國立高雄大學化學工程及材料工程學系
碩士論文
三苯胺與噻吩電化學共聚合行為與其電致色變元件
Electropolymerization of triphenylamine-thiophene
comonomers applied to electrochromic devices
研究生:馮立德 撰
指導教授:楊乾信 博士
謝誌
感謝我的恩師 楊乾信 教授兩年來不厭其煩對我的課業與研究
上的諄諄教誨,生活上的關心,使的我在碩士班這兩年生涯裡,不但
論文順利完成,並且在待人處事上有了相當的成長。此外要感謝口試
委員 楊文都 教授及 林文崇 教授,能在百忙之中撥空前來參與學生
得口試委員,讓論文內容得以更加完善。並感謝系辦的亞貞姊與怡雅
姊,幫助我完成許多系上的事宜。
在兩年的研究過程感謝仕銘學長、漢隆學長、俞均學姊等學長姊
在實驗上的教導與鼓勵,沒有學長姐的指導,我的實驗也不會順利完
成,並感謝碩班培子、泓諭、孟諺等同學們在我碩班生涯的鼓勵與幫
助以及感謝學弟妹在許多地方上的幫忙。感謝高雄醫學大學的王淇佳
小姐在 NRM 儀器上的技術指導與分析 NMR 圖譜上的幫助。
最後,感謝我的親人,無時無刻支持與關心我,並作為我的避風
港,在我失意的時候打氣加油,讓我拾回信心與更相信自己。
目錄
謝誌………..……….……..I 目錄………...………II 表目錄………..………...VI 圖目錄………..……..…VII 中文摘要………1 ABSTRACT………...………...3 第一章 緒論………5 1.1 簡介………...5 1.2 電致變色…..………..………...6 1.2.1 電致變色原理………7 1.2.2 變色材料………..…………..7 1.2.3 電致變色元件的結構………..11 1.2.4 電致變色元件之應用………..12 1.3 文獻回顧……….13 1.3.1 三苯胺……….…….13 1.3.2 三苯胺的電化學性質………..……13 1.3.3 導電性高分子材料………..18 1.3.4 導電機制………..21 1.3.5 電化學聚合………..23 1.3.6 二氧化鈦簡介及特性………..24 1.4 研究動機...262.2 三苯胺衍生物之合成步驟與性質鑑定……….29 2.2.1 三苯胺衍生物之合成步驟………..34 2.2.1.1 Hexyloxybenzene 合成………..…34 2.2.1.2 1-bromo-4-(hexyloxy)benzene 合成………...…34 2.2.1.3 HTPA 合成………35 2.2.1.4 Vilsmeier reagent 製備………..…36 2.2.1.5 4-Formyltriphenylamine 合成………...37 2.2.1.6 TPAR1 合成……….…..…37 2.2.1.7 3CN-TPA 合成………..38 2.2.2 三苯胺衍生物性質鑑定………..…39
2.2.2.1 液態核磁共振光譜儀(Solution state Nuclear Magnetic Resonance, 400 MHz and 500 MHz)………..…39 2.2.2.2 傅立葉紅外線光譜儀(FTIR)鑑定官能基………46 第三章 三苯胺衍生物的原位電化學聚合行為的實驗結果討論………..49 3.1 實驗部分………...49 3.1.1 實驗藥品及儀器………..49 3.1.2 實驗流程………..50 3.1.3 水熱製備銳鈦礦多孔奈米二氧化鈦粉體………..51 3.1.4 多孔奈米二氧化鈦/聚乙二醇複合漿料製備……….52 3.1.5 ITO 基材前處理………...52 3.1.6 奈米二氧化鈦多孔性電極………..…………52 3.1.7 電化學原位共聚合法………..52 3.1.8 交流阻抗量測………..53 3.2 結果與討論……….54
3.2.1 銳鈦礦多孔二氧化鈦奈米粉體之特性與吸附染料後的官能基探討.54 3.2.1.1 銳鈦礦多孔二氧化鈦奈米粉體之表面形貌……….….54 3.2.1.2 奈米二氧化鈦多孔性電極吸附染料前後的官能基探討….….55 3.2.2 三苯胺衍生物電聚合電化學行為……….….56 3.2.3 三苯胺衍生物電共聚合電化學行為……….……….66 第四章 噻吩提升三苯胺衍生物電致變色元件性能………..……77 4.1 實驗部分……….77 4.1.1 藥品與儀器………..77 4.1.2 實驗架構………..…79 4.1.3 水熱製備銳鈦礦多孔奈米二氧化鈦粉體………..80 4.1.4 多孔奈米二氧化鈦/聚乙二醇複合漿料製備……….80 4.1.5 ITO 基材前處理………...81 4.1.6 奈米二氧化鈦多孔性電極………..81 4.1.7 循環伏安法共聚合薄膜………..81 4.1.8 電致變色元件組裝………..82 4.1.9 電致變色特性測試………..82 4.1.10 三苯胺與噻吩衍生物薄膜光學分析………..83 4.1.11 三苯胺與噻吩衍生物薄膜化學分析電子光譜分析……….….83 4.1.12 掃描式電子顯微鏡 (SEM)………...84 4.2 結果與討論……….84 4.2.1 三苯胺與噻吩衍生物薄膜成長………..84 4.2.2 表面形貌………..92
4.2.3.2 三苯胺與噻吩衍生物電聚合薄膜紫外光/可見光(UV-Vis)光譜
分析………...99
4.2.3.3 三苯胺與噻吩衍生物電聚合薄膜 HOMO/Eg/LUMO………..103
4.2.4 三苯胺與噻吩衍生物電聚合薄膜化學分析電子光譜分析…………103
4.2.5 三苯胺衍生物薄膜電致變色元件………..…..107
4.2.5.1 以 UV-Visible 對 PHTPA、PTPAR1 及 P(HDAP-co-TPAR1) 薄 膜電致變色元件之光學性質探討….………...107 4.2.5.2 PHTPA、PTPAR1 及 P(HTPA-co-TPAR1) 薄膜電致變色元件穩 定度探討………112 4.2.5.3 三苯胺衍生物薄膜電致變色元件變色情形……….120 4.2.5.4 結論……….121 4.2.6 噻吩提升三苯胺衍生物薄膜電致變色元件………122 4.2.6.1 以 UV-Visible 對 PEDOT、P(HTPA-co-EDOT)及 P(TPAR1-co-EDOT) 薄膜電致變色元件之光學性質探討…122 4.2.6.2 PEDOT、P(HTPA-co-EDOT)及 P(TPAR1-co-EDOT) 薄膜電致 變色元件穩定度探討………...….127 4.2.6.3 噻吩提升三苯胺衍生物薄膜電致變色元件變色情形……….135 4.2.6.4 結論……….…136 第五章 總結………..138 參考文獻………139
表目錄
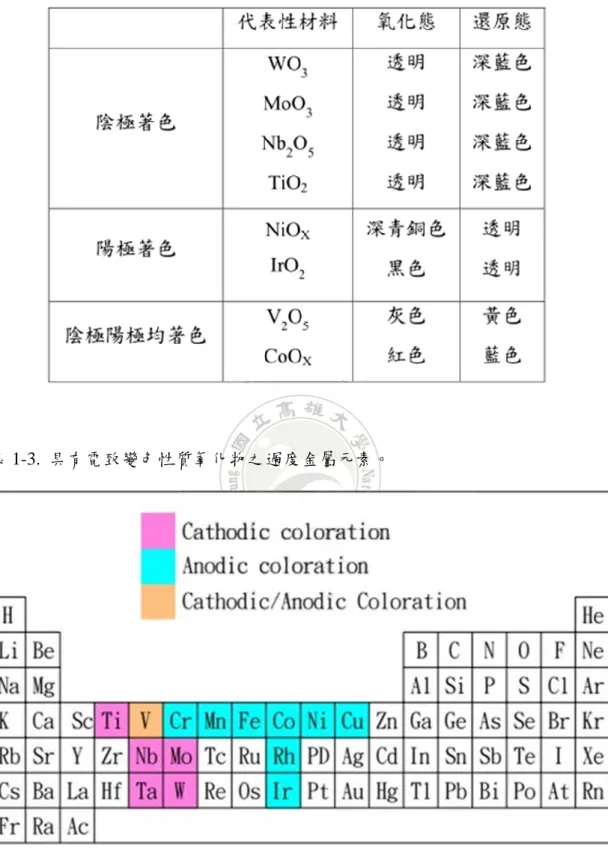
表 1-1. 聚雜環高分子的電致變色參數[1]……….…….6 表 1-2. 無機電致色變材料著褪色變化分類……….……….…....9 表 1-3. 具有電致變色性質氧化物之過度金屬元素……….…….9 表 1-4. 三苯胺衍生物三個對位取代基的電化學特性………....……16 表 1-5. 含氨基取代的三苯胺衍生物的電化學性質……….………...17 表 1-6. 導電高分子種類[25]……….…….……20 表 2-1. 中間產物與最終產物之結構與 IUPAC 命名………..….29 表 4-1. 三苯胺與噻吩衍生物電聚合薄膜的 HOMO/Eg/LUMO………103 表 4-2. PHTPA、PTPAR1 及 P(HDAP-co-TPAR1) 薄膜電致變色元件於不同電位 顏色變化情形………..………..121 表 4-3. PEDOT、P(HTPA-co-EDOT)、P(TPAR1-co-EDOT) 薄膜電致變色元件於 不同電位顏色變化情形………..……...136圖目錄
圖 1-1. WO3材料的電致變色機理[14]……….…8 圖 1-2. 普魯士藍材料的電致變色機理[14]………8 圖 1-3. 紫羅精類化合物氧化還原態轉化(A:二價陽離子二價陽離子,B:單價陽 離子單價陽離子,C:中性態中性態)………..10 圖 1-4. 電致變色元件結構示意圖……….12 圖 1-5. 盛貿科技的大尺寸電致變色智慧窗灰階展示……….13 圖 1-6. 5x10-4 M TPA 包含 0.1M TBAP 在二氯甲烷溶劑中的吸收光譜變化圖 (a)0.92V,(b)0.95V,(d)1.01V。掃描速率:0.1V/s………...…….14 圖 1-7. TPA 的電化學雙聚合反應………...………...15 圖 1-8. 三苯胺衍生物三個對位取代基……….……16 圖 1-9. 含氨基取代的三苯胺衍生物……….17 圖 1-10. 主要的導電性高分子導電度和金屬,半導體與絕緣體做比較[21]……19 圖 1-11. 隨分子大小遞增的 PA,(CH)n其 pMOs 之能階示意圖[24]………...…….22 圖 1-12. Trans-(CH)n 的 soliton 型態[24]……….…23 圖 1-13. 偏極子和雙偏極子能階示意圖[24]………...…….…23 圖 1-14. 三苯胺衍生物 HTPA、TPAR1、和 3CN-TPA 的分子結構式………....27 圖 2-1. HTPA合成步驟………31 圖 2-2. TPAR1 合成步驟………..32 圖 2-3. 3CN-TPA合成步驟………..33 圖 2-4. 1 H-NMR spectrum hexyloxybenzene in DMSO- d6 [27]……….42圖 2-5. 1 H-NMR spectrum 1-bromo-4-(hexyloxy)benzene DMSO- d6 [27]…………42
圖 2-6. 1 H-NMR spectrum 4-(hexyloxy)-N,N-diphenylaniline in DMSO- d6 [27]...………43
圖 2-7. 1
H-NMR spectrum 4-Formyltriphenylamine in DMSO- d6 [26]……….43
