 環氧基可能比例為 1:1.5,此外植入官能基團個數則是參考文獻[31]所示, 文獻中藉由分子模擬探討不同改質比例對介面強度的影響,指出介面強度 隨著表面改質比例的提升有所增加,從文獻中發現當表面改質比例為 10% 時有較高之介面強度,且介面強度在改質比例為 10%時有所收斂,在此表 面改質比例定義為官能基個數除上石墨烯碳原子個數;因此表面改質石墨
21 烯時將依照實驗所估算羥基與環氧基 1:1.5 比例進行建構,並且以亂數的 方式植入羥基與環氧基,改質比例為10%,形成表面改質石墨烯,如圖 3.3(c) 所示。最後將三片表面改質石墨烯均勻放置於模擬室中,使條聚酰亞胺分 子鏈填滿模擬空間,藉此建構出分散改質石墨烯奈米複材分子模型。 經由上述步驟,即可建構初始之群聚與分散石墨烯奈米複合材料、分 散表面改質石墨烯奈米複合材料及單純聚酰亞胺模型,由於初始分子結構 模型的位能(Potential energy)通常很高,導致進行分子動力學模擬運算時產 生能量無法收斂的問題,所以在進行分子動力學模擬運算之前,藉著 Materials Studio 軟體中 Discover 模組進行能量最小化,能量最小化的方法 則是選用牛頓法(Newton) [47],牛頓法是藉著二階導數求得能量極小值,並 微調奈米複合材料模型的結構,使系統的位勢能達到最低且穩定的狀態。 在得到了穩定結構模型之後,我們將結構之原子位置取出,使用 Dreiding 勢能[36]以 DL-POLY 軟體[48]進行模擬,本研究所選用時間步階 (Timestep)皆為 1fs(femtosecond, fs)。在此說明分子模型平衡的過程,首先, 我們在 NVT 系綜下,將溫度定為 1100K 的環境下,並將石墨烯固定凍結 (freeze),進行 400ps 的平衡時間,接著,將溫度降至 600K,且石墨烯固定 凍結,並持續400ps 之平衡時間。在高溫下進行平衡是為了提供高分子足夠 的動能,並且固定模擬室體積,使得高分子能夠均勻分佈在模擬室中;將 石墨烯固定凍結是為了避免在高溫的過程中石墨烯皺褶。接著在NPT 系綜
22 下,我們將溫度降至300K,並且讓石墨烯自由運動,進行 400ps 模擬時間。 最後,在NPT 系綜下使溫度達到絕對溫度 0K,且壓力為 0MPa 的平衡結構, 圖3.4 分別為平衡模擬後所得到之群聚、分散以及表面改質分散石墨烯奈米 複合材料的結構,並將結構進行分析與討論。
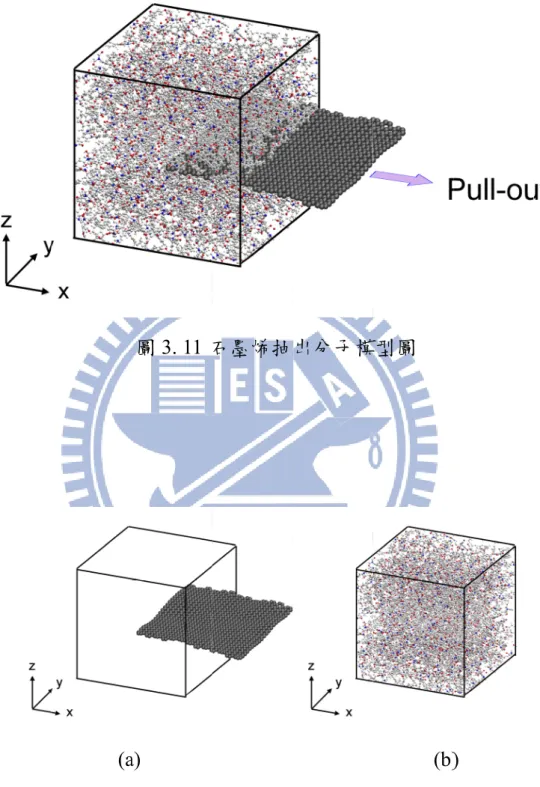
3.1.2 模型 II:表面改質石墨烯抽出模型
為了瞭解表面改質石墨烯對奈米複合材料介面性質的影響,在此,建 構石墨烯與表面改質石墨烯奈米複合材料抽出模型,首先,我們使用 Materials Studio 軟體建構有限長度之石墨烯,其中長為 51.12Å 且寬為 51.66Å,並在石墨烯邊界接上氫原子。表面改質石墨烯則是將羥基與環氧 基植入石墨烯,改質比例為 10%。接著將單層表面改質石墨烯放置於模擬 室中,為了方便模型的建構,我們將模擬室的尺寸設定為60×60×60Å3的正 方形結構,且三方向為週期性邊界條件。最後將12 條聚酰亞胺分子鏈填滿 模擬空間,建構石墨烯奈米複合材料之抽出模型。