由有機金屬化學氣相沈積成長不同長晶溫度之氮化銦奈米點的光學與形貌特性研究
65
0
0
全文
(2) 由有機金屬化學氣相沈積成長不同長晶 溫度之氮化銦奈米點的光學與形貌特性 研究 Study of optical and structural properties of InN nano-dots grown at various temperature by metal organic chemical vapor deposition 研 究 生:戴士凱. Student: Shi-Kai Tai. 指導教授:陳衛國 教授. Advisor: Prof. Wei-Kuo Chen. 國立交通大學電子物理研究所碩士論文. A Thesis Submitted to Institute of Electrophysics College of Science National Chiao Tung University in Partial Fulfillment of the Requirements for The Degree of Master of Physics in Electrophysics August 2007 Hsinchu, Taiwan, Republic of China 中華民國九十六年八月.
(3) 索引 中文摘要..………………………………………………………………i 英文摘要..………………………………………………………………ii 第一章 緒論…………………………………………………………01 第二章 理論背景……………………………………………………06 2.1 半導體中的光激螢光光譜..………………………………06 2.2 Burstein-Moss 效應..…………….………….….…………12 第三章 實驗方法………………………………………………………16 3.1 樣品的製備……………………………………….………16 3.2 原子力顯微鏡(AFM)系統…………………….……...18 3.3 光激螢光(Photoluminescence)系統……………...…....22 第四章 結果與討論……………………………………………………24 4.1 樣品形貌討論…………………………….………………24 4.2 低溫光激螢光光譜……………………….………………34 4.3 變溫光激螢光光譜……………………….………………42 第五章 結論……………………………………………………………50 附錄………………..……………………………………………………52 參考文獻 ………………………………………………………...………55.
(4) 致謝 碩士的生活就要結束了,因為許多人的幫助這篇論文才能完成。 首先要感謝陳衛國老師、李明知老師、周武清老師、張文豪老師們的 悉心教導,讓我能瞭解自身的盲點與與必須補強的地方,在這特別要 感謝指導教授陳衛國老師耐心的指導,並且讓我有機會瞭解作研究該 有的嚴謹態度,並且訓練我如何思考與解決問題。再來要感謝陳京玉 學長在這篇論文中的大力幫助,常常在與學長討論後,對現在的問題 與解決方法又有了進一步的想法。也要感謝柯文博士、古慶順博士、 李寧學長、蔡人文哲學長在研究與實驗上鼎力相助。感謝包家偵、李 啟仁、羅士傑、邱泰鑫、洪維偲、張尚樺、尤書鴻、林峰毅、陳仲威、 傅少甫、戴進吉、楊沛雯、翁嘉駿、陳膺中、楊子德、汪志遠、王仕 銘、王聖予、林家賢、陳威宇、林偉翔陪我度過這難忘的碩士生活。 也謝謝聯華電子的王秋月小姐對論文撰寫與文獻整理所提供的寶貴 意見。 在煎熬與困苦的時候,感謝我的家人的體諒與陪伴。感謝我爸媽 對我的鼓勵與幫助,在這人生的叉路解答我的疑惑,提醒我做人做事 該有的拿捏與分寸。感謝球球對我的包容與帶給我快樂時光,這是我 堅持下去的動力。 要感謝的人太多,因為有你們我才能走完這碩士的路程,也因為 你們我才能有所成長。希望每個幫過我的人都能感受到我的感激。祝 健康、順心 戴士凱,2007.08 新竹交大.
(5) 由有機金屬化學氣相沈積成長不同長晶溫 度之氮化銦奈米點的光學與形貌特性研究 研究生:戴士凱. 指導教授:陳衛國博士. 國立交通大學電子物理研究所. 中文摘要 由流量控制法(flow-rate modulation epitaxy,FME)成長在 550 oC 到 725 oC 的氮化銦奈米點,利用光激螢光光譜(PL)與原子力顯微 鏡(AFM)的量測,研究氮化銦奈米點的光學性質與表面形貌。由 AFM 與 X 光光譜的分析,指出在長晶溫度低於 575 oC 時,有滴狀金 屬銦(In droplet)形成,這是被歸因為在低溫下氨的分解效率低落所 致。而當長晶溫度大於 675 oC 時,氮化銦的成長率快速下降,我們認 為在這長晶的區域內有銦原子的脫逸(In desorption)效應產生。在 光學的部分,我們發現滴狀金屬銦的形成,總伴隨著光學性質的惡 化。而在長晶溫度大於 600oC 沒有形成滴狀金屬銦的樣品上,則可以 看到較好的光學性質。所對應到的光譜峰值能量(peak energy ~ 0.76eV)幾乎不隨長晶溫度改變,而光譜的半高寬隨著溫度的上升也 只有些微的從~71 增加到~74 meV。此外我們也作了變溫光激螢光 光譜的量測,發現成長在 675~725 oC 的氮化銦奈米點樣品有 14~ 20meV 的紅移,這代表我們在高溫成長的氮化銦樣品有很好的品質。. i.
(6) Study of optical and structural properties of InN nano-dots grown at various temperature by metal organic chemical vapor deposition. Student:Shi-Kai Tai . Advisor: Prof. Wei-Kuo Chen. Institute of Electrophysics National Chiao Tung University. Abstract Photoluminescence(PL)and atomic force microscopy(AFM) measurements were used to investigate the optical properties and morphology of InN nanodots prepared by flow-rate modulation epitaxy at 550 ~ 725. o. C. In droplets were observed for samples grown at. temperatures <575oC, as confirmed by both AFM and X-ray diffraction measurements, which can be attributable to the poor cracking efficiency of ammonia at this temperature range. Nevertheless, as the growth temperature is higher than 675oC, rapid declination in growth efficiency. It suggests that desorption of In adatom occurs at such a growth region instead. Concerning the optical properties, it is found that once the In droplets are formed, the optical properties of InN dots deteriorate significantly. As for droplet-free samples grown at temperatures ≥600 oC, good luminescence properties are obtained. The corresponding PL peak. ii.
(7) energy is nearly independent ( ~ 0.76 eV ) of growth temperature, accompanied by a slight linewidth variation from 71 to 74 meV as the temperature varies from to . Furthermore, we also conducted temperature-dependent PL measurement. A clear redshift of 14~20 meV were found in InN nanodot samples prepared at 675~725 oC, suggesting good quality of our high-temperature InN samples.. iii.
(8) 第一章 緒論 氮化銦(InN)是這幾年積極受到重視的氮化物材料,因為近幾年 的研究發現,六角晶形(wurtzite)的氮化銦能隙為0.69eV1-4,而不是早 先所認為的2.0eV5,6。這可能是因為早期長晶技術還未成熟所致。氮化銦 的製備最早是在1938年由Juza和Hahe從InF6(NH4)3得來。70年代氮化銦合 成的方式則是由含有銦的化合物與氨或含氮的復合物反應所得。但是從 上述方法所得到的氮化銦不是粉末就是較小的結晶。到了1972年Hovel 與 Cuomo5 利 用 反 應 式 射 頻 濺 鍍 ( rf-sputtering ) 的 方 法 將 多 晶 (polycrystalline)氮化銦薄膜成長在藍寶石(sapphire)和矽基板上。並 且經由吸收光譜的量測,得知氮化銦薄膜的能隙約為1.9eV。然而到了 90 年 代 之 後 , 由 於 分 子 束 磊 晶 ( MBE ) 與 有 機 金 屬 化 學 氣 相 沈 積 (MOCVD)等長晶技術的成熟,可獲得結晶性較好的樣品以供研究。 所以近年來對之前較大的能隙數值有了大幅的修正,現在一般公認氮化 銦薄膜的能隙約在0.69eV。而這能隙的前後差異可能歸咎於氮化銦帶有 氧化物或者是形成的多晶結構2,3的關係。 由於上述對氮化銦能隙的修正,氮化物成為發展光電元件的重要材 料。因為氮化銦、氮化鎵與氮化鋁的合金的能隙範圍,可以包括從氮化 銦的 0.69eV,氮化鎵的 3.4eV 到氮化鋁的 6.2eV。因此這些氮化物合金 的發光波段,能涵蓋從紅外、可見到紫外光的範圍。所以能廣泛地應用 在微波通訊(1.3~1.55μm)與發光波長從微波到紫外光的光電元件 7,8。 特別是氮化銦鎵(InxGa1-xN)合金的能隙會隨銦組成的增加而從 3.4eV 下降到 0.69eV,所以可以把不同銦組成的氮化銦鎵(InxGa1-xN,x=0~1) 1.
(9) 合金堆疊起來,藉以吸收所處能隙的光子。而由圖 1-1 所示氮化銦鎵的 寬光譜可以涵蓋太陽主要的發光波段,以高效率將光能轉換為電能,因 此相當適合在太陽能電池的應用。 除了上述能隙上的特點外,氮化銦材料還有一些有趣的特性。例如 氮化銦材料的電子有效質量很低,約在 0.042~0.070 個電子質量(m0)9, 所以有較高的漂移速度(4.2×107cm/s),而且氮化銦材料還有熱穩定性 高的特性,使得氮化銦材料在發展高速元件與發光元件上有很好的遠 景。然而在 1990 年以前,因為材料品質不佳,有關氮化物半導體的研 究是與時遞減。最近在晶體品質的改進,以及 P 型摻雜的成功,導致了 另一波氮化物研究的熱潮。 除了上述氮化物的薄膜外,我們可以利用長晶技術創造限制電子在 半導體中運動的結構,近來有許多的團隊投入這類的研究。如果以電子 在半導體中運動的維度來分,可分為零維、一維、二維與三維,其別對 應到的結構為量子點、量子線(或量子柱)、量子井與塊材。由於奈米 點能將電子電洞限制在很小的三維空間中,使電子電洞容易在奈米點中 復合發光。並且由於奈米點中電子運動的三個維度都被侷限,使得電子 能階變得不連續,所以電子能態密度成為δ 函數,進而得到高熱穩定性 與狹窄的譜線寬度10的特性。再加上奈米點具有低門檻電流密度11、減少 缺陷密度,與調變發光波長等特性,使得奈米點結構適合用於雷射二極 體(Laser diode,LDs)與高效率光電元件的發展。 然而在成長高品質的氮化銦材料還是一個重要而困難的挑戰,這是 因為氮化銦成長的溫度範圍很窄。由於氮化銦的熱分解(decomposition) 約在 520oC~540 oC,而銦原子的脫逸現象(In desorption)約發生在 650 2.