圖 2-8. 1 H-NMR spectrum TPAR1 in DMSO- d6 [26]………..……...44
圖 2-9. 13 C-NMR spectrum TPAR1 in DMSO-d6 [26]………...44
圖 2-10. 1 H-NMR spectrum 3CN-TPA in CDCl3……….…45
圖 2-11. 13 C-NMR spectrum 3CN-TPA in CDCl3………45
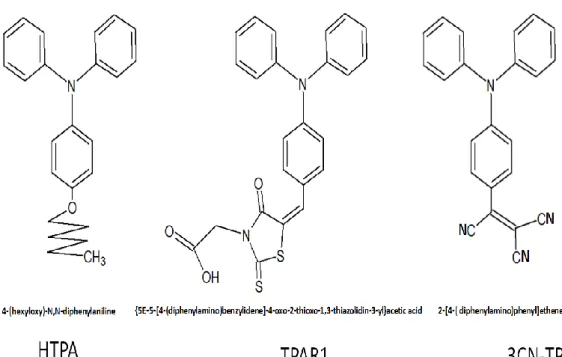
圖 2-12. HTPA 的 FTIR 光譜………..………...47
圖 2-13. TPAR1 與 TPA-CHO 的 FTIR 光譜………...……….47
圖 2-14. 3CN-TPA 的 FTIR 光譜………..……….……..48 圖 3-1. 三苯胺衍生物的循環伏安法聚合程序示意圖……….50 圖 3-2. 三苯胺衍生物的循環伏安法共聚合程序示意圖……….51 圖 3-3. 交流阻抗可能量測之圖形[37]………..53 圖 3-4. P25 和水熱法製備的二氧化鈦粉體之外觀………..54 圖 3-5. 奈米二氧化鈦多孔性電極 FE-SEM 圖……….54
圖 3-6. (a)TPAR1 顆粒 (b)TPAR1 吸附於 TiO2後的 FTIR 光譜圖……….…55
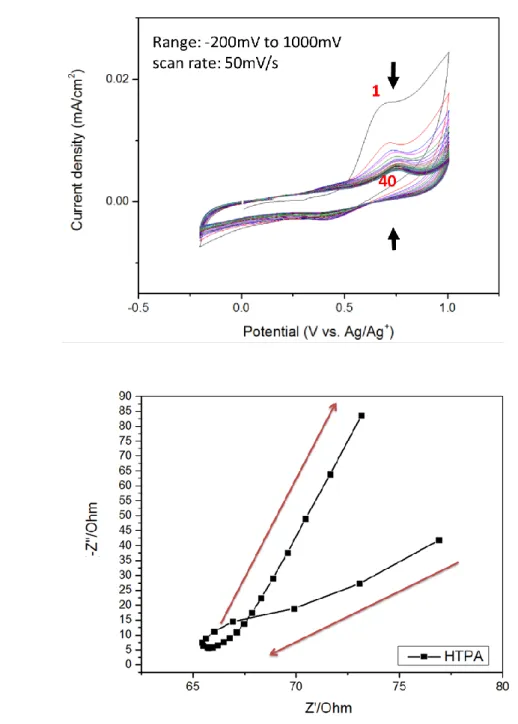
圖 3-7. 奈米 TiO2吸附 HTPA 單體(2x10-3M 溶液)在 1N HClO4酸性水溶液中(a)連 續掃描循環伏安圖; (b)交流阻抗圖………...59
圖 3-8. 奈米 TiO2吸附 HTPA 單體(2x10-3M 溶液)在 1N LiClO4有機溶液中(a)連續 掃描循環伏安圖; (b)交流阻抗圖………...60
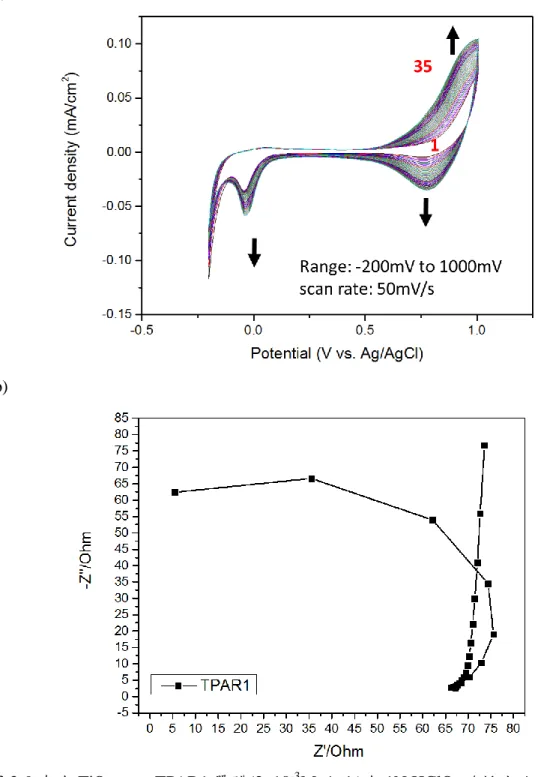
圖 3-9. 奈米 TiO2吸附 TPAR1 單體(2x10-3M 溶液)在 1N HClO4酸性水溶液中(a) 連續掃描循環伏安圖; (b)交流阻抗圖………...61
圖 3-10. 奈米 TiO2吸附 TPAR1 單體(2x10-3M 溶液)在 1N LiClO4有機溶液中(a) 連續掃描循環伏安圖; (b)交流阻抗圖………...62
圖 3-12. 奈米 TiO2吸附 3CN-TPA 單體(2x10-3M 溶液)在 1N HClO4酸性水溶液中
(a)連續掃描循環伏安圖; (b)交流阻抗圖…...………64
圖 3-13. 奈米 TiO2吸附 3C-TPA 單體(2x10-3M 溶液)在 1N LiClO4有機溶液中(a) 連續掃描循環伏安圖; (b)交流阻抗圖……….………...…...65
圖 3-14. 奈米 TiO2吸附 HTPA 和 TPAR1 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N HClO4酸性水溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖....69.
圖 3-15. 奈米 TiO2吸附 HTPA 和 TPAR1 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N LiClO4有機溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖…....70
圖 3-16. 奈米 TiO2吸附 HTPA 和 3CN-TPA 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N HClO4酸性水溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖….71 圖 3-17. 奈米 TiO2吸附 HTPA 和 3CN-TPA 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N LiClO4有機溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖……72
圖 3-18. 奈米 TiO2吸附 3CN-TPA 和 TPAR1 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N HClO4酸性水溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖….73 圖 3-19. 奈米 TiO2吸附 3CN-TPA 和 TPAR1 共混單體(莫爾比例 1:1,濃度 2x10-3M) 在 1N LiClO4有機溶液中(a)連續掃描循環伏安圖; (b)交流阻抗圖…....74 圖 3-20. P(HTPA-co-TPAR1)可能的電化學反應機制………...75 圖 3-21.TPAR1 產生有效通路並與 HTPA 產生電共聚合反應……….75 圖 3-22. P(3CN-TPA-co-TPAR1)可能的電化學反應機制……….76 圖 3-23.TPAR1 產生有效通路並與 3CN-TPA 產生電共聚合反應………..76 圖 4-1. 三苯胺衍生物電致變色元件程序示意圖……….79 圖 4-2. 噻吩加入提升三苯胺衍生物電致變色元件程序示意圖……….79 圖 4-3. 電致變色元件示意圖……….82 圖 4-4. Spectrochronoamperometric 示意圖………82
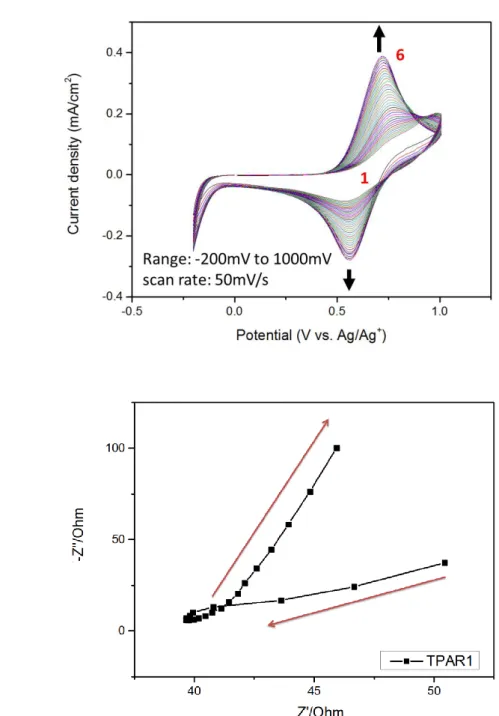
圖 4-5. 7.5x10-3
M HTPA 單體溶於 0.1M LiCl4有機溶液(a)電化學聚合循環伏安曲
線圖;(b) PHTPA 可能的電化學反應機制………...…86
圖 4-6. 7.5x10-3 M TPAR1 單體溶於 0.1M LiCl4有機溶液(a)電化學聚合循環伏安曲 線圖;(b) PTPAR1 可能的電化學反應機制………...87
圖 4-7. 7.5x10-3 M EDOT 單體溶於 0.1M LiCl4有機溶液(a)電化學聚合循環伏安曲 線圖;(b) PEDOT 可能的電化學反應機制………….………88
圖 4-8. 7.5x10-3 MHTPA 與 TPAR1 共單體(莫爾比例 1:1)溶於 0.1M LiCl4有機溶液 (a)電化學聚合循環伏安曲線圖;(b) P(HTPA-co-TPAR1)可能的電化學反 應機制………..89
圖 4-9. 7.5x10-3 M HTPA 與 EDOT 共單體(莫爾比例 1:1)溶於 0.1M LiCl4有機溶液 (a)電化學聚合循環伏安曲線圖;(b) P(HTPA-co-EDOT)可能的電化學反 應機制………..…90
圖 4-10. 7.5x10-3 M TPAR1 與 EDOT 共單體(莫爾比例 1:1)溶於 0.1M LiCl4有機溶 液(a)電化學聚合循環伏安曲線圖;(b) P(TPAR1-co-EDOT)可能的電化學 反應機制………..91 圖 4-11. 奈米二氧化鈦多孔性電極 FE-SEM 圖………92 圖 4-12. PHTPA 電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖………...93 圖 4-13. PTPAR1 電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖…………...93 圖 4-14. P(HTPA-co-TPAR1)電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖…..94 圖 4-15.PEDOT 電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖………..94 圖 4-16.P(HTPA-co-EDOT)電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖…….95 圖 4-17.P(TPAR1-co-TPAR1)電聚合於奈米二氧化鈦多孔性電極 FE-SEM 圖..…95
圖 4-20. 利用 PESA 量測 P(HTPA-co-TPAR1)薄膜 HOMO 值……….…97
圖 4-21. 利用 PESA 量測 PEDOT 薄膜 HOMO 值……….98
圖 4-22. 利用 PESA 量測 P(HTPA-co-EDOT)薄膜 HOMO 值………..…98
圖 4-23. 利用 PESA 量測 P(TPAR1-co-EDOT)薄膜 HOMO 值………99
圖 4-24. 三苯胺衍生物電聚合薄膜(a)吸收光譜;(b)能隙圖………...101 圖 4-25. 三苯胺與噻吩衍生物電聚合薄膜(a)吸收光譜;(b)能隙圖………..102 圖 4-26. PHTPA 薄膜 XPS 全圖譜………...104 圖 4-27. PTPAR1 薄膜 XPS 全圖譜………...…105 圖 4-28. P(HTPA-co-TPAR1)薄膜 XPS 全圖譜…...……….105 圖 4-29. P(HTPA-co-EDOT)薄膜 XPS 全圖譜………...…....106 圖 4-30. P(TPAR1-co-EDOT)薄膜 XPS 全圖譜……..………..…106 圖 4-31. PEDOT 薄膜 XPS 全圖譜………107 圖 4-32. 為 PHTPA 薄膜電致變色元件於不同電位下穿透度隨波長變化,並且±2V 時光學對比為 20.6%,波長為 780nm………..…109 圖 4-33. 為 PHTPA 薄膜電致變色元件於不同電位下穿透度隨波長變化,並且± 2.5V 時最大光學對比為 43.5%,波長為 780nm………..109 圖 4-34. 為 PTPAR1 薄膜電致變色元件於不同電位下穿透度隨波長變化之情形, 並且±2V 時光學對比為 9.6%,波長為 496nm……….…110 圖 4-35. 為 PTPAR1 薄膜電致變色元件於不同電位下穿透度隨波長變化,並且 ±2.5V 時最大光學對比為 35.2%,波長為 780nm…..……….….110 圖 4-36. 為 P(HTPA-co-TPAR1) 薄膜電致變色元件於不同電位下穿透度隨波長 變化,並且±2V 時光學對比為 15.6%,波長為 470nm……...111 圖 4-37. 為 P(HTPA-co-TPAR1) 薄膜電致變色元件於不同電位下穿透度隨波長 變化,並且±2.5V 時最大光學對比為 39.4%,波長為 780nm………..…111
