國立臺灣大學工學院環境工程學研究所 碩士論文
Graduate Institute of Environmental Engineering College of Engineering
National Taiwan University Master Thesis
以石墨烯修飾二氧化鈦粒子及奈米管進行光催化 甲醇溶液產氫之研究
Photocatalytic Hydrogen Production in Methanol Solution using Graphene Composited TiO
2particles
and Nanotubes under Light Irradiation 陸觀一
Kwun-Yat Luk
指導教授:駱尚廉 教授 Advisor: Shang-Lien Lo, Ph. D.
中華民國104年7月
July, 2015
I
II
中文摘要
自 1970 年代石油危機,利用光能生產氫氣作為替代能源的技術逐漸受關注。
起因於 1972 本多藤嶋效應的發現,以半導體受光催化來分解水的研究,成為綠 色能源的新課題。隨後,水分解的異相光催化系統(Heterogeneous photocatalytic system)有賴光催化材料的改善,和犧牲劑的投入得以逐步發展。
在眾多具備光催化活性的半導體中,以二氧化鈦 (TiO2)的歷來研究最為豐 富及深入,加上便宜、無毒、以及化學、光學穩定性好,二氧化鈦是最有潛力投 入工業尺度應用的材料。
近年來,石墨烯受到奈米研究領域的關注。利用氧化減薄法,可以既簡便而 又大量地製備氧化石墨烯 (GO)。理論上,還原氧化石墨烯 (rGO)導電性良好,
加上可以進行奈米尺度的修飾,適合用作改善二氧化鈦的光催化活性。考慮石墨 烯披覆於二氧化鈦的技術各式各樣,本研究分別以水熱還原法 (HrGO)、光還原 法 (PrGO)及熱還原法 (TrGO)製備TiO2/rGO,並發現TiO2/HrGO在紫外光下 (365nm)催化產氫表現最好,1%披覆比例的產氫量比純TiO2提升 2.7 倍。
另外,本研究對二氧化鈦進行同時改質、披覆上石墨烯及進行還原,製備出 二氧化鈦奈米管/還原氧化石墨烯 (TiNT/rGO)。在改質過程中,由於晶型改變使 TiNT 吸收峰藍移,在 0%及 1%披覆比例下產氫量較TiO2差。但由於比表面積大,
當披覆比例提升至 2%,TiNT/rGO 的產氫量比純TiO2提升了 13 倍。
關鍵字:二氧化鈦、石墨烯、奈米管、異相光催化系統、氫氣
III
Abstract
1970s was known to be the time of oil crisis. Since then, technologies of utilizing light to produce hydrogen energy had drawn much attention. Hydrogen produced from water splitting was considered to be one of the potential candidates of alternative energy owing to the discovery of Honda-Fujishima effect. By applying sacrificial reagents and improving photocatalysts, heterogeneous photocatalytic system of water splitting became possible few years later.
Among various semiconductors, TiO2 is probably the most suitable material for industrial scale photocatalysts in future. TiO2 is cheap, nontoxic and stable. Its properties had also been well studied.
Recently, nanomaterial researchers had been concerned about graphene. Oxidative exfoliation allows graphene oxide (GO) to be synthesized in gram-scale through facile method. Theoretically, reduced GO (rGO) possess good conductivity. Also GO is capable to be manipulated under nanometer scale, which is suitable for composition on TiO2 powder to further enhance photocatalytic activity. Considering various existing reduction methods, here in we synthesized TiO2/rGO by hydrothermal reduction (HrGO), photo-assisted reduction (PrGO) and thermal reduction (TrGO).
Under UV irradiation, TiO2/HrGO was discovered to perform best among others, with 2.7 fold hydrogen production for 1%w/w compare with mere TiO2.
In addition, synthesizing TiO2 nanotube (TiNT) with simultaneous composition and reduction of GO had been achieved to produce TiNT/rGO. Crystallinity changed along with nanotube fabrication and subsequent blue shift of absorption edge was observed. As a result, TiNT of 0% and 1%w/w rGO composition possess worse
IV
photocatalytic activities than that of TiO2. For the fact that specific surface area of TiNT is much higher than TiO2, hydrogen production rate of 2% composition of rGO on TiNT increased dramatically by 13 fold compare to mere TiO2.
Key words: Graphene, Nanotube, Heterogeneous photocatalytic system, Hydrogen, Titanium dioxide
V
目錄
中文摘要... II Abstract ... III 目錄... V 圖目錄... VII 表目錄... X
第一章 緒論... 1
1.1 前言 ... 1
1.2 研究目的 ... 2
1.3 研究內容 ... 2
第二章 研究背景... 3
2.1 光催化產氫 ... 3
2.1.1 光伏電解法 ... 3
2.1.2 太陽能集熱法 ... 4
2.1.3 光生物法 ... 4
2.1.4 光電化學法 ... 5
2.2 光催化產氫原理 ... 6
2.2.1 水分解的化學意義 ... 6
2.2.2 光觸媒催化 ... 6
2.2.3 還原電位 ... 10
2.2.4 電荷分離 ... 12
2.3 光催化材料 ... 17
2.3.1 二氧化鈦 ... 18
2.3.2 石墨烯 ... 19
2.3.3 二氧化鈦奈米管 ... 22
第三章 實驗材料與方法... 29
VI
3.1 實驗藥品與設備 ... 29
3.1.1 實驗藥品 ... 29
3.1.2 實驗設備 ... 31
3.2 實驗方法 ... 32
3.2.1 實驗設計 ... 32
3.2.2 製備氧化石墨烯 ... 33
3.2.3 製備 TiO2/rGO... 35
3.2.4 製備 TiNT/rGO ... 37
3.2.5 光催化材料特性分析 ... 38
3.2.6 光催化產氫實驗 ... 40
第四章 結果與討論... 42
4.1 特性鑑定 ... 42
4.1.1 掃描式電子顯微鏡/X 光能量分散光譜儀 ... 42
4.1.2 穿透式電子顯微鏡 ... 44
4.1.3 傅立葉轉換紅外光譜儀 ... 49
4.1.4 X 光粉末繞射圖譜 ... 51
4.1.5 紫外-可見光吸收度圖譜 ... 55
4.2 光催化產氫 ... 59
4.2.1 空白實驗 ... 59
4.2.2 TiO2/rGO... 60
4.2.3 TiNT/rGO ... 62
4.3 rGO 與光催化效率之關係 ... 66
第五章 結論及建議... 68
5.1 結論 ... 68
5.2 建議 ... 69
參考文獻... 70
附錄... 82
VII
圖目錄
圖 2-1、光電化學電池……… 5
圖 2-2、鋰 (Li)的原子軌域組合出能帶……… 7
圖 2-3、能帶結構示意圖……… 7
圖 2-4、Downhill 與 uphill 隨反應進程的自由能變化……… 8
圖 2-5、半導體光觸媒產氫之主要步驟……… 9
圖 2-6、三種不同型態的能帶結構………... 11
圖 2-7(a)、光電化學電池;(b)、異相催化系統………... 12
圖 2-8、接上導體 (右)時半導體 (左)之激發電子流動方向………. 14
圖 2-9、電子電洞各種反應時間……… 15
圖 2-10、犧牲劑的功能……… 16
圖 2-11、半導體觸媒能帶結構與水分解電位的關係……… 17
圖 2-12、二氧化鈦各種晶型之結構……… 18
