國立臺灣大學醫學院生物化學暨分子生物學研究所 碩士論文
Graduate Institute of Biochemistry and Molecular Biology College of Medicine
National Taiwan University Master Thesis
人類胸腺嘧啶核酸激酶與抑制物 YMU1 之複合體結構 解析
Structural Analysis of Human Thymidylate Kinase in Complex with Inhibitor YMU1
郭韋辰 Wei-Chen Kuo
指導教授:詹迺立 博士 Advisor: Dr. Nei-Li Chan
中華民國 102 年 7 月
July 2013
摘 要
在快速分裂的癌細胞中,DNA 複製的速度顯著高於正常細胞,使得癌細胞對 於DNA 的損傷非常敏感,因此 DNA 修復機制的運作與效率尤為重要。先前研究 指出,DNA 修復的效率與修復過程所需之 dNTPs 的濃度高低密切相關。dNTPs 的 生合成是以核苷酸新合成路徑 (de novo pathway)或是回收路徑 (salvage pathway) 所產生的 NDPs 為原料,再經由 ribonucleotide reductase (RNR)催化之還原反應將 NDP 還原成 dNDP,這個步驟在 dNTPs 的合成扮演關鍵的角色。而大部分的 dNDPs 包含dADP、dGDP、dCDP 和 dUDP 都可以經由 RNR 催化 NDPs 的還原而生成,
然而,只有dTDP 的合成是經由 TMPK (thymidylate kinase)所催化,此酵素以 dTMP 為其受質,利用ATP 做為磷酸根的供給者,藉由對 dTMP 的磷酸化以合成 dTDP,
有鑒於 hTMPK 對於 dTTP 合成不可或缺的重要性,因此,抑制 TMPK 或許可作為 治療癌症的手段之一 。
目前許多化療藥物仍被廣泛的用於癌症治療,如doxorubicin 可以造成 DNA 雙 股斷裂,使癌細胞生長停止或是誘發細胞凋亡。然而這些化療藥物的副作用也不可 忽視,病人可能會出現噁心、嘔吐等症狀,亦可能產生心臟毒性。研究發現,當以 siRNA 技術使癌細胞的 TMPK 表達下降,會增強 doxorubicin 的毒殺效果,因此發 展針對TMPK 的專一性抑制物或許有助於降低 doxorubicin 的 IC50,進而減少不必 要的副作用。這個概念在近期使用TMPK 抑制物 YMU1 進行動物實驗後得到進一 步的支持:YMU1 自身並無顯著的細胞毒性,但與低劑量的 doxorubicin 合併使用 則可以使癌細胞凋亡。因此YMU1 應可做為發展抗癌藥物的先導化合物。
為了瞭解 YMU1 如何與 TMPK 交互作用並抑制其活性,我們著手 TMPK 與 YMU1 複合體的結構解析。首先利用大腸桿菌表現大量的人類 TMPK 蛋白,並且 利用液相層析法純化蛋白,最後再以共結晶的方式培養複合體的晶體,另一方面,
我們同樣以共結晶的方式,取得TMPK 與 dTMP 複合體之晶體,之後或許可以將 晶體浸泡於含有 YMU1 的溶液中,取得 TMPK 與 YMU1 的複合體晶體。此結構
亦可能加深我們對TMPK 催化反應機制的認識。
關鍵詞:癌細胞、DNA 修復、dTTP 合成、人類胸腺嘧啶核酸激酶 (TMPK)、
doxorubicin、YMU1
Abstract:
In fast-proliferating cancer cells, DNA replication usually occurs at a higher frequency compared to normal cells. Therefore, cancer cells are in general more sensitive to DNA-damaging agents and rely more heavily on the DNA repair mechanisms. Previous studies have established that the efficiency of DNA repair, which involves synthesis of new DNA at the damage site, is tightly linked to the cellular concentration of dNTPs.
Several enzymes are known to regulate the level of dNTPs pools by participating in the de novo and salvage pathways. Ribonucleotide reductase (RNR)-mediated reduction reactions play a crucial role in dNTPs biosynthesis by producing dNDPs as precursors.
While the majority of dNDPs, including dADP, dGDP, dCDP and dUDP are generated from NDPs by RNR, the synthesis of dTDP is catalyzed by thymidylate kinase (TMPK).
Using dTMP as the substrate and ATP as the phosphate donor, TMPK produces dTDP for the subsequent biosynthesis of dTTPs. It has been suggested that the inhibition of TMPK may be exploitable in cancer therapy.
The drug doxorubicin, which is widely used in anticancer chemotherapy, is highly effective in killing cancer cells by inducing DNA double-strand break (DSB). However, patients receiving doxorubicin may develop nausea, vomiting and irreversible myocardial toxicity. Because TMPK knockdown has been shown to sensitize cancer cells toward doxorubicin treatment, thus the use of TMPK inhibitor may suppress the undesired side effects by reducing the IC50 of doxorubicin. This concept is supported by the finding that YMU1, a specific inhibitor of human TMPK with no obvious cytotoxicity, may be used in conjunction with low dose of doxorubicin to induce cancer cell-specific apoptosis.
Therefore, it appears that YMU1 is a promising lead compound for drug development.
To understand how YMU1 interacts with and inhibits the activity of TMPK, we have initiated structural analysis on the TMPK-YMU1 complex. First, large amount of human
TMPK was obtained using the Escherichia coli expression system, and liquid chromatography was performed for protein purification. Co-crystallization of human TMPK with YMU1 has been performed using highly purified protein. As an alternative approach, we have crystallized TMPK in complexes with dTMP. These crystals will be exposed to various soaking buffers that contain YMU1 to produce the inhibitor-bound crystals for structural determination.
Key words: cancer cells, DNA repair, dTTP synthesis, thymidylate kinase (TMPK), doxorubicin, YMU1
目 錄
口 試 委 員 會 審 定 書… … … . i
中文摘要...ii
英文摘要...iv
目錄...vi
圖目錄...viii
表目錄...x
一、 前言...1
前言...1
二、 材料與方法...8
2-1 實驗材料... ...8
2-2 實驗方法... ... ...8
2-2-1 hTMPK之小量表現測試方法.... ... ... ...8
2-2-2 hTMPK之純化... ... ... ...9
2-2-2-1 hTMPK之大量表現、破菌與萃取.... ...9
2-2-2-2 GSH resin之純化... ... ...10
2-2-2-3 Thrombin之酵素切割... ... ...10
2-2-2-4 陰離子交換管柱... ... ... 11
2-2-2-5 分子篩膠體過濾管柱... ...11
2-2-3 hTMPK與YMU1之共結晶... ... ...11
2-2-4 hTMPK 晶體與 YMU1 之 crystal soaking.... ... ...14
2-2-5 hTMPK晶體之X-ray繞射... .... ... .... ... .... ...14
2-2-5-1 hTMPK晶體之X-ray之資料收集... ...14
2-2-5-2 hTMPK晶體之結構解析... .. ... ... ...15
三、實驗結果... ... ...16
3-1 hTMPK之表現測試... . ... . ... . ... ...16
3-2 hTMPK之純化... ... ... ... ... ... ... ...16
3-3 hTMPK之結晶... ... ... ... ... ... ...17
3-4 hTMPK結晶之整體結構解析... .. ... ... ... ....19
3-5 hTMPK、dUMP、AMPPNP 共結晶之結構解析...20
3-6 hTMPK、dTMP、AMPPNP 共結晶之結構解析...21
四、討論... ... ... ... ...22
4-1 hTMPK之純化... ... ... ... ...22
4-2 hTMPK之結晶... ...22
4-3 hTMPK 與 dTMP 之結構... ... ... ...23
