鄰位硝基甲苯的解離與重排反應與9-芴酮的溶劑效應之量子化學計算研究
95
0
0
全文
(2) 鄰位硝基甲苯的解離與重排反應 及 9-芴酮的溶劑效應之量子化學計算研究. 學生:陳思成. 指導教授:刁維光博士. 國立交通大學應用化學所碩士班. 摘. 要. 本論文的第一部份利用密度泛函方法(Density Functional Theory,DFT)以及高 精確度方法 G2M(cc,MP2)來研究鄰位硝基甲苯(o-nitrotoluene)在氣態下的解離及重 排反應。經由提出可能的反應路徑並找到其反應物、產物及過渡態結構,我們得 以對其整體反應有一個初步的認識。接下來我們使用 DFT 以及 G2M(cc,MP2)兩種 方法對所得的結構進行單點能量計算,以繪製其反應能量曲面,同時對兩種方法 所得到的能量作比較及討論。根據計算所得的能量,我們確認了文獻中所觀察到 的反應在位能面上確實是較為可能發生的反應。 本論文的第二部份是關於 9-芴酮在不同溶劑中展現的溶劑效應。文獻指出, 其螢光光譜峰在非質子性溶劑(aprotic solvents)中隨著溶劑的極性增加而產生紅位 移,同時在質子性溶劑(protic solvents)中則會產生異常大的紅位移。本研究中使用 DFT 方法、單電子激發組態交互作用(Configuration Interaction Single excitation, CIS) 及完整活化空間自洽場(Complete Active Space Self Consistent Field, ACSSCF)方法 取得 9-芴酮在基態及激發態的結構,利用含時密度泛函方法(Time Dependent DFT, TDDFT)求得其吸收及螢光波長,並搭配可極化連續體模型(Polarizable Continuum Model, PCM)來處理 9-芴酮在非質子性溶劑中的溶劑效應。對於質子性溶劑中的溶 劑效應,本研究使用超分子法(supermolecule method, 或稱微溶劑效應)來處理之, 並成功解釋了分子間氫鍵對螢光波長所產生的效應。.
(3) Quantum Chemical Studies of Dissociation and Rearrangement of o-Nitrotoluene and of Solvent Effects of 9-Fluorenone. Student:Szu-Chen Chem. Advisor:Dr. Eric Wei-Guang Diau. Institute of Applied Chemistry National Chiao Tung university. ABSTRACT. The first part of the thesis utilizes Density Functional Theory (DFT) and high-accuracy scheme G2M(cc,MP2) to study the dissociation and rearrangement reactions of o-nitrotoluene. We enumerate possible reaction paths, and determine the geometries of reactants, products and transition states. By calculating the single point energies with DFT and G2M(cc,MP2) methods along specific reaction paths, we plot the potential energy surface (PES). Based on these calculations, we identify the reaction pathways connecting to the three experimental observed products. The second part of the thesis studies the solvent effect of 9-fluorenone (9F) in (protic and aprotic) solvents with different polarity. We use DFT, configuration interaction single excitation and complete active space self-consistent field methods to calculate the ground- and excited-state geometries of 9F. We use polarizable continuum model to treat the solvent effect in aprotic solvents. The calculations reproduce the gradual red-shift in fluorescence spectra with increasing polarity of aprotic solvents, observed experimentally. In protic solvents, our results, based on the supermolecule (microsolvation) method, show that intermolecular hydrogen bonding contribute to additional red-shift in spectrum, which is consistent with experimental observations..
(4) 謝誌 感謝指導老師刁維光教授在這段過程中對我耐心地指導及幫助,以及林明璋 教授提供了許多研究上的經驗與洞見。感謝兩位口試委員:朱超原教授及胡景瀚 教授對論文提供的見解及指教。感謝曾怡仁博士在論文寫作的階段給我的許多意 見與經驗分享。 感謝在這三年中實驗室的伙伴:盧盈志、張智煒、王載德和駱立揚學長在這 三年中給我許多生活上和學業上的幫助,江佳珍同學在過程中為彼此互相打氣, 程士勳、黃建源學弟(謝謝你的腳踏車!)與陳鈺菁、鍾思敏、李睿勻學妹及黃雯妃 學姐平時的鼓勵與幫忙。 在這過程中,如果沒有家人們的信任與支持,我是做不到這些事情的的,非 常感謝父母的栽培與鼓勵。 最後不那麼正式的,感謝所有我在諾瑞斯與中土世界認識的朋友們:你們給 了我許多美好的回憶。很高興能認識各位。.
(5) 內容目錄 第一章 序論. 1. 第二章 理論方法. 9. 2-1. 量子化學的基礎. 9. 2-2. Hartree-Fock 方法. 11. 2-3. 基底函數. 13. 2-4. 密度泛函理論. 15. 2-5. 結構最佳化與振動頻率計算. 16. 2-6. 激發態的計算. 18. 2-7. 高準確度方法. 19. 2-8. 溶劑的處理與自洽反應場. 20. 2-9. 本研究中使用的計算軟體. 23. 第三章 鄰位硝基甲苯之分解與重排反應的量子化學計算研究. 24. 3-1. 預期的反應路徑. 26. 3-2. 反應高階理論計算位能面. 41. 第四章 9-芴酮在質子性溶劑與非質子性溶劑中溶劑效應的量子化學計算研究. 48. 4-1. 氣態計算-(9-芴酮的基本性質). 49. 4-2. 穩態吸收光譜計算. 50. 4-3. 基態位能曲面掃瞄. 52. 4-4. 穩態螢光光譜計算. 54. 4-5. 9F 基態在質子性溶劑中的理論計算. 56. 4-6. 9F 激發態在質子性溶劑中的理論計算. 61. 4-7. 總結. 62.
(6) 圖目錄 圖 1-1 圖 1-2. 硝基甲苯的三種異構物-鄰位(a),間位(b),對位(c)與三硝基甲苯 (d)的結構 Wettermark 提出的反應-右邊結構即為有顏色的中間產物. 圖 1-3. Dacons 等人觀察到的 TNT 分解產物之一 4,6-dinitroanthranil. 2. 圖 1-4. anthranil 的結構. 2. 圖 1-5. Fields 等人提出的反應路徑. 2. 圖 1-6. He 等人提出可能的 anthranil 產生機制. 3. 圖 1-7. 9-芴酮的分子結構與其上的取代位置編號. 5. 圖 1-8. 9-芴酮在不同溶劑中的吸收與螢光光譜. 7. 圖 1-9. 9-芴酮在不同溶劑中的時間解析螢光光譜. 7. 圖 2-1. 分子位能面的示意圖. 16. 圖 2-2. 兩種溶劑空腔的示意圖. 21. 圖 3-1. 預期的反應途徑、結構及其代號. 25. 圖 3-2. 反應位能面,以編號表示各結構. 42. 圖 3-3. 位能面上重要的反應,以編號表示各結構. 43. 圖 4-1. 9F 在基態下最佳化的結構. 49. 圖 4-2. 9F 在氣態下的 HOMO 與 LUMO. 49. 圖 4-3. 基態的 C=O 基團平面外彎曲掃瞄位能圖. 53. 圖 4-4. 9F 加一個甲醇分子最佳化結構. 57. 圖 4-5. 9F 加兩個甲醇分子最佳化結構. 57. 圖 4-6. 9F 加一個乙醇分子最佳化結構. 57. 圖 4-7. 9F 加兩個乙醇分子最佳化結構. 57. 圖 4-8. 9F 加一個異丙醇分子最佳化結構. 58. 圖 4-9. 9F 加兩個異丙醇分子最佳化結構. 58. 圖 4-10. 9F 加一個第三丁醇分子最佳化結構. 58. 圖 4-11. 9F 加一個第三丁醇分子最佳化結構. 58. 1 2.
(7) 表目錄 表 3-1. G2M(cc,MP2)之計算方法. 24. 表 3-2. 不同方法得到的鄰位硝基甲苯中硝基與苯環平面之夾角. 26. 表 3-3. 重要結構的理論計算能量與實驗值之比較. 44. 表 3-4. 重要結構在 G2 方法中各種計算的能量值. 48. 表 4-1. PCM 方法對 9F 結構最佳化的效應. 50. 表 4-2. 不同的密度泛函配合 6-311+G(d,p)所得吸收峰值. 51. 表 4-3. 基態 9F TDDFT 計算的基底函數效應. 51. 表 4-4. 質子性溶劑與非質子性溶劑用 PCM 模型處理的測試. 52. 表 4-5. CAS(8,8)/4-31G 最佳化結構的密度泛函測試. 54. 表 4-6. CIS/6-31G(d)最佳化結構的 TDDFT 密度泛函測試. 55. 表 4-7. 不同的激發態結最佳化方法所得到的重要結構參數. 56. 表 4-8. 不同的結構最佳化方式所得到的氫鍵鍵長 不同的密度泛函/結構最佳化/基底函數對甲醇/9F 系統的 TDDFT 結果之影響 不同數量的各種醇類超分子系統最佳化得到的結構參數 甲醇分子的數量、最佳化方法以及密度泛函對 TDDFT 計算所得 螢光波長的影響. 59. 表 4-9 表 4-10 表 4-11. 60 60 61.
(8) 第一章 序論 帶有硝基的小型分子因為擁有較低的解離反應活化能,因此對於燃燒、爆炸 或是高能分子的反應來說它們是相當有價值的研究題材。在過去有相當多關於這 類分子的研究,證實了帶硝基的小型分子容易進行單分子的解離反應。在這些分 子中,硝基甲苯由於其在結構上與炸藥-三硝基甲苯(2,4,6-trinitrobenzene,TNT,如 圖 1-1c 所示)的相似性,因此成為格外被重視的研究題材。在三種硝基甲苯的異構 物中(圖 1-1a~c 所示),鄰位的異構物由. CH3. CH3 NO2. 於苯環上的甲基與硝基最為靠近,因此. NO2. 有可能會發生取代基間的原子重排反 應,所以我們可以合理推測鄰位的硝基. (a). (b). CH3. CH3 O2N. 甲苯將擁有較另兩種異構物更多樣化 的化學反應。 鄰位硝基甲苯的相關化學反應, 主要可以分成光分解和熱分解兩種。其. NO2. NO2. NO2. (c). (d). 圖 1-1 硝基甲苯的三種異構物-鄰位(a), 間位(b),對位(c)與三硝基甲苯(d)的結構. 二者在機制上最大的差異在於,光分解是利用光子將分子提升到激發態,此額外 的能量可能會誘導產生激發態的光化學反應。而熱分解是加熱分子,以外加的熱 能誘導其反應,因此這個方法不會產生激發態的反應。 早期文獻中關於此類化合物的重要研究,當屬 Wettermark1於 1962 年發表的 工作。該實驗將硝基甲苯溶於於酸性(1N HCl)或鹼性(0.1N NaOH)的水溶液中,利 用閃光燈照射樣品以誘導反應的發生。在該實驗中觀察到一種有顏色的中間產. 1.. Wettermark, G. J. Phys. Chem. 1962, 66, 2560. 1.
(9) 物,此種因為照光而產生顏色變化的現象. CH3 NO2. 稱為光致變色(photochromism)2。該研究群 並提出了可能的反應過程,如圖 1-2 所示, 並且透過測量該中間產物濃度隨時間的變. CH2 OH N O. 圖 1-2 Wettermark 提出的反應-右邊 結構即為有顏色的中間產物 HC O. O2N. 化,認為該反應是可逆反應。. N. 在 1968 年,Fields3等人發現在相同的 條件下,鄰位硝基甲苯的分解反應過程與 另外兩種異構物不同,作者認為是由於甲. NO2 圖 1-3 Dacons 等人觀察到的 TNT 分解產物之一 4,6-dinitroanthranil HC O. N. 基與硝基的近距離產生的反應,如圖 1-5 所示。. 圖 1-4 anthranil 的結構. NO2. NO2. +. -H. CH3. CH2 H N. N O H C O. C O. O N H O C H NH2. O. C O O. 圖 1-5 Fields 等人提出的反應路徑 另外,Dacons4等人於 1970 年直接加熱三硝基甲苯樣品至 200℃,且利用色層 分離法成功的分離並鑑別其中的七種反應產物。圖 1-3 列出其中的一種,此 anthranil 衍生物在之後的研究也被發現是重要的產物。. 2. 3. 4.. Wettermark, G. Nature 1962, 194, 677 Fields, E. K.; Meyerson, S. Tetrahedron Lett. 1968, 10, 1201. Dacons, J. C.; Adolph, H. G.; Kamlet, M. J. J. Chem. Phys. 1970, 74, 3035. 2.