圖 2-13、單層石墨烯具蜂巢狀結構……… 19
圖 2-14(a)、石墨具有層狀結構;(b)、氧化石墨………... 21
圖 2-15、不同溫度及 NaOH 比例生成的產物形態……… 25
圖 2-16、銳鈦礦、金紅石和 P25 以不同條件進行水熱法的產物分布圖……… 25
圖 2-17、準穩定態的奈米管及較穩定的奈米線……… 26
圖 3-1、紫外燈管波長分布………. 31
圖 3-2、本研究實驗設計……… 32
圖 3-3、以分液漏斗靜置 12-24 小時,清洗出氧化石墨烯中的硫酸………… 33
圖 3-4、乾燥後的氧化石墨;呈棕黑色……… 34
VIII
圖 3-5、使用於水熱還原法及奈米管製備之反應氣……… 35
圖 4-1(a)、掃描式電子顯微鏡下的氧化石墨;(b) 、EDS 掃描(a)範圍內得 出之元素構成……… 43
圖 4-2、穿透式電子顯微鏡下的氧化石墨烯……… 44
圖 4-3、穿透式電子顯微鏡下的(a)(b) TiO2/HrGO;(c)(d) TiO2/PrGO; (e) TiO2/TrGO ……… 46
圖 4-4、透式電子顯微鏡下的 TiNT/rGO,放大(a)10 萬倍;(b)25 萬倍;(c)100 萬倍;(d)1000 萬倍……… 48
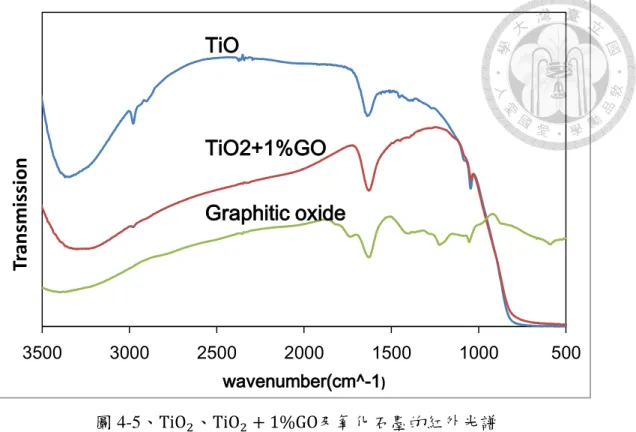
圖 4-5、TiO2、TiO2 + 1%GO及氧化石墨的紅外光譜……… 50
圖 4-6、氧化石墨及水熱還原氧化石墨烯的紅外光譜……… 50
圖 4-7、銳鈦礦標準 X 光繞射圖譜……… 51
圖 4-8、金紅石標準 X 光繞射圖譜……… 51
圖 4-9、石墨及氧化石墨的 X 光繞射圖譜………... 52
圖 4-10、退火對二氧化鈦奈米管晶型的影響……… 54
圖 4-11、各還原法製備TiO2/rGO之 UV-Vis 吸收圖譜……… 56
圖 4-12、水熱還原法下不同 GO:TiO2(w/w) 製備TiO2/rGO之 UV-Vis 吸收 圖譜……… 56
圖 4-13、二氧化鈦粒子和奈米管及各披覆 GO 之 UV-Vis 吸收光譜圖……… 58
圖 4-14、曝氮與不曝氮的空白光照產氫量……….. 59
圖 4-15、不同還原方法製備材料之光催化產氫量……… 61
圖 4-16、TiO2/HrGO以不同 GO:TiO2(w/w)……… 61
圖 4-17、比較二氧化鈦及奈米管之光催化活性……… 63
圖 4-18、TiNT/rGO以不同 GO:TiO2(w/w)製備之材料每小時每克產氫量….... 63
圖 4-19、具代表性材料的累積產氫量對時間作圖……….. 64
圖 4-20、TEM 下 TiNT/rGO 放大 100 萬倍之奈米管和石墨烯邊界……… 65
IX
圖 4-21、TiNT 在光照實驗後 (左)及實驗前 (右)之反應溶液……….. 65 圖 4-22(a)、單層石墨烯附載TiO2;(b)、石墨化構成遮蔽效應及抑制電子流
通……… 66
X
表目錄
表 2-1、製備二氧化鈦奈米管的方法及其優缺點……… 22
1
第一章 緒論
1.1 前言
氫氣是具潛力的理想能源,其能量密度高 (140MJ/kg),無碳排放,而且水 是其燃燒的唯一產物,對環境造成危害之虞甚微。妥適的氫氣生產技術,將在解 決能源問題上具有重大意義。
目前生產氫氣主要來自化石燃料(如天然氣)的蒸氣重組 (Steam-reforming) [1] ,此方法受制於低效率和高成本問題,且不能避免二氧化碳排放,以及消耗 蘊藏量有限的化石燃料,實非長遠計。是以光觸媒 (Photocatalyst)研究自 1980 年代、石油危機後受到重視,研究多試圖讓陽光和水作為氫氣來源,企圖讓產氫 課題成為解決能源困境的大方向[2]。
光觸媒現時所面對的困難,在於缺乏合適吸收波段和穩定的材料[3]。本研 究則嘗試將二氧化鈦披覆於奈米碳材—石墨烯 (Graphene)上,進行光催化產氫測 試。
2
1.2 研究目的
二氧化鈦 (Titanium dioxide)具有良好的化學穩定性,加上價格低廉、對人體 無害等因素,在光催化領域已經有不少商業上的應用,而在光催化產氫中更是最 具投入工業尺度應用潛力的材料。然而由於二氧化鈦的光催化活性位在紫外光 (Ultraviolet radiation,或稱 UV 光)波段範圍,而紫外光僅佔地表太陽輻射能的百 分之三,如此則引伸出重要課題,就是讓二氧化鈦能夠最大程度利用太陽光,包 括提高量子產率 (Quantum yield)以及開發可見光 (Visble light)波段的吸收[2]。
本研究嘗試以石墨烯披覆二氧化鈦、以及二氧化鈦奈米管上,尋找改善二氧 化鈦產氫效率的途徑,並以簡便及產量大的製備方法為主體。經由不同光催化材 料之製備方法,分析披覆過程對二氧化鈦的光催化活性及特性有何影響。
1.3 研究內容
1. 參考現有文獻,製備披覆石墨烯之二氧化鈦 (下稱TiO2/graphene);
2. 各種製備方法於產氫之光催化活性及其特性分析之比較;
3. 探討石墨烯披覆於二氧化鈦奈米管 (TiO2 nanotube)的可行性;
4. 找出TiO2/graphene、TiO2 nanotube/graphene之最佳披覆比例;
5. 探討造成各材料光催化活性差異的原因。
3
第二章 研究背景
2.1 光催化產氫
光催化產氫始於 1972 年本多 (Honda)與藤嶋 (Fujishima)的研究[4],當時使 用二氧化鈦 (Titanium oxide, TiO2)配合鉑黑 (Platinum black)電極成功分解水,
由此二氧化鈦表面受光後與水進行反應生出氫氣和氧氣,此一特性被稱為本多藤 嶋效應 (Honda-Fujishima effect)。
本多藤嶋效應屬於水分解 (Water splitting)的其中一支。水分解為一廣義議題,
在永續能源的觀點而言,水分解概括了各種利用可再生能源獲得氫氣的方法,但 即使方法不同,其最終要促成的反應仍然是相同,其化學反應式如式 1-1 所示:
H2O(l) ⟶ 1/2𝑂2(𝑔) + 𝐻2(𝑔) (1-1)
時至今日,再生能源已是形形式式,族繁不及備載。本文且對使用光能分解 水生成氫氣略作敘述,其主要的方法有以下四種[5, 6],分別為:(1)光伏電解法 (Photovoltaic electrolysis);(2)太陽能集熱法 (Solar-thermal water splitting);(3)光 生物法 (Photobiological water splitting);(4)光電化學法 (Photochemical water splitting)。
2.1.1 光伏電解法
光伏電解法利用光線照射太陽能裝置產生電壓,經外接電路對水進行電解。
現行方法包括鹼性電解 (Alkaline electrolysis)以及質子交換薄膜電解 (Proton exchange membrane electrolysis,又稱 PEM 電解)。鹼性電解行之有年,技術較成 熟;PEM 電解則系統電阻低,容許高電流密度通過,耗能較少、效率較佳,然
4
而酸性溶液會腐蝕器材和觸媒,材料選擇上價格較為高昂。[7, 8]
2.1.2 太陽能集熱法
太陽能集熱法則是把大面積陽光聚焦加熱,為各種水分解系統注入能量。這 些系統會被加溫至700 − 1000℃,根據不同機制茲列舉數種技術:
一、熱化學系統 (Thermochemical cycles),系統中的觸媒如氧化鐵會促成氫 氣生成;
二、蒸氣重組,與一般蒸氣重組類似,但以太陽能加熱代替傳統加熱方法;
三、高溫電解 (High-temperature electrolysis),以高溫輔助電解,提高電能與 氫氣的轉換效率。
這些設計之所以存在,理由在於只考慮把純水加熱至自發分解成氫氣和氧氣,
溫度需要達到2000℃以上,耗能相對龐大[9]。
2.1.3 光生物法
光生物法包括直接光解 (Direct photolysis)、非直接光解 (Indirect photolysis) 以及光發酵 (Photo-fermentation)。直接光解利用含氫化酶 (Hydrogenease)的藻類,
如綠藻 (Green algea)或藍綠菌 (Cyanobacteria),在缺氧環境下通過 PSII
(Photosystem II)受光反應,把水分解成氫氣;非直接光解則利用光合作用生成醣 類後,在暗發酵過程產出氫氣;光發酵嘗試利用光合菌的固氮酶產出氫氣,但離 實際應用仍然有段距離[10]。
5 2.1.4 光電化學法
光電化學法的研究由發現本多藤嶋效應開始,其時主流方法是把單晶觸媒作 為電極,經外接電路連接另一端電極 (e.g.鉑電極)。當系統浸泡於水中,並讓光 觸媒接受入射光,電子受激發後會經外電路傳導至電極,把水還原成氫氣。這類 系統稱為光電化學電池 (Photoelectrochemical cell),一般多以雙槽運行,中間有 選擇性容許離子通過的薄膜分隔,使氫氣和氧氣不至重新混合為水(圖 2-1) [2, 4]。
圖 2-1、光電化學電池
光電化學法中又分出光觸媒反應法 (Photocatalytic reaction),又稱懸浮系統 (Suspension system)或異相光催化系統 (Heterogeneous photocatalytic system)。此 反應系統將粉體觸媒懸浮於水中,光源從水中或水面[11]照射,大幅增加觸媒受 光面積。然而,在缺乏外接電路分開陰陽極的情況下,由於粉體觸媒本身兼有陰 陽極的功能,氧化還原反應中心非常接近,容易發生電子電洞再結合與水分解的 逆反應,故於早期研究中效果不彰[2, 12]。直到 1979 至 1981 年間,川合 (Kawai) 與坂田 (Sakata)先後利用碳[13]、碳水化合物[14]以至甲醇和乙醇[15, 16]將系統 氧化劑消耗,使氫氣產量大幅提高,光催化產氫技術方邁入另一階段。
6
2.2 光催化產氫原理
2.2.1 水分解的化學意義
化學反應有分為自發 (Spontaneous)與非自發 (Non-spontaneous),其所指稱 的是特定溫度與壓力下,反應發生後系統的自由能 (Gibbs free energy,常以 G 表示)是增加抑或減少?如果是減少(i.e. ∆G < 0),則反應為自發;如果是增加(i.e.