並將此模型進行能量最 小化微調模型結構,以得到位勢能最低的初始結構。 將上述所得初始結構模型之原子位置取出後,以 DL-POLY 軟體[48]進 行分子動力模擬。首先,在 1100K 的環境下,將石墨烯固定凍結(freeze), 給定 NVT 系綜的環境,進行 400ps 的平衡時間,接著,將溫度降至 600K 且石墨烯固定凍結,並持續400ps 之平衡時間。在高溫下進行平衡是為了提23 供高分子足夠的動能,使得高分子能夠均勻分佈在模擬室中。接著,我們 在 NPT 系綜下,壓力設定為 0MPa,將溫度依序降至 300K 以及 0K,並且 讓石墨烯自由運動,進行400ps 模擬時間。我們可以得到絕對溫度 0K 且壓 力為0MPa 之平衡結構,三邊長度約為 56.38Å,如圖 3.5 所示,並將所得之 抽出模型進行分析與討論。
3.1.3 分析方法與計算
模型I 中,主要目的為瞭解不同分散性石墨烯以及不同分散程度表面改 質石墨烯添加在基材中,對於高分子的密度分佈、秩序參數、楊氏係數、 熱膨脹係數、玻璃轉換溫度的影響。在此說明模擬分析與計算方法。 1. 密度分佈與秩序參數:為了瞭解添加石墨烯造成高分子鏈之結構與排列 方式的改變,在此對平衡之結構進行物理性質的分析,物理性質包含密 度分佈以及秩序參數。首先,將結構沿著Z 方向,亦為石墨烯出平面方 向分成許多微小區間,如圖 3.6 所示,區間的厚度為 1Å,藉由將平衡 結構之石墨烯出平面(Z 軸)方向切割數個單位區間,以瞭解每個單位區 間高分子的密度以及高分子的方向性。區域密度的計算方式為每塊單位 區間內高分子原子質量的總和(不考慮石墨烯質量)除以單位區間體積。 最後,統計石墨烯出平面(Z 軸)方向不同區間內的密度,進而得到密度 分佈的情況。24 為了充分瞭解添加石墨烯對高分子鏈排列方向性的影響,本研究使 用秩序參數方法量化其結果,選用較為堅硬(幾乎維持平面)的氨基與苯 環作為聚酰亞胺代表向量(Representative vector)亦為 C-N 向量,如圖 3.7 所示;石墨烯出平面(Z 軸)方向設定為參考向量(Reference vector)。參照 於密度分佈的分析方法,本研究將結構之石墨烯出平面(Z 軸)方向切割 數個單位區間,並計算區間內之秩序參數,秩序參數[49]表示如下
1)
φ
cos
(3
2
1
f
2
(3.1.1) 其中,角度為代表向量與參考向量之夾角,如圖 3.8 所示。符號 定 義為單位區間之代表向量與參考向量所夾角度的平均。當秩序參數值 為1 時,表示代表向量平行參考向量;參數值為-0.5 時,表示代表向量 垂直參考向量,亦為高分子鏈平貼於石墨烯上;參數值為 0 時,表示 代表向量不具方向性,沿著 Z 軸方向統計每個單位區塊內秩序參數, 以量化高分子鏈方向性。藉著密度分布及秩序參數兩種分析方法,以 利於我們瞭解石墨烯的添加造成奈米複合材料中高分子結構與排列方 式的影響。 2. 楊氏係數:本研究將不同分散程度之石墨烯奈米複合材料進行拉伸模擬,25 並探討其對於楊氏係數的影響。首先在結構的 X 方向施予 0.5%之拉伸 應變
xx,其餘方向模擬室之長度維持不變,如圖 3.9 所示。應變的定 義如下,
i
x,y,z
L
L
L
i i i ii
0 0
