應用二氧化鈦複合奈米碳管於甲基第三丁基醚(MtBE)光催化分解反應特性研究
127
0
0
全文
(2) 誌 謝 首先感謝指導教授 袁菁老師及洪崇軒老師,不僅給予學生課業 上的指導,更提供良好的研究環境及儀器設備,對於生活上也適時關 懷及建議,使得本論文之完成更加順利。同時感謝口試委員 連興隆 老師與李家偉老師給予本論文題出寶貴之建議,使本論文更趨完善, 於此僅致誠摯謝意。 研究所的時間雖然僅有兩年,但很感謝碩士班同學好友耀增、佳 民、証耀、佳靜、京澤、君傑、琨焯,因為有了你們陪我一起努力走 過這艱辛路程。而實驗室學弟妹瑋婷、永惇、宗諭、炫妤、奕安與尚 豪,不僅給予我研究上的協助,更使得我研究所生涯更加的多采多 姿。最後感謝於這兩年中時常鼓勵我及在學術上給予建議的朋友與學 弟妹們(靜怡、柄橓、俊竹、詠翔、傑雄、登耀、志明等),大家都是 我一生中深刻的回憶與永遠的情誼。 最後,感謝我的父母親順治、美惠,給予我成長過程豐富的資源、 與精神上的資持,使得我順利取得學位,謹致上最深的感激。.
(3) 應用二氧化鈦複合奈米碳管於甲基第三丁基醚(MtBE) 光催化分解反應特性研究 指導教授:袁菁 1、洪崇軒 2 1. 2. 國立高雄大學土木與環境工程學系 國立高雄第一科技大學環境與安全衛生工程學系 學生:黃琬鈴 國立高雄大學土木與環境工程系碩士班 摘要. 本研究旨在探討以溶膠凝膠法製成之 TiO2/CNTs 複合光觸媒,光催化分解甲基第 三丁基醚(methyl tert-butyl ether, MtBE)之反應特性與反應分解途徑。系列實驗係在批分 式光催化反應器中進行,研究中測試觸媒配比、水蒸氣濃度(0~880 μM)、反應溫度(30~90 o C)等實驗參數對 MtBE 光催化降解效能之影響。實驗結果顯示,適度添加奈米碳管之 二氧化鈦光觸媒,其反應速率高於單純二氧化鈦為光觸媒時的反應速率,最高可達單純 二氧化鈦為光觸媒時 MtBE 分解速率之 1.7 倍。促進光觸媒活性的原因,可能與奈米碳 管增加吸附濃縮作用及增生官能基有關;另外,適當的水蒸氣含量,具有促進 MtBE 分 解的作用,但當水蒸氣濃度過高時,因水氣與反應物競爭活化位址,而導致分解效率降 低;在反應溫度影響方面,反應溫度低於 75oC 時,MtBE 反應速率均隨溫度增高而提 升,但較高溫時反而減緩 MtBE 的反應速率,反應溫度的改變,導致整體反應速率限制 步驟的改變,可能為其原因。 此外,對於操作條件與產物種類及選擇性影響方面, MtBE光催化分解之主要有機 產物為甲酸第三丁基酯(tert-butyl formate, TBF)、第三丁基醇(tert-butyl alcohol, TBA)、 丙酮(acetone)。本研究發現,TBF、TBA的生成選擇性TiO2/CNTs配比和水氣濃度提高 而增加,但acetone選擇性隨水氣增加而下降。. 關鍵字:MtBE、溶膠凝膠法、光催化、二氧化鈦、奈米碳管、溫度、水汽. i.
(4) The photocatalytic reaction characteristics of MtBE assisted by TiO2/CNTs composite photocatalysts Advisor: Dr. Yuan, Ching Department of Civil and Environmental Engineering National University of Kaohsiung Co-Advisor: Dr. Hung, Chung-Hsuang Department of Safety Health and Environmental engineering National Kaohsiung First University of Science and Technology Student: Huang, Wan-Ling Department of Civil and Environmental Engineering National University of Kaohsiung ABSTRACT This research was to investigate the properties and degradation pathway of the photodegradation of methyl tert-butyl ether (MtBE) with the titanium dioxide/carbon nanotubes (TiO2/CNTs) composite photocatalyst fabricated by the sol-gel process. Series of experiments were conducted using a batch photocatalytic reactor. During the experiments, several experimental parameters were tested to analyze their effects towards the photodegradation efficiency of MtBE. These experimental parameters included the ratio of photocatalysts, concentration of water vapor (0~880 μM) and reaction temperature (30~90 o C). The experimental results indicated that appropriate addition of CNT to TiO2 photocatalyst would accelerate the reaction rate. The reaction rate with the addition of CNT was higher than the reaction rate with only TiO2 acting as the photocatalyst. Besides, the degradation rate of MtBE with the appropriate addition of CNT was 1.7 times higher than the degradation rate of MtBE with only TiO2. The addition of CNT would promote the activation of photocatalysts because CNT might increase the absorption, concentration and availability of functional groups. Additionally, appropriate amount of water vapor could enhance the degradation of MtBE. However, when the concentration of water vapor was too high, the water vapor might compete with the reactant for binding to the active site. This incident would cause the decrease in degradation efficiency. In the aspect of reaction temperature, the reaction rate would increase proportionally with the temperature when the temperature was below 75oC. However, the reaction rate of MtBE was impeded by higher temperature. It was ii.
(5) suggested that the changes of temperature might cause the changes in overall reaction rate, which inhibited the process of degradation. Moreover, the experimental condition, product and selectivity also had effects on the reaction. During the photodegradation of MtBE, the major organic byproducts were tert-butyl formate (TBF), tert-butyl alcohol (TBA) and acetone. This research revealed that the selective generation of TBF and TBA were enhanced by a higher ratio of TiO2/CNTs and elevated water vapor concentration, but the selective generation of acetone was decreased as the concentration of water vapor was increased. Keyword: MtBE, Sol-gel, Photocatalysis, TiO2, Carbon nanotubes, Temperature, Water vapor. iii.
(6) 目錄 ………………………………………………………………... i. 英文摘要 ………………………………………………………………... ii. 目錄. ………………………………………………………………... v. 表目錄. ………………………………………………………………... vii. 圖目錄. ………………………………………………………………... viii. 摘要. 第一章 緒論 1.1 研究緣起……………………………………………………………... 1. 1.2 研究目的……………………..………………………………………. 3. 第二章 文獻回顧 2.1 甲基第三丁基醚特性………………………………………………... 5. 2.1.1 甲基第三丁基醚來源與性質………………………...…………... 5. 2.1.2 甲基第三丁基醚之危害性…………………………...…………... 5. 2.1.3 甲基第三丁基醚之處理技術………………………...…………... 8. 2.2 二氧化鈦光觸媒特性解析………..…………………………………. 10. 2.2.1 二氧化鈦結構特性……………………………...………………... 10. 2.2.2 二氧化鈦之製備方法……………...……………………………... 12. 2.2.3 二氧化鈦光催化反應原理……………………………………….. 14. 2.2.4 光催化之反應動力模式………………………………………….. 23. 2.3 奈米碳管特性………………..………………………………………. 25. 2.3.1 奈米碳管結構及性質………………...…………………………... 25. 2.3.2 奈米碳管製備……………..…………………………………….... 28. iv.
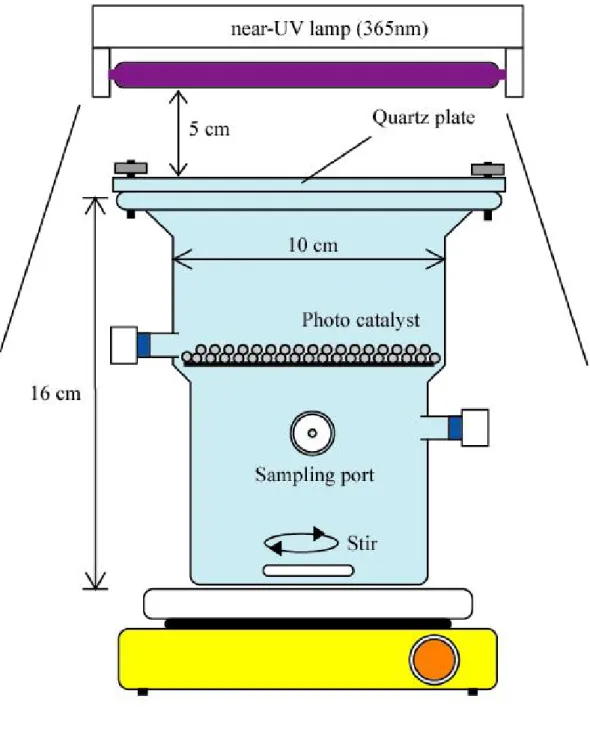
(7) 2.3.3 奈米碳管之純化及應用………………………………..……….... 30. 2.3.4 影響吸附之因素…………………………...……………………... 34. 2.4 複合光觸媒材料製備……………………………………..…………. 36. 第三章 研究方法 3.1 實驗設備………………………………………………..……………. 42. 3.2 實驗材料及製備方法………………………………………………... 43. 3.2.1 實驗材料……………………………...…………………………... 43. 3.2.2 光催觸媒製備方法……………………………………………….. 45. 3.3 實驗方法……………………………………………………………... 47. 3.4 MtBE 及產物分析方法…………………………………………….. 50. 3.5 量子效率……………………………………………………………... 52. 3.6 分析方法之品保品管………………………………………………... 54. 3.6.1 產物與標準品之比對…………………………………………….. 54. 3.6.2 MtBE 檢量線之配製…………………………………………….. 54. 第四章 結果與討論 4.1 複合觸媒 TiO2/CNTs 之表面特性…………………………………... 56. 4.1.1 掃描式電子顯微鏡(SEM)掃描結果…………...……………….... 56. 4.1.2 穿透式電子顯微鏡(TEM)分析結果……………………………... 56. 4.1.3 表面化學成分分析結果………………………………………….. 59. 4.1.4 BET 比表面積檢測結果………………………………………….. 63. 4.1.5 FTIR 之檢測結果…………………………………………………. 63. 4.2 光催化反應系統特性測試…………………………..………………. 65. v.
