國立交通大學
材料科學與工程學系
碩 士 論 文
無電鍍製備鈀系列多元合金薄膜於氧化鋁基材
應用於氫氣分離
Electroless Deposition of Palladium-based
Alloy Membrane on an Alumina Support
for Hydrogen Separation
研 究 生:李孟翰
指導教授:林 鵬 教授
吳樸偉 教授
無電鍍製備鈀系列多元合金薄膜於氧化鋁基材
應用於氫氣分離
Electroless Deposition of Palladium-based
Alloy Membrane on an Alumina Support
for Hydrogen Separation
研 究 生:李孟翰 Student: Meng-Han Li
指導教授:林 鵬 教授 Advisor: Prof. Pang Lin
吳樸偉 教授 Advisor: Prof. Pu-Wei Wu
國 立 交 通 大 學
材料科學與工程學系
碩 士 論 文
A Thesis
Submitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University in Partial Fulfillment of the Requirements
for the Degree of Master in
Materials Science and Engineering
July 2012
Hsinchu, Taiwan, Republic of China
i
無電鍍製備鈀系列多元合金薄膜於氧化鋁基材
應用於氫氣分離
研究生:李孟翰 指導教授:林 鵬 教授
吳樸偉 教授
國立交通大學
材料科學與工程學系 碩士班
摘要
本研究主要是利用無電鍍法沉積鈀合金薄膜於多孔氧化鋁管上,並在高溫環 境下作氫氣和二氧化碳分離。陶瓷封裝部分,由熱重分析儀分析在 600°C 去除有 機黏合物,並由 SEM 觀察在 1150°C 熱處理後,表面有燒結之現象,最後 GC 測 試下沒有發現二氧化碳漏氣,證實本封裝材料及參數可有效作用。 鈀合金薄膜部份選用鈀銀銅鎳多元合金薄膜作透氫材料,在無電鍍於管形基 材前,先無電鍍於緻密氧化鋁片上,求得各金屬之最佳鍍膜參數,並分析各成份 之金屬合金膜。各成分金屬膜厚度都可控制在 2 μm 左右。600°C 退火過後除了 鈀銀銅鎳合金膜出現雜相外,推測鎳銅有二相分離之結果,其而鈀銀、鈀銀銅之 金屬膜為均一。退火後膜表面形貌沒有出現破損,但是鈀銀銅、鈀銀銅鎳合金膜 表面粗糙度上升,推測主要是銅無電鍍時均勻性成核之結果。各合金膜之成份分 析都能約略控制在預期之組成,且可以使用秤重減量法來粗估膜沉積之量。 管形基材部分採用先封裝後鍍膜方式操作,目前以成功製備鈀銀銅鎳多元合 金膜於多孔氧化鋁管上,接著將利用氣相層析儀作氣體分離測試。ii
Electroless Deposition of Palladium-based
Alloy Membrane on an Alumina Support
for Hydrogen Separation
Student: Meng-Han Li Advisor: Prof. Pang Lin
Prof. Pu-Wei Wu
Department of Materials Science and Engineering
National Chiao Tung University
Abstract
In this work, we attempt to fabricate a Pd-based alloy membrane on an alumina
support via a sequential electroless deposition technique. The membrane enables the
separation of hydrogen from a mixture of hydrogen and carbon dioxide at elevated
temperature. We use commercial ceramic paste as a sealing material in which the
debindering and sintering temperatures are identified as 600 and 1150 degree Celsius,
respectively. From GC results, after proper sealing we attain desirable air tightness at
600 degree Celsius.
The electroless deposition is conducted on a flat alumina disk initially in order to
determine suitable processing parameters for targeted alloy composition. Materials
characterization such as phase, composition, and morphology are carried out. The
iii
maintains nicely after annealing at 600 degree Celsius. However, the PdAgCuNi
membrane behaves differently because of phase separation between Cu and Ni. The
roughness is found to increase in PdAgCu and PdAgCuNi as the deposition
progresses, a fact that is attributed to thehomogenous nucleation of Cu during
electroless plating. The expected ratio for Pd-based alloy membrane can be controlled
and estimated after careful weight determination.
In addition, we successfully prepare PdAgCuNi alloy membrane on a porous
alumina tube after proper sealing and the hydrogen permeation test will be performed
iv
誌謝
時間像是忘了上鎖的水龍頭,不斷的流逝著,碩士班二年的時間說長不長, 說短不短,但也一轉眼地來到了盡頭,回想當初剛面臨研究所時的慌恐與不安, 在經歷過二年洗禮及歷練後,現在,我可以很驕傲的說:「我是交大人。」 能完成一件學業,除了靠自己的努力之外,絕大部分還得仰賴一路上伴隨我、 幫助我的貴人。首先,我非常感謝我的二位指導教授-林鵬教授及吳樸偉教授, 二位教授在我的研究指導上不遺餘力,總是給我方向、推薦好的實驗方法,林鵬 教授甚至親自動手設計實驗模具,推薦實驗方式及藥品選擇,使我在實驗方面如 魚得水;吳教授特別注重邏輯的訓練,當我遇到瓶頸時,可以先試著推理,再去 想解決方法,此外吳教授非常注重學生的英文能力,肯犧牲自己的時間訓練學生 英文,實屬難得。二位教授除了關心學生的研究方面外,在生活態度及健康方面 也非常注重,不定時的告知我們人生哲學,讓我們除了做研究之外,更能看清自 己往後的道路;定期的實驗室腳踏車之旅也讓我們做研究之於,也能關心自己健 康。二位教授對學生非常親合,讓我更能夠勇於的與他們分享無論是實驗上、人 生上的種種問題,非常謝謝這二位教授對我的付出,非常榮幸能當您的學生。 此外,非常感謝實驗室學長姐們這二年對我的指導,張玉塵學長、黃苡叡學 長、謝逸凡學長在實驗方面總是給我幫助;林勝結學長、張雲閔學長除了教我實 驗方法外,也會額外花時間陪我運動;謝育淇學長豐厚的電化學知識,替我解決 了不少問題,學長人非常親切,爽朗的笑聲總讓人印象深刻;陳境妤學姐在我的 無電鍍製程上扮演著非常重要的角色,每當我遇到瓶頸時,學姐總是立刻放下手 邊工作與我討論,並推薦我相關的書籍及期刊,讓我受益無窮;廖晨宏學長在 BET 方面對我指導非常多,也很感謝他在我接下材料實驗助教後,對我的教育 訓練,順帶一提,他跟我一樣是台南人,與我非常契合,對我非常熱情且親和力 十足,讓我很快就適應實驗室的生活。對於 CO2組別的學長姐們我也非常感激, 謝謝蔡致芳學長為我們肩負起重責大任,也你離開前也做了非常足夠的交接,使 我不至於手足無措,謝謝你總是願意花時間與我討論實驗的種種問題,常常一通 電話過去,義不容辭的與我討論,讓我非常感動,感謝你的付出及照顧;我也非 常感謝二位學姐周亮余及陳婉瑩教了我許多研究方法,特別感謝陳婉瑩推薦我進 來這個實驗室,非常照顧我,大學同學不是白當的。