圖 4-38. PHTPA 薄膜電致變色元件於±2 V 階梯電位變換下穿透度與電流變化圖 (λ = 780 nm)………...114 圖 4-39. PHTPA 薄膜電致變色元件於±2.5 V 階梯電位變換下穿透度與電流變化圖 (λ = 780 nm)………...115 圖 4-40. PTPAR1 薄膜電致變色元件於±2 V 階梯電位變換下穿透度與電流變化圖 (λ = 496 nm)………...116 圖 4-41. PTPAR1 薄膜電致變色元件於±2.5 V 階梯電位變換下穿透度與電流變化 圖 (λ = 780 nm)………..……….…..117 圖 4-42. P(HTPA-co-TPAR1) 薄膜電致變色元件於±2 V 階梯電位變換下穿透度與 電流變化圖 (λ = 470 nm)………..………...118 圖 4-43. P(HTPA-co-TPAR1) 薄膜電致變色元件於 ±2.5 V 階梯電位變換下穿透 度與電流變化圖 (λ = 768 nm)………..………...119 圖 4-44. 為 PEDOT 薄膜電致變色元件於不同電位下穿透度隨波長變化,並且±2V 時光學對比為 50%,波長為 525nm….………124 圖 4-45. 為 PEDOT 薄膜電致變色元件於不同電位下穿透度隨波長變化,並且 ±2.5V 時最大光學對比為 73.8%,波長為 524nm………....124 圖 4-46. 為 P(HTPA-co-EDOT) 薄膜電致變色元件於不同電位下穿透度隨波長變 化,並且±2V 時光學對比為 46.6%,波長為 780nm……….……125 圖 4-47. 為(HTPA-co-EDOT) 薄膜電致變色元件於不同電位下穿透度隨波長變 化,並且±2V 時最大光學對比為 60.7%,波長為 780nm……….125 圖 4-48. 為 P(TPAR1-co-EDOT) 薄膜電致變色元件於不同電位下穿透度隨波長 變化,並且±2V 時光學對比為 39.1%,波長為 780nm……….…126
圖 4-50. 為 PEDOT 薄膜電致變色元件於±2 V 階梯電位變換下穿透度與電流變化 圖 (λ = 525 nm)………..………..129 圖 4-51. 為 PEDOT 薄膜電致變色元件於±2.5 V 階梯電位變換下穿透度與電流變 化圖 (λ = 524 nm)………..………..130 圖 4-52. 為 P(HTPA-co-EDOT) 薄膜電致變色元件於±2 V 階梯電位變換下穿透度 與電流變化圖 (λ = 780 nm)………..………..131 圖 4-53. 為 P(HTPA-co-EDOT) 薄膜電致變色元件於±2.5 V 階梯電位變換下穿透 度與電流變化圖 (λ = 780 nm)………..……..132 圖 4-54. 為 P(TPAR1-co-EDOT) 薄膜電致變色元件於±2 V 階梯電位變換下穿透 度與電流變化圖 (λ = 780 nm)………..…………..133 圖 4-55. 為 P(TPAR1-co-EDOT) 薄膜電致變色元件於±2.5 V 階梯電位變換下穿 透度與電流變化圖 (λ = 780 nm)………..………..…134
三苯胺與噻吩電化學共聚合行為與其電致色變元件
指導教授:楊乾信 博士 國立高雄大學 化學工程及材料工程學系 碩士班 學生:馮立德 國立高雄大學 化學工程及材料工程學系 碩士班 中文摘要 本研究首先合成三苯胺衍生物(triphenylamine derivative ),然後進行電化學 聚合並應用於電致變色元件。第一部分實驗,以奈米二氧化鈦 (TiO2) 塗佈於 ITO 形成多孔工作電極,浸泡於三苯胺衍生物(單體)溶液一日,取出吸附此單體的電 極後進行電化學聚合。探討三苯胺衍生物單體,與奈米二氧化鈦表面的-OH 官能 基和三苯胺衍生物上的官能基有無產生化學鍵結,並對電化學聚合時的電化學行 為有何影響。實驗結果顯示含醚鏈推電子基的三苯胺(HTPA)和含三個 CN 吸電子 基的三苯胺(3CN-TPA)無法聚合,含 carboxylic acid 吸電子基的三苯胺(TPAR1) 可聚合為 PTPAR1;另一方面,HTPA 和 TPAR1 以及 3CN-TPA 和 TPAR1 則可進 行共聚合反應,但 HTPA 和 3CN-TPA 還是無法作進行聚合反應。第二部分實驗,則以奈米二氧化鈦修飾 ITO 多孔工作電極,經電化學共聚 合三苯胺衍生物成為薄膜,並組裝成電致變色元件。由實驗結果得知,只有三苯 胺衍生物的電致變色元件光學對比小於 40%,長時間操作穩定度不佳。在本研 究想要了解噻吩單體加入三苯胺衍生物單體溶液後,以電聚合的方式共聚合成薄
2
及穩定度。
關鍵字:
三苯胺衍生物、電化學行為、電致變色元件、推電子官能基
Electropolymerization of triphenylamine-thiophene
comonomers applied to electrochromic devices
Advisor(s): Dr. Chien-Hsin Yang
Graduate Program in Chemical and Materials Engineering National University of Kaohsiung
Student: Li-Te Feng
Institute of Chemical and Materials Engineering National University of Kaohsiung
ABSTRACT
Triphenylamine (TPA) derivatives were first synthesized and performed electrochemical polymerization employing to the electrochromic devices in this study. In the first part of the experiment, nano titanium dioxide was spin-coated on an ITO electrode to form a porous working electrode, and then immersed in the triphenylamine derivative (monomer) solution 24 h. Adsorption of triphenylamine derivative monomer on this working electrode was used to study the effect between a functional group of –OH on nano titanium dioxide surface and the functionl groups on triphenylamine derivatives, which is correlated to the electrochemical behavior of electrochemical polymerization. Results showed that electron-donor ether-linked TPA (HTPA) and electron-acceptor cyano-linked TPA (3CN-TPA) cannot be polymerized on the substrate, but electron-acceptor carboxylic acid-linked TPA (TPAR1) can be
4
comonomer of HTPA-3CN-TPA cannot be copolymerized.
The second part of the experiment, the monomers of triphenylamine derivatives were electrochemically copolymerized on a TiO2-coated ITO porous working
electrode. The obtained polymer thin films were assembled into electrochromic devices. Experimental results showed that the optical contrast of these triphenylamine derivative polymer films is less than 40% with poor long-term operation stability. In this study, we tried to know the effects of thiophene-monomer addition into triphenylamine derivative monomers. The copolymer films were also employed to electrochromic devices. Results showed that the addition of the thiophene into triphenylamine derivatives can greatly enhance the optical contrast of electrochromic devices from from 40 to 60% and stability.