(8) 4.2.1 反應器密閉性測試……………………………...………………... 65. 4.2.2 反應系統吸附測試…………………….......................................... 66. 4.2.3 均相光反應測試.............................................................................. 69. 4.2.4 光活性持續性測試………………………...……………………... 69. 4.3 TiO2 與 CNTs 配比對 MtBE 光催化分解速率之影響……………... 72. 4.4 濕度與 MtBE 光催化分解速率關係……………………………….... 80. 4.5 反應溫度與 MtBE 光催化分解速率關係………………………….... 84. 4.6 MtBE 光催化分解之產物分析及反應路徑……………..………….. 89. 第五章 結論與建議 5.1 結論…………………………………………………………………... 96. 5.2 建議事項……………………………………………………………... 97. 參考文獻………………………………………………………………….. 98. 附錄 A MtBE 反應物與各產物之檢量線………………………………. 105. 附錄 B MtBE 與中間產物之圖譜.……………………………………... 108 附錄 C 各操作條件下之實驗值……………………..………………….. 110. vi.
(9) 表目錄 表 2.1 MtBE 主要之物理及化學特性…………………………………. 6. 表 2.2 金紅石與銳鈦礦之物理特性比較……………………………….. 11. 表 2.3 不同觸媒製備方法之優缺點…………………………………….. 13. 表 2.4 單層奈米碳管及多層奈米碳管之比較…………………………... 27. 表 2.5 奈米碳管製程中四種方法之比較……………………………...... 28. 表 2.6 奈米碳管吸附去除有機污染物之比較………………..……...…. 33. 表 3.1 商業奈米碳管之基本理化特性………………………………….. 44. 表 3.2 操作參數條件表………………………………………………...... 50. 表 3.3 GC/FID 分析 MtBE 及產物之滯留時間……………………….. 52. 表 4.1 不同碳管添加比例之 TiO2/CNTs 複合觸媒比表面積檢測結果... 63. 表 4.2 MtBE 光催化降解之操作參數條件及結果……..………………. 73. 附表 C.1 密閉實驗測試………………………………………………….. 111 附表 C.2 反應系統吸附實驗…………………………………………….. 111 附表 C.3 各操作條件下之光催化實驗結果…………………………….. 111 附表 C.4 產物分析……………………………………………………….. 115 附表 C.5 光催化分解產物選擇性……………………………………….. 116. vii.
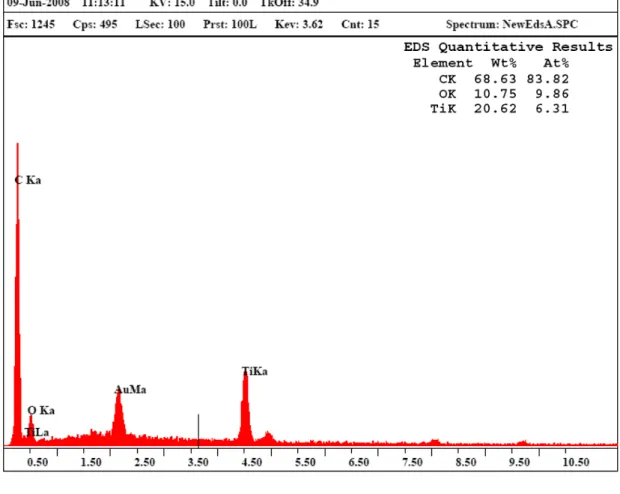
(10) 圖目錄 圖 2.1 光催化分解 MTBE 反應路徑……………………………...……. 9. 圖 2.2 金紅石與銳鈦礦之晶格型態……………………………………. 11. 圖 2.3 受光後光觸媒電子電洞傳遞示意圖……………………………. 13. 圖 2.4 碳之同素異形體…………………………………………………. 26. 圖 2.5 奈米碳管之型態…………………………………………………. 27. 圖 2.6 碳管生長機制……………………………………………………. 29. 圖 3.1 光催化分解 MtBE 研究實驗架構圖………………………….... 40. 圖 3.2 批次式光催化反應系統示意圖…………………………….….... 41. 圖 4.1 溶膠凝膠製備二氧化鈦之 SEM 圖…………………………….... 57. 圖 4.2 溶膠凝膠製備 TiO2/CNTs 複合觸媒之 SEM 圖…………………. 57. 圖 4.3 TiO2/CNTs 複合觸媒之 EDS 元素分析…………………………. 58. 圖 4.4 不同複合比例之 TiO2/CNTs 觸媒 TEM 圖………..……………. 61. 圖 4.5 為 TiO2/CNTs 觸媒之 XRD 分析圖…………………..…………. 62. 圖 4.6 TiO2/CNTs 觸媒之 FTIR 分析圖.................................................... 64. 圖 4.7 批次式光催化反應器之壓力隨時間遞變情形…………………. 67. 圖 4.8 MtBE 於反應系統吸附測試結果…………………….…….….... 68. 圖 4.9 MtBE 均相光分解實驗測試結果………..…………………….... 70. 圖 4.10 MtBE 光活性持續性測試濃度對數曲線隨時間變化之趨勢圖. 71. 圖 4.11 複合觸媒配比與光催化分解 MtBE 之殘餘率影響……………. 75. 圖 4.12 MtBE 光催化分解反應速率常數隨觸媒比例之變化圖………. 76. 圖 4.13 TiO2/CNTs 複合光觸媒光催化反應機制圖……………………. 79. viii.
(11) 圖 4.14 水蒸氣濃度對光催化分解 MtBE 之殘餘率影響………………. 81. 圖 4.15 光催化分解 MtBE 之反應速率隨水蒸氣濃度變化趨勢圖……. 82. 圖 4.16 反應溫度對光催化分解 MtBE 殘餘率之影響…………………. 85. 圖 4.17 光催化分解 MtBE 之反應速率溫度變化趨勢圖………………. 86. 圖 4.18 MtBE 光催化分解反應各產物隨反應時間遞變關係圖……...... 93. 圖 4.19 觸媒配比對 MtBE 光催化分解產物選擇性之影響……………. 94. 圖 4.20 MtBE 光催化分解反應路徑……………………………….……. 95. 圖 A.1 MtBE 檢量線…………………………………………………….. 106 圖 A.2 Acetone 檢量線…………………………………………………... 106 圖 A.3 TBA 檢量線……………………………………………………… 107 圖 A.4 TBF 檢量線……………………………………………………… 107 圖 B.1 MtBE 與中間產物之圖譜……………………………………….. 109. ix.
(12) 第一章 緒論 1.1 研究緣起 自1970年代世界各國陸續採用無鉛汽油以來,為提昇汽油的燃燒效 率,降低燃燒未完全空氣污染物的排放,各類的汽油添加劑也被大量地 採用[環保署,2004]。在所使用的各種汽油添加劑中,甲基第三丁基醚 (Methyl tert-butyl ether, MtBE)為目前使用最為廣泛的汽油添加劑,MtBE 具有製造及運輸儲存容易、製造成本低、摻配於汽油中簡易等特性,對 於降低汽機車尾氣一氧化碳及碳氫化合物排放,有顯著的助益。不過, 也由於MtBE廣泛地使用,尤其在不當的儲存與運作下,雖然解決了早期 汽車尾氣含鉛的問題,但MtBE在環境中流佈的現象,衍生另一個環境危 害 的 問 題 , MtBE 已 成 為 除 了 苯 (benzene) 、 甲 苯 (toluene) 、 乙 苯 (ethylbenzene)、二甲苯(xylene)等揮發性空氣污染物外,另一個因汽機車 所導致危害空氣品質的有害空氣污染物(hazardous air pollutants, HAPs)。 由於MtBE常因為地下儲油庫或貯油槽破裂而滲漏至環境中,因其高水溶 性,MtBE容易隨著地下水流動而污染地下水源,衍生出飲用水污染的問 題。 MtBE在室溫下為無色之低黏度液體,具高揮發性有機物,在高溫環 境下有刺鼻味,且具有高水溶解度。高濃度暴露可能造成頭痛、噁心、 頭昏眼花等症狀[物質安全資料表,2007]。美國EPA將MtBE列對一般動 物具有致癌危害性,導致肝細胞線瘤、腎小管細胞瘤、睪丸癌、血癌或 淋巴腫瘤等癌症的發生[USEPA, 1999]。至於對人體健康之影響,吸入或 接觸MtBE時,除了可能造成刺激或導致眼睛和皮膚灼傷外,MtBE蒸氣. 1.
(13) 也可能會導致暈眩或窒息。MtBE已被證實對動物確實具有致癌性,對於 人體的健康也疑似具危害性。許多國家已陸續訂定相關法規,以減少 MtBE汽油添加劑的使用,例如:美國許多大城市於2001年起,陸續宣佈 限制在汽油中添加MtBE,但是MtBE目前在台灣地區,仍持續被使用中。 如前述,由於 MtBE 具有高揮發性,其可能經由製造、輸送、儲存、 使用等過程中,逸散至大氣環境中,而危害環境,MtBE 的防治技術逐漸 受到重視。由於 MtBE 的生物分解性不高,且不易被吸附所去除,故應 用高級氧化程序來去除 MtBE,被認為較具可行性的控制技術[Carp et al., 2004] 。 在 近 年 來 研 究 中 , 結 合 紫 外 光 照 射 之 光 催 化 氧 化 技 術 (photocatalytic oxidation process),受到普遍的重視,其對於去除中、低濃 度的揮發性有機物,具有不錯的成效。基於此,本研究中探討應用光催 化氧化技術於 MtBE 的可行性並探討其相關的反應特性。 光催化氧化程序係一種利用在特定光源照射,伴隨固體光觸媒下的 氧化程序[Fujishima and Honda, 1972]。所謂光觸媒,其本質上屬於半導體 金屬氧化物,其具有在適當的光能量照射下,可在觸媒表面產生電子和 電洞對(electron-hole pairs)的特性,除了可以直接與吸附於觸媒上的污染 物進行反應外,也可能衍生具高氧化能力的自由基(如:OH·),來破壞分 解有機污染物。 在各種半導體氧化物中,由於二氧化鈦(titanium dioxide, TiO2)具有穩 定性高、活性適合、能階距適當、價格便宜、毒性低等特性,為目前在 光催化反應的相關研究中,最常使用之光觸媒。不過,由於單純TiO2的 電子-電洞能階距介於3.0~3.2間,故必須在近紫外燈光源下,才能夠被激. 2.
