國
立
交
通
大
學
應用化學系碩士班
碩
士
論
文
利用共振腔振盪衰減光譜法研究 CH
3OO 在 C
H 伸展
振動模光區的紅外吸收光譜
Infrared absorption spectra of CH3OO in the CH stretch vibrational
modes region detected with cavity ring-down spectroscopy
研 究 生:林震洋 (Chen-Yang Lin)
指導教授:李遠鵬 博士 (Dr. Yuan-Pern Lee)
利用共振腔振盪衰減光譜法研究 CH3OO 在 CH 伸展振動模光區的紅
外吸收光譜
Infrared absorption spectra of CH3OO in the CH stretch vibrational
modes region detected with cavity ring-down spectroscopy
研 究 生:林震洋 Student:Chen-Yang Lin 指導教授:李遠鵬 Advisor:Dr. Yuan-Pern Lee
國 立 交 通 大 學 應用化學系碩士班
碩 士 論 文
A Thesis
Submitted to M. S. Program, Department of Applied Chemistry College of Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Applied Chemistry June 2011
Hsinchu, Taiwan, Republic of China
I 摘要 利用波長為 193 nm 的雷射光解流動的 CH3C(O)CH3 / O2混合氣體和波 長為 248 nm 的雷射光解流動的 CH3I / O2混合氣體,並以共振腔振盪衰減法 取得其共同產物 CH3OO 的紅外吸收光譜。吾人將 2953.4 cm 1和 3020.7 cm1 的吸收峰分別指派為 CH3OO 的 ν2和 ν9,與黃登瑞等人得到同為氣態環境下 的 CH3OO 低解析度光譜之 ν2和 ν9的位置 2954 cm 1和 3020 cm1一致。此 結果亦和 Nandi 與 Morrison 兩研究組分別在 Ar 間質與 He 奈米液滴環境下 得到ν2和ν9的相對位置一致,並與 B3LYP/aug-cc-pVTZ 計算得到之非簡諧 振動頻率相差在 1%以內。在吾人光譜中並未能指認出可能是 ν1的吸收峰, 可能是 ν1譜線較弱且結構缺乏一明顯的吸收峰所致。此外,吾人以近似對 稱陀螺分子之模式,分析 CH3OO 的轉動譜線結構而得振動基態及 ν2和 ν9 的轉動常數,和 Endo 研究組以及理論計算得到之結果一致。吾人利用 SpecView 光譜模擬程式模擬ν2和 ν9之光譜並與實驗光譜做對照,亦模擬 ν1 並討論其可能的躍遷原點位置。對 ν2而言,模擬光譜大致上和實驗光譜吻 合,但因模擬軟體未考慮 ν2可能受到 coriolis coupling 之影響,導致模擬光 譜和實驗光譜在 29402950 cm1的光區範圍內有所差異。而 ν9的實驗光譜 在 3014 cm1和 3017 cm1出現沒有對應到模擬光譜之譜線,且在 3025 3050 cm1 之光區,實驗和模擬光譜並不一致,吾人推測這些不一致的譜線應是 受到 ν1吸收之影響。經由改變 ν1模擬光譜的躍遷原點並將其和實驗光譜比
II 較,吾人暫時將 ν1 的躍遷原點指派為 3031.7 cm 1 。此外 ν9 的半高寬比 SpecView 模擬程式預測的半高寬為大,應是和 ν1譜線重疊,或是 CH3OO 內轉動運動造成的譜線分裂所致。而此內轉動運動所引起的譜線分裂,亦 是造成 ν1的 Q 分枝和模擬光譜不一致的可能原因。ν2的平行躍遷扭動分裂 極小,因此譜線半高寬和模擬光譜所得之結果一致。
III Abstract
We employed a cavity ringdown spectrometer with a tunable infrared OPO/OPA laser with a bandwidth of 0.02 cm-1 to record the absorption spectra of methylperoxy radicals (CH3OO) in the range 2930-3050 cm
-1
. Methylperoxy radicals were produced by irradiating a flowing mixture of CH3I and O2 with emission at 248 nm from a KrF excimer laser or a flowing mixture of CH3C(O)CH3 and O2 with emission at 193 nm from an ArF excimer laser. Two absorption bands with origins at 2953.4 cm-1 and 3020.7 cm-1 were observed; they are assigned to ν2 (symmetric CH3 stretching) and ν9 (antisymmetric CH2 stretching) modes of CH3OO, respectively. We analyzed the rotational structures of the ν2 and ν9 bands by simply treating CH3OO as a prolate symmetric top, and determined the rotational constants both in the ground state and in the vibrationally excited state. We predicted vibrational wavenumbers and rotational parameters for the upper and lower vibrational states, and the mixing ratio among a-, b-, c-types of bands of CH3OO with the B3LYP/aug-cc-pVTZ density-functional theory. The rotational contours for the ν1, ν2 and ν9 bands of CH3OO were simulated with the SpecView software. For the ν2 band, the simulation agrees satisfactorily with the experimental observations except for the intense peaks with regular spacing about 2.4 cm-1 in the range 2940-2950 cm-1. For the ν9 band, the simulation result is consistent with the experimental observations in the region 3000-3020 cm-1 but not in the region 3020-3050 cm-1. The discrepancy might be due to the interference from the ν1 band. That ν1 band
IV
is unobserved is likely due to its relatively small intensity. We temporarily assigned the ν1 band to be at 3031.7 cm
-1
by matching the simulated spectra with the peaks which do not correspond to the ν9 band.
V 謝誌 從升大三的暑假開始在李遠鵬老師的實驗室當小小專題生,經過四年的 學習,現在終於要畢業了。在這段時間裡,最感謝的當然就是李遠鵬老師。 實驗上老師時常給予我有用的建議,讓我可以順利突破瓶頸,而在寫論文 或研討會摘要時,老師也不惜花費自己的時間幫我訂正了許多錯誤。這些 對於我的學習都有莫大幫助,尤其是在邏輯思考能力的訓練方面更是顯著。 此外,我也要謝謝 Tanaka 和 Matsui 老師教會我許多光譜學和動力學上的知 識,使我分析實驗結果時不會毫無頭緒。當然也必須謝謝清大的朱立岡學 長,無論是課業還是論文方面,都給了我不少建議與啟發。 這四年待在實驗室的生活是非常快樂的,這都要感謝我在實驗室的戰友 們!大學長鄭棋文是教會我架設 CRDS 系統的老師,同時也是打籃球和壘 球的好夥伴。Megan 學姊在我分析光譜時給予了很大的幫助,也特別謝謝 她在我生日時體貼地為我準備奶皇包、啤酒和嘔吐袋,讓我三個願望一次 滿足。李俞範學姐無論在做實驗還是玩小遊戲上都有兩把刷子,她認真不 懈的態度是我學習的榜樣。皇上是攝影的高手,向他請教拍照的技術或相 機的問題總是讓我獲益良多。Momo、巴山塔和芭芭拉是我的英文小老師, 和他們聊天讓我英文因此進步不少。同屆的傅龍和林書毓是我在實驗室的 最佳夥伴,除了討論做實驗的技巧與心得之外,也時常分享心事。在這四 年患難與共的日子裡,每當我們煩悶時就在戶外聽聽「風聲」,聊些以後
VI 工作賺大錢後就要蓋一棟棟「富饒之城」之類的事。多虧他們才使我每次 遭遇挫折時仍能奮勇向前。此外還要感謝實驗室的學弟妹們,Wade 在我做 實驗時幫了不少忙,吳大常常跑來幫我對雷射系統的光,菜哥、喜德和奕 安提供不少他們系統的資訊讓我做實驗時可以有個參考。當然也不能忘了 實驗室助理月貴和建亨在採買實驗耗材與零件上給予相當大的幫助,讓我 能全心全意投入在實驗上。 除了實驗室的大家,刁老師家的巧克力學長和謝明修學弟也帶給我不少 的歡樂,每個禮拜一和他們打籃球是我在交大最開心的時光之一。而在隔 壁家做動力學實驗的陳威宇和我同為竹中人,跟他講話總是特別有親切感。 必須感謝的還有國際化學年認識的夥伴們,和他們遊山玩水的日子裡,不 僅增廣我不少見聞,也讓平日累積的壓力有了宣洩的出口。而從小就認識 的凱文兄、阿福、阿陳、鴨子和學弟,總是在聚會或出遊時不忘找我一起 同樂。此外,我也要感謝系壘夥伴們,與他們一起比賽奮鬥的日子,是我 四年來最美好的回憶之一。最後,我必須感謝在背後默默支持我的家人們, 當我不順遂時給予我安慰與鼓勵,也體諒我時常因為忙碌而無法回家。每 次家裡捎來的關心,總使我內心感到相當溫暖。我想把我這篇論文的完成, 獻給我最親愛的家人們。 林震洋 2012 年 6 月 交大
VII 目錄
第一章 緒論 ... 1
參考文獻 ... 10
第二章 實驗原理與技術 ... 12
2.1 共振腔振盪衰減法(cavity ringdown spectroscopy, CRDS)簡介 ... 12
2.1.1 共振腔振盪衰減法的發展與歷史 ... 12
2.1.2 光子彈模型 (photon bullet model) ... 14
2.1.3 靈敏度 (sensitivity) ... 16
2.1.4 腔模 (cavity modes) ... 17
2.1.5 雷射頻寬效應 (laser bandwidth effect) ... 20