(10) o. C 以上 12,因此限制了氮化銦的成長溫度必須在較低溫的環境下,再加. 上氨要在溫度大於 500oC 時才開始分解 13,造成能夠成長氮化銦的溫度 範圍很窄,也因此增加了成長高品質氮化銦的困難度。除此之外,氮化 銦材料中經常被討論到的 Burstein-Moss 效應,會嚴重影響到氮化銦材 料的光學特性。原因在於 Burstein-Moss 效應指的是簡併(degenerate) 半導體中,載子濃度上升所造成的費米能階位置上升與吸收光譜起始點 的藍移(blue shift)。由於氮化銦材料的電子有效質量很低約為 0.042~ 0.070m0,所以傳導帶中的電子能態密度較低,以致於在成長氮化銦材料 時,常有高濃度非刻意添加的載子容易填到較高的能階上,而造成量測 氮化銦材料能隙與光學性質上的差異。再加上氮的高蒸氣壓. 14,15. ,因此. 成長高品質的氮化銦仍然是一個困難的議題。 截至目前為止討論氮化銦奈米點的論文仍然屬於少數,而對於氮化 銦奈米點的光學性質研究更是少之又少。第一個發表氮化銦奈米點光學 性質的是在 2005 年 Intartaglia16 的文章。Intartaglia 是利用有機金屬氣相 沉積(MOVPE)的方式在二氧化矽上成長氮化銦奈米點,其光激螢光 光譜峰值能量(PL peak energy)並不會隨著不同量測溫度而改變。而 在 2006 年柯文正博士 9 的論文中提及利用氮化銦奈米點大小調變其發光 特性的研究,隨著奈米點的平均高度由~32.4nm 下降到~6.5nm,光激 螢光光譜峰值能量也明顯的從 0.78eV 藍移到 1.07eV。然而對於氮化銦 材料狹窄的長晶溫度範圍,對氮化銦奈米點光學性質的影響,到目前為 止還沒有相關的研究發表。所以在這篇論文中,我們主要討論的議題是 長晶溫度在 550~725oC 的氮化銦奈米點,其表面形貌與光學性質隨長 晶溫度的變化情形。 3.
(11) 在這篇論文中,我們會在第二章中,簡短的介紹兩個相關的理論與 實驗結果,如半導體中的光激螢光光譜與 Burstein-Moss 效應。而第三 章會說明樣品的製備與實驗的儀器,包括原子力顯微鏡(AFM)於光激 螢光(PL)系統。在第四章中,我們會針對不同長晶溫度的樣品形貌、 低溫與變溫光激螢光光譜的實驗結果加以分析討論。而最後我們會在第 五章總結這些實驗結果。. 4.
(12) 圖1-1 太陽光在各波段的發光強度與氮化銦鎵(InxGa1-xN)合金 能隙的關係圖. 5.
(13) 第二章 理論背景 為了能分析實驗結果與深入討論,我們將在這一個章節中描述此樣 品在量測時的相關理論模型及常見的特性。所以本章將就半導體中的光 激螢光光譜與 Burstein-Moss 效應兩個部分個別討論:. 2.1 半導體中的光激螢光光譜 當雷射光打在半導體上時,可能會發出比入射光的波長還長的光 子,而這種光子是藉由雷射光子激發所得,故稱為光激螢光 (Photoluminescense,PL)。這光子的產生原因在於入射雷射光子的能 量被價帶裡的電子吸收,而使得電子從價帶激發到傳導帶。而這激發態 的電子會再次落到基態而放出光子這就是所謂的輻射躍遷(radiative transitions)。這樣的光子能量可能代表的是傳導帶的底部,到價帶頂端 的能量差。如果存在缺陷或雜質,則此能量就可能代表兩缺陷位階 (defect level)的能量差。而這些差異都反映在材料發光的特性上,而 從光激螢光光譜,我們能得到電子躍遷的資訊。然而電子也可能藉由非 輻射的過程(non-radiative recombinations)與電洞復合,因為這樣的復 合不伴隨著光子的產生,因此非輻射的復合會減低發光的效率。 由於光激螢光光譜的分佈與電子電洞對的結合路徑有關,因此光激 螢光光譜的峰值能量(peak energy) 、強度與半高寬(FWHM) ,都受缺 陷與雜質的種類與濃度影響,所以光激螢光光譜可作為樣品光學品質的 量測利器。以下我們會描述一些影響光激螢光光譜的主要輻射與非輻射 復合過程。 2.1.1 輻射過程 6.
(14) (I)帶間躍遷(band to band transition) 帶間躍遷通常是發生在直接能隙(direct band gap)的材料上,例如 III-V 族的半導體。電子由傳導帶躍遷到價帶不牽扯到動量的改變,如圖 2-1-1 所示。 而這樣的電子電洞復合率(R)可以表示為 R = ∫ R(hν )d (hν ) ≈ np ,. 這裡的 n、p 分別代表電子與電洞的濃度,h 是蒲郎克常數(Plank s constant) ,ν 是光子頻率,R(hν )是光子能量在 hν 時電子電洞復合的機 率密度。所以載子濃度越高,復合率越高。 (II)施體與受體對的再結合(Donor acceptor pair recombination) 即是在施體位階上電子與受體位階上電洞的再復合。電子與電洞是 由電中性的施體(Do)與受體(A0)所產生的。當電中性的施體(Do) 與受體(A0)中的電子與電洞復合時會放出光子,而剩下帶正電的施體 +. -. 。 (D )與帶負電的受體(A ) Do +Ao → hν +D +A +. -. 而發出的光子能量為 . e2 hν = E g − ( ED + E A ) + ε × RDAP. ED 與 EA 是施體與受體中載子的束縛能,ε 是材料的介電係數,RDA 是施 體與受體的距離。 2.1.2 非輻射過程 然而輻射躍遷並不是電子電洞對復合的唯一途徑,幾個造成發光效. 7.
(15) 率下降的非輻射過程列舉如下: (I)聲子放射(phonon emission) 經由雷射激發產生的電子電洞對,再復合時放出的能量可能不是以 光子的形式,而是以多個聲子的方式放出能量。所以傳導帶的電子與價 帶的電洞復合後,能量由多個聲子帶走,並且與晶格散射使晶體發熱﹐ 這種過程稱為聲子放射 (Ⅱ) 表面復合(Surface recombination) 在長晶的過程中,表面與界面的斷鍵容易形成缺陷能階,而電子在 移動時可能會被這些缺陷能階所捕捉,使得電子電洞在表面復合並且經 由電子在缺陷周圍的大量振動釋放出熱能。 (III) 歐傑效應(Auger effect) ㄧ般歐傑效應主要分為三種,分別為: 在傳導帶(C)裡的兩個電 子與價帶(H)裡的ㄧ個電洞所進行的復合過程(CCCH),在傳導帶 裡的ㄧ個電子與重電洞帶裡的兩個電洞進行復合(CHHS)、(CHHL)。 如圖2-1-2,CCCH過程主要發生在傳導帶裡的電子1(C)由於庫倫交互 作用力撞擊另ㄧ個電子2(C),電子1本身因為反作用力彈向高能階處1’ (C),另ㄧ顆電子2被撞擊後跑到價帶2’(H)與電洞進行復合(所以 稱為CCCH)。而在高能階處1’的電子由於位能高,容易在傳導帶中將 能量轉換為聲子,而掉至傳導帶的基態,這釋放給聲子的能量稱之為歐 傑之臨界能量(threshold energy)。而CHHL過程與CCCH相反,在價帶裡 的兩個電洞1(H)與2(H)由於庫倫交互作用力,吸引傳導帶的電子1’ (C)與被撞擊的電洞進行復合,而本身帶能量的電洞2’(L)跑至輕電洞 帶(light hold band)(所以稱為CHHL),再轉換能量回到價電帶的基 8.
(16) 態,此轉換能量亦是歐傑臨界能量,此能量與能隙成正比;然而CHHS 與CHHL相似,只是將輕電洞帶換為分離帶。而轉換過程中所放出的能 量亦是歐傑臨界能量,此能量與能隙成正比,與分離帶分離能量(△SO) 成反相關。. 9.
(17) E. Ec 激 發. 放 出 光 子. hν. 復 合. Ev. K. K. 圖 2-1-1 直接能隙半導體材料的帶間躍遷示意圖. 10.
(18) CCCH C. E. 1’ 2 1. K HH 2’ △SO LH. CHHL. CHHS. SO. C. C. E. E. 1’. 1’. 1. 2. 1. K HH △SO. 2. K HH 2’. △SO 2’. LH. LH. SO. SO. 圖 2-1-2 ㄧ般 CCCH、CHHL 與 CHHS 的歐傑過程(Auger process) 。C 代表傳導帶(conduction band) ,HH 代表重電洞帶(heavy hold band) ,LH 為輕電洞帶(light hold band) ,SO 為分離帶(split-off hold band) ,△SO 為分離帶與重電洞帶間的能量差。 11.
(19) 2.2 Burstein-Moss 效應 簡併(degenerate)半導體是指載子濃度很高的情況下,費米能階 (Fermi level)會離開能隙(band gap) ,而跑到傳導帶或價帶中。以 n 型簡併半導體而言,吸收光譜的起始點,會對應到在傳導帶中費米 能階到價帶頂端的能量差。由於費米能階的位置會受電子濃度的影 響,進而改變吸收光譜的起始點。所以 Burstein-Moss 效應指的是電 子濃度增加時,所造成吸收光譜起始點的藍移(blue shift) 。 在氮化銦材料的討論中常會提及 Burstein-Moss 效應,這可能是 因為在氮化銦材料的成長中,常見高濃度非刻意添加的電子,並且氮 化銦材料的電子有效質量很低約為 0.042~0.070m0 9,所以傳導帶中 的電子能態密度較低,以致於電子容易填到較高的能階上,進而影響 氮化銦材料在光學性質的討論上,例如光激螢光譜的分析與吸收光譜 的量測。 電 子 濃 度 與 費 米 能 階 之 間 的 對 應 , 跟 傳 導 帶 結 構 ( band structure,E(k) )有關。以 J.Wu. 17. 對氮化銦材料研究的結果,認為. 氮化銦的傳導帶結構為一非拋物面的曲面,而對於電子濃度與吸收光 譜起始點的計算與實驗的數據如圖 2-2-1。可以明顯看到當電子濃度 增加時,吸收光譜起始點的藍移或費米能階位置向高能量移動。 光激螢光光譜的強度與放射光子能量的關係如下 1: ∞∞. I (hν ) ~ ∫∫ g n ( En ) f n ( En − E Fn )g p ( E p ) f p ( E p − E Fp ) 0 0. × δ ( En − E p − E g − hν )dEn dE p. (2.3.1). gn(En)代表電子能量在En 時,傳導帶電子的能態密度(density of states), gp(Ep)代表電洞能量在Ep時,價帶電洞的能態密度,EFn與 EFp代表傳導帶與價帶的準費米能階(quasi-Fermi levels) ,fn與fp代表 12.
