國 立 交 通 大 學
應用化學系應用化學研究所
碩 士 論 文
利用步進式時域解析霍氏轉換紅外光譜法
研究苯甲醛於 193 nm 及 248 nm 光解產生
一氧化碳之內能分佈
Internal Energy of CO upon Photolysis of Benzaldehyde at 193
and 248 nm Monitored with Step-scan Time-resolved
Fourier-transform IR Emission Spectroscopy
研究生:黃郁琁 (Yu-Hsuan Huang)
指導教授:李遠鵬 博士 (Dr. Yuan-Pern Lee)
摘 要 吾人利用步進式時域解析霍氏紅外放光光譜技術,研究苯甲醛於 193 nm 及 248 nm 之光解動態學,藉由觀測產物 CO 的振轉動放光譜 線,以分析光解產物CO 的內能分佈。苯甲醛經 193 nm 及 248 nm 激 發至高電子激發態後,均經由分子解離途徑產生CO 及 C6H6。 在193 nm 之光解實驗中,可觀測產物 CO 分佈到 v≦2、J≦43 之 放光譜線,其平均轉動能量為 12.6 ± 1.4 kJ mol-1;平均振動能量為5.9 ± 0.5 kJ mol-1。此外,亦觀測到產物HCO 之放光,其轉動溫度約為 1300 K,振動激發態最高分佈至 v = 2。在 248 nm 之光解實驗中,可 觀測產物CO 分佈到 v≦2、J≦43 之放光譜線,其平均轉動能量為 12.4 ± 3.3 kJ mol-1;平均振動能量為7.0 ± 1.2 kJ mol-1。 理論計算方面,利用Gaussian03 程式以 CCSD(T)/6-311G+(3df, 2p)//B3LYP/6-311G+(3df, 2p)密度函數理論方法計算苯甲醛光解之反 應位能圖,並以VTST 理論計算光解後各反應途徑之反應速率常數。 觀測到的CO 內能分佈與理論計算過渡態結構 TS1 所預測之低振動及 低轉動激發相當吻合。
謝 誌
從寫論文開始,我最想最想寫的部分,就是謝誌了!就像做投影片 時會想趕快做到”Thanks for your attention!”這一頁一樣,謝誌就像是 完成整本論文的最後一頁,也為我的碩士生涯劃下句點。 首先,感謝李遠鵬老師讓我可以在這麼好的學習環境下完成我的 碩士學位。我覺得當李遠鵬老師的學生非常幸福,因為老師總是很細 心的替每一位學生著想,並不停的思考學生是否有學習、有進步。雖 然老師的期望是我們最大的壓力來源,但是在這樣的壓力下我們也學 會互相扶持與成長。另外,我得說我是個任性的學生,很感謝老師對 我的信任與耐心,並且給予我很大的自由度(糟糕,寫到這個我想到 的竟是degee of freedom),真的很謝謝老師!此外,也感謝王念夏老 師與林志民老師對本論文的指導。 我也要謝謝我系統的師傅們,momo、雪兒與芝敏,特別是芝敏 當時還得在水深火熱的碩二帶著我這個破壞王學妹,也謝謝最溫柔美 麗的慧芬學姐,在我成為系統孤兒時的幫助。此外,還有實驗室裡其 他的學長姐學弟妹們;謝謝很愛罵靠盃的鋼琴歐吉桑花叔很貼心的照 顧我們,強者學姐小韓在我剛進實驗室的時候教我很多很多,還有我 的好麻吉媽媽桑鄭大頭一起分擔我在實驗室裡的喜怒哀樂,愛拉大姊
小meeting,聚集我們所有碩一的小朋友一起學習,那時候大家一起 努力互相學習的感覺真的很棒!感謝我的好戰友們:很棒很認真又有 目標外表冷冷但是其實個性很可愛的瓊緯、努力到有自虐傾向但是又 依賴又愛哭一直被嗆的笨歪、傻傻天真又直率但內心其實很容易受傷 的TRS Emission 好伙伴鴻菊大姊與腦袋構造不太一樣神出鬼沒的陳 兩津,很開心碩班這兩年能跟你們一起互相砥礪;也謝謝麥克斯在我 忙著畢業的日子裡,協助我和我一起努力,當然也不能忘記可愛的書 毓!雅苓妹妹、海哥、窘達、孔哥、young2、傅龍也一併感謝,也謝謝 實驗室裡的母親-莊”小姐”在生活上及行政上的照顧。 此外,謝謝418.5 所有的好伙伴們,能認識你們真的是在新竹最 大的收穫,希望畢業後大家還是能常相聚。謝謝我的超級秘書魯恩, 除了生活上大大小小的照顧,還要分擔我所有的情緒,這陣子真是辛 苦你了!最後我要謝謝我的家人們,謝謝媽媽在我過去的每一個求學 階段,不管日子多艱苦都堅持讓我接受好的教育,沒有你的付出,我 不知道我自己的能力可以發揮到什麼樣的程度,也謝謝嘴巴很壞但都 很善良的姊姊們,還有負責搞笑的小麻煩林伯宇,以及總是會煮好料 給我吃的小阿姨。謝謝你們!不過也請你們繼續再容忍我四年吧! 郁琁
目 錄 第一章 緒論 ... 1 參考資料: ... 8 第二章 實驗原理 ... 11 2.1 霍氏轉換紅外光譜儀 ... 11 2.1.1 Michelson 干涉儀 ... 12 2.1.2 霍氏轉換紅外光譜儀基本原理 ... 13 2.2 霍氏轉換紅外光譜儀的優點 ... 20 2.3 時域解析霍氏轉換紅外光譜法 ... 22 參考資料: ... 37 第三章 實驗技術與數據處理 ... 39 3.1 實驗裝置 ... 39 3.1.1 雷射系統 ... 39 3.1.2 反應系統 ... 40 3.1.3 偵測系統 ... 41 3.1.4 其他周邊儀器 ... 43 3.2 實驗前準備工作 ... 43
3.2.1 CaF2透鏡組架設與Welsh cell 對正 ... 44
3.2.2 儀器波長響應曲線的量測 ... 46 3.2.3 觸發光解雷射的反應時間 ... 49 3.2.4 偵測器及相關電子儀器之響應時間 ... 50 3.2.5 移動鏡穩定時間 ... 52 3.2.6 樣品之光吸收截面積(cross section)的量測 ... 53 3.3 實驗步驟與條件 ... 54 3.3.1 光解雷射的準備 ... 54 3.1.1 光譜儀之準備與對光 ... 55 3.1.2 周邊儀器之設定 ... 59 3.1.3 進行光解實驗量測產物之放光訊號 ... 60 3.4 數據處理 ... 61 參考資料: ... 81
第四章 結果與討論 ... 82 4.1 C6H5CHO 在 193 nm 光解之放光光譜 ... 82 4.1.1 1900-2300 cm-1光區CO 放射光譜之振轉能階分析 ... 83 4.1.2 連續放光譜帶之分析 ... 88 4.2 C6H5CHO 在 248 nm 光解之放光光譜 ... 91 4.2.1 CO 放射光譜之振轉能階分析 ... 92 4.3 理論計算苯甲醛光分解之過渡態及反應速率 ... 94 4.3.1 C6H5CHO 光解之過渡態與反應途徑 ... 94 4.3.2 VTST 預測之反應速率 ... 95 4.4 討論 ... 96 4.4.1 反應途徑 ... 96 4.4.2 產生 CO 的反應途徑 ... 97 4.4.3 各光解途徑之能量計量 ... 98 4.5 結論 ... 101 參考資料 ... 140
第一章 緒論 芳香族化合物(aromatic compounds)為石化燃料所排放的廢氣中 的重要種類,這些化合物在空氣中會反應產生臭氧[1]及懸浮微粒(或 稱氣膠,aerosol)[2],造成空氣污染。苯甲醛(benzaldehyde, C6H5CHO) 是芳香族化合物在光化學反應中的重要中間產物[3],並且常在空氣 污染的地區被偵測到。甲苯(toluene)與 NOx及空氣混和後照光,即會 反應生成苯甲醛與甲酚(cresol, C7H8O)[4]。大氣中,在有氧氣存在的 條件下,苯甲醛與 OH 自由基或 NO3反應會產生 C6H5C(O)O2,此自 由基再與NO 反應後,會形成 C6H5O (phenoxyl)[5]。此外,苯甲醛經 由 太 陽 光 照 射 後 , 激 發 至 電 子 激 發 態(S1) 再 經 由 系 統 間 轉 移
(intersystem crossing, ISC)到三重態(T1),隨著大氣中氧氣含量的多寡,
會 分 別 經 由 開 環(ring opening) 或 光 化 學 聚 合 作 用 (photochemical polymerization)這兩種反應途徑形成懸浮微粒[6]。
苯甲醛分子中苯基(phenyl group)與羰基(carbonyl group)之間的共
軛形式(conjugation),及其高效率的系統間轉移造成容易產生磷光
(phosphorescence)的特性,使得苯甲醛成為光化學領域研究中有趣的 課題。過去已有許多研究組對於苯甲醛之電子能態進行研究[7-15], 並指出C6H5CHO 分子 n → π*的躍遷(transition),主要會激發與醛基
相關的振動模,而π → π*的躍遷,則會激發與苯基相關之振動模。
根據Molina 及 Merchán[7]理論計算之結果,苯甲醛分子除了基態(S0)
至第一電子激發態(S1)之躍遷為 n → π*的躍遷外,基態至其他高電子
激發態(S2-S5)之躍遷皆為 π → π*的躍遷。而實驗測得電子激發態 S2、
S3及 S5之垂直躍遷能量(vertical excitation energy)分別為 421.12 kJ
mol-1 [9]、497.10 kJ mol-1 [10]及 613.79 kJ mol-1 [11]。因此,本實驗使 用193 nm (619 kJ mol-1)及 248 nm (482 kJ mol-1)做為激發光源,其能 量分別足以激發苯甲醛分子至S5及S2能態。 苯甲醛分子受到光子激發後,若僅考慮各物種之熱焓[16-20],其 可能的反應途徑如下列所示: C6H5CHO→ C6H6+CO Δ0H298 = 8.79 kJ mol-1 (1) C6H5CHO→C6H5CO + H Δ0H298 = 363.59 kJ mol-1 (2)
C6H5CHO →C6H5+ HCO Δ0H298 = 419.66 kJ mol-1 (3)
若產物具有足夠內能,則C6H5CO 及 HCO 可能二次解離: C6H5CO →C6H5+CO Δ0H298 = 120.08 kJ mol-1 (4) HCO → H + CO Δ0H298 = 64.02 kJ mol-1 (5) 根據實驗結果[8, 21-28],苯甲醛分子激發至第一電子激發態(S1, nπ*)後,主要經由磷光放光途徑回到基態,並無光分解反應產生;而 當苯甲醛分子激發至第二電子激發態(S2, ππ*)後,除了經由磷光放光 途徑回到基態外,亦會經由反應途徑(1)分解產生 C6H6與 CO。Berger
等人[8]以 276 nm 激發苯甲醛至第二電子激發態(S2),研究其磷光放