4-4 hTMPK 結合 dTMP 及 dUMP 之差異.. ... ... ...24
4-5 hTMPK 與 YMU1 之交互作用.. ... ... ...25
五、圖... ... ...26
六、表... ... ... ... ... ... ... ... ... ... ... ... ... ....59
七、參考文獻... ... ... ... ... ... ... ... ... ... ... ...61
八、附錄………..….67
圖 目 錄
圖 1:dNTP pool 的新合成路徑(de novo pathway) ...26
圖 2:dTTP 之合成路徑... ...27
圖 3:不同物種 TMPK 之序列分析... ...28
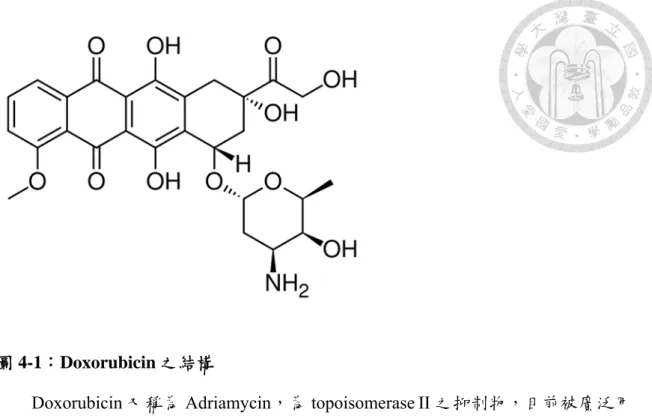
圖 4-1:Doxorubicin 之結構... ...29
圖 4-2:hTMPK 抑制劑 YMU1 之結構... ...29
圖 5:hTMPK 與 YMU1 之模型模擬結構... ...30
圖 6:結晶條件之微調... ...31
圖 7:利用 crystal soaking 製備 hTMPK 與 YMU1 複合體晶體之方法...32
圖 8:GST-hTMPK 融合蛋白之小量表現... ...33
圖 9:hTMPK 之 GSH 純化、thrombin 酵素切割及陰離子管柱純化...34
圖 10:hTMPK 之分子篩膠體過濾管柱之純化... ... ... ... ... ..35
圖 11:hTMPK 之穩定性... ... ... ... ... ... ... ... ... ..36
圖 12:hTMPK 與 YMU1 及 dTMP 條件下結晶出之晶體... ... ... ...37
圖 13:hTMPK、AMPPNP、dTMP 與 MnCl2 之條件所結晶出之蛋白晶體……38
圖 14:hTMPK 與 dTMP 之條件所結晶出之蛋白晶體... ... ... ...41
圖 15:hTMPK、AMPPNP、dUMP 與 MgCl2 之條件所結晶出之蛋白晶體…...42
圖 16:hTMPK、AMPPNP 與 MgCl2 之條件所結晶出之蛋白晶體... ... .43
圖 17:hTMPK、AMPPNP 與 MgCl2 條件的微調………..…44
圖 18:hTMPK、AMPPNP 與 MgCl2 之條件微調長出之晶體………...45
圖19:hTMPK、AMPPNP、dTMP 與 MnCl2 條件之晶體,以 X-ray 繞射實驗檢測 是否為蛋白晶體... ... ... ... ... ... ... ... ... ... ……...46
圖20:TMPK、AMPPNP、dTMP 與 MnCl2 soaking out AMPPNP,16 小時後….47 圖21:Crystal soaking 的實驗並無法完全去除 AMPPNP... ... ... ...48
圖 22:不同條件 hTMPK 之整體蛋白結構... ... ... ... ... ... ...49
圖 23:hTMPK、AMPPNP、dTMP 與 MnCl2 之結構... ... ... ... ...50
圖 24:hTMPK、AMPPNP、dUMP 與 MgCl2 之結構... ... ... ... ...51
圖 25-1:老鼠 TMPK 同樣可以 dUMP 為受質催化反應... ... ... ... ....52
圖 25-2:酵母菌 TMPK 同樣可以 dUMP 為受質催化反應... ... ...52
圖 26:dTMP 中 thymidine 之 C5的甲基與 hTMPK 之間的交互作用... ...53
圖 27:hTMPK 與 dTMP 結構中 P-loop 的結構改變... ... ... ... ...54
圖 28:hTMPK 與 dTMP 之表面結構比較... ... ... ... ... ... ...55
圖 29:hTMPK 與酵母菌 TMPK 之 dTMP 複合體的交互作用比較... ... ..56
圖 30:AMPPNP 與 hTMPK 之交互作用... ... ... ... ... ... ...57
圖 31:hTMPK 與酵母菌 TMPK 中 F105 與 Y102 之差異... ... ... ...58
表 目 錄
表1:hTMPK 表面電性之預測... ... ... ... ... ... ... ... ...59 表2:hTMPK 之受質藥品配製... ... ... ... ... ... ... ... ...59 表3:hTMPK 與 dTMP 複合體、hTMPK 與 dTMP 及 AMPPNP 複合體、hTMPK 與dUMP 及 AMPPNP 複合體之資訊……….…..60
一、 前言
去氧核醣核酸 (DNA) 在自然界中被用來做為遺傳物質,而遺傳訊息則取決於 DNA 鹼基特定的排列組合,DNA 的鹼基有腺嘌呤 (adenine)、鳥糞嘌呤(guanine)、
胞嘧啶 (cytosine) 以及胸腺嘧啶 (thymine)等四種(1、2),這些組成去氧核糖核苷酸 (dNTPs)的原料,在細胞中的合成受到嚴密的調控。dNTP pool 的恆定在細胞中的 重要性已經被報導(3),許多研究結果指出無論在原核或是真核細胞,缺乏特定核 苷酸會造成細胞生長停止甚至死亡,而dNTP pool 的不穩定也會影響 DNA 聚合酶 (DNA polymerase)催化時的正確性,可能造成點突變、模板位移、插入與刪除突變,
也可能會造成DNA 單股或雙股斷裂(2,4),而有些與核苷酸代謝相關的基因缺陷甚 至會造成免疫缺陷的疾病如:adenosine deaminase deficiency 和 purine nucleoside phosphorylase deficiency。因此,在細胞中有許多重要酵素扮演著合成 dNTPs 並且 調控的角色,包含ribonucleotide reductase (RNR)、dCMP 脫胺酶(dCMP deaminase)、
thymidylate synthase (TS)、dCMP 激酶 (dCMP kinase)、thymidylate kinase (TMPK)、
dNDP 激酶 (nucleoside diphosphate kinase)和 dUTP 磷酸水解酶 (dUTPase) (圖 1)。
當細胞將 dNTPs 之前驅物 NDPs 合成完成後,可以經由 RNR 透過還原反應將 核醣(ribose)上位於 C2上的羥基(hydroxyl group)還原為氫原子,合成 dNDPs,再經 由非專一性的dNDP 激酶進行磷酸化(5),合成 dNTPs。然而 RNR 在合成不同種類 之dNDPs 的活性受到複雜的機制所調控(6,7),這對控制 dNTP pool 的恆定性有重 要的影響。RNR 由兩個 α 單元形成的 R1 以及兩個 β 單元形成的 R2 所組成的 α2β2
四聚體(tetramer)。人類細胞中的 RNR 屬於一類含有 iron-tyrosyl radical 的酵素,大 單元R1 之分子量約 90 kDa,小單元 R2 之分子量約 45kDa。在 R1 單元上有兩個
異位調控因子的結合位,其一是利用回饋調控來影響酵素活性的結合位A,可以結
合ATP 以及 dATP,藉由偵測細胞中 ATP / dATP 的比例來調控酵素活性,當此比 例較高時酵素活性上升,比例較低時活性下降,一旦 dATP 結合上 A 位的比例增 加,會促使RNR 形成不具活性的 α6β2的八聚體(octamer) (8,9)。而另一個是受質
專一性結合位S,可以分別與 ATP、dATP、dGTP 及 dTTP 結合,當 ATP 以及 dATP 與S 位結合時,酵素傾向以 UDP 及 CDP 為受質;當與 dGTP 結合時,酵素主要以 ADP 為受質;而當 dTTP 與 S 位結合時,酵素可以與受質 GDP 結合,依此可調控 酵素結合受質的選擇性,維持dNTP pool 的平衡。在 R2 單元上,有主要催化反應 的iron-tyrosyl radical,並且含有受質結合位,受到 R1 單元的異位調控,與不同的 dNDPs 結合並執行催化反應。RNR 除了本身酵素的異位調控外,其表現會也受到 DNA 損傷的誘導與細胞週期的調控,在 S phase 時達到最高,在 G0 phase 時降低 (10)。
然而 dTTP 的合成在哺乳類細胞中與其他 dNTPs 不同,並不是經過 RNR 的催 化。dTTP 主要有兩條合成路徑(圖 2),其一是新合成路徑(de novo pathway),另一 條為回收路徑(salvage pathway)。在新合成路經中,首先被合成的是 dUMP,在細 胞中首先經由dCMP 脫胺酶以及 dUTPase 來合成(11-13),主要是由 dCMP 脫胺酶 所產生(12)。其中 dCMP 脫胺酶催化一不可逆反應將 cytosine 之 C4上的胺基(amino group) 經過脫胺作用變成酮基(ketone group),使 dCMP 轉變為 dUMP,此酵素亦 同樣扮演著調控dNTP pool 的角色,並受到異位回饋機制的調控,能被 dCTP 活化 以及dTTP 的抑制。另一個 dUMP 的來源是從 dUTPase 所催化,當 RNR 及 dNDP 磷酸酶將 dUTP 合成後,細胞內的 dUTPase 會將 dUTP 脫去一個焦磷酸根 (pyrophosphate),水解為 dUMP。值得注意的是,DNA 聚合酶並無法分辨 dUTP 及 dTTP,因此在 DNA 合成時可能會誤將 dUTP 接入 DNA 中,造成基因的不穩定,
因此 dUTPase 去除 dUTP 的活性在哺乳類細胞中是非常有效率的,以至於在細胞 的粗萃取物中幾乎偵測不到 dUTP 的存在。除此之外,當 dCMP 脫胺酶因突變造 成活性缺失之後,細胞只能依靠dUTPase 的作用維持 dUMP 的來源,並且伴隨著 DNA 合成速率下降,細胞生長減緩(14)。dUMP 合成後,可繼續經由 TS 將其轉變 為 dTMP(15,16) , 此 催 化 機 制 為 利 用 亞 甲 基 四 氫 葉 酸 (5,10-methylene- tetrahydrofolate)上的亞甲基(methylene group)作為甲基的提供者,轉移到 dUMP 之
uridine 的 C5上,形成dTMP。另一方面,在回收路徑中,則是經由 TK 所催化(17- 19) ,將細胞內 DNA 代謝分解後的胸腺嘧啶核苷 (thymidine) 作為受質,以 ATP 為磷酸的提供者,在thymidine 中 ribose C5上的hydroxyl 基團進行磷酸化反應形成 dTMP。 無論是由新合成或是回收的路徑所製造出的 dTMP 都必須經由其中的關 鍵酵素-TMPK 的作用(20,21)。TMPK 主要催化的反應是以 ATP 為磷酸提供者,
將dTMP 磷酸化成 dTDP,其反應需要二價金屬離子作為 cofactor。另外,也有報 導指出TMPK 可催化 dUMP 為受質進行反應(22)。如同其他 dNDPs,dTTP 亦會受 到dNDP 磷酸酶催化為 dTTP,作為 DNA 合成的原料。在整個 dTTP 的合成的反 應中,TMPK 位於新合成與回收路徑的交叉口,與其他 dNTPs 的合成比較,除了 dTTP 外,其他的 dNTPs 主要都可藉著 RNR 從 NDPs 還原為 dNDPs 或是經由各自 的dNMP 磷酸酶催化成 dNDP 作為前驅物,然而細胞內 dTTP 的合成受到了更多 酵素的調控,而TMPK 在其中更扮演重要的角色。在人類細胞中的 TMPK (hTMPK) 亦受到細胞周期的調控,譬如出芽酵母菌(S. cerevisiae)的同源基因 CDC8,會在 S phase 的時候被大量活化 (21,23),以提供細胞中 DNA 複製所需之原料。