(14) 發而產生電子-電洞對。因此,許多學者致力於研究改良TiO2的光吸收波 長,希望能夠將可激發TiO2的光源,從紫外光紅移至可見光。相關文獻 中除了添加其它無機離子(如:金屬、氮原子等)於TiO2結晶物中外[Arana et al., 2003;Piera et al., 2003;Di et al., 2002;Bahnemann et al., 2002; Liqiang et al., 2006],結合奈米碳管(carbon neon tubes)的使用,也可能可 行的方式之ㄧ[Vincent et al., 2002]。 由於奈米碳管已被證實具備許多優異之特質,如:機械特性、電極 特性、高度化學及熱穩定性、大比表面積,常應用於複合材料的補強、 電場放射、奈米設備、氣體吸附等用途,也常作為觸媒的載體 [Serp et al., 2003]。基於此,本研究擬利用奈米碳管為TiO2的載體,製備成含奈米碳 管之TiO2複合光觸媒(TiO2/CNTs),期能提高TiO2光觸媒分解有機污染物 的活性。. 1.2 研究目的 本研究以 MtBE 為目標污染物,探討 TiO2/CNTs 複合觸媒催化分解 MtBE 之效能,並測試觸媒配比、溫度、水氣含量等反應參數對 MtBE 分 解效能之影響。另外,研究中也分析光催化分解反應過程中,所衍生的 分解產物,藉此解析其可能的分解途徑。針對本研究之主要研究目的, 彙整如下: 1. 製備與篩選出反應速率較高之 TiO2/CNTs 複合光觸媒。 2. 探討 TiO2/CNTs 複合光觸媒之表面結構特性與 MtBE 光催化分解反應 速率之關係。. 3.
(15) 3. 測試不同複合觸媒配比之光觸媒,反應溫度、溼度等反應參數,對 MtBE 光催化氧化速率之影響。 4. 鑑定與分析 MtBE 光催化分解反應產物,並藉以探討分解途徑與分解 產物之選擇性。. 4.
(16) 第二章 文獻回顧 2.1 甲基第三丁基醚特性 2.1.1 甲基第三丁基醚來源與性質 甲 基 第 三 丁 基 醚 (methyl tert-butyl ether , MtBE) 的 化 學 式 為 (CH3)3COCH3,分子量為 88,在室溫下為無色之低黏度液體,屬於高揮 發性有機物,在室溫時無色,但在高溫環境下有刺鼻味等特性,為目前 最常見的汽油含氧添加劑。MtBE 的製造方法,主要係利用異丁烯和甲醇 以強酸性離子交換樹脂催化下,產生化學反應所成,其反應式如下: CH3 CH3OH+C-CH. CH3 acidic. CH3-O-C-CH3. catalyst. CH3. CH3. 由於 MtBE 是屬於極性分子,具較高的水溶性及低分配係數 (KOW), 故一旦擴散在地下水體中,易造成地下水體的污染。另外,也由於 MtBE 不容易被微生物所分解,故 MtBE 可在地下水環境中的停留時間較長, 表 2.1 為 MtBE 之主要物理及化學特性。. 2.1.2 甲基第三丁基醚之危害性 MtBE 目前仍是使用最普遍之含氧添加劑,由於 MtBE 具有高揮發 性,故其製造、混合、輸送、儲存、使用等過程,皆能使 MtBE 極可能 逸散至空氣中。MtBE 對於水中生物具危害性,其可經由交通工具的排 放、蒸發損失以及加油過程的逸散排放等方式,污染其鄰近區域。. 5.
(17) 表 2.1 MtBE 主要之物理及化學特性 CAS.NO. 1634-04-4. 中文名稱 一般名稱 物理態 pH 值 自燃溫度 蒸氣壓(25℃) 密度(20℃). 甲基第三丁基醚 MtBE;2-Methoxy-2-methl-propane 澄清無色 可能為中性 193 ℃ 245 mmHg 0.7404 g/mol 11 1.04 似醚味 - 109 ℃ 55.2 ℃ -28 ℃(易燃). KOC Log KOW 氣味 溶點 沸點 閃火點 爆炸界限 蒸氣比重(空氣=1) 亨利常數. 2.5-15.1% 3.04 -4 5.5×10 atm m3/mol 0.32-0.47 mg/m3 23576 ppm/4H(大鼠,吸入) 141 g/m3/15M(小鼠,吸入). 嗅覺閾值 LC50. 40 ppm. 八小時日時量平均容許濃度(TWA). 另外,地下儲油槽附近之土壤及地下水,為另一個可能遭受 MtBE 污染的區域。其污染可來自運輸管路之洩漏、儲存槽之洩漏、以及工廠 廢水之排放等。此外,在非點源污染源方面,空氣中之 MtBE 可能因降 雨而被帶入水體中,而都市雨水的地表逕流沖刷亦可能成為非點源污染 源,而水上交通工具使用添加 MtBE 之燃料時,也可能會造成水體污染。 根據美國賓州環境資源部的研究,埋設五年以上的地下儲油槽有 46%會發生洩漏,而埋設時間超過十年,其機率更高達 71%[高雄市環保. 6.
(18) 局,2007]。油品種類及成分相當複雜,一旦洩漏會造成土壤及地下水的 嚴重污染,明顯地改變土壤的正常機能。林春賓[2000]於 10 個加油站周 界環境 MtBE 濃度進行監測,結果顯示加油站周界空氣中 MtBE 濃度日 平均值,約介於 30.3~104.7 μg/m3 間。而一般在車流量或發油量較高的加 油站周界,其 MtBE 濃度值也較高。另外,經調查臺北市道路鄰近地區, 以及加油站空氣中的揮發性有機化合物濃度之結果顯示,公民營加油站 內空氣中 MtBE 濃度介於 61.2~1527.6 μg/m3,而加油站員工個人暴露於 MtBE 濃度,約介於 250.4~3087.1 μg/m3 間[范姜威凱,1999]。 根據MtBE物質安全資料表得知,人體可藉由吸入或接觸等方式,造 成眼睛和皮膚的刺激的傷害,而且過高的MtBE蒸氣會導致暈眩或窒息。 高濃度暴露可能造成頭痛、噁心、頭昏眼花等症狀。在致癌方面,MtBE 已被證實對動物確實具有致癌性。美國各大城市皆已陸續訂定相關法 規,以減少汽油添加劑的使用,例如:美國芝加哥、紐約及加州分別在 2001 年、2004 年、2002 年12月前,禁止在汽油中添加MtBE[環保署, 2001];美國EPA將MtBE列為疑似致癌物,其對一般動物具有致癌之危 害,如肝細胞線瘤、腎小管細胞瘤、睪丸癌、血癌或淋巴腫瘤等[Buckley, 1997]。另外,MtBE也可能導致地下水源的污染,衍生飲用水質不良的問 題[Johnson,1998],USEPA建議,飲用水中MtBE的濃度不要超過20~40 μg/L [USEPA,1999],另外,台灣環保署亦已將 MtBE 列管為第四類毒 性化學物質。. 7.
(19) 2.1.3 甲基第三丁基醚之處理技術 處理MtBE污染之技術可分為生物、物理及化學處理方法,生物處理 方面有:曝氣法、生物降解[陳谷汎,2005]、自然衰減法[郭雅玲,2006]、 活性污泥曝氣、生物滴濾塔法[蘇立群,2005];而物理及化學部分有濾膜 法、活性碳吸附、氣提法、高級氧化法[Sutherland,2003]、抽取法、透 水性反應牆[林財富,2002]、填充床或者電化學法等。這些方法仍有缺失, 例如生物處理法的降解速率慢,活性碳吸附對於MtBE之去除效果不佳。 研發適用於MtBE的去除技術有其必要性。 近年來紫外光結合半導體觸媒光催化技術,能有效地應用於環境污 染的問題,相關於應用光催化氧化技術於 MtBE 分解之研究,彙整如下: 賴瑞明[2003]利用近紫外光結合環型反應器光,探討二氧化鈦光催化分解 MtBE 蒸氣之效能。結果顯示,微量水氣存在時,有利於 MtBE 之光催 化分解;當氧氣濃度小於 5%時,MtBE 的反應速率著氧氣濃度之提高而 增加,但氧氣高於此濃度時,MtBE 反應速率便趨於平緩而維持一定值, 反應為典型異相觸媒反應之反應特性;而在溫度影響方面, MtBE 之反 應速率,皆隨著反應溫度的提高而有增加之趨勢。 Elena 等[2004]以 O2/Ar 80:20 之氣體、TiO2 光觸媒、或合併光催化及 超音波進行處理水中之 MtBE。由研究結果指出,MtBE 之降解速率成一 階反應,而在不同之條件下,由產生中間產物之過程是較占優勢的; Preis 等[2005]探討不同反應溫度下,MtBE 之分解情形。其發現 MtBE 之光催化分解產物有水、丙酮、二氧化碳及一氧化碳,但 MtBE 單純熱 氧化後的產物,則有:2-methyl-1-propene(2-MP)、一氧化碳、二氧化碳. 8.