(20) 準平衡的費米-迪拉克函數(nonequilibrium Fermi-Dirac functions), Eg為能隙。我們將電子電洞的能態密度與濃度隨能量的分佈作圖,並 表示在圖2-2-2。 由方程式(2.3.1)可知,因為光激螢光光譜的強度極大值,會 出現在電子濃度最高的能量與電洞濃度最高的能量之間的躍遷(如圖 2-2-2(a))。由於在氮化銦材料中,電子濃度遠大於電洞濃度,所以 光激螢光光譜的特徵主要受電子濃度分佈影響。當比較圖2-2-2(a、b) 兩圖,可以發現當費米能階上升,電子濃度分佈向高能量移動,所以 電子濃度最高的能量會藍移,使光激螢光光譜的強度極大值產生藍 移。也因為電子濃度分佈變寬,所以光激螢光光譜的半高寬(FWHM) 會變大。由此可知,當電子濃度增加,光激螢光光譜的強度極大值會 產生藍移,而且半高寬會變大。. 13.
(21) 理論計算 實驗數據. 圖 2-2-1 引用 J.Wu 17 的結果,得知霍爾量測得到的電子濃度大小與吸 收光譜起始點的關係。實線是非拋物面的傳導帶結構的理論 計算,而實點是實驗結果。. 14.
(22) E a). b) EFn2. EFn1 Ec PL peak. PL peak Ev. EFp. gn,gp. EFp. gn,gp. 圖 2-2-2 簡併半導體的電子電洞復合簡圖。縱軸是能量,橫軸是 傳導帶或價帶的狀態密度。圖中左半邊為 a) ,代表費米 能階較低的情況(EFn1<EFn2) ,而右半邊為 b) ,代表費米 能階較高的情形。塗滿的部分代表電子或電洞濃度隨能 量的變化。而光激螢光光譜的強度極大值,會出現在電 子濃度最高的能量與電洞濃度最高的能量之間的躍遷。. 15.
(23) 第三章 實驗方法 3.1 樣品的製備 我們的氮化銦量子點為 Wurtize 結構,是由有機金屬化學氣相沈 積(MOCVD)成長。樣品的結構簡圖如圖 3-1-1。首先在藍寶石基板 (sapphire)的(0001)面上,先後成長低溫(520oC、25nm)與高溫 (1080oC、1μm)的氮化鎵。再以流量控制(flow-rate modulated epitaxy,FME)的方法,在成長溫度為 550oC 到 725oC 之間,以 25oC 為間隔,成長 8 個不同成長溫度的氮化銦奈米點。五族的氮來源於氨 (NH3 ),而三族的鎵與銦分別由三甲基鎵(TMGa)與三甲基銦 (TMIn)提供。 流量控制法的簡圖如圖 3-1-2。我們的氮化銦奈米點是成長 6 個週 期,在一個週期中分為四個階段:20 秒讓活性氮原子與活性銦原子 充分化合的氮階段(NH3 step)與 20 秒讓活性銦原子成核的銦階段 (TMIn step) ,而中間分別有 10 秒的緩衝階段。在氮階段中,氨的 流量為 18000sccm。而在銦階段中,三甲基銦的流量為 150sccm。此 外在銦階段中,我們也提供了較少流量的氨 500sccm,其目的在於抑 制已沈積下來的銦再次散逸。因此在銦階段沈積下來的銦,將會在氮 階段中與充足的氮化合,轉換成氮化銦。. 16.
(24) InN QDs 550oC~725oC. HT-GaN 1080oC 1μm LT-GaN 520oC 25nm Sapphire. 圖 3-1-1 樣品結構簡圖。8 片氮化銦量子點樣品分別成長在 550oC、575 . o. C、600 oC、625 oC、650 oC、675 oC、700 oC、725 oC。. flow rate (sccm) 18000 NH3. 10000 500. 20s 10s 20s 10s. 150 TMIn 0 Time ( s ) 圖 3-1-2 流量控制法(FME)簡圖。我們的氮化銦量子點是成長 6 個 週期,在一個週期中分為四個階段:20 秒的氮階段與 20 秒 的銦階段,而中間分別有 10 秒的緩衝階段。. 17.
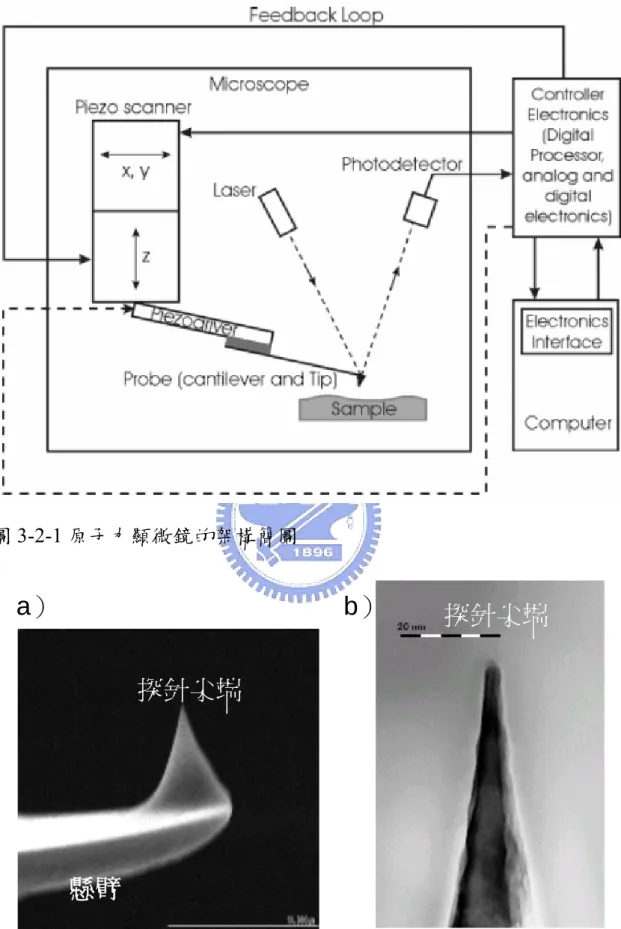
(25) 3.2 原子力顯微鏡(AFM)系統 NT-MDT SOLVER P47H 的原子力顯微鏡系統是用於量測氮化 銦奈米點的表面形貌。其架構簡圖如圖 3-2-1。基本原理在於利用探 針懸臂上探針的尖端(如圖 3-2-2)靠近樣品表面,並且利用打在探 針懸臂前端的雷射被反射後打在光偵測器的位置(如圖 3-2-1) ,藉以 推測懸臂的彎曲量。當探針尖端距離樣品甚遠時,探針尚未受力,探 針懸臂不會偏折,故打在探針懸臂前端的雷射被反射後會打在光偵測 器的中心。當探針尖端接近樣品時,因探針尖端開始受力,探針懸臂 產生偏折,雷射打在光偵測器的位置開始偏移。所以我們可以從控制 雷射打在光偵測器的位置,來控制探針尖端與樣品表面間的作用力。 當我們決定雷射打在光偵測器的位置後開始掃瞄,樣品表面的起伏, 會改變探針尖端與樣品間的作用力,進而改變雷射打在光偵測器的位 置。經由回饋電路(electrical feedback loop)可以將這改變回傳給控 制探針的壓電材料(piezoelectric crystal) ,此時壓電材料會在 Z 方向 調整,使得雷射打在光偵測器的位置,回到我們一開始的設定值(每 個掃瞄點的量測時間小於 0.6ms)。所以我們能得到一個作用力固定 的樣品表面形貌。 一般而言有三種方式能夠掃瞄出樣品的表面形貌,分別是接觸式 (contact mode)、非接觸式(non-contact mode)與輕敲式(tapping mode) 。分別介紹如下: (I)接觸式(contact mode) 顧名思義,這種量測方法是利用探針尖端直接接觸樣品表面,以 得到較強的作用力。利用回饋電路,使得探針尖端的作用力維持固 定,經由軟體計算壓電材料的伸長量變化,而得到一個作用力固定的. 18.
(26) 樣品表面形貌。 NT-MDT SOLVER P47H對探針懸臂彎曲角度(Δθ) 的偵測可以精確到0.1秒(1o/3600),探針懸臂的彈力係數(k)約為 5.5~11.5N/m,懸臂長度(L)約為100~130μm,所以經由下式可推 得探針受力的解析度(ΔF)為2.7~7.3×10-10 N:. ΔF = k × L × Δθ (II)非接觸式(non-contact mode) 雖然接觸式的掃瞄能獲得來自樣品表面較大的作用力,但相對的 也對樣品表面有較大的傷害。所以為了避免探針對樣品的破壞,可以 利用非接觸的方式量測。首先樣品與探針保持一段距離(大約是幾百 奈米) ,將一個交流電壓(~0.1V)加在探針的懸臂上(如圖 3-2-2), 使其在自然頻率(ω0,150~300kHz)下震盪,藉以探測樣品與探針 尖端間的遠距離凡得瓦力(Van der Waal’s force) ,經由下式:. ΔF ~ C × Δθ × ω0 C是與環境阻尼有關的常數,約為~2.9×10-10Ns,可推得探針受力的 解析度(ΔF)為2.1~4.2×10-11 N。雖然非接觸式量測對探針受力的解 析度較高,但距離樣品較遠,所受樣品作用力也相對減少(F~1/r4), 所以為了得到較佳的解析度,必須在真空的環境下量測。 (III)輕敲式(tapping mode) 輕敲式(也稱為半接觸式)是由非接觸式改良而來,經由增加懸 臂的振幅,縮減探針與樣品的距離,以得到較大的樣品作用力,並保 留非接觸式量測法,高受力解析度的優點,所以能得到比非接觸式更 清晰的樣品表面圖像。而且探針尖端與樣品的接觸較接觸式來的少, 所以能避免破壞樣品表面 在我們的研究中是使用輕敲式量測樣品形貌,而不使用會磨損樣. 19.
(27) 品表面的接觸式,與探針受力較小的非接觸式量測方法。 在這篇論文中,我們使用原子力顯微鏡量測氮化銦奈米點的表面 形貌。而所得到的奈米點形狀、奈米點密度和奈米點大小的數據將在 第四章中加以討論。. 20.
(28) 圖 3-2-1 原子力顯微鏡的架構簡圖. a). b). 探針尖端. 探針尖端. 懸臂 圖 3-2-2 掃瞄式電子顯微鏡(SEM)下探針的形貌 a)為探針懸臂於 探針尖端的形貌,而 b)為探針尖端的放大圖 21.