(10) 接下來 1985 年,Gonzalez5等人利用二氧化碳雷射對硝基苯、鄰位二硝基苯、 2,4-二硝基甲苯及硝基甲苯的三種異構物進行熱裂解。在實驗中,鄰位硝基甲苯的 分解速率被發現是這些化合物中最快的,作者同時也引用了 Fields 等人的研究做 為佐證。 1986 年,Tsang6等人利用震波管(shock tube)裝置測量到硝基苯、鄰位硝基甲 苯與對位硝基甲苯的 NO2 解離速率,並且再一次的證實鄰位硝基甲苯的反應速率 是三者中最快的。但是作者也經由分析反應速率的資料提出:在硝基苯與對位硝 基甲苯的例子中,熱分解產生 NO2 雖然是主要的反應,但是並不是佔絕對多數; 硝基的異構化(NO2 轉變成 ONO)也是一個相當重要的過程,其產生的是 NO 解離 的產物。但在鄰位硝基甲苯中,這兩者都不是主要的反應路徑,這兩個反應路徑 的總和只對反應產生了三分之一的貢獻。這顯示鄰位硝基甲苯必定還有在其他兩 者中不會發生的反應,並且扮演相當重要的角色。 1988 年,He7等人一樣利用 CH3. CH2 OH N O. NO2. 震波管裝置進行鄰位硝基甲苯的. H2C. O N OH. 熱分解研究,但這次他們在反應 速率決定步驟. 產物中找到了 anthranil 產物,如. HC O N. 圖 1-4 所示,並對其生成及消失 的反應機制作了初步的研究。(圖 1-6)這個發現解釋了鄰位硝基甲. HC O. +H+OH. N. +H2O. 圖 1-6 He 等人提出可能的 anthranil 產生機制. 苯的反應特別快的原因-其他位置取代的異構物不會有這一條產生 anthranil 的反應 路線,而這個額外的反應使得鄰位硝基甲苯的總體反應速率加快。 5. 6. 7.. Gonzalez, A. C.; Lamon, C. W.; McMillen, D. F.; Golden, D. M. J. Phys. Chem. 1985, 89, 4809. Tsang, W.; Robaugh, D.; Mallard, W. G. J. Phys. Chem. 1986, 90, 5968. He, Y. Z.; Cui, J. P.; Mallard, W. C.; Tsang, W. J. Am. Chem. Soc. 1988, 110, 3754. 3.
(11) 1994 年,Galloway8等人利用真空紫外光解的方法偵測到 NO 離子的訊號,並 且對其轉動能量分佈進行了詳盡的分析。另外該研究群也利用 MP2/6-31G*的方法 進行理論計算,成功的找到了 NO2 異構化反應的產物。 1995 年,Chen9等人對鄰位硝基甲苯的熱裂解反應進行了初步的理論計算。 作者使用 HF/6-31G*的方法進行理論計算,並成功找到了 He 等人提出的中間產物 之結構與能量。但這個研究中並沒有做過渡態的計算。 Il’ichev10等人則於 2000 年對鄰位硝基甲苯的異構化反應進行了相當完整的理 論計算研究,該研究群利用 B3LYP/6-31G(d,p)的方法找到各反應可能中間物和過 渡態的結構,並且用 B3LYP/6-311+G(2d,p)方法與 QCISD(T)/6-31G(d,p)方法計算各 結構的能量,以得到其反應的勢能面。 綜觀以上的文獻,我們目前可以瞭解到硝基甲苯的研究確實有一定的重要 性,實驗的部分證實鄰位硝基甲苯產生其他位置取代的異構物所沒有的反應,而 且此反應佔有重要的角色。但在理論計算的部分,這種大小的分子直到最近幾年 才有辦法進行高階的理論計算研究,本研究的目的就是希望利用高階的計算對於 鄰位甲苯的異構化反應進行研究,並試著補足前人的理論計算或有尚待補充之 處。藉由高階的計算,我們將可以得到高準確度的反應熱及反應活化能,日後將 可以與實驗得到的資料互相比較。 本論文的另外一部份,是關於 9-芴酮(9-fluorenone, 簡稱 9F。結構見圖 1-7) 在溶劑中的吸收與放光行為。此化合物屬於芳香酮類,由於這類化合物的 n-π*和 π-π*激發態的能量相當靠近11 ,因此其最低激發態的性質容易受到外來環境如溶. 8. 9. 10. 11.. Galloway, D. B.; Glenewinkel-Meyer, T.; Bartz, J. A.; Huey, L. G.; Crim, F. F. J. Chem. Phys. 1994, 100 1946. Chen, P. C.; Wu, C. W. J. Mol. Struct. (Theochem) 1995, 357, 87. Il'ichev, Y. V.; Wirz, J. J. Phys. Chem. A 2000, 104, 7856. Murphy, R. S.; Moorlag, C. P.; Green, W. H.; Bohne, C. J Photoch Photobio A 1997, 110, 4.
(12) 劑、取代基的推拉電子特性…等影響。而 O. 根據 Kasha’s rule12,分子的光物理與光化 學性質通常由其最低的激發態決定,因此 9-芴酮和其衍生物成為很好的光化學研究. 8. 2. 6. 3 5. 對象。在溶劑效應方面,早期的研究包括. 1. 7. 9-fluorenone. 4. 圖 1-7 9-芴酮的分子結構與其上的 取代位置編號. Kobayashi 等人13在 1976 年所做的研究。該 研究群利用瞬態吸收光譜(transient absorption spectroscopy)的實驗方法得到 9-芴酮 在不同的極性、非極性溶劑中的能階位置,並認為 9-芴酮在非極性溶液中的 S1 是 屬於 π-π*激發態,而在極性溶液中是 n-π*激發態。 Fuji 等人14則是在 1996 年進行了 9-芴酮在不同溶劑中螢光被淬熄(quench)速率 的研究。該研究群利用混合高低極性的兩種溶劑,並改變其比例來製造出一系列 不同極性的混合溶劑。該研究的特點在於,使用乙醇/環己烷與乙醇/乙腈(acetonitrile) 的混合溶劑進行實驗,並從不同醇類比例溶液中吸收光譜上等位點(isosbestic point) 的存在,推論 9-芴酮在醇類溶液中是以 1:1 的比例與醇類分子組合,而因此改變了 其 S1 與 T3 能階的相對能量。該研究的結論認為:在醇類溶劑和非極性溶劑中,9芴酮的 S1 激發態是為 n-π*類型,而在極性溶劑中為 π-π*類型。該研究群並同時做 了簡單的 CNDO/S 計算來驗證此論點,但這與 Kobayashi 等人得到的結論不同。 1997 年,Biczok 等人15則是觀察到 9-芴酮在醇類和酚類溶劑中,其螢光生命 期會較為縮短(即謂淬熄, quenching)。這表示質子性溶劑能夠透過改變 9-芴酮的能 階相對能量或是與 9-芴酮產生光物理/化學反應來加速其能量緩解的過程。. 12. 13. 14. 15.. 123-129. Kasha, M. Disc. Faraday Soc. 1950, 9, 14. Kobahashi, T.; Kagakura, S. Chem. Phys. Lett. 1976, 43, 429. Fujii, T.; Sano, M.; Mishima, S.; Hiratsuka, H. B Chem Soc Jpn 1996, 69, 1833-1839. Biczok, L.; Berces, T.; Linschitz, H. J. Am. Chem. Soc. 1997, 119, 11071-11077. 5.
(13) 而在最近的 2003 年,Jozefowicz 等人16則是分析了 9-芴酮在一系列混合溶劑 中的吸收與螢光光譜變化。該研究群使用的方式是利用環己烷/ 四氫化呋喃 (tetrahydrofuran)與環己烷/已醇兩種混合溶液,調整其比例並觀察 9-芴酮在不同比 例的混合溶液中,吸收與螢光光譜的變化。該研究群使用的分析模型是 Bakshiev 等人17、Mazurenko18等人提出,以及 Suppan19在 1990 年的文獻回顧中描述的特定 溶劑交互作用(preferential solvation)模型。此模型的概要為:當一個溶質溶在混合 溶劑系統中時,可能會因為彼此的交互作用不同,而使得溶質分子傾向與混合溶 劑中的某一種溶劑成分作用,結果提高該溶劑成分在溶質分子周圍的有效濃度。 Jozefowicz 等人. 16. 的實驗分析結果顯示,在環己烷/四氫化呋喃之混合溶劑系. 統中,9-芴酮並沒有明顯的特定溶劑交互作用之行為;而在環己烷/已醇混合溶劑 中則有明顯的特定溶劑交互作用。由於四氫化呋喃為非質子性溶劑(aprotic solvent) 而乙醇為質子性溶劑(protic solvent),因此該研究的結論是 9-芴酮可以與質子性溶 劑形成氫鍵。. 16. 17. 18. 19.. Jozefowicz, M.; Heldt, J. R. Chem Phys 2003, 294, 105-116. Bakshiev, N. G.; Wolkow, W. P.; Altajskaja, A. W. Opt. Spektrosk. 1970, 28, 51. Mazurenko, J. T.; . Opt. Spektrosk. 1972, 33 1060. Suppan, P. J. Photochem. Photobiol. 1990, 50 293. 6.
(14) Wavenumber /cm 25000. -1. 20000. 15000. O. 1.0 Hexane Pyridine ACN DMSO tBuOH IPA EtOH MeOH. Normalized Intensity. 9-fluorenone 0.8. 0.6. 0.4. A. F. 0.2. 0.0 350. 400. 450. 500. 550. 600. 650. 700. 750. Wavelength /nm. 圖 1-8 9-芴酮在不同溶劑中的吸收與螢光光譜。螢光光譜的激發波長為 375 nm。 1.0. ACN DMSO Pyridine tBuOH IPA EtOH MeOH Hexane. λex405nm,λem500nm. Normalized Intensity. 0.8. 0.6. ACN. 0.4. 0.2 Hexane. 0.0 0. 5. 10. 15. 20. Time /ns. 圖 1-9 9-芴酮在不同溶劑中的時間解析螢光光譜。激發波長為 405 nm,偵 測波長為 500 nm。 鍾思敏20在 2005 年的論文中則是進行了 9-芴酮在不同極性的非質子性溶劑與 數種質子性溶劑中的穩態吸收/螢光光譜的測量,同時也進行了不同溶劑中的時間 20.. 鍾思敏. 芴酮與咔唑衍生物在溶液中之光物理與光化學研究. 國立交通大學, 新竹市, 2005. 7.
(15) 解析螢光光譜測量(圖 1-9,1-10)。由穩態光譜可以清楚看出,各溶劑的吸收光譜 相差不大,但是螢光光譜的峰值會隨著不同的溶劑而有明顯的移動;對非質子性 溶劑而言,溶劑極性越大則放射光譜峰的波長越長。而實驗中 9-芴酮在所有質子 性溶劑中,其吸收峰都比極性最大的非質子性溶劑中的吸收峰位置還要紅位移。 而時間解析螢光光譜上可以看到 9-芴酮在醇類溶液中的螢光生命期遠比極性 的非質子性溶劑還要短,這也就是 Biczok 等人觀察到的螢光淬熄現象。 綜觀以上的文獻可以發現,雖然 9-芴酮此分子在實驗上有相當多與溶劑效應 有關的現象,但是卻相當缺乏利用理論計算方式對溶劑效應進行的研究。本研究 的目標在於用理論計算的方式描述 9-芴酮在質子性溶劑和非質子溶劑中的溶劑效 應,以及這些效應如何影響其光譜特性。. 8.
(16) 第二章 理論方法 本章將簡述此研究中用到的理論計算方法及其背景。 2-1. 量子化學的基礎 量子力學中的基礎乃是 Schrödinger 所提出,以其為名的 Schrödinger 方程式:. Hˆ ψ = εψ. (2.1). 但是一個因為系統通常不只有一個粒子,因此對系統中的第 i 個粒子而言,(2.1)可 以寫成: Hˆ ψ i = εψ i. (2.2). 其中的 Hˆ 稱為 Hamiltonian 運算子,在此式中的物理意義為系統總能的運算子,具 備下列的形式:. Hˆ = TˆN + Tˆe + VˆNN + Vˆee + VˆNe G G Z Z ∇ 2N ∇ e2 = ∑N − + ∑e − + ∑ N ∑ N '( N < N ') G N GN ' 2mN 2me rN − rN '. (2.3). Z 1 + ∑ e ∑ e '( e '<e ) G G − ∑ N ∑ e G N G re − re ' rN − re 其中 TˆN 代表核的動能,Tˆe 代表電子的動能,VˆNN 代表核間的排斥力位能,Vˆee 代表電 子間的排斥力位能, VˆNe 代表電子與核間的吸引力位能。. (2.1)中的 ψ 稱為波函數,描述系統中粒子的波動行為(所以量子力學也稱為波 動力學,因為用波動的概念來描述物質)。對 ψ 用 Hˆ 來操作則可得到一個值 ε,此值 即為系統的能量。在(2.1)中,ψ 稱為 Hˆ 的本徵函數(eigenfunction),ε 稱為 Hˆ 的本徵 值(eigenvalue)。 但是在求解 Schrödinger 方程式時,這個具有 3N 個自由度(N 為粒子數)的偏微 9.