我也要謝謝黃筱琳學姐做事 非常細心,把實驗室管理的有條不紊;黃冠傑學長除了教導我研究方法外,也提 供了很多職場的倫理、面試的技巧,讓我在進入職場前先做準備,也非常謝謝你 時常回交大找我們敘舊;謝謝陳琪學姐在實驗室工作交接上交代非常清楚,讓我 可以立即上手。我也要謝謝賴俊翰學長、王儷瞱學姐、陳儷尹學姐、陳致源學長、 張詠策學長、邱于凡學姐、張立忠學長的教導,雖然在我進來實驗室時,你們都v 快離開實驗室了,但是我真的很開心這段時間的相處,特別謝謝賴俊翰學長在學 習英文方面給了我很多建議,祝你接下來的博班旅程可以順利;謝謝大小儷學姐 及張立忠學長常常回來看我們,實驗室有了你們更增添的歡樂氣氛。最後要謝謝 黃昆平學長,非常照顧我們學弟,除了在研究方面提供方法之外,也會自掏腰包 請我們吃飯,讓我倍感溫馨;我也非常謝謝二位博班學姐:張庭瑜學姐及陳菁華 學姐,謝謝碩二這一年有你們的陪伴。 除了長輩的指導之外,同儕間互助也相當重要,我非常感謝這二年與我朝暮 相處的同學們-邱尊偉、郭哲瑋、許議文及陸意德。尊偉是我進入交大第一個認 識的朋友,這二年中,一起合作很多事情,無論是策劃迎新旅行、尾牙主持、具 有挑戰性的 CO2 計畫報告,過程中難免意見不合,但最終都能達成共識,完成 任務,很高興這二年與你合作;哲瑋除了解決我實驗上的難題外,對生活上的疑 難雜症也會提供我建議、幫我處理,真的很謝謝你願意幫我處理生活上的瑣事; 議文常幫我解決英文及電腦問題,尤其是我的英文,常常都要麻煩你幫我訂正, 謝謝你這二年的指導;意德是實驗室的知識家,在大一修課及實驗理論上都幫了 我很多,我也非常感動他會額外幫我惡補,讓我在非材料系背景的前提下可以快 速的銜接。非常謝謝你們這二年的陪伴。接下來我們都各奔西東,希望大家都有 好的發展,美好的未來。 我也非常感謝碩一的學弟妹們,建程、韋霖、柏翰、依叡、雁汝、欣儀、宥 閔。建程跟柏翰都要留意實驗室財產,維護實驗室工安的韋霖跟欣儀,負責聯絡 大家及會議室預約的雁汝,幫忙採購的依叡及宥閔,謝謝你們這段時間的付出, 有你們的分憂解勞,可以讓我更能專心在實驗上面,這段時間辛苦你們了,祝福 各位明年可以順利畢業,平順的往下個目標前進。最後,特別感謝韋霖跟建程, 謝謝你們在我畢業前夕幫忙 CO2 小組的大小瑣事,非常感謝你們在實驗上的幫 忙,我也私心希望 CO2小組可以更壯大,大家加油! 剛進實驗室的延璋、萱維、晟瑋、孫佑、詠民,雖然我們相處時間不長,但 這段時間下來,覺得大家都是很好相處的人,對於實驗室向心力也很足夠,希望 大家能把握這二年的時間,充分學習,遇到困難時可以彼此討論、解決,相信你 們會獲得很多,預祝各位二年後豐收滿載,順順利利。 我也要謝謝我的口試委員-方冠榮教授,千里迢迢從成大參加我的口試,並 從中給我很多寶貴意見及想法,因為您的建議,讓我的研究更臻完美,謝謝您。 謝謝陳境妤學姐、廖晨宏學長、呂佳凌學姐、張博學學長、羅登元學長、劉育成 學長分別對我的材料分析儀器訓練,使我能分析我的樣品後,做出最好的改進; 謝謝電物所的 lido、金城甫、彭彭、小開、常臨在我做實驗之餘還能約我一起運 動健身、抒發情緒,希望你們未來也有好的發展。
vi 最後我要非常鄭重感謝我的家人,每當實驗不順、遇到瓶頸、心浮氣躁、毫 無思緒時,瞬間的念頭就是「回家吧!」,一個暫時喘息的避風港。謝謝我的家人 總是給我鼓勵,不間斷的加油打氣,每當回家之後身體裡總是滿滿的能量,預備 前往新竹再戰!在經濟上的也是毫無保留地支助,使我能無後顧之憂的專心在課 業上。謝謝我的家人,總是能包容我的脾氣,肩負我的經濟重擔,不吝嗇地給我 鼓勵,使我在碩士班生涯可以一帆風順。 時光匆匆,倏忽即逝,在交大二年的碩士班生涯過去了,非常感謝交大能給 我那麼好的研究環境,在高手雲集的環境下切磋過後,才知道自己其實很渺小, 因此不斷前進是我唯一的使命,即使離開了交大,我也必保持著交大「知新致遠、 崇實篤行」之精神,繼續往下個人生目標邁進。 李孟翰 二零一二年七月 於國立交通大學
vii
目錄
摘要... i Abstract ... ii 誌謝... iv 目錄... vii 表目錄... x 圖目錄... xi 第 1 章 緒論... 1 1.1 研究背景 ... 1 1.2 研究動機 ... 2 第 2 章 文獻回顧... 3 2.1 氫氣分離與純化 ... 3 2.1.1 鈀金屬之氫氣分離膜簡介 ... 3 2.1.2 金屬透氫傳導機制 ... 4 2.1.3 鈀合金複合薄膜製程 ... 7 2.1.4 基材之要求與選擇 ... 9 2.2 鈀膜之性質與無電鍍製程 ... 11 2.2.1 鈀膜氫脆化現象與合金選擇 ... 11 2.2.2 鈀膜及其合金之無電鍍製程 ... 14 2.2.2.1 基材之敏化與活化 ... 14 2.2.2.2 無電鍍簡介 ... 14 第 3 章 實驗方法與步驟... 15viii 3.1 實驗藥品與儀器 ... 15 3.1.1 實驗藥品與氣體 ... 15 3.1.2 實驗分析儀器 ... 17 3.2 實驗流程 ... 18 3.2.1 基材為氧化鋁片實驗示意圖 ... 19 3.2.2 基材為氧化鋁管實驗示意圖 ... 20 3.3 氧化鋁基材前處理 ... 21 3.3.1 氧化鋁基材之清洗 ... 21 3.3.2 氧化鋁基材之敏化及活化 ... 21 3.4 陶瓷封裝製程 ... 22 3.4.1 封裝材料定性分析 ... 22 3.4.1.1 熱重分析儀分析 ... 23 3.4.1.2 SEM 表面分析 ... 23 3.4.2 封裝方法及步驟 ... 23 3.4.3 封裝材料漏氣測試 ... 24 3.5 金屬無電鍍製程 ... 24 3.5.1 無電鍍法製備鈀膜 ... 26 3.5.2 無電鍍法製備鈀銀合金膜 ... 26 3.5.3 無電鍍法製備鈀銀銅合金膜 ... 26 3.5.4 無電鍍法製備鈀銀銅鎳合金膜 ... 27 3.6 氣體測試及分析 ... 29 3.6.1 氣體分離整體裝置設計圖 ... 29 3.6.2 各氣體檢量線建立 ... 30
ix 3.6.3 漏氣測試 ... 31 3.6.4 氫氣測試 ... 32 第 4 章 實驗結果與討論... 33 4.1 封裝材料性質分析 ... 33 4.1.1 封裝材料 TGA 分析 ... 33 4.1.2 封裝材料不同燒結溫度之 SEM 分析 ... 34 4.1.3 封裝材料之 GC 漏氣測試 ... 35 4.2 鈀膜於緻密氧化鋁片之無電鍍製程 ... 40 4.2.1 無電鍍時間及鍍液濃度對鈀膜厚之影響 ... 40 4.2.2 退火溫度對表面形貌之影響 ... 42 4.2.3 鈀膜於氫氣下退火後之 XRD 分析 ... 44 4.3 鈀銀膜於緻密氧化鋁片之無電鍍製程 ... 45 4.3.1 無電鍍時間對銀膜沉積量之影響 ... 45 4.3.2 鈀銀膜於氫氣下退火前後之 XRD 分析 ... 47 4.3.3 鈀銀膜於氫氣下退火前後之 SEM 分析 ... 49 4.3.4 鈀銀膜於氫氣下退火後之 EDS 分析 ... 51 4.4 鈀銀銅膜於緻密氧化鋁片之無電鍍製程 ... 53 4.4.1 無電鍍時間對銅膜沉積量之影響 ... 53 4.4.2 鈀銀銅膜於氫氣下退火前後之 XRD 分析 ... 55 4.4.3 鈀銀銅膜於氫氣下退火前後之 SEM 分析 ... 57 4.4.4 鈀銀銅膜於氫氣下退火後之 EDS 分析 ... 59 4.5 鈀銀銅鎳膜於緻密氧化鋁片之無電鍍製程 ... 61
x 4.5.1 無電鍍時間對鎳膜沉積量之影響 ... 61 4.5.2 鈀銀銅鎳膜於氫氣下退火前後之 XRD 分析 ... 63 4.5.3 鈀銀銅鎳膜於氫氣下退火前後之 SEM 分析 ... 64 4.5.4 鈀銀銅鎳膜於氫氣下退火後之 EDS 分析 ... 65 4.6 鈀合金膜於氧化鋁管型基材之特性分析及氣體測試 ... 68 4.6.1 管形基材之封裝 ... 68 4.6.2 鈀銀銅鎳合金膜 ... 68 4.6.2.1 表面形貌分析 ... 68 4.6.3 氣體測試 ... 71 第 5 章 結論... 73 第 6 章 未來方向... 74 第 7 章 參考文獻... 75
表目錄
表 3.1 實驗藥品清單... 15 表 3.2 實驗氣體清單... 16 表 3.3 實驗材料清單... 16 表 3.4 鹼液配方及操作條件... 21 表 3.5 敏化液配方及操作條件[45] ... 22 表 3.6 活化液配方及操作條件[45] ... 22 表 3.7 鈀無電鍍液配方及操作條件[46] ... 27 表 3.8 銀無電鍍液配方及操作條件[46] ... 28xi 表 3.9 銅無電鍍液配方及操作條件[47-49]... 28 表 3.10 鎳無電鍍液配方及操作條件[50, 51] ... 28 表 3.11 封裝漏氣測試條件 ... 31 表 3.12 各種氣體測試條件... 32 表 4.1:由秤重減重法預估不同銀無電鍍時間之鈀、銀重量比... 52 表 4.2:由秤重減重法預估不同銅無電鍍時間之鈀、銀、銅重量比... 60 表 4.3:由秤重減重法預估不同鎳無電鍍時間之鈀、銀、銅、鎳重量比... 66
