行政院國家科學委員會補助專題研究計畫 ■成果報告
□期中進度報告
含雙螺環官能基膨脹性單體(BSEM)之合成與光化學性質探討及其與壓克力樹脂混合液
經光聚合後之 IPN 結構混成材料之性質研究
Synthesis and Photochemistry of Bis-spiroorthoester Group Containing Expanding Monomer(BSEM) and Properties of IPN-structure Hybrids from Photopolymerization of BSEM/Acrylate Resin Mixtures
計畫類別:■個別型計畫 □整合型計畫 計畫編號:NSC 97-2221-E-011-026-MY2
執行期間: 2008 年 08 月 01 日至 2010 年 07 月 31 日 執行機構及系所:國立台灣科技大學材料科學與工程系
計畫主持人:許 應 舉 共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整報告
本計畫除繳交成果報告外,另須繳交以下出國心得報告:
□赴國外出差或研習心得報告
□赴大陸地區出差或研習心得報告
□出席國際學術會議心得報告
□國際合作研究計畫國外研究報告
處理方式:除列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢 中 華 民 國 九十九 年 十二 月 四 日
中文摘要
光起始劑—(4-octylphenyl)phenyliodonium hexafluroantimonate (OPIA)與光敏感劑—Anthracene 的 混合液,在經紫外光(254 nm)照射後生成一陽碳離子(carbocation)—9-phenyl-9,10-dihydroanthracen- -10-ylium cation (PDAC);將膨脹性單體—cis-2,3-tetramethylene-1,4,6-trioxaspiro[4,4]nonane (I) 加入 PDAC 中 , 使 進 行 陽 離 子 聚 合 反 應 而 產 生 一 立 體 規 則 性 (stereoregular) 聚 醚 酯 (poly(ether ester))—poly(trans-2-oxycyclohexyl butanoate) (-[trans-2-OCB]n-) (II) 。 聚 醚 酯 II 、 聚 合 反 應 中 間 體—1,3-dioxolan-2-ylium carbocation (DOLC)及 PDAC 的化學結構均以光譜加以鑑定,並以三者的關連 性推定
I 之聚合為一區域特異性(regiospecific)的反應機制(mechanism)。相較於 I
以SnCl4 為起始劑進 行區域特異性聚合反應形成 (-[cis-2-OCB]n-)(III)時,體積收縮為 2.49%,I 以 PDAC 為起始劑進行區域特異性聚合反應形成
II 時卻是體積膨脹 2.09%;研究結果證實造成聚醚酯 II 體積膨脹的原因是,單體
I 在進行區域特異性聚合反應時,其在 C-1 及 C-2 之順式(cis-)取代基團變成反式(trans-)形態所致。
關鍵詞:
立體規則性聚合體、區域特異性聚合反應、光起始劑、光敏感劑、陽離子光聚合反應、膨脹性 單體
I
英文摘要
A stereoregular poly(ether ester), poly(trans-2-oxycyclohexyl butanoate) (-[trans-2-OCB]n-) (II), was obtained by cationic polymerization of cis-2,3- -tetramethylene-1,4,6-trioxaspiro[4,4]nonane (I) initiated by the 9-phenyl-9,10-di- hydroanthracen-10-ylium cation (PDAC) obtained by exposing a mixture of (4-octylphenyl)phenyliodonium hexafluroantimonate (OPIA) and anthracene (A) to UV light at 254 nm. The structure of II, the polymerization mechanism of I, and the relationship between volume changes during polymerization and polymer structure were studied. The polymerization mechanism of I proceeded in a regiospecific manner and was determined in terms of the relation among the polymer structure, the steric hindrance sensitized PDAC, and the only intermediate—1,3-dioxolan-2-ylium carbocation (DOLC). Polymer
II exhibited high volume expansion (2.09 ± 0.25%) during polymerization compared to that of -[cis-2-OCB]
n- (III), obtained by polymerization of I using SnCl4 as an initiator, which demonstrated high volume shrinkage (-2.49 ± 0.13%) during polymerization. The volume expansion of II is caused by the conversion of cis substitution at C-2 and C-3 in I to the trans form during polymerization.
Key words: spiroorthoester, anthracene photosensitizer, stereoregular polymer, regiospecific reaction, cationic photopolymerization.