(17) 分方程式將會極難求解。實際的作法是使用適當的近似來簡化此方程式,最重要的 近似是波恩-歐本海默近似(Born-Oppenheimer approximation),它的基本概念是:雖 然波函數 ψ 中包含了電子與原子核的描述,但是因為電子與原子核的質量相差約. 1836 倍,因此在系統中的電子運動時,原子核的運動實質上可以視為靜止。於是乎 (2.3)的 ψ 在波恩-歐本海默近似的假設之下,可以被分解成核的波函數ψ N 與電子的 波函數ψ e 之乘積: K K K Ψ tot = ψ N (rN )ψ e (rN , re ). (2.4). 因此 Hˆ 就可以被分解成核的部分 Hˆ N 與電子的部分 Hˆ N :. Hˆ tot = ⎡⎣TˆN + VˆNN ⎤⎦ + ⎡⎣Tˆe + Vˆee + VˆNe ⎤⎦ = Hˆ N + Hˆ e. (2.5). 所以(2.1)可以被重寫成下面的形式: K K K K Hˆ tot Ψ tot (rN , re ) = ( Hˆ N + Hˆ e )Ψ tot (rN , re ). (2.6). 由 於 波 函 數 經 過 (2.4) 式 去 耦 合 的 處 理 , 所 以 原 來 的 電 子 與 原 子 核 耦 合. Schrödinger 方程式,就可以被分離並個別求解: Hˆ Nψ N = ε Nψ N. (2.7). Hˆ eψ e = ε eψ e ,. (2.8). 其中 m K 2 m −1 2 m K K K Hˆ e = ∑ hˆ(ri ) + ∑ ∑ g (ri , rj ) , i =1. (2.9). i =1 j = i +1. 而 K hˆ(ri ) = −. h2 e2 2 ∇ − i 8π 2 me 4πε 0. N. Z. α ∑ R α =1. (2.10). αi. 且. 10.
(18) gˆ (rˆi , rˆj ) =. e2. 1 。 4πε 0 rij. (2.11). 我們將(2.4)帶入(2.1)中,就可以得到下式: ( Hˆ N + Hˆ e )ψ eψ N = ε totψ eψ N. (2.12). 將(2.12)兩邊同時除以ψ eψ N 可以得到:. 1. ψ eψ N. ( Hˆ N + Hˆ e )ψ eψ N = ε tot. (2.13). 這其中 Hˆ N 與ψ N 只與核的位置有關而與電子的位置無關; Hˆ e 與ψ e 則與兩者的位置 都有關。這表示:. G G ∇ e2ψ eψ N = ψ N ∇ e2ψ e G G ∇ 2Nψ eψ N = ψ e∇ 2N + extra terms. (2.14). 根據 Born-Oppenheimer 的近似,這組額外的項(extra terms)是可以被合理的忽 略的。 2-2. Hartree-Fock 方法 在 Born-Oppenheimer 近似的基礎上,我們面對的問題是該如何求解電子的. Schrödinger 方程式(2.8)。由於(2.3)式中電子排斥項( Vee )耦合系統中所有電子,因此 我們無法得到它的分析解。為了得到一個合理的近似解,根據 Hartree-Fock21方法的 假設,電子的總波函數可以寫成個別電子波函數的乘積。由於電子是一種費米子. (Fermion),並考慮電子自旋的情況下,以一個具有 m 個軌域且填滿的分子而言,我 們可以將波函數寫成以下的 Slater 行列式:. 21.. Fock, V. Z. Phys. 1930, 61, 126. 11.
(19) ψ e = (r1 , s1 , r2 , s2 ,..., r2 m , s2 m ) =. ψ A (r1 )α ( s1 ) ψ A (r2 )α ( s1 ). ψ A (r1 ) β ( s1 ) ψ A (r2 ) β ( s2 ). .... ψ M (r1 ) β ( s1 ) ψ M (r2 ) β ( s2 ). .... .... .... .... .... (2.15). ψ A (r2 m )α ( s2 m ) ψ A (r2 m ) β ( s2 m ) ... ψ M (r2 m ) β ( s2 m ) 其中 r 是電子的位置,S 是電子的自旋,ψ M 代表第 m 個軌域的波函數。這樣的寫 法會自動滿足電子波函數必須要是反對稱的要求。 將(2.15)的電子波函數帶入(2.8)式中,再經過 undetermined Lagrange multiplier 數學處理後,我們將可以得到 Hartree-Fock 方程式: Hˆ iFψ i = ε iψ i. (2.16). K K 其中的 Hˆ iF = hˆ(ri ) + VHF 稱為 Fock 運算子, hˆ(ri ) 的定義如 (2.10) 。 VHF 是所謂的. Hartree-Fock 位能:. (. 2m K K K VHF (r1 ) = ∑ Jˆi (r1 ) − Kˆ i (r1 ) i. ). (2.17). 其中 Jˆi 稱做庫侖運算子,. K K 2 1 K Jˆi (r1 ) = ∫ ψ i (r2 ) dr2 r12. (2.18). 而 Kˆ i 稱做交換運算子,. K K K 1 K K K Kˆ i (r1 )ψ j (r1 ) = ∫ψ i* (r2 ) ψ j (r2 )dr2ψ i (r1 ) r12. (2.19). 我們可以以變分法來求(2.16)中 Hamiltonian( H iF )的能量本徵值 ε i 。最常用的做 法是將分子軌域的波函數,以原子軌域波函數之線性組合來展開: b. ψ i = ∑ csi χ s. (2.20). i. 將此式帶入(2.16):. 12.
(20) ∑c. si. s. Fˆ χ s = ε i ∑ csi χ s. (2.21). s. 兩邊同乘以 χ r* 並積分後會得到 Hartree-Fock-Roothaan 方程式: b. ∑c s =1. si. ( Frs − ε i S rs ) = 0,. Frs ≡ χ r Fˆ χ s ,. r = 1, 2,..., b. (2.22). Srs ≡ χ r | χ s. 為了使(2.22)有非 0 的解,我們必須使 ( Frs − ε i S rs ) 的行列式=0:. det( Frs − ε i Srs ) = 0. (2.23). 通常式(2.23)的解法是先給一組軌域波函數的初始猜測,然後用遞迴的方式來 解出 ε i ,直到每次的能量誤差值小於一個給定的極限為止。這種解法就稱為自洽場. (self-consistent field, SCF)法。 雖然 Hartree-Fock 法已經大幅簡化量子化學計算的難度,但是對較大的系統而 言,此方法仍然需要大量的積分計算,其中最耗時間的就是雙電子的積分。為了更 有效的進行大分子體系的量子化學計算,有些學者就想到把這些雙電子積分以參數 化的形式簡化,這樣的方法就稱為半經驗法則計算。這些參數都是從實驗得到的值 來帶入或是適當的省略,以更有效的進行較大型系統的理論計算。這類型的方法包 括 AM122,PM323等。因為此方法的確包含了電子的描述,因此可以有限度的處理 與電子相關的性質,例如 ZINDO24方法在一般有機分子的光譜預測方面有不錯的準 確度和速度表現。 2-3. 基底函數 在 (2.21) 中 的 波 函 數 ψ , 在 量 子 化 學 計 算 中 則 是 由 所 謂 的 基 底 函 數 (basis. function)來處理,因為直接用真實的原子軌域波函數下去運算,會因為其數學形式. 22. 23. 24.. Dewar, M. J. S.; Zoebisch, E. G.; Healey, E. F.; Stewart, J. J. P. J. Am. Chem. Soc. 1985, 107, 3902. Stewart, J. J. P. J. Comp. Chem. 1989, 10, 209 Bacon, A. D.; Zerner, M. C. Theo. Chim. Acta. 1979, 53, 21 13.
(21) 不利於積分運算而耗費許多時間。因此許多研究者試著用一組統一的數學形式,只 是改變函數中不同的參數來代表不同的波函數。在這之中第一組被發展的是所謂的. Slater type 基底函數25,26,它使用簡單的類氫原子波函數來當作函數原型。但是應用 在 Hartree-Fock 的積分計算時,這種基底函數的原型是指數型態的,在計算上相當 的麻煩。. John A. Pople 等人提出以數個 Gaussian 函數的和來逼近 Slater type 的波函數 27. ,因為 Gaussian 函數的數學特性之一就是積分後會維持原來的型態,因此有利於. 計算上的方便性。這種基底函數被稱做是 STO/nG 型態的基底函數。 這種波函數接下來發展成更有彈性的 STO/k-nlmG28的型態:k 代表核心電子波 函數使用的 Gaussian 函數(被稱做原始基底函數,primitive basis function )數量,其 函數間的係數是固定的;n 代表價電子波函數的第一組 Gaussian 函數數量,這組中 的函數係數也是固定的;l 和 m 分別代表價電子波函數中的第二組和第三組 Gussian 函數數量。這裡 n,l 和 m 間的係數是獨立的。每組係數固定的原始基底函數被視 作是一個收縮基底函數(contracted basis functions)。 另外為了修飾基底函數並給予更大的彈性,J. B. Collins 等人還加入了極化函數. (polarization function)29,這是在每個原子上加入高一層的空原子軌域,以應付遠距 離電子被極化的方向性需求。通常的寫法如 6-31G*或 6-31G(d),這個*或(d)就表示 在碳原子等重原子上加入一層空的 d 原子軌域。6-31G**或 6-31G(d,p)的第二組極化 函數則是在氫原子上面也加入一組空的 p 原子軌域。 又為了要應付電子分佈相當遠的系統(如陰離子),還可以加入所謂的擴散函數. 25. 26. 27. 28. 29.. Clementi, E.; Raimondi, D. L. J. Chem. Phys. 1963, 38, 2686. Clementi, E. J. Chem. Phys. 1964, 40, 1944. Hehre, W. J.; Stewart, R. F.; Pople, J. A. J. Chem. Phys. 1969, 51, 2657. Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724. Collins, J. B.; Schleyer, P. v. R.; Binkley, J. S.; Pople, J. A. J. Chem. Phys. 1976, 64, 5142. 14.
(22) (diffuse function)30,這是在原子上再加入一組徑向分佈較大的軌域,在符號上是用+ 符號代表,如 6-31+G(d)。 2-4. 密度泛函理論 密度泛函理論(Density functional theory, DFT)和一般的 ab-initio 方法是從不同 的角度出發而發展出來的。一開始這個方法是由固態物理學家所研究發展,因為對 固態物理的研究者來說,電子並不是定域化的,而是散佈在整個固體中。這個體系 到後來由 Hohenberg 和 Kohn 兩人集大成並提出所謂的 Hohenberg-Kohn 定理31。此 定理認為系統的能量可以完全由系統中的電子分佈 P(r ) 來決定。 系統的總能運算子 Hˆ 可以拆開成電子動能 Tˆ ,單電子的位能 Vˆ 和電子間的排斥 能 Uˆ : Hˆ = Tˆ + Vˆ + Uˆ. 因此依照 Hohenberg-Kohn 定理,電子的能量可以被寫成:. H [ P] = T [ P] + V [ P] + U [ P] 之後 Kohn 和 Sham 兩人發表了 Kohn-Sham32方程式,其中描述了系統的能量可 以由哪些與電子密度相關的項來決定:. ε e [ P] = υ[ P] + h1[ P] + J [ P] + VXC [ P]. υ[ P] 項是系統的外部位能,通常是 0。 h1[ P] 項是動能與原子核的吸引力, J [ P] 項 是電子間的靜電排斥力, VXC [ P] 是電子交換(exchange)能與相互關係(correlation)能 的和。 在密度泛函理論中,主要的問題都發生在最後一項 VXC [ P] :沒有系統化的方法. 30. 31. 32.. Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. v. R. J. Comp. Chem. 1983, 4, 294. Hohenberg, P.; Kohn, W. Phys. Review 1964, 136, B864. Kohn, W.; Sham, L. J. Phys. Review 1965, 140A, 1133. 15.