∆G > 0),則反應屬於非自發。
水分解成氫氣和氧氣屬於非自發反應,下面讓我們重溫公式(1-1):
H2O(l) ⟶ 1/2𝑂2(𝑔) + 𝐻2(𝑔); ∆𝐺𝑂 = +237.2𝑘𝐽/𝑚𝑜𝑙 (1-1)
在25℃、1atm 下,每莫耳 (mole)的水須要注入237.2𝑘𝐽 自由能方能分解出 氫氣和氧氣。換言之,水分解的發生有賴於從外部對系統做功。
前述的各種水分解方法,都在想方設法把能量投入水中使之進行水分解反應。
而光電化學法,就是通過吸收光能讓觸媒具氧化還原能力,克服水分解反應的自 由能變化。
2.2.2 光觸媒催化
光觸媒之所以能對分解水的反應提供能量,在於光觸媒的主要成分──半導 體 (Semiconductor),能夠藉由吸收光能,激發電子 (e−)從價帶 (Valence band) 躍遷到傳導帶 (Conduction band),並於價帶上留下電洞 (Electron hole, h+)。
能帶 (Energy band)的存在是基於原子或分子軌域 (Atomic or molecular orbitals)重複排列而成,由圖 2-2,鋰原子的電子組態 (Electron configuration)是我 們所熟知的1s22𝑠1,當兩個以上的鋰原子軌域重疊 (Overlap),交互作用會使原
7
子軌域組合成相同數量的分子軌域,包括鍵結 (Bonding)與非鍵結 (Antibonding);
而當系統中原子數量繼續增加,分子軌域也會越來越多,致使能階密度 (Density of states)上升。能階之間幾乎密接在一起,形成能帶結構,隨之衍生出材料導電 性質的差異。
圖 2-2、鋰 (Li)的原子軌域組合出能帶;N 為原子數
圖 2-3 簡單列舉了價電子附近的幾種能帶結構。空白區域為傳導帶,由未填 電子的原子軌域結合而成;斜線則為價帶,由填入電子的原子軌域結合而成。
圖 2-3、能帶結構示意圖;包括絕緣體(a),半導體(b)和導體(c)(d)
8
材料的導電性,是基於電子從價帶躍遷到傳導帶,因此我們可以看到像圖 2-3(a)這種情況,價帶和傳導帶中間稱為能隙 (Band gap)的寬度,在常溫下不足 以讓電子躍遷到傳導帶的,則屬於絕緣體 (Insulator)。而圖 2-3 (c)的情況中,能 帶由未填滿的原子軌域組合而成,使價帶到傳導帶在同一條能帶上,則材料具有 導電性,屬於導體。另外導體中一些金屬如鎂,雖然價帶屬於填滿電子的 2s 軌 域,但因為由 2p 空軌域組合而成的傳導帶與價帶重疊 (圖 2-3 (d)),則依然具有 導電性。
半導體一般是指能隙寬度在 1-4eV (i.e. 159-290kJ/mol)之間的材料 (圖 2-3 (b)),在常溫下不導電,但在外加偏壓、升溫或光照等等特定的情況下則具有特 殊的導電性。由於半導體特殊的導電性,在工業、研發以致一般起居中已被廣泛 應用,而本研究則集中敘述光活性 (Photoactivity)的部分。[17, 18]
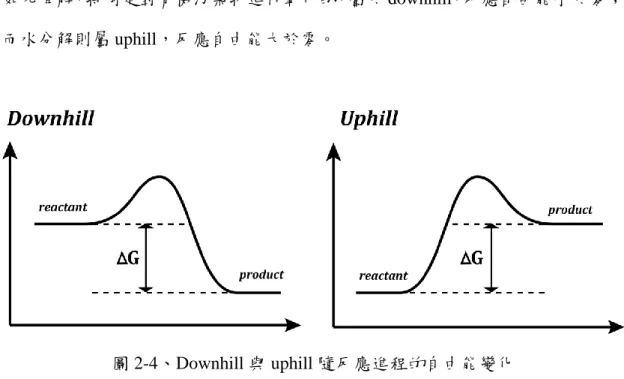
半導體之所以具有光活性,在於能隙的存在與其寬度,可以使電子躍遷,形 成帶有能量的電子電洞對 (Electron – hole pair),與其他反應物進行反應。這些反 應包含 uphill 與 downhill[19],分別代表反應前後自由能增加與減少,一般應用 如光降解,特別是對有機污染物進行氧化的,屬於 downhill,反應自由能小於零;
而水分解則屬 uphill,反應自由能大於零。
圖 2-4、Downhill 與 uphill 隨反應進程的自由能變化
9
Downhill 和 uphill 之間最大的差別,在於 uphill 反應的發生須要確實吸收足 夠光能,而 downhill 則是為了克服活化能 (Activation energy)[20]。換言之,能隙 的寬度須要超過237.2𝑘𝐽/𝑚𝑜𝑙,受激發電子電洞對才具有足夠能量催化水分解。
概括而言,經由半導體光觸媒催化產生氫氣,過程包含三個主要步驟[21]:
(1)能量比半導體能隙更大的光子,被半導體吸收,產生電子電洞對;(2)電荷分 離,即載流子(charge carrier)如電子、電洞的遷移;(3)載流子於觸媒表面發生化 學反應。
圖 2-5、半導體光觸媒產氫之主要步驟
一般情況下,入射光都會以波長表示,半導體能隙所對應的臨界波長,則可 由普朗克關係式(Planck’s relation)計算:
E = h𝑐
𝜆 (2-1) 其中,h 為普朗克常數(Planck constant)=6.626 × 10−34J ∙ s。
c 為真空光速(speed of light)=2.998 × 108m ∙ s−1。 𝜆 為波長(wavelength)/m。
所謂臨界波長,指的是能隙所對應光子的波長。對缺乏雜質的半導體而言,
10
能隙中並沒有可以接收價電子的能階,因此如果入射光子的能量小於能隙,電子 就無法受激發。
以二氧化鈦 (TiO2)為例,價帶和傳導帶的能量差為 3.2eV,此即其能隙。由 上式可算出對應的波長為 387.5nm。由於光子的波長越短,能量越大,因此可以 得知,激發二氧化鈦的價帶電子躍遷至傳導帶,入射光的波長要短於 387.5nm。
2.2.3 還原電位
要有效分解水產生氫氣和氧氣,入射光子必須有足夠能量克服反應自由能,
然而這並非充分條件。如受光激發產生的電子電洞對缺乏足夠氧化和還原能力,
則反應依然不會發生。
廣義的氧化還原,是指物質之間的電子轉移。氧化代表電子被移走,還原則 是得到電子。電子在物質之間的轉移趨勢,可以用標準電位 (Standard potential) 表示;氧化和還原半反應的標準電位相加,則是化學反應的電極電位 (Cell potential),其和反應自由能的關係如式 2-2 所示:
∆G𝑜 = −𝑛𝐹𝐸𝑜 (2-2) 其中,n 為參與反應的電子數(mole)。
F 為法拉第常數(Faraday) = 96485 C ∙ mol−1。 E 為反應的電極電位。
利用上式計算某一氧化還原反應的反應自由能,則可知反應在熱力學的觀點 上是否自發。以水分解為例,在 pH=0 時氧氣還原成水的還原電位是+1.23V,氫 離子的還原電位為 0。
2H++ 2𝑒− ⟶ H2; E0 = 0V (2-3)
−) 1
2O2 + 2H++ 2𝑒− ⟶ H2O; E0 = +1.23V (2-4) H2O ⟶1
2O2+ H2; E0 = −1.23V (1-1)
11
上式列舉了水分解的兩個半反應,將此兩反應式相減,可得反應式(1-1),而 其電極電位則為-1.23V,代入式(2-2)便可得水分解的反應自由能:
∆G𝑜 = −(2𝑚𝑜𝑙)(96485C ∙ 𝑚𝑜𝑙−1)(−1.23𝑉) = +237.2𝑘𝐽/𝑚𝑜𝑙
現在讓我們假設光觸媒的傳導帶 (CB)為一反應物,其氧化還原半反應可以 表示為:
CB + 𝑒− ⟶ CB−; E0 = E𝐶𝐵(2-5) 用同樣方式與氫離子還原半反應相加,可以得到下式:
CB− + 2H+ ⟶ H2 + CB E0 = −2E𝐶𝐵(2-6) 從上式可以看到,電子在觸媒傳導帶的還原電位E𝐶𝐵必須要小於零
(i.e.E𝐶𝐵 < 0),才能還原氫離子並生成氫氣 (i.e.∆G𝑜 = −𝑛𝐹𝐸𝑜< 0)。據此則可以 畫出水的氧化還原電位,與觸媒價帶、傳導帶電位的相對位置,並藉以判斷觸媒 是否具催化水分解的功能:
圖 2-6、三種不同型態的能帶結構
上圖可以看到三種類型的觸媒能帶結構,(a)的能隙非常寬,激發電子電洞 對具有足夠能量克服水的氧化還原,雖然吸收的臨界波長較短,往往須要利用紫
12
外光,但適度的修飾仍然能作為光觸媒產氫,常見例子有TiO2;(b)的能隙則較 小,又具有超過237.2𝑘𝐽/𝑚𝑜𝑙的寬度,但因為E𝐶𝐵大於零,在傳導帶的電子不能 還原水分子,因此無法單獨催化產氫,常見例子有WO3;(c)的能帶結構則先天 屬於較理想的觸媒,既可以還原氫離子,吸收波長也能有效利用可見光,常見例 子有CdS。