目錄
中文摘要………I
英文摘要………..……….………II
目錄………..……...….……….. III
Introduction……….………..…...1
Experimental section……….………..….………2
Results and Discussion……….4
Conclusions………..12
Acknowledgement………12
References……….………13
Introduction
Spiroorthoesters, which act as expanding monomers, undergo cationic double ring-opening polymerization and, theoretically, show volume expansion during polymerization.1–6
However, most reported
spiroorthoester monomers did not show volume expansion during polymerization initiated by a Lewis acid (e.g., BF3·OEt2, CH3OSO2CF3, or SnCl4) or a peroxide, but always exhibited volume shrinkage.7–13 The reason for this shrinkage has rarely been further studied in the literature. Photopolymerization initiated by UV light is a method of rapid polymer production. The reaction is completed, or the change from a liquid to a solid, takes place within a few seconds or even within a fraction of a second.14,15 Cationic photopolymerization has several advantages compared to free radical photopolymerization: it is not inhibited by oxygen, the reaction can continue long after irradiation has ceased, and reaction can take place in areas that the light did not reach. It can be used to polymerize important classes of monomers, including epoxides and vinyl ethers. However, to our knowledge, it has rarely been used in initiating double ring-opening polymerization of spiroorthoesters.13,16,17We have reported that cis-2,3-tetramethylene-1,4,6-trioxaspiro[4,4]nonane (I) (Table 1) underwent cationic photopolymerization initiated by a diphenyliodonium salt after direct exposure to UV light at 254 nm, or by exposure to UV light at 365 nm in the presence of the non-aromatic photosensitizers, isopropylthioxanthone (ITX) and 2,2-dimethoxy-2-phenylacetophenone (DMPA). The polymer, poly[(trans-OCB)x′- -(cis-OCB)x″-(CHO)y], consisting of poly(trans-2-oxycyclohexyl butanoate) ([trans-OCB]x′), poly(cis-2-oxycyclohexyl butanoate) ([cis-OCB]x″), and poly(1,2-cyclohexene oxide) ([CHO]y) segments was produced.17 The polymer thus formed exhibited volume expansion during polymerization, unlike the polymers obtained by conventional cationic polymerization using Lewis acids as an initiator, which demonstrated volume shrinkage during polymerization. This was because of the higher and optimal molar ratio of (trans-OCB)x′/(cis-OCB)x″ segments, and the lower content of the higher density [CHO]y segment in the polymer chain.
Fused-ring aromatic compounds such as anthracene (A) and perylene often show photosensitizing activity and they have been used to photosensitize diaryliodonium (or triarylsulfonium) salts to undergo cationic photopolymerization.18,19 The mechanism and kinetics of the reaction of diaryliodonium salts photosensitized by A have been studied thoroughly.20–22 It could be concluded that A is first excited to the singlet state by absorbing photons, and then undergoes intersystem crossing to the triplet state. The triplet-state A subsequently forms an excited-state complex—an exciplex—with the diaryliodonium salt. In the exciplex, A may undergo electron transfer to the diaryliodonium salt,23 becoming a radical cation. After a series of reactions,21 the radical cation is converted to a 9-phenyl-9,10-dihydroanthracen-10-ylium cation (PDAC), which acts as an active center to initiate the reaction.
In this paper, the cationic polymerization of I initiated by PDAC, obtained by exposing an OPIA/A mixture to UV light at 254 nm, was studied. The novel reaction mechanism of I initiated by PDAC, and the unique stereoregular structure and volume expansion of the polymer thus formed were studied in detail.
Experimental Section Materials
I was prepared by the reaction of cis-1,2-cyclohexene oxide with γ-butyrolactone (γ-BL) in the presence
of BF3·OEt2 in dichloromethane using a previously reported method24(bp: 88–89 °C, 2.2 mm Hg), and was
stored at room temperature under nitrogen. OPIA was used as the photoinitiator. ITX and DMPA were used as non-aromatic photosensitizers, A was used as an aromatic photosensitizer, and SnCl4, CH3SO2OCF3, and BF3·OEt2 were used as conventional cationic initiators. All compounds, except OPIA (Nissho Sangyo) and A (Acros), were purchased from Aldrich Co. and used as received without further treatment. Dry dichloromethane was obtained by refluxing over calcium hydride and distillation.Cationic photopolymerization with an OPIA/A initiating system
In a dry box, 3.89 × 10-2 g (0.062 mmol) of OPIA and 9.76 × 10-3 g (0.065 mmol) of A were dissolved in 1.0 ml of dichloromethane and added to a round Teflon fillister (diameter × depth = 16.5 × 5 mm). A 30-μm-thick transparent polypropylene film was laminated on the top of the fillister to prevent moisture diffusion. The solution was exposed to UV light (λmax: 254 nm) at 25 °C in a BLX-E254 crosslinker box (Vilber Lourmat, Marne-la-Vallée, France) for 5 min with a light intensity of 6.0 mW/cm2 (measured using a UVX-25 radiometer, European Union). Then, 1.00 g (5.44 mmol) of liquid I was immediately added to the fillister and mixed thoroughly with the photoreacted OPIA/A solution with a dry glass rod. After standing for 5 min, 10 μL of trimethylamine were added to the mixture to quench the reaction. After qualitative analysis of the γ-BL by gas–liquid chromatography (GLC), the solution was washed with water and dried with anhydrous MgSO4. The procedure for separating and purifying the product was performed as described in the literature.17 Other experimental procedures, such as the cationic polymerization of I initiated by a Lewis acid and photoinitiator were performed as described in the literature.17
Degree of monomeric conversion
17Homogeneous mixtures of I, initiators, and other additives before and after irradiation were coated on the KBr disks. After the solvent was removed under reduced pressure, the samples were recorded on a Fourier-transform infrared (FTIR). The total area of the νas,C-H and νs,C-H absorptions at 2950 and 2850 cm-1 was considered the internal standard (IS). The change in the peak area due to the spiroester group at 1323 cm-1 before and after the reaction was monitored, and the degree of conversion (DC) was determined using the following equation:
DC (%) = [1-(A1323 /AIS)a /(A1323 /AIS)b] x 100
Where (A1323/AIS)a is the ratio between the area of spiro absorption at 1323 cm-1 and the area of IS absorption after irradiation; (A1323/AIS)b is the ratio between the area of spiro absorption and the area of IS absorption before irradiation.