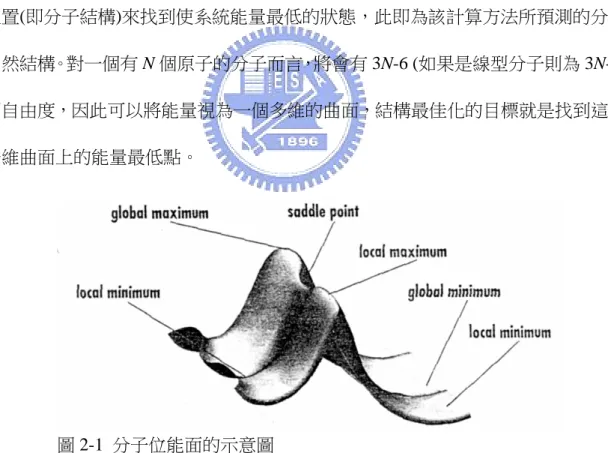
(23) 可以改進這一項,也因此不同的學者會對這一項的式子有不同的看法。在實際應用 時 , 這 一 項 通 常 會 被 拆 開 成 交 換 能 泛 函 (exchange funcitonal) 和 相 互 關 係 泛 函. (correlation functional)。 在這許多的交換能泛函和相關效應泛函中,最常用的是 B3LYP 泛函-由 Becke 提出的三參數交換能泛函33,34與 Lee, Yang, Parr 三人提出的相互關係泛函35所組合而 成。另外,在泛函中包含了 Hartree-Fock 項的被稱為混合型泛函(hybrid functional), 否則就稱為是純泛函(pure functional)。B3LYP 泛函即為混合型泛函。 2-5. 結構最佳化與振動頻率計算 顯而易見的,既然系統能量是原子核位置的函數,那麼就可以藉由調整原子核 位置(即分子結構)來找到使系統能量最低的狀態,此即為該計算方法所預測的分子 自然結構。對一個有 N 個原子的分子而言,將會有 3N-6 (如果是線型分子則為 3N-5) 個自由度,因此可以將能量視為一個多維的曲面,結構最佳化的目標就是找到這個 多維曲面上的能量最低點。. 圖 2-1 分子位能面的示意圖 圖 2-1 是一個分子位能面的示意圖36,我們可以用這張圖簡單的說明一下位能. 33. 34. 35. 36.. Becke, A. D. J. Chem. Phys. 1988, 88, 2547. Becke, A. D. J. Chem. Phys. 1993, 98, 5648. Lee, C.; Yang, W.; Parr, R. G. Phys. Review B 1988, 37, 785. Foresman, J. B.; Friesh, Æ., Exploring Chemistry with Electronic Structure Methods. In 2nd ed.; 16.
(24) 曲面的概念。一個位能曲面會有一個全域最低點(global minimum),對應到分子結 構上就是該分子能量最低的結構。這樣的點滿足的條件有二:一為. δE = 0 ,這表示 δr. 該點是一個靜態點(stationary point);但是靜態點可以是圖中的最低點(minimum)、 鞍點(saddle point)甚至是最高點(maximum)。此時要找到一個最低點還需要第二個條. δ 2E δ 2E 件:在每一個 r 的自由度上 2 > 0 。 2 > 0 在古典力學和量子力學中就代表該方 δr δr δ 2E 向簡諧運動的力常數。因為 r 其實有很多個,這個多自由度 2 構成的矩陣稱為 δr Hessian,也叫做力常數矩陣(force constant matrix): ⎛ ∂ 2U ⎜ 2 ⎜ ∂x1 ⎜ ∂ 2U ⎜ H = ⎜ ∂x2 x1 ⎜ ⎜ ... ⎜ ∂ 2U ⎜⎜ ⎝ ∂x p ∂x1. ∂ 2U ∂x1∂x2 ∂ 2U ∂x2 2 ... ∂ 2U ∂x p ∂x2. ∂ 2U ⎞ ⎟ ∂x1∂x p ⎟ ∂ 2U ⎟ ⎟ ... ∂x2 ∂x p ⎟ ⎟ ... ... ⎟ ∂ 2U ⎟ ... ⎟ ∂x p 2 ⎟⎠ .... 計算一個分子的 Hessian 再加上原子的質量就可以求得自由度上該振動的頻 率,這除了用來預測紅外線光譜外,還可以確認該分子結構是否為位能面的最低點。 同樣的道理,此種能量最小化計算也可以用來尋找反應過渡態的結構:只要使 條件為在一個自由度上的 Hessian 為負值即可,這樣就會得到一個位能面上鞍點的 結構。這樣得到的振動頻率將會是個負值,事實上負的振動頻率是沒有物理意義 的,因此又被稱為是虛頻(imaginary frequency)。 由位能曲面也可以看出來,一個曲面上可以有很多個鞍點。這些鞍點都各自對 應到一個反應過渡態,因此當找到一個反應過渡態之後,必須要確定這個過渡態兩. Gaussian, Inc.: Pittsburgh, PA, 1996; p 39. 17.
(25) 端連接的是想要的反應物與產物。這時候就需要進行反應座標跟隨(intrinsic reaction. coordinate, IRC)37,38的計算。要進行這種計算必須要給一個反應過渡態的結構,然後 計算時會沿著此結構的虛頻方向,朝反應座標的兩端各前進幾個點。之後只要從這 些已經”下坡”後的結構再做最佳化,就可以得到該過渡態連接到的反應物和產物。 2-6. 激發態的計算 上述提到的計算方法都是針對基態的電子結構。如果要計算激發態的性質,必 須要用別的方法。在這之中最簡單的是單電子激發組態交互作用 (configuration. interaction single excitation, CIS)39,可以視做是 Hartree-Fock 方法的激發態版本。這 個方法其實是把基態的電子組態:. ψ A2ψ B2 ...ψ M2 之中的一個軌域換成未填滿軌域(virtual orbital) X:. ψ 1Aψ 1Xψ B2 ...ψ M2 然後剩下的部分就按照 Hartree-Fock 的方式處理。於是事實上,這個計算就包括了 所有單一電子激發的組態。這方法繼承了 Hartree-Fock 方法的優缺點:簡單快速但 能量的準確度不足。此方法有一個低能量的系統誤差,經常可以看到 100nm 以上的 誤差。但是相對來說,CIS 方法得到的結構尚可,既然激發態方法的選擇較少,CIS 經常被當作在激發態做結構最佳化的選擇。 這裡就帶出所謂的組態交互作用極限(CI limit)的概念:只要包含所有可能的組 態,那麼理論上可以算出非常準確的能量。但是簡單的思考之後就可以發現,這樣 的排列組合太多種了,只有最小型的分子有可能用完全組態交互作用(full CI)的方 法計算,對稍大的系統來說都是不切實際的。 37. 38. 39.. Gonzalez, C.; Schlegel, H. B. J. Chem. Phys. 1989, 90, 2154. Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1990, 94, 5523. Foresman, J. B.; Head-Gordon, M.; Pople, J. A. J. Phys. Chem. 1992, 96, 135. 18.
(26) 如果已經對某個系統有瞭解,例如以知某系統的 S1 激發態是一個 π-π*激發態, 那麼就可以人工選擇可能相關的電子與軌域,並在這些電子和軌域之間作 full CI 計 算 。 這 樣 的 方 法 稱 為 完 整 活 化 空 間 自 洽 場 40 , 41 , 42 (complete active space. self-consistent field, CASSCF 或簡稱 CAS),這些被選擇的軌域和電子就稱為活化空 間(active space)。典型的符號表示法如:CAS(8,8),這表示在活化空間中選擇了 8 個電子和 8 個軌域。 這樣的方法雖然可以計算稍大的分子,但是需注意的是:CAS 需要比較多人工 涉入的部分,因此同一個分子可能會因為研究者選擇了不同的活化空間,而得到不 同的結果。活化空間的選擇也沒有一定的準則,需要相當的經驗才能有效的使用這 個計算方法。 密度泛函理論的另一個應用:所謂的含時密度泛函 (time-dependent density. functional theory, TDDFT)43,44方法,也可以用來計算激發態的能量。其計算速度相 當快,並且準確度也不差,唯一的缺點是因為它並沒有 Hessian 的分析解。這意味 著此方法無法做結構最佳化。因此一般的作法是利用 CIS 或是 CAS 方法得到激發 態最佳化的結構,然後使用 TDDFT 方法在已最佳化的結構做單點計算。 2-7. 高準確度方法 前面提到的電子相互關係,雖然其在整體能量中佔的比例不大,但是在需要高 準確度的場合中卻是不可忽略的。有數種高階的方法是被發展來補足這方面的不 足,以下簡單說明本研究有用上的幾種: 微擾理論(perturbation theory)是從 Hamiltonian 運算子的角度來處理電子相互關. 40. 41. 42. 43. 44.. Hegarty, D.; Robb, M. A. Mol. Phys. 1979, 38, 1795. Eade, R. H. E.; M. A. Robb Chem. Phys. Lett. 1981, 83, 362. Yamamoto, N.; Vreven, T.; Robb, M. A.; Frisch, M. J.; Schlegel, H. B. Chem. Phys. Lett. 1996, 250, 373 Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454. Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R. J. Chem. Phys. 1998, 108, 4439 19.
(27) 係。假設我們要處理一個 Hˆ 0ψ i = ε 0ψ i. 的系統,微擾的方法就是在運算子中加入一個”修正項”: Hˆ = Hˆ 0 + λ Hˆ (1). 要詳細的處理微擾理論需要相當多額外的計算,但是 Chr. Møller 和 M. S.. Plesset45發展了一種簡化式的微擾計算方法。簡單來說,這個微擾項就是真實的 Hˆ 與 Hartree-Fock 的 Hˆ F 的差,這個相差的雙電子積分項可以被轉換,但是並不需要做完 整的轉換,而是只做部分轉換。接下來就視做到第幾階的微擾,而稱做 MPn 的計 算方法,如 MP2, MP3…等。 多重組態自洽場(multiple configuration SCF),則是使用一個以上的電子組態(或. Slater 行列式)放在波函數中,所以前面提到的 CIS 和 CAS 事實上都是屬於這類方 法的一種。 另一個得到較精確能量的方法是使用更大的基底函數,但是很明顯的這會使系 統的運算量快速增加。在難以同時使用大型基底函數和高階計算的情況下,一個很 聰明的方式就是使用複合方法。利用高階但較小基底函數和較低階但較大基底函數 的計算,配合實驗值來調整其能量的係數以得到一個高準確度的值。. Gaussian-1(G1)46、Gaussian-2(G2)47、Gaussian-2 with MP2(G2MP2)48等方法都是屬 於這種複合方法。 2-8. 溶劑的處理與自洽反應場 由於大多數的化學反應事實上是在溶液中發生的,因此在計算時,有時必須要. 45. 45. 46. 47. 48.. Møller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618. Pople, J. A.; Head-Gordon, M.; Fox, D. J. J. Chem. Phys. 1989, 90, 5622. Curtiss, L. A.; Raghavachari, K.; Trucks, G. W.; Pople, J. A. J. Chem. Phys. 1991, 94, 7221. Curtiss, L. A.; Raghavachari, K.; Pople, J. A. J. Chem. Phys. 1993, 98, 1293. 20.
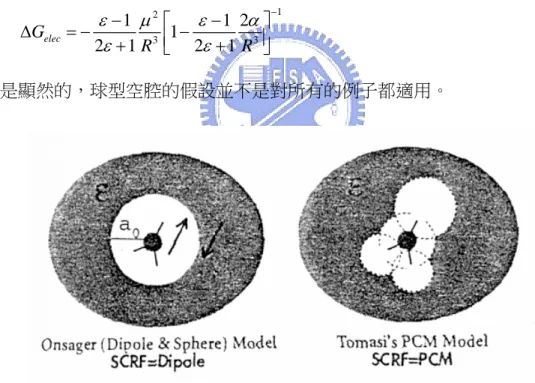
(28) 考慮溶劑的效應,才能得到正確的結果。計算溶劑時有兩種大方向,一是把溶劑看 成是無結構的連續體,另一種是在系統中真的放進個別的溶劑分子49。但是後者很 顯然的會使系統大小快速增加,因此能夠應用的範圍比較有限。 而以連續體處理溶劑的方式,只要在溶質分子周圍放置一個靜電場即可。此時 的問題在於,如何正確的描述溶質分子與溶劑連續體間的靜電關係,以及溶質分子 在溶劑連續體中產生的空腔。 最簡單的模型稱做 Onsager 模型50,這是將一個溶質分子至於球型空腔中,假 設空腔的半徑是 R,溶劑的介電常數是 ε,極化度是 α,溶質的偶極矩 μ,此時溶劑 對溶質造成的穩定自由能為:. ΔGelec. ε −1 μ 2 =− 2ε + 1 R 3. ε − 1 2α ⎤ ⎡ ⎢⎣1 − 2ε + 1 R 3 ⎥⎦. −1. 但是顯然的,球型空腔的假設並不是對所有的例子都適用。. 圖 2-2 兩種溶劑空腔模型的示意圖. Tomasi 51 等人則是發展了一種極化連續體模型 (polarizable continuum model, PCM),其作法是以溶質分子的各原子為中心,放置球型的空腔,各球型空腔的半. 49. 50. 51.. Orozco, M.; Alhambra, C.; Barril, X. J. Mol. Model. 1996, 2, 1. Onsager, L. J. Am. Chem. Soc. 1936, 58, 1486. Cance`s, E.; Mennucci, B.; Tomasi, J. J. Chem. Phys. 1997, 107, 3032. 21.