圖目錄
圖 1.1 氣化複循環發電系統示意圖[1] ... 2 圖 2.1 氫氣於金屬中之傳導機制[4] ... 5 圖 2.2 各種金屬於各溫度之透氫能力圖[10, 17] ... 6 圖 2.3 Force-flow CVD 操作示意圖[4, 19] ... 7 圖 2.4 濺鍍法之實驗示意圖[4, 21] ... 8 圖 2.5 電鍍法實驗示意圖[23] ... 9 圖 2.6 補洞材料顆粒大小與基材孔隙大小之效應 [29] ... 10 圖 2.7 不同溫度及氫氣壓力之氫於鈀之溶解度相圖[4] ... 11 圖 2.8 室溫下相變化晶格差異與鈀銀鎳組成關係圖[4] ... 12 圖 2.9 不同鈀合金含量對透氫率之影響[11] ... 13 圖 3.1 實驗步驟總流程圖... 18 圖 3.2 氧化鋁片為基材之實驗步驟流程圖... 19 圖 3.3 氧化鋁管為基材之實驗步驟流程圖... 20xii 圖 3.4 封裝專用模具圖... 24 圖 3.5 氧化鋁管形基材置入無電鍍裝置圖... 25 圖 3.6 實驗裝置平面示意圖... 29 圖 3.7 實驗裝置實際圖... 30 圖 3.8 檢量線實驗裝置平面示意圖... 31 圖 3.9 漏氣測試實驗裝置平面示意圖... 32 圖 4.1 封裝材料之 TGA 分析圖 ... 34 圖 4.2 封裝材料於不同溫度下燒結後之 SEM 圖 ... 35 圖 4.3 塗佈封裝材料之陶瓷管對接後經高溫燒結後成品圖... 36 圖 4.4 封裝材料經燒結後於 25°C 之二氧化碳漏氣測試圖 ... 37 圖 4.5 封裝材料經燒結後於 600°C 之二氧化碳漏氣測試圖 ... 38 圖 4.6 封裝材料經燒結後於 25°C 之氦氣漏氣測試圖 ... 39 圖 4.7 鈀無電鍍時間及鍍液濃度與膜厚之關係圖... 40 圖 4.8 使用聯胺濃度 0.005 M 於不同時間下鈀無電鍍之 SEM 側視圖 ... 41 圖 4.9 鍍液體積與無電鍍面積比 12.5 ml/cm2之 4 小時鈀無電鍍 SEM 側視圖 ... 42 圖 4.10 不同鈀無電鍍時間退火前之 SEM 俯視圖 ... 43 圖 4.11 不同鈀無電鍍時間於 950°C 退火後之 SEM 俯視圖 ... 44 圖 4.12 鈀膜/氧化鋁退火前後之 XRD 圖 ... 45 圖 4.13 銀無電鍍時間與膜厚之關係圖... 46 圖 4.14 不同銀無電鍍時間於 1 μm 鈀膜之 SEM 側視圖 ... 47 圖 4.15 銀無電鍍 2 小時所得之鈀銀膜熱處理前後之 XRD 圖 ... 48 圖 4.16 銀無電鍍 8 小時所得之鈀銀膜熱處理前後之 XRD 圖 ... 49
xiii 圖 4.17 不同銀無電鍍時間於 1 μm 鈀膜退火前之 SEM 俯視圖 ... 50 圖 4.18 不同銀無電鍍時間於 1 μm 鈀膜退火後之 SEM 俯視圖 ... 51 圖 4.19 銀無電鍍時間 8 小時退火後之 EDS 圖 ... 52 圖 4.20 銅無電鍍時間與膜厚之關係圖... 54 圖 4.21 不同銅無電鍍時間於 1.2 μm 鈀銀膜後之膜厚 SEM 側視圖 ... 55 圖 4.22 銅無電鍍 4 小時所得之鈀銀銅膜熱處理前後之 XRD 圖 ... 56 圖 4.23 銅無電鍍 8 小時所得之鈀銀銅膜熱處理前後之 XRD 圖 ... 57 圖 4.24 不同時間銅無電鍍於鈀銀膜之退火前 SEM 俯視圖 ... 58 圖 4.25 不同時間銅無電鍍於鈀銀膜之退火後 SEM 俯視圖 ... 59 圖 4.26 銅無電鍍時間退火後之 EDS 圖 ... 60 圖 4.27 鎳無電鍍時間與膜厚之關係圖... 62 圖 4.28 不同鎳無電鍍時間於鈀銀銅膜後之膜厚 SEM 側視圖... 62 圖 4.29 鎳無電鍍 1 小時所得之鈀銀銅鎳膜熱處理前後之 XRD 圖 ... 63 圖 4.30 不同時間鎳無電鍍於鈀銀銅膜之退火前 SEM 俯視圖 ... 64 圖 4.31 不同時間鎳無電鍍於鈀銀銅膜之退火後 SEM 俯視圖 ... 65 圖 4.32 銅無電鍍時間退火後之 EDS 圖 ... 67 圖 4.33 為氧化鋁管行基材封裝後成品圖... 68 圖 4.34 多孔氧化鋁管無電鍍鈀、銀、銅、鎳後之退火前後成品圖... 69 圖 4.35 多孔氧化鋁基材無電鍍鈀、銀、銅、鎳經退火後之 SEM 俯視圖 ... 70 圖 4.36 多孔氧化鋁基材之 SEM 俯視圖 ... 70 圖 4.37 多孔氧化鋁基材無電鍍鈀、銀、銅、鎳之 SEM 側視圖 ... 71 圖 4.38 二氧化碳漏氣測試圖... 71
1
第1章 緒論
1.1
研究背景
科技的進步,隨之而來的是人們對於能源之需求也大幅提高,目前主要能源 供給來源仍是以石化燃料為主,但石化能源有朝一日會消耗殆盡,近幾年來,國 際原油價格逐年提高,使得人們不得去發展其他替代能源,其中,氫能廣受人們 矚目,因其產物為無汙染的水,是具有發展潛力之替代能源之一。 除此之外,使用石化燃料也伴隨著大量二氧化碳之排放,造成近年來溫室效 應日益嚴重,因此除了上述發展無污染之替代能源之外,減碳也成為了當今最重 要的議題。整合氣化複循環發電系統(Integrated Gasification Combine Cycle,IGCC)具有 高能源轉化之效率,淨發電效率達 40%。此發電系統是將煤炭在氣化爐還原氣氛 下,將煤炭轉成合成氣(Syngas),其主要產物為二氧化碳及氫氣,若能將二者氣 體分開收集的話,即可避免二氧化碳逸散於大氣中造成溫室效應,氫氣則可以做 燃料電池之動力來源。目前國際上正積極發展之淨煤發電技術,無論從技術未來 應用發展潛力、能源效率及環保性能都非常高,預期未來將被國內推廣應用。圖 1.1 為氣化複循環發電系統示意圖。
2 圖 1.1氣化複循環發電系統示意圖[1]
1.2
研究動機
在氫氣分離膜領域中,主要仍以鈀為主,先前的研究中,在鈀膜中添加其他 金屬除了可以減低成本、抑制毒化之外,也可以增加透氫率,只是大多數鈀合金 薄膜都是以雙元合金為主,如鈀銀合金、鈀銅合金等,只有少數研究團隊研發出 三元合金薄膜,Uemiya 已成功利用無電鍍法製備鈀銀釕三元合金薄膜,且透氫 率比純鈀、鈀銀合金膜來的高[2];Ryi 也利用濺鍍法成功製備了鈀銅鎳三元合金 薄膜,其膜之透氫率比起純鈀膜高出 3 到 4 倍[3],就種種現象來看,鈀三元合 金膜似乎比雙元合金膜更好,因此本研究嘗試利用無電鍍法製備鈀四元合金薄膜, 作為氫氣分離之應用,與文獻比較並驗證其可行性。本研究材料的選用是比照文 獻常用之鈀銀銅合金,第四個組成選擇鎳,原因在於鎳具有氫氣催化效果,可加 快氫氣分解成氫原子,另外鎳也具備將氫原子變成氫質子與電子之能力,可提供 額外之氫離子來源,若未來與陶瓷導氫材料結合,將可形成新穎之質導陶瓷金屬 複合薄膜,在陶瓷導氫離子、金屬導氫原子及電子之雙通道運作下,預估可有更 好之透氫率。 .3
第2章 文獻回顧
2.1 氫氣分離與純化
關於氫氣的分離(separation)及純化(purification)技術,目前最常見的有四種 方式,分別是吸附法(adsorption)、溶劑吸收法(solvent absorption)、低溫回收法
(cryogenic recovery)及薄膜分離法(membrane separation)[4]。
吸附法可分為物理吸附(Physisorption)及化學吸附(Chemisorption)。物理吸附 的原理是吸附體藉由凡德瓦爾力(Van Der Waals Force)而附著在物體表面,由於 凡德瓦爾力在分子間作用力小,吸附能較小,因此吸附體容易脫附。化學吸附涉 及到原子間的電子轉移,吸附體與物體間的吸附能大,因此脫附能需要較高的能 量。利用不同吸附劑的特性結合不同的分離程序可依序分為,變壓吸附法 (Pressure Swing Adsorption,PSA)、變溫吸附法(Temperature Swing Adsorption,TSA) 及變電吸附法(Electrical Swing Adsorption,ESA),其中 PSA 已經被廣泛的應用於 化學工業之中,氫氣分離後的純度可達到 99.99 mol% [5]。溶劑吸收法是利用溶 解度原理的一種物理吸收法,常應用於較高濃度氣體的分離,因為氣體溶在水中 會放熱,因此在高溫中氣體溶解度會下降,因此在高溫下作氣體分離時較不適合。 低溫回收法也是受溫度控制,必須在低溫才能有效的分離,於應用上限制較多。 跟上述三種方法比較下來,薄膜分離法是一種限制較少的分離技術,此方法 具有低操作成本、節能、操作簡單方便、高能源效率等優點[6],除此之外,薄 膜可以大規模的製備,也可與其他產業技術作結合,是工業上不可或缺的分離技 術。但其缺點在於薄膜的耐久性較差,而造成往後分離效率不高,必須使用二階 段以上的分離程序才可達到一定的分離效率。另外薄膜對氫氣的化學穩定性、機 械穩定性及熱穩定性都須考慮。