3.1.2 其中ii為拉伸應變, 0 i L 為平衡結構的原始長度,Li為變形後的長度,下 標i 則代表其方向。在 NVT 系綜設定其溫度為 0K,進行 200ps 的平衡 達到結構的穩定狀態,並計算三方向的應力,所求得的應力稱為維里應 力(Virial stress) [50],其定義如下 x,y,z
i,j
,
r
F
2
1
v
v
M
V
1
σ
α β α αβ j αβ i α j α i α ij
3.1.3 其中、
代表系統內所有的分子,
ij為系統模擬室的應力張量,V 為系 統模擬室體積,M為第
顆原子質量,v 為第i 顆原子i 方向原子速度, j v 為第顆原子j 方向的原子速度,Fi為
與
原子間i 方向的作用力, j r 為
與
原子間 j 方向的距離。方程式3.1.3的第一項為動能項,第 二項為原子間距離改變所造成的能量,若模擬的溫度為,則將動能 項忽略,於是維里應力簡化成 26
x,y,z
i,j
,
r
F
2
1
V
1
σ
α β α αβ j αβ i ij
(3.1.4) 將拉伸應變
xx以及所得三方向應力
xx、
yy與
zz帶入應力應變關係
zz yy xx zz zy zx yz yy yx xz xy xx zz yy xxε
ε
ε
C
C
C
C
C
C
C
C
C
σ
σ
σ
3.1.5 求得勁度矩陣Stiffness matrix中之 Cxx、Cyx與Czx,接著分別施加0.1% 應變於Y、Z 方向,同樣計算在拉伸應變
yy以及拉伸應變
zz下所產生 的三方向應力,並帶入方程式(3.1.5)中求得勁度矩陣,柔度矩陣與勁度 矩陣的關係可表示如下
z y yz x xz z zy y x xy z zx y yx xE
1
E
ν
E
ν
E
ν
E
1
E
ν
E
ν
E
ν
E
1
C
S
1 3.1.6 其中E 與
分別表示楊氏係數與泊松比,下標表示其方向。藉著上述的27 方法,我們可以探討不同分散程度之石墨烯奈米複合材料在三方向楊氏 係數的差異以及添加石墨烯補強材的影響。 3. 玻璃轉換溫度:將分子模型在等壓環境隨著時間降溫,經由溫度變化計 算結構密度,藉此求得分子結構之玻璃轉換溫度。首先採用NσT 系綜 [52],壓力設定一大氣壓,並將溫度從 700K、680K、660K……每階段 溫度減少20K 直至 320K,在每個溫度下各進行 200ps 的平衡。因為控 制系統於指定溫度時,系統溫度會在指定溫度上下5K 的震盪,此時模 擬室的體積隨著溫度震盪有些微改變,所以將每個溫度後 50ps 的體積 取平均作為平均體積。將系統質量除以平均體積,即可計算出不同溫度 下系統的密度,在藉由兩條線性曲線擬合溫度與密度關係圖,而兩條線 性曲線的範圍係計算兩條線性回歸曲線之 R-square[53],當兩條回歸曲 線之R-square 總和最大時,則定義此範圍為最佳範圍,並藉由所得最佳 兩條線性曲線擬合定義其斜率轉折處,此轉折點視為玻璃轉換溫度,如 圖 3.10 所示。藉著此模擬步驟可分析純聚酰亞胺、群聚與分散石墨烯 奈米複合材料的玻璃轉換溫度,瞭解石墨烯的添加以及石墨烯的分散性 對玻璃轉換溫度的影響。 4. 熱膨脹係數:熱膨脹係數模擬的方式與玻璃轉換溫度模擬的方式類似,