關於 hTMPK 的結構方面的研究指出,hTMPK 由 212 個胺基酸所組成,為一 個分子量約24 kDa 的蛋白(24),並且以 coil-coil 方式與另一個 hTMPK 單元交互作 用形成雙體(dimer),其結構是由 9 條 α-helices 環繞著 5 條 β-strands 所組成 β-sheet 中心,其中包含三個重要並且高度保留的loop regions (圖 3),第一個是與核甘酸結 合的保守性序列GX1X2X3X4GKS/T 形成 P-loop 的結構,由第 13 至第 17 號胺基酸 組成,利用P-loop 的蛋白醯胺(amide)主鏈上之氫原子與 ATP 上的 α 及 β 磷酸基團 上的氧原子交互作用,與ATP 的結合有關,並且扮演催化反應的重要角色,其中 P-loop 中 X2位置上含羧基(carboxyl group)的胺基酸支鏈可與 dTMP 中 ribose 上的 hydroxyl 基團形成氫鍵(hydrogen bond)交互作用,在酵母菌與人類的 TMPK 中 X2
的位置都是天門冬胺酸(aspartic acid),將酵母菌 TMPK 此位置之 aspartate 進行突 變後發現酵素催化活性下降約200 倍,證明此胺基酸在催化功能上的重要性(24,25)。
而另一個關鍵的loop 包含 DR(Y/H)保留序列(22),在 hTMPK 中,D96 與鎂離子及 ATP 的結合有關。R97 扮演一個類似於夾鉗的角色,可中和 ATP 與 dTMP 兩者磷 酸根間的負電斥力,使磷酸根供給者ATP 之γ磷酸基團和 dTMP 之α磷酸基團得 以相互靠近並進行反應。最後一個loop 稱之為 Lid 的區域,由第 135 至 150 號胺 基酸所組成,此段區域與其他loop 相較其結構較易彎曲改變,從酵母菌 TMPK 與 dTMP 的複合體結構中(26),這段 loop 的電子密度並不明顯,配合 hTMPK 與 ATP 的類似物(AMPPNP)及 dTMP 的結構可以發現(24),R143 可以和 ATP 的 adenine 形 成cation-π 的交互作用,並且和 A180 及 R16 主鏈上的 carbonyl group 上的氧原子 形成氫鍵,涉及這三個loop 區域之間的交互作用,可以穩定 Lid 區域的結構,並 與ATP 的結合和蛋白結構改變有關。除此之外還有一段由第 178 至 188 號胺基酸 組成的 loop 與 ATP 上的 adenine moiety 的結合有關。從先前的研究裡發現,當 hTMPK 結合不同受質時,會產生結構的改變,當結合上 dTMP 及 ADP 時屬於“P- loop open"的構型;當結合上 dTMP 及 AMPPNP 和 dTMP、ADP 與 AlF3時屬於
“P-loop partially closed"的構型;當結合上 dTDP 及 ADP 和兩個受質的抑制物 TP5A 時則屬於“P-loop closed"的構型。除了這些與催化反應及受質結合相關的 loop,hTMPK 上還有許多其他 的 motifs 如 2 個 D-box(RXXL) 以及 1 個 KEN box 這3 個 motifs 與 anaphase-promoting complex/cyclosome (APC/C) 之 E3 ligase 降解 有關,這也說明了hTMPK 在細胞週期受到的調控。
由於 TMPK 所催化的反應位於新合成與回收路徑的交叉口,對於 dTTP 合成 非常重要,因此無論是在發展抗癌、抑菌或抗病毒藥物方面,研究TMPK 與藥物 的蛋白結構並且篩選找出專一性對TMPK 的抑制物,或許可以應用於疾病的治療。
在抗病毒藥物方面,如HIV 的抑制藥物 AZT (azidothymidine),在進入人類細胞後,
首先會藉由 TK 之磷酸化生成 AZTMP,再由 hTMPK 進行第二次磷酸化成為 AZTDP,最後由 dNDP 磷酸酶催化為 AZTTP,此分子在 HIV 的 reverse trancriptase (RT) 作用時,會被 RT 接入 HIV 的 genomic DNA 中,由於正常 ribose 在 C3的
hydoxyl 基團在 AZT 中被置換為 azido 基團,無法與下一個 dNTP 反應,因此造成 DNA 合成中止,使病毒複製受到抑制,達到抗病毒的效果,然而 hTMPK 對於 AZTMP 的催化效率比 dTMP 小約 60 倍,使 hTMPK 成為速率限制步驟的酵素,
因此有許多從結構的觀點出發的研究嘗試探討兩者催化活性的差異,做為藥物改 良 的 基 礎(26-29) 。 而 在 抑 菌 方 面 的 研 究 , AZTMP 能 抑 制 結 核 分 枝 桿 菌 (Mycobacterium tuberculosis)TMPK 的活性,並且有些結構類似 dTMP 的化合物如 5-methyl isodCMP 同樣能抑制其活性(30,31),另一方面也有關於抑制綠膿桿菌 (Pseudomonas aeruginosa)TMPK 之藥物篩選的相關研究被報導(32)。 在抗癌藥物 的發展方面,由於癌細胞之生長與分裂快速,DNA 複製速率較高,對於 dNTPs 的 需求增加,因此早期已有針對抑制dTTP 合成路徑以期抑制癌細胞生長的研究,在 抑制TS 參與的新合成路徑方面,如抑制二氫葉酸還原酶(dihydrofolate reductase)的 methotrexate 及抑制物前驅物 5FU (5-fluorouracil)或 FdUrd (fluorodeoxyuridine),可 以有效達到抑制癌細胞生長的效果(32,33),而在 TK 方面也有研究指出,在配合利 用複製缺陷的反轉錄病毒(retroviruses)或腺病毒(adenoviruses)來做基因療法(gene therapy),將單純皰疹病毒(Herpes simplex virus)之 TK 基因送入人體,並配合 DNA 合成抑制物前驅物 Ganciclovir (GCV),可以有效殺死惡性神經膠質瘤(Malignant Glioma)(34)。藉由抑制 dTTP 合成以干擾癌細胞生長的想法同樣也被運用在 TMPK 抑制物的發展上(35),然而癌細胞常常會產生抗藥性,例如減少藥物於細胞中的含 量(36)或是基因發生突變使抑制物的結合能力下降(37)。由於這些抑制物許多都屬 於核苷酸的類似物或衍生物,因此對於酵素的專一性並不高,甚至會產生許多的副 作用(38)。而近年由台灣陽明大學團隊所篩選而得之 TMPK 抑制物 YMU1,可以 專一性的抑制hTMPK 的酵素活性,使細胞中 dTTP pool 降低至約 60%,其 Ki 值 約0.22μM,酵素之 IC50約為0.6μM,並且不會影響酵母菌 TMPK 以及人類 TK 之 活性。在細胞實驗中發現YMU1 在 10μM 濃度下對 non-tumorgenic 細胞之生長無 顯著影響,證明其為低細胞毒性、具有開發潛力的候選藥物。
最近的研究報告進一步指出以 YMU1 治療癌症的可能性。Doxorubicin (圖 4- 1) 為一個目前廣泛於臨床使用的抗癌藥物,能與第二型拓樸異構酶(Type II Topoisomerase)作用並誘發 DNA 雙股斷裂,促使癌細胞死亡(39)。當以 siRNA 抑 制癌細胞中的hTMPK 表現時,發現在給予低劑量的 doxorubicin 誘導少量細胞 DNA 雙股斷裂的情況下(40-43),即可造成癌細胞的 DNA 無法有效修復,導致其生長停 止甚至出現凋亡的情形,此現象在利用 hTMPK 抑制劑 YMU1 (圖 4-2) 合併低劑 量doxorubicin 亦有相同的效果,動物實驗中亦證實此種療法可以使腫瘤的重量顯 著減少(43,44)。除此之外,當 DNA 出現雙股斷裂時,可以發現 hTMPK 會從細胞 質進到細胞核中,並且從免疫共染色圖譜中發現 hTMPK 會與 DNA 斷裂處以及 RNR 的 R2 單元有共定位(colocalization) 的現象,意味著當細胞在出現 DNA 雙股 斷裂時,hTMPK 會進入到細胞核中,並在 DNA 損傷處聚集(43,45),但詳細的調 控機制目前仍待釐清。目前所提出的假說指出,癌細胞的細胞週期調控通常較差,
因此由 doxorubicin 造成的 DNA 損傷會在未修復完全的情況下細胞即進入到 S phase,而由於 RNR 的 R2 單元在 S phase 時被大量活化,使聚集在 DNA 雙股斷裂 處的RNR 的活性上升,伴隨著 hTMPK 被 YMU1 抑制後失去了催化活性,使 DNA 損傷處之dUTP/dTTP 比例上升(細胞在正常情況下 dUTP/dTTP 比例約在 0.3% - 3%
左右)(46),由於人類的 DNA 聚合酶無法辨認 dTTP 與 dUTP,因此使 dUTP 接入 DNA 的可能性增加。這種錯誤會以 NER (nucleotide excision repair)或是 BER (base excision repair)的方式予以修復,但由於局部之 dUTP / dTTP 的比例較高,因此仍 有可能繼續將錯誤的dUTP 接入 DNA 中,以此不斷做無效的修補,造成基因的不 穩定,進而造成癌細胞生長停止甚至凋亡。
值得一提的是,YMU1 雖然是 hTMPK 的抑制劑,但從細胞實驗的結果看來並 無顯著的細胞毒性(43),並且可以使癌細胞對於 doxorubicin 的敏感度提高,進而降 低藥物的IC50並減輕負副作用,是一個有潛力的癌症用藥。同樣的,抑制TS 達到 增加癌細胞對化療藥物的敏感度也曾被報導過(32),然而與 YMU1 不同的是,TS
抑制劑或是阻斷核酸代謝路徑的藥物,通常帶有基因毒性,並且同樣也會對其他正 常複製的細胞有影響。對分析 hTMPK 與 YMU1 的交互作用,先前研究以結構模 擬預測,YMU1 可能結合於 P-loop 的 ATP 結合位的地方 (圖 5) (43),然而此藥物 與hTMPK 之真實交互作用與抑制機制仍待釐清,因此本篇論文的主要目的就是以 蛋白結構的角度,了解 YMU1 抑制劑如何與 hTMPK 產生交互作用,從解出藥物 與蛋白的結構中,了解哪些胺基酸與YMU1 的結合有關,以提供訊息來改善其水 溶性、穩定度與結合能力,使其抑制效果能有進一步的改善。
二、材料與方法
2-1 實驗材料
本研究中所使用的 GST-hTMPK 融合蛋白表現載體,是由國立陽明大學 張智 芬 教授的實驗室所提供,此載體使用的是 pGEX-2T(附錄 1),其中含有一個穀胱 甘肽硫轉移酶(Glutathione S-transferase , GST)作為融合蛋白,可用來做下一步純化 使用,並利用一段多肽來連接hTMPK 基因,而這段多肽含有一段凝血酶(thrombin) 切位LVPRGS,可在之後的純化步驟將 GST 蛋白切除留下 hTMPK 蛋白。
2-2 實驗方法
2-2-1 hTMPK 之小量表現測試方法
由於剛取得之 GST-hTMPK 表現載體已經被轉移進大腸桿菌 (Escherichia coli) JM109 菌株中,可以直接用來做為表現之測試。此 pGEX-2T 載體含有 ampicillin 抗 性,因此利用含有50 ug/ml 之 LB 培養盤來進行篩選,並挑選出單一菌落接種進 3 ml LB 中,內含適量 ampicillin,在培養箱中以 37 ℃下 16 至 18 小時用 200 r.p.m.