Volume change test
The densities of monomer (liq.) (ρ1) and polymer (liq.) (ρ2) were determined by means of a pycnometer at 25
°C. Volume change was calculated from (1/ρ2 -1/ρ1)/ (1/ρ1).
Identification of the PDAC structure
PDAC was prepared and identified indirectly as follows. In a round Teflon fillister, 0.1000 g (0.1594 mmol) of diaryliodonium pentafluorophosphate (used to simplify the patterns in the 1H NMR and high-resolution MS (HRMS) spectra) and 0.0283 g (0.1594 mmol) of A were dissolved in 1.0 ml of dry dichloromethane. A 30-μm-thick transparent polypropylene film was laminated on the fillister top. The solution was exposed to UV light (λmax: 254 nm) at 25 °C for 10 min. Then a 10 wt% alcohol solution of sodium boronhydride (0.02 g) was added to react with any PDAC produced to yield 9-phenyl-9,10-dihydroanthracene. After drying under reduced pressure, the residue was purified by column chromatography using silica gel (Acros, 70–80 mesh, 60 Å) as the stationary phase and n-hexane/ethyl acetate (95/5 v/v) as the eluent. 9-Phenyl-9,10-dihydroanthracene25 (Rf (TLC): 0.24) was obtained (<30 mg) after drying under a vacuum at 60 °C.
1H NMR (CDCl3): δ (ppm) = 3.94–4.06 (m, 2H, benzylic hydrogen), 5.36 (s, 1H, benzylic hydrogen), 7.07–7.39 (m, 13H, aromatic hydrogen).
HRMS (EI) calcd for C
20H16 [M]+: 256.1252; found: 256.1239Measurements
Fourier-transform infrared (FTIR) spectra of the samples were recorded on a Digilab FTS-1000 spectrometer with a resolution of 4 cm-1. Samples for measurement were prepared by coating the polymer or homogeneous mixtures on KBr disks followed by vacuum drying (<5 Torr) at 40 °C for 10 h. The 1H and 13C NMR spectra of the polymers were recorded on a Brucker AMR 300 NMR spectrometer (Bruker, Madison, WI, USA) in CDCl3 with TMS as an internal standard. HRMS were recorded on a Finnigan MAT 95 mass spectrometer. UV absorption spectra were recorded using a Cary 100 Conc UV-visible spectrophotometer (Varian Inc., Palo Alto, CA, USA). GLC was used to qualitatively analyze the γ-BL produced by the side reaction. The molecular weight of the polymer was measured using a Jasco PU-980 GPC with a Jasco RI-930 RI detector (Jasco, Easton, MD, USA). A mixed bed column (Jordi DVB gel), 250 × 10 mm in size, was used. DMAc was used as the eluting agent, with polystyrene as a standard.
Calculations
The parameters of the optimized-geometry monomer and polymers were calculated using the Spartan 08 software suite (Version 1.2.0, Wavefunction, Inc., Irvine, CA, USA).The Mullikan net atomic charge on the molecules and the energetically minimized structures of the polymers were determined in the semi-empirical PM3 mode, followed by single energy point calculations using the DFT method (B3LYP with a 6-31G(d) basis set). The steric energies of the molecules were calculated using the ChemBio 3D ultra 11 progam (MM2 method) (CambridgeSoft Corp., Cambridge, MA, USA).
Results and Discussion
Cationic photopolymerization of I with an OPIA/A initiating system
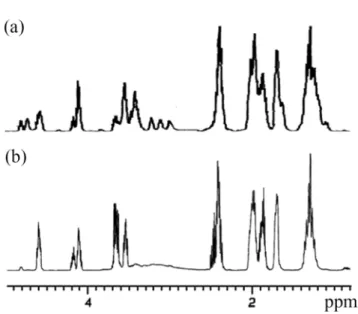
When I was mixed thoroughly with OPIA/A (molar ratio: 1.0/1.05) and the mixture was exposed to UV radiation at 254 nm (λmax), cationic photopolymerization occurred instantly to afford a polymer with an NMR spectrum (Figure 1a) similar to that of the polymer obtained by direct irradiation of I with OPIA.17 By-products such as γ-BL and -[CHO]n- oligomers produced by single ring-opening side reactions were examined. The structure of the polymer was identified as [(trans-OCB)p-(cis-OCB)q-(CHO)s]. An OPIA/A mixture was irradiated with UV of the same wavelength; I was then added immediately, and reaction took place within 3–5 min. The polymer thus formed has a simple 1H NMR spectrum (Figure 1b), quite different from that in Figure 1a. The by-product mentioned above was not detected by GLC or FTIR spectroscopy. This proved that I had undergone a full double ring-opening cationic polymerization without any single ring-opening side reactions.
Figure 1.