2.1.6 模匹配 (mode matching) ... 22
2.1.7 CRDS 技術的優點 ... 22
2.2 可調頻紅外雷射光源 (tunable IR laser) ... 24
2.2.1 混頻(frequency-mixing)作用 ... 24
2.2.2 光學參量振盪器(optical parametric oscillation, OPO) ... 27
2.2.3 光學參量放大器(optical parametric amplifier, OPA) ... 28
參考文獻 ... 34 第三章 實驗系統與實驗步驟 ... 36 3.1 實驗裝置 ... 36 3.1.1 可調頻紅外雷射光源 ... 36 3.1.2 反應系統 ... 37 3.1.3 光解雷射 ... 38 3.1.4 其他周邊儀器 ... 38 3.2 實驗前準備事項 ... 39 3.2.1 可調頻紅外雷射光源的對正 ... 39 3.2.2 振盪衰減腔體的對正 ... 42
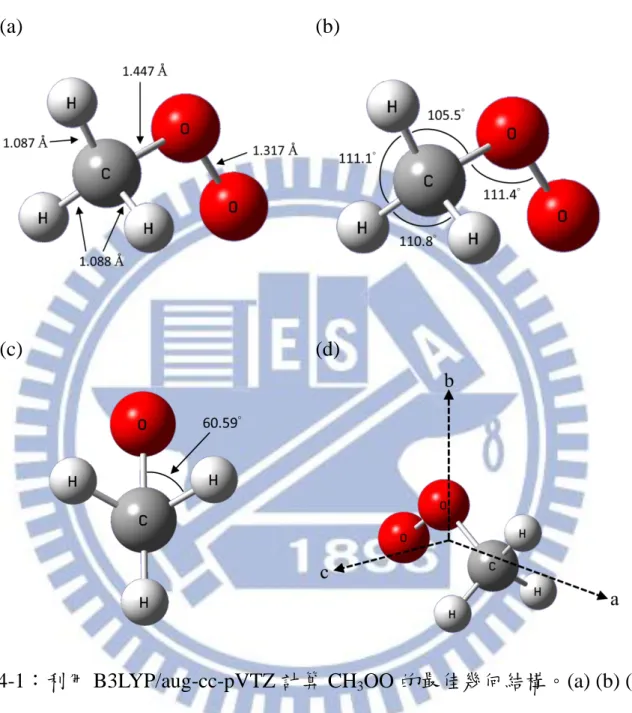
VIII 3.2.3 模匹配之步驟 ... 44 3.2.4 氣體流量校正 ... 45 3.3 實驗步驟 ... 46 3.3.1 周邊儀器時序控制... 46 3.3.2 實驗條件 ... 47 3.3.3 LabView 程式設定 ... 50 參考文獻 ... 57 第四章 結果與討論 ... 58 4.1 理論計算 ... 58
4.2 對稱陀螺剛體轉子(symmetric top rigid rotor)模型 ... 59
4.3 實驗結果與討論 ... 61 4.3.1 ν2振動模的分析 ... 65 4.3.2 ν9振動模的分析 ... 72 4.3.3 ν1振動模的討論 ... 74 4.4 結論 ... 76 參考文獻 ... 106
1 第一章 緒論 有機過氧自由基(ROO,R 代表烷基團或醯基團)是對流層(troposphere) 中碳氫化合物氧化後的初期產物[1, 2, 3]。大氣中諸如 OH 等具有強氧化力 的自由基會抓取碳氫化合物上的氫原子,使之形成有機自由基( R‧),這些 有機自由基會和氧氣快速地進行三體反應而產生過氧化物自由基[1] 。 (1-1) (1-2) 過氧化物自由基和 NO 反應後會產生 NO2,進而生成臭氧(O3): (1-3) (1-4) (1-5) 此外,由於 NO 和碳氫化合物為燃燒後的重要產物,因此反應(1-3)至(1-5) 將導致工業區中臭氧煙霧(ozone smog)的形成[4],此乃都市空氣汙染的元凶 之一。 甲基自由基(CH3)是有機自由基中結構最簡單者,它被氧氣氧化的主要 途徑有三種: kJ mol−1 (1-6) kJ mol−1 (1-7) kJ mol−1 (1-8) 反應(1-6)是一個沒有活化能障礙的三體反應,形成 CH3OO 並放熱 129.3 kJ
2
mol−1[5],其反應速率隨總壓力變化。若 CH3OO 擁有足夠的內能,則可能 越過 57.3 kJ mol−1的能障[6],進行反應(1-7)生成 CH2O OH,或是吸熱 120.1 kJ mol−1,進行反應(1-8) 生成 CH3O O [7]。
Walsh[6]利用 CASSCF/ICCI 方法搭配 ANO 基底函數計算 CH3和 O2的 反應位能面。CH3和 O2( )反應生成 態的 CH3OO,其對稱面上末端氧 的類 2p 軌域只有填一個電子,因此碳上的氫原子可遷移到氧上,分解出 OH 基生成 CH2O。而 CH3和 O2( )生成 態的 CH3OO,有兩個電子填 在末端氧的類 2p 軌域,導致氫原子難以遷移到末端氧上。雖然如此, 態 的 CH3OO 仍可經由交叉點(crossing point)通到 態 CH3OO 的位能曲面, 進而斷 OO 鍵產生 CH2O OH。Jafri 和 Phillips 等人[8]利用擬二級組態作 用法(pseudo-second-order configuration interaction)計算出 CH3OO 的 CO 和 OO 鍵的平衡鍵長分別為 1.454 Å 和 1.355 Å ,以及 CH3OO 斷 CO 鍵生成 CH3 O2 的解離能為 194.1 kJ mol−1。另外他們還利用擬一級組態作用法 (pseudo-first-order configuration interaction)計算出電子基態 ̃ 到第一激發 態 ̃ 的躍遷能量為 86.9 kJ mol−1(7267 cm1)。他們由位能圖推測 CH3OO 的第一電子激發態 ̃ 是束縛態,也指出 CH3OO 在 240 nm 附近的 UV 吸 收峰是電子基態 ̃ 躍遷到第二電子激發態 ̃ 所造成。CH3OO 被激發 至第二電子激發態後,會沿著 態的排斥位能面(repulsive potential energy
3 surface)斷 OO 鍵生成 CH3O O。 關於反應(1-6)至(1-8)的比較,Zhu 等人[9]利用 RRKM 理論計算出在大 氣壓力下,溫度 1500 K 內,CH3OO 是最主要的產物(反應(1-6))。反應(1-6) 在 298580 K 的高壓反應速率常數 和低壓極限反應速率常數 分別為 (以 Ar 為淬熄體): cm3 molecule1 s1, cm6 molecule2 s1。 當溫度提升時,反應(1-7)和(1-8)逐漸變得重要,且兩者之間互為競爭關係。 在溫度 2000 K 以內,主要進行反應(1-7)產生 CH2O + OH,其反應速率常數 為 cm3 molecule1 s1,當溫度 超過 2000 K 時,CH3O + O 的生成比例逐漸增加(反應(1-8)),其反應速率常 數為 cm3 molecule1 s1 (適用 溫度為 10003000 K)。此外他們還提出:CHO + H2O 為熱力學上最穩定的 產物(ΔH = −351.0 kJ mol−1),但因為反應物需要越過高達 199.6 kJ mol−1的能 障才能生成,故以動力學角度觀之,該反應較難進行。 關於 CH3OO 光譜學的研究,最早可追溯到 1967 年 Thomas[10]利用脈 衝輻射分解法(pulse radiolysis)分解水溶液中的 CH3I 產生 CH3自由基,並通 入大量 O2,利用 UV 吸收光譜法在 240 nm 附近發現一寬頻、不具結構之吸 收峰,Thomas 將其指認為 CH3OO 的吸收。隨後氣態 CH3OO 的 UV 吸收光
4 譜(200300 nm)也陸續被觀測到[11, 12, 13]。 CH3OO 的 UV 吸收光譜之所以沒有結構性,主要原因是電子躍遷到的 第二電子激發態 ̃ 為一排斥態(repulsive state),導致無法得到關於分子 結構的訊息[14],因此愈來愈多實驗組開始研究 CH3OO 躍遷至第一電子激 發態 ̃ 的光譜[1519]。Henziker 和 Wendt[15]利用激發態的汞原子碰撞 丙酮產生 CH3,再與 O2反應使生成 CH3OO。他們用相位靈敏偵測技術偵測 CH3OO 躍遷至第一電子激發態 ̃ 的光譜,指認出躍遷譜帶的起始點約在 7375 cm1。此外,他們亦觀測到離起始點約 120 cm1的 CH 3扭動模和 890 cm1的 OO 伸展模。Pushkarsky 等人[16]利用波長 193 nm 的雷射光解丙酮 或以 248 nm 的雷射光解碘甲烷,產生 CH3自由基與 O2反應,再以共振腔 振盪衰減法得到 CH3OO 躍遷至第一電子激發態 ̃ 的光譜,他們得到電 子基態和激發態的轉動常數,並指認譜帶起始位置為 7382.5 0.5 cm1,其 吸收截面積為 2.7 × 10-20 cm3 molecule-1。此外,他們還得到了 CH3OO 的自 我反應速率常數:k =4.9 × 10-13 cm3 molecule-1 s-1。Blanksby 等人[17]利用光 致分離(photodetachment)技術觀測 CH3OO 和 CD 3OO 的光電子光譜,決定 出 CH3OO 躍遷至第一電子激發態 ̃ 的譜帶起始點為:0.914 0.005 eV (7372 40 cm1);而 CD3OO 躍遷至第一電子激發態 ̃ 的譜帶起始點為: 0.913 0.004 eV (7364 32 cm1)。Fu 等人[18]利用波長 193 nm 的準分子雷 射光解丙酮或其氘取代物/氧氣/氦氣的混合物使生成 CH3OO 或 CD3OO,然
5 後藉脈衝式噴嘴將光解產物噴射到真空室中,先用一道紅外光雷射照射噴 射流使 CH3OO 或 CD3OO 由電子基態躍遷到電子激發態的各個振動能態, 再以另一道波長 118 nm 的真空紫外光雷射使其游離成 CH3OO 或 CD3OO , 由於吸收兩個光子的 CH3OO 或 CD3OO 有足夠內能從 CH3OO 裂解為 CH3 或由 CD3OO 裂解為 CD3,故當掃描紅外雷射波長並偵測 CH3或 CD3質 譜訊號之改變,便可得到 CH3OO 或 CD3OO 的 ̃ ̃ 躍遷光譜。他 們分別指派 CH3OO 和 CD3OO 的電子躍遷譜線原點為 7381 和 7371 cm 1。 此外,他們還指派了 CH3OO 第一電子激發態 ̃ 的 ν6振動模(1002 cm 1 ) 和ν7振動模(898 cm 1 ),以及 CD3OO 第一電子激發態 ̃ 的ν3到ν7振動模。 其後,本實驗室的鍾昭宇等人[19]利用共振腔振盪衰減法觀測到許多新的 ̃ ̃ 電振躍遷譜線,分別指派 CH3OO 在激發態 ̃ 的 COO 彎曲 (ν8)和 COO 對稱伸展振動波數(ν7)為 378 cm 1及 887 cm1;而對 CD 3OO 而 言,除了指派 348 cm1和 824 cm1為激發態 ̃ 的 COO 彎曲(ν 8)和 COO 對稱伸展振動模(ν7)外,還指派了 ν5 (954 cm 1,CH 3搖擺)和 ν6 (971 cm 1, COO 非對稱伸展),這些振動波數的指派,均和 UB3LYP/aug-cc-pVTZ 之理 論計算結果相近。至於 CD3OO ν5振動模的活化,他們推測應是和 ν7振動模 混合之結果。上述各個實驗組對於 CH3OO 躍遷至第一電子激發態 ̃ 的 譜帶原點除了精確度不同外,結果是一致的。 CH3OO 的紅外吸收光譜目前有四個研究組觀測到[20, 21, 22, 23]。Ase
6 等人[20]熱解 CH3NNCH3或 CH3I 產生 CH3自由基,CH3再與 Ar 間質中的 O2反應生成 CH3OO,他們觀察到八個 CH3OO 振動模。Nandi 等人[21]利用 兩個噴嘴,分別將 O2和熱解 CH3NNCH3產生的 CH3自由基射束沉積在 20K 的 Ar 間質中,CH3和 O2反應生成 CH3OO,再以傅立葉轉換光譜儀偵測其 偏極化紅外吸收光譜。他們觀測到 CH3OO 十二個振動模當中的十個,其中, 3024 cm1(ν9)和 1434 cm 1(ν 10)為 模,另外八個為 模,而 CH3的擺動(ν11, CH3 rock)和內轉動模(ν12,CH3 torsion)在其光譜中沒有被觀測到。此外, Nandi 等人還偵測了 CH3 18 O18O、13CH3OO 和 CD3OO,藉以定出同位素位移。 這些實驗上得到的振動波數均和 UB3LYP/6-311G(d,p)理論計算結果吻合。 比較 Ase 和 Nandi 兩個實驗組的光譜,可發現 Ase 將 CH3OO 在 2968 cm