2.1.1 鈀金屬之氫氣分離膜簡介
法國學者 Deville 等人,在 1863 年首先發現氫氣可經由擴散通過鉑金屬[7-9], 而在 1866 年英國學者 Graham 發現加熱之鈀金屬也具有透氫效果[9],雖然後來 仍有發現氫氣對鎳、鉑、等金屬有溶解度,且也可被拿來做氫氣分離應用,但相 對於鈀而言,其溶氫量都是小於鈀,在低於 1400°C 時,鈀都具有最大的溶氫量4 [10],因為這個特性也開啟了鈀系列薄膜於氫氣分離領域的應用。
2.1.2 金屬透氫傳導機制
金屬透氫的程序如何進行?在 1951 年科學家 Barrer 的研究中指出氫氣於金屬 中擴散機制是一個多步驟的機制[11]。可分為 7 個步驟,如圖 2.1 所示。 1. 外在擴散: 氫氣分子受自由能影響,會由高濃度往低濃度擴散,達到平衡為止,因此會 接近金屬表面。 2. 吸附並分解: 氫 氣 分 子 在 金 屬 表 面 進 行 可 逆 的 化 學 吸 附 (reversible dissociative chemisorption),並且受金屬的催化作用而分解成氫原子。 3. 溶解: 分解後之氫原子溶入金屬中。 4. 內部擴散: 氫氣原子因兩端氫原子濃度差異,造成自由能不同的影響,而由高自由能往 低自由能驅使而擴散至另一端。 5. 再結合: 氫原子再度結合成氫分子。 6. 脫附: 氫氣分子從金屬表面離開。 7. 外部擴散: 脫附後之氫分子再度受自由能影響而遠離金屬表面。5
圖 2.1 氫氣於金屬中之傳導機制[4]
由上述機制可知,氫氣的透氣通量與氫氣在金屬膜中的擴散係數、氫氣二端
的壓力差、薄膜厚度、金屬對氫的溶解量有關。故氫氣之透氣通量 (J)可用 Fick,s first law (2.1) 來表示:
J=(ρ/L)×( Phn - Pln) (2.1) 其中,ρ 是氫氣之滲透係數(hydrogen permeability),n 為壓力指數,此數值 通常介於 0.5-1 之間,Phn 、Phn分別表示進氣端(高壓端)及滲透端(低壓端)的氣體 分壓。若透氫的速率決定步驟為氫氣表面的吸附及脫附速率時,則 n 值=1,稱為 表面限制(surface control),氫氣的滲透通量與其分壓成正比;若速率決定步驟為 氫原子在金屬中的擴散速率時,則 n 值=0.5,稱為擴散限制(diffusion control), 氫氣的滲透通量與其分壓的平方根成正比,又稱為Sievert’s law,如式子(2.2)所 示: J=(ρ/L) ×(Ph0.5 - Pl0.5) (2.2) ρ= D×S (2.3) 由式子(2.3)可知,ρ 為 D 與 S 的乘積,D 與 S 分別為氫原子在金屬中的擴散 係數及溶解度,此二係數會隨著溫度及金屬膜之組成而改變,擴散係數 D 與粒 子的動能相關,在高溫時粒子具有較高的動能,因此擴散係數較高;而溶解度 S 正好相反,氣體分子溶於金屬中是放熱反應[12],因此溫度越高越不利溶解,故
6 溶解度降低,另一說法是因為分子動能大,不容易被束縛在固體表面。另外組成 也是影響ρ 的因素,以鈀銀合金為例,因為銀原子之晶格比鈀原子大,故添加銀 於鈀金屬中時,可以撐大鈀金屬的晶格,造成晶格體積變大,使整體的溶氫量增 加,但也因為晶格擴大而使氫原子之擴散係數降低[13],故整體的氣體流量為溶 解度與擴散的競爭。 關於壓力指數 n 值,也有學者探討,Holleck[14]發現在鈀膜之膜厚達 1 mm 時,所得到之 n 值為 0.5。Hurlbert[15]也發現當膜厚在 25-154 μm 間時,其 n 值 為 0.68,他認為此現象與氫氣在鈀膜表面吸附與氫原子在鈀膜裡面擴散都有關。 Ilias[16]之研究中顯示當氫氣通過厚度為 8.5 μm 之鈀膜時,其 n 值為 0.778;而厚 度為 12 μm 之鈀膜時,n 值為 0.501,因此他們認為 n 值接近 0.5 時,代表擴散限 制越顯著;而當膜越薄,表面氫氣溶解之過程成了影響氫氣滲透速度之關鍵,理 論上 n 值會接近 1。綜合上述,若不考慮基材因素或鍍膜技術,膜厚越厚,理論 上 n 值越接近 0.5,表示速率決定步驟在於氫原子在膜中之擴散;反之,膜厚越 薄,表示速率決定步驟在於氫氣分子在膜表面之反應,n 值會接近 1。 由圖 2.2 可知只有鈮、釩、鉭其透氫率與溫度成反比,表示其表面反應影響 大於內部擴散;另外在 550°C 以下時,鈮、釩、鉭、鈀都具有非常好之透氫能力, 但再比較下來,鈮、釩及鉭之透氫效果都具有比鈀更好,目前卻只有鈀被廣泛應 用,主要原因在於鈮在 200°C 左右會跟二氧化碳反應,因此不適用於氣體分離系 統;而釩容易氧化變成五氧化二釩,此物質有劇毒;而鉭質地硬、熔點高,因此 不易加工、鍍膜。 圖 2.2 各種金屬於各溫度之透氫能力圖[10, 17]
7
2.1.3 鈀合金複合薄膜製程
目前已有很多種方式來製備鈀或其合金複合薄膜,常見之製程有化學氣相沉 積法、物理氣相沉積法、電鍍法、溶膠凝膠法及無電鍍法。 1. 化學氣相沉積法(CVD) 以金屬薄膜來說,其原理是利用一種或多種易揮發性之前驅鹽,在載流氣體 通過反應源時,讓反應源之飽和氣體產生化學反應,前驅鹽因受分解而產生金屬 原子,進而沉積在基材表面,目前常用之載流氣體為氫氣。最早被報導使用此方 法製備鈀膜的是 Ye[18],他使用氯化鈀當作前驅鹽,將鈀膜沉積在氧化鋁片上。 而在 2005 年,Itoh[19]使用 Force-flow CVD 將鈀膜鍍在管形基材上,其鈀膜 厚度約 2-4 μm,H2/N2之選擇比可達 5000。CVD 法之優點在於可製備厚度小於 2 μm 之薄膜,且厚度掌控非常精準,在工業界常用此技術製備薄膜,但其缺點在 於所沉積上的膜之前驅鹽必須要有揮發性及熱穩定性,且純度要高,製程之環境 要求非常嚴謹,因此操作之成本也較其他製程高。圖 2.3 為 Force-flow CVD 操作 示意圖。 圖 2.3 Force-flow CVD 操作示意圖[4, 19] 2. 物理氣相沉積法(PVD) 物理氣相沉積法與化學氣相沉基法相似,差別在沒有化學分解過程。此技術 原理是在真空條件下,利用加熱、電子束撞擊、離子束撞擊或電弧熔融之方式將 材料氣化後,再於基材表面沉積膜。常見之方式為熱蒸鍍與濺鍍。熱蒸鍍是利用8 材料在真空條件下加熱,使其氣化後因為動能上升而往基材方向擴散,因為基材 溫度較低,氣化之材料會冷凝於基材表面形成薄膜;濺鍍是施加高電壓於濺鍍槍 引發輝光放電效應,讓腔體內之氬氣受電子撞擊形成氬氣離子,氬氣離子受電場 作用撞擊靶材後,使靶材之分子飛出而在基材上沉積,形成薄膜,文獻上 Liu[20] 已使用此方法沉積 100 nm 之鈀膜於多孔鎳基材上。PVD 技術與本研究使用無電 鍍比較起來,此方法較能控制薄膜組成及厚度,但需要高真空之環境,因此設備 成本也相對的高。圖 2.4 為濺鍍法之實驗示意圖 圖 2.4 濺鍍法之實驗示意圖[4, 21] 3. 電鍍法(EPD) 電鍍法是一種電化學沉積方法,涉及氧化還原反應,欲鍍物放在陽極,基材 則放置陰極,欲鍍物氧化後形成金屬離子,金屬離子在液相中受到電場作用而向 陰極移動,最後在陰極被還原後形成金屬而沉積在基材上。Hsieh 也利用電鍍法 製備鈀複合薄膜[22]。電鍍法之優點在於設備簡單,且只要控制好電流大小及電 鍍時間,即可鍍上預期之膜厚,然而其缺點在於基材的限制,基材必須要能夠導 電或者須沉積一層導電層才能操作。圖 2.5 為電鍍法實驗示意圖。
9 圖 2.5 電鍍法實驗示意圖[23] 4. 溶膠凝膠法(Sol-Gel) 此技術是將粒徑大小約 10-100 Å 的膠體粒子均勻的分散在溶液中,除了粒 子本身之布朗運動外,粒子表面會受溶液之酸鹼性影響,產生電雙層作用,因此 不會沉降下來,此稱為 Sol。而膠體粒子受到連續之水合及縮合反應後,流動性 會下降,形成半固態之物質,此時稱為 Gel。薄膜之製備常利用浸入塗佈法 (Dip –coating)、噴霧法(Spray-coating)以及旋轉塗佈法(Spin-coating)等。 5. 無電鍍法: 於 2.2.2 介紹。
2.1.4 基材之要求與選擇