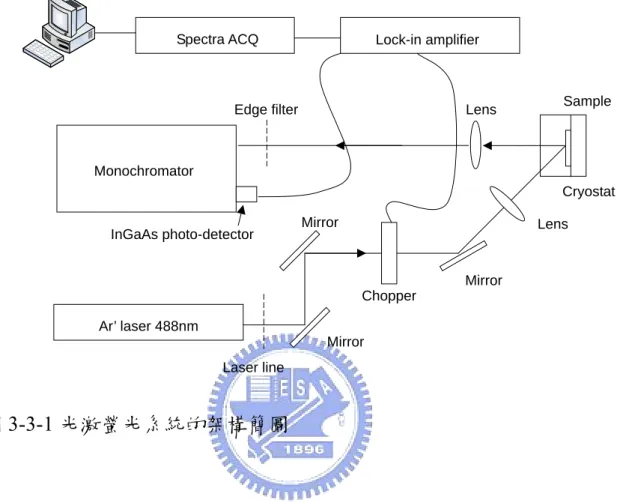
(29) 3.3 光激螢光(Photoluminescence)系統 光激螢光系統的架構簡圖如圖 3-3-1。光激螢光系統是使用工作 波長為 488nm 氬離子雷射(Argon-ion laser)作為激發光源,雷射功 率密度約為~80W/cm2。雷射光經過一個 laser line 之後,利用反射鏡 將雷射光導向樣品,並利用焦距為 15cm 的透鏡將雷射聚焦到樣品表 面。此聚焦光點的直徑約為 0.5mm。而經雷射光激發的螢光,通過焦 距為 5cm 的透鏡後,還會再通過一片波長大於 850nm 可過的濾光片 ( Long-pass edge filter )。 最 後 光 激 螢 光 會 進 入 光 譜 儀 ( mono-chromator )。 這 訊 號 是 利 用 InGaAs 的 光 子 偵 測 器 (photo-detector)來偵測波長從 1000nm 到 2200nm 的光子。並且經 由鎖相放大器(lock-in amplifier)放大後,由 Spectra ACQ2 處理分析。 對於低溫與變溫的光激螢光光譜量測,樣品是放置在封閉回路的 低溫恆溫器中,低溫是維持在 20K,而變溫的量測則是改變溫度從 20K 到 300K。. 22.
(30) Spectra ACQ. Lock-in amplifier. Edge filter. Lens. Sample. Monochromator Cryostat Mirror. InGaAs photo-detector. Lens. Mirror Chopper Ar’ laser 488nm Mirror Laser line. 圖 3-3-1 光激螢光系統的架構簡圖. 23.
(31) 第四章 結果與討論 無庸置疑的,長晶溫度是一個相當關鍵的長晶參數,無論是成長 薄膜或者是奈米點。所以我們在本章中,會就成長溫度為 550oC、 575oC、600oC、625oC、650oC、675oC、700oC 與 725oC 等八片樣品, 作表面形貌、低溫與變溫光激螢光光譜的實驗,並對其結果加以分析 討論。在形貌上我們分析了氮化銦奈米點的密度、高寬與晶形,而在 光激螢光光譜的實驗中,我們將討論不同長晶溫度下,氮化銦奈米點 的光學性質。 4.1 樣品形貌討論 觀察這一系列改變成長溫度樣品的表面形貌,我們可以歸納成三 種結構,分別是滴狀金屬銦(In droplet 圖 4-1-1 a) 、梯形六角錐的氮 化銦奈米點(Truncated pyramid,如圖 4-1-1 b)與六角平台的氮化銦 奈米點(Flat-top,圖 4-1-1 c) 。 滴狀金屬銦在形貌上的特徵為半球型結構,沒有明顯晶形,類似 水滴狀的形貌,高寬比約為~0.36。這種結構只在長晶溫度為 550oC 和 575oC 的低溫才會出現,我們認為他可能是由金屬銦組成。原因在 於由圖 4-1-2 X 光光譜中,我們會看到只有在長晶溫度為 550oC 和 575oC 時,會有從金屬銦[1,0,1]或[1,1,0]面反射出來的訊號(在 θ=33.4o or 38.7o 處) ,而滴狀金屬銦的形貌也只出現在這低溫的兩個樣品,所 以合理地推斷,金屬銦的訊號來自滴狀金屬銦的結構。而就長晶的觀 點來說也是合理的,因為低溫成長的氮化銦奈米點通常會受限於氨的 分解量不足,所以使多餘的銦形成滴狀金屬銦的形貌,這部分的論述 會在之後作說明。. 24.
(32) 而梯形六角錐與六角平台的氮化銦奈米點,在形貌上的特徵分別 為: 1.從俯視圖中皆可看到明顯的六角晶形(見圖 4-1-1 b) 、c) ) 。而由截 面圖則可看出,兩種結構都是平頂(見圖 4-1-1 e) 、f))。 2.斜邊晶面與底面(氮化鎵[0,0,0,1]面)的夾角在梯形六角錐為 28o, 而在六角平台則為 20o,由此推測梯形六角錐的斜邊晶面是對應到 氮化銦的[1,0,-1,2]面,而六角平台的斜邊晶面是對應到氮化銦的 [1,0,-1,3] 面。 這兩種類型的結構,可以在每一個成長溫度的樣品中看到。而在 X 光光譜(圖 4-1-2 )中,而且在所有成長溫度下,都會有從氮化銦[0,0,0,2] 面反射的訊號(在 θ=31.86o 處) ,所以可以得知這兩種類型的結構, 為氮化銦奈米點。這兩種類型的氮化銦奈米點,在不同成長溫度下的 高寬分佈表列在表 4-1-1。可以看到長晶溫度在 550oC~725oC 之間的 樣品中,梯形六角錐氮化銦奈米點的高度約在~35nm 左右,直徑約 在~210nm。而六角平台氮化銦奈米點的高度則約在~18nm,直徑約 為~210nm。 在圖 4-1-3 氮化銦奈米點密度對成長溫度的關係中,可以明顯分 成三區:低溫區,長晶溫度在 600oC 以下,氮化銦奈米點的密度從 550oC 的 7.2×107cm-2 迅 速 增 加 超 過 一 個 數 量 級 到 600 oC 的 8.6×108cm-2。中溫區,長晶溫度在 600oC~650oC,可以發現隨長晶溫 度的增加,氮化銦奈米點的密度幾乎沒有改變,都維持在~1×109cm-2 左右。高溫區,長晶溫度在 675oC 以上,氮化銦奈米點的密度從 675 o. C 的 9.2×108cm-2 快速減少將近十倍到 725 oC 的 1.0×108cm-2。 首先我們討論低溫區的情況。如之前所提到的,在低溫區中,長. 25.
(33) 晶溫度為 550oC 和 575oC 的樣品上發現滴狀金屬銦的形成,這是因為 長晶溫度在 600oC 以下,從氨中所分解出來活性的氮原子數量較少 13. ,所以沒有與氮化合成氮化銦的多餘銦原子,就形成滴狀金屬銦。. 就長晶的角度而言也是合理的,因為氨的分解量不足,造成活性氮原 子與活性銦原子化合的氮階段(NH3 step)中沒有足夠的活性氮原子 從氨中分解出來,以致於在銦原子成核的銦階段(TMIn step)中沈 積下來的銦,來到氮階段時,沒有足夠的活性氮原子能使銦化合成氮 化銦。所以沒有化合成氮化銦的多餘銦原子,就會形成為半球型結 構,沒有明顯晶形的滴狀金屬銦。 也因此在低溫區(長晶溫度小於600oC)內,隨著長晶溫度逐漸 增加,活性氮原子的提供量也快速增加,化合成氮化銦的奈米點密 度,從550 oC的7.2×107cm-2迅速增加到600 oC的8.6×108cm-2,而滴狀金 屬銦的密度也從550 oC的9.2×107cm-2急遽減少到600 oC已看不到滴狀 金屬銦出現。 一般在改變溫度成長的奈米點,在底溫區中的奈米點密度會有較 高的現象18,通常解釋為低溫下原子移動能力較低所造成。但是我們 氮化銦奈米點在低溫區(長晶溫度為550~575oC)的情況卻是相反 的,原因可能是受限於III族或V族原料熱分解量的影響。又因為上述 在低溫區內有發現滴狀金屬銦的形成,故推論是受限於氨熱分解量的 可能性較大。為進一步說明在低溫區中,較低的氮化銦奈米點密度並 不受限於銦原子的提供,我們計算了不同長晶溫度下,單位面積內沈 積下來的銦原子莫耳數,如圖4-1-4。這銦的莫耳數是包括氮化銦奈 米點,與滴狀金屬銦內的銦原子莫耳總數。銦原子莫耳數的估計,是 利用原子力顯微鏡作表面形貌的分析,而得到每一個氮化銦奈米點與. 26.
(34) 滴狀金屬銦的體積,再利用莫耳數=(質量密度×體積)/分子量的公 式,可換算出這些體積所含有的銦原子莫耳數。在這裡我們所使用的 參數為,氮化銦與金屬銦的質量密度為6.81與7.31g/cm3,而分子量分 別為128.82與114.82g/mole。而對於取得氮化銦奈米點與滴狀金屬銦 體積的方法,詳細描述在附錄。 由上述圖4-1-4中,我們可以清楚看到,長晶溫度從550 oC~650 oC 之間,單位面積內沈積下來的銦原子莫耳數,都維持在~ 4×10-8mole/cm2,沒有明顯改變。也就說明了在銦原子成核的銦階段 (TMIn step) ,沈積下來的銦原子總量是固定的,這與一般認為三甲 基銦在~400 oC就已經完全熱分解的結果是相符合的,所以在低溫區 (長晶溫度在550 oC與575 oC)中三甲基銦應該是完全熱分解的。換 句話說,在低溫區中氮化銦奈米點密度較低的現象,並不是受限於銦 原子的提供。 除此之外,我們從沈積下來的銦原子莫耳數對長晶溫度倒數的關 係圖(圖4-1-4)中發現,在長晶溫度550 oC時,滴狀金屬銦所提供的 銦 莫 耳 數 , 約 在 ~ 4.22×10-8mole/cm2 , 到 了 575 oC 也 還 有 ~ 2.98×10-8mole/cm2。但是到了長晶溫度為600oC的樣品,滴狀金屬銦卻 突然消失,並且無論是從表面形貌或者是從X光的量測,都沒有看到 滴狀金屬銦的形成。同時我們也發現,在成長率與長晶溫度倒數的關 係圖中(圖4-1-4),低溫區氮化銦的成長率,隨長晶溫度的倒數成 指數關係(因為圖中,成長率的對數與長晶溫度的倒數,在低溫區是 成線性的關係)。這就對應到V族或III族原料的熱分解效應,而如之 前所討論的結果,在低溫區內的氮化銦奈米點密度變化,並不受限於 三甲基銦的熱分解,較有可能是與氨的熱分解有關。所以我們認為,. 27.