震盪培養後,再取出1 ml 菌液接種進 100 ml 含 ampicillin 之 LB 中,在 37 ℃下 以 200 r.p.m. 培養菌液至 OD600為 0.4 至 0.5 後,利用同樣條件以最終濃度為 0.1 mM 之 isopropyl-thiogalactopyranoside (IPTG)進行表現誘導,分別取出培養 2 小時 (誘導前)做為控制對照組、誘導後 4.5、5.5、6.5、7.5、9.5 小時之菌液做為實驗組,
觀察蛋白表現情形。
控制組與實驗組分別取出 1.5 ml 菌液以 10000 r.p.m. 離心一分鐘後,去除上清 液後加入1 ml 破菌緩衝溶液 (50mM Tris-HCl pH 7.5、500 mM NaCl、1% Tritone X-100、0.5 mM CHAPS、2 mM EDTA、1 mM PMSF 及 1mM DTT)進行回溶,再利 用超音波震盪的方式進行破菌 (intensity 21 %、震盪 5 秒後停止 10 秒,連續 5 次 循環直到完全破菌),全程必須在冰浴下進行以避免蛋白受高熱變性。破完菌的粗 萃取液以14000 r.p.m. 在 4 ℃下離心 30 分鐘,取得樣品上清液 20 μl 與等比例樣
品緩衝液混合,將樣品加熱煮沸 10 分鐘後讓蛋白完全變性,再以 12 %的 SDS- PAGE 進行電泳分析。方法如下所示:
(1) 將與緩衝液混合好的樣品加熱 10 分鐘
(2) 把樣品加入配置好的 SDS-PAGE 凹槽中,每個凹槽加入 20 μl,並加入 5 μl 的 marker
(3) 以 90 伏特電壓進行 30 分鐘的電泳,直到樣品到達聚焦膠體 (stacking gel)與分 離膠體 (separating gel)之間。
(4) 再以 150 伏特電壓進行 50 分鐘電泳直到樣品到達底部
(5) 將聚焦膠體和分離膠體切開,把分離膠體放入 staining buffer 中染色 30 分鐘。
(6) 染色後將膠體放入 destaining buffer 進行退染,直到背景接近透明。
(7) 最後將膠體放入 ddH2O 保存。
2-2-2 hTMPK 之純化
2-2-2-1 hTMPK 之大量表現、破菌與萃取
將含有 pGEX-2T-hTMPK 質體的大腸桿菌 JM 109 從- 80 ℃冰箱取出,將冷凍 菌液稍微解凍後塗在含有適量ampicillin 的 LB 培養盤上,放入培養箱裡以 37℃培 養16 至 18 小時,在從中挑選單一菌落,接種進 100 ml 含適量 ampicillin 的 LB 中 以37 ℃、200 r.p.m. 培養 16 至 18 小時,最後以百分之一的比例,分別加入 10 ml 的菌液接種進1 L 含適量 ampicillin 的 LB 中,以 37℃、200 r.p.m. 培養至 OD600約 為0.4 到 0.5,再以最終濃度為 0.1 mM 的 IPTG 進行誘噵,以 37 ℃、200 r.p.m. 誘 導4.5 小時候收菌。一次純化大約需要 8 到 10 L 的菌液,約略可以得到 40 mg 的 蛋白,視情況而調整。誘導後的菌液以 4750 r.p.m. 在 4 ℃下離心 15 分鐘收集菌 體,將上清液去除乾淨後,加入適量破菌緩衝溶液將菌體均勻回溶,可以保存在- 80 ℃冰箱或是直接進行破菌。
本實驗所用的破菌方式為超音波震盪破菌法 (intensity 30 %、震盪 10 秒後停
止10 秒冷卻,震盪所需時間約 30 到 40 分鐘,直到完全破菌),全程必須在冰浴下 進行以避免蛋白受高熱變性。破菌時可再額外加入最終濃度1 mM PMSF 及 1 mM EDTA 防止蛋白在破菌期間受蛋白酶降解。
完全破菌後的菌液,使用 18000 至 20000 r.p.m. 在 4 ℃下高速離心 2.5 小時,
將殘餘不可溶的固體分離。離心後取上清液進行接下來的純化。
2-2-2-2 GSH 樹脂(resin) 純化
pGEX-2T-hTMPK 質體上上含有 GST 融合蛋白的基因,因此適用來做為第一 步的純化。而本實驗所使用的是glutathione-4B sepharose ( Amersham pharmacia ),
在使用前必須先用 ddH2O 以 10 倍體積清洗兩次,去除保存在 resin 上的 20 %酒 精。沖洗後的resin 取出 5 ml 的體積加入離心後的蛋白粗萃取液,在冰浴下溫和攪 拌2 至 4 小時讓蛋白完全與 resin 結合。之後再將蛋白粗萃取液以 1500 r.p.m. 在 4
℃下離心10 分鐘,將上清液去除留下 resin,加入 10 倍體積以清洗緩衝液 (50 mM Tris-HCl pH 7.5、500 mM NaCl、1% Tritone X-100、2 mM EDTA、30% Glycerol、
1 mM PMSF 及 1 mM DTT)溫和搖晃直到 resin 重新懸浮在緩衝液中,再以 1500 r.p.m. 在 4 ℃下離心 10 分鐘,將 resin 離心沉降,去除上清液,重複三次後,去除 非專一性的蛋白結合。 之後利用 PBS 緩衝液 ( 20 mM potassium phosphate buffer pH 7.4 及 150 mM NaCl ) 沖洗二次後再加入與 resin 等體積的 PBS 緩衝液約 5 ml,
進行接下來的步驟。
2-2-2-3 Thrombin 之酵素切割
保存在 PBS 緩衝液中的 resin 上目前含有 hTMPK 與 GST 的融合蛋白,為了 要去除掉純化所使用的 GST,必須在 PBS 緩衝液的條件下用 thrombin (Sigma- Aldrich, USA ) 切除之間連接的多肽。因此接下來需加入適量的 thrombin (約 1 mg : 10 U,5 ml 膠體微粒約需 250 U)在 4 ℃下進行反應,溫和搖晃 16 至 18 小時直到
大部分的hTMPK 被切割分離到 PBS 緩衝液中,以 1500 r.p.m. 在 4 ℃下離心 10 分 鐘,取出上清液,再利用0.22 μm 的濾膜去除大顆粒的雜質,進行下一步的純化。
2-2-2-4 陰離子交換管柱
為了進一步將蛋白純化以及去除上一步所加入的 thrombin,因此利用離子交 換管柱來達成。將前一步驟所得到的上清液以QA 緩衝液 ( 50 mM Tris-HCl pH 7.5、
1 mM EDTA、1 mM PMSF 及 1 mM DTT )稀釋至 10 倍體積約至 50 ml 降低鹽濃度 以利接下來離子交換管柱之純化。純化所使用的 Q column (GE) 為強的陰離子交 換管柱,再使用前先使用3 倍管柱體積的 QA 緩衝液進行平衡。由於 hTMPK 表面 帶正電(表 1),因此不會與管柱結合,所以收集這些流出未結合的樣品,以 SDS- PAGE 分析挑選純度較高部分,繼續做下一步的純化。而結合在管柱上的雜蛋白以 QB 高鹽緩衝液( 50 mM Tris-HCl pH 7.5、1 M NaCl、1 mM EDTA、1 mM PMSF 及 1 mM DTT )沖提出來一起進行 SDS-PAGE 分析。
2-2-2-5 分子篩膠體過濾管柱
進行膠體過濾法 (Gel filtration) 之前須先縮小體積,為了達到這點,上一步所 純化的蛋白必須先將其濃縮,由於hTMPK 其分子量約為 24 kDa,因此利用 10 kDa cutoff 濃縮離心管 (Amicon Ultra) 以 2500 r.p.m. 在 4℃下離心直到蛋白溶液體積 約為 5 ml 再注入管柱中。本實驗使用的是 Superdex 200 (SD200),使用前先利用 SD200 緩衝液 ( 50 mM Tris-HCl pH 7.5、100 mM KCl、1 mM EDTA 及 1 mM DTT ) 平衡 1.25 倍管柱體積,再將濃縮後的蛋白樣品注入進行層析,純化後的蛋白收集 後,使用SDS-PAGE 進行分析。
2-2-3 hTMPK 與 YMU1 之共結晶
在進行結晶實驗之前,為了能了解需要多少濃度的蛋白較為適合結晶,我們先
利用Pre-Crystallization Test (PCT) kit (Hampton Research, USA)來測試,利用四種不 同試劑(A1、A2、B1、B2)與不同濃度的蛋白溶液以 1μl:1μl 的方式混和滴在表面 經過矽化處理之蓋玻片上,利用真空膠完全密封於裝有200 μl 不同試劑的圓孔中,
隔絕外面空氣完全密封。並在室溫條件下(約 25℃) 觀察 30 分鐘後蛋白沉澱的情 形,之後再放置在4 ℃下隔天再觀察一次,最後根據 PCT kit 所提供的表格來決定 何種濃度適合用來結晶。蛋白濃度是利用Bradford assay 來測定,先將 5 倍 protein assay reagent (Bio-red)用 ddH2O 稀釋至 1 倍,並分裝 1 ml 在微量離心管,之後加 入不同濃度小牛血清蛋白 (bovine serum albumin, BSA) 配製 1 mg / ml、2mg / ml、
4 mg / ml、6 mg / ml、8 mg / ml、10 mg / ml 樣品,利用分光光度計測量 OD575之 吸光值,計算出標準曲線 (R2 > 0.995),之後將 1 μl 蛋白溶液加入稀釋後的試劑,
測出吸光值再利用標準曲線算出蛋白濃度。
為了能得到與其抑制劑 YMU1 的複合體晶體,可由(1)共結晶和(2)crystal soaking 兩種方式取得:
(1 )在共結晶的部分,將藥物加入蛋白溶液中進行結晶實驗,試圖直接取得複合體 晶體。在這邊配置了兩種條件,根據先前研究結構模擬的結果,YMU1 可能結合於 ATP 結合位,因此配置了(I) hTMPK 與 YMU1 (II)hTMPK、YMU1 與 dTMP 這兩 種條件,希望可以得到蛋白與藥物共結晶的晶體。製備用來與其抑制物YMU1 進 行共結晶實驗的蛋白溶液,必須先將YMU1 藥物溶解至 DMSO (dimethyl sulfoxide, Sigma-Aldrich, USA)中,由於 YMU1 溶解度不好,最多只能溶解到 8 mM,在共結 晶時希望能使藥物與蛋白的結合達到飽和,因此加入YMU1 對 hTMPK 之 Ki 值 10 倍的最終濃度,約為0.2 mM,DMSO 最後濃度為 2.5 %,以此溶液進行結晶。
(2)在 crystal soaking 的部分,必須先製造出不含藥物的蛋白結晶後,利用加入藥物 的緩衝液進行擴散的方式,使藥物進入晶體中而取得複合體晶體。另一方面,為了 同步取得不含YMU1 的蛋白晶體,用來做 crystal soaking,配置了六種不同條件的 蛋白溶液如下:
(I) 不加入任何受質 (II) 含 2 mM dTMP (III) 含 2 mM dUMP
(IV) 含 5 mM AMPPNP 與 5 mM MgCl2
(V) 含 5 mM AMPPNP、2 mM dTMP 與 5 mM MnCl2
(VI) 含 5 mM AMPPNP、2 mM dUMP 與 5 mM MgCl2
配置完畢後,將所有條件的蛋白溶液進行大量的結晶條件掃描,而這些篩選多 是利用商業的套裝成品(Hampton Research, USA 和 Sigma-Aldrich, USA)來進行篩 選,利用蒸氣擴散法的原理(Vapor diffusion)配合手動懸吊式 Manual hanging drop) 和自動化坐立式 (Robot sitting drop)的方式使蛋白結晶。本實驗兩種方式都有使用 如下所示:
(1) 手動懸吊式:將特定濃度的蛋白溶液以 1 μl:1 μl 的方式與 1 倍、1 / 2 倍、1 / 3 倍的試劑混合,滴在矽化處理的蓋玻片上,利用真空膠完全密封於裝有 200μl 不同試劑的圓孔中,放置在4 ℃下培養。
(2) 自動化坐立式:一種試劑分為三種條件,
(I) 以 0.2 μl 蛋白溶液加入 0.2 μl 試劑
(II) 以 0.2 μl 蛋白溶液加入 0.2 μl 試劑再加入 0.2 μl 的 ddH2O
(III) 以 0.2 μl 蛋白溶液加入 0.2 μl 試劑再加入 0.4 μl 的 ddH2O,每個孔洞中含有 20 μl 試劑做為沉澱劑,最後利用透明膠帶完全密封,放置於 4 ℃及 22 ℃下培養。
當初步的掃描有得到晶體或是類似晶體的條件時,將其條件進行後續的微調,
主要是改變其pH 值以及沉澱劑濃度,也可以改變其鹽濃度,做不同種類的排列組 合,以篩選到的條件為中心點上下調整pH 值 0.2 的範圍,沉澱劑濃度調整約 2 % 的範圍(圖 6),以此希望可以使晶體產生機率及品質提升,用來做為之後 X-ray 晶 體繞射的實驗。
2-2-4 hTMPK 晶體與 YMU1 之 crystal soaking
從先前的研究指出,將 YMU1 以模型模擬的方式放進 hTMPK 蛋白結構中發 現,YMU1 可能結合的位置是原本與 ATP 結合的區域 (P-loop),而非 dTMP 結合 的地方,因此為了得到只含dTMP 之晶體,除了利用共結晶的方式外,我們利用晶 體浸泡擴散 (crystal soaking)的方式先將 hTMPK 與 AMPPNP、dTMP、MnCl2之複 合體晶體內的AMPPNP 擴散出來,其做法是以該晶體長出條件的緩衝液,提高約 2 到 5 %的沉澱劑濃度並加入約 15 至 20%甘油,防止晶體在擴散時溶解,再額外 加入1 mM 的 EDTA 和 2 mM 的 dTMP,維持 dTMP 的濃度並利用二價離子螯合劑 EDTA 將 Mn2+或Mg2+從晶體中擴散出來,降低AMPPNP 與 hTMPK 的結合,使其 能擴散出來。將晶體撈起浸泡於此緩衝液 1 天,之後再將此緩衝液之 EDTA 置換 成0.2 mM 之 YMU1,將 AMPPNP 已擴散完全的晶體,再用含 YMU1 之緩衝液浸 泡1 天,使 YMU1 擴散進入晶體內,以得到與 YMU1 之複合體晶體,進行結構解 析(圖 7)。
2-2-5 hTMPK 晶體之 X-ray 繞射
2-2-5-1 hTMPK 晶體之 X-ray 之資料收集
從最初步的掃描可以得到許多不同條件的晶體,為了確定這些晶體的品質夠 好,並且符合我們預期的將這些受質或抑制物產生共結晶,因此我們須將這些晶體 做X-ray 繞射的實驗,收集數據進行結構解析。方法如下所示:
(1) 用適當大小的 loop 將晶體從溶液中小心撈起。
(2) 將晶體撈起後,放入保護晶體之抗凍緩衝液(Cryoprotectant),本實驗所使用的 是paraffin oil ( Hampton Research, USA ),將晶體完全覆蓋並去除原本殘留在 loop 上的蛋白溶液。
(3) 將保護好的晶體放入液態氮中急速冷卻,並保存於液態氮中,等待進行 X-ray 繞射實驗。
(4) 將晶體存放在液態氮中保存,並帶到新竹同步輻射中心 (National Synchrotron Radiation Research Center, HsinChu, Taiwan)以光束線 15A1 (hTMPK 與 TMP , hTMPK、AMPPNP 與 dUMP)以及 13C1 (hTMPK、AMPPNP 與 TMP)收集數據,從 液態氮取出後,放置於 X-ray 光束線上進行 X-ray 繞射實驗收集數據,最後利用 HKL2000 軟體算出晶體之晶格排列規律,進行資料彙整。
2-2-5-2 hTMPK 晶體之結構解析
將 HKL2000 所彙整的數據以 PHENIX 軟體以先前研究所解出的 hTMPK、
AMPPNP 與 TMP 之結構做為同源模型 (Protein Data Bank code 1E9C),利用 Auto Molecular Replacement 方式將結構之相位角 (phase) 解出,並利用 Auto Building 步 驟將hTMPK 之蛋白序列輸入算出來的電子密度圖中,再利用 Ligand Fit 步驟,將 受質放進可能存在位置的差異電子密度圖中,之後將所有算出來的數據利用 Coot 軟體手動調整胺基酸之位置,使數據進行refinement,再將 Mg2+或Mn2+以及H2O 放入電子密度圖,放入後,再重複進行幾個步驟的refinement,使 Rwork以及Rfree
值下降,使整體結構更為正確。
三、實驗結果
3-1 hTMPK 之表現測試
從 hTMPK 的小量表現測試中,取出 4.5、5.5、6.5、7.5、9.5 小時所取出的樣 品離進行超音波震盪破菌後,離心取出上清液在每個孔洞注入 5 μg 樣品以 12 % SDS-PAGE 進行分析 (圖 8),可以發現與控制組相比,在 37℃ 下以 0.1 mM IPTG 誘導4.5 到 6.5 小時所產生的 GST-hTMPK 融合蛋白 (約 50 kDa)表現量最多,並且 產量足以用來做接下一步的純化實驗,但隨著時間增長,蛋白表現量慢慢遞減消失,
因此約略在誘導4.5 小時左右收集菌液最佳。
3-2 hTMPK 之純化
將 8 至 10 公升的菌液,經過收集、破菌及離心後的粗萃取液,與 GSH resin 進行結合2 至 4 小時後,從 SDS-PAGE 分析發現 (圖 9-1),經過沖洗緩衝液清洗 三次之後,許多雜蛋白可以被去除,之後置換到PBS 緩衝液條件中與 thrombin 作 用,在4℃下切割 16 至 18 小時後,離心後取出之上清液,可以取得大量的 hTMPK 蛋白 (圖 9-2),但仍有許多其他蛋白參雜其中,在之後的步驟利用 Q column 進行 純化,而由於hTMPK 蛋白存在於 PBS 緩衝液中,為了避免 PBS 中鹽離子的濃度 太高影響純化,因此需將上清液的鹽離子降低至50 mM 以下,而降低鹽離子濃度 可以利用透析的方式、desalting column 或是直接用低鹽的緩衝液進行稀釋,根據 蛋白特性不同可以有不同做法。由於 hTMPK 蛋白相對穩定,因此我們直接使用 QA 緩衝液將 5 ml 含 hTMPK 的 PBS 溶液緩慢稀釋到 50 ml,降低 NaCl 濃度約至 15 mM,此步驟直接稀釋的過程並無顯著蛋白沉澱的情形,因此可以不用透析的方 式,直接稀釋即可。從預測分析得知hTMPK 之等電點(Isoelectric point) 約為 8.73,
當pH 值為 7.0 時 hTMPK 淨電荷約為 3.30 (表 1),因此預期在 Q column 純化時,
hTMPK 的蛋白並不會與管柱結合。而從陰離子管柱的層析圖譜中可以發現 (圖 9- 1),hTMPK 蛋白確實不會與陰離子管柱結合,並從 SDS-PAGE 的分析可以看到 (圖
9-2) ,從 A2 到 A8 流出的片段,在 20 kDa 和 30 kDa 之間有大量且高純度的 hTMPK 蛋白,而從100 % QB 高鹽緩衝液的片段 B4 和 B5 可以看到有少許的 hTMPK 及 其他雜蛋白的流出,因此我們收取A2 至 A8 的片段濃縮蛋白至 5 ml,進行下一步 的分子篩管柱層析。
從分子篩管柱層析圖可以看到有兩個分子量的 A280峰值 (圖 10-1),一個約在 82 ml 的位置,另一個約在 110 ml 的位置,由於 hTMPK 分子量大小約 24 kDa , 因此以SDS-PAGE 分析 21 和 34 之間的片段發現 (圖 10-2),在 20 kDa 和 30 kDa 之間有大量且高純度的hTMPK 蛋白,其分子量大小正確,且無明顯沉澱之情形,