28 採用NσT 系綜[52],壓力設定一大氣壓,溫度從高溫降溫至 400K。在 此探討結構在玻璃態時熱膨脹係數,並觀察結構溫度 500K 至 400K 時 體積之變化,並計算系統體膨脹係數,其公式如下
ΔT
ΔV
V
1
γ
0
(3.1.7) 其中γ為體膨脹係數,V0為每個溫度階段下系統的平均體積,ΔV 為體 積變化,ΔT 為溫度變化。並計算分子模型在玻璃轉換溫度至 320K 時 體積變化,將體積變化與溫度變化帶入方程式(3.1.7),求得體積熱膨脹 係數,然而較多實驗文獻所測量之膨脹係數皆為線膨脹係數,因此我們 在此將體積熱膨脹係數轉換至線膨脹係數並與實驗進行比較。 由於我們模擬膨脹係數時採用 NσT 系綜[52],故分子模型之模擬 室角度呈現非90°情形,因此無法觀察特定方向之長度變化,故在此假 設分子模型在溫度變化下其三方向應變量相同,並參考文獻[54]線膨脹 係數與體積膨脹係數之關係求得線膨脹係數,其關係如下所示ΔT
ΔV
V
1
γ
λ
3
0
(3.1.8) 其中λ為線膨脹係數,由方程式(3.1.8)可知體膨脹係數為線膨脹係數的 三倍,並可將體膨脹係數轉換求得線膨脹係數。29 模型 II 主要藉由石墨烯抽出模擬,瞭解表面改質石墨烯對奈米複合材 料介面間之互動能量及剪切應力的影響。首先,取消 X 方向之週期性邊界 條件,其餘兩方向仍維持不變,並將石墨烯視為剛體(rigid body),給予石墨 烯X 方向 0.5Å 之位移,由於石墨烯與高分子基材間具有約 2.71Å 之凡得瓦 空隙(vdW gap),此時石墨烯仍在凡得瓦空隙間,不發生因移動石墨烯造成 其穿過高分子鍵結之情況。接著在 NVT 系綜下設定溫度 0K,將石墨烯移 動後之奈米複合材料結構,進行10ps 的平衡時間,平衡過程中讓石墨烯與 聚酰亞胺分子自由運動進行位置的調整,當結構達穩定的狀態,計算此抽 出距離時奈米複材結構模型的能量。奈米複材結構模型之互動能量可表示 如下[55] PI graphene total eraction int
E
E
E
E
(3.1.9) 其中Etotal為聚酰亞胺/石墨烯奈米複合材料結構模型全部的能量,如圖 3.11 所示;石墨烯部分的能量 Egraphene,由奈米複合材料單位結構中聚酰亞胺高 分子移除後計算得到,如圖 3.12(a)所示;同樣地,將奈米複合材料單位結 構中石墨烯部分原子移除,如圖 3.12(b)所示,求得聚酰亞胺高分子部分的 能量 EPI;Einteraction 則是聚酰亞胺高分子和石墨烯之間交互作用的凡得瓦能30 量,在此稱為互動能量,當互動能量越高時,石墨烯的表面及聚酰亞胺高 分子的內表面之間存在非鍵結的能量就越高。最後重複上述模擬過程,直 到石墨烯完全抽出與高分子基材分離,便可以求得不同抽出距離時奈米複 合材料結構之互動能量。當石墨烯完全抽出時,由於奈米複合材料結構模 型的能量等於石墨烯的能量與聚酰亞胺高分子的能量相加,故在石墨烯完 全出抽時結構之互動能量為零。 藉著抽出模擬來探討石墨烯與基材間相對剪切應力,首先計算石墨烯 抽出能量Epullout,其定義如下[56] 1 2 total_ _ total pullout
E
E
E
(3.1.10)其中 Etotal_2為石墨烯完全抽出時奈米複合材料的能量,Etotal_1為石墨烯完全