光,並利用氣相層析法(gas chromatography)偵測 C6H6及CO,以研究
其光解產率(dissociation yield)。實驗結果顯示,C6H6及CO 之光解產 率在低壓的情況下(0.2-0.6 Torr)趨近於 1,並隨著氣體總壓力上升而 下降,而其磷光放光率則隨著光解產率下降而上升。Berger 等人[8] 並於反應中加入氧氣,發現其光解產率並沒有改變,證明產物 C6H6 及 CO 是經由反應途徑(1)產生,而不是經由自由基途徑(radical channel)二次解離產生。而 Bruhlmann 等人[22]利用 276 nm 激發以 1:1 比例混和之氘取代的苯甲醛(C6D5CDO)與苯甲醛(C6H5CHO),僅偵測 到產物 C6D6與 C6H6,更證明了產物 C6H6及 CO 是經由反應途徑(1) 產生。
Silva 及 Reilly[25]利用雙色雷射游離質量光譜法(two-color laser ionization mass spectroscopy),以 257-284 nm 激發苯甲醛分子束至 S2
電子態上不同的振動能階(vibronic state),再以 157 nm 游離並偵測 (probe)產物分子,以研究其解離動力學與動態學。他們在實驗中觀察 到 苯 甲 醛 離 子 訊 號 強 度 隨 著 時 間 呈 現 雙 對 數 形 式 的 衰 減 (biexponential decay),故推斷苯甲醛激發後會經由兩個不同生命期的
生成速率與苯甲醛離子的消失速率皆與激發能量大小呈現正相關性, 故作者推斷此能態為一預解離能態(predissociation state),並從其生命 期推斷其為三重態,命之為 T*。對於生命期較短(τ>10-6 s)之能態, 由於該能態與苯離子的生成無相關性,作者推斷其為磷光放光途徑的 主要能態T (nπ*)。因此,Silva 及 Reilly 提出苯甲醛激發至 S2能態後, 會以kST > 1012 s-1之速率進行系統間轉移至預解離能態T*,而在 T* 能態後則有三個可能的途徑,分別為(1)解離產生 CO 與三重態的苯, (2)內轉移至 T (nπ*)能態後,以磷光放光的形式弛緩至基態,(3)經由 放光或非放光途徑弛緩至基態。然而,此反應機制卻無法說明在實驗 中偵測得到之電子基態的苯分子,因此作者認為可能還有另外一個未 知的反應途徑(entrance channel)。 Zewail 研究組[26-28]以 266 nm 激發苯甲醛分子束,利用超快電 子繞射(ultrafast electron diffraction, UED)方法,以 1 皮秒(ps)的時間解
析度及 0.01 Å 的空間解析度觀測分子的電子繞射圖譜,並藉由理論
計算結果輔助,研究分子結構隨時間的變化,進而瞭解激發態分子之 非放光(nonradiative)反應途徑。由實驗得知,苯甲醛由 266 nm 光子 激發至第二電子激發態後,經由內轉移(internal conversion, IC)至第一 電子激發態(S1, nπ*),其 S2 →S1衰減時間(decay time)為 250 飛秒(fs),
自S1能態快速的系統間轉移(1/kISC = 42 ps)至三重態(T2, ππ* )後,弛緩
至 T1(nπ*)能態,再緩慢的分解產生 CO 與三重態的苯或是藉由磷光
放光回到基態;若分子具有較少的可用能量(Eexcess = Eavi – S1),則主
要會經由磷光放光途徑回到基態。(2.)氫原子藉由一個能量障礙 Ebarrier 相當於102 kJ mol-1的反應途徑與苯環結合形成中間產物IS,其形成 的反應速率常數ki = 1.6×1010 s-1,之後便分解產生 CO 與基態的苯分 子(kd = 9.4×1010 s-1)。比較實驗量測到之苯甲醛分子形成中間產物 IS 的生成速率常數 ki,與利用 RRKM 理論計算的反應速率常數,發現 實驗值較計算值大三個數量級,作者經由此差異推測反應物分子內能 振動重分佈(intramolecular vibrational erergy redistribution, IVR)並不
完全。此外,作者無法確認產物 CO 為中間產物 IS 於電子激發態時 直接解離產生,還是中間產物IS 內轉移至基態位能面後才解離產生, 但經由理論計算得到中間產物 IS 於基態位能面時即會快速的解離產 生CO,作者推測於基態解離的可能性較大。 Shin 等人[29]研究氘取代的苯、苯乙炔及苯甲醛分子經由吸收兩 個243.2 nm 光子後之光解反應,利用兩個 243.2 nm 光子將光解後的 產物氫及氘原子激發至 2s 能態,藉由觀測其原子放光光譜的杜普勒 加寬效應(Doppler broadening)得到原子移動能之資訊,以探討其光解
能分別為70.3 kJ mol-1及70.8 kJ mol-1,C6H5CCD 其氫及氘的原子移
動能分別為72.8 kJ mol-1及 73.5 kJ mol-1,而 C6H5CDO 其氫及氘的原
子移動能則分別為74.2 kJ mol-1及82.7 kJ mol-1,由此可推測C6H5CDO
分子其解離途徑與前兩者相異;此外,C6H5CCD 光解產物之氫/氘比
值為5,顯示其符合統計分解(statistical dissociation),然而,C6H5CDO
光解產物之氫/氘比值為 1.5,因此作者認為 C6H6及 C6H5CCD 係經由
內轉移至基態後,緩慢的進行單分子解離(unimolecular dissociation),
而 C6H5CDO 的光解機制則有別於上述兩個分子,為斷鍵形成具有高
內能的C6H5自由基及 HCO 自由基,之後 HCO 再二次分解(secondary
dissociation)得到 CO 及 H。作者並利用 MP2/6-31G**計算出 HCO 分 解產生CO 及 H 的能障及解離能分別為 89.5 kJ mol-1及52.1 kJ mol-1。 此外,Zhu 及 Cronin[30]分別使用 280 nm、285 nm 及 308 nm 雷射光 解 苯 甲 醛 分 子 , 利 用 共 振 腔 振 盪 衰 減 光 譜 法(cavity ringdown spectroscopy, CRD)偵測 HCO 之光譜,以研究苯甲醛分子經由反應途 徑(3)產生 HCO 之光解產率,由實驗結果得知,苯甲醛分子於 280 nm, 285 nm 及 308 nm 之 HCO 光解產率分別為 0.32±0.05, 0.45±0.05, 0.29±0.05,且其光解產率與氣體總壓力無相關性。他們發現利用 308 nm 光解時,無法觀測到 C6H5CO 之光譜(612-670nm),亦即無法觀測 到反應途徑(2)。然而,根據反應途徑(3)之反應焓 Δ0H298 = 419.7 kJ
mol-1,285 nm (Ehν = 418.5 kJ mol-1)及 308 nm (Ehν = 387.2 kJ mol-1)之
激發光源均無法光解產生 HCO,而 Zewail 研究組[28]也指出 Zhu 及
Cronin[30]觀測之 HCO 應為苯甲醛分子經由多光子吸收所造成。 綜合以上所述,對於苯甲醛分子單光子之光解反應,過去的研究
組主要觀測經由反應途徑(1)產生 C6H6與 CO 之反應機制。對於產物
的能量分佈或其他的反應途徑之觀測卻沒有文獻詳細報導。由於時間 解析霍氏轉換光譜法(time-resolved Fourier-transform spectroscopy, TR-FTS)具有同時觀測多重波長的優點,因此可同時偵測各種瞬態
物種之各個振動模在不同時域下的紅外放光。本實驗組利用 TR-FTS
技術研究過鹵化乙烯(vinyl halide,CH2CHX,X=F、Cl、Br)[31, 32]、
2-氯-1, 1-二氟乙烯(2-chloro-1, 1-difluoroethene,CF2CHCl)[33]、2-氯
丙烯(2-chloropropene, C3H5Cl)[34]、氟化苯(fluorobenzene,C6H5F)[35]、
鄰與對-氟化甲苯(ortho- and para- fluorotoluene)[36]、乙二醯氯(oxalyl chloride, C2O2Cl2)[37]及苯酚(phenol)[38]等分子於 248 nm 或 193 nm
光解後產物分子(HX 或 CO)之放射光譜,分析得到產物分子之初生態 振轉態佈居數及其內能分佈,並探討其光解動態學和可能之反應機制。
因此,吾人利用TR-FTS 技術觀測苯甲醛於 193nm 及 248 nm 光解後
參考資料:
1. J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics (John Wiley & Sons, Inc., New York, 1998).
2. J. R. Odum, T. P. W. Jungkamp, R. J. Griffin, R. C. Flagan, and J. H. Seinfeld, Science 276, 96 (1997).
3. E. Christophy, K. Myli, T. R. Viegut, J. A. Rzepiela, and J. M. Hossenlopp, J. Photochem. Photobiol. A Chem. 110, 229 (1997). 4. B. Klotz, S. Sorensen, I. Barnes, K.H. Becker, T. Etzkorn, R.
Volkamer, U. Platt, K. Wirtz, and M. Martin-Reviejo, J. Phys. Chem. A 102, 10289 (1998).
5. F. Caralp, V. Foucher, R. Lesclaux, T. J. Wallington, and M. D. Hurley, Phys. Chem. Chem. Phys. 1, 3509 (1999).
6. S. N. Dubtsov, G. G. Dultseva, E. N. Dultsev, and G. I. Skubnevskaya, J. Phys. Chem. B 110, 645 (2006).
7. V. Molina and M. Merchán, J. Phys. Chem. A 105, 3745 (2001). 8. M. Berger, I. L. Goldblatt, and C. Steel, J. Am. Chem. Soc. 95, 1717
(1973).