2.2.4 電荷分離
圖 2-7(a)、光電化學電池
圖 2-7(b)、異相催化系統
13
電荷分離在異相光催化系統 (Heterogeneous photocatalytic system)中扮演了 重要角色。
自 1972 年本多藤嶋效應的發現,到 80 年代初為止,光催化產氫的主流研究 都要以觸媒作為電極,並經外電路把激發電子分離到另一槽中進行還原反應 (圖 2-6(a)),粉體二氧化鈦於水中往往沒有顯著產氫量[2]。
對異相系統 (圖 2-6(b))的研究可以追溯至 1977 年 Schrauzer 與 Guth 的嘗試 [12],他們分別以純二氧化鈦,以及摻雜氧化鐵的二氧化鈦進行實驗,結果卻不 盡如人意[13, 22]。早期的後續研究包括 Sato 與 White[22]、Duonghong[23]、
Borgarello[24]、Magliozzo[25]等,利用導體或共觸媒 (Cocatalyst)的披覆分離電 荷,而川合和坂田則提出使用犧牲劑[13, 14, 15, 16],下文茲就共觸媒披覆及犧 牲劑兩種電荷分離技術,對其原理略作敘述。
2.2.4.1 共觸媒披覆
光催化的原理是利用入射光子,激發價帶的電子到傳導帶,形成電子電洞對。
電子電洞有可能經歷電荷分離,與目標物進行氧化還原反應,完成催化反應;也 有可能在觸媒中互相結合,其能量以光或熱的形式逸散,是為再結合
(Recombination)。
電荷分離和再結合是影響光催化效率的競爭機制。在異相光催化中,再結合 的快慢在很多研究裡被用作詮釋觸媒效果的好壞,例如把催化效果好的觸媒歸因 於其設計有效抑制再結合。然而甚麼條件會影響再結合速率、以致如何影響,仍 然是需要深入探討的課題[26]。
14
圖 2-8、接上導體 (右)時半導體 (左)之激發電子流動方向
於粉體觸媒上披覆其他材料,是常用作協助電荷分離的技術。以本研究所用 的二氧化鈦為例,在其表面披覆上貴金屬是常用於改善光催化效率的方法,Bard 認為此類系統可視為一「短路」的光電化學電池[20]。此乃由於鉑等貴金屬的費 米能級 (Fermi level, E𝑓)較半導體低,當材料互相接觸時電子便會從半導體流向 金屬,達到電荷分離的效果。
不少研究都間接證明,二氧化鈦半導體的激發電子確實會傾向移動到表面披 覆的貴金屬[27, 28, 29]。但此結果卻與常用於金屬-半導體界面的蕭特基勢壘模 型預測不符[30],使得抑制再結合的詮釋缺乏通用模型。
圖 2-9[30]是 Schneider 等人整理二氧化鈦受光激發後,電子電洞的形成、遷 移與再結合的反應時間,可使我們對光催化有更深入了解:
15
圖 2-9、電子電洞各種反應時間[30]
2.2.4.2 犧牲劑
1979 年,川合和坂田提出犧牲劑的研究,其研究刊載於自然雜誌[13],文中 指出粉體觸媒異相系統之所以無法有效對純水進行分解,在於氫氣與氧氣的逆反 應發生。對此,該研究在觸媒粉體中加入碳粉,使之與水進行下列反應:
2H2O + C ⟶ 2H2+ CO2; ∆G𝑜= +63𝑘𝐽/𝑚𝑜𝑙(2-7) H2O + C ⟶ H2+ CO; ∆G𝑜= +92𝑘𝐽/𝑚𝑜𝑙(2-8) 此系列反應藉由犧牲碳,除了大幅降低自由能,也消耗了觸媒電洞和其生成 的氧氣。自此,在異相光催化產氫系統中加入犧牲劑漸成主流。圖 2-10 簡單敘 述了犧牲劑消除氧化中心的機制:
16
圖 2-10、犧牲劑的功能
實際上,犧牲劑主要功用在於去除觸媒上的電洞。犧牲劑本身為電子提供者 (Electron donor),可以通過自身的還原提供電子,消除觸媒的電洞,減少電子電 洞再結合的機率[14, 15, 16]。
另外,犧牲劑如 EDTA 和Na2S也具備抑制氧氣生成的功能[31]。電洞促成氫 氧自由基生成,是光降解中重要的技術,氫氧自由基又會結合成氧氣[32, 33]:
H2O + ℎ𝑣𝑏+ ⟶ OH ∙ +H+; (2-9) OH ∙ +OH ∙⟶ H2O +1
2O2; (2-10) 雖然氧氣與氫氣經逆反應生成水需時不短[23],但在披覆的共觸媒表面,如 鉑等催化劑,如果跟氫氣和氧氣接觸則很有可能加速反應發生[34],在情非得已 要使用能催化逆反應的共觸媒時,適當的犧牲劑也可權當完善系統的措施之一。
另外,甲醇為常用的犧牲劑,其產氫機制如下式所示[16]:
CH3OHℎ𝜈,cat.→ HCHO + H2 (2-11) HCHO + H2Oℎ𝜈,cat.→ HCOOH + H2 (2-12) HCOOHℎ𝜈,cat.→ CO2+ H2 (2-13)
17
2.3 光催化材料
上文總結了一些完成光催化產氫的必要條件,圖 2-11 提供各種半導體對應 的能隙及能帶電位,卻並非所有能還原水的半導體就等於是好的光催化材料。如 CdS、CdSe、ZnO 等半導體受光後會被自身的電洞氧化腐蝕[19],鎘離子的釋出 更是劇毒。因此選擇半導體的種類時,還是應該盡可能根據研究的方向作判斷。
本研究所採用的則是二氧化鈦,其優點包括便宜、無毒、以及化學、光學穩 定[35],加上歷來研究豐富,在設計上相對具有彈性。
圖 2-11、半導體觸媒能帶結構與水分解電位的關係[19, 36]
18 2.3.1 二氧化鈦
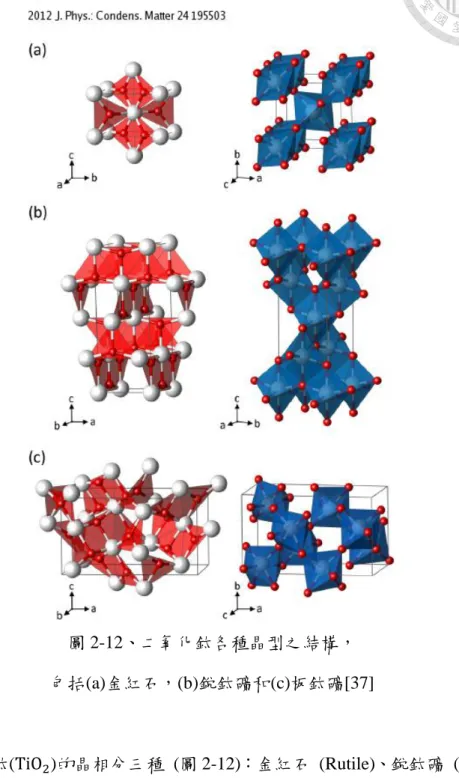
圖 2-12、二氧化鈦各種晶型之結構,
包括(a)金紅石,(b)銳鈦礦和(c)板鈦礦[37]
二氧化鈦(TiO2)的晶相分三種 (圖 2-12):金紅石 (Rutile)、銳鈦礦 (Anatase) 以及板鈦礦 (Brookite)。其中以金紅石結構最為穩定,其次為銳鈦礦,板鈦礦則 遠較前兩者來得不穩定,在高溫環境下會發生不可逆反應,轉變成金紅石結構。
在半導體應用上,對二氧化鈦的討論大都圍繞金紅石與板鈦礦兩種晶相。
19
以半導體特性而言,銳鈦礦的能隙為 3.2 eV[38, 39, 40],金紅石則為 3.0 eV[41, 42, 43]。很多研究──包括理論模型及光學實驗都指出,銳鈦礦的光催化能力比 金紅石較佳[40, 41, 44],而造成此一結果的可能原因包括表面晶向與光活性的關 係[45]、電子電洞對的生期 (Life-time)[46, 47]以及結構是否容許較深層載流子被 激發[48]等等。
然而,除了單一晶型外,常用的二氧化鈦更多的是混合晶型,例如 Degussa 的 Aeroxide® P25 二氧化鈦奈米粒子,其銳鈦礦與金紅石平均比例為 78 比 14,
另有少量無定形體 (Amorphous)以非均勻方式分布[49]。在很多情況中,P25 的 光催化活性都高於單一晶型。之所能以表現出優異的特性,得歸因於銳鈦礦和金 紅石的能帶並列。當受光激發後,電子傾向從金紅石遷移至銳鈦礦[50, 51]。金 紅石能隙較窄,可吸收更多光子;銳鈦礦的電子生期長,則有利於電荷分離。此 兩者協同效應遂增加了二氧化鈦的光催化效率。
2.3.2 石墨烯
圖 2-13、單層石墨烯具蜂巢狀結構
20
石 墨 烯 (Graphene) 的 發 現 可 以 追 溯 到 1962 年 , Boem 曾 將 氧 化 石 墨 (Graphite oxide)加熱剝離,並經還原製備而成[52]。然而真正將石墨烯分離出來 並證實其獨特的電子特性,則要等到 2004 年,Novoselov 利用思高膠帶法 (Scotch tape technique)[53, 54]將石墨烯從石墨中剝離並進行分析。
石墨烯是具有單原子厚度的二維平面,碳原子以sp2混成軌域(hybridized orbital)與其他碳原子組成σ鍵結,型成蜂巢狀平面結構 (圖 2-13)。垂直於σ鍵的π
鍵和π∗鍵,則分別構成了價帶和傳導帶。在能帶結構上,石墨烯具有零能隙半導
體 (Zero bandgap semiconductor)的特性,價帶和傳導帶連接在狄拉克點 (Dirac point) 上 , 靠 近 狄 拉 克 點 的 價 電 子 行 為 類 似 零 質 量 狄 拉 克 費 米 子 (Massless Dirac-Fermion),可以106𝑚𝑠−1的極高速度移動,使石墨烯電阻極低,傳導能力 非常好[55]。