一般用於鈀複合膜之基材之選擇,必須要有下列幾種特性: 1. 化學穩定性高 2. 熱穩定性高 3. 機械強度高10 4. 高孔隙度或高質導率 5. 價格低廉 因此基材主要可分為陶瓷材料及金屬材料,陶瓷材料之優點為兼具熱及化學 穩定性,機械強度也高,其代表為多孔之氧化鋁;金屬材料因為加工性佳,且其 熱膨脹係數與金屬膜接近,因此與金屬膜間黏合性較好,常見金屬基材為多孔不 鏽鋼管,但因為金屬基材在高溫會與金屬膜發生互擴散效應 (Inter diffusion -effect),因此在鍍膜前必須先長一層氧化層當作阻抗層(Barrier layer),目前阻抗 層材料有使用氧化鉻[24],或者直接在基材在空氣中加熱使成金屬氧化物[25, 26]。 基材之性質,如表面粗糙度、孔隙大小、孔隙率等因素也會影響到鍍膜之品 質,若基材表面孔隙或粗糙度大,則需要有更厚之膜來覆蓋,才能降低漏氣率, Yolcular 及 Olgun 已使用無電鍍法在多孔氧化鋁管上鍍上鈀膜,其厚度為 9 μm, 在 375°C 時,H2/N2之選擇比為 5500[27],而 Ma 也在多孔不鏽鋼管上鍍上 19.1 μm 厚之鈀銅合金膜,在 450°C 更是沒有發現氦氣漏氣[28],雖然增加膜厚可以有效 的減少漏氣量,但花費成本也高,因此,在鍍金屬膜之前,可以針對基材之表面 孔隙結構加以修飾及改質。 關於基材表面改質之研究,Oyama[29]指出可在基材上多加一層緩衝層來降 低表面粗糙度,補洞材料之顆粒大小與基材表面孔隙大小相關,如圖 2.6 所示, 補洞材料如果大於基材表面之孔隙時,並無法有效完全覆蓋孔洞,若補洞材料顆 粒小於基材表面之孔隙時,也會有膜破損而滲透氣體,因此最理想之方式為按照 補洞材料之顆粒大小,由大至小循序的覆蓋基材,將可以達到完整且均一的覆蓋。 補洞材料方面,Ma 是使用氧化鋁漿料塗佈在多孔不鏽鋼基材表面,其鈀合金膜 膜厚可以減少至 10 μm[30]。 圖 2.6 補洞材料顆粒大小與基材孔隙大小之效應 [29]
11
2.2 鈀膜之性質與無電鍍製程
2.2.1 鈀膜氫脆化現象與合金選擇
儘管純鈀膜具有非常好之透氫效果,但在氣體分離方面還是有些先天上的問 題: 1. α-β 相變化: 由圖 2.7 可知,當溫度低於 298°C、氫氣壓力 20 大氣壓以下時,鈀隨著溶氫 量的增加會從原本之α 相逐漸轉成 β 相,雖然二相皆為 FCC 結構,其晶格常數 卻不一樣,室溫下的α 相晶格常數為 0.389 nm,而 β 相為 0.410 nm[31],當鈀膜 於不同之溫度、氫氣壓力反覆操作時,鈀膜會產生相變化,體積的膨脹或收縮都 會造成晶格扭曲,導致膜破損而漏氣。 圖 2.7 不同溫度及氫氣壓力之氫於鈀之溶解度相圖[4]12 2. 鈀膜氫脆化: 當鈀膜持續的曝露在氫氣氣氛中時,會失去其原本金屬延展性,稱作氫脆化, 此現象會讓膜出現裂損,造成漏氣[32]。 3. 鈀膜毒化: 若鈀膜操作在含有 CO、H2S 環境之下,將會使鈀膜不具有透氫效果,此稱 為鈀膜毒化現象[33]。以 H2S 為例,鈀易在表面與 H2S 反應成 Pd4S,然而 Pd4S 不具有透氫效果,且又會佔據鈀膜表面氫氣催化之位置,造成整體之流量降低, Miller 研究指出,在含有 1000 ppm 之 H2S 環境下,其透氫率為純氫氣的十分之 一[34]。 Uemiya、Bryden、Jun 等人分別發現添加銀、鐵、鎳與鈀形成合金,可以有 效的解決上述純鈀膜之問題[35-37]。 針對鈀膜 α-β 相變化問題,Uemiya[38]提出若添加 1B 族晶格較鈀大之金屬 時,可以降低其操作溫度。例如在鈀加入銀形成鈀銀合金時,因為銀之晶格常數 比鈀大,因此溶入鈀中可以撐大鈀之晶格至 0.396 nm,相變化時晶格差異變小, 因此膜破損可降低,尤其若銀添加至 23 wt%時,其操作溫度可降至室溫[39]。 圖 2.8 為室溫下相變化晶格差異與鈀銀鎳組成關係圖。 圖 2.8 室溫下相變化晶格差異與鈀銀鎳組成關係圖[4]
13 另外 Morreale 發現鈀銅合金可以有效的抗 H2S 毒化及減少氫脆化[40],當銅 含量佔鈀銅合金 30 wt%時,在 1000 ppm 之 H2S 氣氛下操作,其透氫率只下降了 10%。最新研究指出純鈀膜也在 1000 ppm 之 H2S 氣氛下操作時,其鈀膜表面會 生成厚度約 6.6 μm 之 Pd4S,若是銅為 47 wt%之鈀銅合金時,其表面會生成約 3 nm 之鈀銅硫複合膜,雖然說此膜對氫無催化效果,也無法透氫,但可以形成一層保 護膜,避免整體被毒化,且在氫氣下可以移除[41]。 鈀膜合金化除了上述幾個優點外,也有研究發現合金化在特定比例下可以使 整體之透氫效果增加[42, 43]。以鈀銅合金為例,添加銅在 40 wt%之前,其鈀銅 合金膜之透氫率是下降的,因為銅會佔據鈀膜表面之氫催化位置,使溶氫量下降, 直到銅為 40 wt%時,可以得到最高之透氫率,主要原因是此時之鈀銅合金由 FCC 相轉成 BCC 相,在 BCC 相中氫原子是以四面體位(Tetrahedral site)擴散,氫原子 易移動,擴散係數提高。 圖 2.9 為不同鈀合金含量對透氫率之影響。實際上滲透係數與溶解度、擴散 係數乘積有關,但二者是成反比關係,因此透氫率都會有一個最大值存在,例如 鈀銀合金在銀含量為 23 wt%時之透氫率約是純鈀膜的 1.7 倍。 圖 2.9 不同鈀合金含量對透氫率之影響[11]
14
2.2.2 鈀膜及其合金之無電鍍製程
2.2.2.1 基材之敏化與活化
一般無活性基材如陶瓷、玻璃進行無電鍍前,需要有一道表面活化處理,先 將具有活性之晶種散佈在基材表面上,之後無電鍍之金屬即可藉由晶種而沉積上 去。目前常用之敏化、活化方法是交替式的浸入敏化液及活化液中,常用之敏化 液及活化液為酸性之氯化亞錫溶液及氯化鈀溶液。 整個基材活化之反應如式子(2.4): Sn2+ + Pd2+ → Sn4+ + Pd (2.4) Charbonnier[44]研究指出基材在經過敏化之後,表面上會先吸附一層亞錫離 子,而在活化過程中,亞錫離子會與鈀離子發生氧化還原反應,鈀離子會被還原 成鈀金屬核,而吸附於基材表面,其核即為晶種,之後無電鍍之前驅鹽被還原後, 將透過晶種成膜於基材表面。2.2.2.2 無電鍍簡介
無電鍍只要在鍍液中添加入還原劑,還原劑會放出電子給鍍液中之金屬離子, 金屬離子一旦被還原即可在基材上成膜,而鍍上之金屬膜又可以催化此無電鍍反 應,讓更多金屬離子被還原。因整個過程只涉及鍍液中之氧化還原反應,不須額 外提供電力,因此無電鍍又可稱為化學鍍(Chemical plating)。 無電鍍法具有下列幾個優點: 1. 基材限制少: 不像電鍍要求基材需導電或塗佈導電材料,在無電鍍中非導體仍可鍍膜,只 要進行敏化、活化步驟即可。另外若基材為複雜之形狀,仍可藉由無電鍍法沉積 一均勻鍍層。 2. 設備簡單: 不需要高規格之鍍膜系統、真空設備。 3. 操作溫度低:15
第3章 實驗方法與步驟
3.1 實驗藥品與儀器
3.1.1 實驗藥品與氣體
表 3.1 實驗藥品清單 藥品名稱 濃度 廠牌 SnCl2 ˙2H2O 99 wt% Alfa Aesar PdCl2 Pd:59.8 wt% Seedchem HCl 37 wt% SHOWA NaOH 95 wt% SHOWA Na2EDTA˙2H2O 100 wt% JT Baker N2H4 98 wt% Alfa AesarAgNO3 99.8 wt% Sigma ALDRICH
CuSO4
˙
5H2O 95 wt% ScharlauTritonX-100 98 wt% MERCK
NiSO4 ˙ 6H2O 99 wt% SHOWA
Na3C6H5O7˙ 2H2O, 99 wt% J.T Baker
16 表 3.2 實驗氣體清單 實驗氣體名稱 純度 廠牌 He 99.995 mol% 建發氣體 H2 99.9 mol% 建發氣體 N2 99.995 mol% 建發氣體 CO2 99.99 mol% 建發氣體 Ar 99.99 mol% 建發氣體
H2/Ar 5 mol%/95 mol% 建發氣體
Air 99.99 mol% 建發氣體 表 3.3 實驗材料清單 材料名稱 廠牌 緻密陶瓷管 高等工業 緻密陶瓷管轉接頭 高等工業 陶瓷封裝膠 傑地 多孔氧化鋁管 廣瀚
17
3.1.2 實驗分析儀器
1.掃描式電子顯微鏡( Field-Emission Scanning Electron Microscope
; SEM) FESEM JSM-6500 和 JEOL-JSM-6700,用來觀察薄膜表面形貌及橫切面
之厚度。
2.能量散佈光譜儀(Energy Dispersive Spectrometer; EDS)
做初步之成分分析及比例分析,比例分析涵蓋重量比及原子比。
3.X 光繞射儀(X-ray Diffractometer; XRD)
用來觀察薄膜之結晶性,判斷有無成相,可定性分析。
4.熱重分析儀(Thermogravimetric Analyzer; TGA)
用來得到封裝材料之去除黏合物(binder)最佳溫度。 5.氣相層析儀(Gas Chromatography; GC) 進行氣體分離測試、氫氣滲透測試、漏氣測試。 6.酸鹼測定計(pH meter) 用來測定溶液之酸鹼性。 7.管狀高溫爐管(Tubular Furnace) 進行高溫封裝及退火。 8.真空幫浦(Pump) 無電鍍時結束時使用,目的讓薄膜更緻密。 9.電子秤 用來精秤樣品之重量。 10.氣體流量計 用來控制氣體流量,單位為 sccm。
18
3.2 實驗流程
以下為本研究之實驗步驟總流程圖,本研究一共分成四個部分進行,分別是 氧化鋁基材前處理、鍍膜製程、陶瓷封裝製程、薄膜特性分析,而我們所使用之 氧化鋁基材可分為氧化鋁片及氧化鋁管,流程有些許不同,至下節討論。 圖 3.1 實驗步驟總流程圖基材前處理