(29) 徑中心原子的種類來決定,如圖 2-2 所示52。之後將組合後的空腔表面切成一小片 一小片(這步驟稱做 tessalation),然後對每一小片表面進行計算。PCM 方法的優點 在於,將所有小球型空腔結合起來,就可以得到一個能隨分子結構調整的空腔,因 此在做結構最佳化的同時,可以在每一個中間結構都產生一個相對應的空腔。 還有其他定義空腔的方式,例如等密度表面可極化連續體模型 (isodensity. surface polarizable continuum model, ISPCI)等等的方法,都是設計來產生更好的溶劑 空腔。但是這類的方法大多需要繁重的計算。 這類型的模型有兩個主要的問題。第一個是來自於空腔這個作法。事實上溶質 分子的電子雲有少部分會穿透到空腔介面之外,這部分的相互作用就不容易處理。 這個現象稱為外部電荷(outlying charge)。 另一個是來自於將溶劑視為無結構的連續體本身。這顯然只能處理和結構無關 的溶質-溶劑交互作用,所以對於像氫鍵之類的交互作用,這類的連續體模型就無 法處理了。 大部分溶劑對溶質分子的效應可以用靜電模型來處理,也就是說必須要把計算 的溶質分子放在一個靜電場當中。這個方法稱做自洽反應場(self-consistent reaction. field, SCRF)。這事實上是基於微擾理論:. H rf = H 0 + H1 其中 H0 是沒有溶劑時的 Hamiltonian,H1 是描述溶質偶極運算子與外加反應場間耦 合的微擾項:. K H1 = − μˆ ⋅ R 此反應場正比於溶質分子的偶極矩:. 52.. Foresman, J. B.; Friesh, Æ., Exploring Chemistry with Electronic Structure Methods. In 2nd ed.; Gaussian, Inc.: Pittsburgh, PA, 1996; p 237. 22.
(30) K K R = gu 將溶劑視為連續且無結構,然後在溶劑中挖一個空腔以放置溶質分子。對於一 個簡單的球型空腔而言,這個比例常數 g 會與該溶劑的介電常數 ε 和該空腔的半徑 有關:. g=. 2(ε − 1) (2ε + 1) R 3. 2-9. 本研究中使用的計算軟體及硬體 在本研究中所有的計算都是使用 Gaussian 03 rev. C0253,54軟體進行,並在交通 大學分子科學研究中心的 16-nodes 叢集電腦系統上進行運算。. 53.. 54.. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; J. A. Montgomery, J.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Daniels, A. D.; Farkas, O.; Rabuck, A. D.; Raghavachari, K.; Ortiz, J. V. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; J. A. Montgomery, J.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision C. 02, Gaussian, Inc.: Pittsburgh PA, 2003. 23.
(31) 第三章 鄰位硝基甲苯之分解與重排反應的量子化學計算研究 鄰位硝基甲苯因為系統較小但要求較高的能量準確度,因此我們使用密度泛函. B3LYP/6-311G(d,p)方法來做結構最佳化及頻率計算,並且利用最佳化之後的電子能與零 點能總和,做初步的位能面描繪。 所有的靜態點結構都做了頻率計算以確定是否是為能量最低點或是過渡態,並且所 有的過渡態都使用反應座標跟隨計算來確認其連接到的反應物與生成物。 至於高精確度的能量計算,本研究中則使用 Mebel 等人55修改的 Gaussian-2 方法來 計算,並將所得的能量與密度泛函得到的能量作一個比較。我們在此將使用該研究群所 提出的 G2M(cc,mp2)方法來做計算,因為是使用複合式方法,因此每個能量都是經由數 個計算得到的。詳細的計算方式如下: 表 3-1 G2M(cc,MP2)之計算方法 計算方法 能量 MP4(SDTQ)/6-311G(d,p) Ebas=E[PMP4/6-311G(d,p)] MP4(SDTQ)/6-31G(d,p) Δ’E(CC)=E[CCSD(T)/6-31G(d,p)]-E[PMP4/6-31G(d,p)] CCSD(T)/6-31G(d,p) ΔE(+3df2p)=E[MP2/6-311+G(3df,2p)]-E[MP2/6-311G(d,p)] MP2/6-311+G(3df,2p) ΔE(HLC,CC6)a=-5.05nβ-0.19nα E[G2M(cc,MP2)]=Ebas+ΔE(+3df2p)+Δ’E(CC)+ ΔE(HLC,CC6)+ZPEc (a) nβ 和 nα 分別代表 α 自旋與 β 自旋的價電子數量,nα≥nβ。(b)ZPE(零點能量)為 B3LYP/6-311G(d,p)方法計算所得到。 每一個結構的原子位置座標請見附錄 1,密度泛函的能量與 G2M 複合能量計算細 節請見附錄 2。. 55.. Mebel, A. M.; Morokuma, K.; Lin, M. C. J. Chem. Phys. 1995, 103, 7414. 24.
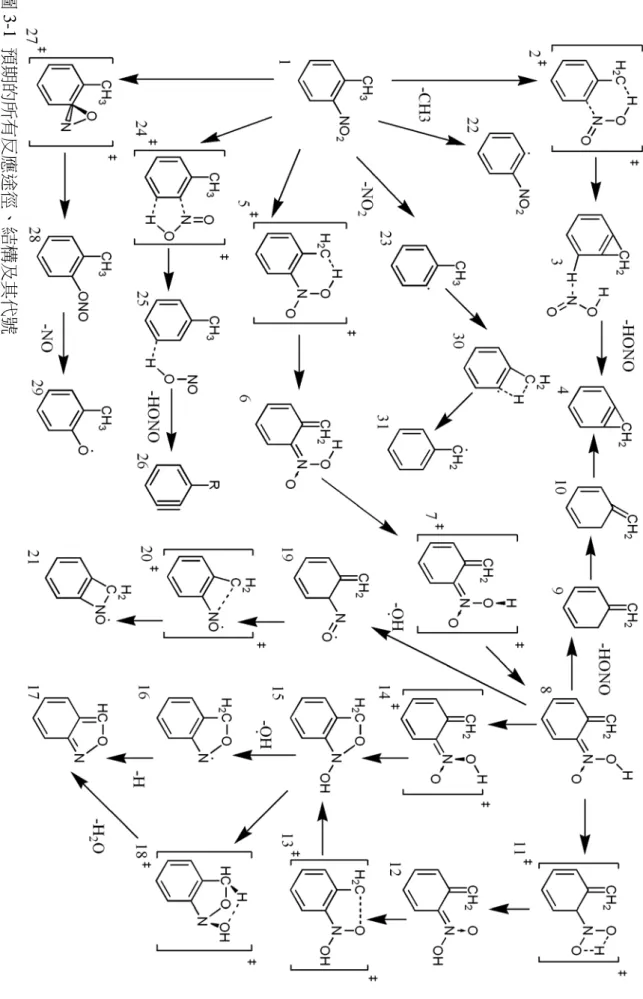
(32) 圖 3-1 預期的所有反應途徑、結構及其代號. 25.
(33) 3-1. 預期的反應路徑 圖 3-1 列出了本部分研究中,所預期的所有可能反應路徑及其結構示意圖。本 圖並在所有結構下附上編號,以利文中參考。以下詳述計算中所觀察到的細節。文 後的結構將列出原子編號及較重要的結構資訊:鍵長以 Å 為單位,鍵角與立體角以 度為單位。. O(17)-N(15) N(15)-O(16) N(15)-C(2) C(3)-C(11) O(17)-N(15)-C(2) O(16)-N(15)-C(2) N(15)-C(2)-C(3) C(2)-C(3)-C(11) O(17)-N(15)-C(2)-C(3) C(2)-C(3)-C(11)-H(13). 1.225 1.224 1.481 1.508 118.1 117.6 121.5 125.3 -21.0 -169.6. 結構 1 (鄰位硝基甲苯) 表 3-2 不同方法得到的鄰位硝基甲苯中硝基與苯環平面之夾角 B3LYPb B3LYPc B3LYPc MP2(FC)e 實驗值 f 計算方法 B3LYPa -21.0º -13.7º -13.0º -24.8º -38º -38º 角度 (a)本研究。B3LYP/6-311G(d,p) (b)文獻 56。B3LYP/6-31G(d) (c)文獻 10。B3LYP/6-31G(d,p) (d)文獻 10。B3LYP/6-311+G(2d,p) (e)文獻 56。MP2(FC)/6-31G(d) (f)文獻 56。電子繞射實驗 所有的反應都是從鄰位硝基甲苯(1)開始,此結構經 B3LYP/6-311G(d,p)計算所 得到的硝基與苯環平面之立體角 C(17)-C(15)-C(2)-N(3)=-21.0º。在 Shinshkov56等人 於 1998 年進行的計算中,利用 MP2(FC)/6-31G(d)與 B3LYP/6-31G(d)方法,分別得 到此立體角為 34.7º 和 12.9º,並以電子繞射的方式測得此立體角約為 38º。而該研 究群此現象的解釋是:實驗時測得的並不是位能最低點的結構,而是許多個因熱能 而旋轉的 NO2 基團之平均結構(表 3-1)。類似的現象也在硝基苯的電子繞射實驗結 果57中發現:理論計算所得的到的 NO2 基團與苯環是接近同平面的,而實驗所測得. 56. 57.. Shishkov, I. F.; Vilkov, L. V.; Kovacs, A.; Hargittai, I. J Mol Struct 1998, 445, 259-268. Domenicano, A.; Schultz, G.; Hargittai, I.; Colapietro, M.; Portalone, G.; George, P.; Bock, C. W. Struct. 26.
(34) 的立體角是 13º。 結構 2 是一個過渡態,可以視作是 HONO 同步解離的過渡態-後面將提到另一 條逐步解離的路徑。在這結構中的特點是較長的 C(5)-N(1)鍵長(2.31Å),大幅彎曲 的甲基-苯環鍵:C(11)-C(7)-C(5)為 92.5º,以及從甲基轉移到硝基上面的氫原子:. C(11)-C(4)為 1.547Å,O(3)-O(4)為 1.120Å。從這個結構朝兩個方向分別做 6 步 IRC 計算後再做結構最佳化(此步驟文後簡稱為 IRC 計算),可以分別得到結構 1 與結構. 3。結構 3 是一個中間產物,很明顯的 HONO 已經與苯環部分拉開了一段距離。而 HONO 離開後的苯環部分形成了一個雙環的結構,原來甲基上的兩個氫位於與苯環 幾乎垂直的位置:C(16)-C(11)-C(5)-C(7) =106.6º,C(15)-C(11)-C(5)-C(7)=106.6º。三. N(1)-C(5) C(5)-C(7) C(11)-C(7) O(3)-H(4) C(11)-H(4) O(2)-N(1) N(1)-O(3) C(11)-C(7)-C(5) N(1)-C(5)-C(7) H(16)-C(11)-C(7)-C(5) H(15)-C(11)-C(7)-C(5). 2.311 1.332 1.490 1.120 1.547 1.193 1.295 92.5 134.7 -110.2 110.2. H(4)-C(5) C(11)-H(4) N(1)-C(5) C(11)-C(5) C(11)-C(7) C(7)-C(5) N(1)-O(3) C(11)-C(5)-C(7) C(11)-C(7)-C(5) H(16)-C(11)-C(5)-C(7) H(15)-C(11)-C(5)-C(7). 2.370 2.439 3.733 1.522 1.495 1.346 1.418 62.5 64.5 106.5 -106.5. 結構 2‡. 結構 3 Chem. 1990, 1, 107. 27.
(35) 元 環 的 部 分 鍵 角 為 接 近 正 三 角 形 的 角 度 : C(11)-C(5)-C(7)=62.5º ,. C(11)-C(7)-C(5)=64.5º。 從結構 3 進一步的將 HONO 拉開,可以得到這條路徑最終的產物-分離的結構. 4 與 HONO。結構 4 與結構 3 中的苯環部分相當類似,就僅列出結構參數而不多加 描述了。 結構 5 也是一個過渡態,由它做 IRC 計算可以分別得到結構 6 和結構 1。結構. 5 與結構 2 類似的地方在於甲基上的一個氫原子轉移到硝基的氧原子上,但是結構 5 中的 C(4)-N(1)鍵短的多-只有 1.39Å,因此這個結構可以視作是單純氫原子轉移反. C(4)-C(5) C(4)-C(11) C(11)-C(3) C(4)-C(3) C(2)-C(3) C(4)-C(3)-C(2) C(5)-C(4)-C(3) C(11)-C(4)-C(3) C(4)-C(3)-C(11) C(3)-C(4)-C(11)-H(12) C(3)-C(4)-C(11)-H(13). 1.377 1.503 1.503 1.348 1.377 124.4 124.4 63.4 63.4 106.5 -106.4. N(1)-C(4) C(6)-C(4) C(6)-C(11) H(16)-C(11) H(15)-C(11) H(7)-C(11) H(7)-O(3) C(4)-N(1)-O(3) N(1)-C(4)-C(6) C(4)-C(6)-C(11) H(15)-C(11)-C(6) H(16)-C(11)-C(6) O(3)-N(1)-C(4)-C(6) C(4)-C(6)-C(11)-H(15). 1.392 1.441 1.415 1.085 1.089 1.502 1.135 118.3 119.7 123.1 118.8 116.6 -8.4 52.9. 結構 4. 結構 5‡ 28.