9. C. R. Silva and J. P. Reilly, J. Phys. Chem. 100, 17111 (1996). 10. D. G. Leopold, R. J. Hemley, V. Vaida, and J. L. Roebber, J. Chem.
Phys. 75, 4758 (1981).
11. K. Kimura and S. Nagakura, Theor. Chim. Acta. 3, 164 (1965). 12. G. Thiault, A. Mellouki, G. Le Bras, A. Chakir, N.
Sokolowski-Gomez and D. Daumont, J. Photochem. Photobiol. A Chem. 162, 273 (2004).
13. N. Ohmori, T. Suzuki, and M. Ito, J. Phys. Chem. 92, 1086 (1988). 14. M. Koyanagi and L.Goodman, Chem. Phys. 39, 237 (1979).
15. J. E. Ridley and M. C. Zerner, J. Mol. Spectrosc. 76, 71 (1979). 16. Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL,
(1997).
17. W.D. Good and N.K. Smith, J. Chem. Eng. Data 14, 102 (1969). 18. S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin,
and W. G. Mallard, J. Phys. Chem. Ref. Data 17, S 1 (1988).
19. R. K. Solly and S. W. Benson, J. Am. Chem. Soc. 93, 1592 (1971). 20. M.W. Chase. Jr., J. Phys. Chem. Ref. Data, Monograph 9, 1 (1998). 21. U. Bruhlmann and J. R. Huber, Chem. Phys. Letters 66, 353 (1979). 22. U. Bruhlmann, M. Nonella, P. Russegger, and J. R. Huber, Chem.
Phys. 81, 439 (1983).
23. T. Itoh, T. Takemura, and H.Baba, Chem. Phys. Lett. 40, 481 (1976). 24. T. Itoh, H. Baba, T. Takemura, Bull. Chem. Soc. Jpn. 51, 2841
(1978).
25. C. R. Silva and J. P. Reilly, J. Phys. Chem. A 101, 7934 (1997). 26. R. Srinivasan, J. S. Feenstra, S. T. Park, S. Xu, and A. H. Zewail.,
Science 307, 558 (2005).
27. J. S. Feenstra, S. T. Park, and A. H. Zewail, J. Chem. Phys. 123, 221104 (2005).
28. S. T. Park, J. S. Feenstra, and A. H. Zewail, J. Chem. Phys. 124, 174707 (2006).
29. S. K. Shin, H. L. Kim, and C. R. Park, Bull. Korean Chem. Soc. 23, 286 (2002).
30. L. Zhu and T. J. Cronin, Chem. Phys. Lett. 317, 227 (2000).
31. S. R. Lin, S. C. Lin, Y. C. Lee, Y. C. Chou, I.C. Chen, and Y. P. Lee, J. Chem. Phys. 114, 160 (2001).
32. S. R. Lin, S. C. Lin, Y. C. Lee, Y. C. Chou, I.C. Chen, and Y. P. Lee, J. Chem. Phys. 114, 7396 (2001).
33. C. Y. Wu, C. Y. Chung, Y. C. Lee, and Y. P. Lee, J. Chem. Phys. 117, 9785 (2002).
34. C.-M. Chang, Y.-H. Huang, S.-Y. Liu, Y.-P. Lee, M. Pombar-Pérez, E. Martínez-Núñez, and S. A. Vázquez, J. Chem. Phys. 129, 224301 (2008).
35. C. Y. Wu, Y. J. Wu, and Y. P. Lee, J. Chem. Phys. 121, 8792 (2004). 36. S. K. Yang, S. Y. Liu, H. F. Chen, and Y. P. Lee, J. Chem. Phys. 123,
224304 (2005).
37. C.-Y. Wu, Y.-P. Lee, and N. S. Wang, J. Chem. Phys. 120, 6957 (2004).
38. C. M. Tseng, Y. T. Lee, M. F. Lin, C. K. Ni, S. Y. Liu, Y. P. Lee, Z. F. Xu, and M. C. Lin, J. Phys. Chem. A. 111, 9463 (2007).
第二章 實驗原理 氣態分子具有移動、振動及轉動三種運動方式,其中分子的振動 躍遷能量約等於紅外光子的能量,因此,我們可以藉由紅外光譜來研 究不同分子之振動躍遷情形。由於不同分子的紅外光譜如人的指紋般 各不相同,故紅外光譜常用於化學分子的定性分析。此外,亦可利用 紅外光譜譜線強度的相對關係,做為分子之定量分析。 紅外光譜儀依照其工作原理可分為兩種:(1)利用光柵(grating)或稜 鏡(prism)分光的傳統光譜儀及(2)利用干涉效應(interference effect)的 霍氏轉換光譜儀。相較於傳統光譜儀,霍氏轉換光譜儀具有高解析度、 高靈敏度、較短的偵測時間、以及容易和其他儀器搭配使用等優勢, 近年來已逐漸取代傳統光譜儀[1]。 雖然一般的霍氏轉換光譜儀僅能擷取連續性的訊號,並不適用於 瞬態的偵測,但是改進訊號的擷取方式後,可利用具有時間解析的霍 氏轉換紅外光譜法(time-resolved Fourier-transform spectroscopy)研究 生命期短的不穩定化合物或穩定化合物的激發能態,並進而對相關的 氣態或液態之分子光譜學、化學動力學[2-4]與動態學[5]提供進一步 的探討,使得研究領域不再侷限於鑑定穩定分子的結構。
原理為藉由光程差使入射光產生干涉現象以紀錄其干涉圖譜
(interferogram),再利用理論推算將干涉圖譜轉換成一般傳統光譜,但
受限於當時技術有限且計算程序非常粗略而未被廣泛使用。西元1949
年,Fellgett 等人[7]首次經由精確計算將干涉圖譜轉換成傳統光譜。 至西元1965 年,Cooley 和 Tukey 等人[8]提出快速霍氏轉換(fast Fourier transform,FFT)數學演算法,使計算時間減少,提升干涉圖譜 的轉換效率。其後,由於微電腦與小分子氣體雷射兩大技術的發展, 藉由使用氦氖雷射(He-Ne laser)[9]精確地計算干涉儀中移動鏡造成的 光程差,並結合新進的微電腦處理數據改善原本複雜又費時的霍氏轉 換法,使得FTIR 技術變為方便可行。西元 1980 年後,商業化之霍 氏轉換紅外光譜儀逐漸取代利用分光技術的傳統紅外光譜儀。 2.1.1 Michelson 干涉儀
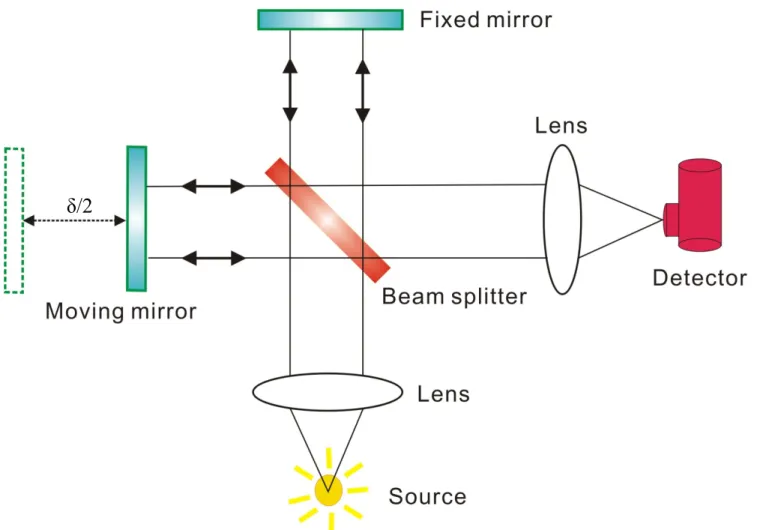
Michelson 干涉儀主要由分光片(beam splitter)、移動鏡(moving mirror)及固定鏡(fixed mirror)所組成,如圖 2-1 所示。光源經聚光後, 形成平行光進入干涉儀並導向分光片。理想情況下,分光片將入射平 行光平均分成強度相同的兩道光束,其中一道光束穿透分光鏡,經由 固定鏡反射後再經由分光片導向偵測器,另外一道光束則由分光片反 射至移動鏡,之後再經由移動鏡反射回分光片並導向偵測器。當移動 鏡沿著光軸移動時,匯集於偵測器之兩道光束所經過的光程便會不同,
造成相位差(phase difference)的改變,因而產生干涉現象。 對於單色光而言,當光程差為半波長的偶數倍(δ = n × λ/2;n = 0, ±2, ±4,…)時,兩道光束抵達偵測器時為同相位(in phase),形成建設性 (constructive)干涉,此時匯集之光強度最強;若光程差為半波長之奇 數倍(δ = n × 1/2λ;n = ±1, ±3,…),則兩道光束抵達偵測器時為反相位 (out of phase),形成破壞性(destructive)干涉,光束強度最弱。因此當 移動鏡以定速(v)來回移動時,由於光程差的改變,匯集之光束重複 地經過建設性及破壞性干涉,而光程差之變化率為時間之函數(δ = 2vt),因此可由偵測器測得一隨時間變化之干涉圖譜。 2.1.2 霍氏轉換紅外光譜儀基本原理 光是電磁波的一種,因此可利用電磁波的電場變化函數來表示:
( )