(20) (CH3)3-OCH3 MTBE. OH.. OH. CH3OC(CH3)2CH2.+ H2O. (CH3)3C-OCH2.+ H2O O2. O2 CH3O(CH3)2CCHO. (CH3)3C-OCHO2.. MMP. OH. (CH3)3COCHO + H2O.. OH.. (CH3)3COCO.+ H2O. TBF. O2/H+ H2O CH3COCH3 + HCHO + CO2. (CH3)3COH + HCOOH. Acetone. TBA. 圖 2.1 光催化分解 MTBE 反應路徑 [Park et al., 2003] 及水。而圖 2.1 為光催化分解 MtBE 之反應路徑[Park et al., 2003]。 Orlov 等[2006]將傳統之 TiO2 添加金改質後,進行 MtBE 之光催化分 解實驗,其發現添加金的光觸媒比原本觸媒,增加三倍的反應速率; Kuburovic 等[2006]比較光解、光催化及生物復育等程序,對淨化地下水 中 MtBE 的能力,發現僅需 5g/L 之 TiO2,便能在 150 分鐘內降解 91% 之 MtBE,為三種程序中反應速率最快者;Arana 等[2007]利用 P-25 TiO2 與改質之 Cu-TiO2,結合螺旋式連續流反應器,進行 MtBE 之去除研究, 結果發現複合觸媒 Cu-TiO2 比商用 TiO2 之降解效率更佳,添加銅後,可 提高 TiO2 光觸媒活性及污染物分解後的礦化率。. 9.
(21) 2.2 二氧化鈦光觸媒特性解析 2.2.1 二氧化鈦結構特性 在 許 多 異 相 光 催 化 反 應 程 序 的 研 究 中 , 常 以 二 氧 化 鈦 (Titanium dioxide, TiO2)為測試的光觸媒(photocatalyst),其原因與TiO2具有穩定性 佳、能階距(energy gap)及活性適切、價格不高、毒性低等特性有關。TiO2 本質上屬於n型半導體,其分子結構呈閃鋅晶格,為一以鈦原子為中心, 周圍環繞6個氧原子的八面體結構,Ti以外圍3d軌域4個價電子,與氧原 子形成共價鍵。一般而言,TiO2在常溫下是無晶型結構,經鍛燒溫度至 200℃以上時,易呈現Anatase晶型,若再加熱至約400~500℃左右,Anatase 晶型將有部份轉移為Rutile 晶型,上述兩晶型之詳細物理特性列於表2.2。 如前述,常見的TiO2結晶型態,一般可分為銳鈦礦(Anatase)、金紅石 (Rutile)、板鈦礦(Brookite)等形態,而銳鈦礦與金紅石晶型之TiO2最常應 用於光催化反應研究中。銳鈦礦晶型態所形成的八面體結構,較為歪斜, 結晶對稱性不若金紅石,而其結晶中的Ti-Ti間距,則較金紅石為寬;但 其Ti-O的間距,則較金紅石為窄[Burdett , 1987]。由於銳鈦礦和金紅石結 晶結構(見圖2.2)的差異,使得擁有不同的費米能階,兩者的能階分別是 3.2與3.0 eV。銳鈦礦的導電帶高於金紅石的導電帶,而其價電帶低於金 紅石的價電帶。銳鈦礦二氧化鈦的電子-電洞對再結合速率,較金紅石 二氧化鈦為低,故一般以銳鈦礦具有較強的氧化能力 [Hashimoto, 1997]。 如前述,TiO2的能隙值約在3.2 eV左右,其可依據 λ= 1240/Eg關係來 推估之。其中,λ為入射波長(nm),Eg為能隙值。據此推估,激發TiO2成 為光觸媒的臨界波長約為380 nm,其屬於近紫外光的範圍[吳炳佑,1997]。. 10.
(22) 圖2.2 金紅石與銳鈦礦之晶格型態[Linsebigler et al., 1995]. 表 2.2 金紅石與銳鈦礦之物理特性比較[Linsebigler et al., 1995] 物理特性. 金紅石. 銳鈦礦. 密度. 4.25 g/cm3. 3.89 g/cm3. 折射率. 2.52. 2.71. 硬度. 5.5~6.0. 6.0~7.0. 能隙. 3.0 eV. 3.2 eV. 等電點. 5.6. 6.1. 溶點. 1858℃. 轉變為高溫金紅石. 光活性. 次之. 優. 11.
(23) 2.2.2 二氧化鈦之製備方法 文獻中常見之 TiO2 製備方法,主要包括:溶膠凝膠法(sol-gel)、化學 氣 相 沈 積 法 (chemical vapor deposition, CVD) 、 含 浸 法 (impregnation method)、陽極氧化法(anodic oxidation)等,表 2.3 比較前述各種觸媒製備 方法的優缺點,並針對上述製備方法的原理簡述如下: 1. 溶膠凝膠法:溶膠(Sol)的定義為極小的膠體粒子,在凡得瓦爾力(Van der Waals’ attraction)及電雙層作用下,均勻分散於液相中,此時即稱 為「溶膠」 。溶膠凝膠法常利用高純度鈦原料,與特定的醇類溶劑與 酸觸媒,經進行水解縮合反應後生成。將前述溶膠態溶液,進一步 烘乾脫水,即可得到凝膠態溶液,如此可製備均勻性高且粒徑微細 之觸媒。不過,製備過程中所使用之鈦原料、溶劑及酸觸媒等種類 皆會直接影響溶液中 TiO2 的粒徑特性。例如;文獻中,Kato 等 [1993] 以烴氧化物溶液製備 TiO2,其研究結果發現 TiO2 的結晶型態、粒徑 大小與比表面積,皆與鍛燒溫度有關,當鍛燒溫度介於 400~600oC 時,結晶型態以單一的銳鈦礦為主,而當鍛燒溫度達 700oC 以上時, 金紅石結晶型態逐漸形成;洪雨利[2004]利用四異丙基化鈦與正丙醇 製備 TiO2,其指出 TiO2 比表面積及孔隙直徑,隨鍛燒溫度增加而降 低。 2. 化學氣相沈積法:係利用氣相化學反應方式,將反應物於特定氣相環 境中進行反應,並沉積於基材表面的一種薄膜技術,目前為許多半 導體製程常採用的薄膜製作技術。TiO2的製備的方式,係將含鈦物質 經由加熱蒸發,再與水蒸氣於高溫環境下進行反應。文獻中,Hung. 12.
(24) 表2.3 不同觸媒製備方法之優缺點 觸媒製備方法 溶膠凝膠法. 化學氣相沈積法. 優. 點. 1.較大之比表面積 2.由溫度可控制觸媒活性 3.參數易控制 1.觸媒膜厚度均勻 2.觸媒顆粒大小孔徑較能 控制. 含浸法. 1.製備過程簡單 2.不須特殊設備 3.觸媒改質容易. 陽極氧化法. 1.效率高 2.製作容易 3.觸媒膜厚度均勻、且控 制容易. 缺. 點. 1.製備過程複雜 2.耗費時間長 3.需要高溫設備 1.設備較複雜且麻煩 2.須高溫蒸鍍設備 3.基版材質受到限制 4.成本高 1.膜厚均勻性較差 2.附著能力差、易脫落 3.較不宜的金屬比例較高 4.覆膜液使用 1.需電解設備 2.表面觸媒濃度相差大. 等[1998]在以UV/TiO2程序處理三氯乙烯氣體研究中,即採用化學氣相 沈積法來製備觸媒。其步驟係利用蒸鍍設備將Ti(OC3H7)4溶液加熱至 60oC,然後與水蒸氣接觸,再以氮氣當攜行氣體將含TiO2氣流導入反 應器中,直接在反應器上生成TiO2,反應器外部保持300oC加熱,最後 TiO2薄膜再以550oC高溫鍛燒,其研究顯示所製備TiO2,具有不錯的催 化活性;Lars等[1998]在利用CVD法製備TiO2的研究中發現,溫度與 TiO2薄膜成長速率有密切關係,當溫度為950oC時,薄膜成長速率可達 5 μm/hr。 3. 含浸法:係製備觸媒最簡單的方法之ㄧ,製備時,先調配好適當配比. 13.
(25) 之金屬化合物與觸媒溶液,然後將載體浸漬其中,進行拉提動作,再 經過烘乾、鍛燒、再浸漬等程序,即成。一般而言,以含浸法製備觸 媒的方法又可區分為兩種,其一是將載體浸入溶液中,不同量的載體 使用不同組成濃度之溶液;另一種方式則是將載體與適當的溶液混合 成泥漿狀。 4. 陽極氧化法:利用電化學原理,將鈦基材置入溶液中進行電解反應, 於陽極上生成 TiO2 薄膜,影響氧化膜生成的因素包括:電解質種類、 施加電壓、反應溫度、電解質濃度、電解時間及施加電流密度等。此 類鈦金屬氧化膜常具有孔洞多、厚度小於 10 μm、附著力強等特性。 相關文獻中,David 等[1994]將 Ti(0001)陽極氧化於 0.1N 之 H2SO4 溶 液當中,以每秒 0.1 mV之速率加電壓至 9 V 觀察其氧化膜的形成。 其發現,所獲得的 TiO2 大多呈銳鈦礦結晶形態,而氧化 Ti(1100)也得 到同樣結果,但氧化 Ti(1120)所得到結晶中銳鈦礦及金紅石晶型各佔 一半。 5. 市售之二氧化鈦以Degussa P-25 TiO2已為目前光催化反應研究所廣泛 採用。P-25 TiO2其製備係在氧氣和氫氣存在下,於1,200oC高溫火燄下 水解TiCl4而形成,其主要結晶型態為銳鈦礦,約佔75%,其它則為金 紅石,其比表面積和平均粒徑分別約為50 m2/g和20 nm。. 2.2.3 二氧化鈦光催化反應原理 2.2.3.1 二氧化鈦光催化原理 在TiO2光催化反應系統中,當入射光子的能量超越其能階隙時,電 子會由價電帶提升至導電帶,在導電帶上形成具活性電子之激發態,並 14.