現 時 最 能 夠 便 宜 、 大 量 生 產 石 墨 烯 的 方 法 是 氧 化 減 薄 法 (Oxidative exfoliation),其原理為使用強氧化劑,於石墨的層狀結構中間進行插層氧化,使 層與層之間存在帶負電的氧化官能基,克服石墨層間的范德瓦力 (van da Waals forces),並通過水分子的插層,大幅增加層間距離,使氧化石墨烯的剝離更容易。
氧化石墨烯則可進一步通過使用還原劑,製備出石墨烯[56]。
21
圖 2-14(a)、石墨具有層狀結構
圖 2-14(b)、氧化石墨;紅色箭頭為氧化官能基電斥力,藍色箭頭為范德瓦力
雖然石墨烯的研究熱潮是最近十年的事情,但令人意外的是,對石墨進行插 層 氧 化 的 技 術 卻 早 至 150 年 前 經 已 存 在 。 其 先 驅 者 包 括 Brodie[57] 、 Staudenmaier[58]和 Hummers[59],由於氧化石墨烯親水性強,這些技術在早期奈 米材料的研究中也有使用[60, 61, 62, 63, 64]。被用以製備石墨烯,則歸功於 Stankovich[65]的研究。當時雖然存在化學或者加熱插層以從石墨中剝離石墨烯 的技術,但其厚度及穩定性卻不盡如人意,而將 Hummers 氧化法投入剝離技術,
不只是能生成接近 2nm 厚度的石墨片,其穩定性也得以大幅改善。
本研究就氧化減薄法製成氧化石墨烯,通過同時還原及披覆於二氧化鈦上,
期望利用其優異導電性改善光催化效能。
22 2.3.3 二氧化鈦奈米管
除了利用一般商業可購買的二氧化鈦材料,本研究也嘗試利用奈米管形態的 二氧化鈦,將石墨烯披覆其上,並作光活性測試和特性分析。
二氧化鈦奈米管 (TiO2 nanotube),指的是由二氧化鈦構成,並在微觀上具 有管徑達到奈米尺度 (i.e. 10−9𝑚)的管狀結構材料。廣義而言,二氧化鈦奈米管 具有以下特性:(1)高比表面積及孔洞體積;(2)離子交換能力[66];(3)能快速並 長距離運送電子[67];(4)因為結構而改變光吸收[68]。
現行主要製備二氧化鈦奈米管的方法包括摸板法[69]、電鍍法[70]及鹼性水 熱法[71]。Ou[72]則就此三種方法整理出表 2-1,並探討了這些方法的優缺點。
本研究基於其簡便經濟,遂採用鹼性水熱法。
表 2-1、製備二氧化鈦奈米管的方法及其優缺點[72]
Fabrication method Advantages Disadvantages TNT features
Template–assisted method
(1) The scale of nanotube can be moderately controlled by applied template
(1)Complicated fabrication process
(2)Tube morphology may be destroyed during fabrication process
Ordered arrays (powder form)
Electrochemical anodic oxidation
method
(1) More desirable for practical applications (2) Ordered alignment with high aspect ratio (3) Feasible for extensive applications
(1)Mass production is limited
(2)Rapid formation kinetics is subjected to the utilization of hydrofluoric acid
(3)Highly expense of fabrication apparatus
Oriented arrays (thin film)
Hydrothermal treatment
(1) Easy route to obtain nanotube morphology (2) A number of modifications can be used to enhance the attributes of titanium nanotubes (3) Feasible for extensive applications
(1)Long reaction duration is needed (2)Highly concentrated NaOH must be added (3)Difficult in achieving uniform size
Random alignment (powder form)
23
鹼性水熱法的基本操作步驟包括:(1)二氧化鈦與濃鹼於高溫高壓中反應;(2) 以稀釋酸性溶液清洗;(3)高溫退火。下面茲就這些步驟對奈米管的型成及產物 的影響因素作序述。
2.3.3.1 奈米管的型成
二氧化鈦從顆粒到奈米管狀結構,其發生的機制仍然具有一定爭議性。譬如 早期的推測提出酸洗是造成奈米管的關鍵步驟[71, 73, 74];另一些研究[75, 76]
卻發現,即使沒有進行酸洗,XRD 和 SEM 仍然能檢測出管狀結構的存在。
現時較廣為接受的假說包含四個階段[77]:(1)二氧化鈦於濃鹼中分解,伴隨 Ti − O − Ti鍵的斷裂;(2)型成奈米層片 (Nanosheet)結構的鈦酸鹽 (Titanate);(3) 奈米層片的剝離;(4)奈米層片經捲縮型成奈米管。
一般製備使用的濃鹼為氫氧化鈉 (NaOH),在高濃度的氫氧化鈉中,
Ti − O − Ti鍵的斷裂有賴於OH−的攻擊,使二氧化鈦型成鈦酸鹽層狀結構,此時 二氧化鈦依然保留了TiO6配位,因而帶有負電荷,吸引鈉離子進行插層[78]。另 一方面,隨著奈米層片結構的成長,會帶來一定程度的不穩定,使奈米層片傾向 捲縮[79, 80],型成奈米管。
奈米層片捲縮的動力來源又有不同假說,包括層片在成長時,邊緣原子的懸 吊鍵 (Dangling bonds)累積造成二維結構的不穩[81];或如 S. Zhang[82, 83]就 Na𝑥H2−𝑥Ti3O7 進行的理論模擬,指出多層重疊的奈米層片的情況中,表面層片 其中一面的氫原子與基質接觸,濃鹼會與氫原子反應,造成 hydrogen-deficiency 的現象,另一面與基質卻沒有直接接觸,如此兩表面的張力不相等,以致剝離並 捲縮;Bavykin[84]對多層重疊的奈米層片,則提出層片成長速度不均等,是造成 捲縮的重要因素。
24 2.3.3.2 奈米管產物的影響因素
鹼性水熱法中影響生成產物的因素眾多,本研究就以下八項,通過參考文獻 整理資料並略作敘述,當中包括:(1)鹼源;(2)鈦源;(3)鹼源濃度;(4)反應溫度;
(5)反應時間;(6)鈦源及鹼源比例;(7)酸洗濃度;(8)退火溫度。
2.3.3.2.1 鹼源
鹼性水熱法中最常用的鹼源為氫氧化鈉,其在製備過程中擔當重要角色,使 用其他氫氧化物並無法取代,如以氫氧化鋰 (LiOH)為基質進行鹼性水熱法,只 會生成奈米粒子 (Nanoparticles);使用氫氧化鉀 (KOH),則生成實心的奈米棒 (Nanorods)或奈米塊 (Nanoplates)[75]。另外使用 NaOH/KOH 混合物也可以製備 出奈米管,但溫度及混合比例會很大程度影響產物種類,Bavykin 便就這兩項參 數繪畫了分佈圖 (圖 2-15)[85]。
25
圖 2-15、不同溫度及 NaOH 比例生成的產物形態[85];
紅色區域為奈米層片,青色區域為奈米管;黃色區域為奈米線
圖 2-16、銳鈦礦、金紅石和 P25 以不同條件進行水熱法的產物分布圖[86]
26 2.3.3.2.2 鈦源、鹼源濃度及反應溫度