( 0) ( 2 ~ φ0) 0 φ 0 ,t =E ek⋅r− t+ =E ek⋅r− c t+ r E ω π ν (2-1) 其中k 為波向量(wave vector)、r 為位置向量(position vector)、ω 為角 頻率(angular frequency)、t 為時間點(time)、φ0為初始時間下的相位 (phase)、c 為光速、ν~為波數(wavenumber),而光束的強度 I 為 E( )
r,t 2。 以一固定波數ν~的單色光源為例,當其經由分光片平分成兩道光束時, 各個光束的電場變化函數就改變為( )
( 2 ~ φ0) 0 2 1 ,t = E ek⋅r− c t+ r E π ν ,又光程為 t c d = ⋅ ,故上式可改寫為( )
( 2 ~ φ0) 0 2 1 ,d = E e k⋅r− d+ r E πν 。因此,經由分光片分( )
( )
(
) (
)
( ) ( )(
)
[
]
( )
( ) (
πδ)
πδ δ ν π ν π 2 cos 0 2 1 0 2 1 2 cos 1 2 1 2 1 2 1 , , , 2 0 2 φ ~ 2 0 φ ~ 2 0 2 2 1 2 0 2 0 1 I I E e E e E d r E d r E d r E I d r k d r k + = + = + = + = = + − ⋅ + − ⋅ (2-2) 其中光程差δ d d 。即單色光的干涉圖譜為一個向上平移的餘弦 函數,其隨光程差變化的頻率為ν = c⋅ν~。圖 2-2 為不同光源及其對 應的干涉圖譜,圖 2-2 (a)為單色光源的干涉圖譜,為一簡單的餘弦 波;圖 2-2 (b)為兩道頻率相近之單色光源的干涉圖譜;圖 2-2 (c)為 一連續光源之干涉圖譜。 利用霍氏轉換方式即可將干涉圖譜轉換成傳統光譜,其數學式如 下:( )
( )
( ) (
)
( ) (
)
( )
ν( )
ν δ δ ν π δ δ δ ν π δ δ δ ν πνδ ~ Im ~ Re ~ 2 sin ~ 2 cos ~ 2 ~ i d I i d I d e I B i + = + = =∫
∫
∫
∞ ∞ − ∞ ∞ − ∞ ∞ − (2-3) (2-3)式之實數部分可描述干涉圖譜經霍氏餘弦轉換所得到之傳統光 譜,如下表示:( )
ν I( ) (
δ πνδ)
dδ B∫
∞ ∞ − = cos2 ~ ~ (2-4) 理想之干涉圖譜應為一左右對稱圖形,因此可將(2-4)式改寫成兩 倍的δ
= 到 +∞ 積分表示: 0( )
ν I( ) (
δ πνδ)
dδ B =∫
∞ 0 ~ 2 cos 2 ~ (2-5)移動鏡移動的距離有限,光程差無法達到無限大。假設實驗上只能得
到
δ
= − 到L L之間的干涉圖譜,以I′( )
δ 表示,則該干涉圖譜如同理想之干涉圖譜I
( )
δ 受到一匣式截斷函數(boxcar truncation function) ( )Dδ
的作用[10],而 ( )Dδ
的定義如下:( )
( )
L D L D > = ≤ = δ δ δ δ 當 當 0 1 (2-6) 因此,偵測器所測得的光束強度隨光程差的變化函數可改寫成下列的 式子:( ) ( ) ( )
δ I δ Dδ I′ = ⋅ (2-7) 即傳統光譜B′( )
ν~ 為( )
ν I( ) ( ) (
δ Dδ πνδ)
dδ B′ =∫
∞ 0 ~ 2 cos 2 ~ (2-8)根據霍氏分析卷積定理(the convolution theorem of Fourier analysis), 兩個函數之乘積的霍氏轉換為此兩個函數個別霍氏轉換後之卷積 (convolution)。匣式截斷函數 ( )D
δ
作霍氏餘弦轉換後為一sinc 函數( )
ν~f ,而此函數稱為儀器譜線形狀函數(instrumental line shape function, ILS),其數學表示式如下:
( )
(
L)
L c(
L)
f πν ν π ν π ν~ = sin 2~~ =2 sin 2 ~ (2-9) 而I
( )
δ
作霍氏餘弦轉換後為B( )
ν~ ,因此,理想傳統光譜和儀器譜線形 狀函數卷積的結果為:( )
( ) ( )
( ) ( ) (
δ δ πνδ)
δ ν ν ν d D I f B G∫
∞ = ∗ = 0 ~ 2 cos 2 ~ ~ ~ (2-10)( )
ν~ G 為實驗所得到的真實光譜,*表示卷積。對於單色光ν~1而言,上 式可簡化為:( )
( ) ( )
( )
c[
(
)
]
L LB f B G ν ν π ν ν ν ν ~ ~ 2 sin ~ 2 ~ ~ ~ 1 1 − = ∗ = (2-11) 因此,如圖 2-3 所示,原本應為單一波數ν~1且無限窄頻寬的圖譜,由 於移動鏡無法移動至無限遠處而經匣式截斷函數修正,使得譜線變寬, 主峰之半高寬(full width at half maximum,FWHM)為0.605L ,此半高
寬常被用來表示霍氏紅外光譜的理論解析度(theoretical resolution)。此 外,經匣式截斷函數修正後亦會在主峰兩側產生額外的側波;側波最 大振幅值(side lobe amplitude maximum,SLAM)與主峰高度的比值為
21.7% s m H H = 。 當主峰附近有其他微弱訊號則易與此側波混淆,為了除去匣式截 斷函數造成的側波干擾,可用其他函數取代匣式截斷函數,其作用彷 彿削去主峰旁的足部一樣,故稱此類函數為削足函數(apodization function)。表 2-1 列出幾種簡單的削足函數[11],從中可發現削足函 數雖然可以降低測波之干擾,但卻也導致主峰的頻寬增加。因此,如 果頻寬不是重要的考量,則可選擇 s m H H 值較小的削足函數;反之,若
頻寬是主要的考量因素,則可選用 FWHM 較小的削足函數。除了這 些考慮因素外,還要考量解析度與譜線密度,再選擇適合該實驗條件 的削足函數。 本實驗使用的削足函數為 Hamming(又稱 Happ-Genzel)函數,其 定義如下:
( )
( )
L A L L A > = ≤ ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ + = δ δ δ πδ δ 當 當 0 cos 46 . 0 54 . 0 (2-12) 經由霍氏轉換之後,其儀器譜線形狀函數的數學表示式如下:( )
(
2 2)
3~(
3)
8 ~ 2 ~ 2 sin ~ 64 . 0 08 . 1 ~ ν π ν π ν π ν ν L L L L L f − − = (2-13) Hamming 函數與其儀器譜線形狀函數如圖 2-4 所示,其 FWHM 為 0.908 L ,而 ms 0.69% H H = 。 由於電子濾波器、光學元件及不當取樣等因素會造成相位差 (phase error),影響干涉圖譜之對稱性。以電子濾波器為例,其對於不 同頻率的光會產生不同的相位延遲(θ( )
ν~ ,phase lag)效應,因此,必 須利用相位修正(phase correction)來修正此誤差。亦即(2-4)式必須加 上θ( )
ν~ 以進行相位修正,才能描述真實之干涉圖譜:( )
( )
[
( )
]
( ) (
ν[
πνδ)
(
θ( )
ν)
(
πνδ)
(
θ( )
ν)
]
ν ν ν θ δ ν π ν δ ~ ~ sin ~ 2 sin ~ cos ~ 2 cos ~ ~ ~ ~ 2 cos ~ d B d B I∫
∫
∞ ∞ − ∞ ∞ − + = − = (2-14)以
δ
= 對稱之干涉譜變得稍不對稱。如果只是以餘弦霍氏轉換將會0 導致光譜上的誤差,因此,(2-3)式中,將干涉圖譜進行霍氏餘弦及正 弦轉換後即可得相位角:( )
( )
( )
ν ν ν θ ~ Re ~ Im arctan ~ =− (2-15) 最後,可進一步得到修正後之傳統光譜。 此外,不當取樣也會影響所量測之干涉圖譜。當理想的干涉圖譜 對稱於δ
= ,但如果第一個取樣點並非於0δ
= ,而是在0δ
= − 時,ε
實際之干涉圖譜應修正表示為[8]:( )
∫
∞( )
[
(
)
]
− = 0 ~ ~ 2 cos ~ πν δ ε ν ν δ B d I (2-16) 此因素所造成的誤差也和有相位差類似。因此,無論是不當取樣或濾 波等因素所產生干涉譜的相位誤差均可以相位修正之數學步驟加以 修正,以避免光譜轉換後所得之傳統光譜發生嚴重的誤差。本實驗所 使用的相位修正方法為Mertz method[12][13]。通常實驗僅擷取單邊之干涉圖譜(single side interferogram),以節 省掃瞄時間及縮短霍氏轉換運算量及時間,故相位校正程序相當重要。
實驗時於干涉圖譜
δ
= 左側多取 n 個數據點,得到一個含 2n 個數據0點的雙邊干涉圖譜,如圖 2-5 所示,再將對稱區域進行 FFT 轉換,
以取得相位誤差資訊,作為相位校正。而n 值的大小取決於實驗時所
一般的霍氏轉換紅外光譜儀有三組不同光源之干涉儀,三組干涉 儀共用分光鏡和移動鏡,三組光源包括內部連續波長的紅外光源、氦 氖雷射以及連續白光光源,分別做為偵測樣品光譜、測量取樣之相對 光程差、定義零光程差點之用途,圖 2-6 為其干涉圖譜及傳統光譜。 氦氖雷射可提供頻率極為穩定之單色光源(波長= 632.8 nm),故其干 涉圖譜為一餘弦函數。如圖 2-7 所示,餘弦波每段波長有兩個零交叉 點,其固定間隔為316.4 nm,霍氏轉換紅外光譜儀以氦氖雷射干涉圖 譜的零交叉點(zero-crossing)做為定位點,建立一個固定光程差的量度 法,並以之作為取樣的間隔。由於此餘弦波之頻率與移動鏡速率成正 比,若移動鏡速率稍有變動時,則餘弦波之頻率亦隨之改變。因此, 電腦隨時調整取樣時間,才能確保每一個取樣點之光程差的準確性。 由於氦氖雷射只能定位移動鏡位移每段距離的相對位置,故利用白光 光源干涉圖譜的最高點作為零光程差位置的訂定。如圖 2-6 (c)所示, 連續波長的白光,其干涉圖譜在