(26) 在價電帶生成「電洞」(如圖2.3所示)。導電帶的電子與價電帶的電洞皆 可能會參與反應。導電帶上之電子具有還原能力,價電帶上之電洞則具 有氧化分解觸媒上污染物的能力。. 圖2.3 受光後光觸媒電子電洞傳遞示意圖 [Linsebigler, 1995] 相對地,當光觸媒表面沒有可資參與氧化還原反應的物種存在時, 導電帶電子及價電帶電洞再重新結合(recombination),並釋放出光及熱 能。此外,除了電子-電洞對可能直接參與氧化還原反應外,光催化反應 會因電子-電洞對與水分子、氧氣或者有機物之相互反應,因而衍生許多 高反應性自由基,如:‧O2-、‧O3-、‧O3-3、‧O、‧H、‧OH-、‧HO2、‧Cl 等 [Chiang et al.,1966;Cundall et al., 1976;Meriaudeau et al., 1976]。 Fox[1993]及Kamat[1993]經由實驗發現二氧化鈦光催化分解有機 物,有二條主要路徑,分別是以價電帶電洞(h+)直接氧化反應,以及以氫. 15.
(27) 氧自由基為主之間接氧化反應。電洞氧化吸附於二氧化鈦表面的水分子 形成氫氧自由基{TiIVOH}+,為一可分解有機物的強氧化劑。從Fukuzawa 和Kwan [1968]的研究指出,‧O2-的形成是吸附於TiO2 表面的O2 捕集及 導電帶電子後所產生;‧OH-則為TiO2 表面水合官能基被價帶帶電動或 水分子激發電子攻擊所致,Cundall等[1976]在添加H2O2 之液相光催化 2-propanol 之實驗中,提出以下自由基之反應機制: OH-(surface)+ hvb+ → ‧OH. (2.1). O2(ads)+ ecb- → O2-(ads). (2.2). O2-(ads)+ H2O → OH- + ‧O2H. (2.3). 2HO2‧ → O2 + H2O2. (2.4). H2O2(ads)+ OH- + ecb- + hvb+ → OH- + 2‧OH. (2.5). 另外,Okamoto等[1985]指出氫氧自由基的來源除電洞外,還可由電 子還原獲得,其反應過程如下: etr- + O2 → O2-‧. (2.6). O2-‧ + H+ → HO2‧. (2.7). 2HO2‧ → H2O2 + O2. (2.8). H2O + hν → 2OH‧. (2.9). H2O2 + O2-‧ → OH‧+ OH- + O2. (2.10). 這些活性極高之自由基,除了可以促使更多氧化還原連鎖反應的進 行外,亦提供更多吸附於光催化觸媒上有機污染物分解之途徑,達到去 除有機污染物之效果。. 16.
(28) 2.2.3.2 光催化反應機制 光催化反應的進行,應包括下列四個步驟[Schiavello and Sclafani, 1989]: 1. 光激發觸媒表面形成電子-電洞對。光觸媒接受適當光能後,價電帶電 子受激發後,躍過能隙到達導電帶,價電帶因缺乏電子而形成電洞, 此時導電帶與價電帶則形成所謂的電子-電洞對,電子-電洞對具有高 電子傳送能力,可促使許多氧化還原反應的進行。 2. 電子-電洞對與觸媒表面上之吸附物,發生氧化還原反應。此過程決定 反應路徑與反應產物,而且電子-電洞對也可與氧氣或水氣相互反應, 形成高反應性的自由基,此類自由基常可加速反應的進行。 3. 電子-電洞對的再結合。上述的電子-電洞對若未能與光觸媒表面上之 吸附物,產生氧化還原反應,則電子與電洞將再結合,同時以熱或光 的形式,釋放出先前被觸媒吸收的光能。 4. 反應產物脫附離開光觸媒表面。此為維持光觸媒的活性關鍵的要素, 若產物脫附不易,將逐漸佔據觸媒表面的活性位置,遂造成觸媒的毒 化現象。 上述四個步驟說明了光催化反應之基本反應路徑,實際上光催化反 應之進行,除了與光觸媒種類、反應物種、以及所使用的光源波長有關 外,反應物被光觸媒吸附的難易、電子-電洞對相對應之氧化還原電位 等,也常是影響反應是否能順利進行的重要關鍵。. 17.
(29) 2.2.3.3 影響光催化反應之因子 影響光催化反應速率及效率因子很多,主要包括: 1. 光催化劑 (1) 粒徑與比表面積:催化劑的粒子越小,在溶液中能分散的單位質量 粒子也就越多,且增加對光的吸附干擾效果,光吸收不易飽和。不 過,較大的比表面積,反應面積增大,有利於吸附等,皆有利於反 應速率及效率相對提升。不過對於奈米微粒而言,根據量子效應的 影響,光觸媒的粒徑愈小,導電帶及價電帶的能隙反而增加,使得 導電帶與價電帶再結合速率降低,相對的光催化活性也就好。例如, Anpo 等[1997]曾提出粒徑與光催化反應量子產率之關係,其指出, 具量子尺寸大小的二氧化鈦顆粒,對水解反應的活性明顯增加,尤 其當粒徑小於 10 nm 時,量子產率迅速提高。 (2) 表面羥基:由於催化劑表面存在之羥基,會與電洞反應生成過氧化 物,具有複合中心之作用,故表面羥基數目愈少,其活性就愈高。 一般可將催化劑進行熱處理,羥基減少的同時,複合中心也減少。 (3) 混晶效應:近來光催化之研究中,發現銳鈦礦與金紅石的混晶(非機 械混合),反而會具有較高的催化活性。其原因係由於銳鈦礦晶體表 面生長了金紅石結晶薄層。因晶體結構之不同,具有效促進銳鈦礦 晶體電子與電洞分離作用。 2. 光源與光強度 由光電壓譜分析顯示,由於 TiO2 表面雜質及晶格缺陷之影響,使 其在較小波長範圍時具有光催化活性。許多研究證實,光催化效率與光. 18.
(30) 強度成正比關係,即光強度越強,反應效率越好。例如, Dibble 等[1992] 與 Raupp 等[1993]利用流體化床反應器,進行三氯乙烯光催化分解實 驗,發現氧化速率隨著光強度增加而上升,而轉化率與紫外光強度成正 比;Peterson 等[1991]的研究中指出,光觸媒之活性並非完全與光強度 成正比,在紫外光強度較低照射下,光催化反應速率與光強度的一次方 成正比;而在較高光強度照射下,其反應速率與光強度的 1/2 次方成正 比;Bahanemann 等[1991]在降解三氯甲烷實驗中發現,光催化反應降解 -5. 速率與光強度的平方根成線性關係,但在光強度大於 6×10. Einstein.. L 1.s 1 時,不具光催化效果。故光強度過高,其光催化效果不一定好。 -. -. 劉安治[1997]及吳永俊[1996]等分別研究四氯乙烯及三氯乙烯之光 催化反應,發現轉化率隨著紫外光之強度增強而上升,略成曲線,而當 光強度介於 0.45~0.65 mW/cm2 時,其反應速率及轉化率的增加幅度較 大。當反應速率與光強度呈一階線性關係,表示光催化所產生之電子電 洞消耗速率大於再結合之速率,若反應呈 1/2 階時,則是電子電洞重組 主導反應之進行;Peill 等[1996]利用光纖反應器光催化分解五氯苯酚的 研究中發現,量子效率與入射光強度有關,但過高的光強度,其量子效 率可能反而會降低。 3. 污染物濃度 光催化氧化反應速率可以用 Langmuir-Hinshelwood 動力學方程式描 述:. r=. kKC 1 + KC. (2.11). 式中 r:反應速率 19.
(31) C:反應物濃度 K:表觀吸附平衡常數 k:發生於光觸媒表面活性位置的表面反應速率常數 低濃度時,KC<<1,則上式可簡化為: r = kKC = K’C. (2.12). 即是反應速率與污染物濃度成正比。初始濃度越高,降解速率越大。而 在高濃度範圍時,反應速率則與污染物濃度無關。 4. 外加催化劑 光催化反應若要有效進行,就需減少電子電洞對之再結合,由於氧 化劑為導電帶電子有效的捕獲劑(Electron captured agent),可以有效的捕 獲光催化產生的電子,增加與電洞分離機會,可提高光量子產率。許多 研究中指出,光催化氧化速度及效率在 O2、H2O2、過硫酸鹽、高碘酸 鹽存在時明顯提高的現象。 5. 反應溫度 根據反應動力學,反應溫度之提升有助於分子碰撞機率,進而增加 反應速率。Fu等[1996]在以Pt/TiO2為光催化劑之乙烯分解研究中發現, 反應溫度的增加有助於提昇乙烯的轉化率,例如:當溫度由32℃提昇至 90℃時,乙烯之轉化率由9 % 提昇至接近100 %,由於Pt本身即是一種 熱觸媒(thermal catalyst),故Pt除了可增進TiO2的光催化反應的效能外, 同時可以在提高反應溫度的情形下,利用本身的熱觸媒特性,增進有機 污染物的破壞率,亦即可會有所謂的協同作用(synergistic effect)發生, 對整體反應速率的提昇,往往會大於個別的Pt及TiO2之催化能力。. 20.