1H NMR spectra of polymers obtained by (a) UV exposure of mixture of I and OPIA/A at wavelength of 254 nm; and (b) UV exposure of OPIA/A at 254 nm followed by addition of I.Polymer structure
The correlation of the protons of the polymer with the simple 1H NMR spectrum (Figure 1b) was achieved using DQF-COSY NMR spectroscopy (Figure 2). The absorption of the -CH2CO2- at 2.42 ppm was not disturbed by any other absorption of the polymer and its proton area is almost one-eighth of the total protons area.12 This indicates that the repeating unit in the polymer contains 16 protons and has the same hydrogen numbers as I. It is therefore reasonable to deduce that the structure of the repeating unit is 2-oxycyclohexyl butanoate. It must be emphasized that the chemical shift at 4.60 ppm is the methine hydrogen Ha of the 2-oxycyclohexyl butanoate unit due to deshielding of the ester group26 at the trans position. The structure of the polymer is thus identified as a stereoregular poly(ether ester), (-[trans-2-OCB]n-) (II). The stereochemistry of the repeating unit was proved by the 1H NMR spectrum of the pure model isomer trans-2-hydroxycyclohexyl butanoate.17
Figure 2. DQF-COSY NMR spectrum of II.
Figure 2. DQF-COSY NMR spectrum of II.
The stereo-structure of I
Before investigating the polymerization mechanism of I, the stereo-structure of the monomer was first identified. cis-2,3-Tetramethylene-1,4,6-trioxaspiro[4,4]nonane, comprising a five-membered cyclic acetal (2,2-disubstituted-1,3-dioxolane) and a cyclic ether (2,2-disubstituted tetrahydrofuran),27
has two possible
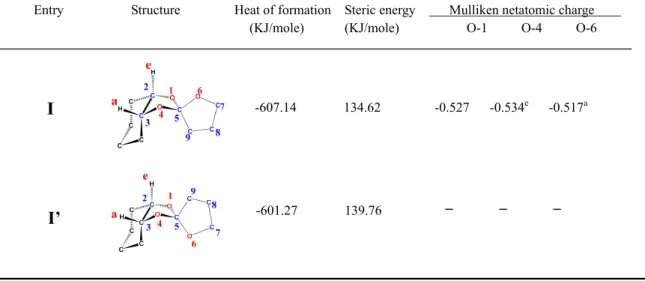
stereoisomers, designated I and I′. The heats of formation and steric energies of I and I′, estimated using PM3 and MM2 computations, are listed in Table 1.27–29 Inspection of the data in Table 1 reveals that the heat of formation and steric energy of I are lower than those of I′. However, it was confirmed by 1H and 13C NMR spectra that the synthesized monomer has only one type of stereo-structure (data not shown). This energy difference could be evidence that the stereo-structure of the monomer is I, not I′.Table 1. Heats of formation, steric energy, and B3LYP/6-31G(d) Mulliken net atomic charges of I.
Entry Structure Heat of formation Steric energy Mulliken netatomic charge (KJ/mole) (KJ/mole) O-1 O-4 O-6
UV irradiation of OPIA
The exposure of OPIA to UV light at different wavelengths, in the presence or absence of A, was studied. 2-Phenylquinoline was used to examine whether or not the photoacid (i.e., H+SbF6-), which initiates the cationic polymerization of I, was produced during irradiation. Once the 2-phenylquinoline has been protonated by the photoacid (or proton), the 2-phenylquinolinium ion formed shows a significant bathochromic shift in its UV absorption spectrum.30 Figure 3 shows the UV absorption of 2-phenylquinoline after addition to the OPIA being exposed to UV light under various conditions. It was found that, in contrast to direct irradiation of OPIA on its own at a wavelength of 254 nm, which causes the UV absorption of the 2-phenylquinolinium ion to undergo a significant bathochromic shift (curves a-c), the UV irradiated OPIA/A mixture shows no bathochromic shift after the addition of 2-phenylquinoline (curve d). When an OPIA/A mixture was UV irradiated at 365 nm, the added 2-phenylquinoline was converted to the 2-phenylquinolinium ion and thus showed a significant bathochromic shift in its UV absorption (curve e). These experimental results reveal that when OPIA/A is irradiated at 365 nm, electron transfer from the excited A to the OPIA in the singlet A/OPIA exciplex yielding a photoacid occurs.31 In contrast, when an OPIA/A mixture is exposed to UV at 254 nm, no photoacid is produced; the abovementioned reaction does not occur. The only reaction may be transfer of an electron from the excited A to the OPIA in the triplet A/OPIA exciplex to yield a PDAC,21,23 which can initiate the cationic polymerization of I.
