國 立 交 通 大 學
應用化學研究所
博 士 論 文
活性聚合具有環境刺激響應共聚高分子與
分析其組裝結構與環境敏感智能行為
Facile Synthsis of Well-defined Environment
Stimuli-responsives Coploymers and Investigation of its
Assembly Architecture and “Smart” Behaviors
研 究 生:黃 承 鈞
指導教授:張 豐 志 教授
中 華 民 國 九 十 九 年 七 月
活性聚合具有刺激響應共聚高分子
與分析其組裝結構與環境敏感智能行為
Facile Synthsis of Well-defined Stimuli-responsives Coploymers and
Investigation of its Assembly Architecture and “Smart” Behaviors
研 究 生:黃承鈞 Student:Cheng-Jyun Huang
指導教授:張豐志 Advisor:Feng-Chih Chang
國 立 交 通 大 學
應 用 化 學 研 究 所
博 士 論 文
A DissertationSubmitted to Department of Applied Chemistry College of Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy
In
Applied Chemistry July 2010
Hsinchu, Taiwan, Republic of China
I
活性聚合具有刺激響應共聚高分子
與分析其自組裝結構與環境敏感智能行為
學生:黃承鈞 指導教授:張豐志
國立交通大學應用化學研究所 博士班
摘
要
隨著奈米科技的發展以及對能源與能源應用上的需求,具有精確結構設計的功能性軟物 質材料已被廣泛的研究。近年來,活性高分子聚合技術的發展,使得我們可以得到具有 可控制分子量分佈、複雜結構與控制組成比例的高分子材料。自然界生物為了維持生命 與生理機能,生物體內的高分子與細胞必須隨著外在環境的變化來改變其化學性質與結 構。由此概念所發展出具有刺激響應的仿生高分子材料已運用在許多生醫方面的應用。 自然界生物為了維持生命與生理機能,生物體內的高分子與細胞必須隨著外在環境的變 化來改變其化學性質與結構。由此概念所發展出具有刺激響應的仿生高分子材料已廣泛 運用在生醫材料 本研究中,我們利用活性聚合出具有刺激響應共聚高分子,並且加以研究這些高分 子在固態或液態中的自組裝結構: (1) 活性聚合聚胜肽嵌段式共聚高分子 poly(N-isopropylacrylamide)-b-poly(Z-L-lysine) (PNIPAm-b-PZLys) 及其性質研究: 聚胜肽(polypeptide)嵌段共聚高分子是利用具有雙官能基起始劑,一端進行原子轉移自 由基聚合(atom transfer reversible polymerization)具有溫度響應的軟鏈段聚異丙基丙烯醯II 胺poly(N-isopropylacrylamide),另一端則進行開環反應聚合硬鏈段聚胜肽polylysine高分 子。藉由此種雙親性嵌段式共聚高分子的親疏水鏈段在極性上的差異,我們發現可以藉 由改變共聚高分子的組成以及共溶劑極性,在形成各種型態熱力學穩定的高分子微胞。 利用小角度(SAXS)、廣角度X光繞射儀(WAXS)與穿透式電子顯微鏡(TEM)觀察此種嵌段 共聚物的微相自組裝相分離結構。在移除胺基保護基苄氧羰基後,我們利用核磁共振儀 觀察此共聚物在改變環境溫度與pH值的刺激響應行為。 (2) 利用逐層組裝與點擊化學製備共價鍵穩定超薄殼層溫度敏感微膠囊中空球 本研究中,我們利用原子轉移自由基聚合聚合出具有疊氮以及炔基官能基的熱敏感聚異 丙基丙烯醯胺共聚物,接著利用表面改質疊氮官能基的二氧化矽微米球當作基板進行逐 層組裝並同時進行1,3-偶極環加成點擊化學反應形成共價鍵穩定的多層薄膜結構。利用 稀釋氫氟酸水溶液移除模板,我們可得到一個穩定型態具有可逆溫度響應的高分子中空 囊球。進一步藉由控制多層薄膜的交聯度以及反應溫度可以得到不同厚度及表面粗糙度 的高分子膠囊中空球。 (3) 利用逐層組裝與點擊化學製備共價鍵穩定雙刺激響應超薄高分子液胞 傳統上高分子液胞(polymer vesicles)是利用兩性嵌段共聚物在水溶液下相分離所產生的 自組裝結構。我們以第二部分研究作為基礎,進一步導入酸鹼敏感聚丙烯醯丙氨酸共聚 物,與熱敏感聚異丙基丙烯醯胺共聚物交替進行交替逐層組裝反應。我們藉由此技巧制 備出具有類似高分子液泡結構,並可控制尺度大小與殼-核層薄膜厚度。利用共軛焦顯 微鏡(CLSM)、TEM與原子力顯微鏡(AFM)可觀察此高分子液泡對溫度與酸鹼值之刺激響 應行為。
III
Facile Synthsis of Well-defined Stimuli-Responsives
Coploymers and Investigation of ts Self-Assembly
Architecture and “Smart” Behaviors
Studenet:Cheng-Jyun Huang Advisors:Dr. Feng-Chih Chang
Institute of Applied Chemistry
National Chiao Tung University
ABSTRACT
Applications for advanced functional soft materials that possess precisely engineered properties and functional groups have been expanding significantly with the development of nanotechnology and the growing need to address resource, health, and energy issues. Recent advances in living/controlled polymerization techniques have facilitated access to (co)polymers with controlled molecular weights, complex architectures, and precisely positioned functional groups. To sustain life and maintain biological function, nature requires selectively tailored molecular assemblies and interfaces that provide a specific chemical function and structure, and which change in their environment. Synthetic materials that change properties in response to local environmental stimuli with very similar attributes are often prepared for a broad range of biomedical applications.