(32) 在室溫條件下,Pt/TiO2對於乙烯之轉化能力低於TiO2,而適當提高 溫度後,反而以Pt/TiO2對於乙烯之轉化能力較TiO2為高;Hung等[2000] 研究發現,當反應溫度由45℃增至80℃時,苯的轉化率可由64.7%提昇 至接近80%,直至反應溫度提昇到150℃時,轉化率仍維持著80%,爾後 則隨反應器溫度的增加而再逐漸增加,當200℃時苯之轉化率為100 %。 根據由前述文獻中溫度效應對光催化分解有機物的研究發現,溫度 的提升對反應速率的影響,可能會產生促進、抑制、或先促進後抑制等 三種不同情況。溫度對類似PBDE、苯、甲苯等帶有苯環類的有機物, 於氣相光催化中有類似的反應趨勢,即在常溫下光催化反應速率不高, 但當反應溫度增加至一定程度時,反應速率提昇。 針對改變反應溫度對光催化分解反應之效應,其至少可以分別從影 響光催化劑之活性,以及影響光催化分解反應機制等兩個方面,來加以 歸納。對於光催化劑活性之影響,改質型的光催化劑(如:Pt/TiO2),可 能會有結合熱觸媒與光觸媒的協同作用,故可能會提昇反應速率;至於 對於反應機制之影響,則可能會因為反應溫度的改變,導致改變部份反 應產物的選擇性(selectivity),進而影響原始反應物的分解速率。 6. 水氣含量 水分子在光催化反應中扮演非常重要的角色,根據過去文獻,水氣 含量增加,可能提升或抑制光催化反應速率,但亦可能在某適當溫溼度 下,反應速率達到最大值。當水分子吸附於觸媒表面後,若經水解作用 及光能之激發後,即生成具活性的氫氧自由基(OH.),此OH.會氧化 吸附於觸媒表面之化合物而生成中間產物,並佔據觸媒表面之活性位置. 21.
(33) (activated sites),進而降低觸媒活性,還原反應速率因而受到限制。另 一種情況係根據Selloni等[1998],研究水分子在觸媒表面的吸附行為指 出,當水氣含量添加過量時,水分子雖然未被觸媒所吸附,但觸媒周圍 之水分子量累積過多,則可能會阻隔反應物與觸媒之間的吸附,影響觸 媒活性;劉安治等[1997]研究光催化氧化三氯乙烯(PCE)和四氯乙烯 (TCE)實驗結果發現,當相對濕度增加至某一濕度以上,對於氧化反應 速率和轉化率將會有明顯之降低;顧洋等[1997]研究光催化還原二氧化 碳實驗當中發現添加水氣含量至相對濕度55%以上,產物CH4之生成濃 度是有被抑制之現象;而在相對濕度55%以下,CH4之生成濃度則隨濕 度之增加而逐漸增加;洪佑良[2005]研究光催化氧化MtBE及MBDE實驗 中,結果顯示發現氧化速率隨濕度之增加而增加;但是當濕度高於165 μM時,其氧化速率隨濕度之增加而降低。 7. 氧氣濃度 光催化反應在有氧氣存在的環境中,由於氧氣為一相當良好之電子 捕捉劑,吸附在二氧化鈦表面的氧,會參與反應並消耗掉電子,使得電 子電洞重組受到抑制,因而促進光觸媒的氧化能力[Miller, 1979]。Dibble 及Raupp[1992]發現當氧濃度較低時,反應成一階反應線性關係,而當 氧氣高時,則反應成零階反應關係,此時光催化劑對氧氣的吸附應已達 飽和;Rauup[1993]在光催化分解丙酮研究中,也發現氧化速率隨著氧 濃度增加而增加,直到超過10%(莫耳比)時才有趨於定值的現象。 洪佑良[2005]研究氧濃度對MtBE光催化分解之影響,當反應的氧濃 低於5%的狀況下,反應速率隨著氧濃度提高而增加,當反應之氧氣濃. 22.
(34) 度大於5%時,MtBE 反應速率便趨於平緩。故光催化氧化反應為一典 型的異相觸媒反應,反應必需有氧氣參與,而在較低氧濃度條件下,有 機物氧化速率隨氧濃度之增加而增加,反應速率與氧氣濃度成一階反 應;隨著氧氣濃度逐漸增加後,當氧氣增加至飽和吸附濃度時,則反應 速率與氧氣濃度關係不大,分解速率與氧濃度成零階反應。. 2.2.4 光催化之反應動力模式 Langmuir-Hinshelwood (L-H)反應關係,常被應用於氣-固表面反應機 制之描述,亦常被用來闡述光催化半導體觸媒反應動力。L-H 動力關係 基本假定光催化催化劑對反應物的吸附能力較產物為強,且遵循 Langmuir 等溫吸附關係,亦即: 1. 可吸附之表面積是有限的,且其為單分子層吸附。 2. 吸附為可逆反應,最初吸附速率大於脫附速率,最後達平衡狀態。 式2.13為Langmuir-Hinshelwood (LH)單分子吸附分解動力模式:. r=. K LH KC 1 + KC. (2.13). 式中 r:反應速率 (μM/s) KLH:反應速率常數 (μM/s) C:反應物濃度 (μM) K:反應物吸附係數 (1/μM). 23.
(35) 另外,在光催化反應中,由於污染物與水分子會競爭TiO2表面上相 同的活化位置,故也可利用一般典型之雙分子反應動力式來闡述其反應 動力關係,如下:. r=−. K O2 CO2 K M C M KW CW dC M ] [ ] = K LH [ × (1 + K M C M + KW CW ) 2 (1 + K O2 CO2 ) dt. (2.14). 式中 r:光催化反應速率 (μM/g -s) KLH:反應速率常數 (μM/g -s) KM:反應物之吸附係數 (1/μM) KW:水分子吸附係數 (1/μM) KO2:氧分子吸附係數(1/μM) C:反應物之濃度 (μM) CW:水分子濃度 (μM) CO2:氧氣濃度 (μM) L-H 雙分子反應動力模式須符合以下假設條件: 1. 動力模式主要描述氣-固反應機制,假設無均相反應發生,所有的 反應皆為在TiO2表面的異相反應。 2. 假設水分子和反應物競爭TiO2 觸媒上之相同活性位址。 3. 氧氣之吸附位置與反應物、水不同,彼此不互相競爭。 4. 忽略反應產物和中間產物對反應速率之影響。 另外L-H 反應動力模式中,其反應速率常數(k)和吸附平衡常數(K), 皆為反應溫度之函數。一般而言,反應速率常數遵守Arrhenius Law [Levenspiel, 1995; Forgler, 1999; 吳榮宗, 1985],其方程式表示如下: 24.
(36) k = k ' exp(. − Ea ) RT. (2.15). 式中 k:反應速率常數(μM/s) k’:不隨溫度變化之反應速率常數(μM/s) Ea:反應活化能(kcal/mol) R:理想氣體常數(1.99×10-3kcal/mole-K) T:反應溫度(K). 2.3 奈米碳管特性 2.3.1 奈米碳管結構及性質 碳物質早在 1970 至 1980 年間即被發現,可在高溫協同金屬觸媒的 環境下,反應生成碳薄膜,此碳薄膜的分子粒徑可能低於 10 μm。另外, 經由雷射激光,使得石墨蒸發成碳灰,並以質譜儀分析之,可發現 C60 與 C70 等類似籠狀結構的物質,當時稱之為富勒希(Fullerene)或巴克球 (Bukminster)。此後,直至 1991 年,飯島澄男(Iijima, 1991)在 Nature 報告 中宣佈成功合成出一種新的碳結構-奈米碳管時,引起當時研究單位的密 切注意,紛紛將研究重心轉移到奈米碳管上,圖 2.4 顯示碳之不同同素異 形體與構造的差異。 奈米碳管為一具有奈米級直徑與長寬高比的石墨管,碳管內徑可從 0.4 nm 至數十奈米,碳管外徑則由 1 nm 至數百奈米,長度則由數微米至 數十微米間,可由單層或多層的石墨層捲曲形成中空管柱狀結構,其結. 25.
(37) 圖2.4 碳之同素異形體(a)石墨(b)鑽石(c)C60(d)CNT[Iijima,1991] 構如圖2.4所示。所形成的中空管柱狀結構主要可分為單層(single-wall carbon nanotubes, SWCNTs)碳管及多層碳管(multi-wall carbon nanotubes, MWCNTs)兩大類。依據奈米碳管截面所形成的邊緣形狀,單層奈米碳管 可分為:扶椅型(armchair)、鋸齒型(zigzag)、對掌型(chiral)等三類奈米碳 管(結構見圖2.5),其結構對稱性極高,且缺陷較少。多層奈米碳管則由 多層碳管捲起而成之同軸碳管,橫切面如同樹的年輪,各層間距為0.34 nm,層與層之間以凡得瓦爾力鍵結,結構中缺陷較多,而表2.1為單層及 多層奈米碳管性質之比較。 碳管的特性決定於石墨層的寬度與捲曲的方向,不同的捲曲方向可 以表現出碳管金屬、半金屬、半導體等特性,依碳管捲曲方向的不同, 可將奈米碳管的形態區分為三類。因不同捲曲方式所造成的碳管螺旋 性,會使管壁上的六圓環有不同的扭曲程度,而造成六圓環上未飽和雙 鍵間電子傳導的阻力。其中,不同的螺旋性,則可能造成了奈米碳管間 26.
(38) (a)單層奈米碳管. (b)多層奈米碳管 圖 2.5 奈米碳管之型態[Odom,1998;Dresselhaus,1995]. 表 2.4 單層奈米碳管及多層奈米碳管之比較 項 目. 單層奈米碳管(SWNTs). 多層奈米碳管(MWNTs). 直徑. <2 nm. 0.7~50 nm. 長度. <1 μm. >1 μm. 比表面積. >600 m /g. 40~300 m2/g. 層間距離. -. 0.34 nm. 1.3~1.4 g/cm3. ~2.1 g/cm3. 電導係數. 10-16~104 ohm-1cm-1. 104 ohm-1cm-1. 熱導係數. 2300 W/m K. 2800 W/m K. 密度. 2. 27.