(35) 滴狀金屬銦所提供的銦莫耳數,在長晶溫度從575oC到600oC之間,迅 速降到零的原因,是來至於氨熱分解量在低溫區內,隨長晶溫度倒數 成指數上升的結果。 在中溫區(長晶溫度在600oC~650oC)的部分,從氮化銦奈米點 密度對長晶溫度關係圖(圖4-1-3)中可以看到,長晶溫度從600 oC增 加到650 oC,氮化銦奈米點一直維持在約~1×109cm-2左右的高密度。 而在成長率對長晶溫度關係圖(圖4-1-4)中,中溫區內的成長率也 是比其他兩個區域來的高,約在~3.6nm/mins左右。這是因為在中溫 區內,有足夠的活性氮原子數量從氨中分解出來,能與銦化合成氮化 銦。而且銦的脫逸(In desorption)效應也要在675oC以上才比較明顯。 這部分的論述將在下一段的文章中詳述。 在高溫區(長晶溫度在675oC以上)的部分。我們可以從氮化銦 奈米點密度對長晶溫度關係圖(圖4-1-3)中看到,氮化銦奈米點的 密度從675 oC的9.2×108cm-2迅速減少到725 oC的1.0×108cm-2,將近有一 個數量級的減少。我們認為這氮化銦奈米點密度,在高溫區驟降的現 象,與銦的脫逸(In desorption)效應有關。 原因在於從上圖中可以看到,氮化銦奈米點密度開始驟降的溫 度,約在650oC~675oC之間,並且從氮化銦成長率對長晶溫度關係圖 (圖4-1-4)中,也可以看到成長率在650oC~675oC之間開始驟降。而 E. Dimakis在論文12,19中,認為銦原子脫逸的溫度,約在650oC~670 oC 之間。這與我們的結果相近,所以推論在高溫區中,氮化銦奈米點密 度與成長率,隨長晶溫度快速下降的原因,與銦的脫逸有關。 比較S.Keller的論文20,21,他們的氮化銦薄膜同樣也是由有機金屬 氣相沈積(MOVPE)成長,長晶溫度從400oC~700oC。其氮化銦成. 28.
(36) 長率在620oC開始驟降。他們將這成長率的驟降的現象,歸因於氮化 銦的分解(InN decomposition),並且隨著溫度的上升,表面上會出 現滴狀金屬銦。這結果與我們是不同的,因為我們氮化銦成長率開始 驟降的溫度,是介在650~675oC之間,並且從X光光譜分析得知,我 們的樣品在高溫區並沒有金屬銦的訊號。這差異可能是因為我們的氮 化銦奈米點在高溫成長時,銦原子的脫逸速率大於氮化銦的分解速 度。氮化銦分解時氮化銦中的氮原子,會迅速的脫離出來,可能形成 氮氣分子(N2)而離開表面,留下銦的原子。而銦原子的脫逸效應則 是銦原子從樣品表面脫逸的現象。所以如果我們的氮化銦奈米點在高 溫成長時,銦原子的脫逸速率大於氮化銦的分解速度,就有可能發生 成長率急遽下降,並且沒有在樣品表面上留下滴狀金屬銦。. 29.
(37) (d). 50 0. 50 40 30 20 10 0. (c). (e). nm. nm. 100. (b). 0. 150. 300. 450. 600. nm. 50 40 30 20 10 0. (f). nm. (a). 0 50 100 150 200 250 300 350 400. 0 50 100 150 200 250 300 350 400. nm. nm. 圖 4-1-1 樣品表面結構的形貌與截面圖。a) 、d)是滴狀金屬銦,b)、 e)是梯形六角錐的氮化銦奈米點,c) 、f)六角平台的氮化 銦奈米點。. GaN. Al2O3 (0006). InN In(101). In(110) 0. Intensity (a.u.). 550 C 0. 575 C 0. 600 C 0. 625 C 0. 650 C 0. 675 C 0. 700 C 0. 725 C. 28. 30. 32. 34. 36. 38. 40. 42. 44. TH/2TH (θ) 圖 4-1-2 二環 X 光光譜。由圖中可清楚看到,長晶溫度在 550oC 與 575oC 時,有金屬銦在(101)面的訊號。. 30.
(38) 10. 10. -3. density(cm ). InN dot In droplet. 9. 10. 8. 10. 1.00. 1.05 1.10 1.15 -1 1000/Tg(K ). 1.20. 1.25. 圖 4-1-3 氮化銦奈米點密度對長晶溫度倒數的關係圖。實心方塊(◆) 代表氮化銦奈米點,半空心圓(. )代表滴狀金屬銦。由. 圖中虛線(600oC 與 650oC)可分為三區。低溫區:長晶溫 度為 550oC~575oC;中溫區:長晶溫度為 600oC~650oC; 高溫區:長晶溫度為 675oC~725oC 31.
(39) 2. 10. -8. 1. 10. Growth rate(nm/mins). In mole fraction per area(mole/cm ). -7. 10. Total In mole InN dot In droplet. 1.00 1.05 1.10 1.15 1.20 1.25 -1. 1000/Tg(K ) 圖 4-1-4 沈積下來的銦原子莫耳數對長晶溫度倒數的關係圖。實心方 塊(◆)代表氮化銦奈米點,半空心圓(. )代表滴狀金. 屬銦,空心正方形(□)則代表銦原子的總莫耳數,包含氮 化銦奈米點,與滴狀金屬銦。. 32.
(40) 梯形六角錐. 六角平台. o. 滴狀金屬銦. o. 長晶溫度( C) 高(nm) 寬(nm) 長晶溫度( C) 高(nm) 寬(nm) 長晶溫度(oC) 高(nm) 寬(nm) 550. 35.9. 240.5. 550. 18.5. 194.3. 550. 138. 322. 575. 24.8. 191.7. 575. 13.0. 168.4. 575. 107. 406. 600. 41.5. 251.8. 600. 20.3. 272.8. 625. 33.7. 197.1. 625. 18.1. 204.7. 650. 34.7. 228.3. 650. 19.3. 220.7. 675. 36.3. 206.8. 675. 18.2. 213.8. 700. 34.9. 205.1. 700. 15.7. 211.3. 725. 39.0. 196.5. 725. 17.4. 208.0. 表 4-1-1 兩種結構的氮化銦奈米點與滴狀金屬銦,在不同長晶溫度下 的平均高、寬。. 33.
(41) 4.2 低溫光激螢光光譜 改變長晶溫度從 550oC~725 oC 的這一系列樣品,量測在低溫 (20K)下的光激螢光光譜,其結果表示在圖 4-2-1a) ,聚焦在表面的 雷射功率密度(power density)為~80W/cm2。每一個長晶溫度的樣 品,其光譜的峰值能量(peak energy)與光譜的半高寬(FWHM), 也整理在圖 4-2-1b) 。其中在低溫成長的樣品(長晶溫度為 550oC 與 575oC) ,有較差的光學性質。我們可以發現光譜的半高寬(FWHM)是 偏高的~100meV,並且光譜的峰值能量是較高的~0.8eV,這與近年 來,一般公認氮化銦的能隙約在~0.69eV1-4 有明顯的差異,這樣的差 異可能來自於嚴重的的 Burstein-Moss 效應。而所謂的 Burstein-Moss 效應指的是電子濃度增加時,所造成吸收光譜起始點的藍移(blue shift) ,或費米能階向高能量移動。 Burstein-Moss效應在氮化銦材料中,是經常被討論到的現象,原 因在於氮化銦的材料中,常見非刻意摻雜的高濃度電子(>1018cm-3) , 並且氮化銦材料的電子有效質量很低約為0.042~0.070m09,所以傳導 帶中的電子能態密度較低,以致於電子容易填到較高的能階上,以致 於材料中的費米能階,被提升到傳導帶內。並且隨著電子濃度的增 加,費米能階也會隨之升高,所以光譜的峰值能量,與光譜的半高寬, 會隨著電子濃度的增加而增加(不同電子濃度的簡併半導體,其電子 電洞復合簡圖,表示在圖2-2-2)。為了定量的分析樣品中的電子濃 度,我們利用光激螢光光譜的譜線圖形模型(line shape modal)做估 算。而擬合的模型,是參考自由能階到受縛能階的復合模型 (free-to-bond recombination model)1。 首先我們可以將光激螢光光譜的強度,隨螢光能量的分佈,表示. 34.
(42) 如下: ∞∞. I (hν ) ~ ∫∫ g n ( En ) f n ( En − EFn )g p ( E p ) f p ( E p − EFp ) 0 0. × δ ( En − E p − Eg − hν )dEn dE p. (1). gn(En)代表電子能量在En 時,傳導帶內電子的能態密度(density of states) , gp(Ep)代表電洞能量在Ep時,價帶內電洞的能態密度。,EFn 與EFp代表傳導帶與價帶的準費米能階(quasi-Fermi levels) ,fn與fp代 表準平衡的費米-迪拉克函數(nonequilibrium Fermi-Dirac functions) , Eg為能隙。由於打在樣品上的雷射功率只有約~80W/cm2,所以激發 的電洞濃度約在~1016cm-3,相較於電子的濃度(~1018cm-3),是相 當微小的。所以在擬合的模型中,我們忽略了由雷射光激發所產生的 電洞在價帶頂端的分佈。所以(1)式可以改寫為: ∞. I (hν ) ~ ∫ g n ( En ) f n ( En − E Fn ) × δ ( En − E g − hν )dEn. (2). 0. 因此譜線圖形就近似成電子在傳導帶的分佈。 由於電子在傳導帶的分佈,在低能量半邊與傳導帶中的電子能態 密度有關,而電子能態密度可以計算如下:. g n ( En ) =. k 2 ( En ). π2. ×. dk dEn. (3). k是電子的波數,En(k)是電子能量與波數的關係,也就是傳導帶的 能帶結構(band structure) 。在此我們引用J. Wu在論文17中,用以模擬 氮化銦的非拋物線(non-parabolic)傳導帶結構,寫成:. h2k 2 1 h2k 2 2 En (k ) = E g + 2 + ( E g + 4 Emomentum × 2 − Eg ) 8π m0 2 8π m0. (4). h為Planck常數,m0為真空中的電子質量,Emomentum為電子動量矩陣元 35.
(43) 素(momentum matrix element)所對應的能量參數,在J. Wu的論文中 認為10eV是與實驗接近的數值。由上述非拋物線的傳導帶結構,可 以利用下式推導出在電子濃度為零(k=0,因為在電子濃度趨近於零 時,傳導帶上的電子會在傳導帶的底部,也就是k=0的地方)時,氮 化銦材料等效電子質量(m*):. 2π 2 d 2 E 1 =( ) × 2 m* h dk. (5). 經由計算得到氮化銦材料等效電子質量(m*):為0.065m0,這與一般 所認為的0.042 m0~0.070m0是相符合的。在高能量的半邊,則是受 Fermi-Dirac分佈函數主導。由於光激螢光光譜所描述的是一個非熱平 衡的狀態,所以同時存在電子與電洞的準費米能階,而在此所討論的 電子在高能量半邊的分佈,是受電子的準費米能階主導。所以我們可 以藉由兩個擬合參數,能隙(Eg)與電子的準費米能階(EFn) ,擬合 光激螢光光譜的譜線圖形。 我們擬合的曲線與低溫光激螢光光譜的譜線,整理比較在圖 4-2-2。可以看到實驗所量測到的光譜譜線(空心圓)與擬合曲線(紅 線)有很好的對應,尤其在高能量的半邊,兩者是十分符合的。因此 在準費米能階位置(決定光譜譜線高能量半邊的強度分佈)的擬合是 具有代表性的。 得到電子的準費米能階(EFn)後,可藉由下式得到熱平衡時的 電子濃度。 ∞. n = ∫ g n ( En ) f n ( En − EFn )dEn − n photon . (6). 0. nphoton代表受雷射激發的電子濃度,約在~1016cm-3。所以擬合結果所 36.