1 的吸收指認為ν9,實際上應為ν2。Morrison[22]等人熱解 di-tert-butyl peroxide 得到 CH3,CH3再和 O2反應後形成 CH3OO,利用 IR-OPO 產生紅外雷射光 掃描 CH3OO 在 He 奈米液滴環境下 CH 伸展振動模的紅外吸收光譜。得到 CH3OO 的 ν1、ν2和ν9分別為 3034.7 cm 1、2955.5 cm1和 3024.5 cm1。上述 實驗組都是在間質中得到 CH3OO 的紅外光譜,由於 CH3OO 在低溫的間質 中難以轉動,故無法得知不同振動模的轉動譜線。此外,間質造成的微擾 亦使得振動譜線位置和氣態光譜略有差異。為了得到 CH3OO 的氣態紅外光 譜,本實驗室的黃登瑞等人[23]利用步進式掃描傅式轉換紅外吸收光譜法, 研究氣態 CH3OO 在電子基態的振動模。他們以 248 nm 的雷射去光解 CH3I
7 產生 CH3,CH3再和大量的 O2氣體碰撞生成 CH3OO,他們指認出 CH3OO 的ν1至ν5,ν9和ν10振動模,其中ν1、ν2及ν9的 CH 振動模吸收相互重疊, 係藉由 SpecView 光譜模擬軟體適解實驗光譜而得到其振動波數。這些觀測 到的振動波數和 Nandi 等人[21]在 Ar 間質下以及 Morrison[22]等人在 He 奈 米 液 滴 環 境 下 觀 測 到 的 位 置 十 分 接 近 。 此 外 , 他 們 也 利 用 B3LYP/aug-cc-pVTZ 之理論計算預測 CH3OO 的振動波數和其相對強度,其 結果亦與實驗觀測相當吻合。上述各研究組得到之結果整理如表 1-1。 關於 CH3OO 於振動基態下的研究,Endo[24]等人已利用 microwave 光 譜法得到其振動基態的轉動常數為 A" = 1.730 cm1、B" = 0.379 cm1和 C" = 0.330 cm1。 CH3OO 的自我反應(self reaction)是造成其濃度衰減的主要原因之一, Lightfoot 等人[25]利用閃光光解法(flash photolysis)研究 CH3OO 在溫度 248573 K 間的自我反應,有下列三種可能的途徑: (1-9) (1-10) (1-11) 反應(1-9)和(1-10)之間呈現高度競爭的情形,且其分支比和溫度息息相關。 他們提出 CH3OO 的自我反應速率常數為 k = cm 3 molecule1 s1(適用溫度為 248573 K),反應分支比為 k(1-9) / ( k(1-10)+ k(1-11)) = (適用溫度為 388573 K)。根據 2005 年的文獻回顧[26],
8 CH3OO 在 300 K 時的自我反應速率常數為 k =3.49 × 10 -13 cm3 molecule-1 s-1, 和 Lightfoot 等人的結果一致,而反應分支比為 k(1-9) : k(1-10) : k(1-11) = 0.375 : 0.625: 0。溫度愈高則反應(1-9)愈重要,而反應(1-11)的貢獻極小可被忽略。 除了上述三種反應途徑之外,Benson[27]等人提出 CH3OO 亦可能自我反應 生成 CH2O2。 (1-12) 只是在實驗上並未真正觀測到 CH2O2的生成[28]。 由於目前 CH3OO 的氣態紅外光譜只有黃登瑞研究過[23],但其實驗解 析度只有 4 cm1,並無法清楚解析 CH 3OO 各個振動模的轉動譜線,因此吾 人利用共振腔振盪衰減法技術搭配高解析度的可調變式紅外光雷射(tunable IR laser)掃描其光譜,以得到轉動解析的振動光譜。受限於紅外光源的掃瞄 範圍,吾人僅針對 CH3OO 的 CH 伸展模光區做探討。
9
表 1-1 文獻中 CH3OO 之比較。單位:cm
1。
mode mode description Ar matrix Ar matrix FTIR He nanodroplet
ν1 CH3 stretch 3032 3033 3034.7 ν2 CH3 symmetric stretch 2968a 2954 2954 2955.5 ν3 CH3 symmetric deformation 1453 1448 1453 ν4 CH3 umbrella 1414 1410 1408 ν5 CH3 rock OO stretch 1183 1180 1183 ν6 CH3 rock OO stretch 1112 1109 1117 ν7 CH3O2 stretch 902 902 ν8 CH3OO bend 492 492 ν9 CH3 stretch 3024 3020 3024.5 ν10 CH3 antisym. deformation 1440 1434 1441
ν11 CH3 rock not observed
ν12 CH3 torsion not observed
reference Ase et al.[20] Nandi et al.[21] Huang et al.[23] Morrison et al.[22] a
10 參考文獻
[1] P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat, and F. Zabel, Atmos. Environ. 26A, 1805 (1992). [2] S. Madronich, J. Greenberg, and S. Paulson, in Atmospheric Chemistry and
Global Change, edited by G. P. Brasseur, J. J. Orlando, and G. S. Tyndall (Oxford University Press, New York, 1999), pp. 325.
[3] G. L. Bras, in Chemical Processes in Atmospheric Oxidation (Springer, Berlin, 1997), Vol. 3, pp. 13.
[4] S. B. Bertman, J. M. Roberts, D. D. Parrish, M. P. Buhr, P. D. Goldan, W. C. Kuster, F. C. Fehsenfeld, S. A. Montzka, and H. J. Westberg, Geophys. Res. 100, 22805 (1995).
[5] I. R. Slagle and D. Gutman, J. Am. Chem. Soc. 107, 5342 (1985). [6] S. P. Walch, Chem. Phys. Lett. 215, 81 (1993).
[7] J. M. W. Chase, C. A. Davies, J. J. R. Downey, D. J. Fruirip, R. A. McDonald, and A. N. Syverud, J. Phys. Chem. Ref. Data 14, Suppl. 1 (1985).
[8] J. A. Jafri and D. H. Phillips, J. Am. Chem. Soc. 112, 2586 (1990). [9] R. Zhu, C. C. Hsu, and M. C. Lin, J. Chem. Phys. 115, 195 (2001). [10] J. K. Thomas, J. Phys. Chem. 71, 1919 (1967).
[11] D. A. Parkes, D. M. Paul, C. P. Quinn, and R. C. Robson, Chem. Phys. Lett. 23, 425 (1973).
[12] C. Anastasa, I. V. M. Smith, and D. Parkes, J. Chem. Soc., Faraday Trans. 1 74, 1693 (1978).
[13] H. Adachi and N. Basco, Int. J. Chem. Kinet. 14, 1125 (1982).
[14] E. N. Sharp, P. Rupper, and T. A. Miller, Phys. Chem. Chem. Phys. 10, 3955 (2008).
11
[16] M. B. Pushkarsky, S. J. Zalyubovsky, and T. A. Miller, J. Chem. Phys. 112, 10695 (2000).
[17] S. J. Blanksby, T. M. Ramond, G. E. Davico, M. R. Nimlos, S. Kato, V. M. Bierbaum, W. C. Lineberger, G. B. Ellison, and M. Okumura, J. Am. Chem. Soc. 123, 9585 (2001).
[18] H. B. Fu, Y. J. Hu, and E. R. Bernstein, J. Chem. Phys. 125, 7 (2006).
[19] C.-Y. Chung, C.-W. Cheng, Y.-P. Lee, H. Y. Liao, E. N. Sharp, P. Rupper, and T. A. Miller, J. Chem. Phys. 127, 14 (2007).
[20] P. Ase, W. Bock, and A. Snelson, J. Phys. Chem. 90, 2099 (1986).
[21] S. Nandi, S. J. Blanksby, X. Zhang, M. R. Nimlos, D. C. Dayton, and G. B. Ellison, J. Phys. Chem. A 106, 7547 (2001).
[22] A. M. Morrison, J. Agarwal, H. F. Schaefer, and G. E. Douberly, J. Phys. Chem. A (2012) (unpublished).
[23] D.-R. Huang, L.-K. Chu, and Y.-P. Lee, J. Chem. Phys. 127, 7 (2007). [24] Private communication with Prof. Y. Endo.
[25] P. D. Lightfoot, R. Lesclaux, and B. Veyret, J. Phys. Chem. 94, 700 (1990). [26] D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M.
J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker, and J. Warnatz, J. Phys. Chem. Ref. Data 34, 757 (2005).
[27] S. W. Benson and P. S. Nangia, Acc. Chem. Res. 12, 223 (1979).
[28] H. Niki, P. D. Maker, C. M. Savage, and L. P. Breitenbach, J. Phys. Chem. 85, 877 (1981).