(36) 應的過渡態。值得注意的是氫原子轉移後的甲基變成接近平面三角形構造,這可以 從 H(15)-C(11)-C(6)=118.8º 和 H(16)-C(11)-C(6)=116.6º 看出來:兩者的角度都接近. 120º。此外,此平面三角結構與苯環成 52.9º 的夾角。 由結構 5 做 IRC 計算可以分別得到結構 1 和結構 6。在結構 6 中,氫原子的轉 移已經完成,可以從 H(7)-O(3)的鍵長接近一般的 H-O 鍵長看出來。另外一個重要 的特點是 HONO 取代基和 CH2 取代基因為彼此的立體障礙而稍微偏離平面,這可 以從 C(4)-N(1)-O(3) -H(7)=19.8º 和 C(4)-H(6)-C(11)-H(15)=8.8º 得知。由這個結構可 以合理的推測,存在一個低能量的 N(1)-O(3)單鍵旋轉過渡態,使 H(3)旋轉到較遠 離 CH2 基團的位置,藉由減少者間的立體障礙而釋放能量。. C(4)-N(1) N(1)-O(3) H(7)-O(3) H(15)-C(11) C(6)-C(11) C(6)-C(4) H(7)-C(11) C(4)-N(1)-O(3) C(4)-C(6)-C(11) C(6)-C(11)-H(16) C(4)-N(1)-O(3)-H(7) C(4)-C(6)-C(11)-H(15). 1.350 1.401 0.978 1.080 1.365 1.473 2.119 119.2 127.3 118.9 19.8 8.8. H(7)-O(3)-N(1)-C(4) H(15)-C(11)-C(6)-C(4) O(3)-N(1)-C(4)-C(6). 76.304 3.191 -1.575. 結構 6. 結構 7‡. 7 29.
(37) 結構 7 就是這個 N-OH 單鍵旋轉的過渡態,由結構 7 做 IRC 計算可以分別得到 結構 6 與結構 8。在結構 7 中,由 H(7)-O(3)-N(1)-C(4)=76.3º 可以看出氫原子是處 在 O-N-C 平面外的位置上,而且由 H(15)-C(11)-C(6)-C(4)=3.2º 和 O(3)-N(1)-C(4)-C(6). =-1.6º 可以知道這個結構的 CH2 與 HONO 基團已經相當接近苯環平面了。 結構 8 是 N-OH 鍵旋轉過去以後的產物,由結構參數列出的四組立體角中,可 以顯示整個分子幾乎完全是平面的。這個結構在整個反應路徑上是很重要的,因為 它連接到四條反應路徑,如圖 3-1 所示。 由結構 8 連接出去的四條反應路徑中,我們首先討論結構 9。它是從結構 8 脫 離一個 HONO 基團的產物。因為是單純的斷鍵反應,因此這個步驟並沒有明確的. C(4)-C(6)-C(11) N(1)-C(4)-C(6) C(6)-C(4)-N(1)-O(3) H(7)-O(3)-N(1)-C(4) H(15)-C(11)-C(6)-C(4) H(16)-C(11)-C(6)-C(4). 127.9 125.4 0.0 180.0 0.0 180.0. C(7)-C(3) C(3)-C(1) C(2)-C(1) C(4)-C(2) C(8)-C(4) C(8)-C(5) C(3)-C(5) C(3)-C(1)-C(2) C(4)-C(2)-C(1) C(8)-C(4)-C(2) C(8)-C(5)-C(3) C(1)-C(3)-C(5) C(8)-C(4)-C(2)-C(1) C(5)-C(8)-C(4)-C(2) C(5)-C(3)-C(1)-C(2). 1.369 1.441 1.395 1.394 1.424 1.364 1.448 117.6 118.0 121.4 118.1 120.6 21.3 -5.0 11.53. 結構 8. 結構 9 30.
(38) 過渡態。這個結構有一個很特殊的地方,就是苯環本身是變形的:從側面看的話整 個苯環稍微扭曲成類似環己烷的船型結構,這顯示在 C(5)-C(4)-C(2)-C(1)=21.3º 這 個異常的立體角上。 繼續縮小 CH2 與苯環的鍵角就會得到結構 10 這個過渡態,由結構 10 做 IRC 計算可以分別得到結構 9 與結構 4。結構 10 與結構 2 有一些相似之處,就是它們都 有大約 100º 的 C(7)-C(3)-C(1)鍵角,同時 CH2 基團上的兩個氫原子也離開平面,這 點由兩組立體角:H(11)-C(7)-C(3)-C(1)=-38.6º 與 H(11)-C(7)-C(3)-C(1)=136.6º 可以 看出。從結構 10 經由 IRC 計算可以連接到結構 4,也就是說這是另一條產生結構 4 的反應路徑。由於在這條路徑中是 HONO 先脫離得到結構 9 然後才經由過渡態得 到結構 4,因此我們可以將這條路徑視為是 HONO 的逐步脫離反應-在這個路徑中. C(7)-C(3) C(1)-C(3) C(1)-C(2) C(1)-C(3)-C(7) C(3)-C(1)-C(2) H(11)-C(7)-C(3)-C(1) H(12)-C(7)-C(3)-C(1). 1.400 1.388 1.401 102.2 118.8 -38.6 136.6. H(17)-O(15) H(17)-O(16) N(14)-O(16) O(15)-N(14) C(4)-N(14) C(11)-C(3) N(14)-C(4)-C(3) C(11)-C(3)-C(4) C(3)-C(4)-N(14)-O(16) H(13)-C(11)-C(3)-C(4). 1.284 1.264 1.328 1.329 1.316 1.356 122.4 125.3 0.0 0.0. 結構 10‡. 結構 11‡ 31.
(39) HONO 並不是從結構 1 直接脫離的,而是先經過數個步驟重排成結構 8,才從結構 8 脫離。 結構 11 則是另一個從結構 8 連接過來的過渡態,由結構 11 做 IRC 可以分別連 接到結構 12 與結構 8。由於在結構 11 中氫原子是介於兩個氧原子之間,因此它很 明顯的是氫原子從硝基的一個氧原子轉移到另一個氧原子上之過渡態。從結構參數 上可以看出整個分子完全是平面的,氫原子與兩個鄰近的氧原子距離也相當接近. (1.284Å vs. 1.264Å),兩個基團也因為立體阻礙稍微偏離對方。 結構 12 是氫原子轉移過後的產物。與結構 8 和 9 類似的是整個分子大約成平 面構造,所以這個氫原子轉移反應是在平面上完成的。H(17)-O(15)的長度(0.97Å) 也在一般 H-O 鍵的長度範圍內。. N(14)-O(16) N(14)-O(15) H(17)-O(15) C(4)-N(14) C(3)-C(11) C(3)-C(4)-N(14) C(4)-C(3)-C(11) C(3)-C(4)-N(14)-O(16) C(4)-C(3)-C(11)-H(13). 1.243 1.419 0.970 1.332 1.359 120.2 125.9 0.0 0.0. O(15)-H(17) O(15)-N(14) O(16)-N(14) C(4)-N(14) C(11)-C(3) N(14)-C(4)-C(3) O(16)-N(14)-C(4) C(11)-C(3)-C(4) C(3)-C(4)-N(14)-O(16) H(13)-C(11)-C(3)-C(4) H(17)-O(15)-N(14)-C(4) O(15)-N(14)-C(4)-C(3). 0.96 1.450 1.274 1.378 1.416 114.0 116.3 119.6 11.7 -62.6 -163.4 -123.7. 結構 12. 結構 13‡ 32.
(40) 結構 13 是一個過渡態,與結構 12 比較可以發現幾個重要的特點:O(16)與 C(11) 的距離縮短了,CH2 基團也旋轉到離開苯環平面的位置,同時 N(14)-C(4)-C(3)的鍵 角也縮小。這暗示了反應可能是一個合環反應。另一個特點是硝基上的 OH 基團離 開了苯環平面,這可以從結構參數的最後兩組立體角看出來。 結構 14 則是另一個從結構 8 連接過來的過渡態。這個過渡態的特徵是離開平 面的 HONO 基團,可以視作是 C-N 單鍵旋轉的一個過程。但是除了 HONO 基團旋. O(14)-N(13) C(4)-N(13) C(3)-C(4) C(11)-C(3) C(11)-O(14) C(11)-H(12) C(11)-H(15) O(14)-N(13)-C(4) C(3)-C(4)-N(13) O(14)-C(11)-C(3) H(12)-C(11)-C(3) C(3)-C(11)-H(15) H(12)-C(11)-C(3)-C(4) H(15)-C(11)-C(3)-C(4). 1.378 1.363 1.411 1.491 1.458 1.095 1.095 106.8 112.9 103.2 114.3 114.3 -116.8 116.7. O(14)-N(13) C(4)-N(13) C(3)-C(4) C(11)-C(3) C(11)-O(14) O(14)-N(13)-C(4) C(3)-C(4)-N(13) C(11)-C(3)-C(4) O(14)-C(11)-C(3) C(11)-O(14)-N(13) H(12)-C(11)-C(3). 1.393 1.325 1.441 1.372 1.335 104.5 112.1 102.9 109.7 110.9 134.1. `結構 16. 結構 17. 33.
(41) O(16)-H(17) O(16)-N(4) H(11)-O(16) H(11)-C(7) H(11)-C(7)-O(8) C(7)-C(2)-C(1)-C(3) C(3)-C(1)-N(4)-O(8). 0.965 2.284 1.585 1.203 92.6 171.4 167.6. O(2)-N(1) N(1)-C(3) C(8)-C(13) C(8)-C(3) O(2)-N(1)-C(3) C(8)-C(3)-N(1) C(13)-C(8)-C(3) O(2)-N(1)-C(3)-C(8) C(3)-C(8)-C(13)-H(14). 1.223 1.421 1.394 1.439 115.7 116.7 122.7 179.0 0.8. 結構 18‡. 結構 19 轉外,CH2 基團也跟著旋轉到苯環平面之外,這一點與結構 13 類似,一樣是暗示 了一個合環反應的結果。 結構 15 是結構 13 和 14 做 IRC 計算後得到的合環反應產物,此結構除了末端 的 OH 基團外,整個分子大約是成平面的。 結構 15 已經相當接近 anthranil 了,如果讓 H 和 OH 分兩次逐步脫離的話,就 可以變成 anthranil。結構 16 就是 OH 從結構 15 脫離之後的結果。因為這是單純的 斷鍵反應,因此中間並沒有過渡態。再從結構 16 上拿掉一個 H 原子就變成 anthranil。 結構 17 就是預期中的 anthranil 產物。整個分子是平面的,具有雙環結構。 但是要從結構 15 到結構 17 還有另一種可能,就是同時脫去 H2O。結構 18 就. 34.
(42) 是這個路徑的過渡態,其結構上的特點是偏離平衡位置許多的 OH 基團,同時 H(12) 也往 O(16)的方向移動。比較值得注意的是 O(16)-N(4)距離已經拉長到 2.283Å 了。 結構 19 則是最後一個從結構 8 連接出來的反應中間物。OH 基團從結構 8 上脫 離,產生結構 20,是一個單純的斷鍵過程。就結構參數上來看,整個分子大約是成 平面的。 因為 N 原子上有未鍵結的電子,而旁邊的 CH2 基團有空軌域可以容納電子, 因此假設它們之間可以生成化學鍵是相當合理的。這個反應將會是一個合環反應, 其過渡態如結構 20 所示。 結構 20 則是從結構 19 連接來的過渡態。結構上的特徵是較小的 N(14)-C(3)-C(4) 與 C(11)-C(4)-C(3)鍵角,以及偏離平面的 CH2 與 NO 基團:由 H(13)-C(11)-C(4). N(14)-O(15) N(14)-C(3) C(3)-C(4) C(11)-C(4) C(3)-N(14)-O(15) N(14)-C(3)-C(4) C(11)-C(4)-C(3) C(4)-C(11)-H(13) H(12)-C(11)-C(4) H(13)-C(11)-C(4)-C(3) O(15)-N(14)-C(3)-C(4). 1.221 1.414 1.400 1.458 122.4 103.8 106.8 118.7 122.1 71.2 -154.2. N(14)-O(15) N(14)-C(3) C(4)-C(3) C(11)-C(4) N(14)-C(11) C(3)-N(14)-O(15) O(15)-N(14)-C(3)-C(4) C(11)-C(4)-C(3)-C(2). 1.251 1.408 1.404 1.526 1.521 135.8 180.0 179.9. 結構 20‡. 結構 21 35.