(44) 得到的費米能階,與經由(6)式換算所得的電子濃度,表列在表 4-2-1。我們可以清楚看見低溫區(長晶溫度在550oC與575oC)的電子 濃度是在較高的4.0~4.2×1018cm-3。相較於中溫區(長晶溫度在600oC ~650oC)則是在較低的2.2~2.3×1018cm-3。到了高溫區(長晶溫度在 675oC~725oC) ,隨著長晶溫度從675 oC提升到725 oC,電子濃度有微 幅的下降從~2.2×1018cm-3到~1.7×1018cm-3。由此可知,低溫區中較 差的光學性質(峰值能量在~0.8eV,半高寬為~100meV),可能是 來至於Burstein-Moss效應。 關於上述在產生滴狀金屬銦,並且有較高電子濃度的低溫區中, 光學性質較差的成因,還沒有一定的論述。然而在C.Kruse22的文章中 有進一步的討論。文中在三族較充裕(Ga-rich)的環境下,成長氮化 鎵(GaN)薄膜,並且在表面形成滴狀金屬鎵(Ga droplet) 。他們發 現在滴狀金屬鎵下方,有氮化鎵的黃光放射(yellow emission) 。他們 將這結果歸因於,滴狀金屬鎵所引發的缺陷,如氮的空缺(N vacancy) 或點缺陷(point defects) ,而其他類似的論文中,也猜測可能與堆疊 錯誤(stacking faults)有關。由此推估,在我們樣品的低溫區中,光 學性質較差的成因,與滴狀金屬銦引發的缺陷有關。 由低溫光激螢光光譜的結果(見圖 4-2-1(a) 、 (b)) ,可以看到 當長晶溫度提升到 600oC 以上時,氮化銦奈米點的光學性質有了明顯 的提升。光激螢光光譜的強度,相較於低溫區,大幅增加了~10 倍, 而且光譜的峰值能量降到較低的~0.77eV,光譜的半高寬(FWHM) 也降到~71meV。在整個中溫區(長晶溫度在 600oC~650oC 之間) 中,氮化銦奈米點都有如此好的光學性質。 然而當長晶溫度升高到高溫區(長晶溫度在 675oC~725oC 之間) 37.
(45) 時,雖然如 4.1 節所討論的有明顯的銦脫逸效應,但是從低溫光激螢 光光譜的結果(見圖 4-2-1(a) 、(b) ),可以看到在高溫區所成長的 氮化銦奈米點,仍然有相當好的光學性質。我們觀察到光譜的峰值能 量,隨著長晶溫度從 675 oC 提升到 725 oC,有微幅的下降從~0.77eV 到~0.76eV。而且光譜的半高寬約為~75meV,也只比中溫區的光譜 半高寬(~71meV)些微上升。即使到了我們長晶溫度的最大值(725 o. C) ,光譜的峰值能量仍只有~0.76eV,光譜的半高寬也在~75meV。. 這樣氮化銦奈米點的光學品質,並不亞於其他也是用有機金屬氣相沈 積(MOVPE) ,在低溫(長晶溫度在 530oC~600oC 之間)所製備的 氮化銦薄膜 23,24。然而從低溫光激螢光光譜的結果(見圖 4-3-1(a)、 (b)),在高溫區中所觀察到的光譜強度下降,光譜的峰值能量的微 幅下降,以及光譜半高寬的些微上升,可能與氮化銦奈米點在高溫區 中,所受的銦脫逸效應有關。可能在銦脫逸吸附時會留下銦的空缺(In vacancies) ,因而產生一些非輻射復合中心。這部分我們將在 4.4 節 中討論。. 38.
(46) a). 20K PL 6. Intensity (a.u.). 2.0x10. 0. 550 C 0 575 C 0 600 C 0 625 C 0 650 C 0 675 C 0 700 C 0 725 C. 6. 1.5x10. 6. 1.0x10. 5. 5.0x10. x5 x5. 0.0 0.6. b). 0.7. 0.8. 0.9. 1.0. Energy (eV) 105 Peak energy FWHM. 90. 0.80 75 0.78 60. FWHM(meV). Peak Energy(eV). 0.82. 0.76 550 600 650 700 o Growth Temperature( C). 45. 圖 4-2-1 a)一系列長晶溫度從 550oC 到 725oC 的樣品,量測在低溫 (20K)下的光激螢光光譜。b)則是光譜的峰值能量(peak energy)與光譜的半高寬(FWHMs) 對不同長晶溫度的關係. 39.
(47) 1.0 0.5. 0.5. 0.0. 0.0 0.6. Normalize PL intensity(a.u.). 1.0. o. 550 C. 0.8. 1.0. 0.6. 0.8. 1.0. o. 1.0. o. 650 C. 575 C. 1.0 o. 675 C. 0.5. 0.5 0.0. 0.0 0.6. 0.8. 1.0. 1.0. 0.6 1.0. o. 600 C. 0.5. 0.5. 0.0. 0.0 0.6. 0.8. 1.0. 1.0. 0.5. 0.0. 0.0 0.8. o. 0.6. 0.5. 1.0. 1.0 700 C. 0.8. 1.0. o. 625 C. 0.6. 0.8. 1.0 o. 725 C. 0.6. 0.8. 1.0. Energy(eV). 圖 4-2-2 低溫所量測的光激螢光光譜譜線(空心圓)與擬合曲線(紅 線)的比較。在高能量的半邊,兩者是十分符合的。因此電 子準費米能階位置的擬合是具有代表性的。. 40.
(48) 長晶溫度(oC). 550 575 600 625 650 675 700 725. 費米能階(meV). 111. 115. 78. 81. 80. 78. 70. 64. 電子濃度(1018cm-3). 4.0. 4.2. 2.2. 2.3. 2.3. 2.2. 1.9. 1.7. 表 4-2-1 利用自由能階到受縛能階的復合模型,所擬合得到的電子準 費米能階位置(以傳導帶底部為原點)與對應的電子濃度。. 41.
(49) 4.3 變溫光激螢光光譜 為了進一步瞭解氮化銦奈米點的光學性質,我們做了變溫光激螢 光光譜的量測。量測溫度從 20K 到室溫,聚焦在表面的雷射功率密 度,與之前低溫的光激螢光光譜量測一樣是~80W/cm2。改變長晶溫 度從 550oC~725 oC 的這一系列樣品,光譜的峰值能量與量測溫度的 關係,表示在圖 4-3-1。其中我們有一片成長溫度為 650oC,厚度為 500nm 的氮化銦薄膜作為對照。在變溫光激螢光光譜的量測中,我們 可以清楚看到,這氮化銦薄膜的光譜峰值能量,從 20K 到室溫有~ 20meV 的紅移(red shift) 。如果討論量測溫度增加時,因為晶格常數 的擴張與電子-聲子的交互作用所造成的能隙縮減效應,我們可以利 用 Varshini 的經驗公式,擬合光激螢光光譜的峰值能量,與量測溫度 的關係曲線:. αT 2 E (T ) = E0 − β +T 擬合的結果,α~1.4×10-4eV/K,β~222K。然而對於我們這一系列不 同長晶溫度的氮化銦奈米點而言,在包含滴狀金屬銦的低溫區(長晶 溫度為 550oC 與 575 oC)內,改變量測溫度從 20K 到室溫,可以看到 光激螢光光譜的峰值能量,有些微的藍移(blue shift)約為 3~8meV。 而中溫區(長晶溫度為 600oC~650 oC)的樣品,則可以看到光譜的 峰值能量,不隨量測溫度的改變而移動(光譜峰值能量的移動小於 2meV) 。但是在高溫區(長晶溫度為 675oC~725 oC)中,我們看到 類似氮化銦薄膜的紅移,而且可以清楚地看到紅移的量約為 14~ 20meV。每一個長晶溫度下的氮化銦奈米點,其光譜峰值能量在 20K 與 300K 的偏移量表列在表 4-3-1。. 42.
(50) 上述在高溫區所看到的紅移現象,對於奈米尺度的氮化銦結構來 說,其實是相當罕見的。到目前為止,多數的論文是在厚度大於350nm 的氮化銦薄膜上,看到隨量測溫度的上升,峰值能量有紅移的現象。 而與我們的氮化銦奈米點高度相近(20~40nm)的薄膜,或者是氮 化銦的奈米結構,我們目前還未看到有論文提及隨量測溫度的上升, 峰值能量有紅移的現象。而有關上述因為晶格常數的擴張與電子-聲 子的交互作用所造成的能隙縮減效應,我們是採用A. A. Klochikhin 25 所作的理論計算結果,認為從4.2K到室溫之間會有55-65meV的能隙 縮減。以A. A. Klochikhin的結果與我們在高溫區(長晶溫度為675oC ~725 oC)內成長的氮化銦奈米點比較,發現在高溫區成長的氮化銦 奈米點,從20K到室溫的光譜峰值紅移量(於為14~20meV),是明 顯小於J.Wu的結果。這反映出在高溫區成長的氮化銦奈米點,除了上 述的能隙縮減效應,造成的55-65meV的紅移量外,還有一個機制使 得這奈米尺度的氮化銦奈米點產生藍移。所以對照A. A. Klochikhin 的結果,假設能隙縮減效應會造成約~60meV的紅移量,由此我們可 以估算在三個不同長晶溫度區間的”淨”藍移量:在包含滴狀金屬銦的 低溫區(長晶溫度為550oC與575 oC)內,約有62~68meV的淨藍移量。 而中溫區(長晶溫度為600oC~650 oC)的淨藍移量則為~61meV;到 了高溫區(長晶溫度為675oC~725 oC),淨藍移量則下降到40~ 46meV。每一個長晶溫度下的氮化銦奈米點的淨藍移量表列在表 4-3-1。 一般在氮化銦鎵量子井中所觀察到的藍移,通常是歸因於侷限 態(localized states)與帶尾能態(band-tail states)的影響26,27,或者 是認為與組成不均勻,所照成的位能擾動(potential fluctuation)有關. 43.