埋至基材時奈米複合材料的能量,兩者能量的差值即為石墨烯抽出能量 Epullout,亦為石墨烯抽出所做的功,並假設石墨烯抽出時剪切應力
τ
i為定值。 藉由抽出能量Epullout與剪切應力τ
i建立一關係[31]如下所示: 2 i i L x 0 x L x 0 x i pulloutAτ
dx
2h(L
x)
τ
dx
hτ
L
E
(3.1.11) 其中參數 A 表示石墨烯接觸於高分子基材的表面積,h 與 L 分別代表石墨31 烯的寬與長。將石墨烯上剪切應力
τ
i與石墨烯接觸面積 A 相乘即為石墨烯 上的作用力,而石墨烯上的作用力與石墨烯抽出的距離進行積分即為石墨 烯抽出能量Epullout,將方程式(3.1.11)整理後如下所示 2 pullout ihL
E
τ
(3.1.12) 由石墨烯抽出能量 Epullout 帶入方程式(3.1.12)式求得剪切應力τ
i,當抽出能 量 Epullout越大時剪切應力也越大。Cheng 等人[31]說明主要有兩個因素會影 響介面剪切應力,一為改質石墨烯與高分子基材間之互動能量,當能量越 高時,介面間非鍵結能量越高則剪切應力越大;二表面改質的程度使石墨 烯在奈米尺度下之不同粗糙程度,進而影響到剪切應力。本研究藉著石墨 烯抽出模擬瞭解表面改質石墨烯對相對剪切應力及互動能量的影響,比較 石墨烯與表面改質石墨烯添加於高分子基材其機械/物理性質之差異。3.2 石墨烯/樹脂奈米複合材料模型建構與模擬
在上一節中,我們針對結構較為簡單之聚酰亞胺高分子進行模擬與分 析,而本章節進一步分析當分子鏈較為複雜時,其奈米複材的機械/物理性 質,此節所選用的高分子為交叉鍵結樹脂(Cross-linked epoxy),是由硬化劑 TETA與樹脂 EPON862兩種分子進行鍵結所構成,其化學式分別如圖32 3.13(a)及 3.13(b)所示,補強材方面選用石墨烯。
3.2.1 建立樹脂分子模型
Yu 與 Choi 等人[54, 57]使用了兩種方法來建構樹脂結構,第一種方法 在模擬室當中放置12 條 EPON862樹脂以及4 條硬化劑 TETA高分子鏈, 其中文獻設定當 TETA高分子鏈中標示紅色氮原子,如圖 3.13(a)所示,與 EPON862樹脂中標示紅色的碳原子,如圖3.13(b)所示,在模擬的過程中, 若彼此距離小於4Å 時兩原子產生鍵結,形成交叉鍵結樹脂,但文獻說明此 方法較適用於分子模型較小的情況,當模型較大時模擬較為複雜與耗時; 第二種方法是將三條 EPON862樹脂與一條 TETA高分子鏈鍵結在一起, 建構出交叉鍵結樹脂代表結構,如圖 3.14 所示,其中紅色線條代表分子樹 脂與硬化劑之鍵結,在模擬過程中樹脂彼此間不考慮鍵結問題。此方法較 適用於較大原子模型中,本研究參考作者的第二種方法建構出交叉鍵結樹 脂代表結構,設定其初始模型大小為 50×50×50Å3的正方形結構,使 24 條 交叉鍵結樹脂代表分子鏈填滿模擬室,且三方向為週期性邊界條件,建構 出單純交叉鍵結樹脂分子模型。將此模型使用Compass 力場[37, 38]進行分 子動力學模擬。首先在1100K 的環境下給定 NVT 系綜,進行 200ps 的平衡 時間,接著將溫度降至600K,並持續 200ps 之平衡時間。在高溫下進行平 衡是為了提供高分子足夠的動能,使得高分子能夠均勻分佈在模擬室中。33 接著,我們在NPT 系綜下,壓力設定為 0MPa,將溫度依序降至 300K 以及 1K,分別進行 200ps 模擬時間,即可得到單純交叉鍵結樹脂穩定結構。