Keywords: triphenylamine derivative, electropolymerization, electrochromic device ,electron donor, electron acceptor
第一章 緒論
1.1. 簡介 隨著國家科技發展,能源使用量逐漸增加,人類對於石化燃料(石油、煤、 天然氣等)的依賴度相對提高,尤其是在 1970 年爆發能源危機之後,人們才意識 到石化燃料終有消耗殆盡的一天,也使人們了解到開發自然能源的重要性。加上 近十年來,因燃燒石油所產生的 CO2 所造成的全球溫室效應問題,使得各先進 國家紛紛開始積極開發高效益替代能源,有效的應用能源被視為重要的課題,節 能減碳的綠色商品成了現今科技發展研究的熱點,而變色材料便是以此發展起來 的新型功能性材料。 世界上大約有 30~40 % 的能源被用於室內的調溫及照明,由於太陽光中的 熱有 60 % 是由紅外線,35 % 為可見光,5 % 的熱為紫外光所帶來,兼顧採 光和節能上的平衡,人們研發出可以變色的材料,使材料產生顏色及穿透率的變 化,調節不同入射波長電磁輻射的入射量來阻隔光與熱,可達到同時控制室內照 明與溫度的影響。 變色材料是因為吸收外部能量產生顏色變化,根據不同的能量來源一般可分 成五大類型:電致變色 (electrochromism) 、光致變色 (photochromism) 、熱致 變色 (thermochromism) 、化學反應變色、物理作用變色。但電致變色控制性較 高,可藉由改變電壓大小來控制材料型態以變化光學特性,電致變色技術可以調 節穿透率(transmittance)、反射率(reflectance)、吸收率(absorptance)等特性,可應 用於建築物、汽車的玻璃,達到節能、調節光線及控制熱輻射。原理為:當室外 陽光強,只要控制電位或電流可改變玻璃窗於特定波段的穿透率,可隨意的控制 陽光通過窗戶的強度且阻擋大達到調節熱輻射之目的,且由於其特殊的顏色記憶 效果,可以不需持續給予電流,加上系統本身耗用的能量極低,對於節能減碳方6 1.2.電致變色 電致變色是指在外接電壓或者電流的驅動下,物質發生電化學氧化還原反應 而導致顏色變化。在外加電場作用下,物質的化學性能 (穿透率、反射率等) 在 可見光波長內產生穩定的可逆變化。其主要特點有以下幾點:(I) 電致變色材料 中電荷的輸入與輸出通過外界電壓或電流的改變而方便地實現,輸入或輸出電荷 的多少直接決定了材料的致色程度,控制電壓、電流可以改變電致變色材料的顏 色變化, (II) 不同電壓的極性可以方便地實現著色及褪色, (III) 已著色的材料 在關閉電流而不發生氧化還原反應的情況下,可以保持著色狀態,即具有記憶功 能。表 1-1 [1]為幾種較常應用於電致變色元件製作的導電性高分子與其物性。 polythiophene 其顏色變化可從紅色轉變為藍色,持續數個小時,具有作為光學 記憶元件的用途;同樣地,polypyrrole 含有三種的不同顏色(黃綠、暗褐和藍色), 許多針對 polyaniline 變色研究報導指出,polyaniline 由淡黃色變為至綠色或藍 色,由提供的電位決定,而氧化還原的改變,則直接影響到顏色的變化,光譜電 化學的研究可以清楚瞭解高分子膜顏色的變化特性,以得到更確切的電致變色特 性。諸如此類,高分子的電致變色性質可以應用於製作多種不同的電致變色元件 [2] 。 表 1-1.聚雜環高分子的電致變色參數[1]。
1.2.1. 電致變色原理 何謂電致變色,就是利用電化學反應使之產生顏色變化的材料,稱之為電致 變色。針對導電性高分子而言,顏色變化的多樣性可歸咎於材料和結構改變。導 電性高分子經過氧化摻雜形成另一狀態,造成能階的改變,依照分子的結構或摻 雜程度,這些狀態我們稱它為 polarons 或 bipolarons,這些變化讓光譜特性改變, 新的吸收波峰也就產生[3-5],造成顏色變化,人類眼睛看見這些顏色改變,主要 是因為這些材料能階的改變,使得導電性高分子呈現不同顏色變化。由於導電高 分子因能階的不同,可顯示不同的顏色變化,高分子能階的改變可經由結構修飾 達到,因此可藉由不同官能基的鍵結,使得高分子結構產生改變,以得到不同顏 色變化特性。 1.2.2. 變色材料 電致變色層是電致變色器件的核心層,是變色反應的發生層。電致變色材料 按照特性可分為:無機電致變色材料和有機電致變色材料。 (1) 無機電致變色材料 無機電致變色材料多是過渡金屬氧化物或其衍生物,人類發現電致變色現象 時就是非晶態 WO3 薄膜的變色。過渡金屬 3d、4f 電子層不穩定,有未成對電 子存在。過渡金屬元素離子一般都具有顏色,且基態與激發態能量差較小,在一 定的條件下價態發生可逆變化,形成混合價態離子共存狀態。隨離子價態和濃度 的變化,顏色也會發生相應的變化,為過渡金屬氧化物含電致變色的原因。常見 的無機變色材料根據其發生氧化還原的原理不同,又可以細分為陽極著色材料和 陰極著色材料。
8
受和應用的機制[6],並且 WO3材料的電致變色機理是在變色過程中由於電場變
化,使薄膜從透明轉為藍色。
圖 1-1. WO3材料的電致變色機理[14]。
陽極變色材料主要是Ⅷ族及 Pt 族(Pt、Tr、Os、Pd、Rh、Ru 等)金屬氧化物 或水合物,如 NiO、IrOx、CoO3、Rh2O3、MnO 等。其中 NiO 有較大的著、褪色
區域,具有良好的循環壽命等優點成為一種研究最多的陽極變色材料。NiO 是一 種具有 NaCl 結構的 3d 過渡金屬氧化物,晶體中顯現鎳空位或過氧的情況,這 導致 NiO 成為一種 P 型半導體。因此,NiO 晶體中經常顯現空位、缺陷以及摻 雜的情況。 另有一些材料不是嚴格的陰極和陽極變色特徵,可以歸類為無機電致變色材 料。這類無機電致變色材料和過渡金屬氧化物不同,可以實現多種顏色變化。以 普魯士藍為代表,它是一種同時含有 Fe2+和 Fe3+混合價配合物。普魯士藍也是通 過鹼金屬離子 (一般為 K+ ) ,使 Fe3+還原變為 Fe2+而著色,可以在無色、藍色、 綠色的顏色轉變[7]。 圖 1-2. 普魯士藍材料的電致變色機理[14]。
表 1-2. 無機電致色變材料著褪色變化分類。
10 (2) 有機電致變色材料 有機型電致變色材料為現今發展電致變色之大宗,因此系列材料在離子摻雜 與去摻雜能力較為迅速,可提供材料快速的著、褪色變化;並且可以藉由合成改 變接枝官能基團而賦予多樣化的顏色變異,迄今為止有機電致變色材料可分為 (1) 有機小分子、(2) 導電高分子。 A. 有機小分子 有機小分子變色材料典型的代表就是 viologen 全名 1,1'-disubstituted- 4, 4'-bipyridinium,該物質在氧化還原過程中會出現顏色變換,屬於氧化還原型化 合物。一般情況下,中性態的 viologen 由於自身結構不同,分子內部電子遷移或 系間遷移受到禁阻,因此顏色較淺 (結構如圖 1-3. C) 。隨著施加電位的上升, 中性態結構逐漸向部分氧化態轉變,生成穩定的二價陽離子形式,該狀態下呈現 無色 (結構如圖 1-3. A) 。viologen 的顯示顏色具有可改變性,可以通過鍵結不 同的烷基或者苄基進行顏色的調節。當取代基為短鏈時,單價陽離子 Viologen 為藍色;隨著烷基鏈的增長,分子間二聚作用加劇,單價陽離子則顯示顏色較深 的紅色[7]。 圖 1-3. 紫羅精類化合物氧化還原態轉化(A:二價陽離子二價陽離子,B:單價陽 離子單價陽離子,C:中性態中性態)。
B. 導電高分子 共軛聚合物被小分子摻雜後呈現高導電度,摻雜劑種類和摻雜濃度除決定導 電性外還支配其顏色變化。因此人們研究導電聚合物的電致變色行為,以芳香化 合物為高分子材料,主要有聚苯胺、聚吡咯、聚噻吩、聚呋喃等。電子佔據的最 高能階和未佔據的最低能階之間的能頻寬(Eg)決定材料的光電性質,並且利用摻 雜和去摻雜來控制材料的光學性質。並且控制電壓的大小,藉由電壓決定摻雜程 度的不同,從而導致可見光區的吸收不同,顯示出顏色的變化,導致電致變色現 象。 1.2.3.電致變色元件的結構 電致色變元件係指可因施加不同的電壓而產生顏色變化之三明治結構。依其 構造的不同,主要可分為三種型態,薄膜型、溶液型以及混和型元件,本研僅針 對薄膜型電致色變元件進行探討。 典型的薄膜型元件,如圖 1-4 所示,此多層結構含有透明導電氧化物的玻璃 基材,如常用的透明 ITO 及 FTO 玻璃,作為電致色變元件的端電極,近年來 的研究趨向製作可撓曲變色元件,基板改採用高分子基板。包含電致色變材料的 電極是以薄膜型態固定於透明導電氧化物上,另一端透明相對電極,使用互補變 色特性之薄膜(如 PEDOT:PSS)作為工作電極之共軛氧化還原對(conjugate redox pair)及離子儲存層(ion storage film)。在兩薄膜電極間注入膠態或固態電 解質,電解質在選擇方面,通常會選擇小型氫離子(H+)以及鋰離子(Li+)作
為電荷傳遞,另一方面較小型的離子,移動快,可使電致色變元件呈現較快的應 答時間[9]。
12 圖 1-4.電致變色元件結構示意圖。 1.2.4.電致變色元件之應用 電致變色元件的應用非常廣泛,整體來說可分為四大項。(a)非發射光的資 訊顯示器,然而使用壽命不高,隨著液晶顯示器的出現,已經慢慢式微了,然而 此系列的研究還是有人繼續進行中[9-12],因為電致變色材料加入陰極射線管內, 可大幅提昇其光學對比性質[13]。(b)反射式鏡子,應用於汽車。(c)電致變色 材料最廣為應用“智慧窗”,此種技術於建築應用上,可阻擋輻射線的進入以及光 線的散失,對於未來綠色環保的建築,將是一個節省能源的技術,如圖 1-5 所示。 (d)這個技術與傳統電致變色元件的應用稍有出入,但其最終應用目的仍非常 好,提供另一種研究的方向,為調節表面發散的熱,對於航太工業的研究很有用, 例如可控制太空梭因溫度驟變所造成的熱散失,維持艙內的溫度調節等。