金屬無電鍍薄膜製程 陶瓷膠定性分析 封裝漏氣測試 薄膜氣體分離測試 薄膜漏氣測試薄膜特性分析
鹼液清洗及乾燥 基材表面敏化及活化陶瓷封裝製程
SEM EDS XRD Pd Ag Cu Ni19
3.2.1 基材為氧化鋁片實驗示意圖
基材為氧化鋁片的部分,主要以薄膜特性分析為主,我們先求得各金屬無電 鍍之參數,並將沉積上去之金屬薄膜作特性分析,所製備之金屬薄膜分別有純鈀 薄膜、鈀銀合金薄膜、鈀銀銅合金薄膜以及鈀銀銅鎳合金薄膜,薄膜製備是使用 連續性無電鍍之製程,主要依各金屬之還原電位大小來決定鍍膜優先順序,而針 對各類薄膜使用 XRD、SEM、EDS 作特性分析,就氧化鋁片之實驗流程如下: 圖 3.2 氧化鋁片為基材之實驗步驟流程圖氧化鋁基材前處理
金屬無電鍍薄膜製程
薄膜特性分析
鹼液清洗及乾燥 基材表面敏化及活化 SEM EDS XRD Pd Pd Pd Pd Ag Ag Cu Cu Ni Ag20
3.2.2 基材為氧化鋁管實驗示意圖
基材為氧化鋁管時,偏應用取向,主要應用於氫氣分離系統,因此在這部分, 我們將前者氧化鋁片之無電鍍製程參數稍做修改後,直接成膜在氧化鋁管上,而 我們主要目標是做鈀銀銅鎳四元合金薄膜,重量比為 60:5:30:5,另外在鍍膜之前, 我們會先進行封裝製程,原因討論於 4.1 章節,封裝製程會先分析封裝材料,得 到最佳之封裝參數。封裝過後即無電鍍各金屬於基材上,接著作氣體測試及薄膜 分析。整體實驗流程如下: 圖 3.3 氧化鋁管為基材之實驗步驟流程圖氧化鋁基材前處理
金屬無電鍍薄膜製程
薄膜特性分析
鹼液清洗及乾燥 基材表面敏化及活化 SEM Pd Ag Cu Ni 陶瓷膠定性分析 封裝漏氣測試陶瓷封裝製程
TGA 陶瓷封裝 SEM 氣體分離測試 漏氣測試21
3.3 氧化鋁基材前處理
3.3.1 氧化鋁基材之清洗
1.將氧化鋁浸於鹼液中,以超音波震盪 20 分鐘。 2.以去離子水沖洗後,用氮氣吹乾。 3.置於丙酮中,以超音波震盪 60 分鐘。 4.將氧化鋁基材置於烘箱中,以 120°C 乾燥一個小時。 5.完成清洗動作。 表 3.4 鹼液配方及操作條件 NaOH,g/L 4 Temperature,°C 253.3.2 氧化鋁基材之敏化及活化
1.分別配製敏化液、活化液並加入磁石,放置於加熱板上使其充分均勻。 2.將氧化鋁基材置於敏化液中,敏化 5 分鐘。 3.用去離子水潤洗氧化鋁基材表面。 4.將氧化鋁基材置於活化液中,活化 4 分鐘。 5.用 0.01M 之鹽酸潤洗氧化鋁基材表面。 6.用去離子水潤洗氧化鋁基材表面。 7.重複步驟 2-6,次數 8-10 次。 8.氧化鋁基材表面發現有明顯的暗色產生,及完成敏化、活化步驟。22 9.置烘箱 120°C 乾燥 2 小時。 表 3.5 敏化液配方及操作條件[45] SnCl2
˙
2H2O,g/L 1 HCl(37%),ml/L 1 Temperature,°C 20 Duration,min 5 表 3.6 活化液配方及操作條件[45] PdCl2,g/L 0.1 HCl(37%),ml/L 1 Temperature,°C 20 Duration,min 43.4 陶瓷封裝製程
3.4.1 封裝材料定性分析
本研究使用之陶瓷封裝材料為商用陶瓷膠,成份為二氧化矽-氧化鋁。起初 我們遵循使用手冊上的說明進行陶瓷封裝,發現氣密性不佳而造成大量漏氣,因 此特別針對此陶瓷膠的性質做了以下分析。23
3.4.1.1 熱重分析儀分析
1.在氧化鋁片上塗佈陶瓷膠,於空氣下乾燥 1 個小時。 2.剝落氧化鋁片上之陶瓷膠,重量介於 2 至 10 mg,此範圍為 TGA 適用重量。 3.於空氣下做 TGA 分析。3.4.1.2 SEM 表面分析
1.在氧化鋁片上塗佈陶瓷膠,於空氣下乾燥 1 小時。 2.陶瓷膠做不同溫度之熱處理,選擇溫度分別為 950°C、1050°C、1150°C、 1250°C,並於特定溫度之下持溫燒結 1 小時。 3.SEM 表面觀察。3.4.2 封裝方法及步驟
此主要針對陶瓷膠經過熱處理之後,以其透氣性做測試。本研究參考未來封 裝的製程,封裝完畢後進行漏氣測試,漏氣量可由檢量線計算得出。檢量線之量 測方式及參數敘述於下小節中。本節就封裝方法及步驟敘述如下: 1.取二根緻密氧化鋁陶瓷管、一個緻密氧化鋁轉接頭,並將陶瓷膠塗佈於二 根緻密陶瓷管一端之外管,以及轉接頭內管塗滿,進行對接封裝。 2.空氣下乾燥 1 小時。 3.於管型爐中在 600°C 持溫 1 小時去除有機黏合物,再經高溫下燒結一個小 時,高溫燒結溫度由前者陶瓷膠 SEM 表面形貌之優劣決定,目前溫度決定為 1150°C 4.使用氣相層析儀分別做 25°C、600°C 之封裝漏氣測試,並由檢量線得漏氣 量。 而針對氧化鋁管形基材之封裝步驟如前述,不過我們封裝之形式為中間為多 孔氧化鋁管,二旁對接緻密陶瓷管,並塗布封裝材料,並上高溫燒結,達到密封 效果。圖 3.4 為封裝專用模具圖,我們使用橫向封裝,緻密陶瓷管及管形基材放24 置於套件上,套件都能自由滑動,目的是為了封裝平直。 圖 3.4 封裝專用模具圖
3.4.3 封裝材料漏氣測試
將於 3.6 節漏氣測試中詳細敘述3.5 金屬無電鍍製程
本研究金屬鍍膜方式採用無電鍍法,像三明治方式層列式的將金屬膜鍍上, 這種方式比混合式共析鍍法較能掌握鍍率及組成,金屬膜之先後順序是依照各金 屬之還原電位大小來排列,還原電位大之金屬優先鍍,若優先鍍上還原電位較小 之金屬,會跟還原電位大之金屬有置換反應,組成不易控制。 本研究所使用的氧化鋁基材分別有緻密氧化鋁片及多孔性氧化鋁管。先將金 屬膜無電鍍在氧化鋁片上,調整鍍膜參數及利用 SEM、EDS、XRD 等儀器分析25 其金屬膜性質後,依同樣配方無電鍍於多孔氧化鋁管,進行氣體測試。 理論膜厚之計算是以秤重減重法來計算膜厚,公式如式子(3.1)所示: 理論膜厚= ΔW / (A×D) (3.1) ΔW:無電鍍於氧化鋁基材之金屬重(g)。 A:氧化鋁基材之表面積(cm2) 。 D: 無電鍍於氧化鋁基材之金屬密度(g/cm2 ) 本實驗所使用之氧化鋁片為 5cm×5cm,使用前會利用鑽石刀切割成 1cm× 1cm 之氧化鋁片進行無電鍍,因此理論之氧化鋁片面積為 1 cm2。 無電鍍氧化鋁片部分,我們是將基材直接放入燒杯中無電鍍,而針對管形基 材,我們使用之無電鍍設備為特製壓克力製之模具,從一端將封裝好之基材放入, 最後再用鐵環將 O 環鎖緊,並用水測試會不會漏水。緻密陶瓷管二端用特製塞 子塞入,並纏上 PTFE 膠帶及 parafilm 避免滲水。最後再用 PTFE 膠帶包住二端 轉接頭部份,即可進行無電鍍。圖 3.5 為氧化鋁管形基材置入無電鍍裝置圖。
26
3.5.1 無電鍍法製備鈀膜
本研究嘗試將鈀金屬薄膜分別無電鍍於氧化鋁片、多孔氧化鋁管,以下統稱 氧化鋁基材。 1.配製鈀無電鍍液,配方如表 3.7,加熱至 50°C。 2.將敏化、活化過之氧化鋁基材浸於無電鍍液中,預溫至 50 °C。 3.使用聯胺為還原劑,添加至無電鍍液中進行無電鍍。若無電鍍於氧化鋁管 上時,則多一道抽真空步驟。 4.無電鍍結束後,以清水洗淨,放置 120°C 烘箱乾燥。 5.秤重量計算量得理論鈀膜厚度。氧化鋁管免此步驟。 6.進行熱處理,接著薄膜分析。3.5.2 無電鍍法製備鈀銀合金膜
1.配製銀無電鍍液,配方如表 3.8,加熱至 50°C。 2.將鈀無電鍍過之氧化鋁基材浸於銀無電鍍液中,預溫至 50 °C。 3.使用聯胺為還原劑,添加至無電鍍液中進行無電鍍。若無電鍍於氧化鋁管 上時,則多一道抽真空步驟。 4.無電鍍結束後,以清水洗淨,放置 120°C 烘箱乾燥。 5.秤重量計算量得理論銀膜厚度。氧化鋁管免此步驟。 6.進行熱處理,接著薄膜分析。3.5.3 無電鍍法製備鈀銀銅合金膜
1.配製銅無電鍍液,配方如表 3.9,加熱至 50°C。 2.將鈀、銀無電鍍過之氧化鋁基材浸於銅無電鍍液中,預溫至 50°C。27 3.使用聯胺為還原劑,添加至無電鍍液中進行無電鍍。若無電鍍於氧化鋁管 上時,則多一道抽真空步驟。 4.無電鍍結束後,以清水洗淨,放置 120°C 烘箱乾燥。 5.秤重量計算量得理論銅膜厚度。氧化鋁管免此步驟。 6.進行熱處理,接著薄膜分析。
3.5.4 無電鍍法製備鈀銀銅鎳合金膜
1.配製鎳無電鍍液,配方如表 3.10,加熱至 65°C。 2.將鈀、銀、銅無電鍍過之氧化鋁基材浸於鎳無電鍍液中,預溫至 65°C。 3.使用聯胺為還原劑,添加至無電鍍液中進行無電鍍。若無電鍍於氧化鋁管 上時,則多一道抽真空步驟。 4.無電鍍結束後,以清水洗淨,放置 120°C 烘箱乾燥。 5.秤重量計算量得理論鎳膜厚度。氧化鋁管免此步驟。 6.進行熱處理,接著薄膜分析。 表 3.7 鈀無電鍍液配方及操作條件[46] PdCl2,g/L 0.36 Na2EDTA,g/L 67 N2H4(0.1 M),ml/L 50 NH4OH,ml/L 650 Temperature,°C 5028 表 3.8 銀無電鍍液配方及操作條件[46] AgNO3,g/L 2.45 Na2EDTA,g/L 67 N2H4(0.1M),ml/L 50 NH4OH,ml/L 650 Temperature,°C 50 表 3.9 銅無電鍍液配方及操作條件[47-49] CuSO4