(51) 28. 。而在這一系列改變成長溫度的氮化銦奈米點中,我們認為還有兩. 個可能的原因,是與氮化銦表面的電子聚集效應有關。討論表面電子 聚集效應,對氮化銦奈米點而言是相當重要的。因為經由薄膜的量 測,已經測得氮化銦的表面電子密度(sheet charge density)約為~ 2.5×1013cm-2,而且分佈的厚度約在~6nm以內29。如果將這些表面電 子平均分佈到40nm的厚度(因為我們的氮化銦奈米點高度約在20~ 40nm)中,則可以得到體密度約為~6.25×1018cm-3,這與在4.2節中 利用光譜圖形模型,所擬合得到的電子濃度約為1.7~4.2×1018cm-3是 相近的。所以在我們的氮化銦奈米點中,多數的電子濃度是來自於表 面電子。而關於上述兩個可能造成光譜峰值藍移的原因,其一認為跟 表面電子聚集所造成的電子電洞高度空間分離有關。其二則指向表面 電子密度會隨量測溫度而改變。 有關第一原因,我們是引述C.H.Shen的文章30。他們認為由文章 中在低溫與室溫的能帶圖(如圖4-3-2) ,可以看到表面電子聚集所造 成的能帶彎曲(band bending) ,進而造成電子集中在表面,而電洞則 傾向遠離邊界的結果。因為他們的樣品是n型的氮化銦奈米柱,所以 電洞是少數載子,電子電洞復合主要受電洞分佈影響。故在低溫時, 電洞傾向遠離邊界,所以電子電洞復合發生在遠離邊界的地方,放出 的光子能量較低。而到了室溫,因為電洞的能量升高,電洞的空間分 佈變寬,使得電子電洞的復合,傾向發生於電子聚集的表面,所以放 出能量較高的光子。因此隨著量測溫度的上升,光激螢光光譜的峰值 能量會隨之藍移。 而第二個原因,則是認為與表面電子濃度會隨量測溫度的上升而 大幅增加有關。在C.H. Swartz31的論文中討論到,量測成長在氮化鎵. 44.
(52) 薄膜上,厚度為7.5μm與5.3μm的兩片氮化銦薄膜,發現在升高量測溫 度(從25K升高到300K)時,表面電子濃度都是由約~4×1012 cm-2快 速升高到約~5×1013cm-2,有超過一個數量級的增加。而相對於薄膜 的電子濃度(bulk electron concentration)則分別維持在2~3×1017與3 ~5×1017cm-3。原因可能來自於當升高量測溫度時,許多被限制的電 子,會因為熱效應而激發到傳導帶中,而這些熱激發的電子,會造成 費米能階的位置往高能量移動,進而使變溫光激螢光光譜的峰值能 量,隨量測溫度的上升而藍移。 C.H. Swartz所量測到的表面電子濃度變化,對我們的氮化銦奈米 點可能是有影響的。因為如果將C.H. Swartz的論文中所量測到的表面 電子平均分佈到40nm的厚度(因為我們的氮化銦奈米點高度約在20 ~40nm)中,則可以得到量測溫度從25K升高到300K時,體密度約 從~1×1018 增加到1.2×1019cm-3 ,增加了將近~1.1×1019cm-3 的電子濃 度,而在此所估計低溫下的電子濃度,與4.2節利用光譜圖形模型, 所擬合得到的電子濃度約為1.7~4.2×1018cm-3是相近的。這表示C.H. Swartz所觀察到表面電子濃度隨溫度上升而大幅增加的效應,很可能 影響我們這一系列不同長晶溫度(550oC~725 oC)的氮化銦奈米點, 在20K與300K光譜峰值能量的藍移。 如同之前所估算的,在高溫區(長晶溫度為675oC~725 oC)成長 的 樣 品 , 相 較 於 中 溫 區 與 低 溫 區 , 有 較 小 的 淨 藍 移 量 約 為29 ~ 35meV。我們認為這現象與高溫區的銦脫逸效應有關。如之前4.1節 所述,當長晶溫度大於675oC時,氮化銦奈米點的密度與氮化銦的成 長率,會受銦脫逸效應的影響而減少。在銦脫逸吸附後,可能產生一 些缺陷,很有可能是銦的空缺(In vacancy)32,並且隨著長晶溫度的. 45.
(53) 增加,銦空缺的濃度也會增加。由於銦空缺屬於受體(acceptor)缺 陷,所以在4.2節中利用光譜圖形模型,所擬合得到的電子濃度在高 溫區逐漸下降的現象(675oC時的電子濃度~2.2×1018cm-3,725oC時 的電子濃度~1.7×1018cm-3) ,可能是銦空缺與自由電子的互補效應。 如果熱激發的表面電子,是造成氮化銦奈米點產生淨藍移的成 因,則銦空缺與自由電子的互補效應,將使得自由電子的密度下降, 所以這些熱激發的電子,所造成的費米能階往高能量移動的現象,將 被緩和,因此造成光譜峰值能量淨藍移量的降低。 所以總結以上討論,可以推測在光譜的峰值能量與量測溫度的關 係圖(圖4-3-1)中,這一系列長晶溫度從550oC~725 oC的氮化銦奈 米點,在低溫與室溫所量測到峰值能量的偏移,是由一組紅移與藍移 的機制所影響,紅移的產生可能與晶格常數的擴張與電子-聲子的交 互作用所造成的能隙縮減效應有關,而紅移量約為~49meV。藍移的 原因很多,對於這一系列的氮化銦奈米點而言,可能與氮化銦表面的 電子聚集效應有關。藉由計算淨藍移量,可以發現在長晶溫度為650 oC 以下的樣品,淨藍移量約為50~60meV,但是在高溫區(長晶溫度為 675oC~725 oC)中,淨藍移量卻是較低的29~35meV。這淨藍移量的 減少,可能與高溫區內銦脫逸吸附後,產生的銦空缺與自由電子的互 補效應有關。. 46.
(54) Peak Energy (eV). 0.80. 0.76. 0.72. 0.68. 0. 550 C 0 575 C 0 600 C 0 625 C 0 650 C. 0. 0. 675 C 0 700 C 0 725 C InN bulk. 50 100 150 200 250 300. Temperature (K) 圖 4-3-1 不同長晶溫度的樣品,其光譜強度的峰值能量與量測溫度的 關係。. 47.
(55) Tg(oC). PL peak shift. band-gap shrinkage. Blue shift. (meV). (meV). (meV). 550. 8.4. -60. 68.4. 575. 2.6. -60. 62.6. 600. 0.1. -60. 60.1. 625. 0.9. -60. 60.9. 650. 1.7. -60. 61.7. 675. -19.9. -60. 40.1. 700. -14.0. -60. 46.0. 725. -15.0. -60. 45.0. bulk. -18.0. -60. 42.0. 表 4-3-1 第一欄(PL peak shift)為不同長晶溫度下的氮化銦奈米點, 其光激螢光光譜峰值能量在 20K 與 300K 的偏移量;第二欄 (band-gap shrinkage)為 A. A. Klochikhin 25 由理論計算所得 到到能隙縮減效應的偏移量;第三欄(Blue shift)為我們所 推論的淨藍移量,等於第一欄與第二欄的偏移量的差。. 48.
(56) 圖 4-3-2 引述 C.H.Shen 的文章 30 中,(a)在低溫與(b)室溫下的能 帶圖. 49.
(57) 第五章 結論 本論文主要利用原子力顯微鏡(AFM)與光激螢光光譜(PL) 來探討成長溫度對氮化銦奈米點的表面形貌與光學性質的影響。這些 樣品是由有機金屬化學氣相沈積(MOCVD)所成長,利用流量控制 法(flow-rate modulated epitaxy)在長晶溫度為 550oC~725 oC 之間成 長氮化銦的奈米點。 由氮化銦奈米點密度與成長溫度的關係,可以將長晶溫度分為三 區。長晶溫度在 550 oC~575oC 之間為低溫區,長晶溫度在 600 oC~ 650oC 之間為中溫區,當長晶溫度超過 675 oC 則為高溫區。 在低溫區內,由表面形貌的分析與 X 光光譜的結果,指出有滴 狀金屬銦(In droplet)在樣品表面形成。推測是因為在成長溫度較低 時,從氨中所分解出來活性的氮原子數量較少,所以沒有與氮化合成 氮化銦的多餘銦原子,就形成滴狀金屬銦。中溫區的部分,氮化銦奈 米點一直維持在約~1×109cm-2 左右的高密度。成長率也是比其他兩 個區域來的高,約在~3.6nm/mins 左右。而在高溫區氮化銦奈米點的 密度從長晶溫度為 675 oC 到 725 oC,有將近一個數量級的減少。我們 認為這與銦的脫逸(In desorption)效應有關。 三個區域中所成長的氮化銦奈米點,在光學性質上也有明顯的差 異。在低溫光激螢光光譜的量測中,發現在包含滴狀金屬銦的低溫區 (長晶溫度在 550 oC~575oC 之間)內成長的樣品有較差的光學性 質。光譜峰值能量(peak energy)為較高的~0.8eV,半高寬(FWHM) 也是偏高的~100meV,並且經由譜線圖形模型(line shape modal) 所擬合出的電子濃度也是偏高的 4.0~4.2×1018cm-3。對於滴狀金屬銦. 50.
(58) 的存在造成光學性質的降低,可能與滴狀金屬銦所引發的缺陷有關。 而在沒有滴狀金屬銦的中、高溫區,則可以看到較好的光學性質。光 譜峰值能量為較低的~0.77eV,半高寬下降到~75meV,電子濃度也 降低到 1.7~2.3×1018cm-3。 在變溫光激螢光光譜的量測中,低溫區與中溫區的樣品,其光譜 峰值能量有些許藍移與不隨量測溫度改變的現象,但是到了高溫區 (長晶溫度在 675 oC~725oC 之間)卻可以明顯看到約~17meV 的紅 移,這在奈米尺度的氮化銦結構中是少見的。這紅移的現象被解釋為 受體能階與自由電子的互補效應。. 51.
(59) 附錄 對於取得氮化銦奈米點與滴狀金屬銦體積的方法,敘述如下。我 們利用圖PS-1 a)的示意圖表示。利用原子力顯微鏡作表面形貌的分 析時,會得到高度對x、y的分佈h (x,y),這高度的零點是設在整個掃 瞄範圍的最低點。而氮化銦奈米點與滴狀金屬銦的體積可計算如下:. Vnano−dot + VIn−droplet = Vtotal − Vplane. (1). Vnano-dot代表氮化銦奈米點的體積(圖PS-1 a)中灰色的部分), VIn-droplett代表滴狀金屬銦的體積(圖PS-1 a)中斜線的部分) ,Vplane代 表高度的零點設在最低點時平坦處的體積(圖PS-1 a)中白色的部分。 因為樣品表面必然有高有低,所以在取最低點為原點時,平坦處的平 均高度不為零),而Vtotal則是代表高度的零點設在最低點時,包含氮 化銦奈米點、滴狀金屬銦與平坦處的總體體積。 總體積(Vtotal)可以利用h (x,y)的面積分求得。而一般平坦處體 積(Vplane)的計算,是經由取幾個平坦處的高度作平均(have) ,再將 平坦處的平均高度乘以總面積所得到。然而為了增加平坦處平均高度 所具有的的代表性,採樣作為平坦處高度平均的採樣點個數是重要的 指標。所以為了增加平坦處高度平均的採樣點個數,我們必須使用一 個方法,分辨平坦處與氮化銦奈米點或滴狀金屬銦的不同,在此我們 是利用高度作為分辨的方法。 由第四章表4-1-1可知,氮化銦奈米點的高度約比平坦處高20~ 40nm,而滴狀金屬銦的高度比平坦處高超過100nm,但是在100μm2 的掃瞄範圍內,平坦處的高低落差皆小於10nm,也就是說氮化銦奈 米點與滴狀金屬銦的高度,皆比平坦處的最高點來的高。所以經由 NT-MDT的分析軟體NOVA,設定一個門檻高度h0(如圖PS-1 b) ) , h 52.