12 第二章 實驗原理與技術
2.1 共振腔振盪衰減法(cavity ringdown spectroscopy, CRDS)簡介
共振腔振盪衰減法(CRDS)是一極為靈敏的偵測技術[1],不但可以用來 偵測穩定分子的微弱吸收譜線,還可以用來偵測濃度很小的自由基分子的 吸收。此技術除了可以用來偵測大氣中一些微量物質,研究其含量和分子 光譜之外,亦可用來量測分子反應的反應速率[2],或者是分子的運動速度 [3]。其後,利用共振腔振盪衰減法的研究領域也漸漸地被拓展開來,包括 液相[4]、薄膜[5]以及表面[6]的分子光譜。 2.1.1 共振腔振盪衰減法的發展與歷史 共振腔振盪衰減法的發展可追溯到 1980 年代由 Herbelin 等人[7]所開創 的以連續式雷射(cw-laser)為光源的共振腔衰減相位差法(cavity attenuated phase shift, CAPS)。CAPS 是利用鎖相放大器(lock-in amplifier)來測量光在經 過由兩片高反射率鏡片所組成的腔體之後,其光相位的改變來決定鏡子的 反射率。此技術的主要限制在於無法很準確地測量光的相位角(phase angle), 其相位不準度在 之間,因此造成所能偵測的最小吸收度為 100 ppm 左右。
13
射注入腔體之中,並以 Pockels cell 當作光開關,待腔體內能量達到一定的 誘發門檻(triggering threshold)時,便通入高電壓使得 Pockels cell 關閉,讓雷 射不再進入腔體內。此技術不再是測量光的相位差,而是測量腔體內光強 度隨時間的變化情形,所得的衰減時間稱之為振盪衰減時間(ringdown time)。 因為 Anderson 等人所用的偵測器之反應時間在數百奈秒(nanosecond)範圍, 因此提高了偵測的靈敏度,其相對的最小可偵測的吸收度為 5 ppm 左右。 若 將 此 一 技 術 和 CAPS 來 比 較 , 此 一 技 術 的 優 點 為 因 為 模 匹 配 (mode-matching)所造成的問題可大為減少,但是其缺點為當利用不同的波 長來掃描分子的吸收光譜時,由於各個波長在腔體內的誘發門檻不同,因 此無法做連續式的掃瞄。
1988 年 O’ Keefe 及 Deacon[9]改以脈衝式雷射(pulsed laser)作為光源, 同樣是偵測其振盪衰減時間,其吸收度的偵測極限達到了小於 1 ppm 的程 度。他們認為主要原因是所利用的脈衝式雷射其同調長度(coherence length) 短,當光子在共振腔體來回振盪時不會發生自我干涉現象(self-interference), 亦即注入共振腔體所有頻率的光都可以離開腔體,與共振腔體的長度無關, 因此便提升了振盪衰減時間的穩定性。1997 年 Romanini 等人[10]以連續式 二極體雷射(cw-diode laser)作為光源,並利用壓電晶體(piezo transducer, PZT) 去微調共振腔體長度以符合縱模(longitudinal mode)共振頻率,其偵測的靈 敏度最佳可達到 cm1。
14 2.1.2 光子彈模型 (photon bullet model)
吾人可以利用光子彈模型[11]簡單說明共振腔振盪衰減法的原理,如圖 2-1 所示。當脈衝雷射光進入由兩面高反射率鏡片所組成的共振腔時,大部 分的光子會在鏡子間來回反射,僅有一小部分光子會穿透反射鏡到達偵測 器。當光子在長度為 L 的腔體內多次反射時,各次到達偵測器的光子時間 上相隔 2L / c,以長度為 63 cm 的共振腔為例,其在共振腔內來回時間間隔 (roundtrip time, tr)為: (2-1) 故對吾人實驗所用脈衝時間(pulse duration)約為 12 ns 的紅外雷射光而言, 到達偵測器的光脈衝是無法一個個被分開的。 當入射強度 的雷射進入到兩面相同鏡片(反射率 ,穿透率 )組成的 共振腔中,第一發直接穿透兩面鏡片到達偵測器的光強度是 ,接 下來每次到達的光,會因為比前一次多經過兩面鏡子之反射而使強度衰減 為 倍,故光在腔體內來回 次後,偵測器測得的光強度為: (2-2) 當 值極趨近於 1 時,即鏡片反射率極高時, [ ] ,又因為光在腔體內來回行經的次數是:
15 (2-3) 故式子(2-2)可表示如下: (2-2) (2-4) (2-5) 其中𝜏為共振腔於真空下的振盪衰減時間(ringdown time): (2-6) (2-6) 式 中 的 代表光在真空下每通過一次鏡片所損失的強度比例 (fractional loss per pass)。例如, 時,漏失比例為 300 ppm。由(2-5) 式可以清楚瞭解,偵測器量測到的是光強度隨時間呈現單一指數形式衰減 的訊號,因此振盪衰減時間便可藉由簡單的指數衰減適解得到。假使共振 腔內放有吸收樣品,其吸收係數為α,且該樣品在腔體內的有效吸收長度為 ,則(2-4)式應改寫成: (2-7) 而振盪衰減時間為: (2-8) 利用(2-6)和(2-8)式,可得到吸收樣品造成光每次通過腔體損失的強度比 例:
16 ( ) (2-9) 如果知道樣品的有效吸收長度 ,由(2-9)式便可以獲得樣品的吸收係數 (cm−1 )。 2.1.3 靈敏度 (sensitivity) 共振腔振盪衰減法藉由量測光強度的衰減速率來測量樣品吸收強度, 所以振盪衰減時間量測得愈準,靈敏度就愈高。根據(2-9)式,可以知道如 果共振腔振盪衰減法能夠測得的最小時間差為 ,則: ( ) (2-9) ( ) (2-10) ( ) (2-11) ( ) (2-12) ( ) (2-13) 以反射率 99.97 % 的鏡子為例,假如 誤差為 1 %,那麼最小可以偵測 到的吸收度即為 3 ppm。(2-13)式說明靈敏度和 息息相關,當鏡子反射 率愈差或者樣品吸收愈強時,振盪衰減時間愈短,量測的準確度亦會降低, 導 致 靈 敏 度 變 差 。 一 般 共 振 腔 振 盪 衰 減 法 量 測 的精確度大約在 之間,偵測靈敏度可達 1ppm 以下[11]。以本實驗系統
17 而言,鏡子反射率 R = 99.97 %,腔體長度 L = 63 cm,𝜏 = 7 s,由於Δ 𝜏min 大約在 0.010.03 s 之間,因此估計偵測靈敏度為 ,大約 和世界上以脈衝式雷射作為偵測光源的共振腔振盪衰減法所能達到的靈敏 度相當。 2.1.4 腔模 (cavity modes) 光子彈模型只是簡單描述共振腔振盪衰減光譜法,無法完整解釋其他 複雜的光學現象。例如:模拍頻(mode beating)現象。因此吾人需要以另一 種方法來說明共振腔振盪衰減法的原理。 根據光學原理,兩片曲率半徑為 的高反射率鏡片,相距為 ,若當 或 時 , 則 所 形 成的 共 振 腔 為 一 穩定 共 振 腔 (stable resonator),其本徵頻率(eigenfrequencies)可表示成: [ (√ )] (2-14) 其中 為縱模模數(longitudinal mode index), 和 為橫模模數(transverse mode indices)。當 時,所形成的模稱為 TEM00 模,TEM 係 transverse electromagnetic waves 之縮寫。由(2-14)式可知縱模間距,又稱自 由光譜範圍(free spectral range)為:
(2-15)
18 ( √ ) (2-16) 當入射光進入到共振腔時,其電場會和腔模產生耦合,因此腔體內的總電 場可以寫成腔模的線性組合[12]: ∑ ∑ (2-17) 其中 是入射光電場空間分布 和腔體橫模 之耦合常數, 是 入 射 光 頻 譜 結 構 和 腔 體 頻 率 響 應 函 數 (cavity response function) 之耦合常數。 ∫ ∫ ( ) (2-18) (2-19)
對於傅立葉轉換極限的同調雷射(fourier-transform limited coherent laser) 而言,頻寬愈窄,則脈衝時間愈長。根據 Zalicki 和 Zare[11],假如脈衝時 間遠大於光在腔體內來回一次的時間,雷射頻寬會遠小於腔體的自由光譜 範圍。當掃描雷射波長時,雷射若是沒有和腔模耦合便無法離開腔體到達 偵測器,導致光譜失真。由此可知若要取得正確的光譜資訊,雷射至少要 包含兩個以上的腔模。脈衝時間若遠小於光在腔體內來回一次的時間 , 光子便不會發生自我干涉現象。此時,因為腔體的自由光譜範圍遠小於雷 射線寬,所以多個腔模將會被激發而產生模拍頻現象,其解釋如下[13]: 假設入射雷射光包含了兩個腔模,其頻率分別為 和 ,則到達偵測
19 器的總電場可表示成: ̃ ̃ (2-20) 而光強度為: ∬| | (2-21) ∬| ̃ ̃ | (2-22) (2-23) 其中 、 和 分別為: ∬| ̃ | (2-24) ∬| ̃ | (2-25) ∬ ̃ ̃ (2-26) (2-23)式已假設 的高頻率項可被偵測器平均掉,故省略。所以偵測 器收到的光強度為 dc 部分( 和 項)加上 ac 部分( 項): [ ] (2-27) (2-27)式中的 即為模拍頻之頻率。上述說明是考慮雷射線寬僅包含 兩個腔模,實際上模拍頻訊號通常為多重模造成之結果。由於模拍頻會使 振盪衰減時間的量測精準度下降,進而降低靈敏度,因此實驗上需盡量避 免。 通常要掃描氣態分子振轉動躍遷(vibration-rotation transitions)的高解析 光譜,便需要頻寬僅幾百 MHz 的雷射,其傅立葉轉換極限的脈衝時間約為
20 十幾個奈秒。由於脈衝時間遠大於或小於光在腔體內來回一次的時間皆不 容易得到精確的光譜資訊,因此脈衝時間通常會和光在腔體內來回一次的 時間差不多,這也就是為何共振腔體的長度通常會設計在一至二公尺以內 的原因。以吾人的系統而言,可調頻紅外雷射的頻寬為 600 MHz,其脈衝 時間為 12 ns,雷射腔體的長度為 63 cm。此設計足以掃描氣態分子振轉動 躍遷的高解析光譜,不會有光譜失真的問題或是受到模拍頻現象嚴重的干 擾。
2.1.5 雷射頻寬效應 (laser bandwidth effect)
上述的光子彈模型是在符合 Beer-Lambert law 的情況下所推論出來的 結果,1994 年 Zalicki 和 Zare[11]認為若是雷射線寬大於吸收物質的線寬, 則光強度隨時間的衰減情形就不適合以 Beer-Lambert law 來描述。因為在雷 射線寬內,僅有一小部份頻率的光會被物質吸收,導致較快的衰減速率, 其餘頻率的光則衰減較慢,是故整個振盪衰減訊號(ringdown signal)是呈現 雙指數率減(bi-exponential)的情形。所以如果用單一指數函數去適解振盪衰 減訊號,會得到較小的吸收強度。Zalicki 和 Zare 估計若只適解振盪衰減訊 號的前面時間部分,則結果會比較正確。 1996 年,Hodges 等人[14]利用兩種不同線寬的雷射,去測量 O2 的 躍遷之 吸收峰,雷射線寬越窄所得到的衰減曲線會越趨