圖 1-5.盛貿科技的大尺寸電致變色智慧窗灰階展示。 (由材料世界網下載 http://www.materialsnet.com.tw) 1.3.文獻回顧 1.3.1.三苯胺 三苯胺(triphenylamine,TPA)熱穩定度佳,電傳導度良,具有易加工性質, 含可逆電化學性質,容易合成且價格便宜,應用廣泛。在光電元件應用廣泛,包 含發光二極體,有機太陽能電池和電致變色等方面。在分析化學方面,也有文獻 研究三苯胺衍生物應用於修飾電極及生物感測的性質。所以三苯胺衍生物不論材 料應用或生物分析都備受重視,針對三苯胺衍生物進行性質的分析與檢測很重要, 以電化學、光譜電化學對三苯胺衍生物進行研究則是一種簡單的方法[14]。 1.3.2.三苯胺的電化學性質 最早以電化學方法針對三苯胺進行研究的文獻,是 1963 年由 D. N. Stamires 和 J. Turkevich 所發表的文獻[16],爾後,三苯胺化合物以電化學方法進行研究 的文獻就陸續問世。1966 年,R. F. Nelson 和 R. N. Adams 等人,發表三苯胺電 化學性質的文獻[17]。以電化學搭配光譜電化學互相比對,了解物質在反應發生 時,用光譜的變化佐證電子轉移,圖 1-6 為三苯胺的光譜電化學及循環伏安圖(CV)
14
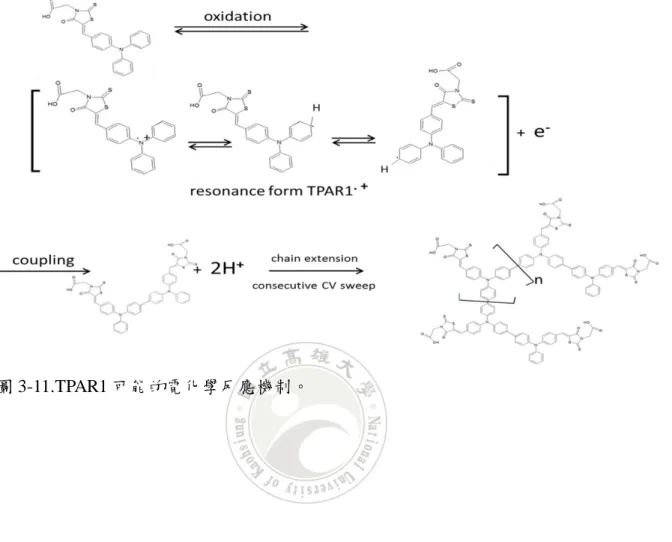
接著在第二圈的掃描可以看到一個新的氧化峰電位 Epa=+0.8V,為 TPA 氧化形成
TPA+.,兩個 TPA+形成雙聚物 TPB(tetraphenylbenzidine),雙聚合反應機制如圖
1-7 所示: 圖 1-6.5x10-4 M TPA 包含 0.1M TBAP 在二氯甲烷溶劑中的吸收光譜變化圖 (a)0.92V,(b)0.95V,(d)1.01V。掃描速率:0.1V/s,參考文獻[17]。 圖 1-6 以光譜電化學對照 CV 圖的結果,將電位提高,吸收光譜最大吸收波 峰在 300nm 不斷下降,而在 484、696 及 1384nm 有新的最大吸收波峰不斷上升, 這是 TPA 雙聚形成 TPB 後二次氧化的光譜,由文獻[17]得之 300nm 為 TPA 最大 吸收波,484 及 1384nm 為 TPB+.的最大吸收波,696nm 則是 TPB2+的最大吸收波, 從文獻與實驗結果得知,TPA 氧化後的不可逆反應及 TPB 可逆反應機制如圖 1-7 所示[18]:
圖 1-7.TPA 的電化學雙聚合反應。 三苯胺由中心氮原子及外圍三個苯環結成,由於苯環的對位活性最佳,因此 在化學反應或電化學反應進行,對位易形成陽離子的自由基(cation radical)。若三 苯胺的苯環對位接上不同的官能基,便會使電化學性質造成很大的影響。從過去 的文獻可以得知,三苯胺的苯環對位若接上推電子基(electro-donating groups), 會使得化合物容易被氧化形成自由陽離子,電化學性質中可逆的氧化還原對與不 可逆的氧化波都會有電位提升,使氧化峰電位降低[16]。圖 1-8 是三苯胺衍生物 三個對位取代基並且表 1-4 是三苯胺苯環對位鍵結不同官能基的氧化電位,可由 表 1-4 中得知接上推電子基(-OCH3與-CH3)的氧化電位提前的較多。
16 圖 1-8.三苯胺衍生物三個對位取代基。 表 1-4.三苯胺衍生物三個對位取代基的電化學特性。 文獻記載中,因為氨基強推電子能力,會讓 E1/2提前的能力更勝甲基氧基與 甲基,也顯示三苯胺上的氨基會讓化合物更容易氧化[17,19]。圖 1-9 為其他具胺 基的三苯胺化合物[17],將圖 1-9 中化合物的對化學性質列表於表 1-5,胺基的數 目不但會讓氧化電位更提前,會增加不可逆的氧化波數。根據文獻中的 CV 圖推 測,第一對 redox couple 是一個電子的移動,第二對 redox couple 也是一個電子
的轉動。其中,amino group 在氧化時容易失去質子呈現不穩定的狀態,在光譜 電化學的研究中,雖然第一個電子移動的可逆性質良好,但是第二個電子移動則 呈現較不穩定的狀態,長時間氧化後較不易回覆。具氨基(-NH2)的三苯胺衍生物 容易氧化,在較高的電位下狀態是不穩定,除了會出現不可逆的氧化波,推測有 可能出現雙聚反應或電聚合反應。 圖 1-9.含氨基取代的三苯胺衍生物。 表 1-5.含氨基取代的三苯胺衍生物的電化學性質。
18 1.3.3.導電性高分子材料 電子、光電產業中,高分子工業仍能以其大範圍的基本物性如:硬度、強度、 易於製造等性質,和其它材料交互應用,發揮其特點。近幾年內把高分子和光電、 電子產業結合,為科技的發展注入新動力。1977 年一項突破性的發現改變人們 的迷思,高分子的導電度可以近似金屬[20],而美國科學家 Alen Heeger、Alan MacDiarmid 和日本科學家 Hideki Shirakawa 也因此項發現,而獲得 2000 年諾貝 爾化學獎。高分子由於機械強度及易塑性等性質,使高分子做為材料的各項用品, 成為日常生活用品之一。若將高分子材料應用至高科技,如:飛機材料或電線方 面,須對高分子的結構加以修飾,而高分子的性質及特性,將會有所變化。科學 家希望能夠製造一種高分子,不僅能夠改變傳統高分子材料無法導電的性質,並 且期望導電度能達金屬水準,或完全取代之。導電性高分子這門領域已經至今累 積相當豐碩的研究成果。圖 1-10 是金屬,半導體與絕緣體的導電度分佈,由圖 中可明顯看出導體與半導體之間為導電性高分子的導電度,範圍非常廣。結構最 簡單的導電性高分子為 polyacetylene,由於具有許多的 π 電子,在空氣中極易 被氧氣氧化而失去導電性,再加上共振的結果,使得整個主鏈就好像一光滑的棒 子,無法以熔化或溶劑來加工處理,為改善此缺點,有人合成環狀的導電性高分 子,如聚苯胺、polyprrole 及 polythiophene 等,為了增加加工性,而在主鏈上便 以共價鍵接上長的側鏈,此類高分子的導電度不穩定,需以強氧化劑(或還原劑) 取代(或供給)主鏈電子,形成電洞(電荷)導電。
20
1.3.4.導電機制 共軛導電高分子結構由主鏈上之 π 電子軌域重疊而成一連續的分子軌域,其 分子軌域π-π*能隙隨著 π 電子共軛的長度減少而增加,此種電子結構類似前述之 半導體結構,因此 1980 年代初期就有學者開始借用物理學中的偏極子,雙偏極 子及孤立子 (soliton) 之觀念來解釋導電高分子的電子結構。 如圖 1-11 當電子從共軛高分子 polyacetylene 的上方被移走,會產生電子空 缺,稱之為電洞或自由基陽離子。由古典能階來看,空缺為部份非定域化,而此 部份非定域化將之擴展至幾個單體單位,造成高分子的結構改變,電子在高分子 的骨幹上任意移動而不影響能量,稱之為孤立子。soliton 依照其電荷分成三種形 式:圖 1-12 所示,S0 為電中性(圖 1-12.a), 是由 A 相轉至 B 相時,兩相邊界處 碳原子上含一個不成對電子,帶 1/2 自旋。S0若少一個π 電子時會形成 S+而帶正 電(圖 1-12.b),若是含兩個 π 電子時,則形成 S-帶負電且無自旋(圖 1-12.c)。 而自由基陽離子 (電洞) 為不安定性的鍵結軌域,所以其能階會比價鍵帶的 能階高,相當於一個電子從一個填滿的鍵結分子軌域被移離後提升其能階,此種 自由基陽離子造成之新的誘導能階在固態物理上稱為偏(正)極子 [22],如圖 1-13.(a)。 Bipolaron[22]是因為在較高能階含有一對電洞且在較低能階有一對造成晶 格扭曲變形,其形成之原因偏極子同一能階再失去第二能個電子,形成(正)雙偏 極子 (positive bipolaron) 能階如圖 1-13. (b)所示。偏極子和雙偏極子皆可由共軛 系統中單雙鍵重排而沿分子鏈移動,如果較多雙偏極子形成 (於高摻雜態時), 其軌域重疊而產生能隙較窄之雙偏極子能階,如圖 1-13.(c) [24]
22
圖 1-11. 隨分子大小遞增的 PA,(CH)n其 pMOs 之能階示意圖[24]。
(a)
(c) 圖 1-12. Trans-(CH)n的 soliton 型態[24]。 圖 1-13. 偏極子和雙偏極子能階示意圖[24]。 1.3.5.電化學聚合 導電高分子合成法中,較為常用的方法便是電化學聚合法,利用電化學聚合 法緻密的導電高分子膜,藉由循環伏安掃瞄聚合過程中,探討高分子氧化還原與 聚合特性。溶劑的選擇也是一項重要的要素,溶劑在反應發生時不會有分解現象。 電解質的選擇對於最佳膜的生成是需要考慮的,因電化學聚合反應的進行過程經 自由基陽離子中間體,為非常活性親電子基,特別傾向親核劑。因此,非質子溶
24 通常標準的電化學聚合裝置含工作電極、相對電極及參考電極的三極電槽。 電極選擇以避免可能在電聚合時,造成不必要的氧化腐蝕破壞。例如:鋁、鐵和 銀應用於高分子聚合是不適合的陽極材料,因為這些材料在反應進行前已被氧化 掉[23]。白金和導電玻璃 (ITO) 是被使用較廣泛的陽極材料,因為這兩種電極非 常穩定,於導電性高分子成長的電位範圍較不會影響到高分子的氧化/還原對, 這二種電極材料常應用於電化學聚合的研究。 電化學合成方法大致可分為三種: (I) 定電流法 (II) 循環伏安法:利用不 斷的循環伏安掃瞄,導電性高分子因為氧化還原進行聚合反應,沈積電極表面上, 得到高分子膜,並且在聚合過程中即時監測氧化還原的變化,進而知道高分子的 特性。(III) 光譜電化學法:電化學方法的優點是簡單且快速,只需要給待測的 物質一個電壓或電流,可得到一個回應。光譜學的研究分析,對於結構鑑定是非 常重要的,所以結合電化學與光譜的解析(如 UV-Vis 及 FTIR),對於研究實在是 一項非常好的工具。臨場光譜電化學法(in-situ spectrochronoamperometric)提供很 好的選擇去探討導電高分子的電聚合機制,光譜電化學法所使用的電化學槽是採 用透光性較好的玻璃或石英製成,工作電極則採用導電玻璃(ITO),參考電極為 Ag/AgCl 或 Ag/AgNO3以及相對電極白金絲。電化學分析的部分可使用定電位元 法或循環伏安法,利用定電位法記錄不同電位下,光譜的改變情形,為研究電致 變色必做的研究。而循環伏安法則是研究變色動力學工具,改變不同的掃瞄速率, 記錄光譜的改變情形,再予以設定參數回歸,得到動力學關係式。 1.3.6 二氧化鈦簡介及特性 鈦是一種化學元素,化學符號是 Ti,原子序為 22,為銀白色的過渡金屬, 其特徵為高強度、耐高低溫、穩定的物理及化學性質、具金屬光澤,也有良好的 抗腐蝕性能力等特性,在自然界中多以其氧化物存在,也就是二氧化鈦。許多固 體材料皆因其原子具有規則性的排列模式而有其結晶特性,二氧化鈦也不例外, 其晶相結構主要包含三種,分別為正方晶系的金紅石 (rutile) 、銳鈦礦 (anatase)