並且計算其總產量約為80 mg 左右。以 2 mg / ml 的濃度保存在 4 ℃保存一個月 後,再次將 hTMPK 重新進行分子篩管柱層析的分析 (圖 11),發現仍保持完好,
其穩定度、純度與產量已足夠我們進行下一步驟的結晶實驗。
3-3 hTMPK 之結晶
將高純度的蛋白溶液濃縮至不同濃度來進行 PCT 的測試,發現在約 15 至 20 mg / ml 的濃度適合用來結晶,因此之後的實驗都是以 20 mg / ml 的濃度來進行。
為了配製不同條件的蛋白溶液,我們試著直接將不同受質從配好的濃縮液 (表 2) 直接加入在濃縮後的蛋白溶液中,發現以這樣的方式會使蛋白嚴重的沉澱 ,因此 我們改變了加入這些受質以及抑制物YMU1 的方法。以受質來說,先將蛋白濃縮 至40 mg / ml 之後,再將與蛋白溶液等量體積之 SD200 緩衝液,內含兩倍受質濃 度 (AMPPNP 為 10 mM、dTMP 為 4 mM、dUMP 為 4 mM、MgCl2為 10 mM、
MnCl2為10 mM)予以混合,使最終濃度可以達到 (AMPPNP 為 5 mM、dTMP 為 2 mM、dUMP 為 2 mM、MgCl2為5 mM、MnCl2為5 mM)依照不同條件分別加入不 同受質,充分混勻後,放置在冰上16 至 18 小時後,使受質與蛋白能完全達到飽和 後,以 14000 r.p.m. 在 4 ℃下離心約 30 分鐘,去除固體雜質,同樣也可以使用 0.22 μm 孔徑之濾膜過濾蛋白溶液,此步驟有效的改善蛋白加入受質時沉澱的現
象。之後再將配置好的溶液,再做一次蛋白定量,以確保蛋白濃度坐落於15 到 20 mg / ml 間的範圍,進行結晶條件的篩選。然而,由於 YMU1 之溶解度太低 (溶於 DMSO 中約 8 mM),為了避免 DMSO 濃度過高而影響蛋白穩定以及結晶,並且又 必須使YMU1 達到飽和,實驗中使用 YMU1 對 hTMPK 的 10 倍 Ki 值 (約為 0.2 mM)之濃度來進行結晶篩選。然而在實驗中 DMSO 濃度太高加入濃縮後的蛋白溶 液會造成hTMPK 沉澱,因此改良後的做法是,先將蛋白以 SD200 緩衝液稀釋至 2 mg / ml 後,再將溶於 DMSO 之 YMU1 加入 2.5% (v / v)使 YMU1 的最終濃度可以 達到0.2 mM,同樣靜置在冰上 16 到 18 小時使其達到飽和後,先用 0.22 μm 孔徑 之濾膜過濾蛋白溶液,之後將此混合液濃縮至15 到 20 mg / ml,再以 14000 r.p.m.
在4 ℃下離心約 30 分鐘後,才開始進行結晶篩選。
在這六種不同條件的蛋白溶液中,有些條件發現有晶體的產生,包含 (1) YMU1 (圖 12)
(2) AMPPNP、dTMP 與 MnCl2 (圖 13)
(3) dTMP (圖 14)
(4) AMPPNP、dUMP 與 MgCl2 (圖 15) (5)AMPPNP、MgCl2 (圖 16)
然而不含受質之 hTMPK 與含 dUMP 之條件並無晶體的產生,因此我們必須進一 步確認這些晶體是屬於hTMPK 的晶體或是鹽的結晶,假如一個結晶條件中有許多 晶體的產生,可以在最初步的確認中利用物理的方式確認,使用針頭將晶體輕微碰 觸,假如晶體異常堅硬,則有很大的可能是鹽類的晶體,或是利用偏光鏡來觀察蛋 白晶體偏光的情形。另外,也可以用小分子染劑對晶體染色來確認,或是直接將晶 體撈起,以Cryoprotectant 進行清洗後,去除殘餘在晶體及 loop 上的蛋白溶液,之 後再溶解於SD200 緩衝液中,以 SDS-PAGE 分析確認,然而我們也會同步將這些 晶體撈起並且利用X-ray 進行繞射實驗 (圖 19),分析此晶體是否為蛋白之晶體。
並將這些確定為蛋白晶體的條件進行微調,以提升晶體的品質,使解析度提升。其
中含有 AMPPNP 與鎂離子之 hTMPK 樣品的結晶條件經微調後可得蛋白晶體(圖 17,18),然而其繞射解析度偏低、有待繼續調整。而其他晶體經過這些確認與分析 後,含有YMU1 的晶體以及 YMU1、dTMP 的晶體,經過 X-ray 繞射實驗測試,
大部分的晶體,其繞射點較少且強度高,且在低解析度的位置並無繞射點存在,大 多位於高解析度的區間,符合鹽類晶體的特徵,分析後大部分為鹽的結晶,然而也 有一些類似晶體的物質並無繞射點,並且經過微調後仍無法產生新的晶體,所以目
前仍不確定是否為蛋白結晶。目前已確定可產生 hTMPK 晶體的樣品組成包含
(1)AMPPNP、dTMP 與 MnCl2 (2) AMPPNP、dUMP 與 MgCl2 (3) dTMP (4) AMPPNP 與MgCl2之條件,另一方面,將(2)條件的晶體經過 crystal soaking 的過程,試圖去 除晶體中的AMPPNP,並以結構解析來確認其 AMPPNP 的結合狀態,然而經過了 1 天的 crystal soaking out 的實驗,發現晶體出現明顯裂痕,並且碎成兩個部分(圖 20),我們之後將比較大的片段拿去做 X-ray 繞射實驗做進一步的分析,然而將數 據收回來並算出的電子密度圖,從fo-fc 的 difference map 中看到(圖 21),儘管 Mn2+
的電子密度似乎不存在了,但AMPPNP 仍與蛋白緊密結合。
3-4 hTMPK 結晶之整體結構解析
將 hTMPK 與不同受質結合的晶體帶到新竹同步輻射中心進行 X-ray 繞射實驗 後,有4 個條件有繞射點,其解析度分別為
(I) AMPPNP、dTMP 與 MnCl2 -1.83 Å (II) AMPPNP、dUMP 與 MgCl2 -1.96 Å (III) dTMP -1.966 Å
(IV) AMPPNP 與 MgCl2 - 約 4.0 Å
從HKL2000 軟體將數據彙整後,找到(I) 、(II) 、(III)條件之晶格排列 (space group) 為
P4
32
12,與先前研究解出之晶格排列一致 (24),然而(IV)條件的之晶體細小並且
分散,其解析度約在4.0 Å 左右,但是目前尚無法確任晶格參數,因此無法進行後續的結構解析,之後一方面利用微調的方式試圖找到更好的結晶條件,同時也利用 這些小晶體做為晶種,試圖提高晶體體積,改善晶體品質,提昇其解析度再做分析。
而另外三組數據其解析度良好,以 PHENIX 軟體進行結構解析可以分別得到以上 三個結構 (圖 22)。從這三個的整體結構可以看到,hTMPK 由 9 條 α-helices 環繞 著5 條 β-strands 所形成的 β sheet 中心,整體看來為一球蛋白結構,然而在我們的 結構中hTMPK 並不是以 dimer 存在,而是以 monomer 存在,從先前得。從 AMPPNP 與 dTMP 以及 AMPPNP 與 dUMP 的結構可以看到 ATP 結合位的保守序列 GX1X2X3X4GKS/T 所形成的 P-loop (G13 到 S20)與 AMPPNP 結合,而 dTMP 及 dUMP 則位於 P-loop、DR motif (D95 到 Y97)與 Lid region (F135 到 F151)所形成的 結合空間中,與先前研究所解出之結構大致相同(圖 23) (24)。
3-5 hTMPK、dUMP、AMPPNP 共結晶之結構解析
然而在與 dUMP 和 AMPPNP 的複合體結構中,相較於 hTMPK、dTMP 與 AMPPNP 之複合以結構,Lid region 有比較不穩定的情形,其電子密度比較不清楚,
但總體來說是差異不大的,然而我們卻意外在P-loop 的結構發現一個差別(圖 24),
在AMPPNP 與 dUMP 的結構中 P-loop 的 D15 可以與 dTMP 的 C3的hydroxyl 基團 形成氫鍵,然而在AMPPNP 與 dTMP 的結構中 P-loop 的 D15 離得較遠,相差約 1.9Å 左右,但是其改變的原因仍有待探討。另一方面,與 dUMP 的結構可以提供 hTMPK 在受質選擇性上額外的訊息,根據先前的研究指出在小鼠與酵母菌之 TMPK 不只可以催化 dTMP 形成 dTDP (圖 25),同樣也對 dUMP 有催化的活性。
而dUMP 及 dTMP 之差別只在於 pyrimidine 環上的 C5位置上的差別,在dTMP 上 為甲基;dUMP 上為氫原子。因此這個甲基或許扮演著 hTMPK 對於受質選擇性的 關鍵。從兩者結構中的比較可以看到(圖 26),dTMP 上的甲基能與 F42、P43 以及 S100 的支鏈產生 Van der Waals 作用力,然而在 dUMP 上缺乏此甲基,因此或許可 以解釋為何hTMPK 對兩受質的選擇性不同,造成催化反應上的差異。
3-6 hTMPK、dTMP、AMPPNP 共結晶之結構解析
另一個值得注意的是,hTMPK 與 dTMP 之結構第一次被解出,發現在其 P- loop 的位置從原本 loop 的結構轉換成 1 號α-helix 的延伸,並將原本與 ATP 結合 的位置完全佔據(圖 27,28),而 dTMP 的磷酸基團所旋轉的方向也與含 AMPPNP 之 結構不同,這個結構與先前研究之酵母菌TMPK 與 dTMP 之結構 (PDB code 1TMK) 進行疊合(superposition)相比發現 (圖 29),在酵母菌 TMPK 結構中,P-loop 仍維持 原本loop 的結構,其 R15 利用 guanyl 基團的一端與 Lid region 上的 E154 形成離 子鍵,而D14 可與 Y102 以及 dTMP 核糖 C3上的hydroxy 基團形成氫鍵。 然而在 人類的結構中可以看到,其P-loop 上的 R16 一端以 guanyl 基團與 dTMP 之磷酸基 團形成離子鍵,並同時與Lid region 上的 E152 也形成離子鍵,而 D15 則是朝向與 dTMP 反方向的位置,並無明顯的交互作用,這些變化將整個 P-loop 扭轉向 dTMP 的方向,形成α-helix 的延伸。而與這次實驗所解出之 hTMPK、AMPPNP 與 dTMP 之結構分析可以發現 (圖 27),當含有 AMMPNP 存在時 P-loop 主鏈上的 amide 基 團可以與AMPPNP 的 α 及 β 磷酸根交互作用,使 AMPPNP 可以與 P-loop 結合,