21 近單一指數的衰減,且不會因為選取適解的時間,強度有太大的變化。在 1999 年,Newman 等人[15] 利用霍式轉換紅外光譜(Fourier-transformed infrared spectrometer) 及 共 振 腔 振 盪 衰 減 光 譜 法 測 量 O2 的 吸收譜帶,用來證明 Zalicki 及 Zare 的理論。此共振腔振 盪衰減法的實驗條件為雷射線寬為 0.25 cm-1,O2 的都卜勒線寬(Doppler linewidth)為 0.098 cm-1,若去適解振盪衰減曲線前段的時間(0-0.5 s),則 共振腔振盪衰減光譜法所得之 O2 吸收譜帶的積分譜帶 強度(band strength)與用霍式轉換紅外光譜儀(其解析度為 0.0015 cm-1 )所得 之積分譜帶強度只有相差約 2%。 若是雷射線寬包覆了物質的許多吸收譜線,振盪衰減訊號將呈現多重 指數衰減(multi-exponential)模式。Zalicki 及 Zare 認為此時仍可適用光子彈 模型,前提為吸收物質所造成的漏失強度要遠小於反射鏡所造成的漏失強 度 ( 2-28 ) 在此條件下,振盪衰減訊號仍可被視為遵循單一指數衰減型式。以反射率 R = 99.99 %的鏡子為例, 需遠小於 ,假使振盪衰減時間測量的相對誤 差為 1 %左右,根據(2-13)式,最小能偵測的吸收強度是 ,如此一來, 偵測範圍僅在兩個數量級以內。
22 2.1.6 模匹配 (mode matching)
依據近軸近似理論(paraxial approximation) [16],假設雷射光束為高斯光 束(Gaussian beam),則光束大小 ,以及光束波前(wavefront)的曲率半徑 ,可表示為: { ( ) } (2-29) { ( ) } (2-30) 其中 z 為光束離腔體中心的距離,當 z = 0 時, 定義為光束腰(beam waist), 其曲率半徑為無限大。為了避免模拍頻現象,就必須利用一望遠鏡組以及 針孔使原來非 TEM00模的雷射光變成 TEM00模,與共振腔的腔體模(cavity mode)作匹配的動作,如圖 2-2 所示。而所謂的模匹配是指利用一望遠鏡組 將雷射光聚焦至共振腔體的中心,此時雷射光束的大小及曲率半徑與利用 (2-29)及(2-30)式所推算出來的結果一致。 2.1.7 CRDS 技術的優點 共振腔振盪衰減光譜法的優點如下: (1) 直接得到物質之絕對濃度 由(2-9)式可知,扣除背景值後,利用 CRDS 技術可得到分子之吸收強 度,如吸收截面積為已知,則可以直接得到分子之絕對濃度。而對於頻率
23
調變光譜法(frequency modulation spectroscopy)[1]來說,其原理是利用調頻 器將單模雷射的頻率作調變,且利用鎖相放大器與偵測器結合以偵測通過 樣品槽的光強度之變化情形,此方法無法直接由訊號的變化得到物質的絕 對濃度。 (2) 不受雷射光源強度變化的影響 CRDS 技術是偵測光在共振腔體內光強度的衰減速率,而不是偵測光 強度的差異,故外在的雷射強度變化並不會造成所測量的分子吸收強度之 改變。一般的吸收光譜技術則是藉由測量光強度變化而得到分子的吸收度, 因此光源強度的不穩定便會造成分子吸收度測量上的誤差。 (3) 有效吸收光徑長 因為光在腔體中來回地反射,其吸收光徑很長,在衰減時間內(當光強 度衰減至原來強度的 1/e 所需時間)可達到數十公里,而傳統上利用多重反 射所能達到的吸收長度則遠小於此。以多重吸收槽為例(white cell),其能達 到的吸收光徑長度通常在數百公尺以內[1]。 (4) 偵測靈敏度高 一般藉由直接量測光強度變化的雷射吸收光譜技術受到光源穩定度的 限制,所能測得的最小吸收度大約為 10−3[1],而共振腔振盪衰減光譜法因 為測量分子吸收度時不受雷射強度變化影響,且有效吸收光徑長,因此可 測到的最小吸收度達到 10−7[1],靈敏度遠比一般雷射吸收光譜技術好。
24 2.2 可調頻紅外雷射光源 (tunable IR laser)
本實驗利用脈衝式釔鋁石榴石雷射(neodymium-doped yttrium aluminum garnet laser, Nd:YAG laser)激發光學參量振盪器/光學參量放大器(optical parametric oscillator/optical parametric amplifier, OPO/OPA) , 經 過 混 頻 (frequency-mixing)作用後產生可調頻紅外雷射光。混頻作用和 OPO/OPA 的 原理如下: 2.2.1 混頻(frequency-mixing)作用 當光與介質產生交互作用時,會對介質產生極化,該極化作用強度和 入射電場強度相關,可表示如下[17]: (2-31) 其中 為介質的 n 階極化率, 為電場。光強度很弱時,只有 項需 要考慮,此時極化強度和電場強度呈線性關係。若是光強度很強或是介質 高階項極化率很大時,(2-31) 式中的高次項便不可忽略。 當兩道光穿越一個非中心對稱結構的非線性晶體時,電場會與二階極 化率作用而產生混頻光。假設兩道入射光的電場分別為 和 , 則經過非線性晶體的電場為兩道入射光電場之和為: (2-32)
25 (2-33) 其中 cc 代表的是共軛複數(complex conjugate)項。將(2-33)式之電場帶入 (2-31)式,則二階極化率之項為: (2-34) ∑ [ ] (2-35) (2-35)式為加總不同的( )組合之結果,例如:(2-35)式展開後 的其中一項為 [ ],對應到的 為 。混頻後產生的第三道光頻率為: (2-36) ( )共有四種組合,分別對應到四種混頻光: (1) (1, 2, 0, 2, 0) 之二倍頻 (2) (1, 0, 2, 0, 2) 之二倍頻 (3) (2, 1, 1, 1, 1) 和 之和頻 (4) (2, 1, 1, 1, 1) 和 之差頻 上述式子省略了電場的位置向量,比較正確的電場表示方法應寫成: [ ⃑ ] (2-37) 其中 為位置向量, ⃑ 為波向量,波向量之值為: | ⃑ | (2-38) 是介質對頻率 的光的折射率。此時(2-35)式應改寫成:
26 { [ ⃑ ⃑ ]} (2-39) 混頻光的波向量為: ⃑ (2-40) ⃑ ⃑ (2-41) 若滿足(2-41)式則表示達到相匹配(phase matching)。 (2-36)式和(2-41)式可分別視為遵守能量守恆和動量守恆。當入射光夾 的角度愈大,光徑重疊的區域愈小,混頻的效果會愈差,因此通常參與混 頻過程的三道光波要幾乎互相平行,以提升轉換效率。此時(2-41)式可以改 寫成: (2-42) 和(2-36)式比較後,可知: (2-43) 只有具有雙折射率的介質才能滿足(2-43)式。所謂的雙折射率為介質對於 ordinary 波(ordinary wave, o-wave)和 extraordinary 波(extraordinary wave, e-wave)有不同的折射率,分別為 和 。對於單光軸非線性晶體(uniaxial nonlinear crystal)來說,通常 之大小和光波行進的方向無關,而 的值和 電場以及波向量方向息息相關。若 ,則稱正雙折射性(positively refringence);若 ,則稱負雙折射性(negatively refringence)。當掃描 雷射波長時,為了要滿足相匹配,晶體相對於入射光的夾角必須要改變,
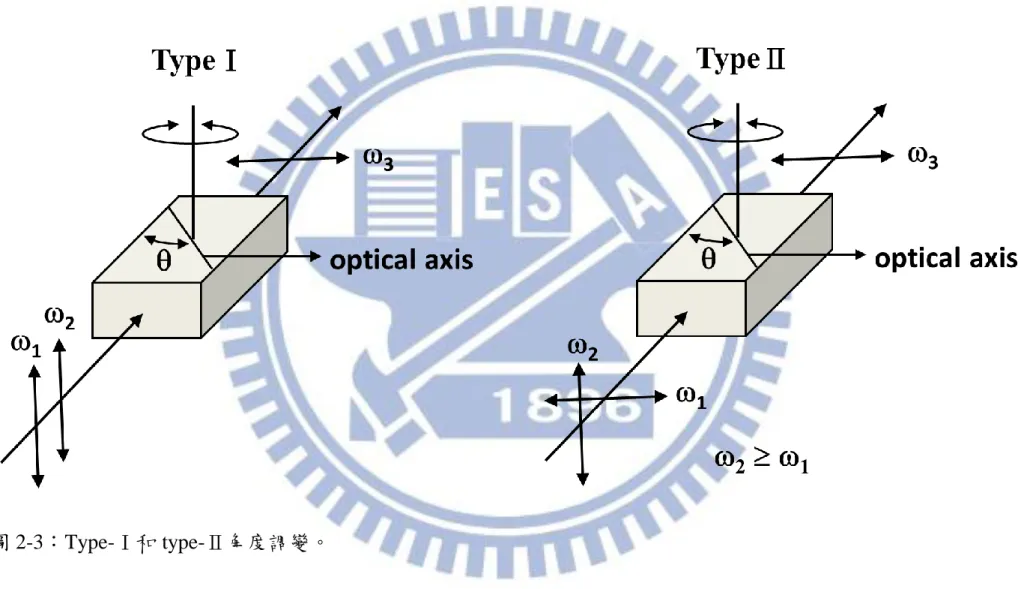
27 此過程稱為角度調變(angle tuning)。依據入射光和產生的混頻光電場極化方 向對應到 ordinary 波和 extraordinary 波的情況不同,角度調變可分成四種, 如圖 2-3 所示: (1) Type-I(適用於正雙向性單光軸晶體):兩道入射光皆為 extraordinary 波, 產生的混頻光為 ordinary 波。 (2) Type-I(適用於負雙向性單光軸晶體):兩道入射光皆為 ordinary 波,產生 的混頻光為 extraordinary 波。 (3) Type-II: (適用 於正 雙向性 單光軸晶體 ):兩 道入射 光中 頻率高 者 為 extraordinary 波,另一道入射光和產生的混頻光為 ordinary 波。 (4) Type-II: (適用 於負 雙向性 單光軸晶體 ):兩 道入射 光中 頻率高 者 為 ordinary 波,另一道入射光和產生的混頻光為 extraordinary 波。
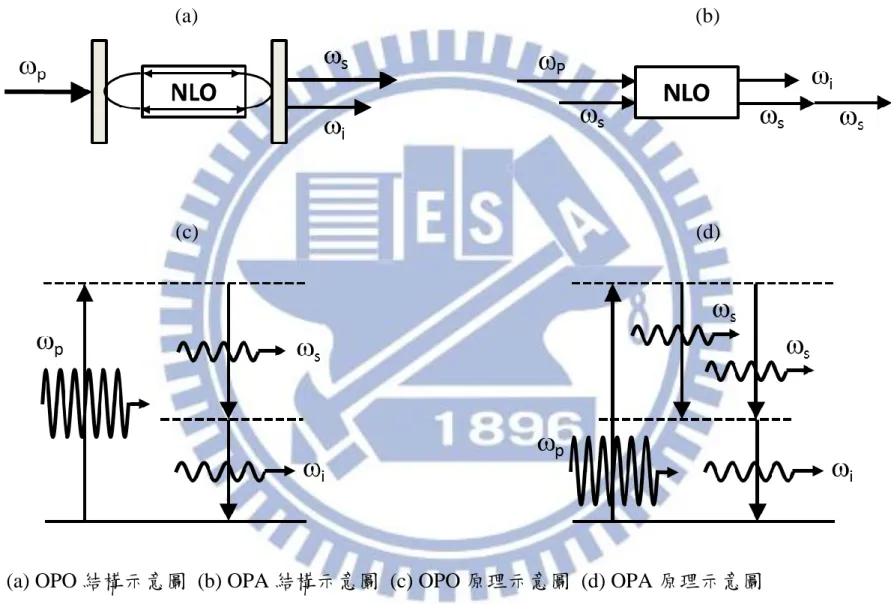
2.2.2 光學參量振盪器(optical parametric oscillation, OPO)
如圖 2-4 所示,光學參量振盪器是藉由高能量的脈衝雷射光和非線性晶 體進行參量作用(parametric interaction),把能量為 的光子轉換成能量分
別為 和 的兩個光子[17],可以視為三波混頻的過程。其中頻率 的
光波為 signal 波,頻率 的波則為 idler 波。根據能量守恆, 、 和 之 間的關係為:
28
(2-44)
Signal 和 idler 波的放大倍率(gain)和入射光的強度以及晶體的有效非線性係 數(有效二階極化係數)相關。單位光徑的參量放大係數(parametric gain coefficient per unit pathlength) 定義為:
或 (2-45)
| | | | (2-46)
| | (2-47)
(2-47) 式中, 是入射光強度,| |是有效非線性係數 ,當 和 相同 時,上述的參量放大係數即為二倍頻的參量放大係數。光學參量振盪器的 出光門檻(threshold)發生在放大率等同於振盪器內 signal 和 idler 波的漏失比 例乘積時。所以振盪器的鏡子反射率愈高,signal 和 idler 波的漏失愈小, 出光門檻就愈容易達到。
2.2.3 光學參量放大器(optical parametric amplifier, OPA)
如圖 2-4 所示,當頻率為 和 的兩道光經過非線性晶體並進行參量
轉換,若 和 共線且波向量方向相反,則會使頻率 的光強度增大為兩
倍,並產生另一道頻率 的光,且 、 和 之間的關係為:
29 新產生的這道光稱為 idler 波。
本實驗的可調頻紅外雷射是串接 OPO 和 OPA 兩個部分而成,其內部 架構詳述於第三章。
30
圖 2-1:以光子彈模型圖說明共振腔振盪衰減法之示意圖。脈衝雷射光在共振腔體內來回反射,其光強度會隨時間 和所走之距離呈單一指數之型式衰減,到達共振腔的光被偵測器接收後被送至示波器,得到的振盪衰減訊號為光強 度隨時間呈單一指數型式衰減的波形。
31 圖 2-2:模匹配之示意圖。其中 L1和 L2為凸透鏡,M1和 M2為高反射率鏡組,0和(z)分別為光束腰和距離腔體中 心 z 之光束大小。
z
(z)
0M
1M
2L
1L
2Iris
32 圖 2-3:Type-Ⅰ和 type-Ⅱ角度調變。
33
(a) (b)
(c) (d)
34 參考文獻
[1] K. W. Busch and M. A. Busch, Cavity-Ringdown Spectroscopy: An Ultratrace-Absorption Measurement Technique. (American Chemical Society, Washington, DC, 1999).