(39) 導電性質的差異。因此,全由碳原子所組成的奈米碳管,可能只因結晶 結構細微的差異,便有成為導體或半導體的不同材質。奈米半導體材料 的研究範疇已成為炙手可熱的研究題材,奈米材料的其他之特殊性質, 尚可能包括:機械特性、電極特性、高度化學及熱穩定性、大量比表面 積,目前已被應用於複合材料補強[Wagner, 1998]、電場放射[Wang, 1998]、奈米設備[Collins, 1997]、氣體吸附[Dillion, 1997]、以及作為觸媒 載體[Planeix, 1994]等用途。. 2.3.2 奈米碳管製備 針對奈米碳管之合成方法有四種:弧光放電法、雷射氣化法、太陽 能法、催化劑化學氣相沉積法,不同之合成方法與合成條件會產生不同 微結構的奈米碳管,表2.5為常見奈米碳管製備方式之優缺點比較[Lu, 2004]。. 表2.5 奈米碳管製程中四種方法之比較 製程. 優點. 缺點. 弧光放電法. 產率高. 生成時雜質多. 雷射氣化法. 高純度碳管. 費時、產率低. 產率高. 費時. 太陽能法. 催化劑化學氣相沉積法 高純度、便宜、製程溫度低、產率高. 以常見的有機氣體化學氣相沈積法(chemical vapor deposition, CVD) 為例,奈米碳管製造時,其條件包括碳原子的供給、金屬觸媒的應用、 以及成長溫度的控制等,通常多層奈米碳管其成長過程有下列三個步驟. 28.
(40) (生長機制見圖2.6)︰ 1. 有機氣體分子(如:乙炔、乙烯等)經受熱後,在金屬觸媒顆粒上裂解, 由於金屬顆粒上存在許多不同取向的晶面,故每一個面對裂解的碳氫分子之吸附與活化能力均可能不相同。當碳-氫分子(CnHm)與金屬觸 媒表面接觸後,即行斷鍵,同時碳向金屬顆粒內部擴散,而氫則由表 面逸出。對不飽和的碳氫分子而言,這個過程為極強之放熱反應,因 而快速的增加金屬觸媒表面吸附位置的溫度,也同步增加金屬觸媒表 面對碳分子的溶解度。 2. 經由表面擴散進入金屬觸媒顆粒中的碳,超過飽和濃度時,即會在表 面上穩定地析出,此時碳分子以形成管狀,且相互間取得力平衡的方 式析出。由於析出的過程為吸熱反應,故常於碳進入與析出的金屬觸 媒顆粒等過程中,以溫度梯度的方式,使得後續的碳能藉此熱驅動力, 逐漸擴散穿越整顆金屬觸媒粒子。 3. 不過,若觸媒粒子表面過度的積碳,使其擴散速率不足,或超過碳奈 米管成核及成長速率時,其表面即會因為碳封閉或堆積,而停止後續 的成長。. 圖2.6 碳管生長機制[Ruoff,1993]. 29.
(41) 直立陣列奈米碳管與一般奈米碳管的基本成長機構類似,但根據其 成長的形貌及直立的方式來看,在沈積初期奈米碳管的生長應是均向性 的,但經過一段時間後只有向上成長的奈米碳管才能順利獲得後續碳分 子的補充,得以維持其成長。但其他方向的奈米碳管,則因無法取得足 夠的碳分子,生長情形受阻,故常僅留下直立陣列的奈米碳管。另外, 若碳奈米管的成核密度高,成長過程中由於相互推擠,不易往側向成長, 因此也有助於奈米碳管長成直立陣列的模式。 單層奈米碳管成長核心之先形成許多類似半個C60的半圓形蓋子,此 核為觸媒表面所支撐,並由不斷碳源之提供,單層奈米碳管成長成束狀 碳管。當碳源供給停止或觸媒表面失去活性時,單層奈米碳管成長將不 再繼續成長。. 2.3.3 奈米碳管之純化及應用 2.3.3.1 奈米碳管之純化 由化學氣相沉積法合成的奈米碳管,常常伴生有大量雜質(例如奈米 碳顆粒、石墨碎片以及催化劑小顆粒等),故奈米碳管需要進一步加以純 化。奈米碳管的純化工作有兩部分:一是去除小顆粒催化劑,另一個是 去除雜質碳。大部分的小顆粒催化劑,經酸浸泡可被除去,但有部分催 化劑由於被包裹在奈米碳管內部,或是被奈米碳小顆粒包覆其外,故有 時即使經酸浸泡,也很難保證管內的催化劑被徹底的清除,這也是一般 奈米碳管純化時,常遭遇的問題。 此外,對於碳雜質的去除,其方式有化學方法及物理方法兩種,化. 30.
(42) 學方法是利用氧化劑對奈米碳管,與碳奈米顆粒等碳雜質之間不同的氧 化速率來完成的,常用的氧化劑有氧氣(或空氣)、二氧化碳、硝酸、混合 酸、重鉻酸鉀等;物理方法則常利用超音波,經降解、離心、沉積、過 濾等程序,來達到雜質碳與奈米碳管分離的目的,從而獲得潔淨的奈米 碳管。. 2.3.3.2 應用奈米碳管去除有機污染物 在奈米碳管被發現後,其高比表面積之優異特性,奈米碳管將成為 最具優勢之吸附材質。故新近的研究中,也嘗試將奈米碳管應用於環境 污染物的去除。 Long等[2001]以奈米碳管吸附劑去除戴奧辛,其奈米碳管係以甲烷為 碳源,以化學氣相沈積(CVD)合成,生長之多壁奈米碳管,再經硝酸在 400℃下溶解觸媒粒子一小時,其BET比表面積為155 m2/g。其所製得的 多壁奈米碳管,具有可吸附戴奧辛的化合物鍵,其吸附能力且優於一般 活性碳的2.6倍;Li等[2001]利用序列奈米碳管(aligned carbon nanotubes, ACNTs)吸附水中氟化物,研究發現此ACNTs在前60分鐘的吸附速率相當 快,吸附容量可達3.0 mg/g,並於180分鐘後,逐漸達吸附平衡。當氟化 物濃度為15 mg/L時,中性環境中,ACNTs的最大吸附能力為4.5 mg/g; Peng等[2003]以奈米碳管吸附水中1,2-二氯苯,經實驗證明生長奈米碳管 (As-grown CNTs)於40分鐘對20 mg/L之1,2-二氯苯溶液,平衡吸附量可達 30.8 mg/g,經熱力學分析顯示其吸附反應自發性高親和力且為吸熱反應。 Li 等[2004]使用已純化後之奈米碳管(purified multi-walled carbon. 31.
(43) nanotubes, PMWCNTs),來吸附揮發性有機化合物(VOCs),其奈米碳管係 經由甲烷經觸媒分解所得。實驗結果指出 PMWCNTs 對空氣中的 VOCs 具有直接吸附效果,且可搭配吹氣捕捉系統來濃縮水樣中之 VOCs。其原 因,乃由於所製備的碳管擁有特殊的孔洞構造,故貫穿體積(BTVs)比相 同比表面積的 Carbopack B 或石墨碳黑來的高;Agnihotri 等[2005]測量單 壁奈米碳管(SWNTs)吸附甲苯、甲乙基酮(MEK)、乙烷、環己烷等 VOCs 之吸附容量,實驗發現在 25、37 及 50oC 之恆溫條件下,SWNTs 對 VOCs 的吸附量,可符合 Freundlich 等溫吸附行為,其吸附熱為蒸發作用的 1-4 倍,為一物理吸附型態。 Lu等 [2005]利用商用奈米碳管吸附自來水中三鹵甲烷,其奈米碳管 的外徑介於10~30 nm,內徑介於5~10 nm,比表面積則介於225~295 m2/g。研究發現,對三鹵甲烷的飽和吸附量可達2.41 mg/g,吸附達平衡 時間約為150~180分鐘,其吸附能力較傳統的粉末活性碳吸附能力(1.20 mg/g)來的高;袁等[2006]研究將奈米碳管,經酸液純化處理後,可有效 去除表面殘留之催化劑與非晶碳等雜質。例如,以3M硝酸在120℃迴流2 小時後,比表面積可高達122.19 m2/g,較純化前的奈米碳管的比表面積, 約可提升約1.5倍。經以氯苯(初始濃度8.0~65.3 mg/L)之等溫吸附實驗確 認,奈米碳管對氯苯之飽和吸附量介於18.0~322.2 mg/g,其吸附達平衡 時間約12小時。 表 2.6 彙整文獻中奈米碳管吸附去除有機污染物之相關製備條件、奈 米碳管比表面積、有機污染物吸附劑量、以及相關的重要研究結果。. 32.