另外,奈米管的生成和鈦源、氫氧化鈉濃度、反應溫度有密切關係,以發明 鹼性水熱法的 Kasuga 為例,他所採用的二氧化鈦是金紅石,氫氧化鈉濃度為 5 − 10M,反應溫度110℃為時 20 小時[71]。而 Morgan 等人則就銳鈦礦、金紅石 和 P25 三種常見的二氧化鈦,以氫氧化鈉濃度及反應溫度生成的產物形態和比例 繪出分佈圖 (圖 2-16)[86]。
2.3.3.2.3 反應時間
Dong 等人[87]就反應時間進行研究,發現在130℃的反應溫度下,4 小時反 應產物存在微量奈米管和大量前驅物;12 小時反應產物則存在大量奈米管和奈 米層片;24 小時反應產物形態以奈米管佔大宗,伴隨少量奈米層片;36 小時反 應產物則幾乎全為奈米管。長時間的鹼性水熱反應有利高純度的奈米管產出,但 一 些 研 究 [80, 85] 進 一 步 指 出 , 奈 米 管 在 鹼 性 水 熱 反 應 中 屬 於 準 穩 定 態 (Metastable)產物 (圖 2-17),當反應時間延長超過 72 小時或反應溫度提升,均會 使產物變成相對穩定的奈米線 (Nanofibre)或奈米帶 (Nanoribbon)。
圖 2-17、準穩定態的奈米管及較穩定的奈米線[85]
27 2.3.3.2.4 鈦源及鹼源比例
奈米管的形成有賴於基質中過量的濃鹼,故此濃鹼和二氧化鈦的數量比也會 影 響 產 物 的 形 態 。 Sreekantan 發 現 [88] 當 TiO2/NaOH 的 莫 爾 比 例 為 13 (32.5mL 10M NaOH per 1g TiO2),產物難以構成層狀結構,缺乏捲縮成奈米管的 必要條件,致使鈦酸鹽以奈米粒子形態組合成奈米棒 (直徑 50nm),而當 TiO2/NaOH的莫爾比例超過 27 (67.5mL 10M NaOH per 1g TiO2),則能維持穩定 的奈米管產物輸出,管徑可至 10nm。Bavykin 也發現TiO2/NaOH莫爾比例的增 加會使產物管徑增加、比表面積減少[84]。
2.3.3.2.5 酸洗
前述曾經提及,早期研究視鹼性水熱法的後續酸洗處理為奈米管成形的關鍵 步驟,這一論述雖然尚具爭議,但酸洗在奈米管製備的重要性仍是不容忽視。經 過鹼性水熱處理的奈米管具有鈦酸鹽結構,並包含插層的鈉離子。酸洗的主要功 能就在於置換鈉離子,以及將多餘的氫氧化鈉清洗。以稀酸溶液的質子置換鈉離 子,可以減低鈦酸鹽的熱穩定性,使晶相更容易轉化為銳鈦礦[89, 90, 91]。因為 在缺乏離子交換的情況下,鈦酸鈉即使在800℃高溫中退火,晶型依然不會改變 [66],由於鈦酸鹽並不具備良好的光活性[92],使酸洗在製備過程中有其必要性。
另外,當酸洗濃度過高 (i.e. pH<1),也會破壞奈米管結構[93]。
2.3.3.2.6 退火
鈦酸鹽奈米管經高溫退火,可以回復銳鈦礦晶型[94]。就經過酸洗置換出鈉 離子的質子化鈦酸鹽 (Protonated titanate)而言,當溫度越高,銳鈦礦的比例也會
28
提高。首先,插層間的結晶水會在180℃左右被排除[95],接近350℃時所有可交 換的質子都會被排除,並在450℃左右轉化成銳鈦礦。另外,有研究[96]指出當 退火溫度超過600℃,較穩定的金紅石晶體結構會出現,但也有研究[97]指出即 使溫度提升至800℃仍然沒有檢測出金紅石,準穩定態的銳鈦礦轉化成穩定的金 紅石要發生在接近900℃的高溫以上[98]。特別需要注意的是,雖然高溫退火能 使鈦酸鹽回復至具有光活性的晶相,但其奈米管形態於500℃左右便會開始瓦解 [97]。
29
第三章 實驗材料與方法
本研究嘗試以三種不同方法製備披覆還原氧化石墨烯 (Reduced graphene oxide, rGO)的二氧化鈦 (TiO2/rGO),包括水熱還原 (Hydrothermal reduction)、光 還原 (Photo-assisted reduction)及熱還原(Therml-reduction),並以甲醇溶液進行光 催化產氫作為一光活性指標。另外,本研究也會嘗試以水熱還原法將已製備的二 氧化鈦奈米管披覆上還原氧化石墨烯,再以同一條件進行光催化產氫測試。
3.1 實驗藥品與設備
3.1.1 實驗藥品
(1) 試劑水:Elix MilliPore
(2) 石墨:純度 99.9995%,100mesh,AlfaAesar (3) NaNO3:純度>99.5%,EMSURE
(4) H2SO4:濃度 95.0-97.0%,SigmaAldrich (5) KMnO4:純度>99.0%,J.T.Baker
(6) H2O2:濃度 34.5-36.5%,SigmaAldrich (7) TiO2:純度>99.5%,Degussa P25。
(8) NaOH:純度>98%,SigmaAldrich (9) HCl:濃度>37%,SigmaAldrich (10) CH3OH:HPLC 級別,Macron
(11) CH3CH2OH:純度>99.8%,Sigma-aldrich。
(12) N2:純度 99.99%,臺北清豐公司提供。
30 (13) H2:純度 99.99%,臺北清豐公司提供。
(14) O2:純度 99.99%,臺北清豐公司提供。
(15) Ar:純度 99.99%,臺北清豐公司提供。
31 3.1.2 實驗設備
1. 磁攪拌器: HMS-102,Fargo Instrument Ltd。
2. 分液漏斗:玻璃製,最適容積 1L。
3. 夾層玻離瓶:夾層外有循環水進出口,容許控制容器溫度。
4. 高溫釜:不銹鋼製,內襯鐵氟龍瓶,容積 250mL。
5. 離心機:Hermle-Z32HK。
6. 熱風循環式烘箱:DO-30,Dengyng Instruments Co., Ltd。
7. 高溫爐:Thermo Scientific™ Lindberg/Blue M™。
8. 冷凝式循環水槽:D-620,Dengyng Instruments Co., Ltd。
9. 氣密針:0.5 mL,側孔針,Hamilton。
10. 燈管外殼:夾層外殼,外有循環水進出口,石英製。
11. 光催化反應槽:氣密不銹鋼瓶,容積 1180mL。
12. 紫外光汞燈:HPA 400S,功率 400W,Philips。
圖 3-1、紫外燈管波長分布
13. 氣相層析儀-熱導偵測器 (GC-TCD):Personal GC 1000,管柱型號:MS-5A,
China Chromatography Co., Ltd。
32
3.2 實驗方法
3.2.1 實驗設計
圖 3-2、本研究實驗設計
33 3.2.2 製備氧化石墨烯
以鱗片石墨作為石墨烯的前驅物,參考 Perera 等人使用的改善 Hummers 法 [59, 99]:先將 0.5g 石墨、0.5g NaNO3和 23mL 98%H2SO4與0℃ 水浴中攪拌 15 分鐘;然後緩慢加入 4.0g KMnO4,使反應混合物中原位生成作為氧化劑的Mn2O7, 此時顏色位紫綠色;接著升溫至40℃,持續攪拌 90 分鐘,生成深棕色黏稠物;
此時以滴定管緩慢加入 50mL DI 水,再攪拌 10 分鐘;然後緩慢加入 6mL 35%
H2O2,使溶液產物變為金黃色;再加入 50mL DI 水,離心去除上澄液。
此時氧化石墨含有大量硫酸,極難清洗。Dimiev[100]指出因為反應時濃硫 酸會對環氧基 (Epoxy group)進行親核攻擊 (Nucleophilic attack),形成硫酸酯,
其半生期有可能接近 14-16 小時[101],使硫酸一直通過水解釋出,故此以大量清 水短時間清洗並無法完全去除強酸。
本研究嘗試將產物移至分液漏斗,加入 1L DI 水,靜置 12-24 小時,待產物 沉澱,去除上澄液;再重新加入 1L DI 水;如此重複清洗 3-4 次,可成功使上澄 液pH > 6,然後將產物移至烘箱以65℃烘乾 1 天。
圖 3-3、以分液漏斗靜置 12-24 小時,清洗出氧化石墨烯中的硫酸
34
產物在水體中完全分散前稱為氧化石墨 (Graphitic oxide),分散後則是氧化 石墨烯 (Graphene oxide)。如非特別指稱,下文中 GO 均指氧化石墨烯。