[2] T. Yu and M. C. Lin, J. Am. Chem. Soc. 115, 4371 (1993). [3] A. P. Yalin, Surla, V. Opt. Lett. 30, 3219 (2005).
[4] A. J. Hallock, E. S. F. Berman, and R. N. Zare, J. Am. Chem. Soc. 125, 1158 (2003).
[5] S. Logunov, Appl. Opt. 40, 1570 (2001).
[6] J. Antonietti, M. Michalski, U. Heiz, H. Jones, K. H. Lim, N. Rösch, A. Del Vitto, and G. Pacchioni, Phys. Rev. Lett. 94, 213402 (2005).
[7] J. M. Herbelin, J. A. Mckay, M. A. Kwok, R. H. Ueunten, D. S. Urevig, D. J. Spencer, and D. J. Benard, Appl. Opt. 19, 144 (1980).
[8] D. Z. Anderson, J. C. Frisch, and C. S. Masser, Appl. Opt. 23, 1238 (1984). [9] A. O'Keefe and D. A. G. Deacon, Rev. Sci. Instrum. 59, 2544 (1988).
[10] D. Romanini, A. A. Kachanov, and F. Stoeckel, Chem. Phys. Lett. 270, 538 (1997).
[11] P. Zalicki and R. N. Zare, J. Chem. Phys. 102, 2708 (1995).
[12] J. T. Hodges, J. P. Looney, and R. D. van Zee, J. Chem. Phys. 105, 10278 (1996).
[13] A. E. Siegman, Lasers (University Science, Mill Valley, Calif., 1986). [14] J. T. Hodges, J. P. Looney, and R. D. van Zee, Appl. Opt. 38, 3951 (1999). [15] S. M. Newman, I. C. Lane, A. J. Orr-Ewing, D. A. Newnham, and J.
Ballard, J. Chem. Phys. 110, 10749 (1999).
[16] A. Yariv, Quantum Electronics, 3rd ed. (John Wiley & Sons, New York, 1975).
35
Spectroscopy, Dynamics and Applications (John Wiley & Sons, Chicester, U.K., 2007).
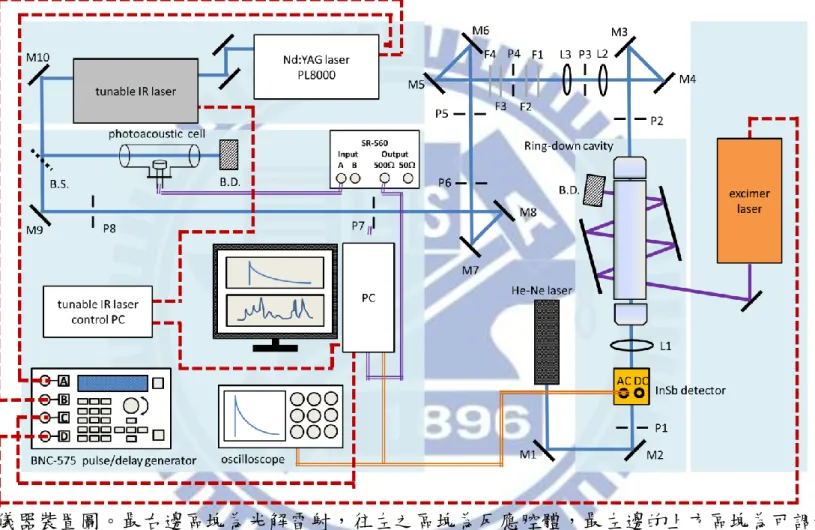
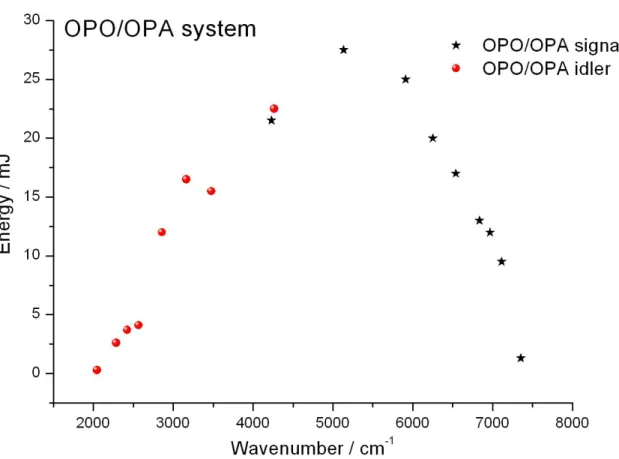
36 第三章 實驗系統與實驗步驟 3.1 實驗裝置 實驗系統主要是由可調頻紅外雷射光源、反應系統、光解雷射三個部分 以及周邊儀器所組成,如圖 3-1 所示。分別說明如下: 3.1.1 可調頻紅外雷射光源 可調頻紅外雷射光源的產生是利用內部導入種子雷射(Continuum, SI-2000,頻寬< 5kHz)光的 Nd:YAG 雷射(Continuum,Powerlite DLS 8000) 產生單一縱模(single longitudinal mode, SLM)、波長為 1064 nm 的光去激發 可調頻紅外雷射(Laser Vision)。紅外雷射內部主要分為兩大部分:光學參量 振盪器(OPO)和光學參量放大器(OPA)。首先,1064 nm 的入射光被分光片 分成兩道,其中一道經過 KDP 晶體倍頻作用產生 532 nm 的綠光,此綠光 進入光學參量振盪器後,經過參量作用轉換成近紅外光的 signal 波(near infrared,出光範圍為 712880 nm)和中紅外光的 idler 波(intermediate infrared, 出光範圍為 1.352.50 m),接著 idler 波會和另一道被分光片分出的 1064 nm 的光在參量放大器處會合,經過參量作用產生新的 signal 波和 idler 波。在 參量放大器產生的 signal 波為原本來自光學參量振盪器的 idler 波,其強度 會被放大,不同波數下的出光能量如圖 3-2 的 signal 波所示。而新產生的 idler
37 波即為本實驗所用之紅外光源,其頻寬為 0.02 cm1,波長可調變範圍為 2.135.00 m (47002000 cm1),在不同波數下的出光能量如圖 3-2 的 idler 波所示。 3.1.2 反應系統 O2 氣體由高壓鋼瓶流出後,分成兩路:一路為大流量經過流量計及針 閥組,從反應槽的左右兩邊注入;另一路則經流量計及針閥組到達體積約 4900 cm3的預混槽,和由樣品瓶流進的 CH3I 蒸氣混合後,從反應槽的下方 注入。O2氣體除了當作反應物外,亦做為淬熄體(quencher)。本實驗的反應 槽即為共振衰減腔體。它是由一個長度 19.8 cm,高度 5 cm,寬度 3 cm 的 不銹鋼材質的長方形腔體,再加上兩個內部裝有反射率為 99.97 % 的反射 鏡(Los Gatos Research,適用範圍為 30003300 nm,鏡子直徑為 1",曲率半 徑為 1 m)之振盪衰減鏡座所構成,共長 63 cm,內容積大約為 430 cm3
。其 中腔體的兩面挖空,利用材質為 S1UV、大小為 2.5 cm × 15 cm × 0.5 cm 的 石英光窗封住腔體,並使得 193 nm 或 248 nm 光解雷射能夠通過此腔體的 中心,光解反應之前驅物。反應槽上方連接到機械幫浦(dual stage rotary vane vacuum pump,Alcatel 2063C,抽氣速率為 63 m3 h-1),將反應後的氣體抽走。 反應槽之壓力以電容式壓力計(MKS Baratron,type 626,1000 Torr)測量。 當可調頻紅外雷射光源進入到共振腔,光子會在兩面反射率為 99.97 % 的
38
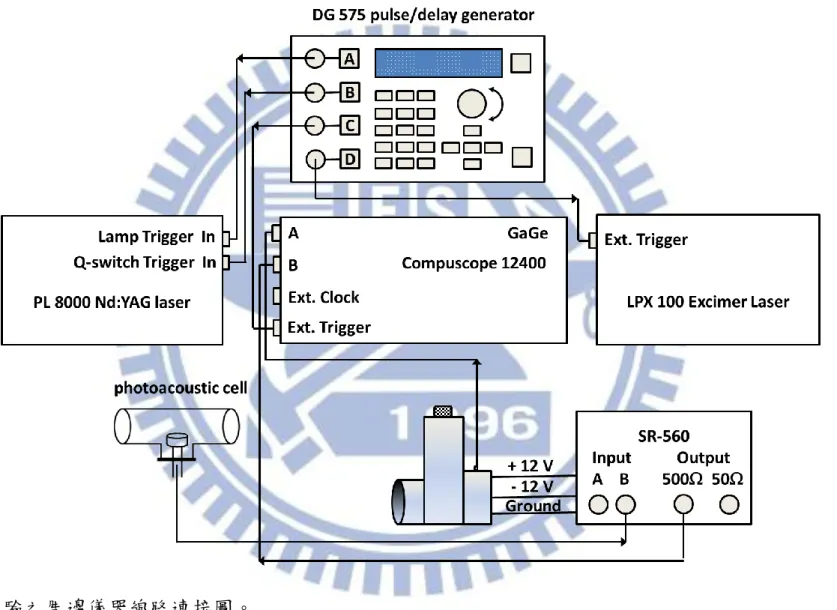
鏡片間來回反射,產生之振盪衰減訊號由 InSb 偵測器(Kolmer Technologies, KISDP-1-LJ2)偵測。 3.1.3 光解雷射 本實驗利用氟化氬準分子雷射(Lambda Physik,model LPX100)以及氟 化氪準分子雷射(Lambda Physik,model LPX100)光解不同反應前驅物。並 用兩面圓柱透鏡(cylindrical lens)調整光解截面積,以控制光解通量(fluence)。 為了增加光解效率,吾人利用兩片反射鏡使光解雷射來回通過反應腔體。 3.1.4 其他周邊儀器 如 圖 3-3 所 示 , 吾 人 利 用 脈 衝 訊 號 產 生 器 (Berkeley Nucleonics Corporation,BNC575)觸發 Nd:YAG 雷射的 flash lamp 和 Q-switch、控制 GagGe 取樣板(GaGe Applied, model 12400, A/D 解析度 12 bit,雙通道最大 取樣速度 200 MHz)進行數據擷取,以及觸發光解雷射。Nd:YAG 雷射的出 光激發可調頻紅外雷射後產生紅外光,吾人以分光片將其分成兩部份,分 別導入共振衰減腔體和 photoacoustic cell。photoacoustic cell 內裝有數十 Torr 的甲烷(CH4),其光聲訊號(photoacoustic signal)用來同步校正紅外雷射波長。 振盪衰減之訊號和 photoacoustic cell 得到的訊號分別傳送至 GaGe 取樣板的 兩 個 頻 道 , 並 以 Labview(Laboratory Virtual Instrument Engineering