δ
= 時,為完全建設性干涉,強度0 最大,而δ >0時強度迅速減弱,故可產生一個強而窄的訊號,以此定 位取樣的起始點。然而,本實驗所使用的 Bruker FTIR 是利用步進式 馬達來驅動移動鏡,可精準的定位移動鏡的位置,因此僅使用兩組干 涉儀,不需使用連續白光干涉儀的部分。在實驗正式擷取數據前,先波峰相對之光程差位置,作為零光程差的參考基準點,以確保每個干 涉圖譜擷取訊號的起始點一致,才可多次掃瞄光譜而不失真。 2.2 霍氏轉換紅外光譜儀的優點 霍式轉換光譜儀相較於傳統分光光譜儀有下列之優點: 1. 高光通量之優點(throughput advantage): 由於傳統分光儀的解析度受到狹縫開口的限制,而干涉儀無須使 用光狹縫及光柵等裝置,只是利用光圈來限制光的散射角,因此光通 量遠高於傳統分光儀。相較於傳統分光儀,偵測器所測得訊號較強, 靈敏度較高,也有更好的訊雜比,此優點由 Jacquinot 提出,故又俗 稱Jacquinot 優點[14]。 2. 多重波數之優點(multiplex advantage): 干涉儀可以在同一時間內偵測整個光區的光譜,而傳統的分光儀 僅能在同一時間內偵測單一波長。因此,對於相同光譜範圍及相同解 析度之光譜,FTIR 所需的偵測時間遠小於傳統的分光儀。此外,在 相同時間下,干涉儀可藉由多次掃瞄平均來提高訊雜比。如果雜訊是 以隨機的形式出現,則該訊雜比(signal to noise ratio,SNR)與掃瞄次
數N 的關係如下:
N
SNR∝ (2-17) 此優點由Fellgett 提出,亦稱 Fellgett 優點[7]。
傳統分光儀無法直接利用光柵的轉動角度來準確地對應實際選 擇的波數,必須利用標準樣品產生的已知譜線來校正其絕對波數位置。 而干涉儀則是利用氦氖雷射精確地標定光程差,經由霍氏轉換干涉圖 譜得到波數準確度可達 0.001cm-1的傳統光譜。因此,干涉儀在波數 的準確度上遠高於傳統分光儀,且不需要另外進行波數的校正工作。 此優點由Connes 提出,又稱 Connes 優點[15]。
4. 高解析度之優點(high resolution advantage):
傳統分光儀的解析度主要是受到光柵刻痕密度的限制,一般傳統 分光儀其解析度為0.1 cm-1。而干涉儀的解析度是與最大光程差
δ
max成 反比,關係式如下: max 2 1 1 ~ ~ δ ν ν ν = − = Δ 其中ν~1、ν~2恰為兩道可完全解析的波數,Δ 即為完全解析度[8]。而ν
最大光程差δ
max即為移動鏡移動距離的兩倍,因此,解析度亦可與兩 倍的移動鏡移動距離成反比。目前市售 FTIR 之最大解析度可達到 0.001 cm-1的解析度,遠較於傳統分光儀高。 5. 抑制散逸光之優點(stray-light control): 與單光儀波長設定不同波長之光子穿越入口狹縫或光圈後,理論 上不會由出口狹縫射出,但因為光學零件之不完美,可由出口狹縫散言,欲降低此散逸光並不容易。然而就干涉儀而言,對於每一波數ν~ 的單光光源,若移動鏡的移動速率為v,則偵測器可測得餘弦干涉訊 號頻率為f = 2vν~。選用適當的電子濾波器將其他頻率範圍的訊號過 濾除去,則偵測器僅能測到特定波段的訊號,便可有效抑制其他波段 的散逸光。 6.靈活且應用廣泛(versatile)之優點: 只要選擇適當的光源、分光鏡、鏡片及偵測器等光學元件,便可 將霍氏轉換光譜儀應用在遠紅外光、中紅外光、近紅外光、可見光或 紫外光區的測量。此外,若再對光譜儀搭配其他元件,例如:氣相層 析儀(GC)、液相層析儀(HPLC)、質譜儀(MS)與多重反射吸收槽(White cell)等,即可應用於其他定量、定性的分析。 2.3 時域解析霍氏轉換紅外光譜法 一般的霍氏轉換紅外光譜儀取樣模式並不適用於觀測瞬態訊 號,因此對一些生命期較短的物種,例如:自由基、反應中間物、分 子離子、微弱鍵結分子及高激發態的分子等,都無法進行鑑定。為了 改進此不足,目前已發展出許多技術,使得霍氏轉換紅外光譜儀具有 時間解析的功能。常見的各種方法介紹如下: 一. 連續式掃瞄模式(continuous-scan mode): 對於霍氏轉換紅外光譜儀取樣模式不做任何的改變,僅改變與反
應系統之間的連結。連續式掃瞄模式又可分成幾種的掃瞄技術: 1. 氣流管法(flow tube method):
氣流管裝置是以氣體流動的形式進行化學反應,藉由調整反應氣 體開始混合到被偵測的距離,即可改變反應時間,所以利用霍氏轉換 紅外光譜儀偵測不同反應距離的光譜,即可得到不同反應時間的光譜。 然而,此方法的時間解析度僅在毫秒(ms)範圍,對於更快的反應變化, 則無法偵測,而且每一次測量只能得到一個時間點的光譜[16-19]。 2. 快速掃瞄 (rapid scan): 直接利用移動鏡快速掃瞄一次或數次的時間為時間的解析度,故 時間的解析度主要受限於移動鏡的移動速率。而移動鏡在快速移動中, 其穩定度也間接限制了移動速率,因此利用快速掃瞄可達到的時間解 析度也只能有數十毫秒(ms)範圍[20],且訊雜比通常受到限制。 3. 同步式掃瞄(synchronous scan): 移動鏡需保持固定速率持續的移動,通過零交叉點時送出脈衝以 觸發反應,產生瞬態放光,並同時在固定延遲時間擷取訊號[21-24]。 以一般市售之FTIR 為例,其移動鏡的最小移動速率為 0.05cm s-1,則 每秒會通過 3161 個零交叉點。對目前進行光解的高能量脈衝雷射來 說,是很難產生如此高重複頻率但強度足夠又穩定的之雷射光;而且
但不易有幫浦可滿足如此高抽氣速率的條件。此外,在同步式掃瞄模 式下,移動鏡移動速度是否能長時間維持其穩定性,亦是造成誤差的 大問題之一。 4. 非同步式掃瞄(asynchronous scan): 非同步式掃瞄是利用移動鏡反覆穩定地掃瞄,光解雷射以另一重 複頻率觸發。每次觸發雷射後,在固定延遲時間擷取訊號,經過多次 掃瞄之後,將訊號點集合在一起所成的干涉圖譜轉換成傳統光譜。所 以雷射之觸發與干涉圖譜掃瞄之間並非同步進行,即氦氖雷射干涉圖 譜和反應起始時間並沒有關連。此方法的優點是反應觸發無須與移動 鏡到達零交叉點的時間同步,可避免同步式掃瞄對光解雷射的高重複 頻率之要求。然而,非同步式掃瞄的缺點是每一次實驗只能得到某單 一時間下的光譜,無法一次得到所有觸發後不同時間下的光譜[25]。 二. 步進式掃瞄模式(step-scan mode): 步進式掃瞄模式即移動鏡並非連續式地移動,而是利用電子儀器 控制移動鏡精準地定位在氦氖雷射零交叉點上,等待移動鏡穩定後才 觸發反應,開始擷取時間解析的訊號。在此一特定的移動鏡位置(定 位點)上,可累積多次的訊號加以平均後,再移動到下一個定位點擷 取訊號,待完成所有的擷取訊號程序後,重新組合並轉換成光譜,即 可得到不同時間下的光譜[26-30]。當每次移動鏡移動到下一個停留點
時,需要時間待其穩定靜止,此時間稱為定位時間(settling time),定
位時間與移動鏡所走距離有關,通常在20-100 ms 之範圍內。待移動
鏡穩定後,反應隨即被觸發並開始擷取訊號。
步進式掃瞄模式示意圖如圖 2-8。當移動鏡穩定停在 x1位置,待
反應觸發之後,每隔固定的時間間隔擷取訊號,並觸發多次反應,累 加多次訊號作平均,即可得到訊號序列I(x1,t1)、I(x1,t2)、I(x1,t3)、……、 I(x1,tm)。接著,當移動鏡移動到下一個位置 x2,並取得訊號序列I(x2, t1)、I(x2,t2)、I(x2,t3)、……、I(x2,tm);重複此方式至擷取完所有 的訊號序列,將這些訊號序列組合成訊號陣列。最後,重組訊號序列, 例如:在同一時間 tk下的訊號序列I(x1,tk)、I(x2,tk)、I(x3,tk)、……、
I(xn,tk)即相當於在時間 tk下所測到的干涉圖譜,再轉換成傳統光譜, 即可得到相當於時間tk之傳統光譜;對所有之時間點tj作相同的轉換, 即可得到時間解析的光譜。 與同步式掃瞄模式相比較,步進式掃瞄可以經由掃瞄一次得到各 個不同反應時間下的光譜;取樣過程中,在每個訊號擷取點可進行多 次訊號的累計以提高訊雜比,且對於能量不穩定的雷射脈衝亦有平均 其脈衝強度的效果,亦不受限於掃瞄速度快慢的影響;也不需要高重 複頻率之雷射光或高效率抽氣幫浦。
若移動鏡在每一個零交叉點停留取樣,則能偵測波段範圍為 15802 cm-1的訊號,即可量測光譜範圍為 0-15802 cm-1、15802-31604 cm-1… 等,由於紅外光區的波段範圍約 100-13000 cm-1,因此本實驗之FTIR 所量測到的光譜範圍應在15802 cm-1之內。為了節省取樣時間,此時 就可使用跳點取樣(undersampling)的方法來進行掃瞄,即移動鏡不在 每一個零交叉點都停留取樣,而是固定間隔數個零交叉點才取樣。舉 例來說,若每兩個零交叉點才停留取樣,其偵測波段範圍為7901 cm-1, 則可取最大光譜範圍為0-7901 cm-1或7901-15802 cm-1;每三個零交 叉點才停留擷取訊號,能偵測波段範圍為 5267 cm-1,可量測 0-5267 cm-1、5267-10534 cm-1或 10534-15082 cm-1等光譜訊號,以此類推。 因此,在相同的解析度下,欲測量的光譜範圍越窄,則其可跳過的零 交叉點越多,使得取樣點減少,故所需的時間也就越少。但使用跳點 取樣必須注意在偵測訊號時,不能有非偵測光區的光線進入偵測器, 所以必須加入濾光片(optical filter)將欲偵測光區以外的光源濾掉,以 免造成偵測光區以外的光源訊號疊合(folding)或失真(aliasing),造成 不必要的譜線干擾。 除了訊號的再現性、偵測器本身的雜訊干擾會影響光譜資訊之精 確度之外,進行步進式掃瞄時,受限於移動鏡位置之準確度與穩定性, 其取樣定位點的誤差會影響光譜的訊雜比。即移動鏡位置的不準確度
將造成訊雜比降低,其關係式如下: max ~ 4 SNR ν δ⋅ Δ = (2-18) 其中Δ (cm)為移動鏡位置的誤差,