然而不含AMPPNP 存在時,其 P-loop 部分形成 1 號 α-helix 的延伸,這個延伸出 來的部分與 AMPPNP 之分子有重疊的情形,將整個結合位置佔據。在 Lid region 的差異上,AMPPNP 存在時,Lid region 可以有需多交互作用來穩定 loop 結構,其 中可以 R16 可以與 Lid region 之 A140 和 R143 主鏈上的 carbonyl 基團形成氫鍵 (圖 30),Lid region 上的 R143 可以與 AMPPNP 的 adenine 形成 cation-π 交互作用。
然而只與dTMP 結合的晶體結構中發現,失去了 R143 與 AMPPNP 上 adenine 的 cation-π 作用力,並且 R16 轉而向 dTMP 交互作用,使得 Lid region 結構上變得非 常不穩定,整段電子密度圖非常破碎,無法正確地將胺基酸放進電子密度圖中。
四、討論
4-1 hTMPK 之純化
hTMPK 在純化上並無明顯問題存在,並且純化後的蛋白其穩定性也較高,然 而在分子篩管柱層析圖譜上發現,hTMPK 並不是如先前研究報導的以 dimer 的形 式存在,這或許和SD200 緩衝液的條件可能相關。另一方面,從分子篩管柱層析 圖譜上可以發現有一個較小的波峰在110 ml 處流出,這個波峰目前為止不清楚是 什麼物質,從 SDS-PAGE 上最小的 marker (14.4 kDa)以下仍無法看到有蛋白的存 在,而這個波峰所流出的物質是什麼,目前有兩種假設,一種是分子量更小的多肽,
可能是原本存在於細菌中本身的多肽,或是由蛋白酶分解而來;另一個可能是殘留 在hTMPK 上的核苷酸,由於核苷酸本身也有 A280的吸光值,在先前研究所解出的 結構中(24),常常因為一些核苷酸的殘留,發現單一晶體中能有不同的構型變化,
結合受質的比例也會有所不同,這些殘餘在蛋白內的核苷酸有機會能影響晶體的 結構,可能會干擾晶體的排列,因此之後我們會進一步去確認這個波峰的物質,希 望可以盡量使蛋白純度更高,排除任何干擾結晶的可能性。
4-2 hTMPK 之結晶
雖然我們仍然未取得 YMU1 與 hTMPK 之晶體,但已經成功找到了能結晶出 不含AMPPNP 之條件,並且將其結構解出,之後可以利用微調的方式,試圖將晶 體大量製造且提升其品質,並利用crystal soakong 的方式取得與 YMU1 之晶體,
然而當我們將晶體放入含YMU1 的 cryoprotectant 中,晶體都出現融化消失的情形,
目前仍未找到一個適合的cryoprotectant 可以使 YMU1 擴散進去,還要進一步調整 條件,之後仍同步試著找尋直接利用共結晶的方式來取得YMU1 的晶體,以此來 證明crystal soaking 的正確性,而非人工改變的結果。而得到與 YMU1 之複合體晶 體,提供我們之後進一步了解其抑制之機制。另一方面,我們結晶出了hTMPK 與 dTMP 的晶體以及 hTMPK 與 AMPPNP 的晶體,這兩種晶體或許可以在 hTMPK 酵
素催化的反應機制上,增加一些訊息,關乎於hTMPK 對這兩種受質是否有結合的 先後順序,或是有異位調控的可能性,然而hTMPK 與 AMPPNP 的晶體目前解析 度不佳,且找不到其晶體排列,因此無法將結構解出,目前已進行微調以及將先前 較小的晶體做seeding 的實驗,試圖可以取的品質較好的晶體供我們做下一步的實 驗,將hTMPK 催化機制的拼圖在補上一塊。
4-3 hTMPK 與 dTMP 之結構
從結構中我們能清楚看到在 hTMPK 與 dTMP 的條件下,P-loop 的構型改變,
從 loop 的構造轉變成一個 α-helix 的延伸,這樣的改變第一次在所有的 TMPK 中 發現,無論是在酵母菌或是肺結核分支桿菌的TMPK 與 dTMP 結構中,這個保留 性的P-loop 結構都並未發生顯著的改變。而為什麼只在 hTMPK 發現這樣的改變,
可能的原因從人類與酵母菌的TMPK 與 dTMP 進行比較可以發現 (圖 31),雖然在 dTMP 結合的 P-loop 及 DR(Y) motif 結構上,其保留性是非常高的,在人類與酵母 菌中並無顯著差異,但發現在P-loop 上的 aspartate 的交互作用有一個關鍵性的不 同,在酵母菌中,D14 可以與 Y102 的 hydroxyl 基團形成氫鍵 (2.8 Å),並且還可 以與dTMP 上 ribose 的 C3之hydroxyl 基團形成氫鍵 (3.2 Å , 3.3 Å),但是在 hTMPK 中取而代之的F105 而非 Tyrosine,缺乏了 hydroxyl 基團可以和 D15 形成氫鍵,使 D15 並無顯著傾向 dTMP 的位置,使 R16 的 guanyl 基團有機會與 dTMP 之磷酸基 團形成穩定離子鍵(2.8 Å , 2.8Å),並且改變了 dTMP 磷酸根的角度 (約 16.8 - 18.1 度),使磷酸根遠離 P-loop 的位置,而這樣的改變或許說明了為何在 hTMPK 中 P- loop 的結構改變,而這樣的改變或許會改變 hTMPK 在與不同受質(dTMP 或是 ATP) 結合時的順序,甚至是改變與另一個受質的結合的交互作用,然而先前也有研究發 現當 hTMPK 中 F105Y 突變會造成其催化活性會下降,約為正常 hTMPK 活性的 23%,但其機制還不是很清楚。從先前的研究探討 hTMPK 的催化機制,認為是屬 於random bi-bi 的方式進行催化反應(23),然而由於缺乏 hTMPK 不含受質以及只
結合單一受質的蛋白結構,因此無法對於其受質結合先後順序能有更多的了解,但 在我們得到只含dTMP 的結構中可以發現,原本結合 ATP 或 ADP 的位置被 α-helix 的延伸所佔據,因此似乎先結合上dTMP 會干擾與 ATP 的結合,然而在正常生理 條件下,細胞中都是含有ADP 和 ATP 的,因此這種構型在細胞中是否會出現仍是 未知數,或許在細胞周期都某個期間,其ATP 的含量較低,有機會使α-helix 延伸 的這種結構的出現。另外,雖然目前已取得只含AMPPNP 的晶體,但與不含受質 的結構,還未解出。因此這樣的結構在酵素與受質結合的步驟上仍然是未知的。另 一方面,從先前的研究指出,hTMPK 在催化的活性中心比起 AMPK 及 UMPK,
少了兩個催化的arginine,而 arginine 在這邊的角色是中和兩個受質磷酸基團的負 電性,並且穩定中間產物的形成而降低活化能,在hTMPK 中,只有 R16、K19、
R45 及 R97 在這邊扮演中和負電性催化反應的角色,並且再分離純化出來的 hTMPK 中發現其催化的活性較低 (turnover number 約 0.7 sec-1,酵母菌TMPK 為 35 sec-1),因此被懷疑或許有其他轉譯後修飾或其他調控能影響催化反應。
4-4 hTMPK 結合 dTMP 及 dUMP 之差異
在細胞中合成 dTTP 的過程中,RNR 將 UDP 還原為 dUDP 再經過非專一性的 dNDP kinase 進行磷酸化為 dUTP,再由 dUTPase 催化為 dUMP,或是經由 dCMP deaminase 催化 dCMP 為 dUMP,最後經由 TS 合成為 dTMP 成為 hTMPK 之受質,
然而其中間產物dUMP 與 dTMP 之結構非常類似,而 hTMPK 是如何去選擇受質,
可以從我們的結構中得到解釋(圖 26),可以看到 dTMP 上多出來的甲基可以與旁 邊的F42、P43 及 S100 形成疏水性作用力,然而 dUMP 卻無甲基與旁邊的胺基酸 支鏈形成交互作用,使的結合的能力低於dTMP 約 10 倍左右,從先前的研究也可 以輔助證明 (圖 25-1、25-2),然而 dUMP 是否也可以使 hTMPK 產生如 dTMP 結 合時的P-loop 構型改變,目前仍不清楚,我們也同步對於 hTMPK 與 dUMP 的條 件做結晶條件的篩選。
4-5 hTMPK 與 YMU1 之交互作用
由於先前研究模擬的結果認為,YMU1 可能結合的方式是佔據 ATP 結合的 P- loop 的位置(48),然而真正的情況仍然是未知的。然而,從得到的三種結構可以發 現其晶格排列相同,為
P4
32
12,這意味著利用 crystal soaking 的方式應該是可行的,
但是當試圖將 AMPPNP 擴散出來時,可以發現晶體有明顯的裂痕產生 (圖 20),