39 Workbench)語言撰寫的程式處理之。其中振盪衰減波形是利用單一指數衰 減函數去適解得到振盪衰減時間(𝜏get),代入 (其中 為腔體長 度, 為光速)即可得知漏失比例,而 photoacoustic 的訊號則是積分數十個 s 的訊號得到一強度值。由 photoacoustic 訊號得到的 CH4 光譜,可利用 HITRAN2008 資料庫的 CH4吸收譜線位置做波長校正,藉以得到紅外雷射 正確的出光波長。當要掃描光譜時,先利用程式指定光譜的掃描起始波數 和終止掃描的波數,使可調頻紅外雷射的出光和指定波數相同。接下來指 定光譜每個數據點的波數間隔、每個數據點波形平均的次數,以決定可調 頻紅外雷射的平均掃描速率和總掃描時間。若掃描 30003060 cm1之光譜, 且每一數據點波數間隔設定為 0.1cm1。若平均 50 次振盪衰減波形後記錄一 點,則 60 cm1的掃瞄範圍須記錄 600 點,以擊發頻率為 10 Hz 的雷射為例, 即每隔五秒記錄一點,可調頻紅外雷射的平均掃描速率為 0.02 cm1,總掃 描時間為 3000 秒(50 分鐘)。 3.2 實驗前準備事項 3.2.1 可調頻紅外雷射光源的對正 可調頻紅外雷射的內部光學元件架設如圖 3-4 所示。在以下討論中,光 的偏振方向和光學桌面平行者定為水平偏振光,偏振方向和光學桌面垂直 者為垂直偏振光。
40 (1) 在可調頻紅外雷射系統入口前架設兩面 1064 nm 反射鏡 M1 和 M2,將 Nd:YAG 雷射產生波長為 1064 nm 的入射光導入紅外雷射系統內部。並 在 M2 及紅外雷射入口之間架設一片 polarizer 將垂直偏振光過濾掉, 使 1064 nm 入射光為水平偏振。在導光之前,可在 Nd:YAG 雷射出口 架設一光圈 A1,然後關光圈 A1 使光點變小,以利於接下來光路的對 正。由於入射光能量太強會將光圈 A1 打壞,故須確保其能量不可高於 200 mJ。 (2) 調整反射鏡 M1 的角度使入射光對準靠近入口處的光圈 A3 中心,再調 整反射鏡 M2 角度讓入射光對準光圈 A4 的中心,重複調整 M1 和 M2 的角度,直到光束能同時對準 A3 和 A4 的中心為止,然後在 M1 和 M2 之間架設光圈 A2,完成後可發現入射光已大致對準在光學參量振 盪器和光學參量放大器的晶體中心(分別為 KTP 和 KTA 晶體)。 (3) 完成步驟(2)之後,接著調整反射鏡 M1 使綠光對準光圈 A5,調整反射 鏡 M2 使綠光對準光圈 A6。完成後應可看到部分綠光反射回光圈 A2 中心,若是偏離則需微調 output coupler 的角度。由於調動 output coupler 的角度會讓導向光圈 A6 的綠光稍微偏移,需調整 end mirror 角度修正 之。
(4) 將入射光能量調至約 570 mJ,然後把從光學參量振盪器逸散出的綠光 導到雷射出口外約三公尺處,並在靠近和遠離雷射出口處分別架設光
41 圈 A7 和 A8 以定位光徑。 (5) 將光學參量振盪器前的 waveplate 轉至 0°使進入到光學參量振盪器的能 量達到最小,並將光學參量放大器前的 waveplate 轉至約 45°,此時進 入光學參量放大器的能量達最大。光學參量放大器的晶體會因非線性 光學作用產生少量倍頻光,調整 1064 nm 反射鏡 1 和 2 使綠光對準至 光圈 A7 和 A8 的中心。 (6) 打開控制晶體馬達位置的軟體,調整 motor #2 位置使光學參量振盪器 內的晶體達到相匹配角度,此時可用白色紙卡看見光學參量振盪器產 生的 near infrared。微調 tuning mirror 的高低角度讓 near infrared 和綠 光重合(可導到三公尺外的光圈 A8 確認位置)。
(7) 繼續以軟體調整 motor #2 位置,使光學參量振盪器的出光達到最強。 接著在光學參量振盪器和光學參量放大器之間架設 silicon plate 把 near infrared 導入 etalon,並藉 CCD 將干涉影像呈現出來。此時應只有一兩 個雷射模在競爭能量,藉由調整供給 piezoelectric actuator 的電壓以微 調 end mirror 的位置,讓光學參量振盪腔體內的能量集中在其中一個模 上。 (8) 以軟體控制 motor #36,旋轉光學參量放大器內晶體的角度使整體出 光能量達到最強。在出光口前架設 silicon polarizer 濾掉水平偏振的 intermediate infrared,只讓垂直偏振 mid infrared 出光。
42 3.2.2 振盪衰減腔體的對正 利用氦氖雷射(Uniphase, model 1107P-0622)來做振盪腔體的對正,如圖 3-1 所示,其步驟如下: (1) 首先將振盪衰減腔體(不包含振盪衰減鏡座)固定在光學桌上,盡量與光 學桌平行。 (2) 在共振腔體的兩端放入兩個中心有小孔的壓克力圓形板,往復調整 M1 及 M2 反射鏡使氦氖雷射通過此兩小孔以決定振盪衰減腔體的中心位 置。接下來將兩個振盪衰減鏡座固定於振盪衰減腔體上,調整兩個振 盪衰減鏡座的角度使氦氖雷射亦通過振盪衰減鏡座的中心。 (3) 放入 P1 及 P2 可調式光圈(iris),使氦氖雷射通過其中心。其目的在於 利用 P1 及 P2 標定出通過腔體中心之光軸。 (4) 利用 M3 及 M4 反射鏡將氦氖雷射導至 M5、M6 反射鏡處,以光圈 P3 和 P4 定出光軸。接著放入 L2 凸透鏡(材質為 BaF2,焦距為 50 mm)使 氦氖雷射通過 L2 和 P3 中心,並確認部分反射回去的光是否通過 P2 中 心,再放入 L3 凸透鏡(材質為 BaF2,焦距為 100 mm)使氦氖雷射通過 L3 和 P4 中心,然後確認部分反射的光使否通過 P2 中心。調整 L2 和 L3 之間的距離,使得當紅外雷射光聚焦至腔體中心時,其光束腰與 (2-29)式所算出來的結果一致。調整 P3 的位置使其位在凸透鏡 L3 的焦
43 點上,以便接下來將紅外雷射光源過濾成 TEM00模。 (5) 用 M5 和 M6 將光導入 M7 和 M8 處,以 P5 和 P6 定位光軸;用 M7 和 M8 將光導入 M9 和 M10 處,以 P7 和 P8 定位光軸,此時氦氖雷射要 大致在可調頻紅外雷射出口處。關閉氦氖雷射並讓可調頻紅外雷射出 光,反覆調整 M9、M10 的角度讓紅外光通過 P7 和 P8 的中心。 (6) 在 M9 和 M10 之間加入分光片 B.S.將部分紅外光導入 photoacoustic cell 做波長校正用。再微調 M9、M10 的角度讓紅外光通過 P7 和 P8 的中 心。調整 photoacoustic cell 位置使紅外光能穿透 cell 中心。
(7) 關小光圈 P3,讓紅外雷射光源過濾成 TEM00模。若聚焦至腔體中心的 光束大小與(2-29)式算出之結果不一致,則需微調 L2 的前後距離。 (8) 加入濾光片 F1-F4(光區範圍分別為 20918430 cm1、21804470 cm1、 24605000 cm1和 28805880 cm1),以確保沒有雜光進入到共振衰減 腔體。 (9) 關閉紅外雷射並打開氦氖雷射,將靠近 P2 端的振盪衰減鏡片裝上,微 調振盪衰減鏡座的角度使氦氖雷射被鏡子部分反射的光點能通過 P1 中 心。再裝上靠近 P1 端的振盪衰減鏡片,微調鏡座角度讓氦氖雷射反射 點穿過 P1 中心。在振盪衰減腔體和 P1 之間加入凸透鏡 L1(材質為 CaF2, 焦距為 5 公分),用以收集進入到偵測器的紅外雷射光。 (10) 將振盪衰減腔體抽真空,把 InSb 偵測器放置在 xyz 軸精密平移台。關
44
閉氦氖雷射然後將可調頻紅外雷射光導入振盪衰減腔體,把 InSb 偵測 器訊號線接至示波器,接著調整平移台的 xyz 軸使 InSb 偵測器能收到 振盪衰減訊號。微調振盪衰減鏡座的角度和精密平移台的各軸位置以 得到最佳振盪衰減訊號。
(11) 將 InSb 偵測器的訊號線接至 GaGe 取樣板,並利用 LabView 軟體撰寫 的程式處理訊號。以單一指數衰減函數分別去適解振盪衰解波形的前 段和後段,得到兩個振盪衰減時間,若數值不同,則需微調振盪衰減 鏡座的角度直到兩者完全相等為止,此時便完成了振盪衰減腔體的對 正。 3.2.3 模匹配之步驟 為了避免模拍頻現象,需利用一望遠鏡組及針孔使原來非 TEM00膜的 可調頻紅外雷射光變成 TEM00膜,與共振腔的腔體膜作匹配之動作。此時 雷射光束的大小和曲率半徑需與(2-29)式和(2-30)式所推算出來的結果一致。 由於共振腔反射鏡的曲率半徑為一公尺,實驗使用的波長大致為 3333 nm, 根據(2-30)式即可導出共振腔中心的光束腰 為 0.7 mm,故雷射光束在腔 體中心的直徑大小需為 1.4 mm。而共振腔反射鏡離腔體中心為 315 mm,根 據(2-29)式,即可導出光振腔反射鏡處的雷射光束需為 1.7 mm。可調頻紅外 雷射光的光束直徑大小為 3.6 mm,使其先後通過焦距分別為 100 mm 及 50