及斜方晶系的板鈦礦 (brookite) ,其中最常見而廣泛應用的是金紅石及銳鈦礦兩 種晶相結構。 金紅石及銳鈦礦兩種晶相結構都是為一變形的八面體結構所堆疊而成,八面 體結構則是一個鈦原子及其周圍環繞著六個氧原子所組成,八面體結構中兩個位 於尖錐的氧原子與其中心的鈦原子形成長鍵,另外四個氧原子則是跟鈦原子形成 短鍵並環繞其周圍。 銳鈦礦晶相結構中,八面體結構是由邊上氧原子共用的方式所堆疊形成的, 而稱為共邊結構。金紅石及板鈦礦兩種晶相結構中其八面體結構皆是以夾角 90 度,並沿著[110]方向的方式共用其長鍵及短鍵上的氧原子,而稱為共角結構。 金紅石晶相結構中其單位晶胞有兩個鈦原子及四個氧原子,其 a = b = 4.594 Å ,c = 9.15 Å ,理論密度為 4.27 g/cm3;銳鈦礦晶相結構中其單位晶胞有四個鈦 原子及八個氧原子,其 a = b = 3.783 Å ,c = 9.15 Å ,理論密度為 3.90 g/cm3;板 鈦礦晶相結構中其單位晶胞有八個鈦原子及十六個氧原子,其 a = 5.449 Å ,b = 9.184 Å ,c = 5.145 Å ,理論密度為 4.13 g/cm3。 金紅石晶相結構因其為單一結晶型態,相較於銳鈦礦及板鈦礦晶相結構來得 對稱,因而其性質較為穩定。文獻[28,29]可說明晶相結構的變型或扭曲,並間接 地影響其能階狀態,因此近年來有學者為了使其吸收範圍從紫外光區轉移至可見 光區,進而提升其照光活性,而摻雜金屬等材質以改變其晶相結構的研究。 銳鈦礦晶相結構其催化活性較優於金紅石晶相結構,是因為兩者能隙具有差 異性的關係。銳鈦礦晶相結構其能隙為 3.2 eV,金紅石晶相結構為 3.0 eV,經由 紫外光照射後會產生電子-電洞對,由於金紅石晶相結構其能隙較小,故電子容 易掉回價帶行再結合作用,而銳鈦礦晶相結構其能隙較大,電子較不易掉回價帶, 因而其催化效果較優於金紅石晶相結構。
26
1.4.研究動機
過 去 的 二 十 年 間 , 導 電 高 分 子 電 致 變 色 元 件 (conducting polymer electrochromic devices, CPECDs) ,由於其驚人電化學及光學性質,使其在後視 鏡、智慧櫥窗及顯示器上的應用上受到很大的關注,然而 CPECDs 存在著某些 缺點,使得它在現實上的應用產生極大的阻礙,包括元件的響應速度、長期穩定 度及壽命等問題,其中元件長期穩定度不佳之缺點為 CPECDs 尚未在市場上成 為主流最主要的原因。 至今,有機型電致變色材料為電致變色之大宗,此系列材料在離子摻雜與去 摻雜能力較為迅速,可提供材料快速的著、褪色變化,並且可以藉由合成方式改 變接枝官能基團而賦予多樣化的顏色變異,並測試能否提高光學對比和長期的穩 定度壽命問題。 在本研究中合成三種三苯胺衍生物,如圖 1-14 三苯胺衍生物 HTPA 的分子 結構式,含己醚鍵推電子官能基團。TPAR1 的分子結構式,接上 Rhodanine-3-acetic acid 拉電子官能基團。3CN-TPA 的分子結構式,接上 TCNE 強拉電子官能基團。 三苯胺衍生物接上不同的官能基團,在多孔質 TiO2電極上做電化學聚合反應,
探討 TiO2和三苯胺衍生物分子間的化學作用,和電聚合反應的關係。
三苯胺衍生物電化學聚合後,應用於電致變色元件。並且嘗試加入噻吩單體 到三苯胺衍生物單體溶液,以電化學共聚合成薄膜,並組成元件後希望噻吩能提 升三苯胺衍生物的電致變色元件的光學對比和穩定度。
28
第二章 三苯胺衍生物單體合成步驟與儀器檢測
2.1.實驗藥品及儀器 藥品
1.Acetone (95 %),CH3COCH3:友和貿易股份有限公司出品。
2.Isopropyl alcohol (98 %),(CH3)2CHOH:友和貿易股份有限公司出品。
3.N,N-dimethylformamide (100%),C3H7NO:J. T. Baker 公司出品。
4. Phosphorus oxychloride (99%),POCl3:Riedel-deHaen 公司出品。
5.Triphenylamine (99%),C18H15N:ACROS 公司出品。
6.Rhodanine-3-acetic acid (98%),C5H5NO3S2:Alfa Aesar 公司出品。
7.Ammonium acetate (98%),CH3COONH4:J. T. Baker 公司出品。
8.Tetracyanoethylene(98%),C6N4:Alfa Aesar 公司出品。
9.Phenol(99.5%),C6H6O:Sigma-Aldrich 公司出品。
10.1-Bromohexane (99%),C6H13Br:ACROS 公司出品。
11.Diphenylamine (99%),C12H11N:ACROS 公司出品。
12.Sodium tert-butoxide (98%),C4H9NaO:company pty.公司出品。
13.O-xylene(99%),C8H10:Grand 公司出品。
14.Magnesium sulfate (99.8%),MgSO4:J. T. Baker 公司出品。
15.Palladium(II) acetate (98%),Pd(OAc)2
16.Tri-Tert-Butyl phosphine (98%),[(CH3)3C]3P 儀器 1.核磁共振儀:德國 Bruker 公司出品之 AV-400 型產品。 2.真空減壓濃縮機:日本 EYELA 公司出品之產品。 3.電子天平:瑞士 Precisa 公司出品之 XS365M 型產品。 4.水流式抽氣機:日本 Eyela 公司出品之 A-1000S 型產品。 5.超音波洗淨器:台灣 Delta 公司出品之 DC-400 型產品。 6.磁石加熱攪拌器:美國 CORNING 公司出品之 PC-420D 型產品。 7.傅立葉紅外線光譜儀:美國 Agilent 公司出品之 Cary 630 型產品。
2.2.三苯胺衍生物之合成步驟與性質鑑定 表 2-1.中間產物與最終產物之結構與 IUPAC 命名。 中間產物與最終產物之結構與 IUPAC 命名 簡稱 hexyloxybenzene HB 1-bromo-4-(hexyloxy)benzene HB-Br 4-(hexyloxy)-N,N-diphenylaniline HTPA TPA-CHO
30 {5E-5-[4-(diphenylamino)benzylidene]-4-oxo-2-thioxo-1,3-thiazolidin -3-yl}acetic acid TPAR1 2-[4-(diphenylamino)phenyl]ethene-1,1,2-tricyano 3CN-TPA
32
34
2.2.1.三苯胺衍生物之合成步驟 (1) 三苯胺 HTPA 合成
2.2.1.1.Hexyloxybenzene 合成
(1)取 phenol 0.24mole 和 5M,140ml 的 NaOH 溶液,放入三頸瓶內開始磁石 攪拌。 (2)調節加熱溫度至 80℃,把 1-Bromohexane 0.36mole 置於恆壓漏斗中,緩慢滴 加入三頸瓶內。 (3)滴完 1-Bromohexane 後,持續加熱 2 小時,收反應並降至室溫。 (4)加入大量去離子水分出水相和有機相,以乙醚萃取有機相,並加入無水 MgSO4 除水。 (5)以重力過濾法過濾雜質後可得淡黃色液狀產物。 (6) 測1H-NMR (圖 2-4.). 1H-NMR(400MHz, in DMSO-d6): δ7.24~7.29 (t,2H,ArH) ;δ6.88~6.92(t, t ,3H,ArH) ; δ3.92~3.95 (t,2H, -CH2-) ;δ1.66~1.73 (t,2H, -CH2-) ; δ1.41~1.44 (t,2H, -CH2-) ;δ1.25~1.39 (t,t,4H, -CH2-) ; δ0.78~0.89 (t,3H, -CH3) :yield 90%。 2.2.1.2. 1-bromo-4-(hexyloxy)benzene 合成 (1)取三頸瓶並架設好反應裝置,先以氮氣填充 1 小時反應裝置。
DMF 倒入三頸瓶內,並磁石攪拌。
(3)取 N-Bromosuccinimide(NBS) 0.308mole 溶於 140g 的 dry DMF 中,置於恆 壓漏斗內。 (4)將 NBS 溶液緩慢滴入三頸瓶內。 (5)調節加熱溫度至 60℃,架設回流裝置反應 24 小時。 (6)關氮氣並加入大量去離子水終止反應。 (7)以乙醚萃取有機相,並單獨取有機相,使用常壓蒸餾法去除乙醚。 (8)加入無水 MgSO4 除水,並以重力過濾法過濾雜質後可得酒紅色液狀物。 (9) 測1H-NMR (圖 2-5.). 1H-NMR(400MHz, in DMSO-d6): δ7.28~7.53 (d,2H,ArH) ;δ6.82~6.87 (d,2H,ArH) ; δ3.92~3.95 (t,2H, -CH2-) ;δ1.66~1.73 (t,2H, -CH2-) ; δ1.41~1.44 (t,2H, -CH2-) ;δ1.25~1.39 (t,t,4H, -CH2-) ; δ0.78~0.89 (t,3H, -CH3) :yield 75%。 2.2.1.3.HTPA 合成 (1)取三頸瓶並架設好反應裝置,先以氮氣填充 1 小時反應裝置。
(2)秤取 diphenylamine 0.06mole、Sodium tert-butoxide 0.48mole 和溶劑 O-xylene 260ml 倒入三頸瓶內,並磁石攪拌。最後在秤取 1-bromo-4-(hexyloxy)benzene 0.06mole 並倒入三頸瓶內。