(44) 表 2.6 奈米碳管吸附去除有機污染物之比較 作者. CNT 之製備. Li et al., 2001. 使用濃硝酸及氫氟酸酸洗 CNT,溶解其 190 m2/g 表面上之催化劑粒子。 以硝酸在 400℃下溶解觸媒粒子 1 小時。 155 m2/g. Long et al., 2001. 比表面積. 吸附劑量. 研究結果. 14.9 mg/g. Al2O3/CNTs 吸附能力高於活性碳 15 倍,在 Al2O3/CNTs 寬 廣 pH 範圍及高吸附能力適合應用在去除水中氟化物。 多壁奈米碳管吸附戴奧辛,有很強吸附戴奧辛化合物鍵, MWCNT 吸附劑對戴奧辛吸附具高效率吸附能力且優於活 性碳吸附 2.6 倍 吸附氟化物平衡濃度為 15 mg/l 時在 pH 7.0 ACNTs 達最大吸 附能力為 4.5 mg/g。. -. 序列奈米碳管係用二茂鐵(Ferrocene)觸 媒溶於二甲苯當碳源至於石英管,且通 入氫氣及氬氣溫度達 800℃催化分解合 成。 CNT以在丙烯-氫(C3H6:H2=2:1)及使 用鎳觸媒粒子在750℃下催化熱解生 長。. 74 m2/g. 3.0 mg/g. 134 m2/g. 30.8 mg/g. 經熱力學分析顯示其吸附反應自發性高親和力且為吸熱反 應。. Li et al.,2003. 在於900℃環境下通入氫氣24小時,再 以6M鹽酸回流24小時進行純化。. 98 m2/g. 4.75×108 l/g. 碳管特殊的孔洞構造,其貫穿體積(BTVs)要比同比表面積的 Carbopack B 及石墨碳黑來的高,而奈米探管脫附回收率接 近 100%,且可對於試驗中之化合物不可逆吸附省略。. Lu, 2005. 奈米碳管經濃硝酸及硫酸分別浸泡 24 小時溶解觸媒粒子。. 295. 2.41 mg/g. 在吸附三鹵甲烷實驗中發現,其吸附達平衡時間約 150-180 分鐘,其吸附能力達 2.41 mg/g 比粉末活性碳(PAC)吸附能力 1.20 mg/g 高於二倍。. Agnihotri et al., 2005. 經濃硝酸及硫酸分別浸泡 24 小時溶解 觸媒粒子。. EA95:500 m2/g CVD80:609 m2/g. 100-150 mg/g 100-180 mg/g. SWNTs 為異質吸附劑且符合 Freundlich 等溫吸附方程式之 描述,且吸附熱為蒸發作用的 1-4 倍,此為物理吸附型態。. 袁菁等, 2006. 商用奈米碳管用 3M 硝酸在 120℃迴流 2 小時以去除表面雜質。. 122.19. 18.0 - 322.2 mg/g. 奈米碳管對於氯苯溶液之吸附量隨著初始濃度增加而提昇。. Li et al., 2003. Peng et al., 2003. 33.
(45) 2.3.4 影響吸附之因素 奈米碳管對於環境中污染物主要之去除機制仍為吸附反應,所謂吸 附(adsorption)係指某一相中的離子或分子,在另一相的表面發生凝聚或 濃縮的現象,此現象會發生在下列之兩種界面間:液相-液相,氣相- 液相,氣相-固相,液相-固相。被濃縮或吸附的物質稱為吸附質 (adsorbate),而用來吸附的物種稱為吸附劑(adsorbent)。吸附現象是吸附 劑表面對吸附質分子之親和力作用使其附著於固體表面上,吸附劑表面 具有許多活性位置(active site),為進行吸附作用。影響吸附之因素,就吸 附劑而言包括: 1. 比表面積:因吸附皆發生於表面,所以當比表面積愈大,所能提供 的吸附位置也愈多,其吸附能力相對也就愈強。 2. 粒徑大小:粒徑小,質傳阻力將較小,可能提高吸附速率。 3. 孔隙大小及分布:孔隙大小分佈會影響吸附容量及吸附速率,當在 低濃度狀態時,通常會產生單層分子吸附的方式,較有利於具有微 小孔隙之吸附劑;反之,若在較大濃度狀態下時,則會引起多層分 子的吸附,此時具有孔隙度大的吸附劑便較有空間可容納。 4. 表面官能基及極性:吸附劑上有各種官能基,不同之官能基針對不 同有機物,吸附效果也有所差異,而官能基之多寡亦會影響吸附之 效果。以活性碳為例,其表面官能基為非極性,不易吸附極性的官能 基。另外,表面官能基也可能與吸附質產生化學性鍵結,形成不可逆 吸附。. - 34 -.
(46) 其次,就吸附質本身特性對吸附反應之影響而言: 1. 分子大小:若分子太大以至於無法擴散進入孔隙時,吸附效果將受 到影響;一般分子大小接近吸附劑孔隙大小時,被吸附之分子愈 大,吸附效果愈好;若孔隙大小剛可容許分子進入,吸附效果較佳; 若分子頗小,小分子之移動性較佳,可使其擴散較快,則可能在較 大分子進入吸附劑的微孔隙前,先進入微孔隙中。 2. 沸點:吸附主要為孔隙表面吸附質凝結現象,因此沸點愈高,愈容 易凝結,被吸附的潛能也就愈大。 3. 極性:由於吸附劑表面與吸附質間之特定作用,吸附劑之表面化學 會影響吸附量及吸附效率,吸附劑表面所存在之官能基,會影響與 吸附質間的化學親和力。 4. 溶解度大小:溶解度大小一般是隨著吸附質之分子量減少而增加, 而使吸附質的有效尺寸增加,導致被吸附量降低,即所謂 Lundeliu’s 定律。 5. 離子化:溶解性之離子化吸附質擴散速率比中性溶質慢,所以具離 子化之吸附質隨溶解度增加其吸附量減少。 另外,環境條件對吸附反應也具有影響性,例如: 1. 攪拌:吸附速率可由膜擴散或孔擴散控制,而膜擴散或孔擴散視受 到之攪拌速率而定,攪拌快,可增進膜擴散速率決定步驟。 2. pH值:吸附進行時之pH 值對吸附程度有很大的影響。部份原因為 氫離子本身被吸附的能力極強,另一個原因則是pH值改變電離的程 度,改變吸附質的表面特性。. - 35 -.
(47) 3. 溫度:一般來說,溫度提高會增加吸附速率,但由於吸附大部份為 放熱反應,所以吸附程度反而會下降。. 2.4 複合光觸媒材料製備 TiO2 為光催化相關研究中,最常用的光觸媒,除了因其能隙有3.2 eV,導致必須以近紫外光才能加以激發的缺點外,尚有電子-電洞會快速 的重組,使其量子產率偏低之問題,故近年來的研究,多著重於光觸媒的. 改質。 Linsebigler等[1995]指出,TiO2光觸媒經由表面改良的優點包括:(1) 促進電荷分離,減少電子電洞再結合的機率,以增進光催化效率,提昇 量子產率、(2)增加波長可吸收之範圍、(3)改變對反應物之選擇性或者能 使反應產生特定之產物。 文獻中,改良的方法包括有表面敏化、添加金屬、與奈米材料相結合 等,茲彙整如下。 1. 表面敏化(surface sensitization) 利用染料分子作為光敏劑(photosensitizer),可使表面TiO2進行光敏化 作用(photosensitization),進而增加TiO2對光源之吸收率,增加所激發出 量子數。當染料分子受激發時所需之氧化能階較TiO2導電帶能階高時, 則光敏劑受激發後所激發出之電子可傳遞到TiO2之導電帶上,而擴大 TiO2所能吸收之波長,可有效地降低能源的使用。 光敏化過程之主要特色,在於可用能量較小之可見光,來激發TiO2, 節省高能光源之使用量。Cho等[2001]研究指出,當染料分子附著於TiO2. - 36 -.
(48) 表面時,利用可見光使染料受激發後,經由光敏化反應所激發出之電子 會傳遞到TiO2之導電帶上,而後進行氧化還原作用以破壞污染物,而 TiO2本身並未受到激發,於整個過程中並無光電洞的產生,故無電子電洞再結合之憂慮。 2. 金屬之添加 利用表面塗佈或初濕含浸法,將過渡金屬添加於TiO2上,可增進激 發過程中對電子的捕捉,抑止電子-電洞再結合速率。Paola等[2002]分 別將多種過渡金屬離子(Fe、Mo、V、W、Co、Cr、Cu)塗佈於TiO2 之 顆粒表面上,並針對對-硝基酚、甲酸、乙酸、苯甲酸等四種酸性有機 物進行光催化反應。其結果顯示,塗鈷之TiO2/Co粉末對甲酸,較純TiO2 顆粒有較佳之降解效率;Sclafani等[1998]在其研究中指出,附著於之 Pt/TiO2上之Pt,會捕捉電子,並吸引電洞而成為再結合中心。 複合觸媒除了抑制電子-電洞再重組外,金屬之添加亦可使觸媒於 可見光下進行利用。Wilke等[1999]在二氧化鈦合成過程中添加Cr3+ 及 Mo5+的金屬離子,在添0.1mol%金屬離子時二氧化鈦的能階從原本大概 3.2 eV(380 nm)能量縮短至Mo5+的2.85 eV(435 nm)及Cr的2.00 eV(620 nm),係藉著金屬離子添加劑來改質二氧化鈦的光吸收特性;白崢鈺等 [2007]其研究中利用以初溼含浸法製得Ag/TiO2及Cu/TiO2觸媒,其中Ag 佔整體觸媒重量比2 wt.%,結合UVA和UVC處理甲苯廢氣,而去除率均 能達95%;謝哲隆等[2007]以Ag/TiO2及Cu/TiO2觸媒(Ag、Cu佔整體觸媒 重量比2 wt.%)對二氧化碳進行還原,其主要之產物為甲醇,結果顯示 可針對溫室效應氣體問題能有效處理。. - 37 -.
數據

![圖 2.1 光催化分解 MTBE 反應路徑 [Park et al., 2003]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8789607.219688/20.892.198.697.120.731/圖21光催化分解MTBE反應路徑Parketal23.webp)

+7





相關文件
Feng-Jui Hsieh (Department of Mathematics, National Taiwan Normal University) Hak-Ping Tam (Graduate Institute of Science Education,. National Taiwan
2 Department of Educational Psychology and Counseling / Institute for Research Excellence in Learning Science, National Taiwan Normal University. Research on embodied cognition
Department of Computer Science and Information
Department of Computer Science and Information
Department of Computer Science and Information
2013 Workshop on Nonlinear Analysis, Optimization and Their Applications, De- partment of Mathematics, National Kaohsiung Normal University, Kaohsiung, Tai- wan, December 30,
Professor of Computer Science and Information Engineering National Chung Cheng University. Chair
2 Department of Materials Science and Engineering, National Chung Hsing University, Taichung, Taiwan.. 3 Department of Materials Science and Engineering, National Tsing Hua