δ
ν~max(cm-1)為光譜最大的波數[14]; 當Δ 越大時,所得到的訊雜比越差。目前技術使得移動鏡停留的位δ
置的準確度可達到 ±1.1 nm [15],若ν~max= 4000cm-1,則 SNR = 9090。 所以吾人目前實驗之訊雜比並不受限於移動鏡停留位置的不準度,而 是受限於移動鏡位置之穩定性、訊號的再現性以及偵測器本身的雜訊 干擾。 另外,在步進式掃瞄模式中,移動鏡並非來回移動使偵測器偵測 到光源ν~經調制(modulate)後的頻率 f = 2vν~,而是停留在固定位置上, 故偵測器所測得的頻率僅與訊號隨時間之變化有關。因此,對瞬態放 光物種隨時間變化之研究來說,其偵測反應時間之上限只受限於偵測 器、放大器及干涉儀數位化電子元之響應時間(response time),而偵 測反應時間之下限則受限於偵測器之RC 電路(RC circuit)充放電之時 間週期。此外,在實驗前應利用相同的偵測系統及相同的實驗條件下, 量測偵測器及其他相關電子儀器之響應時間,實驗取得有時間解析之 光譜後,才能精確地外推到有效的紅外光訊號之起始時間,關於本系 統電子儀器之響應時間的量測,請參見本論文第三章第二節。圖 2-1. Michelson 干涉儀之示意圖。其主要由分光片(beam splitter)、移動鏡(moving mirror)及固定鏡(fixed mirror) δ/2
圖 2-2. 不同光源之傳統光譜(右側)及其對應之干涉圖譜(左側)。 (a) 單色光源,(b) 強度相同,波數相近之兩單色光源,(c) 連續光源。
(a) 0.605/L Hm Hs 2L 1/L (a) f( )
ν
~ (cm-1) ν ~ (b) 1/L ν1 ~ ν~1 2L 1 (cm-1) ν ~ ν~1 2L 1 B( )ν~ (b) 圖 2-3. (a) 匣式截斷函數進行霍氏轉換後之圖譜,其波形為 sinc 函數; (b) 在移動鏡之有限位移±L 下,單色光波數ν
1之干涉圖譜以匣式截斷 函數進行霍氏轉換後之圖譜。( )
L c(
L)
f ν~ =2 ⋅sin 2πν~( )
LB( )
c[
(
)
L]
Bν~ =2 ν~1 sin 2π ν~1−ν~( ) 0.54 0.46cos( ) A L L L
πδ
δ
= + 當 − ≤ ≤δ
-L
L δ
A( )δ 0霍 氏 轉 換
逆 霍氏 轉 換
( )
(
)
3 3(
)
2 2 ~ 8 ~ 2 ~ 2 sin ~ 64 . 0 08 . 1 ~ ν π ν π ν π ν ν L L L L L f − − =f
( )
ν
~
(cm-1)ν
~0
圖 2-4. Hamming 函數與其經霍氏轉換後所得的儀器譜線形狀函數。 L 0.908 Hm Hs 0.69% s m H H =圖 2-5. 干涉圖譜之取樣示意圖。實驗擷取單邊之 N 點干涉圖譜,並
以零光程差點為中心,相位校正時,左右各取n 個點數以進行相位校
正。
n
n
N points full precision
interferogram
2n points double-sided interferogram
ZPD
圖 2-6. 干涉圖譜及其對應之傳統光譜。(A)連續波長的紅外光源;(B) 氦氖雷射;(C) 連續白光光源。
(A)
(B)
圖 2-7. 氦氖雷射之干涉圖譜。圖中實心方格為零光程差點,實心圓 點為零交叉點。 -1000 0 1000 2000 3000 4000 0 path difference (nm) Int ens ity
zero path difference
圖 2-8. 步進式時間解析掃瞄模式取樣示意圖。X 為光程差,t 為反應 時間,I為訊號強度。
intensity of the interferogram
optical path difference of the interferometer (Xn)
T
X
t
time evolution of the process (tm)
表 2 光程 0 .4 2 + 0 .5 c 0.54 2-1 幾種簡單 程差δ 的範圍為 Apodizati Blackman ) co s( 0 .0 8 L πδ + Hamming 4 0.46 cos( L πδ + Bartlett 1 L δ − Welch 2 2 1 L δ − Uniform 1 單削足函數對儀 為-L≦δ≦L。 ion Function 2 8 co s( ) L πδ ) δ 儀器譜線形狀 。而 s m H H 為側 狀函數的影響 側波最大振幅值 Instru 。Δ
ν
1 2為主峰 值H (side lobS ument Function 1/ 2 ν Δ = 1/ 2 ν Δ = 1/ 2 ν Δ = 1 / 2 ν Δ = 1/ 2 ν Δ = 峰之半高寬(f e amplitude m n Ins 2.30 2L = 1.82 2L = 1.77 2L = 1.59 2L = 1.21 2L = full width at h maximum)與主 strument Functi half maximum 主峰高度H 之mion Side lobes
0.11% s m
H
H
= 0.69% s m H H = 4.72% s m H H = 8.61% s m H H = 21.72% s m H H = m),實驗可測得 之百分比絕對 % 得之 對值。參考資料:
1. B. C. Smith, in Fundamentals of Fourier Transform In Fourier Transform Infrared Spectroscopy, (1996).
2. P. Y. Chen, R. A. Palmer, and T. J. Meyer, J. Phys. Chem. A, 102, 3042 (1998).
3. P. Y. Chen, R. A. Palmer, Appl. Spectrosc., 51, 580 (1997).
4. J. Eberhard, P. S. Yeh, and Y.-P. Lee, J. Phys. Chem., 107, 649 (1997). 5. G. V. Hartland, D. Qin, and H.-L. Dai, A. Simon and M. J. Anderson,
Rev. Sci. Instrum. 63, 3261 (1992).
6. A. A. Michelson, Phil. Mag., Ser. 5, 31, 256 (1891). 7. P. B. Fellgett, J. Phys. Radium 19, 187 (1958).
8. P. R. Griffith and J. A. de Haseth, Fourier Transform Infrared Spectroscopy (John Wiley & Sons, Inc., New York, 1986).
9. A. Javan, W. R. Bennet, Jr., and D. R. Herriott, Phys. Rev. Lett. 6, 106 (1961).
10. Jyrki Kauppinen, and Jari Partanen, in Fourier Transforms in Spectroscopy”, 1st edition (Berlin, Germany, 2001).
11. http://bbs.sachina.pku.edu.cn/Stat/Math_World/math/a/a279.htm. 12. L. Mertz, Transformations in Optics, Wiley, New York (1965). 13. L. Mertz, Infrared Phys. 7, 17 (1967).
14. P. Jacquinot, Rep. Progr. Phys. 23, 267 (1960).
15. J. Connes, and P. Connes, J. Opt. Soc. Am. 56, 896 (1966). 16. D. J. Donaldson and J. J. Sloan, J. Chem. Phys. 82, 1873 (1985). 17. E. Arunan, G. Manke II, and D. W. Setser, Chem. Phys. Lett. 207, 81
18. N. I. Butkovskaya, and D.W. Setser, J. Chem. Phys. 106, 5028 (1996).
19. N. I. Butkovskaya, and D.W. Setser, J. Phys. Chem. 102, 9715 (1998).
20. L. Mertz, Astron. J. 70, 548 (1965).
21. E. L. Woodbridge, T. R. Fletcher, and S. R. Leone, J. Phys. Chem.
92, 5387 (1988).
22. P. W. Seakins, and S. R. Leone, J. Phys. Chem. 96, 4478 (1992). 23. P. W. Seakins, E. L. Woodbridge, and S. R. Leone, J. Phys. Chem.
97, 5633 (1993).
24. C. A. Carere, W. S. Neil, and J. J. Sloan, Appl. Opt. 35, 2857 (1996). 25. K. Masutani, H. Sugisawa, A. Yokota, Y. Furukawa, and M. Tasumi,
Appl. Spectrosc. 46, 560 (1992).