(43) -C(3)=71.2º 和 O(15)-N(14)-C(3)-C(4)= -154.2º 可以得知。 結構 21 則是從結構 20 來的產物。由結構可以證明此反應是一個合環反應,所 以得到的是一個雙環的結構。由結構參數可以知道整個分子大約是平面的,但是因 為四元環的結構能量比較高,因此可以預期這個結構在熱力學上並不會很穩定。. O(12)-N(11) N(11)-O(13) N(11)-C(2) C(3)-C(2) C(4)-C(3) C(4)-C(3)-C(2) C(3)-C(2)-N(11) O(12)-N(11)-C(2) O(13)-N(11)-C(2) O(13)-N(11)-C(2)-C(3) O(12)-N(11)-C(2)-C(3) C(4)-C(3)-C(2)-N(11). 1.219 1.219 1.486 1.368 1.372 124.3 121.1 116.7 116.7 -90.3 90.2 180.0. C(1)-C(2) C(2)-C(3) C(11)-C(3) C(6)-C(1)-C(2) C(1)-C(2)-C(3) C(4)-C(3)-C(2) C(11)-C(3)-C(2) C(6)-C(1)-C(2)-C(3) C(1)-C(2)-C(3)-C(4) C(1)-C(2)-C(3)-C(11). 1.375 1.378 1.511 116.7 127.0 114.8 123.3 0.0 0.0 180.0. 結構 22. 結構 23 從結構 1 的鄰位硝基甲苯也可能會發生簡單的解離反應,結構 22 和 23 就分別 是解離 CH3 和 NO2 之後的產物。因為是簡單的斷鍵反應,所以這兩個反應過程並 不會有明顯的過渡態。 結構 22 可以看出苯環部分成平面狀,但是 NO2 基團很明顯的是在平面外,而 36.
(44) 且大約是在垂直於苯環平面的位置上。這點可以由 O(13)-N(11)-C(2)-C(3)=-90.2º 和. O(12)-N(11)-C(2)-C(3)=90.3º 得知。至於 NO2 基團的部分整個是對稱的,這可以由 相同的 N(11)-O(13),N(11)-O(12)鍵長和 O(12)-N(11)-C(2),O(13)-N(11)-C(2)鍵角得 知。 結構 23 就參數來看可以知道分子成平面形,但是結構上特殊的地方是 C(2)的 鍵角顯然特別大 -C(1)-C(2)-C(3) 是 127.0º ,大於一般的 120º 。另外 C(2)-C(3) 與. C(2)-C(1)兩組鍵長也比一般苯環上的鍵長稍短。 我們已經看過有兩種方式可以從結構 1 上面脫離 HONO,這兩種方式都是從甲. O(3)-N(1) O(2)-N(1) O(2)-H(4) C(6)-H(4) C(5)-N(1) H(4)-O(2)-N(1) C(5)-C(6) C(5)-C(7) C(10)-C(7) C(5)-C(7)-C(10) C(6)-C(5)-C(7) C(8)-C(6)-C(5) C(6)-C(5)-C(7)-C(10) H(4)-O(2)-N(1)-O(3). 1.180 1.355 1.000 1.991 2.343 101.1 1.268 1.369 1.500 129.0 139.2 114.3 180.0 180.0. O(2)-H(4) O(2)-N(1) O(3)-N(1) H(4)-O(2)-N(1) H(4)-C(6) N(1)-C(5) C(5)-C(7) C(6)-C(5)-C(7) C(8)-C(6)-C(5) C(7)-C(5)-C(6)-C(8) H(4)-O(2)-N(1)-O(3). 0.974 1.413 1.172 102.7 2.426 3.608 1.383 131.4 124.5 0.0 180.0. 結構 24‡. 結構 25. 37.
(45) 基上面抽取一個氫原子。但還有另一個方式,就是直接從苯環本身抽取一個氫原 子。這個反應的過渡態如結構 24 所示,是 NO2 從 C(6)上面抽取一個氫原子所形成 的,其中較重要的是 N(1)-C(5)=2.343Å,C(6)-H(4)=1.991Å 兩個距離。還有一個特 殊的地方:C(6)-C(5)-C(7)=139.1º,這很明顯比一般苯環的 120º 來的大上許多。但 是整個苯環大約還是成平面形狀。. C(10)-C(3) C(2)-C(3) C(2)-C(1) C(6)-C(1) C(2)-C(3)-C(10) C(1)-C(2)-C(3) C(6)-C(1)-C(2) C(1)-C(2)-C(3)-C(10). 1.501 1.389 1.243 1.384 127.2 128.1 127.3 180.0. N(11)-C(1) O(12)-C(1) N(11)-O(12) O(12)-N(11)-O(13) C(2)-C(1) C(6)-C(1) C(6)-C(1)-C(2) C(14)-C(2)-C(1)-C(6). 1.704 1.798 1.312 121.0 1.397 1.386 125.3 178.4. 結構 26. 結構 27‡ 結構 25 則是從結構 24 做 IRC 計算所得到的反應中間體。事實上這個 HONO 已經離原來的苯環相當遠了,H(4)-C(6)的距離長達 2.426Å。整個分子成平面狀,和 結構 24 相比 C(6)-C(5)-C(7)=131.4º 雖然仍然比正常值大一些,但是已經比結構 25 的的角度 139.2º 來的小。. 38.
(46) C(1)-O(12) N(11)-O(13) N(11)-O(12) N(11)-O(12)-C(1) O(12)-N(11)-O(13) O(13)-N(11)-O(12)-C(1) C(6)-C(1)-O(12)-N(11). 1.383 1.150 1.530 111.2 110.3 -177.7 77.1. O(15)-C(1) C(6)-C(1) C(2)-C(1) C(2)-C(11) O(15)-C(1)-C(6) C(2)-C(1)-O(15) C(2)-C(1)-C(6) C(11)-C(2)-C(1)-C(6) C(5)-C(6)-C(1)-O(15). 1.250 1.452 1.467 1.497 121.3 121.0 117.7 180.0 180.0. 結構 28. 結構 29 結構 26 是 HONO 完全脫離之後的產物。這個化合物是 benzyne 的甲基衍生物, 其 C(1)-C(2)應具有三鍵的性質。這一點我們可以從結構參數中的 C(1)-C(2)特別短 得到確認-只有 1.243Å,比一般苯環的 1.4Å 短上許多,而比較接近一般有機化合物 中三鍵的鍵長。C(1)-C(2)-C(3)和 C(6)-C(1)-C(2)也比一般的 120º 來的大。整個分子 大約成平面狀。 結構 27 則是一個異構化的反應過渡態。Wing Tsang 等人 6 在 1986 年的實驗中 曾經發現有 NO 的分解產物,這點在序論中已經提到過了。結構 27 可視作是 N(11) 遠離 C(1)而同時 O(12)接近 C(1)的反應過渡態,可以從圖上清楚的看到整個 NO2 基 39.
(47) 團大約是垂直於苯環平面的,其中 C(1)、O(12)、N(11)三個原子形成一個類似三元 環的過渡態結構。苯環本身是維持平面的。 結構 28 則是異構化的產物-從 NO2 基團轉變成 ONO 基團。但是這個結構有一 個 很 特 殊 的 地 方 - 雖 然 說 是 ONO 基 團 , 但 是 N(11)-O(12) 的 距 離 明 顯 的 大 於. N(11)-O(13)的距離。同時 N(11)-O(12)兩個原子也偏離了苯環與 O(12)構成的平面。 這暗示了 NO 可以很容易的從這個結構脫離。 結構 29 是 NO 從結構 28 脫離後的產物。整個分子成平面形,和結構 28 比較 起來的特點是 C-O 鍵稍微變長了,這可能是因為 NO 脫離後電子重新分佈的關係。 另外,結構 23 應可以經由重排反應而形成 C6H5CH2 自由基(benzyl radical)。由 於此能量已有實驗值58,因此我們對這個重排反應做了計算,並在下一節討論其能 量。此重排反應的過程為: H. H2 C. H2C. CH2 H. 23. 30. 31. 圖 3-2 自由基重排反應示意圖. C(11)-H(12) C(11)-H(13) C(11)-C(3) H(8)-C(4) H(8)-C(11) C(3)-C(11)-H(12) C(3)-C(11)-H(13) C(11)-C(3)-C(4) C(11)-C(3)-C(4)-C(5). 結構 30 58.. McMillen, D. F.; Golden, D. M. Ann. Rev. Phys. Chem. 1982, 33, 493. 40. 1.090 1.090 1.501 1.438 1.427 118.7 118.7 98.7 180.0.
(48) 結構 30 是重排反應的過渡態,可視作是一個氫原子從甲基轉移到苯環的自由 基中心上。較值得注意的是 C(11)-C(3)-C(4)的角度=98.4º,顯示整個 CH3 基團往苯 環上的自由基中心拉近,同時氫原子與兩端碳原子的距離分別為 1.438 和 1.427Å。. H(13)-C(11) H(12)-C(11) C(3)-C(11) C(3)-C(11)-H(13) C(3)-C(11)-H(12) C(2)-C(3)-C(11)-H(12) C(4)-C(3)-C(11)-H(13). 1.083 1.083 1.404 121.0 121.0 0.0 0.0. 結構 31 結構 31 則為 benzyl 自由基,其結構為 C2V 對稱,自由基中心在 CH2 基團上。 此 平 面 結 構 和 Satink 等 人 59 於 2005 年 計 算 得 到 的 類 似 : 該 研 究 群 利 用. B3LYP/D95(d,p)計算此結構的紅外線光譜,並得到與實驗相當吻合的結果。 3-2. 反應高階理論計算位能面 圖 3-2 列出了以上所有結構的能量,以反應物鄰位硝基甲苯(1)的能量為零。每 個結構都有兩個能量值,一個是利用 G2M(cc,MP2)方法所得到的能量,另一個括號 中的數值是利用 B3LYP/6-311G(d,p)得到的能量總和(包括電子能與零點能)。能量值 的高度標示是以 G2M 方法得到的為準。為了避免太過雜亂,位能面上的各結構只 用編號表示。 由於 Il-ichev 等人 10 曾經對鄰位硝基甲苯的反應位能面做過計算,因此我們比 較了本研究及該研究群的研究中得到的能量值,其結果表列於表 3-1 當中。. 59.. Satink, R. G.; Meijer, G.; Helden, G. v. J. Am. Chem. Soc. 2003, 125, 15714. 41.