-607.14 134.62 -0.527 -0.534e -0.517a
I
-601.27 139.76
I’
250 300 350 400 0.0
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0
Absorbance
Wavelength (nm) (a)
(b)
(c)
(e) (d)
Figure 3. UV spectra of 2-phenylquinoline (a), and 2-phenylquinoline in solutions of (b) perchloric acid, (c)
OPIA after irradiation at 254 nm, (d) OPIA/A after irradiation at 254 nm, and (e) OPIA/A after irradiation at 365 nm.However, when an OPIA/A mixture was irradiated at 254 nm in the presence of I, it seems that the formation of a triplet exciplex from triplet A and OPIA was interfered with to some extent. Some OPIA will be photodecomposed to produce a photoacid during irradiation to induce polymerization of I. A surplus A residue was thus found in the reaction system (data not shown). It was therefore concluded that when an OPIA/A mixture is exposed to UV at 254 nm in the absence of I, no photoacid is produced. The only initiating species, PDAC, could be isolated, and its structure was further confirmed by 1H NMR and HRMS (see experimental section).25
Polymerization mechanism
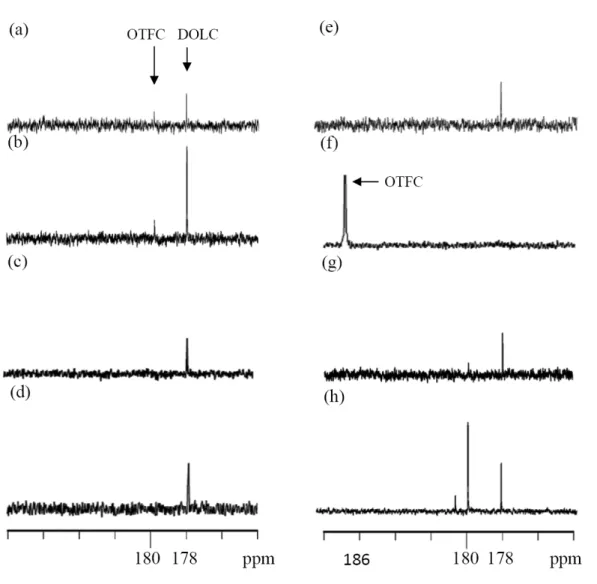
The cationic polymerization mechanism of I initiated by PDAC to afford II with a stereoregular structure is worth studying. The mechanism was determined in terms of the relation among the structures of I, II, PDAC, and the activated cationic propagating chain end, and is shown in Eq. 1 of Scheme 1. It was deduced that the PDAC attacked the cyclic ether oxygen (O-6) of I to afford one type of 1,3-dioxolan-2-ylium cation (DOLC) (Figure 4e).17,32 The subsequent ring-opening polymerization took place regiospecifically by backside attack by the cyclic ether oxygen of I at C-2 or C-3 of DOLC to yield
II. It seems that PDAC, a bulky and sterically sensitive species, only attacks oxygen atoms, either the
cyclic ether oxygen (O-6) or the acetal oxygen (O-1 or O-4), with less steric hindrance. The experimental results shown in Figure 4 could prove this theory. Figure 4a shows the 13C NMR spectrum ofof the 2-oxytetrahydrofuran-2-ylium cation (OTFC)32 (obtained by attack of the PDAC on the acetal oxygen of I) at ca. 180 ppm are both found. Figure 4b shows the spectrum of 2-methyl-1,4,6-trioxaspiro[4,4]nonane (MTN) after reaction with PDAC. The DOLC absorption at ca.
178 ppm is much higher than that of the OTFC, which is only a trace absorption at 180 ppm. Figures 4c and 4d show the spectra of cis- and trans-2,3-dimethyl-1,4,6-trioxaspiro[4,4]nonanes (cis- and trans-DMTNs), respectively, after reaction with PDAC. Only the peaks at ca. 178 ppm, corresponding to DOLC are found, and no OTFC absorption at 180 ppm occurs. The substituted methyl groups at C-2 and C-3 of the DMTNs may prevent the PDAC from attacking the adjacent acetal oxygen because of steric hindrance. It is therefore reasonable to infer that the attack on PDAC by the cyclic ether oxygen of I (a more complicated structure) is a steric-directing reaction. In addition to the effect of its bulky volume, the resonance stabilization effect of PDAC may play an important role in the regiospecific polymerization of I. The Mulliken net atomic charges on the oxygen atoms in I were calculated by computer simulation (Table 1).33 The charges on the oxygen atoms in the cyclic acetal ring (O-1 and O-4) are higher than that on the cyclic ether ring oxygen (O-6). Theoretically, the cation (or Lewis acid) should predominantly attack the cyclic acetal oxygen to afford the OTFC. For example, SnCl4 is a weaker Lewis acid34 and only attacks the cyclic acetal oxygen to afford the OTFC (Figure 4f); the OTFC then propagates the reaction (Eq. 2 of Scheme 1) of I to yield III, a stereoregular poly(ether ester), the structure of which was identified by its DQF-COSY NMR spectrum (Figure 5). However, when I undergoes conventional cationic polymerization initiated by BF3·OEt2 or CH3SO2OCF3 (both are stronger Lewis acids than SnCl4),35,36 or cationic photopolymerization in OPIA/ITX or an OPIA/DMPA initiating system, both DOLC and OTFC cations are produced (Figures 4g and 4h). No regiospecific reaction producing the stereoregular poly(ether ester) occurs. The reason is perhaps that the corresponding initiating species such as BF3, CH3+, H+, and C6H5C(OCH3)2+17,37 thus formed are so energetic that they can attack the oxygen atoms in both the cyclic ether and the acetal without any selectivity with regard to the polymerization route (Eq. 1 or 2 in Scheme 1)17. Although C6H5C(OCH3)2+ cation might be greatly stabilized by two methoxy and a phenyl groups and bulky enough to increase its selectivity in the reaction , but the free energy of the transition state of [C6H5C(OCH3)2+..O-6-I] (O-6-I: cyclic ether oxygen of I) (141.08 kJ/mol) is ca. 16.16 kJ/mol (thermodynamic calculation using PM3 method)16,38 lower than that of [C6H5C(OCH3)2+..O-1-I (or O-4-I)] (O-1-I: cyclic acetal oxygen of I) (157.69 kJ/mol).