In this study, we synthesized well-defined stimuli-responsive copolymers by controlled/living polymerization and investigated their assembled nanostructures in the
IV
solid-state or in solution:
(1) Polypeptide diblock copolymers: syntheses and properties of poly(N-isopropylacrylamide)-b-polylysine:
A hydrolysis-resistant amide-linkage hetero-functional initiator was synthesized and used successfully for polymerization of well-defined rod-coil block copolymers poly(N-isopropylacrylamide)-b-poly(Z-L-lysine) (PNIPAm-b-PZLys) by combination of atom
transfer radical polymerization (ATRP) and amine hydrochloride mediated ring-opening polymerization (ROP). These amphiphilic block copolymers are able to form universal micelle morphologies of spherical micelles, wormlike micelles, and vesicles by varying the polymer compositions and the helicogenic common solvents. From synchrotron SAXS, WAXS, and TEM results, the PNIPAm-b-PZLys microphase self-assembly morphology in solid state is a hierarchical lamellar-in-hexagonal structure. After removing the protective ε-benzyloxycarbonyl group, the dual stimuli-responsive behaviors of the PNIPAm-b-PLys investigated by nuclear magnetic resonance spectroscopy in aqueous solution resulted in either coil-to-helix or coil-to-globule transition by changing the environmental condition of elevating the temperature or increasing the pH value.
(2) Using click chemistry to fabricate ultrathin thermoresponsive microcapsules through direct covalent layer-by-layer (LbL) assembly
We report the syntheses of azido- and acetylene-functionalized PNIPAm copolymers and their use in the fabrication of ultrathin thermoresponsive microcapsules through direct covalent LbL assembly using click chemistry. These clickable copolymers were prepared through ATRP at 0 °C using a synthesized dansyl-labeled initiator and the CuBr/Me6TREN
catalyst complex in 2-propanol. These clickable PNIPAm copolymers assemble alternately onto azido-modified silica particles in aqueous media through click reactions catalyzed by copper sulfate and sodium ascorbate. After removing the template, the microcapsules
V
remained stable because of the presence of the covalently bonded triazole units; the microcapsules exhibited thermoresponsive and thermo-reversible swelling/de-swelling behaviors upon changing the temperature of the medium. Adjusting the number of clickable functionalities resulted in changes to the degree of cross-linking, thereby allowing control over the surface morphology and thickness of the covalently stabilized PNIPAm multilayer thin films. The microcapsules fabricated close to the lower critical solution temperature of PNIPAm exhibited extremely low surface roughnesses and thick multilayer films as a result of their compact chain conformation in aqueous solution, leading to tighter packing of the PNIPAm structure.
(3) Fabrication of vesicle-like dual-responsive click capsules by direct covalent LbL assembly
We report a click chemistry approach for the consecutive LbL assembly of thermo and pH-sensitive clickable copolymers on silica particles and the subsequent formation of a vesicle-like dual-responsive click capsules. This click capsules exhibit both thermo and pH-responsive behaviors by elevating the solution temperature and incubating in acidic or basic solutions respectively. These stimuli-responsive behaviors were examined by using confocal laser scanning microscopy (CLSM), TEM, and atomic force microscopy (AFM). This approach provides potential applications in preparing well-defined vesicle-like capsules with covalent stabilization and flexibility in introducing a range of new materials including different functional polymers.