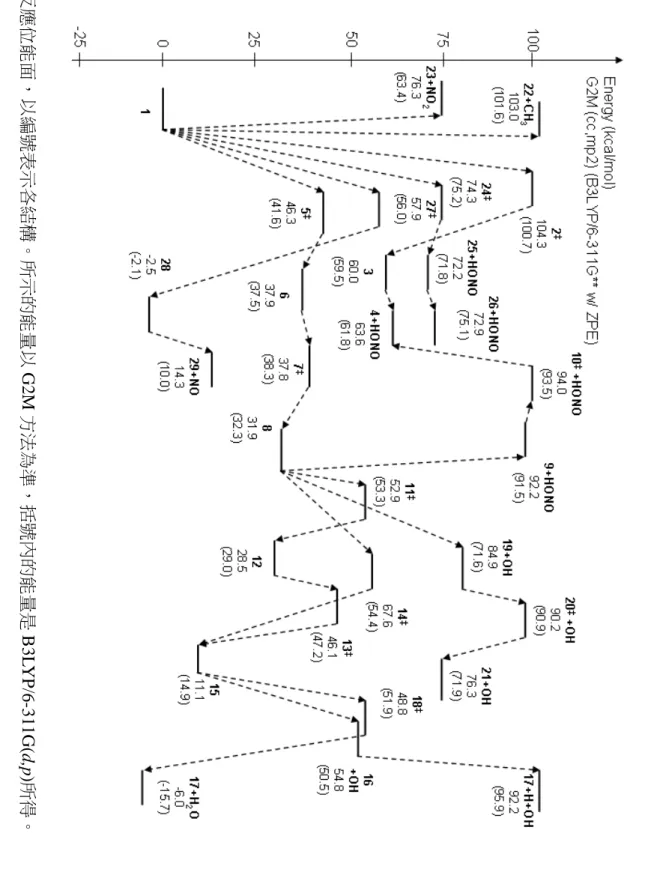
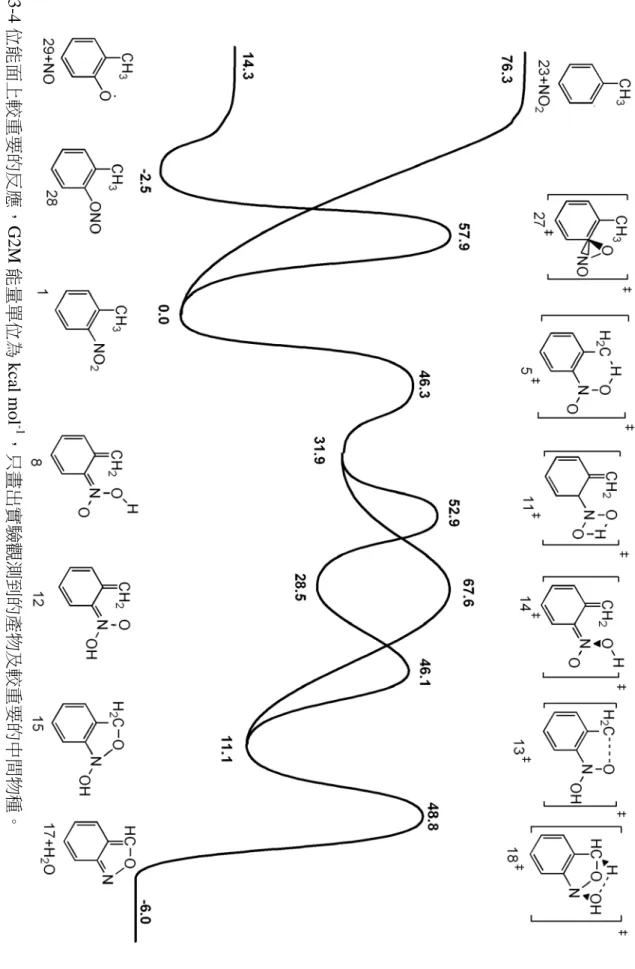
(49) 圖 3-3 反應位能面,以編號表示各結構。所示的能量以 G2M 方法為準,括號內的能量是 B3LYP/6-311G(d,p)所得。. 42.
(50) 圖 3-4 位能面上較重要的反應,G2M 能量單位為 kcal mol-1,只畫出實驗觀測到的產物及較重要的中間物種。. 43.
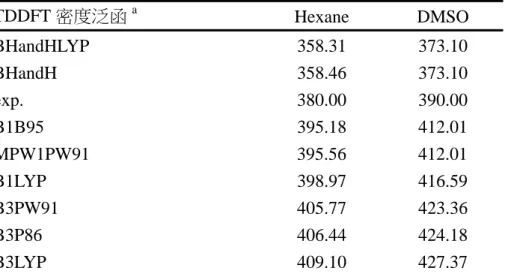
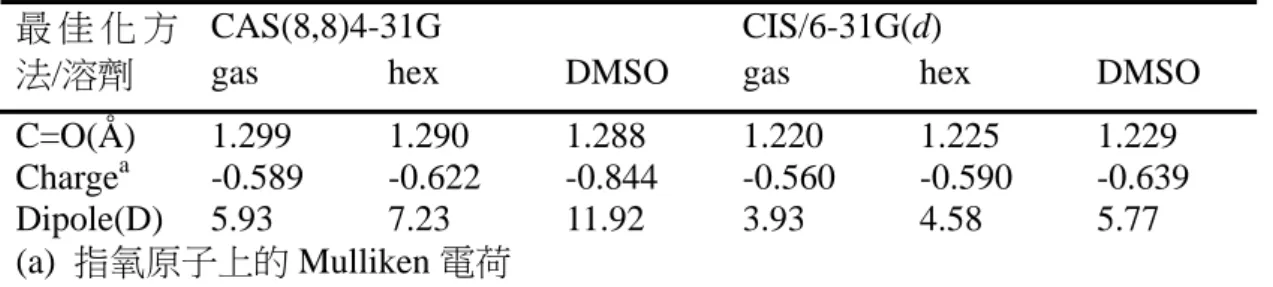
(51) 從表 3-1 中可以找到幾個趨勢。第一,G2M(cc, MP2)方法與 B3LYP/6-311G(d,p) 方法所得能量互有高低,在大部分的情形下差異約在 1~4 kcal mol-1 之間。這可以被 認為是由於 G2M 方法中使用了 MP2、MP4 等考慮電子相關作用(electron correlation) 的計算方法所導致的差異。另一方面,對於同是使用 B3LYP 方法的計算來說,. Il’ichev 等人. 10. 使用的基底函數較本研究中使用的基底函數大,而對於表中所有的. 結構來說,其得到的能量都比本研究中使用 DFT 方法得到的能量低大約 0.5~1.5 kcal. mol-1。其中值得注意的是 NO2 解離反應的產物:實驗值為 65.5±2.0 kcal mol-1,但 是要考慮的是該實驗在 110 torr 壓力下進行,其測得的反應熱很可能會因為壓力的 效應而產生誤差。 表 3-3 重要結構的理論計算能量與實驗值之比較 (kcal mol-1) DFTa G2Mb HFc DFTd 物種 5 41.6 46.3 41.2 6 37.5 37.9 36.4 7 38.3 37.8 36.6 8 32.3 31.9 43.1 31.4 11 53.3 52.9 51.7. 12 13 15 16+OH 17+H2O 23+NO2. 29.0 47.2 14.9 50.5 -6.0 63.4. 28.5 46.1 11.1 54.8 -15.7 76.3. 實驗值. 49.5e 51.5f. 27.4 45.8 13.4 42.8h -16.4 65.5±2.0g. (a) B3LYP/6-311G(d,p) in this work (b) G2M (cc, MP2) in this work (c) Chen, P. C.; Wu, C. W. J. Mol. Struct. (Theochem) 1995, 357, 87. HF/6-31G(d)//HF/6-31G(d) (d) Il’ichev, Y. V.; Wirz, J. J. Phys. Chem. A 2000, 104, 7856 B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d) Including ZPE scaled by 0.9806 (e) Tsang, W.; Robaugh, D.; Millard, W. G. J. Phys. Chem. 1986, 90, 5968. Pressure is 4 atm. (f) Matveev, V. G.; Dubikhin, V. V.; Nazin, G. M. Izv. Akad. Nauk SSSR, Ser. Khim. (in Russian) 1978, 474 (g) Gonzalez, A. C.; Lamon, C. W.; McMillen, D. F.; Golden, D. M. J. Phys. Chem. 1985, 89, 4809. Pressure is 110 torr. (h) Includes a leaving OH group in the structure. 對於一些具有未成對電子的物種(自由基)而言,G2M 與 DFT 方法所得到的能 量間就有比較大的差異,可以大到超過 10 kcal mol-1。為了瞭解在 G2M 複合方法中 44.
(52) 造成此現象的原因,我們列出 G2M 中各計算所得到的能量,並與 DFT 以及組合後 的 G2M 能量作一個比較。其結果如表 3-4 所列。 表 3-4 重要結構在 G2M 方法中各種計算的能量值 a. Geom.. DFTb. 5 6 7 8 11 12 13 15 16+OH 17 23+NO2. 41.6 37.5 38.3 32.3 53.3 29.0 47.2 14.9 50.5 -6.0 63.4. MP2b 83.7 44.7 48.1 42.7 67.2 39.4 56.1 17.0 91.4 -21.8 74.7. MP2c. MP4b. 82.5 42.2 45.1 38.8 63.2 35.1 52.5 15.1 94.5 -17.7 86.1. 64.8 43.9 44.8 38.9 63.3 36.0 52.8 16.2 60.8 -12.3 90.3. MP4d 64.9 42.4 43.8 38.8 63.9 35.8 52.6 17.0 63.1 -4.7 35.4. CCSDe 58.2 42.9 43.2 36.6 59.7 33.6 51.4 11.9 55.9 -14.8 75.3. G2M. S**f. 54.5 37.9 37.8 31.9 52.9 28.4 46.1 11.1 53.6 1.3709 -6.0 76.3 1.3789. (a) 所有能量皆以 kcal mol-1 為單位。若無註明,系統為自由基時列出 spin-projected unrestricted 方法所得能量 (b) PMP4(SDTQ)/6-311G(d,p),分別取 MP2 和 PMP4 能量 (c) MP2/6-311+G(3df,2p) (d) PMP4(SDTQ)/6-31G(d,p) (e) CCSD(T)/6-31G(d,p)。自由基時則為 UCCSD(T) (f) MP4(SDTQ)/6-311G(d,p)。僅列出自由基物種 幾乎對所有在表 3-4 中列出的物種來說,G2M 使用的方法都高估了能量,而在 這些方法中以 CCSD(T)與最後的 G2M 能量最為接近。在系統為一般分子時,G2M 使用各方法所得到的能量之間,除了 MP2/6-311G(d,p)與其他方法所得能量有較明 顯的差異外,大多數方法得到的能量與平均值的差異都在 9 kcal mol-1 之內。 而在自由基系統中,這些能量存在明顯的差異:在結構 16+OH 的情形中,兩 種 MP2 方法所得的能量較 G2M 的值高了約 30 kcal mol-1,而兩種 MP4 方法所得能 量則是高了 7~10 kcal mol-1。CCSD(T)所得能量與 G2M 能量的差異最小,約只高了. 1.5 kcal mol-1。 在結構 23+NO2 的情形中,除了 CCSD(T)之外,卻是以 MP2/6-31G(d,p)的能量 45.
數據
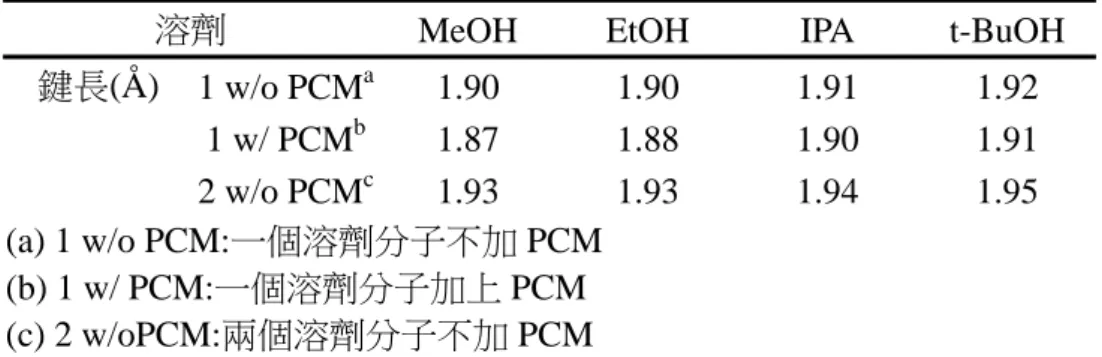
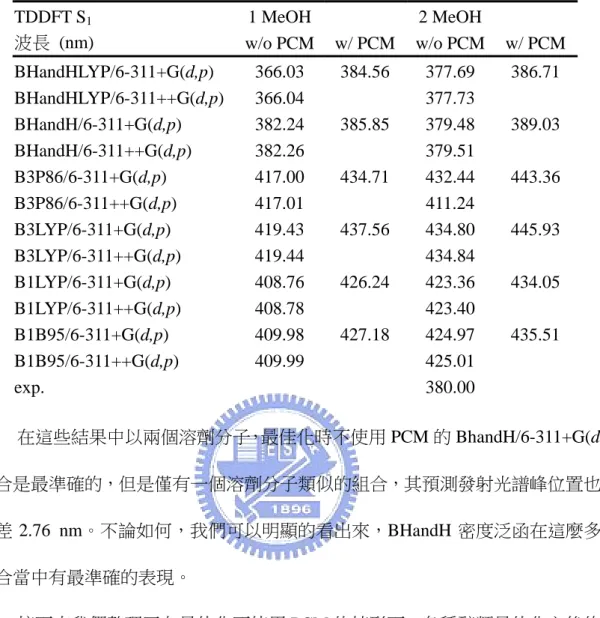
+7
Outline
相關文件
為降低藥品安全性與有效性試驗的成本與其耗費的時間, 合併第一期
LED。Wii remote 裏的光學感應器,可以根據這些 LED 成像的
本實驗測得的 pH 值與酸鹼度計測得的 pH 值差異不大,約 1%,證明了我們 也可以利用吸光值,來推算出溶液中不同分子的濃度,進而求得
因電導值與溶液中的離子濃度有關,滴定過程中溶液內的離子濃
觀念學習一 速率公式的變化 對應能力指標. 6-n-08
2-1 化學實驗操作程序的認識 探究能力-問題解決 計劃與執行 2-2 化學實驗數據的解釋 探究能力-問題解決 分析與發現 2-3 化學實驗結果的推論與分析
範圍:下學期第二次段考 科目:物理..
本研究為了將結構物內的牆以不同單位重來做比較,在計算每棟