36 (6)使用減壓濃縮法除 O-xylene。 (7)加入無水乙醇析出雜質,並以重力過濾法過濾雜質。 (8)再次使用減壓濃縮法除無水乙醇,得固狀產物。 (9)產物倒入甲醇中,成為飽和溶液,放入冰箱內且使用再結晶法純化,並取杯 壁上的產物。 (10)置於真空烘箱內趕溶劑後可得深咖啡色固狀產物。 (11) 測1H-NMR (圖 2-6.). 1H-NMR(400MHz, in DMSO-d6): δ7.12~7.34 (d,4H,ArH) ;δ6.88~7.12 (d,4H,ArH) ; δ6.72~6.88 (d,6H,ArH) ;δ3.77~4.07 (s,2H, -CH2-) ; δ1.56~1.86 (s,2H, -CH2-) ;δ1.20~1.57 (t,t,t,6H, -CH2-) ; δ0.86~1.05 (d,3H, -CH3) :yield 60%。 (2) 三苯胺 TPAR1 合成 2.2.1.4. Vilsmeier reagent 製備 (1)取三頸瓶並架設好反應裝置,先以氮氣填充 1 小時反應裝置。 (2)秤取 2mole 的 dry DMF 置入三頸瓶內,於冰浴中冷卻至 0℃,並磁石攪拌。 (3)秤取 0.1mole 的 phosphorus oxychloride 置於恆壓漏斗內。
(4)將 phosphorus oxychloride 溶液緩慢滴入三頸瓶內。
(5)滴定完後使混合物溫度回升至室溫,磁石攪拌 1 小時,即可得 Vilsmeier reagent。
2.2.1.5.4-Formyltriphenylamine 合成
(1)之前所得的 Vilsmeier reagent,接續反應下去。
(2)將 Vilsmeier reagent 置於冰浴中冷卻至 0℃,並磁石攪拌。 (3)將 0.1mole 的 triphenylamine 緩慢加入 Vilsmeier reagent 中。 (4)室溫下,反應 24 小時。 (5)關氮氣並且將三頸瓶置於冰浴中冷卻至 0℃,並秤取 3M,200ml 醋酸鈉溶液 置於恆壓漏斗內。 (6)將醋酸鈉溶液緩慢滴入三頸瓶內。 (7)滴定完畢後,以抽氣過濾法過濾,並加入大量去離子水除雜質後取濾餅。 (8)置於真空烘箱內趕水氣後可得淡綠色固體。 (9)測1H-NMR (圖 2-7.). 1H-NMR(400MHz,in DMSO-d6):
δ6.88(d,J=8.8Hz,2H,ArH) ; δ7.18~7.24(q,6H,ArH) ; δ7.42(t,J=8.0Hz,4H,ArH) ; δ7.71(d,J=8.4Hz.2H,ArH);δ9.76(s,1H,-CH=O) :yield 90%。
2.2.1.6. TPAR1 合成
(1)取三頸瓶並架設好反應裝置,先以氮氣填充 1 小時反應裝置。
38 (5)以抽氣過濾法過濾,持續加入大量去離子水除醋酸後取濾餅。 (6)把濾餅上的產物倒入甲醇中,成為飽和溶液,放入冰箱內且使用再結晶法純 化,並取杯壁上的產物。 (7)以抽氣過濾法過濾,持續加入大量去離子水除甲醇後取濾餅。 (8)置於真空烘箱內趕水氣後可得橘紅色固體。 (9)測1H-NMR (圖 2-8.). 1H-NMR(400MHz, in DMSO-d6):
δ4.70(s,2H,-CH2COOH) ; δ6.90(d,J=8.0Hz,2H,ArH) ; δ7.1 5~7.22(q,6H,ArH) ;
δ7.40(t,J=7.6Hz.4H,ArH) ; δ7.46(d,J=8.4Hz,2H,ArH) ; δ7.73(s,1H,-CH=) : yield 70%。
(3) 三苯胺 3CN-TPA 合成 2.2.1.7. 3CN-TPA 合成
(1)取三頸瓶並架設好反應裝置,先以氮氣填充 1 小時反應裝置。
(2)將 0.02mole triphenylamine 和溶劑 50ml 的 dry DMF 置於三頸瓶內,並磁石 攪拌後,秤取 0.023mole Tetracyanoethylene 倒入三頸瓶內。 (3)室溫下,反應 12 小時。 (4)關氮氣並將溶液倒入冰塊中而析出,以抽氣過濾法過濾,持續加入大量去離 子水去除雜質後取濾餅。 (6)使用正己烷:去離子水雙溶液(正己烷:去離子水=1:5)來純化,取正己烷液相層, 以減壓濃縮法趕溶劑,可得深紫色固狀產物。 (7)測1H-NMR (圖 2-.). 1H-NMR(400MHz, in CDCl3): 7.72-7.70 (d, 2H, He), 7.50 (s, 1H, Hf), 7.38-7.34 (t, 4H, Hc), 7.23-7.20 (t, 2H, Ha), 7.19-7.17 (d, 4H, Hb), 6.94-6.91 (d, 2H, Hd):yield 69%。
2.2.2.三苯胺衍生物性質鑑定
2.2.2.1. 液態核磁共振光譜儀(Solution state Nuclear Magnetic Resonance,400 MHz and 500 MHz)
1
H-NMR 光譜中分裂形式 (splitting pattern) 的標記代號定義如下:s,單峰 (singlet);d,雙峰 (doublet);t,三重峰 (triplet);q,四重峰 (quartet);quint, 五重峰;m,多重峰 (multiplet)。J 為偶合常數(coupling constant),單位是 Hz。
光譜數據之紀錄次序為化學位移 (chemical shift),分裂型式,偶合常數,氫 數。以四甲基矽為內部標準,化學位移以 δ 值表示。樣品製備如下:取適量的 樣品(約 5mg)置入 NMR tube 中,再以(Methyl sulfoxide)-d6 溶解(溶液約
為 0.5 ml),放入核磁共振光譜儀中,並完成參數設定後,進行掃瞄,再經由圖 譜處理後即可得所需之 1 H-NMR 及 13C-NMR 光譜圖。本文中的 1H-NMR 是 使用 400 MHz 液態核磁共振光譜儀,而13 C-NMR 則是使用 500 MHz。 (1)hexyloxybenzene(中間體 HB)的1H-NMR 光譜: 中間體 HB 的結構相對位子與質子峰積分位子由圖 2-4. 所示,在化學位移 δ 值從 6.88 至 7.29 ppm 處的峰群,皆是苯環結構上的氫質子峰的訊號,以苯環 之間位上氫的質子峰當作積分為 1,苯環上鄰位和對位的氫質子峰相對積分為 1.5, 另外在化學位移δ 值 0.78 至 3.95 ppm 之間(扣除了在化學位移 δ 值 2.5 ppm 附 近的為 DMSO-d6 溶劑之訊號與 3.31 ppm 附近則為 H2O 分子之訊號)即是接 在苯環上的己醚鏈上甲基與亞甲基的質子峰訊號,相對積分為 1、1、1、2、1.5 , 總積分數為 9 ,代表著有 18 個質子峰,中間體 HB 的總積分數為 18 且與氫數 相符合,可以證明中間體 HB 的確合成成功[27]。 (2) 1-bromo-4-(hexyloxy)benzene(中間體 HB-Br)的1H-NMR 光譜: 中間體 HB-Br 的結構相對位子與質子峰積分位子由圖 2-5.所示,在化學位移 δ 值從 6.82 至 7.53 ppm 處的峰群,皆是苯環結構上的氫質子峰的訊號,另外 在化學位移δ 值 0.78 至 3.95 ppm 之間(扣除了在化學位移 δ 值 2.5 ppm 附近
40 1.5 , 總積分數為 8.5,代表著有 17 個質子峰,比起原先 hexyloxybenzene 上的 18 個質子的峰群表現少了 1 個質子峰,中間體 HB-Br 的確在苯環對位上有一個 溴基取代,可以證明中間體 HB-Br 的確合成成功[27]。 (3) 4-(hexyloxy)-N,N-diphenylaniline(產物 HTPA)的 1H-NMR 光譜: 產物 HTPA 的結構相對位子與質子峰積分位子由圖 2-6.所示,在化學位移 δ 值從 6.72 至 7.34 ppm 處的峰群,皆是苯環結構上的氫質子峰的訊號,另外在化 學位移δ 值 0.86 至 4.07 ppm 之間(扣除了在化學位移 δ 值 2.5 ppm 附近的為 DMSO-d6 溶劑之訊號與 3.31 ppm 附近則為 H2O 分子之訊號)即是接在苯環上 的己醚鏈上甲基與亞甲基的質子峰訊號,以最接近己醚鍊上氧的亞甲基質子峰訊 號當作積分為 1,其餘相對積分為 3、2、2、1、3、1.5 ,產物 HTPA 的總積分 數為 13.5,代表著有 27 個質子峰且氫數相符合,可以證明產物 HTPA 的確合成 成功[27]。 (4) 4-Formyltriphenylamine 的 1H-NMR 光譜: 中間體 TPA-CHO 的結構相對位子與質子峰積分位子由圖 2-7.所示,在化學 位移δ 值從 6.86 至 7.72 ppm 處的峰群,皆是苯環結構上的氫質子峰的訊號, 在化學位移δ 值 9.76 ppm 即是接在苯環上的一支醛基的質子峰訊號當作基分數 為 1,其餘相對積分為 1、2、4、6、2,中間體 TPA-CHO 的總積分數為 15,代 表著有 30 個質子峰且氫數相符合,可以證明中間體 TPA-CHO 的確合成成功 [26]。 (5) 產物 TPAR1 的 1H-NMR 光譜: 產物 TPAR1 的結構相對位子與質子峰積分位子由圖 2-8.所示,在化學位移 δ 值從 6.89 至 7.51 ppm 處的峰群,皆是苯環結構上的氫質子峰的訊號,原本在 9.79 ppm 處之醛基質子峰與 Rhodanine-3-acetic acid 上的亞甲基(-CH2)質子峰,因 進行縮合反應而消失,並在 7.73ppm 多了-CH=的質子峰,而在 4.70 ppm 處的質 子峰則為 Rhodanine-3-acetic acid 上的-N-CH2-COOH。在 13.50ppm 處也有一支
Rhodanine-3-acetic acid 上的羧酸基(-N-CH2-COOH)質子峰,但羧酸上的氫在