VI
誌 謝
終於到了寫致謝詞的這一刻,這意味著研究所生涯即將正式的畫上句點。大學四年級上 學期甄試上交大應化研究所後,非常幸運的能進入張豐志老師的實驗室進行研究,開啟 了我對高分子研究的興趣。在研究所求學時期,非常感謝張豐志老師提供我們學生良好 的研究環境與實驗室互助合作的氣氛讓我能在研究上全力以赴,老師不但培養了我具備 邏輯與獨立思考的能力與自動自發的精神,讓我的研究成果能一點一滴的堆砌出今日的 成果。老師的耐心與愛心著實令學生備感溫馨,不僅讓學生在研究上得到豐富的知識, 亦是促使整體研究動力的來源,在此,學生要向您說聲謝謝。 論文口試期間,感謝口試委員: 謝國煌教授、段葉芳教授、吳震裕教授、黃華宗教 授與陳志勇教授在學生的論文上提供了非常寶貴的建議,讓學生的論文的內容更加詳實 與嚴謹,在此致上最誠摯的謝意。 在研究的過程中,首先要感謝的是小呆學姐帶我走入高分子合成與高分子自組裝的 領域,讓我能順利的找到我博士班的研究主題,讓我的研究順利步上軌道。還有跟我同 梯一起堅持到最後的小堅,高分子物理的疑難雜症通通全部都幫我解決。還要感謝實驗 室許多的學長姐,所謂前人種樹,後人乘涼,實驗室能有今天的良好的研究氣氛以及研 究設備也都是各位學長姐的心血結晶,希望我在畢業後也能對實驗室繼續有貢獻讓這傳 統能夠持續的維持下去,讓實驗室更加茁壯。也感謝小叮噹學妹在實驗上的幫助,以及 實驗室學長與學弟妹大家共同維持實驗室的運作以及整潔,讓這我們這團隊彼此能相互 合作,可以互相扶持,讓我能順利的度過豐富及多采多姿的研究生生涯。也要感謝各個 學校裡的貴重儀器技術人員,幫助我各式各樣的實驗,讓我有豐富的實驗數據來完成我 的實驗與論文寫作。 還要感謝陪我一起走過博士班四個年頭的女友曉詩,有了妳,讓我有更明確奮鬥的 目標,有妳的陪伴讓我有了奮發向上的精神,而妳也總是支持著我,給我最大的鼓勵。 希望未來我們也能在人生的路上繼續相互扶持當對方的心靈支柱。最後,要感謝的是我 的爸爸、媽媽與姊姊在學業與生活上對我全力的支持,讓我能順順利利的完成我的學 業,感謝我的爸爸、媽媽給了正確的人生態度,這是我最大的資產。 僅以這份論文,獻給在這一路上關心與照顧我的各位,謝謝大家。VII
Table of Contents
Pages
Chinese Abstract...I English Abstract ...III Acknowlegement...VI Table of Contents ... VII List of Schemes ...XI List of Tables... XII List of Figure ... XIII
Chapter 1 Introduction
1-1 Recent advances in the design of functional polymers from controlled/living radical
polymerization...1
1-2 Controlled/living radical polymerization (CLRP)...2
1-2.1 Atom transfer radical polymerization (ATRP) ...3
1-2.2 Reversible addition fragmentation chain transfer (RAFT) ...5
1-2.3 Nitroxide-mediated polymerization (NMP)...6
1-3 Click chemistry reactions employed in polymer science...8
1-3.1 Copper-Catalyzed Azide−Alkyne Cycloadditions (CuAAC) ...10
1-4 Polymeric capsules... 11
1-4.1 Polymersomes ...12
1-4.2 Polypeptide-based vesicles...15
1-4.3 Layer-by-layer vesicles ...17
1-5 Stimuli-responsive polymer materials...20
1-5.1 Stimuli-responsive solutions ...21
1-5.2 Temperature and pH responses...22
1-5.3 Electromagnetic-responsiveness ...23
1-5.4 Multiple-responsive systems...26
References ...27
Chapter 2 Polypeptide Diblock Copolymers: Syntheses and Properties of Poly(N-isopropylacrylamide)-b-Polylysine Abstract...36
VIII
2-1 Introduction ...37
2-2 Experimental Section ...40
2-2.1 Materials...40
2-2.2 Synthesis of 2-bromo-N-(2-hydroxyethyl)-2-methylpropionamide...40
2-2.3 Synthesis of phthalimidoethyl 2-bromo-2-methylpropionamide ...41
2-2.4 Preparation of phthalimide end-capped poly(N-isopropylacrylamide) by ATRP ...42
2-2.5 Hydrazinolysis of phthalimide end-capped PNIPAm to primary amine and amine hydrochloride-functionalized PNIPAm ...42
2-2.6 General procedure for synthesis of poly(N-Isopropylacrylamide-b-peptide) block copolymer by ROP polymerization ...43
2-2.7 Deprotection of the ε-benzyloxycarbonyl (Cbz-group) side chains in PNIPAm-b-PZLys...43
2-2.8 Preparation samples of block copolymer micelles assembled in water ...44
2-2.9 Preparation of polymer film ...44
2-2.10 Characterizations...44
2-3 Results and Discussion...46
2-3.1 Synthesis of amide linkage hetero-functional ATRP initiator ...46
2-3.2 Preparation of phthalimide end-capped poly(N-isopropylacrylamide)...46
2-3.3 Hydrazinolysis of phthalimide end-capped PNIPAm ...49
2-3.4 Synthesis of Diblock Copolymer ...50