It seems thatthe difference of free energy is not large enough to prevent the ring-opening reaction of [C6H5C(OCH3)2+..O-1-I (or O-4-I)] (Figure 4g). For the PDAC, the energy could be also greatly stabilized by its resonance behavior, and might be lowered enough to enable selection of the reaction route and exclude side reactions. Theoretically, PDAC should select the cyclic acetal oxygen of I to attack because of the higher Mulliken atomic charge on that oxygen (Table 1), but it only attacks the cyclic ether oxygen (Eq. 1 in Scheme 1) to afford II. The result of thermodynamic calculation shows that the free energy of the transition state of [PDAC..O-6-I] (11.38 kJ/mol) is ca. 144.15 kJ/mol lower than that of [PDAC..O-1-I (or O-4-I)] (155.53 kJ/mol). The difference is large enough to let PDAC attack the cyclic ether oxygen of I (Figure 4e) in regiospecific manner to afford II. It is therefore reasonable to conclude that the polymerization of I initiated by PDAC is controlled by steric factors rather than by electronic factors.
trans-OCB
n
cis-OCB
n SbF6
O
O O
1 2
4 3 5 7 6
8 9
R
R: H or C8H17
O O
O SnCl4
O O O
O O
O
I O
O O
n O
O O
O O
O I
R
Scheme 1. Polymerization mechanisms of I initiated by DPAC and SnCl
4,respectively.I C8H17
SbF6
A
OPIAPDAC Ⅰ
DOLC
Ⅱ
backside attacks
(Eq.1)
(Eq.2)
Ⅲ OTFC
O
O O
n
Figure 4.
13C NMR spectra (25 °C, CDCl3) of carbocations produced in the systems (a) TN/DPAC, (b) MTN/DPAC, (c) trans-DMTN/DPAC, (d) cis-DMTN/DPAC, (e) I/DPAC, (f) I/SnCl4, (g)I/OPIA/DMPA, and (h) I/OPIA/ITX; PDAC was obtained by UV exposure of OPIA/A at 254nm
without isolation. The molar ratio of spiroorthoester and OPIA/A was 1:1.Figure 5. DQF-COSY NMR spectrum of III.
Volume changes during polymerization
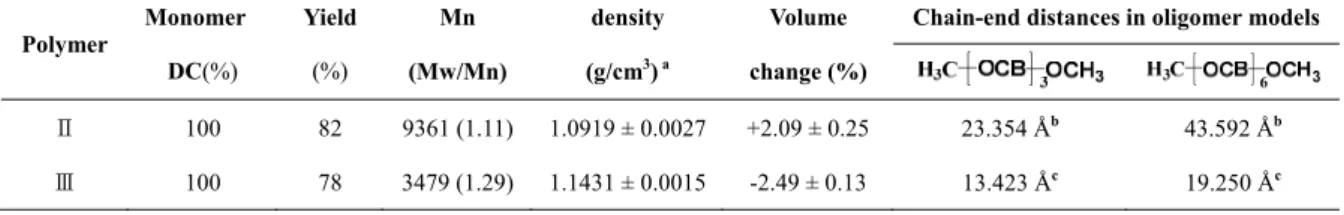
Most spiroorthoester monomers exhibit shrinkage during polymerization.17 In our study of the cationic polymerization of I, however, it was found that the volume of II expands by 2.09%, and the volume of III shrinks by 2.49%, during polymerization (Table 2). Methoxyl-capped oligomer models of II and III were designed, and a series of computational simulation studies was undertaken to further understand the macromolecular structures of II and III, to attempt to explain these experimental results. Sequential calculations using the semi-empirical PM3 and DFT (B3LYP with 6-31G(d) basis set) methods were used to obtain the optimized-geometry configurations of the model oligomers (Figure 6). It was found that the methoxyl-capped six-subunit model oligomer (H3C-[cis-OCB]6-OCH3) of III collapses to a more compact configuration with the methyl chain ends 19.250 Å apart (Figure 6a). In contrast, the corresponding chain ends in the model oligomer of II were found to be 43.592 Å apart (Figure 6b). By comparing the chain-end distances of the two models (Table 2), it is reasonable to deduce that II has a highly extended structure, but III collapses to a more compact and coiled configuration.39,40 The radius of gyration, which is proportional to the chain-end distance,40 of II is much higher than that of III. The volume of the former is thus larger than that of the latter because when I undergoes polymerization initiated by PDAC, the cis substitution at C-2 and C-3 is converted to the trans substitution (Eq. 1 in Scheme 1) during the polymerization.