˙
5H2O,g/L 3.1125 Na2EDTA,g/L 20.1 NaOH,g/L 20 TritonX-100,mg/L 25 N2H4(1M),ml/L 10 Temperature,°C 50 表 3.10 鎳無電鍍液配方及操作條件[50, 51] NiSO4˙
6H2O,g/L 42.05 Na3C6H5O7˙ 2H2O,g/L 58.82 NaOH,g/L 6.8 N2H4(1 M),ml/L 10 Temperature,°C 6529
3.6 氣體測試及分析
3.6.1 氣體分離整體裝置設計圖
氣體整體之實驗裝置設計圖如圖 3.6,系統中所使用之載流氣體為氮氣,測 試氣體因測試類型有所不同,測試類型於下小節討論,另外,載流氣體所使用之 流量計規格最大流量為 1000 sccm,測試氣體之流量計之規格最大流量為 50 sccm, 而實際流量也依照測試類型而做流量調整。圖 3.7 為氣體測試實驗裝置實際圖。 圖 3.6 實驗裝置平面示意圖30 圖 3.7 實驗裝置實際圖
3.6.2 各氣體檢量線建立
檢量線之建立主要是依照與氮氣不同相對濃度下調整各氣體之流量,整體流 量為 50 sccm。建立之檢量線一共有氫氣、氦氣、二氧化碳 3 種類型。操作方法 我們是將檢量線氣體與氮氣在石英管中混合過後,接著送入 GC 測試,並依照不 同濃度可以得不同波鋒面積,建立波鋒面積與濃度之關係圖即我們之檢量線。圖 3.8 為檢量線建立之平面圖。 高溫爐 氣體分離系統 GC31 圖 3.8 檢量線實驗裝置平面示意圖
3.6.3 漏氣測試
漏氣測試可分為封裝材料及金屬合金薄膜漏氣測試,主要是將測試樣品放入 石英管中,在石英管中通入測試氣體,測試氣體分為氦氣跟二氧化碳,而在陶瓷 管內通入氮氣當作載流氣體,如封裝材料、金屬薄膜有破洞的話,偵測氣體即可 滲透進去而被 GC 偵測,接著我們在對檢量線算出其濃度,再換算成流量,即可 得到漏氣率,測試溫度我們分為 25°C 與 600°C。表 3.11 為漏氣測試條件,圖 3.9 為漏氣測試平面示意圖。 表 3.11 封裝漏氣測試條件Temperature Detecting gas Carrier gas Application
25°C He N2 He leakage test
600°C He N2 He leakage test
25°C CO2 N2 CO2 leakage test
32 圖 3.9 漏氣測試實驗裝置平面示意圖
3.6.4 氫氣測試
氫氣測試一共分成二部份:純氫之滲透率及氫氣、二氧化碳分離選擇比,並 且於 350°C、400°C、450°C、500°C 下進行,載流氣體與偵測氣體分別各 50 sccm, 實驗裝置如圖 3.6,另外為了避免氫氣並非經由薄膜滲透,我們額外用氦氣測漏, 並用氫氣流量扣掉氦氣流量,得到真正經由薄膜通過之量。表 3.12 為各種氣體 測試條件。 表 3.122 各種氣體測試條件Temperature Detecting gas Carrier gas Application
300-600°C H2 N2 Pure H2 Permeation
300-600°C He N2 He leakage test
300-600°C H2+CO2 N2
H2+CO2 Separation
33
第4章 實驗結果與討論
4.1 封裝材料性質分析
4.1.1 封裝材料 TGA 分析
本研究使用之封裝材料為二氧化矽-氧化鋁陶瓷膠,主要經高溫燒結過後, 可以使內部之玻璃質成份燒結在一起,在燒結步驟前,會有一道去除有機黏合物 (binder)之步驟,稱之為 De-binder process,為了燒掉所有的黏合物,必須要有一 道最佳之去黏合劑溫度,因此我們使用 TGA 來測得此陶瓷膠的去黏合物溫度。 圖 4.1 為封裝材料之 TGA 分析圖,大部分黏合物都是一些有機分子所組成, 在空氣的環境下,溫度在 500°C 以前就可將大部分的有機黏合物燒光,為了確定 能將所有有機黏合物驅趕走,我們將去黏合物溫度設在 600°C,並由 TGA 驗證。 由 TGA 之測試結果顯示,在 300°C 以前樣品的重量大幅下降,這是因為有機黏 合物與氧氣結合後變成碳化合物離開樣品,是樣品重量減輕的最主要因素,因此 做陶瓷膠高溫燒結時,300°C 以前之升溫速率不可過快,以免大量的黏合物燃燒 後快速的離開樣品而造成大量的破洞,達不到預期的封裝效果。而在 300°C 至 600°C 之間,仍有黏合物逸散,只是速率較 300°C 前和緩許多。到 600°C 時有持 溫,重量也不再變化,故將最高去黏合物溫度設定在 600°C,以確保所有的黏合 物可以跟氧氣結合而逸散。34 圖 4.1 封裝材料之 TGA 分析圖
4.1.2 封裝材料不同燒結溫度之 SEM 分析
本研究使用之封裝材料為二氧化矽-氧化鋁陶瓷膠,二氧化矽是玻璃質材料, 在高溫燒結時,玻璃粒子可以黏合,達到密封效果。本章節就依不同之燒結溫度 後之陶瓷膠,用 SEM 來觀察其表面玻璃粒子黏合的效果,進而得到最佳之燒結 溫度。圖 4.2 分別是不同溫度燒結後,封裝材料表面玻璃粒子之形貌。 由圖可以發現在溫度 1000°C 以下燒結時,陶瓷膠表面還是有很多孔洞分佈, 且洞的大小在 10 μm 以上,研判此結構下對氣體的氣密性較差,反觀溫度升至 1000°C 以上時,陶瓷膠表面之玻璃質有燒結成團的現象,使孔洞的分佈及大小 有明顯的改善,由圖中也發現燒結至 1150°C 的效果最好,玻璃粒子有燒結成塊 的現象,洞的大小低於 10 μm;而溫度達到 1250°C 時,發現孔洞率反而增加了, 推測因為溫度過高,導致玻璃粒子間有熔融的現象,洞的大小約略在 10 μm,在 高溫燒結過後內部微結構孔洞變多、變大,推測此微結構密封效果不大,因此陶 瓷膠無法至 1250°C 溫下燒結,最後我們選擇 1150°C 為最高燒結溫度。35 圖 4.2 封裝材料於不同溫度下燒結後之 SEM 圖 (a)950°C (b)1050°C (c)1150°C (d)1250°C。
4.1.3 封裝材料之 GC 漏氣測試
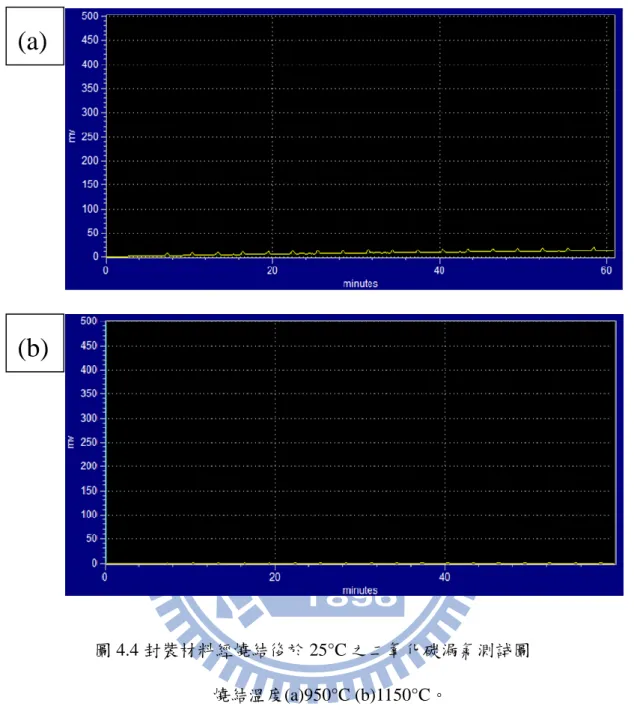
我們主要測試在經過 1150°C 燒結後,封裝材料的密閉效果,因為本研究主 要是作氫氣與二氧化碳分離膜,因此在封裝上最理想的情況是可以完全的隔絕氫 氣與二氧化碳漏氣,而在文獻上指出漏氣測試通常使用氦氣來取代氫氣,其原因 可大致推估為氦氣粒子較氫氣小,若氦氣可達到氣密效果,則可確定氫氣達到完 全密封。 在作氣體測試之前,本實驗先建立起氦氣及二氧化碳之檢量線。我們測試的 溫度分別為 25°C 及 600°C,接著進入 GC 量測,在積分波鋒面積後,計算其漏(a)
(b)
(c)
(d)
36 氣量。我們也額外做了在 950°C 燒結之對照組,比較與 1150°C 燒結之差別。圖 4.3 為陶瓷管與轉接頭塗佈封裝材料並經過 1150°C 燒結後之成品圖。 圖 4.3 塗佈封裝材料之陶瓷管對接後經高溫燒結後成品圖 (a)整體部分 (b)接頭部份 圖 4.4 為陶瓷膠分別在 950°C、1150°C 燒結後,使用 GC 於 25°C、二氧化碳 氣氛下之漏氣測試圖。因為二氧化碳之熱導率較氮氣小,而我們 GC 使用的載流 氣體是氮氣,因此二氧化碳的波峰應為負值,由圖可知,無論在 950°C 或 1150°C 之測量結果,都沒有發現負波峰,代表這二個燒結參數於室溫下都可以有效的阻 抗二氧化碳漏氣;之後我們將測試溫度上調至 600°C,並觀察其波峰變化。圖 4.5 為陶瓷膠分別在 950°C、1150°C 燒結後,於 600°C 下、二氧化碳氣氛中之 GC 測 量圖,由圖我們也未發現負波峰產生,這代表陶瓷封裝製程在 600°C 下也可以有 效的阻抗二氧化碳滲透,另外,在 600°C 測量中二者圖中都有強度 10 mv 的波峰 出現,推測是在分離系統中卡榫沒有鎖死,造成些為空氣進入形成干擾,不過其 訊號弱,對分離系統不會造成太大影響。
(a)
(b)
37
圖 4.4 封裝材料經燒結後於 25°C 之二氧化碳漏氣測試圖 燒結溫度(a)950°C (b)1150°C。
(a)
38 圖 4.5 封裝材料經燒結後於 600°C 之二氧化碳漏氣測試圖 燒結溫度(a)950°C (b)1150°C。 接著,我們再進行氦氣測漏,氦氣熱導率高,因此在載流氣體為氮氣的中, 其波峰為正值。測試的樣品及參數也如同二氧化碳漏氣測試,我們比較室溫下氦 氣之漏氣。圖 4.6 為室溫下之氦氣漏氣測試。由室溫測試的結果得知,950°C 下 燒結在氦氣測漏中,強度達 1200 mv;反之在 1150 度下燒結之氦氣強度大約 900 mv,因此在 1150°C 下燒結可有較高之氦氣阻抗能力,其結果與 SEM 圖中預算 符合。 我們接著經由檢量線計算氦氣漏氣百分比,陶瓷膠在 950°C 下燒結後,其漏 氣量約 9.43%;而 1150°C 下燒結後,其漏氣量為 6.25%,其值表示我們的陶瓷 封裝製程尚未能完全的阻抗氦氣漏氣,但是針對二氧化碳而言,我們已可有效的 阻抗。因此,針對陶瓷封裝氫氣漏氣問題,我們打算用氦氣測漏,接著再用氫氣