26. R. E. Murphy, F. H. Cook, and H. Sakai, J. Opt. Soc. Am. 65, 600 (1974).
27. R. A. Palmer, C. J. Manning, J. A. Rzepiela, J. M. Widder, and J. L. Chao, Appl. Spectrosc. 43, 193 (1989).
28. G. V. Hartland, W. Xie, and H.-L. Dai, Rev. Sci. Instrum. 63, 3261(1992).
29. D. E. Heard, R. A. Brownsword, D. G. Weston, and G. Hancock, Appl. Spectrosc. 47, 1438 (1993).
30. G. V. Hartland, D. Qin, and H.-L. Dai, J. Chem. Phys. 100, 7832 (1994).
第三章 實驗技術與數據處理 將雷射導入反應槽以光解反應物,光解後的產物如在振動激發態, 會自發性的放射紅外光,而放光訊號再經由反應槽內的Welsh cell 收 集並導入霍氏轉換紅外光譜儀進行訊號擷取。完成光譜擷取後,將不 同時序下的光譜進行振-轉動能階佈居數分佈之分析,可得到光解產 物的內能分佈,進而了解反應的動態學。 3.1 實驗裝置 本實驗系統裝置主要分成四個部分:雷射系統、反應系統、偵測 系統及其他周邊儀器。上述的前三項系統如圖 3-1 所示。 3.1.1 雷射系統
利用氟化氬雷射(ArF excimer laser, Gam laser, EX100H/60),產生 波長為193 nm 的無偏極性雷射光,光束截面為 11.5 mm (長) × 4.2 mm (寬)的長方形。其最快重複頻率為 60 Hz,每發雷射的最高能量為 100 mJ。在電腦中輸入不同的電壓值會改變光束輸出能量大小,並可在 雷射出口處利用能量計(power meter)量測能量。使用雷射前,需預先 配置好雷射氣體(premix),雷射氣體以氖氣(Ne)做為緩衝氣體(buffer gas),氟氣(F2)、氦氣(He)及氬氣(Ar)的氣體濃度分別為 0.17 %、1 % 及6 %。
200)產生波長為 248 nm 的無偏極性雷射光,光束截面為 11 mm (長) × 33 mm (寬)的長方形。其最快重複頻率為 100 Hz,每發雷射的最高能 量為250 mJ。 3.1.2 反應系統 如圖 3-2 所示,反應槽為一個六通的不銹鋼腔體。在 x 軸方向的 兩面腔體封蓋上各裝上材質為S1UV 的 2 吋光窗(λ≧180 nm,穿透度 ≧90%),使光解雷射可以入射腔體。在 y 軸方向的腔體內部裝置兩 組已切割之鍍金球面鏡(Welsh mirror,直徑= 2 吋,焦距= 4 吋,兩鏡 組之距離= 4 吋),光解產物的放光在這兩組球面鏡間作至少 8 次的來 回反射,理論上可以使收集的訊號強度增加 7.4 倍[1, 2]。所收集之放 光再經由材質為CaF2的 2 吋光窗(5000≧σ≧800 cm-1,穿透度≧95%) 射出反應槽,導入光譜儀中。z 軸方向為氣體流動方向,反應氣體從 反應槽下方經一狹長隙縫(4 cm × 0.1 cm)流入;狹縫的擺設位置需靠 近Welsh cell 之聚焦點(即接近 M1、M2 中央,見 3.2.1),並與雷射光 徑平行,以提高放光訊號的收集效率。由於室溫下苯甲醛之蒸氣壓約 為1.2 Torr,因此實驗時以水浴加熱反應物以提高其蒸氣壓,並以加 熱帶包覆加熱氣體管路以避免反應物凝聚。腔體內部的氣體管路並加 裝恆溫式加熱系統,其裝置主要由加熱帶、K 型熱電偶(thermocouple)、 電源供應器、溫度控制器(Brainchild,C91)所組成。此外,亦經由針
閥於光解雷射入口的光窗一端加入少許氦氣進入反應槽,用以清洗 (purge)光窗,避免反應物經雷射光解後所產生之碳化物吸附在光窗上, 降低光窗之穿透度使雷射能量下降。 反應槽上方的管路連接至乾式真空幫浦(dry pump,TAIKO,model BEH-1800,抽氣速率為 25000 L min-1),幫浦與反應槽之間用蝶形真 空閥(butterfly valve)調整抽氣速率。反應槽側面分別接一熱對流式壓 力 計(convectron gauge, model 275071) 及 一 電 容 式 壓 力 計 (MKS Baratron, model 122AA, 10 Torr),用來量測反應槽內的壓力。
3.1.3 偵測系統 本 實 驗 使 用 步 進 式 霍 氏 轉 換 紅 外 光 譜 儀(Fourier-transform spectrometer,Bruker,model IFS-66 v/s),其移動鏡為步進式移動, 為了保持移動鏡的穩定度,需將之獨立擺放在具有隔離振動作用 (vibration-free)的光學桌上,且光譜儀之抽氣幫浦的前段抽氣管亦需以 重物將之固定於地上,以避免幫浦的振動藉由管路傳至系統。此FTIR 之最佳解析度為0.13 cm-1。 在反應槽與光譜儀之間架設一組 CaF2透鏡組(直徑 2 吋,焦距分 別為 4 吋及 6 吋),可將 Welsh cell 收集之產物放光有效率地引導至 FTIR。放光訊號經過透鏡組及光圈(aperture)後導入 FTIR,並先後經 由干涉儀、光圈、濾光片(optical filter),最後到達偵測器。
本實驗選用之FTIR 內部相關光學元件如下:
1 偵測器:InSb 紅外光偵測器(Kolmer,model KISDP-1-LJ2,rise time
220 ns),偵測範圍為 1666-10000 cm-1。偵測器內置一個前置放大 器,將電流訊號放大並轉換為電壓訊號。偵測器可輸出 ac 與 dc 耦合兩種訊號,此實驗選用ac 耦合輸出。 2 分光片:材質為 CaF2,可穿透之光區為1200-10000 cm-1。 3 光圈: FTIR 光譜儀在特定的解析度下,可使用的最大光圈直徑 為 S R F 2 D= × × (3-1) 其中 D 為光圈直徑(cm),F 為光譜儀中的光源集光拋物面鏡之焦 距(cm),R 為光譜解析度(cm-1),S 為欲觀測光譜之最大波數(cm-1)。 本實驗中,F = 15 cm,R = 0.4 cm-1,S = 2330 cm-1,故光圈直徑 為0.39 cm。 4 濾光片:因實驗中僅需測量小段光區之光譜,為節省掃瞄時間, 本實驗使用跳點取樣方式。為避免訊號摺疊(folding)的現象,需加 入適當的濾光片,只讓欲偵測光區的光線通過。本實驗所使用的 濾光片其光區為1670-2325 cm-1(OCLI,W05200-6X)。
3.1.4 其他周邊儀器
利用脈衝產生器(digital delay pulse generator,Stanford Research System,model DG535)產生一道脈衝以觸發光解雷射。由於雷射經觸 發後需要經過延遲時間(delay time)才會放出雷射光抵達反應槽,因此 經延遲時間後必須再產生另外一道脈衝至光譜儀觸發放光訊號之擷 取。訊號經由偵測器偵測後,利用偵測器的內置放大器將電流訊號放 大並轉換為電壓訊號,於交流電耦合模式(ac-couple mode)下,再利用 電壓放大器(low noise preamplifier,Stanford Research System,model SR560)以適當倍率進行二次放大。放大後的訊號再傳送到 FTIR 的類 比/數位轉換器(analogue to digital converter,ADC)進行數據擷取程序。 FTIR 內部配置的類比/數位轉換器,其 A/D 解析度為 16 bit,時間解
析度為5 μs,輸入訊號上限為 10V。但本實驗另外於電腦主機板上插 置一類比/數位轉換器,其 A/D 解析度為 12 bit,時間解析度為 25 ns, 輸入訊號上限為 1V。本實驗亦利用示波器(oscilloscope,Tektronix, TDS220)觀測訊號強度及時間分佈,以及雷射輸出能量之強度。 3.2 實驗前準備工作 實驗前的準備工作可分為下列幾個部分:CaF2 透鏡組架設與 Welsh cell 對正、儀器波長響應曲線的量測、儀器之響應時間的量測、 移動鏡穩定時間及樣品之光吸收截面積的量測。
3.2.1 CaF2透鏡組架設與Welsh cell 對正 主要原理為利用光的可逆性之行為,將光束由偵測器位置導向反 應槽,以對正反應槽內的Welsh cell,並調整光徑通過之相關光學原 件的位置。對正步驟如下: 1. 架設 Welsh 球面鏡組:準備兩組鍍金球面鏡(直徑 2 吋,焦距 4 吋), 其中一組中間切去5 mm 間隙,可得到鏡面 M1 及 M2,另一組中 間切去2.5 mm 間隙,得到 M3 及 M4,如圖 3-3 所示。將 M1 及 M2 固定於腔體內靠近 CaF2光窗處,M1 及 M2 之間相隔 5 mm。 與M1 及 M2 鏡面組相隔 4 吋距離處架設 M3 及 M4 鏡面組,其中 M3 及 M4 之間相隔 2.5 mm。鏡座上均有調整鈕,可調整兩球面 鏡之角度。 2. 架設鹵素燈:完成光譜儀的準備及對光(請參見本文 3.1.1),利用 OPUS 軟體將偵測器位置轉換至 DTGS,調整 DTGS 前方的拋物 面鏡,使入射的NIR 光束為一對稱圓形,且均勻的聚焦於偵測器 之光窗,並根據DTGS 偵測器所偵測到的訊號大小微調拋物面鏡, 調整到最佳訊號後移走 DTGS 偵測器。架設鹵素燈,使鹵素燈之 燈絲的中心位於NIR 光束焦點,並使燈絲與聚焦光徑垂直。理論 上應將光源架設於 InSb 之位置,但是自 DTGS 至 InSb 之光徑並 不會影響此對正程序,因此可不必移動InSb,將鹵素燈光源架設