45 mm 的 BaF2凸透鏡(凸透鏡相距約 150 mm)而聚成光束直徑大小約為 1.8 mm。 將光圈放置在第一面 BaF2凸透鏡的焦距處,把針孔關小使紅外雷射盡量被 過濾成 TEM00膜,為了避免繞射現象,光圈所開孔洞的直徑大小 D 至少需 為: (3-1) 其中 為第一面 BaF2凸透鏡的焦距 100 mm, 為可調頻紅外雷射的波長, 大致為 3333 nm, 為紅外雷射在第一面 BaF2凸透鏡上的光束腰 1.8 mm。 由(3-1)式可推出 為 0.1 mm。接下來微調第二面 BaF2凸透鏡相對於第 一面 BaF2凸透鏡的前後距離,使可調頻紅外雷射光到達共振腔反射鏡處和 腔體中心的光束大小分別為 1.7 mm 和 1.4 mm,雷射光通過鏡組後之焦距 f 可由下式求得: (3-2) 其中 、 分別為凸透鏡之焦距, 為兩凸透鏡的距離。完成後即達成模匹 配。 3.2.4 氣體流量校正 由於本實驗需量測氣體樣品的流量,因此需要校正流量器以利後續的 定量分析。本實驗使用定壓下體積對時間之變化率( )校正方法來校正 流量計,其步驟如下:利用氣泡校正法(bubble method),將想要測量的氣體
46 (無毒性且不溶於水)先流經裝有蒸餾水的錐形瓶,再導入含有肥皂液的玻璃 瓶,玻璃瓶上端出口裝置一支有體積刻度的玻璃管,調整肥皂水液面與玻 璃管底部相隔小於 5 mm 的距離。打開氣體閥門,旋轉針閥到某特定刻度讓 氣體以特定流速流經錐形瓶及玻璃管。此時擠壓玻璃滴管的吸球使玻璃管 底部產生肥皂膜,測量定壓下,氣體在該流速所造成肥皂液液面上升體積 與時間的變化率(dV/dt),依下式計算在標準狀態下的氣體流量( ): (3-3) 其中 為流量的測量值(cm3 s-1), 為標準狀態溫度(273.15 K), 為室溫(K), 為當時氣壓(Torr), 為室溫下水的蒸氣壓(Torr), 為 標準狀態壓力(760 Torr)。改變流速反覆此步驟數次,將測量所得的數值與 流量計的讀數作一關係圖。 3.3 實驗步驟 3.3.1 周邊儀器時序控制
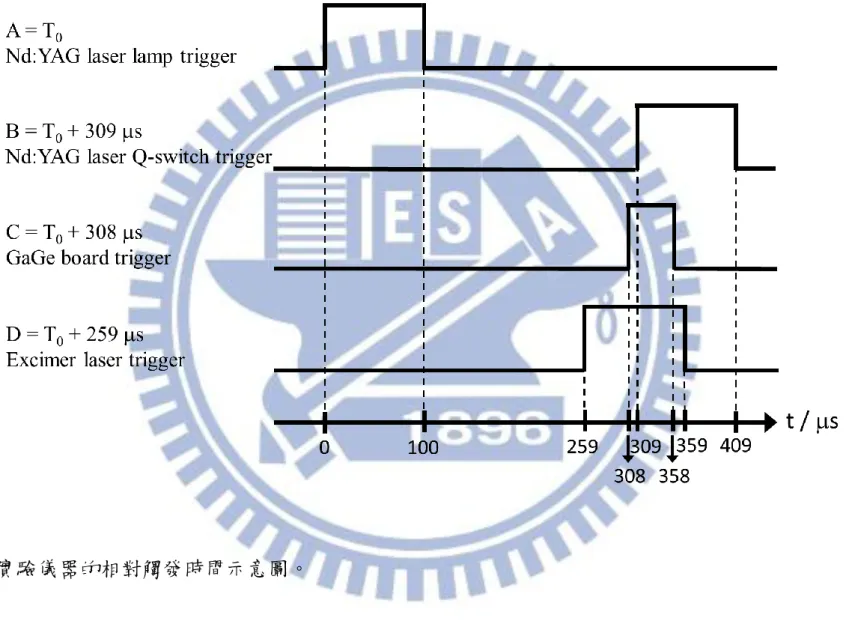
吾人以脈衝產生器(Berkeley Nucleonics Corp., model 575)產生 TTL 波觸 發 Nd:YAG 雷射、準分子雷射以及 GaGe 訊號取樣板。其相對觸發時間如 圖 3-5 所示,設定如下:
週期:10 Hz
47 B = T0 + 309 s:觸發 Nd:YAG 雷射之 Q-switch,width = 100 s; C = T0 + 308 s:觸發訊號取樣板,width = 50 s; D = T0 + 259 s:觸發光解雷射,width = 100 s。 B 和 D 可用以調整可調頻紅外雷射和光解雷射的出光時間,其時間差是依 據反應物被光解後,經過多少時間能產生最大的光解產物訊號來決定。此 外,以本實驗系統而言,利用 phtodiode 可量測實際上光解雷射比紅外雷射 到達反應槽的時間早 49.8 s,略小於頻道 D 和通道 B 的觸發時間差(50 s) 。 3.3.2 實驗條件 (A) 光解效率的評估
本實驗所用的樣品為碘甲烷(CH3I,99.5%,KANTO CHEMICAL CO.) 或是丙酮(CH3C(O)CH3,99.8%,J.T. Baker)以及 O2,所有樣品皆直接使用, 未做進一步純化。實驗使用碘甲烷和 O2時,光譜掃描較低解析度(0.4 cm 1 ) 的實驗中,碘甲烷的分壓為 0.5 Torr,而在光譜掃描較高解析度(0.14 cm1 ) 的實驗中,碘甲烷的分壓則為 0.3 Torr,兩實驗的總壓皆約 150 Torr,反應 溫度為 298 K,其在標準狀態下的總流速為 32 STP cm3 s-1。碘甲烷在波長 248 nm 的光解截面積(cross section)為 cm2 molecule1[1]。由於 光解雷射面積為 2 cm2 (4 cm × 0.5 cm),能量為 340 mJ,假設量子產率
48 (吸收一 248 nm 的光子產生一個 CH3自由基),則光解效率為: (3-4) 為了增加總光解區域,吾人利用兩面 248 nm 反射鏡使光解雷射來回通過反 應腔體 3 次,光解總體積約為 18 cm3 (12 cm × 0.5 cm × 3 cm),考慮反應腔 體的體積為 430 cm3,產生 CH 3自由基的濃度在光譜掃描較低解析度的實驗 時為: (3-5) 而在光譜掃描較高解析度的實驗時為: (3-6) 實驗使用丙酮和 O2時,丙酮的分壓為 0.3 Torr,總壓約 150 Torr,反應 溫度為 298 K,其在標準狀態下的總流速為 32 STP cm3 s-1。丙酮在波長 193 nm 的光解截面積為 cm2 molecule1 [2]。由於光解雷射面積為 2 cm2 (4 cm × 0.5 cm),能量為 105 mJ,其量子產率 (平均每吸收一 193 nm 的光子產生 1.92 個 CH3自由基)[3],則光解效率為: (3-7) 吾人利用兩面 193 nm 反射鏡使光解雷射來回通過反應腔體 3 次,光解總體 積約為 18 cm3 (12 cm × 0.5 cm × 3 cm),可推估產生 CH3自由基的濃度為:
49 (3-8) (B) 預估 CH3OO 生成的時間 CH3 O2形成 CH3OO 屬於三體反應(termolecular reaction): (3-9) [ ] [ ][ ][ ] (3-10) 其中 M 為第三體(third body), 為低壓極限下之三級反應速率常數。本實 驗中的 O2除了一方面當作反應物與 CH3反應生成 CH3OO,一方面亦可當 成淬熄體,將 CH3OO 多餘的能量帶走,延長 CH3OO 的存活時間並減少光 譜中熱譜帶的干擾。O2壓力約為 150 Torr,由理想方程式 可知在實驗 溫度 298 K 下約等於 molecules cm-3,由文獻中[4]可大約估計此 溫度與壓力下的速率常數尚在 fall-off region,但非常接近高壓極限。在 fall-off region 的速率常數並不像在高壓極限時與第三體壓力無關。吾人將 (3-10)式改寫成: [ ] [ ][ ] (3-11) [ ] [ ] [ ] (3-12) 其 中 [ ] 為 CH3 的 起 始 濃 度 , [ ] , 利 用 修 正 過 的 Lindemann-Hinshelwood 公式[5, 6, 7]及其相關參數[4]即可求得在 298 K、[M]
50 = molecules cm-3時的速率常數: [ ] [ ] { [ [ ] ] } (3-13) 其中 為溫度 T K 時的低壓極限速率常數, 為溫度 T K 時的高壓極限 速率常數, 為 center broadening factor。由於文獻中[4]僅有[M] = N2之 及 ,若假設 O2與 N2淬熄效果相同,則: , , ([M]=N2)。 將上述參數帶入(3-13)式便可求得 , 把 和[O 2] = molecules cm -3代入(3-12)式可推出在 後,已有 90%的 CH3OO 生成。吾人藉由脈衝產生器改變光解雷射和紅外雷 射的出光時間差並掃描光解產物的紅外吸收光譜。當光解雷射到達反應槽 約 50 s 後,光解產物有最大的吸收強度。 3.3.3 LabView 程式設定 點選 Goto start Wn 頁面,設定光譜掃描起始波數為 2930 cm1,執行程 式使可調頻紅外雷射藉著轉動內部非線性晶體的角度,而讓出光為指定波 數。接著按下 Read Wn 按鈕並執行,讓可調頻紅外雷射回傳雷射波數,確
51 認是否和指定值相同。再來進入 Scan setup 頁面,設定掃描終止波長為 3060 cm1,掃描解析度為 0.4 cm1的光譜時,相鄰取樣點的波數間距為 0.2 cm1, 每平均 50 張振盪衰減訊號記錄一點。由於脈衝產生器的觸發頻率為 10Hz, 故掃描一張光譜約需 54 分鐘。而在掃描解析度為 0.14 cm1的光譜時,相鄰 取樣點的波數間距為 0.07 cm1,掃描一張光譜約需 2.6 小時。吾人先後各取 一張不開光解雷射和打開光解雷射的光譜,再將後者減去前者即可得差異 光譜。