Table 2. Properties of Polymers II and III.
Polymer Monomer DC(%)
Yield (%)
Mn (Mw/Mn)
density (g/cm3) a
Volume change (%)
Chain-end distances in oligomer models
Ⅱ 100 82 9361 (1.11) 1.0919 ± 0.0027 +2.09 ± 0.25 23.354 Åb 43.592 Åb
Ⅲ 100 78 3479 (1.29) 1.1431 ± 0.0015 -2.49 ± 0.13 13.423 Åc 19.250 Åc
a: The density of I is 1.1147 ± 0.0014 g/cm3; b: trans form; c: cis form.
Figure 6. Methoxyl-capped six-subunit model oligomers of B3LYP/6-31G(d)-derived geometry-optimized
structures of polymers II and III. The computed distance between the oligomer chain ends (methyl–methyl) is shown.Conclusions
I underwent cationic polymerization initiated by the PDAC, which was obtained by exposing an
OPIA/A mixture to UV light at 254 nm. The PDAC was shown to attack the cyclic ether oxygen of I to yield the only intermediate, DOLC, which propagates the reaction regiospecifically to afford II. The stereoregular structure of II was identified by its DQF-COSY NMR spectrum. II exhibited high volume expansion (2.09%) during polymerization; in contrast, III, obtained using SnCl4 as an initiator, demonstrated high volume shrinkage (-2.49%) during polymerization. The volume expansion of II is caused by the conversion of the cis substitution at C-2 and C-3 of I to trans substitution during polymerization. PM3 and B3LYP/6-31G(d) computationally geometry-optimized oligomer models of II and III were designed to explain the experimental result.Financial support for this work by the National Science Council of the Republic of China under Grant No. NSC 97-2221-E-011-026-MY2 is gratefully acknowledged.
12
References
1. Robert, F.; Brady, J. R. J. Macromol. Sci. Rev., Macromol. Chem. Phys. 1992, 32, 135–181.
2. Takata, T.; Endo, T. In Expanding Monomers: Synthesis, Characterization, and Application, Sadhir, K. R., Luck, R. M., Eds.; CRC: Boca Raton, 1992.
3. Kume, M.; Maki, Y.; Ochiai, B.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 7040–7053.
4. Kume, M.; Hirano, A.; Ochiai, B.; Endo, T. J. Polym. Sci., Part A: Polym. Chem.
2006, 44, 3666–3673.
5. Bailey, W. J.; Sun, R. L. J. Am. Chem. Soc., Div. Polym. Chem. Prepr. 1972, 13, 284.
6. Endo, T.; Sudo, A. J. Polym. Sci., Part A: Polym. Chem.
2009, 47, 4847–4858.
7. Endo, T.; Bailey, W. J. J. Polym. Sci., Polym. Lett. Ed. 1980, 18, 25–27.
8. Endo, T.; Okawara, M.; Yamazaki, N.; Bailey, W. J. J. Polym. Sci., Polym. Lett. Ed. 1981, 19, 1283–1286.
9. Pan, C. Y.; Wang, Y.; Bailey, W. J. J. Polym. Sci., Part A: Polym. Chem. 1988, 26, 2737–2747.
10.Trathnigg, B., Hippmann, G., Angew. Makromol. Chem. 1982, 105, 9–14.
11. Chikaoka, S.; Takata, T.; Endo, T., Macromolecules (Washington, DC, U.S.) 1991, 24, 6557–6562.
12. Chikaoka, S.; Takata, T.; Endo, T. Macromolecules (Washington, DC, U.S.) 1992, 25, 625–628.
13. Bolln, C.; Frey, H.; Müihaupt, R. Macromolecules (Washington, DC, U.S.) 1996, 29, 3111–3116.
14. Decker C., Prog. Polym. Sci. 1996, 21, 593–650.
15. Fouassier, J. P. Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications;
Hanser: New York, 1995.
16. Ge, J.; Trujillo, M.; Stansbury J. W. Macromolecules (Washington, DC, U.S.) 2006, 39, 8968–8976.
17. Hsu, Y. G.; Wan, Y. S. J. Polym. Sci., Part A: Polym. Chem.
2009, 47, 3680-3690.
18. Yağci, Y.; Reetz, I. Prog. Polym. Sci. 1998, 23, 1485–1538.
19. Kasapoglu, F.; Yagci, Y. Macromol. Rapid Commun. 2002, 23, 567-570
20. DeVoe, R. J.; Sahyun, M. R. V.; Schmidt, E.; Serpone, N.; Sharma, D.K. Can. J. Chem. 1988, 66, 319–324.
21. Nelson, E. W.; Carter, T. P.; Scranton, A. B. Macromolecules (Washington, DC, U.S.) 1994, 27, 1013–1019.
22. Nelson, E. W.; Carter, T. P.; Scranton, A. B. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1993, 34, 705.
23. Nelson, E. W.; Carter, T. P.; Scranton, A. B. J. Polym. Sci., Part A: Polym. Chem.
1995, 33, 247–256.