3.2.2 模型 I:不同分散程度石墨烯奈米複合材料模型
此小節主要目的在建構石墨烯與樹脂奈米複合材料模型,其建構方法 與聚酰亞胺奈米複合材料類似,先建構 x 方向(Armchair)與 y 方向(Zigzag) 之長度分別為26.47Å與26.81Å的石墨烯模型,並於石墨烯邊界接上氫原子, 接著將上述建構之石墨烯以群聚與分散兩種不同方式放置,群聚石墨烯放 置方式是將八層石墨烯以 A-B-A 的方式群聚堆疊成石墨板,相鄰層與層間 距為 3.4Å;分散石墨烯放置則將三層石墨烯均勻放置於模擬室中,彼此的 間 距 為 18Å,群聚與分散石墨烯所占重量百分比為 18%, 最 後 藉 由 Amorphous Cell 工具,分別將 96 條以及 36 條分子鏈填滿模擬空間,建構群 聚與分散石墨烯奈米複材。 此外為了瞭解表面改質石墨烯對奈米複合材料機械性質的影響,在此 將建構分散改質石墨烯奈米複材分子模型,我們將氫氧基團與環氧基團以 亂數的方式植入石墨烯,改質比例約為 10%,形成表面改質石墨烯,其中 氫氧基團與環氧基團以1:1.5 之比例植入石墨烯。最後將三片表面改質石墨 烯均勻放置於模擬室中,36 條分子鏈填滿模擬空間,藉此建構出分散改質 石墨烯奈米複材分子模型。最後將群聚石墨烯、分散石墨烯以及分散改質34 石墨烯奈米複材模型進行能量最小化。 將上述結構模型使用 Compass 力場[37, 38],進行分子動力學模擬。在 1100K 的環境下,將石墨烯固定凍結(freeze),給定 NVT 系綜,進行 200ps 的平衡時間,接著,將溫度降至600K 且石墨烯固定凍結,並持續 200ps 之 平衡時間。在高溫下進行平衡是為了提供高分子足夠的動能,使得高分子 能夠均勻分佈在模擬室中。接著,我們在NPT 系綜下,壓力設定為 0MPa, 將溫度依序降至 300K 以及 1K,並且讓石墨烯自由運動,進行 200ps 模擬 時間。我們可以得到溫度1K 且壓力為 0MPa 之平衡結構,圖 3.15 分別為平 衡模擬後所得到之群聚、分散以及表面改質分散石墨烯奈米複合材料的結 構,並將結構進行分析與討論。
3.2.3 模型 II:表面改質石墨烯抽出模型
此小節建構石墨烯與表面改質石墨烯奈米複合材料抽出模型,選用有 限長度之石墨烯,其中長為 38.34Å 且寬為 36.9Å,並在石墨烯邊界接上氫 原子。而表面改質石墨烯則是將羥基與環氧基植入石墨烯,改質比例為10%。 接著將單層表面改質石墨烯放置於模擬室中,模擬室的尺寸設定為 50×50× 50Å3的正方形結構,且三方向為週期性邊界條件,最後將 24 條樹脂填滿模 擬空間,建構單層改質石墨烯奈米複合材料之單元結構。將所得結構進行 分子動力模擬,模擬的流程與3.2.2 小節模擬過程相同,最後即可得到單層35 石墨烯與單層表面改質石墨烯奈米複材抽出模型。
3.2.4 分析方法與計算