經過X-ray 繞射實驗後,雖然其解析度依舊還有 1.8 Å 左右,但是從結構中的 fo - fc 的電子密度圖中可以發現仍有明顯的 AMPPNP 的電子密度(圖 21),因此之後就 直接用只含dTMP 的晶體進行 crystal soaking,但由於 P-loop 結構的改變,實際上 YMU1 能否成功擴散進入晶體內,仍然未知,又或者是 YMU1 的抑制機制是結合 或穩定P-loop 轉變為 α-helix 延伸的結構,等待進一步的實驗證明,而目前所知道 的是,YMU1 不會抑制酵母菌 TMPK 的活性,並且在酵素活性實驗上發現,當 hTMPK 先與 dTMP 及 YMU1 混和後再加入 ATP 進行反應,可以使酵素活性被抑 制(IC50 = 06μM),然而 hTMPK 先與 ATP 及 YMU1 混和後再加入 dTMP 催化反應 時,YMU1 並無法抑制反應的催化,因此 P-loop 轉變為 α-helix 延伸的結構對於 YMU1 的抑制或許是重要的。另一方面,先前研究發現 hTMPK 在 DNA 雙股斷裂 進行修補時,hTMPK 會被帶進細胞核中,並且從免疫共染色的圖中可以看到與 RNR 的 R2 單元有重疊之情形,RNR 已知會在 DNA 雙股斷裂時利用 R1 單元的 C 端 與 在 修 補 區 域 之 Tip60 蛋 白 複 合 體 有 交 互 作 用 。 Tip60 為 一 histone acetyltransferase,和 DNA 的修補有關,而 RNR 則與在 DNA 斷裂處製造修補所需 的 dNTPs 有關,然而目前仍不清楚 hTMPK 會與哪個蛋白產生交互作用,或是如 何被帶到DNA 雙股修補區域製造 DNA 修復所需的原料。因此我們之後也會進一 步純化GST-hTMPK 的融合蛋白,並且與加入 0.1μM doxorubicin 之 HEK293T 細 胞粗萃取液混合進行pull down 的實驗,試圖尋找能與 hTMPK 進行交互作用之蛋 白,了解整個DNA 修補的機制。
五、圖
(Reichard 1988)
圖 1:dNTP pool 的新合成路徑(de novo pathway)
從圖片中可以看到關於 dNTP pool 新合成路徑的酵素,其中 dTTP 合成的前驅 物dUMP 需要經過 dCMP deaminase 以及 dUTPase 來催化,dUMP 再經由 TS 催化 成dTMP,之後由 hTMPK 催化為 dTDP,最後由(d)NDP kinase 催化成 dTTP,以供 給DNA 的合成。
(Rampazzo, Ferraro et al. 2004)
圖 2:dTTP 之合成路徑
從圖中可以看到細胞中 dTTP 的合成,經由 TS 的 de novo pathway 以及 TK1 的 salvage pathway 合成的 dTMP 都必須經由 TMPK 的磷酸化,最後才能生合成 dTTP。
TS
TMPKdNDP kinase
圖 3:不同物種 TMPK 之序列分析
從序列分析比較人類、小鼠及酵母菌之 hTMPK,其催化相關的三個 loop 序列 中可以發現P-loop 與 DR motif 有較高的保留性,而 LID region 保留性較低,根據 物種由不同胺基酸組成。
圖 4-1:Doxorubicin 之結構
Doxorubicin 又稱為 Adriamycin,為 topoisomerase II 之抑制物,目前被廣泛用 於癌症的治療上,但其副作用可能會造成噁心嘔吐及不可逆之心肌損傷,因此癌症 病人終其一生能所使用的劑量有限。
圖 4-2:hTMPK 抑制劑 YMU1 之結構
YMU1 為 hTMPK 之專一性抑制物,經實驗證實無顯著細胞毒性,並且能增加 癌細胞對於doxorubicin 的敏感度。
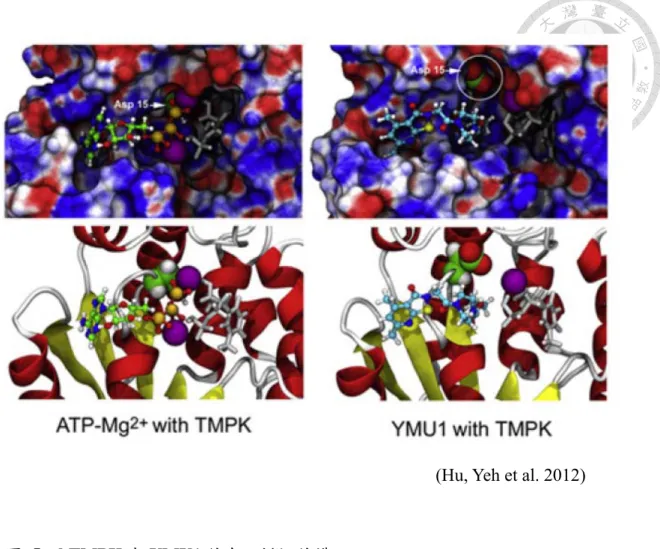
(Hu, Yeh et al. 2012)
圖 5:hTMPK 與 YMU1 結合之模擬結構
結構模擬的結果指出,原本與 ATP 結合的 P-loop 亦可能參與 YMU1 與 hTMPK 的交互作用,因此推論YMU1 可能會影響到 D15 與 Mg2+在催化上作用,達到抑制 hTMPK 的效果。
圖 6:結晶條件之微調
從初步掃描到的結晶條件中,些微調整其沉澱劑濃度、pH 值或是鹽離子濃度,
沉澱劑每次調整約上下2 %左右,而 pH 值約上下調整 0.2 左右,鹽濃度則看條件 而定,找到適合長晶體的條件後,可以改變其中心點位置,進行進一步的微調,直 到可以得到品質夠好且大量的晶體提供接續的實驗使用。
圖 7:利用 crystal soaking 製備 hTMPK 與 YMU1 複合體晶體之方法
我們可以利用crystal soaking 的方法來解決使用共結晶無法得到之 hTMPK 與 YMU1 之晶體,利用擴散的原理,在不劇烈改變晶格排列的前提下,維持晶體的完 整,將目標分子擴散進晶體的方式取得,在我們的實驗中,在取得 AMPPNP 與 dTMP 之晶體後,先將晶體浸泡在不含 AMPPNP 及 1 mM EDTA 的 cryoprotectant 中,將Mn2+及AMPPNP 給擴散出來,再放入含有 YMU1 及 Mn2+之cryoprotectant 使YMU1 能夠擴散進蛋白中,再將此晶體保存於液態氮中,做進一步的 X-ray 繞 射實驗。
圖 8:GST-hTMPK 融合蛋白之小量表現
根據 SDS-PAGE 的分析,M 代表 marker、C 為 control、4.5 至 9.5 表示誘導的 時間,可以發現在4.5 到 6.5 小時 GST-hTMPK 的蛋白表現量最多,從 7.5 小時後 慢慢被降解,因此之後大量表現就以誘導4.5 小時後收菌。
M C 4.5 5.5 6.5 7.5 9.5
97 kDa
66 kDa
45 kDa
30 kDa
20 kDa
14.4 kDa
GST-hTMPK 約 50 kDa
圖 9-1
圖 9-2
圖 9:hTMPK 之 GSH 純化、thrombin 酵素切割及陰離子管柱純化
從圖 10-1, 10-2 中看到,TCL 表示 total cell lysate、W1 至 W3 為 GSH resin wash 的次數、TD 為 thrombin digestion 後的上清液、A2 至 A8 為 Q column 的 flow through、B4 和 B5 是高鹽緩衝液沖提的片段。整個純化流程中,hTMPK 不與 Q column 結合,大都在 A2 到 A8 流出,並去除大部分的雜蛋白,有效的純化出高品 質的hTMPK 蛋白,因此我們就收取 A2 至 A8 來進行分子篩管柱的分析。
Q column 20120726 TMPK002:10_UV1_280nm Q column 20120726 TMPK002:10_Conc Q column 20120726 TMPK002:10_Fractions
0 500 1000 1500 2000 mAU
0 20 40 60 80
%B
0 50 100 150 ml
A1 A2 A3 A4 A5 A6 A7 A8 A9 A10 A11 A12 B2 B3 B4 B5 B6 B7
圖 10-1
圖 10-2
圖 10:hTMPK 之分子篩膠體過濾管柱之純化
從圖 11-1 的純化圖中看到,約 84 ml 的位置可以看到一個 A280的峰值,推測其 分子量約26 kDa,與 hTMPK 24 kDa 相近,判斷 hTMPK 應該是以 monomer 形式 存在,並收取片段21 至 34 之間的樣品進行 SDS-PAGE 分析,從圖 11-2 可以看到 在20 kDa 與 30 kDa 之間的位置有大量且純度高的 hTMPK 蛋白足以讓我們接下去 進行結晶條件篩選的實驗。
SD200 TMPK 20120726:10_UV1_280nm SD200 TMPK 20120726:10_Fractions
0 500 1000 1500 2000 mAU
60.0 80.0 100.0 120.0 ml
123 4 56 7 8 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39 41 44 46 48 50 52 54 56 58 60 62 64 66 Waste
97 kd 66 kd 45 kd 30 kd 20 kd
14.4 kd
Fraction 21‐34
圖 11:hTMPK 之穩定性
從圖中可以發現,當 hTMPK 以 2 mg / ml 保存在 4 度下保存 26 天後,再將蛋 白濃縮通過分子篩膠體管柱,可以發現 hTMPK 依舊保持 monomer 的形式,並無 沉澱之情形,這說明在目前的條件中hTMPK 是可以穩定存在一段時間的。
SD200 hTMPK 20120821(after 26 days)001:10_UV1_280nm SD200 hTMPK 20120821(after 26 days)001:10_Fractions
0 500 1000 1500 2000 mAU
40.0 50.0 60.0 70.0 80.0 90.0 100.0 110.0 ml
5 6 7 8 9 101112131415161718192021222324252627 Waste
圖 12-1 Membfec 24 圖 12-5 Index 81
圖 12-2 Index 89 圖 12-6 Index 68
圖 12-3 CSI 6 圖 12-7 PEG / Ion2 H12
圖 12-4 Salt Rx E7 圖 12-8 Salt Rx C6
圖 12:hTMPK 與 YMU1 及 dTMP 條件下結晶出之晶體
這些晶體經過測試,大部分為鹽的晶體,只有圖 13-3 及圖 13-4,經過 X-ray 繞射實驗發現沒有繞射點,不確定是否為蛋白晶體。
圖 13-1 3D structure E6 圖 13-5 Index F8
圖 13-2 Index D10
圖 13-6 Index F9
圖 13-3 PEG / Ion1 H6
圖 13-7 Index G1
圖 13-4 Index F7 圖 13-8 Index H1