24. Trathnigg, B.; Hippmann, G.; Junek, H. Angew. Makromol. Chem. 1982, 105, 1–7.
25. Kamata, K.; Kasai, J.; Yamaguchi, K.; Misuno, N. Org. Lett. 2004, 6, 3577–3580.
26. Silverstein, R. M. Spectrometric Identification of Organic Compounds, 6th Edition; John Wiley & Sons:
New York, 1998; Chap. 4.
27. Chikaoka, S.; Takada, T.; Endo, T. J. Polym. Sci., Part A: Polym. Chem. 1990, 28, 3101–3106.
28. Harris, C. D.; Holder, A. J.; Eick, J. D.; Chappelow, C. C. Cryst. Growth Des. 2003, 3, 239–246.
29. Takata, T.; Sanda, F.; Ariga, T.; Nemoto, H.; Endo, T. Macromol. Rapid Commun. 1997, 18, 461–469.
30. Pohlers, G.; Virdee, S.; Scaiano, J. C.; Sinta, R. Chem. Mater. 1996, 8, 2654–2658.
31. Kura, H.; Fujihara, K.; Kimura, A.; Ohno, T.; Matsumula, M.; Hirata, Y.; Okada, T. J. Polym. Sci., Part B:
Polym. Phys.
2001, 39, 2937–2946.
32. Matyjaszewski, K, J. Polym. Sci., Part A: Polym. Chem. 1984, 22, 29–40.
33. Taranto, A. G.; Carneiro, J. W. de M.; Araujo, M. T. Bioorg. Med. Chem. 2006, 14, 1546–1557.
34. Smith, M. B.; March, J. March’s Advanced Organic Chemistry, 6th Edition; Wiley-Interscience: Hoboken, NJ, 2006; pp. 356–394.
35.Laszlo, P.; Teston. M. J. Am. Chem. Soc. 1990, 112, 8750–8754.
36. Deters, J. F.; McCusker, P. A.; Pilger, Jr., R. C. J. Am. Chem. Soc. 1968, 90, 4583–4585.
37. Yonet, N.; Yagci, Y.; Ochiai, B.; Endo, T. Macromolecules 2003, 36, 9257-9259.
38. Ariga, T.; Takata, T.; Endo, T. Macromolecules 1997, 30, 737-744.
39. Jensen, T. R.; O’Donnell III, J. J.; Marks, T. J. Organometallics 2004, 23, 740–754.
40. Flory, P. J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, 1953; pp. 399–431.
國科會補助專題研究計畫成果報告自評表
請就研究內容與原計畫相符程度、達成預期目標情況、研究成果之學術或應用價 值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)、是否適 合在學術期刊發表或申請專利、主要發現或其他有關價值等,作一綜合評估。
1. 請就研究內容與原計畫相符程度、達成預期目標情況作一綜合評估 達成目標
未達成目標(請說明,以 100 字為限)
□ 實驗失敗
□ 因故實驗中斷 ■ 其他原因
說明:
1.本計畫以改進膨脹性單體起始反應系統達到高分子產品體積膨脹的效果,將原本希望以合 成新單體來達到使產品體積膨脹的策略略做修改。
2.本計畫原擬合成含雙螺環官能基膨脹性單體(BSEM)使之在進行陽離子光聚合反應時,所產 生的聚醚酯(polyether ester)高分子的體積有膨脹的效果。然而,該類膨脹性單體於合成過程 中產生嚴重轉移重組(rearrangement)現象,導致無法順利合成出來;因此,不得不另闢蹊徑。
3.本實驗室發現一陽離子光聚合起始系統,可以使含雙螺環官能基膨脹性單體進行區域特異
性聚合反應(regiospecific polymerization),可更有效的使所合成之高分子的體積達到膨脹的效 果。本研究成果已刊登於Macromolecules 期刊上(Macromolecules, 2010, 43(20), 8430-8435)。
2. 研究成果在學術期刊發表或申請專利等情形:
論文:■已發表 □未發表之文稿 □撰寫中 □無 專利:□已獲得 □申請中 □無
技轉:□已技轉 □洽談中 □無 其他:(以 100 字為限)
3. 請依學術成就、技術創新、社會影響等方面,評估研究成果之學術或應用價 值(簡要敘述成果所代表之意義、價值、影響或進一步發展之可能性)(以 500 字為限)
1.本實驗室發現一新型陽離子光聚合起始系統,可以使含雙螺環官能基膨脹性單體進行區 域特異性聚合反應(regiospecific polymerization),可有效的使所合成之聚醚酯(polyether ester)高分子的體積達到高度膨脹的效果,體積膨脹的原因可經電腦模擬的方式給證實出 來。本研究成果已刊登於 Macromolecules 期刊上(Hsu YG, Wan YS, Lin WY, Hsieh WL,
Macromolecules, 2010, 43(20), 8430-8435)。
2. 本研究成果具學術及實用價值,可簡單及有效的解決單體因聚合而造成其 產品體積收縮的現象,使產品不致於在聚合過程中因體積收縮而有龜裂、
變形及尺寸誤差等情事出現。本方法可應用於光電、牙科綴補、塗裝及精 密射出成型等材料的合成上。