模型I 中,主要目的為瞭解不同分散性石墨烯以及不同分散程度表面改 質石墨烯添加在基材中,對於高分子的密度分佈、秩序參數、楊氏係數、 熱膨脹係數、玻璃轉換溫度的影響。在此說明模擬分析與計算方法。 1. 密度分佈與秩序參數:此分析方法 3.1.3 方法相同。將結構沿著 Z 方向, 亦為石墨烯出平面方向分成許多微小區間,區間的厚度為 1Å,並計算 每個單位區間高分子的密度以及高分子的方向性。區域密度的計算方式 為每塊單位區間內高分子原子質量的總和(不考慮石墨烯質量)除以單位 區間體積。最後,統計石墨烯出平面(Z 軸)方向不同區間內的密度,進 而得到密度分佈的情況。方向性的統計則是用秩序參數方法量化其結果, 選用本環上碳原子與碳原子作為交叉鍵結樹脂之代表向量,如圖 3.16 所示;石墨烯出平面方向為參考向量。藉由秩序參數計算每個區間內的 方向性,當秩序參數值為1 時,表示代表向量平行參考向量;參數值為 -0.5 時,表示代表向量垂直參考向量,亦為高分子鏈平貼於石墨烯上; 參數值為0 時,表示代表向量不具方向性。藉著密度分布及秩序參數兩 種分析方法,以瞭解石墨烯的添加造成奈米複合材料中高分子結構與排 列方式的影響。36 2. 楊氏係數:計算結構模型的楊氏模數可藉由 Discover 模組中 Analysis 方法進行分子結構的分析,其分析過程分為四個過程,首先將分析結構 進行能量最小化,微調原子結構位置,使整體結構的位能達到最穩定的 狀態,第二步驟對三方向分別施予0.05%之拉伸應變,並保持其他方向 應變為0,第三步驟將分別對變形後結構做能量最小化之處理,使變形 後之結構達到能量穩定的狀態,最終分別計算變形前後的應力與應變, 應力計算方式採用維里應力(Virial stress)[50],將三方向應力與應變帶入 方程式(3.1.5)中,即可求得結構的勁度矩陣,並藉著勁度矩陣與柔度矩 陣關係,如方程式(3.1.6)所示,求得出結構三方向之楊氏係數。 3. 玻璃轉換溫度:此分析過程與先前模擬過程相同,但由於樹脂的玻璃轉 換溫度較聚酰亞胺低,為了節省模擬所需的時間,在此將溫度的範圍設 定為600K 至 200K,在 NσT 系綜壓力設定一大氣壓,溫度從 600K 遞 減,每階段溫度減少20K 直至 200K,在每個溫度下各進行 100ps 的平 衡。最後由溫度與密度關係圖找出玻璃轉換溫度,來瞭解石墨烯的添加 以及石墨烯的分散性對玻璃轉換溫度的影響。 4. 熱膨脹係數:首先採用 NPT 系綜,壓力設定一大氣壓,高溫降溫並計 算400K 至 300K 時體積之變化,藉著方程式與方程式分別
37 計算系統體膨脹係數及線膨脹係數。 模型II 主要藉由石墨烯抽出模擬,首先取消 X 方向之週期性邊界條件, 其餘兩方向仍維持不變,並將石墨烯視為剛體(rigid body),給予石墨烯 X 方向0.5Å 之位移。接著在 NVT 系綜下設定溫度 1K,將石墨烯移動後之奈 米複合材料結構,接著在 NVT 系綜下設定溫度 1K,將石墨烯移動後之奈 米複合材料結構,進行5ps 的平衡時間,平衡過程中讓石墨烯與聚酰亞胺分 子自由運動進行位置的調整,當結構達穩定的狀態,計算此抽出距離時奈 米複材結構模型的能量,接著重複此步驟,直至石墨烯抽離高分子基材, 藉著方程式(3.1.9)計算石墨烯在不同抽出距離下的互動能量;相剪切應力則 是分析石墨烯抽出所需要的能量並帶入方程式(3.1.12)中求得。藉著石墨烯 抽出模擬瞭解表面改質石墨烯對相對剪切應力及互動能量的影響。