使用前,氧化石墨烯會先以 4g/L 的濃度,用超聲波震盪 1.5 小時均勻分散 於水中,再以離心機篩選出粒徑較小的氧化石墨烯。據文獻所述[102],3000rpm 離心 45 分鐘可以有效將大面積、多層重疊的氧化石墨烯沉澱。舉例而言,在 500rpm 轉速下離心,上層液中的氧化石墨烯平均厚度為 7.3 層,直徑3.5μm;當 離心轉速增加至 3000rpm,則上層液中的氧化石墨烯平均厚度可降至 3.2 層直徑 1μm。
圖 3-4、乾燥後的氧化石墨;呈棕黑色
35 3.2.3 製備 TiO2/rGO
製備TiO2/rGO,包含了建立二氧化鈦和氧化石墨烯的交互作用,以及還原 氧化石墨烯兩個步驟。
現行主流的還原方法可大致上分成熱還原和化學還原[103],化學還原中最 常見的還原劑是聯胺 (Hydrazine),其和一般金屬氫化物還原劑不同,金屬氫化 物會與水產生劇烈反應,不利於以水作溶劑的氧化石墨烯,而聯胺則可避免這問 題。但同時間要注意的是,聯胺屬於劇毒化合物,不便於處理,故本研究另行參 照其他化學還原法。
總 括 而 言 , 本 研 究 實 踐 了 水 熱 還 原 法 ( TiO2/HrGO )[104] 、 光 還 原 法 (TiO2/PrGO)[105]及熱還原法 (TiO2/TrGO)[106],其添加比例以 GO: TiO2之重 量比計算。
圖 3-5、使用於水熱還原法及奈米管製備之反應器;
包括鐵氟龍瓶及高溫釜
36 3.2.3.1 水熱還原法 (Hydrothermal reduction)
水熱還原法利用密閉空間中的高溫高壓,以乙醇作還原劑,於水中對氧化石 墨烯進行還原,並將二氧化鈦披覆其上。
首先將 100mL DI 水與 50mL 乙醇混合;再加入一定比例的氧化石墨烯,然 後以超聲波震盪 1 小時;接著加入 1g TiO2,並攪拌 2 小時使之分散均勻;此混 合液將移至移至鐵氟龍瓶,再放入高溫釜加熱120℃、3 小時;反應完成後,灰 色產物以 DI 水清洗,再置於65℃吹乾。
3.2.3.2 光還原法 (Photo-assisted reduction)
光還原法屬於化學還原的一支[103],以純乙醇作為反應基質及還原劑,經 下列反應式,同時還原並披覆石墨烯於二氧化鈦上:
TiO2+ ℎ𝜐 → TiO2(ℎ + 𝑒)C→ TiO2H5OH 2(𝑒) +∙ C2H4OH + H+ (3-1) TiO2(𝑒) + GO → TiO2+ rGO (3-2) 首先將氧化石墨烯以一定比例加入 400mL 乙醇;以超聲波震盪 1 小時,再 加入 1g TiO2;於黑暗中攪拌 1 小時,並持續打入氮氣;待反應物分佈均勻後,
以 400W 紫外汞燈照射 2 小時,氮氣氣泡仍持續打入;反應完成後,灰藍色產物 以 DI 水清洗,再置於65℃吹乾。
3.2.3.3 熱還原法 (Thermal reduction)
熱還原法除了通過高溫移除氧化官能基,還可以高速升溫,使氧化官能基以 CO 或CO2氣體快速釋出,在層間累積氣壓,進一步剝離出石墨烯[107]。
首先,將一定比例的氧化石墨烯加入 200mL DI 水中,以超聲波震盪 1 小時;
37
再加入 1g TiO2,並攪拌 2 小時使均勻混合;置於65℃吹乾後,放入高溫爐,以 100℃/min的速度升溫至300℃,維持 2 小時後獲得灰藍色產物。
3.2.4 製備 TiNT/rGO
根據 Perera 等人的研究[99],我們可以利用濃鹼同時還原氧化石墨烯及製備 二氧化鈦奈米管 (TiO2 nanotube,簡稱 TiNT)。此法係根據 Rourke 等人的研究 [108],他們通過以強鹼混合氧化石墨烯,並分析其中上澄液和沉澱,提出氧化 石墨烯的結構中存在約 30%重量比的氧化碎片。和一般氧化官能基平均分佈於石 墨層的模型不同,Rourke 等人認為氧化石墨烯中存在氧化程度低的類石墨烯 (長 度約=1μm)和氧化程度高、分子量較小 (長度約=1nm)的氧化碎片 (Oxidative debris)。氧化碎片能幫助石墨烯穩定分散水中,但在強鹼環境中,氧化碎片會與 石墨烯分離並經酸洗洗出,留下石墨烯。
TiNT/rGO的製備過程為:首先製備 100mL NaOH,並加入一定比例的氧化 石墨烯;此時濃鹼會使帶負電的氧化石墨烯聚集成塊,需要超聲波震盪 1.5 小時,
使之完全分散;然後加入 1g TiO2,再攪拌 1 小時使混合物分散均勻;接著將混 合物移至鐵氟龍瓶,再放入高溫釜,加熱 120℃、24 小時;產物以 0.1M HCl 酸 洗(每次約 100mL DI 水/1g TiO2),經 6000rpm 離心 2 分鐘去除上澄液,重複 4 次;再以 DI 水清洗,經 6000rpm 離心 2 分鐘去除上澄液,重複至 pH=6;獲得 產物以65℃吹乾。
二氧化鈦奈米管方法同上,但過程不加入氧化石墨烯。
38 3.2.5 光催化材料特性分析
1. 掃描式電子顯微鏡 /X 光能量分散光譜 儀 (Scanning Electron Microscope/
Energy Dispersive Spectrometer,SEM/EDS)
以蒸鍍法將導體如鉑、金等覆蓋材料表面。然後透過電子束聚焦於樣品上,
通過入射電子與導體電子間的交互作用,可收集其發出的各種二次電子訊號,
分析材料表面形態以及元素含量。
型號:JEOL JSM6510
2. 200kV 場發射槍穿透式電子顯微鏡 (Field Emission Gun-Transmission Electron Microscope,FEG-TEM)
先將樣品於液相中分散,使樣品顆粒變小變薄,再將之滴於 400mesh 鍍碳 銅網上,銅網的范德瓦力會將顆粒固定到網格中,此時將多餘樣品吸乾,再 置銅網於真空中乾燥,即可上機。測量時樣品在高真空狀態,電子束會聚焦 到銅網上,通過接收穿透電子訊號,可以了解樣品內部結構,包括晶格、厚 度等等,一般解析度較 SEM 高。
型號:FEI Tecnai G2 F20
3. 傅立葉轉換紅外光譜儀 (Fourier Transform Infrared Spectrometer,FTIR) 分子鍵的伸縮震動 (Stretching)和彎曲震動 (Bending)頻率接近紅外光,故具 有特定結構以致震動模式 (Vibration mode)的分子鍵,會吸收特定波長的紅 外光。因此通過測量樣品的紅外光吸收,可以掌握樣品中存在的化學鍵種類,
特別適用於有機物如 H、C、N、O 等原子間鍵結。送測前樣品以 KBr 稀釋 100 倍,打成片狀再上機掃描。
型號:PerkinElmer Spectrum 100
39
4. X 光粉末繞射儀 (X-Ray powder diffractometer,XRD)
X 光繞射多用於長程有序 (Long-range ordered)的物質。由於物質內部排列具 有重覆性,原子間距離也會有固定模式。X 光以特定的入射角照射時,會由 於這些原子間距離產生相長干涉,而滿足此條件產生繞射,則可由 Bragg’s law 計算:
2d sin 𝜃 = nλ (3-3) 型號:PANalytical; X' Pert PRO
5. 紫外光-可見光光譜儀 (UV-Vis spectrophotometer)
當分子吸受紫外光-可見光時,能量會以電子激發的形式被吸收。對於研究 光催化材料而言,紫外光-可見光吸收的資料,可以幫助我們了解物質的吸 收波長範圍,進而判斷材料對入射光的利用效率。粉體樣品會先用 KBr 稀 釋 50 倍,並壓成片狀再送測。
型號:Hitachi U3900
6. BET 表面積分析 (Brunauer–Emmett–Teller surface area analysis)
利用氣體吸附於固體表面的特性,測量樣品的表面及孔洞體積、大小。
型號:ASAP 2020 V4.02
40 3.2.6 光催化產氫實驗