(a)
(b)
39 通量減掉氦氣測漏量,得到氫氣實際上經由薄膜之滲透量。 圖 4.6 封裝材料經燒結後於 25°C 之氦氣漏氣測試圖 燒結溫度(a)950°C (b)1150°C。
(a)
(b)
40
4.2 鈀膜於緻密氧化鋁片之無電鍍製程
4.2.1 無電鍍時間及鍍液濃度對鈀膜厚之影響
為了得到較好之鍍膜參數,我們先試著將鈀膜無電鍍於表面與之後應用於氧 化鋁管相似粗糙之氧化鋁片上,由秤重減重法計算理論之膜厚,再由 SEM 驗證。 圖 4.7 是利用秤重減重法計算理論之膜厚所得到的結果,可看出不同鍍膜時 間及聯胺濃度對膜厚之影響,聯胺濃度為 0.001 M 及 0.005 M,鍍膜時間分別是 2、4、6、8 小時,由圖可觀察到,二種濃度聯胺的還原之下,厚度都隨時間成 長,0.005 M 之聯胺因為濃度大因此鍍膜速度較快,且厚度呈穩定性的成長,而 0.001 M 下之聯胺相較之下鍍率較慢,且 6 小時過後,鍍率有變慢的趨勢,是與 0.005 M 聯胺之最大差別,推測為雖然 0.005 M 含有較多之聯胺,反應速度較快, 但整體反應並無出現鈀粒子在溶液中均勻性成核(Homogenous nucleation)的現象, 鈀粒子都沉積於氧化鋁基材上,因此儘管提供的聯胺再多,但是反應面積有限, 而造成膜厚可以穩定增加的主因;反之,0.001 M 之聯胺的量較少,且反應過程 中也有些許聯胺蒸發逸散,導致 6 小時後鍍率減緩。圖中發現使用 0.005 M 之聯 胺經過 2、4、6、8 小時後可分別得到厚度為 1、2、3、4 μm,而使用 0.001 M 聯胺 8 小時最多只能得到 1-1.5 μm 後之薄膜,在 0.001 M 聯胺之參數是較不符合 時間成本,因此接下來之鈀鍍膜參數,我們都採用 0.005 M 聯胺進行還原。 圖 4.7 鈀無電鍍時間及鍍液濃度與膜厚之關係圖41 我們使用 SEM 來驗證理論膜厚計算的結果。圖 4.8 分別為聯胺濃度 0.005 M 分別無電鍍 2、4、6、8 小時之結果,可觀察到其厚度與秤重減重法相差不遠, 其誤差造成的主因為秤重時因機況所造成的誤差。 圖 4.8 使用聯胺濃度 0.005 M 於不同時間下鈀無電鍍之 SEM 側視圖 (a)2 小時 (b)4 小時 (c)6 小時 (d)8 小時 無電鍍製程之膜厚控制,除了與反應物之濃度、溫度相關外,也與單位面積 所反應的無電鍍液體積有關。上述之鈀無電鍍之鍍液體積與無電鍍面積比為 50 ml/cm2,此比例在 2 小時後即可得到 1 μm 之鈀膜,但是絕大部分之鈀粒子仍在 鍍液中未被沉積,因此造成浪費,我們也曾考慮過將原鍍液繼續無電鍍下一個氧 化鋁基材,但考慮到花費時間成本而作罷,最後我們選擇以鍍液體積與無電鍍面 積比為 12.5 ml/cm2做為無電鍍參數,並且經實驗證實無電鍍 4 小時後可以得到 膜厚為 1 μm 之鈀膜,此鍍膜效率可提升,且大幅降低鍍液成本。 圖 4.9 為鍍液體積與無電鍍面積比為 12.5 ml/cm2做 4 小時無電鍍之 SEM 側 視圖,由圖可知膜厚約在 1 μm 左右,與我們預期之厚度相符,此後若要鍍 1μm
(a)
(b)
(c)
(d)
42 之鈀膜,我們將選用此參數製程。 圖 4.9 鍍液體積與無電鍍面積比 12.5 ml/cm2之 4 小時鈀無電鍍 SEM 側視圖
4.2.2 退火溫度對表面形貌之影響
本研究之製程為先封裝再鍍膜,主要考量在於鈀膜是否能忍受 1150°C 之高 溫,因為先前封裝參數為 950°C 燒結,故我們做鈀膜高溫測試時是在 950°C 下 進行,若 950°C 下出現破損,可推測在 1150°C 時鈀膜無法負荷。 測試的鈀膜同樣是在 0.005 M 下之聯胺分別在 2、4、6、8 小時還原下得到, 測試參數為在 H2/Ar 為 5/95 之莫耳比例、950°C 下進行。圖 4.10 為測試前之鈀 膜表面形貌之 SEM 圖,可觀察到在四個不同鍍膜時間下所得到之鈀膜都非常平 整,表面沒有孔洞,鈀膜也無聚集成團之現象,表面均勻性一致。圖 4.11 為經 過 950°C 高溫測試後之 SEM 圖,發現在膜厚低於 3 μm 時,鈀膜出現聚集的現 象,而造成膜破損,因為在 950°C 時接近鈀的熔點,因此鈀會有熔融產生,為了 降低與基材的表面能,表面張力會讓整個鈀膜內聚,而造成孔洞產生;鈀膜厚度 達到 4 μm 時,雖然底下鈀膜內縮,但上方之鈀可覆蓋破損面,因此由 SEM 觀43 察到幾乎沒有破洞產生,故若要在 950°C 下操作時,鍍膜時間至少要 6 小時。本 研究選擇先封裝再鍍膜之原因在於,我們封裝最後決定在 1150°C,因此鈀膜勢 必要更厚才能承受此高溫,然而膜厚會影響氫氣流量,二者是成反比,因此為了 得到較高之氫流量,我們希望膜厚在於 1-2 μm 間,因此,我們選擇先封裝再鍍 膜。 圖 4.10 不同鈀無電鍍時間退火前之 SEM 俯視圖 (a)2 小時 (b)4 小時 (c)6 小時 (d)8 小時
(c)
(d)
(b)
(a)
44 圖 4.11 不同鈀無電鍍時間於 950°C 退火後之 SEM 俯視圖 (a)2 小時 (b)4 小時 (c)6 小時 (d)8 小時。
4.2.3 鈀膜於氫氣下退火後之 XRD 分析
本研究退火的過程在 600°C、還原氣氛下操作 12 小時,Takuji Ikeda 研究指 出,鈀銀/氧化鋁複合薄膜在氫氣環境下,氧化鋁會被還原成鋁[13],進而與鈀合 金薄膜產生互擴散(inter-diffusion)效應,使鈀合金膜膜產生雜質而降低氫氣滲透 量。為了驗證實驗過程是否產生氧化鋁被還原現象,我們在退火過後用 XRD 來 確認,圖 4.12 為鈀膜/氧化鋁退火前後之 XRD 圖。 由 JCPDS 之資料顯示,XRD 圖中鈀金屬之波峰為 2θ 為 40.2°、46.7°、68.0°、 82.3°、86.7°為鈀 FCC 結構之特性峰,可知退火前後波峰出現位置沒有改變,也 沒有雜相產生,代表氧化鋁沒有被還原成鋁金屬而與鈀形成合金,也說明了在 600°C、氫氣氣氛中,鈀薄膜可以安定的存在。(b)
(a)
(c)
(d)
45 圖 4.12 鈀膜/氧化鋁退火前後之 XRD 圖 (a)退火後 (b)退火前
4.3 鈀銀膜於緻密氧化鋁片之無電鍍製程
4.3.1 無電鍍時間對銀膜沉積量之影響
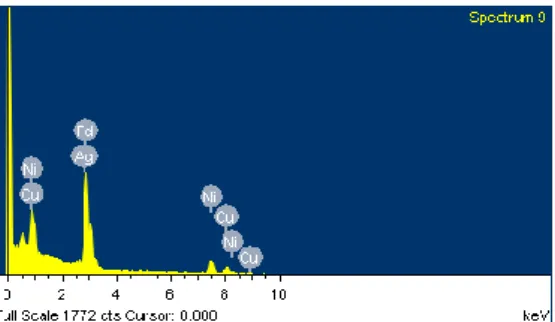
本研究之鈀合金製備方式,是連續性地將金屬一道道的無電鍍上去,最後再 用熱處理的方式使成為合金。鍍銀的製程中,我們以 1 μm 厚之鈀膜當成模板 (template),再將銀無電鍍上去,最後退火形成合金。 我們使用之無電鍍主要建立在無電鍍液沒有產生均勻性成核(Homogenous- nucleation)的前提之下,其優點為可以避免大量的前驅鹽直接在溶液中反應造成 浪費,也較能控制厚度。預期的無電鍍鈀銀合金複合薄膜成份為鈀銀重量比例 是:77:23、91:9、93:7,分別用來製作鈀銀合金(77:23)、鈀銀銅合金(68:7:25)、鈀 銀銅鎳合金(60:5:30:5)複合薄膜,因此時間控管相當重要。 為了得到鈀銀合金薄膜預期之成份,我們嘗試在已鍍上鈀膜之氧化鋁片上進 行不同沉積時間之無電鍍銀製程,時間分別為 2、4、6、8 小時,並由秤重減重46 法量測無電鍍前後之重量差,計算理論膜厚,最後用 SEM 觀察並驗證。 圖 4.13 為不同無電鍍銀之沉積時間與其經秤重減重法計算理論膜厚之關係 圖,由圖可觀察到隨著鍍膜時間增加,膜厚也隨著穩定成長,但 4 個小時過後可 發現鍍率明顯下滑,研判是銀的前驅鹽與聯胺因為反應後濃度下降,造成反應速 度降低,即使 4 小時後鍍率降低,尚可鍍上我們預期銀的厚度。 圖 4.13 銀無電鍍時間與膜厚之關係圖 理論計算厚度之後,接著使用 SEM 觀察實際的厚度,由於我們是將銀無電 鍍在鍍有鈀之基材上,因此在厚度的量測上尚要再加鈀膜的厚度才可推估,鈀膜 的厚度平均控制為 1 μm 左右,因此在加上銀之厚度後,銀鍍膜時間 2、4、6、8 小時之鈀銀膜的理論厚度分別為 1.25 μm、1.5 μm、1.7 μm 及 1.9 μm 左右。 圖 4.14 為不同無電鍍銀鍍膜時間之 SEM 側視圖,因為我們是鍍在粗糙之氧 化鋁片上,造成膜表面有些為起伏,但由圖可觀察到實際膜厚與理論膜厚相近, 顯示對鈀銀合金膜之膜厚可先用秤重減量法先初估膜厚。
![圖 2.1 氫氣於金屬中之傳導機制[4]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8598757.190031/20.892.141.757.125.857/圖21氫氣於金屬中之傳導機制4.webp)