(6) 產物 TPAR1 的 13C-NMR 光譜: 由13C-NMR 光譜圖 2-9 我們可以知道在化學位移 δ 值為 167.54 ppm 處為四 級 碳 之 羧酸質子峰,在化學位移 δ 值為 45.14 ppm 處為二級碳的質子峰 (-N-CH2-COOH),δ 值為 193.03 ppm 及 166.62 ppm 為四級碳的質子峰,共有十 四個碳原子吸收峰,可以證明產物 TPAR1 的確合成成功[26]。 (7) 產物 3CN-TPA 的 1 H-NMR 光譜: 產物 3CN-TPA 的結構相對位子與質子峰積分位子由圖 2-10 所示,在化學位 移δ 值從 7.26 至 7.454 ppm 處的峰群和從 6.943 至 6.966ppm 處的峰群,皆是 苯環結構上的氫質子峰的訊號,但從 6.994ppm 至 7.251ppm 的峰群皆為未反應完 成的 TPA 苯環結構上的氫質子峰的訊號,並且因為 3CN-TPA 上的 cyano 鍵結並 未含有氫元素,故無法判斷 3CN-TPA 是否合成成功。 (8) 產物 TPAR1 的 13C-NMR 光譜: 由 13C-NMR 光譜圖 2-11 我們可以知道在化學位移 δ 值為 127.076ppm 及 130.184 ppm 處為四級碳之質子峰,在 117.949ppm、126.932ppm、132.246ppm 及 147.862ppm 為二級碳的質子峰,在 120.193ppm 和 154.472ppm 為一級碳質子峰, 並且 81.798ppm 和 144.254ppm 為一級碳之乙烯質子峰,在 112.962ppm、 113.151ppm 和 114.144ppm 為一級碳之 cyano 質子峰,共有 23 個碳原子吸收峰, 可以證明產物 3CN-TPA 的確合成成功。並在其餘化學位移 δ 值為未反應完成之 TPA 的質子峰。
42
圖 2-4.1
H-NMR spectrum hexyloxybenzene in DMSO- d6 [27]。
圖 2-5.1
圖 2-6. 1
44
圖 2-8. 1
H-NMR spectrum TPAR1 in DMSO- d6 [26]。
圖 2-9. 13
圖 2-10. 1
46 2.2.2.2.傅立葉紅外線光譜儀(FTIR)鑑定官能基 從傅立葉紅外線光譜儀可分析三苯胺衍生物所含有的官能基,並且可更進一 步證實所合成的產物確實有三苯胺衍生物的特徵吸收峰。 (1)HTPA 三苯胺衍生物 FTIR 圖譜 由圖 2-12 (a)TPA (b)HTPA 的合成前後官能基變化,並可發現在圖 2-12(b)的 1250cm-1和 1020cm-1位置上為 HTPA 所鍵結的己醚鏈之 C-O 特性伸縮吸收峰, 因此可判斷所合成的產物確實有己醚鏈並且為 HTPA。 (2) TPAR1 和 TPA-CHO 三苯胺衍生物 FTIR 圖譜
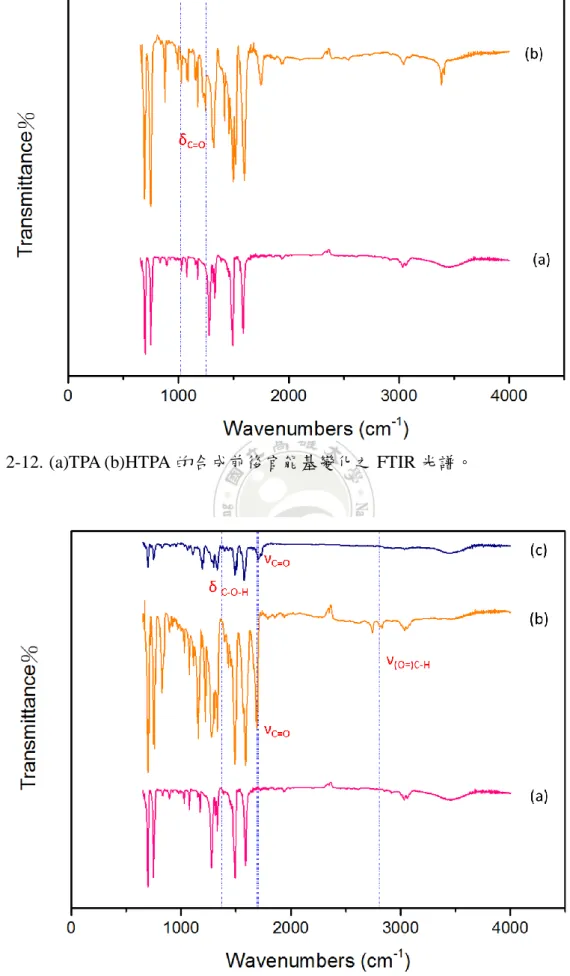
由圖 2-13 (a)TPA (b)TPA-CHO (c)TPAR1 的合成前後官能基變化。可發現圖 3-13(b)的 1690cm-1和 2800cm-1位置上為中間產物 TPA-CHO 所鍵結的醛基之 C=O
彎曲特性吸收峰與(O=)C-H 彎曲特性吸收峰,因此可判斷所合成的中間產物確實 有 醛 基 並 且 為 TPA-CHO ; 圖 3-13(c) 的 1710cm-1 和 1360cm-1 位 置 上 為
Rhodanine-3-acetic acid 上羧酸鏈的 C=O 和 C-O 特性吸收峰,並在 1690cm-1和 2800cm-1 位 置 上 的 C=O 與 (O=)C-H 特 性 吸 收 峰 消 失 。 因 此 可 證 實 Rhodanine-3-acetic acid 確實有和中間產物 TPA-CHO 的醛基進行縮合反應,並產 生為產物 TPAR1。
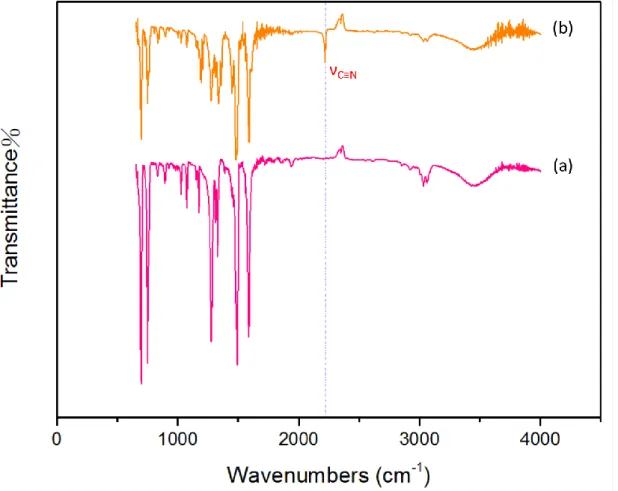
(3) 3CN-TPA 三苯胺衍生物 FTIR 圖譜
由圖 2-14 (a)TPA (b)3CN-TPA 的合成前後官能基變化。可發現 2221cm-1位置 上為 3CN-TPA 所鍵結的 cyano 鏈之 C≡N 彎曲特性吸收峰。因此可證實反應物 TCNE 確實和反應物 TPA 進行反應,並產生為產物 3CN-TPA。
48
第三章 三苯胺衍生物的原位電化學聚合行為的實驗結果討論
在一般的循環伏安法電聚合為同時進行氧化還原反應並且沉積薄膜於電極 表面,因為持續的進行沉積而導致薄膜厚度增加、電阻變大,需要更高的能量來 做氧化還原反應,造成氧化還原峰會往更高電位方向偏移,並且氧化還原峰電流 值會增加。 但在本研究中,先把奈米 TiO2多孔性電極加熱至 130℃恆溫 30 分鐘後,快 速浸泡於單體溶液中,吸附單體於奈米 TiO2孔隙中。浸泡單體溶液一天時,三 苯胺衍生物單體上鍵結的官能基與 TiO2表面的氫氧鍵有無化學鍵結而影響電化 學聚合行為。當我們取出三苯胺衍生物吸附於奈米 TiO2多孔性電極後利用循環 伏安法進行電化學聚合。當雙聚反應發生時,因只有奈米 TiO2孔隙間的吸附單 體進行電化學聚合反應,故其膜厚不會增加,電阻不會變大,因此氧化還原峰不 會改變電位值[26],但可由氧化還原峰電流值是否會增加來判斷有無高分子聚合 反應發生。 3.1. 實驗部分 3.1.1. 實驗藥品及儀器 藥品 1.Acetonitrile (99 %):J. T. Baker 公司出品。 2.Acetone (95 %),CH3COCH3:友和貿易股份有限公司出品。3.Isopropyl alcohol (98 %),(CH3)2CHOH:友和貿易股份有限公司出品。
4.Lithium perchlorate,LiClO4:Alfa 公司出品。
5.Nitric acid (70 %),HNO3 :J. T. Baker 公司出品。
6.Polyethylene glycol (Mw = 20,000),H(OCH2CH2)nOH:係 Fluka 公司出品。
7.Poly (ethylene oxide) (Mw = 400,000),C2nH4n+2On+1:係 Sigma-Aldrich 公司出品。
8.Sodium hydroxide,NaOH: Sigma-Aldrich 公司出品。 9.Titanium (Ⅳ) oxide,TiO2, P25:Degussa 公司出品。
50 儀器 1.電化學儀:荷蘭 AutoLab 公司出品之 PGSTAT 320 型產品。 2.精密熱風循環烘箱:德國 Binder 公司出品之 FD-23 型產品。 3.酸鹼度計:韓國 Istek 公司出品之 pH-220L 型產品。 4.水流式抽氣機:日本 Eyela 公司出品之 A-1000S 型產品。 5.離心機:台灣 Fudata 公司出品之 FG16-DT 型產品。 6.電子天平:瑞士 Precisa 公司出品之 XS365M 型產品。 7.高溫分解爐:台灣 Fudata 公司出品之 DF-40 型產品。 8.旋轉塗佈機:台灣 Swienco 公司出品之 PM-490 型產品。 9.超音波洗淨器:台灣 Delta 公司出品之 DC-400 型產品。 10.磁石加熱攪拌器:美國 CORNING 公司出品之 PC-420D 型產品。 11.傅立葉紅外線光譜儀:美國 Agilent 公司出品之 Cary 630 型產品。 3.1.2.實驗流程 圖 3-1. 三苯胺衍生物的循環伏安法聚合程序示意圖。
圖 3-2. 三苯胺衍生物的循環伏安法共聚合程序示意圖。 3.1.3.水熱製備銳鈦礦多孔奈米二氧化鈦粉體 Step 1.以 3 克 P25 加入 100 毫升的 NaOH 於鐵氟龍罐,以磁石攪拌器攪拌 10 分 鐘,使其均勻。 Step 2.攪拌均勻後,將其放入壓力釜中,置於熱風循環烘箱反應,反應溫度為 130 ℃,反應時間為 20 小時。 Step 3.壓力釜冷卻至室溫後,取出鐵氟龍杯,取出上層液,保留下層物 (中間產 物 X)。 Step 4.將中間產物 X 以大量蒸餾水清洗,再倒入過濾網,以水流抽氣機輔助過 濾,於過濾網上得到中間產物 Y。 Step 5.以 0.1 N HNO3調節中間產物 Y 酸鹼值到 10,並置於鐵氟龍杯,以磁石攪 拌器攪拌 10 分鐘,使其均勻。 Step 6.攪拌均勻後,將其放入高壓釜中,置於烘箱反應,反應溫度為 240 ℃,反 應時間為 12 小時,自然冷卻至室溫。 Step 7.壓力釜冷卻至室溫後,取出鐵氟龍杯,將上層液取出,保留下層物。 Step 8.將下層物放置於 50 mL 的離心管,以離心機進行分離反應,轉速為 8000 rpm,時間為 40 分鐘。
52 3.1.4.多孔奈米二氧化鈦/聚乙二醇複合漿料製備 將沉澱於離心管的奈米二氧化鈦粉體,加入二氧化鈦 1.5 倍重的蒸餾水以及 0.15 倍重的聚乙二醇 (Mw = 20,000),完全混和後即得二氧化鈦/聚乙二醇複合漿 料。 3.1.5 .ITO 基材前處理 ITO 基材 (電阻值為 8 Ωcm-2) 裁切成 3 × 25 cm 大小,以清潔劑、去離子水、 異丙醇及丙酮,超音波震盪各 20 分鐘,以空氣槍除丙酮。 3.1.6.奈米二氧化鈦多孔性電極
本實驗中 TiO2 薄膜製備[31,33]是採用刮刀法(Doctor-blade Method),在乾
淨的 ITO 導電玻璃的導電面上用 3M 膠帶圍出 1 x 1 cm2的面積,再用刮刀將 自製的奈米二氧化鈦漿料塗於此面積上,待自然乾燥 10 min 後,將 3M 膠帶 撕除並置於高溫爐中,於 45 分鐘內加熱至 450 ℃,再持溫 30 分鐘,最後再降 至室溫,即可製成奈米二氧化鈦多孔性電極。 3.1.7.電化學原位共聚合法 本研究探討三苯胺衍生物鍵結不同的官能基對電化學聚合的影響,藉由三苯 胺衍生物吸附於 TiO2多孔性電極,於 0.1 M HClO4的酸性水溶液和 0.1 M LiClO4
的 acetonitrile 溶液中分別進行電化學循環伏安法聚合,並檢視三苯胺衍生物鍵結 不同的官能基對電化學聚合的影響。並且當我們使用循環伏安法來作原位共聚合 反應時,可以知道外加電位達到單體的反應電位時,氧化或還原反應隨即發生, 此時電流將隨著電位的變化而改變。而電流的大小是由物質到電極表面的擴散速 率所控制,所以當擴散速率大於外加電位的變化速率,表示電極表面發生氧化還 原反應,此時電流會增加;並且擴散速率小於外加電位的變化速率,表示電極表 面未發生氧化還原反應或電極表面的反應已趨近完全時,此時電流會衰退[26]。 使用循環伏安法的控制參數,電位掃描範圍為 -200 至 1000 mV,以 50 mVs-1掃描速率做連續掃描電聚合,同時紀錄循環伏安曲線圖。
![圖 1-11. 隨分子大小遞增的 PA,(CH) n 其 pMOs 之能階示意圖[24]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/7419981.105230/37.892.232.665.105.393/圖111隨分子大小遞增的PACHn其pMOs之能階示意圖24.webp)