本研究以光催化材料加入甲醇水溶液,進行光催化產氫測試,作為材料的光 活性指標。其中光源燈管波長峰值為 365nm、功率 400W;甲醇溶液 750mL,體 積比 20%;並添加 0.2g 光催化材料;反應混合物中會放置磁攪拌子,並以固定 轉速將混合物持續攪拌均勻;反應槽為一氣密空間,除去燈管之總容積為 1180mL;
外圍有循環水將反應槽溫度固定於25℃,以酒精溫度計測溫確保每次實驗溫度相 同。
實驗步驟略述如下:
1. 製作檢量線:將高純度氫氣,以給定體積 (1 − 8μL,間隔 1μL)打入氣相 層析儀,並記錄訊號面積。然後繪畫出檢量線,找出氣體體積及氣相層析儀 訊號的關係式。氮氣及氧氣也以同樣步驟製作檢量線,唯給定體積較大。
2. 測試前:把 0.2g 光催化材料均勻分散於 750mL 20%v/v 甲醇溶液後,將 混合液移入不銹鋼反應槽,並關緊反應槽蓋。然後於反應槽內打入氮氣,並 置於黑暗中以磁攪拌子攪拌 1 小時,期間反應槽外循環水維持25℃。
3. 光催化反應測試:打開 400W 紫外燈管,同時記錄此時間點,定義為 t=0。
然後在 t=0, 30min, 60min, 120min, 180min 五個時間點上,抽取反應槽內氣體 0.3mL 打入氣相層析儀,並記錄氫氣、氧氣和氮氣之面積。
4. 總氣體量換算:反應槽內容積為 1180mL,扣除甲醇溶液,氣體體積為 430mL。由於反應前以高純度氮氣進行曝氣,使其氧氣總體積小於 1mL,故 可合理假定 t=0 時 430mL 全為氮氣,並因反應槽溫度已固定於25℃,現設 槽內氣壓為 1atm,則其氮氣莫爾數:
n𝑇,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛 = PV
RT= 1atm∙0.43L
(0.0821atm∙L∙mol−1∙K−1)∙(298K)= 0.01758𝑚𝑜𝑙 (3-4) 另外假設反應過程沒有氣體洩漏,並且槽內氣體均勻混合,則槽內各點空氣
41
的氫氮比均相同,此時根據我們取樣並注入氣相層析儀所獲得的數據,我們 知道取樣的氫、氮氣量,則可得下式:
n𝑇,ℎ𝑦𝑑𝑟𝑜𝑔𝑒𝑛
n𝑇,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛 =n𝑆,ℎ𝑦𝑑𝑟𝑜𝑔𝑒𝑛
n𝑆,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛 (3-5)
其中,n𝑆,ℎ𝑦𝑑𝑟𝑜𝑔𝑒𝑛=取樣氫氣量
n𝑆,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛 =取樣氮氣量 n𝑇,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛 =0.01758𝑚𝑜𝑙 n𝑇,ℎ𝑦𝑑𝑟𝑜𝑔𝑒𝑛=總氫氣量
由於n𝑆,ℎ𝑦𝑑𝑟𝑜𝑔𝑒𝑛、n𝑆,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛和n𝑇,𝑛𝑖𝑡𝑟𝑜𝑔𝑒𝑛為已知,總氫氣量便可由式(3-5)求
得。
42
第四章 結果與討論
4.1 特性鑑定
4.1.1 掃描式電子顯微鏡/X 光能量分散光譜儀
由於掃描式電子顯微鏡無法看到內部結構,加上披覆上石墨烯的二氧化鈦以 及二氧化鈦奈米管,在形成粉體的乾燥過程中會聚集成塊,沒法分辨彼此,缺乏 詮釋意義,因此本研究只呈現偵測氧化石墨的微觀影象。圖 4-1(a)可以看到插層 氧化使氧化石墨的層狀結構浮現。此時的氧化石墨尚未於水中分散,故仍然保持 重疊結構。然而從後續穿透式電子顯微鏡當中,可以看到當氧化石墨烯經過超聲 波震盪分散於水中後,便會呈現僅數層重疊的結構。
另外,通過簡稱 EDS 的 X 光能量分散光譜儀 (圖 4-1(b)),可以大略知道圖 像中的元素構成。雖然負載樣品的基台上貼上雙面碳膠帶,使碳元素比例不能參 考,但通過氧元素和硫的比例,以及文獻調查化學氧化法製備氧化石墨的碳氧比 約為 79:20 至 65:33[109, 110, 111],可以推測硫的含量約占 1-2%的數量比,說明 硫酸非常難去除,佐證 Dimiev 提出氧化過程硫酸酯形成的理論[100]。
43
圖 4-1(a)、掃描式電子顯微鏡下的氧化石墨
圖 4-1(b)、EDS 掃描(a)範圍內得出之元素構成
44 4.1.2 穿透式電子顯微鏡
4.1.2.1 氧化石墨烯
圖 4-2 為氧化石墨烯的 TEM 圖像,可以觀察到其片狀半徑約為 200nm。氧 化石墨烯的成品大小,與前驅物及製備條件息息相關,為了控制氧化石墨烯的大 小,本研究在使用前會先通過 3000rpm、45 分鐘離心,讓大面積、多層重疊的 氧化石墨烯沉澱,並取回上層分散程度較好的氧化石墨烯 (見 3.2.2)。根據文獻 所述,經篩選過的氧化石墨烯平均重疊層數為 3.2 層、直徑 1000nm[102]。
然而從 TEM 圖像看來,氧化石墨烯的仍然不能算非常薄。這可能歸因於 TEM 送測前,樣品需要負載於鍍碳銅網上,並通過范德瓦力懸浮於網格中間。由於經 篩選的氧化石墨烯直徑較小,較薄的樣品缺乏足夠的范德瓦力附著銅網上,容易 在負載的過程中流失,於是只餘下較厚的樣品 (i.e. 范德瓦力較強)供送測。實際 上從下面TiO2/rGO的樣品中,我們可以往往可以看到較薄石墨烯。
圖 4-2、穿透式電子顯微鏡下的氧化石墨烯
45 4.1.2.2 TiO2/rGO
圖 4-3 顯 示 了 三 種 還 原 方 法 分 別 製 備 的 TiO2/rGO , 包 括 水 熱 還 原 法 (TiO2/HrGO)、光還原法 (TiO2/PrGO)和熱還原法 (TiO2/TrGO)。其披覆比例為 1%重量比的 GO: TiO2,可以看到普遍石墨烯厚度較單獨氧化石墨烯的測量結果 薄,推測原因為二氧化鈦附著於石墨烯上,使材料整體的范德瓦力增加,比較不 會因為石墨烯的厚薄而從負載銅網上流失。
另外,我們可以從比較中發現,只有水熱還原法的二氧化鈦較能附著於石墨 烯的表面;特別是光還原法的產物中,二氧化鈦顆粒多附著於石墨烯邊緣。此結 果可以從 Gao 等人的研究[112]中推論,他們通過理論計算分析了石墨烯上幾種 氧化官能基的穩定程度,發現氧化石墨烯邊緣的羥基遠較表面上的穩定,從熱力 學的觀點而言,這會使邊緣的氧化官能基的密度較高。由於在光還原的情況中,
石墨烯和二氧化鈦的交互作用是通過二氧化鈦的激發電子對氧化石墨烯進行還 原。換言之,在氧化官能基集中在石墨烯邊緣的情況下,二氧化鈦累積在邊緣也 有其合理性。
46
圖 4-3、穿透式電子顯微鏡下的(a)(b) TiO2/HrGO;(c)(d) TiO2/PrGO;
(e) TiO2/TrGO
47 4.1.2.3 TiNT/rGO
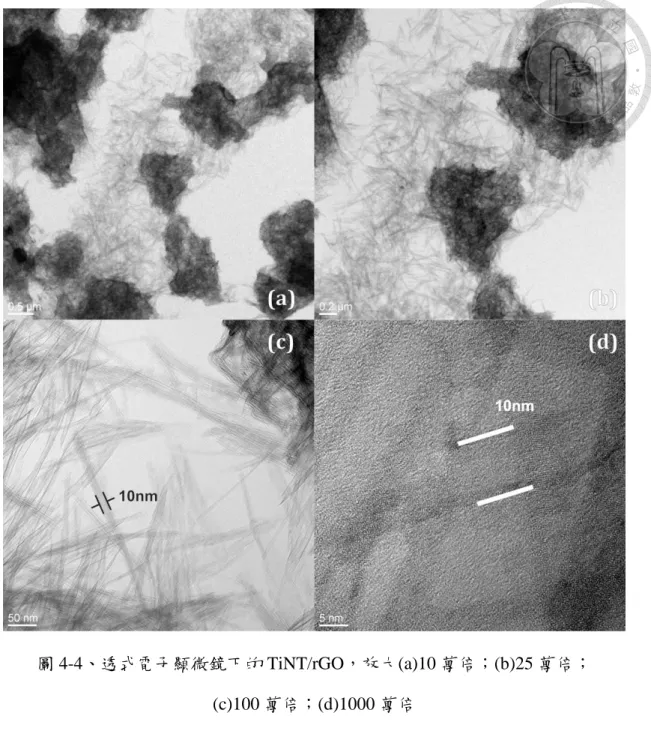
圖 4-4 為披覆還原氧化石墨烯的二氧化鈦奈米管 (TiNT/rGO),披覆比為 1%,
放大倍數從 10 萬倍到 1000 萬倍。從各圖左下角的量尺可以看到二氧化鈦奈米管 的管徑極細,直徑約為 10nm,不容易被偵測,故解析度較低的掃瞄電子顯微鏡 便無法提供清晰圖像說明樣品是否具管狀結構。
以鹼性水熱法合成之二氧化鈦奈米管,無法如電鍍法或模板法一樣整齊排列,
而是以互相重疊、隨機排列的方式分佈,高度差異大,使 TEM 的電子束不易聚 焦。但除此以外,就 TEM 的成像依然可以看到以 Perera 等人的方法[99],在鹼 性水熱條件下同時合成二氧化鈦奈米管並披覆上石墨烯,並未有明顯破壞兩者的 形態結構。Perera 等人所製備的奈米管管徑為 9nm,也與本研究相若。
48
圖 4-4、透式電子顯微鏡下的 TiNT/rGO,放大(a)10 萬倍;(b)25 萬倍;
(c)100 萬倍;(d)1000 萬倍

![圖 2-15、不同溫度及 NaOH 比例生成的產物形態[85];](https://thumb-ap.123doks.com/thumbv2/9libinfo/9603260.629648/37.892.234.745.106.643/圖215不同溫度及NaOH比例生成的產物形態85.webp)