於DTGS 即可。關閉 NIR 光源,並開啟 OPUS 軟體將光源轉換至 emission,使光源選擇鏡轉換至外在光源位置。開啟鹵素燈光源, 調整 FTIR 內部樣品槽中的光圈大小,以調整鹵素燈出光強度大 小。 3. 架設 CaF2透鏡組:開啟鹵素燈光源後,光束通過干涉儀組件,逆 向地導出FTIR,並聚焦於距離出光口約 1 吋的位置。在此位置放 置一光圈,使鹵素燈光束的聚焦點通過光圈正中央,如圖 3-1 中 的 A1 所示。此光圈位置的精準度將影響高解析光譜的訊號強度 (因為若精準度不佳,使用較小的光圈直徑時則可能減弱訊號強 度)。在距離光譜儀出光口 5 吋(即光圈後方 4 吋)的位置放置一 CaF2 透鏡(直徑= 2 吋,焦距= 4 吋),L1,使鹵素燈光束通過 L1 後形成 平行光。調整L1,使光束平行進入反應槽,並位於 welsh cell 之 中間。擺置一平面鏡於L1 前並與入射光徑成 45 度,將平行光束 導向至少1 公尺處,藉由觀察光束是否平行(即:光束之大小不隨 距離改變)來微調 L1 的架設位置。移去平面鏡,在 L1 與反應槽之 間放置另一CaF2透鏡,L2(直徑= 2 吋,焦距= 6 吋),調整 L2 之 位置,使通過 L1 之平行光再經由 L2 聚焦於 M1 及 M2 之間中央 處,且使光束在M3 上呈現一個完整且對稱的圓形,並分佈在 M3 的中央,如圖 3-3 中的圖(B)所示。
4. 微調 welsh cell:微調 M3 上的控制鈕,使反射光聚焦於 M1 靠內 側之垂直中心位置,形成一個聚焦光點。微調 M1 上的控制鈕, 使 M1 的反射光完整的覆蓋至 M4 中央;同理,微調 M4 上的控 制鈕,使反射光聚焦於 M2 靠內側之垂直中心位置;微調 M2 上 的控制鈕,使 M2 的反射光完整的覆蓋至 M3 中央。依照上述程 序,反覆調整 M1-M4,直至 M1 與 M2 上各呈現 4-5 個以水平排 列的聚光點,各點亮度均勻、亮度一致且置於鏡面中央等高處, 如圖 3-3 中的圖(B)左圖所示,即完成 Welsh cell 之對正。 3.2.2 儀器波長響應曲線的量測 待測光束必需藉由許多光學元件才能導入偵測器,但每個光學元 件對於不同波長的光之穿透率及反射率不盡相同,且所使用的偵測器 對於不同波長的靈敏度也不同,因此在測量光譜之前,必須使用一個 標準的黑體輻射光源進行校正,以得到所有元件對於不同波長的響應 曲線,才能反映真正的放光強度。雖然黑體輻射校正程序並無法測量 Welsh 球面鏡之反射響應值,但根據測試報告可得知,在測量光區內 球面鏡的反射率幾乎為定值,故其對實驗的影響可忽略不計。黑體輻 射校正程序如下: 1. 架設鹵素燈光源於 DTGS 偵測器位置,如本文 3.2.1 之步驟 2 所 述。
2. 移去反應槽,將黑體輻射校正儀(Graseby,model SR20)擺設至原 反應槽的位置。微調黑體輻射校正儀的位置,使鹵素燈光源經 CaF2透鏡組聚焦後,其聚焦點位置正好在黑體輻射源的輸出小孔, 並固定黑體輻射校正儀的位置。 3. 關閉鹵素燈,開啟 OPUS 軟體將偵測器位置更改為 InSb。 4. 設定黑體輻射源溫度,使其逐漸增溫至 1273 K 並穩定之(約費時 1.5 小時)。
5. 利用 FTIR 對正程序,觀測黑體輻射源的強度(ADC counts),再調 整黑體輻射源的出射光束之方向及輸出光圈大小,使訊號最佳化, 但應避免訊號過飽和(saturate)而造成誤差。 6. 掃瞄全光區(以 InSb 偵測器為例,其偵測光區範圍為 1666-10000 cm-1)光譜,不放任何濾光片,先做低解析度(8 cm-1)放光光譜量測。 在熱平衡之條件下,黑體輻射源其單位波數(cm-1)區間內之輻射密 度ρ 為:
(
)
(
3 1)
T k ν~ 100hc 3 8 cm m J 1 ~ πhc 10 8 T , ~ B − − − × = e ν ν ρ (3-2) 其中ν~為放射光波數(cm-1)、T 為溫度(K)、c 為光速(2.9979×108 m s-1)、h 為 Planks 常數(6.626176×10-34 J s)。因此,可計算黑體輻射 源在1273 K 時,其光譜曲線最大值約在 2500 cm-1附近。若光譜 曲線的最大值小於2500 cm-1,表示黑體輻射光源位置不佳,偵測器量測到部分低溫之放光(可能為黑體輻射源出口的金屬轉盤之 低溫所致)。則必須調整黑體輻射源的位置,使放光譜線的最大值 約在2500 cm-1附近。
7. 將 FTIR 內部抽真空,以實驗所需的解析度,進行全光區之一般 模式掃瞄(normal scan),如圖 3-4 中的 Curve A 所示。
8. 因為實驗中有些光徑並未能抽真空,因此前步驟所量測的光譜可 能含有水及二氧化碳之吸收,故利用軟體將此吸收峰去除後,即 可得到如圖 3-4 中的 Curve B 所示。理論上已知溫度的黑體輻射 曲線可直接經由電腦軟體計算出來,但是實驗利用紅外光偵測器 所得到的是輻射率L (radiance):
(
)
(
2 1 1)
T k ν~ 100hc 3 2 8 T k ν~ 100hc 3 8 cm sr m W 1 e ~ hc 10 2 4π c 1 ~ πhc 10 8 Ω c T , ~ L B B − − − − × = × − × = × = ν e ν ν ρ (3-3) 其中Ω為立體角(solid angle,sr)。而在實驗上最後要知道的是每 個能階之佈居數,和放出的光子數目有關,因此必須將偵測器量 測之能量訊號轉換為光子數目之訊號;故在計算黑體輻射曲線時 必須採用光子/波數分佈(photon/wavenumber distribution)的方式, 即光子通量密度Φ (photon flux density),其表示式如下:1 e ~ c 10 2 ~ 100hc radiance E radiance T k ~ 100hc 2 6 photon B − × = = = Φ ν ν ν (3-4) 利用 OPUS-NT 軟體內建的黑體輻射光子通量密度理論曲線,並
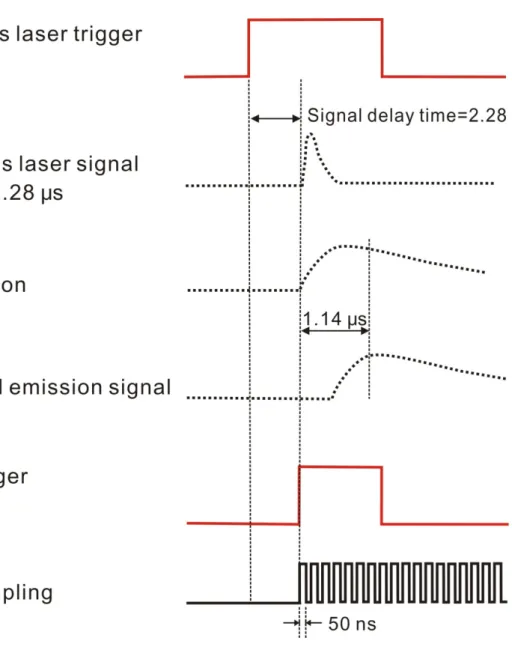
設定黑體輻射溫度為 1273 K 後,即可得之曲線如圖 3-4 中的 Curve C 所示。 9. 將 Curve B 除以 Curve C,即可得到反應系統之儀器響應曲線。 10. 欲量測不同的濾光片所造成之波長響應關係時,可將濾光片放置 於光徑中進行黑體輻射源之放光量測;也可以利用 FTIR 內部光 源測量濾光片之穿透度,如圖 3-4 中的 Curve D 所示。因此,若 實驗中加入濾光片,將全光區之黑體輻射源放光光譜除以特定溫 度下之黑體輻射理論曲線後,再乘以濾光片之儀器響應曲線即為 完整之儀器響應曲線。 3.2.3 觸發光解雷射的反應時間 由於雷射經觸發後,需經過延遲時間後才會抵達反應槽,故利用 一快速響應光電二極體量測此延遲時間。步驟如下: 1. 完成脈衝產生器的設定:內部觸發、23Hz;A 輸出端:high impedance、TTL、normal、A=T0。 2. 將 DG535 之 T0輸出端連接至光解雷射之外部觸發輸入端、A 輸 出端連接至示波器之頻道 A,並將偵測雷射光的光電二極體 (photodiode)連接至示波器之頻道 B。設定示波器受到頻道 A 的觸 發。
3. 將光電二極體置於反應槽的入口光窗旁,量測光解雷射進入反應 槽時,由光窗所反射的散射光。觀察示波器上頻道A 與頻道 B 訊 號之時間間隔,即為觸發光解雷射的反應時間。本實驗所使用的 光解雷射,受到觸發後,需經過2.28 μs 後才會輸出雷射光抵達反 應槽,其時序示意圖如圖 3-5 上半部所示。本實驗系統中,雷射 出口至反應槽之距離約為2 公尺,故吾人估算光行走之時間大約 為 ~6ns ms 10 3 m 2 1 8 − × ,因此,雷射經觸發後抵達反應槽之延遲時間, 主要來自於雷射中Thyratron 放電所造成之延遲時間。 3.2.4 偵測器及相關電子儀器之響應時間 在本實驗中,訊號經由偵測器偵測後,經過兩個前置放大器才進 入PAD board 進行訊號擷取,因此,必須考慮所有儀器所需的響應時 間。吾人將一道短脈衝的 IR 雷射光導入光譜儀中,並利用本實驗所 使用的偵測系統進行偵測,即可測得偵測系統的響應時間,從而精確 地判斷瞬時放光訊號之起始時間。 測量之相關裝置連接情形如圖 3-6 所示,利用 Nd-YAG 雷射
(Spectra Physics,Lab 170)激發染料雷射(Spectra Physics,PDL-3),產
生波長λ=830 nm 之可見光,並導入內裝有約 15 bar H2的拉曼位移 槽,產生一個拉曼移頻之IR 光( 1 IR 12048 4155.2 7892.8cm ~ = − = − ν ),再將 此紅外光減弱後導入反應槽,IR 光之散射光在 Welsh cell 內作多次反
射後,經CaF2透鏡組被引導至FTIR 進行偵測。偵測器所記錄之訊號
經過其內置放大器放大後,再傳送到SR560,將訊號以 100 倍率放大
(SR560 之設定需與實驗條件一樣),最後傳送至數位式示波器觀測並
存檔,且將此訊號傳送至FTIR 之 PAD 1232 A/D converter 以擷取光
譜 , 由 此 時 域 解 析 光 譜 可 導 出 真 實 之 儀 器 響 應 函 數 (instrument response function,IRF)。
藉由脈衝產生器控制各儀器間之先後觸發順序,利用快速響應的
光電二極體量測雷射光束抵達反應區之延遲時間,觸發 Nd-YAG 雷射
及FTIR 取樣之時序關係如下:
T0 觸發 Nd-YAG 雷射之 flash lamp
A= T0 + 182 μs 觸發 Nd-YAG 雷射之 Q-switch
B= A + 344 ns 觸發 FTIR 開始取樣
其中,Nd-YAG 雷射之 Q-switch 在 DG535 觸發 flash lamp 後約 182 μs
才啟動。而產生之IR 光抵達反應槽之延遲時間為 344 ns,此為 FTIR
取樣之時間零點,即PAD 1232 A/D converter 之延遲時間為 B= A + 344 ns。
根據所量測的時域解析光譜其訊號隨時間之變化,可得到相關電 子儀器之響應函數(IRF),如圖 3-7 所示,利用 Gaussian 函數適解 之,