(60) (x,y)>h0 的範圍是氮化銦奈米點加平坦處的體積(圖PS-1 b)中灰色 的部分) ,或滴狀金屬銦加平坦處的體積(圖PS-1 b)中斜線的部分) , 而h (x,y)<h0 的區域則是單純的平坦處的體積(圖PS-1 b)中白色的 部分) 。而從圖PS-1 c)可以看到實際對長晶溫度在575 oC的樣品所作 的表面形貌分析,左圖為樣品的表面形貌,右圖中白色區域代表h (x,y). >h0,黑色區域代表h (x,y)<h0。經由比較左右兩圖,可以發現利用 門檻高度h0,可以篩選出絕大多數氮化銦奈米點與滴狀金屬銦的區域 (門檻高度設定的人為誤差,造成的體積差異在10%以下) ,所以將h (x,y)<h0 的區域內每個點的高度作平均,能合理近似為掃瞄範圍內平 坦處平均高度(have) ,進而推算平坦處體積(Vplane) 。 在沒有發現滴狀金屬銦的樣品中,利用(1)可以直接求出氮化 銦奈米點的總體積。然而在有滴狀金屬銦的樣品上,則需先利用形貌 特徵分辨h (x,y)>h0 的範圍中(見圖PS-1 b) ) ,何者為氮化銦奈米點 (明顯的六角晶形且為平頂結構)加平坦處的體積,何者為滴狀金屬 銦(半球型結構,沒有明顯晶形,高寬比約為~0.36)加平坦處的體 積。最後扣掉平坦處的體積就得到氮化銦奈米點與滴狀金屬銦個別的 體積。. 53.
(61) a). Z. In droplet InN nano-dot h(x,y) plane. b) Z. plane X.Y. In droplet InN nano-dot threshold h0 plane. plane X.Y. c). 圖 PS-1 使用 NT-MDT 分析軟體 NOVA,估計氮化銦奈米點與滴狀 金屬銦體積的方法示意圖。a)表面形貌簡圖,b)藉由設定 門檻高度 h0,可以分辨 h (x,y)>h0 的範圍是氮化銦奈米點加 平坦處的體積(灰色的部分),或滴狀金屬銦加平坦處的體 積(斜線的部分),而 h (x,y)<h0 的區域則是單純的平坦處 的體積(白色的部分) 。c)左圖為長晶溫度在 575 oC 樣品的 表面形貌,右圖中白色區域代表 h (x,y)>h0,黑色區域代表 h (x,y)<h0。 54.
(62) 參考文獻 1. B. Arnaudov, T. Paskova, P. P. Paskov, B. Magnusson, E. Valcheva, B. Monemar, H. Lu, W. J. Schaff, H. Amano, and I. Akasaki, Physical Review B 69, - (2004).. 2. S. Gwo, C. L. Wu, C. H. Shen, W. H. Chang, T. M. Hsu, J. S. Wang, and J. T. Hsu, Applied Physics Letters 84, 3765-3767 (2004).. 3. T. Matsuoka, H. Okamoto, M. Nakao, H. Harima, and E. Kurimoto, Applied Physics Letters 81, 1246-1248 (2002).. 4. J. Wu, W. Walukiewicz, K. M. Yu, J. W. Ager, E. E. Haller, H. Lu, W. J. Schaff, Y. Saito, and Y. Nanishi, Applied Physics Letters 80, 3967-3969 (2002).. 5. H. J. Hovel and J. J. Cuomo, Applied Physics Letters 20, 71-& (1972).. 6. T. L. Tansley and C. P. Foley, Journal of Applied Physics 59, 3241-3244 (1986).. 7. S. Nakamura, M. Senoh, and T. Mukai, Japanese Journal of Applied Physics Part 2-Letters 32, L8-L11 (1993).. 8. S. Nakamura, M. Senoh, S. Nagahama, N. Iwasa, T. Yamada, T. Matsushita, H. Kiyoku, and Y. Sugimoto, Japanese Journal of Applied Physics Part 2-Letters 35, L74-L76 (1996).. 9. W. C. Ke, C. P. Fu, C. Y. Chen, L. Lee, C. S. Ku, W. C. Chou, W. H. Chang, M. C. Lee, W. K. Chen, W. J. Lin, and Y. C. Cheng,. 55.
(63) Applied Physics Letters 88, - (2006). 10. O. Moriwaki, T. Someya, K. Tachibana, S. Ishida, and Y. Arakawa, Applied Physics Letters 76, 2361-2363 (2000).. 11. D. L. Huffaker, G. Park, Z. Zou, O. B. Shchekin, and D. G. Deppe, Applied Physics Letters 73, 2564-2566 (1998).. 12. E. Dimakis, G. Konstantinidis, K. Tsagaraki, A. Adikimenakis, E. Iliopoulos, and A. Georgakilas, Superlattices and Microstructures 36, 497-507 (2004).. 13. M. Mesrine, N. Grandjean, and J. Massies, Applied Physics Letters 72, 350-352 (1998).. 14. O. Ambacher, M. S. Brandt, R. Dimitrov, T. Metzger, M. Stutzmann, R. A. Fischer, A. Miehr, A. Bergmaier, and G. Dollinger, Journal of Vacuum Science & Technology B 14, 3532-3542 (1996).. 15. N. Dietz, M. Strassburg, and V. Woods, Journal of Vacuum Science & Technology A 23, 1221-1227 (2005).. 16. R. Intartaglia, B. Maleyre, S. Ruffenach, O. Briot, T. Taliercio, and B. Gil, Applied Physics Letters 86, - (2005).. 17. J. Wu, W. Walukiewicz, W. Shan, K. M. Yu, J. W. Ager, E. E. Haller, H. Lu, and W. J. Schaff, Physical Review B 66, - (2002).. 18. J. Porsche, M. G. A. Ruf, and F. Scholz, Journal of Crystal Growth 195 591-595 (1998).. 19. E. Dimakis, E. Iliopoulos, K. Tsagaraki, T. Kehagias, P. Komninou, and A. Georgakilas, Journal of Applied Physics 97, - (2005).. 56.
(64) 20. A. G. Bhuiyan, A. Hashimoto, and A. Yamamoto, Journal of Applied Physics 94, 2779-2808 (2003).. 21. S. Keller, I. Ben-Yaacov, S. P. DenBaars, and U. K. Mishra, Proceedings. of. the. International. Workshop. on. Nitride. Semiconductors (IWN’ 2000), Nagoya, Japan, September 24–27, 2000, IPAP conference series 1, p.233. 22. C. Kruse, S. Einfeldt, T. Bottcher, D. Hommel, D. Rudloff, and J. Christen, Applied Physics Letters 78, 3827-3829 (2001).. 23. P. Ruterana, M. Morales, F. Gourbilleau, P. Singh, M. Drago, T. Schmidtling, U. W. Pohl, and W. Richter, Physica Status Solidi a-Applications and Materials Science 202, 781-784 (2005).. 24. K. Sugita, H. Takatsuka, A. Hashimoto, and A. Yamamoto, Physica Status Solidi B-Basic Research 240, 421-424 (2003).. 25. A. A. Klochikhin, V. Y. Davydov, V. V. Emtsev, A. V. Sakharov, V. A. Kapitonov, B. A. Andreev, H. Lu, and W. J. Schaff, Physical Review B 71, - (2005).. 26. P. G. Eliseev, P. Perlin, J. Y. Lee, and M. Osinski, Applied Physics Letters 71, 569-571 (1997).. 27. S. W. Feng, Y. C. Cheng, Y. Y. Chung, C. C. Yang, Y. S. Lin, C. Hsu, K. J. Ma, and J. I. Chyi, Journal of Applied Physics 92, 4441-4448 (2002).. 28. Y. T. Moon, D. J. Kim, J. S. Park, J. T. Oh, J. M. Lee, Y. W. Ok, H. Kim, and S. J. Park, Applied Physics Letters 79, 599-601 (2001).. 29. H. Lu, W. J. Schaff, L. F. Eastman, and C. E. Stutz, Applied. 57.
(65) Physics Letters 82, 1736-1738 (2003). 30. C. H. Shen, H. Y. Chen, H. W. Lin, S. Gwo, A. A. Klochikhin, and V. Y. Davydov, Applied Physics Letters 88, - (2006).. 31. C. H. Swartz, R. P. Tomkins, T. H. Myers, H. Lu, and W. J. Schaff, physica status solidi (c)2,2250-2253 (2005).. 32. A. Pelli, K. Saarinen, F. Tuomisto, S. Ruffenach, and O. Briot, Applied Physics Letters 89, - (2006).. 58.
(66)
數據




+7




Outline
相關文件
吳佳勳 助研究員 國立台灣大學農經博士 葉長城 助研究員 國立政治大學政治學系博士 吳玉瑩 助研究員 國立台灣大學經濟博士 陳逸潔 分析師 國立台灣大學農經系博士生 林長慶
臺中榮民總醫院埔里分院復健科 組長(83年~今) 中山醫學大學復健醫學系職能治療 學士.. 南開科技大學福祉科技與服務管理研究所
論文寫作指導 釋覺興 佛光山國際學部英文佛學院 佛教史、英文、國文 釋覺培 南華管理學院比較宗教研究所 佛教經典研究、佛門禮儀 釋覺機
(1)針對具有中子研究專長者,具備下列要件之 一:①物理、化學、核工系所博士畢業,具 二年以上中子研究經驗;執行中子散射、繞
本案件為乳癌標準化化學藥物治療與個人化化學治 療處方手術前化學治療療效比較之國內多中心研 究,於 2008 年 8 月 1 日由
在1980年代,非晶矽是唯一商業化的薄膜型太 陽能電池材料。非晶矽的優點在於對於可見光
威夷大學社會心理學博士。曾任 國家科學委員會特約研究員。榮 獲國家科學委員會優良研究獎、美國東
港大學中文系哲學碩士、博士,現 任香港中文大學人間佛教研究中心