2-3.5 Self-Assembly behavior of PNIPAm-b-PZLys diblock copolymer in aqueous solution ...52
2-3.6 Hierarchical Self-Assembly structure of PNIPAm-b-PZLys rod-coil block copolymer in solid state...54
2-3.7 Stimuli-responsive behavior of PNIPAm-b-PLysdiblock copolymer in aqueous solution ...55
2-4 Conclusion...56
References ...57
Chapter 3 Using Click Chemistry to Fabricate Ultrathin Thermoresponsive Microcapsules through Direct Covalent Layer-by-Layer Assembly Abstract...75
3-1 Introduction ...77
IX
3-2.1 Materials...82
3-2.2 Synthesis of Propargyl 2-Bromo-2-methylpropionamide ...83
3-2.3 Synthesis of 5-Dimethylaminonaphthalene-1-sulfonic Acid (3-Azidopropyl)amide (Dansyl-N3)...83
3-2.4 Synthesis of Dansyl-Labeled ATRP Initiator ...84
3-2.5 Synthesis 3-Azidopropylacrylamide ...84
3-2.6 Trimethylsilyl-Protected Acetylenic Acrylamide Copolymers {Poly[NIPAm-co-(trimethylsilyl)propargyl Acrylamide]} ...85
3-2.7 Acetylenic Acrylamide Copolymers {Poly(NIPAm-co-propargyl Acrylamide} ...85
3-2.8 Azido-Functionalized PNIPAm Copolymers {Poly(NIPAm-co-3-azidopropyl Acrylamide} ...86
3-2.9 Synthesis of 3-μm Azido-Modified Silica Particles...86
3-2.10 General Procedure for Direct Covalent LbL Assembly of PNIPAm Multilayer Thin Films on Azido-Modified Silica Particles ...87
3-2.11 Post-Functionalization of Multilayer Thin Films ...88
3-2.12 Characterizations...88
3-3 Results and Discussion...90
3-3.1 ATRP Synthesis and Characterization of Clickable Thermoresponsive PNIPAm Copolymers ...90
3-3.2 Click Reactions for the Direct Covalent LbL Assembly of Multilayer Thin Films on Silica Particles ...92
3-3.3 Effect of Cross-Linking Degree on Multilayer Thin Films Thickness and Morphology ...93
3-3.4 Tailoring Multilayer Thin Film Morphologies by Adjusting the Reaction Temperature ...95
3-3.5 Post-Functionalization of PNIPAm Clicked Thin Films Through Click Reactions With a Fluorogenic Small Molecule and Preliminary Permeability Study...96
3-4 Conclusion...99
References ...100
Chapter 4Fabrication of Vesicle-like Dual-responsive Click Capsules by Direct Covalent Layer-by-Layer Assembly Abstract... 118
X
4-1 Introduction ... 119
4-2 Experimental Section ...121
4-2.1 Materials...121
4-2.2 Synthesis of 2-(2-(2-azidoethoxy)ethoxy)ethanol...122
4-2.3 Synthesis of 2-(2-(2-azidoethoxy)ethoxy)ethyl methanesulfonate ...122
4-2.4 Synthesis of 2-(2-(2-(2-azidoethoxy)ethoxy)ethyl)isoindoline-1,3-dione ....122
4-2.5 Synthesis of 2-(2-(2-azidoethoxy)ethoxy)ethanamine ...123
4-2.6 Synthesis of N-(2-(2-(2-azidoethoxy)ethoxy)ethyl)acrylamide ...123
4-2.7 Preparation of Alkyne-functionalized Acrylamide Random polymer, Poly[NIPAm-r-propargyl acrylamide] (PNIPAm-r-PPAm) ...124
4-2.8 Preparation of Azido-functionalized Acrylamide Random polymer, Poly[NIPAm-r-N-(2-(2-(2-azidoethoxy)ethoxy)ethyl) acrylamide] (PNIPAm-r-PEOAm) ...125
4-2.9 Synthesis of Poly(N-acryloylalanine)-r-Poly(propargyl acrylamide) (PAAL-r-PPAm) ...126
4-2.10 Synthesis of poly(N-acryloylalanine)-r-Poly[N-(2-(2-(2-azidoethoxy)ethoxy)ethyl) acrylamide] (PAAL-r-PEOAm)...126
4-2.11 Procedure for Layer-by-Layer Multilayer Coating of Azide Modified Silica Particles ...127
4-3 Results and Discussion...128
4-4 Conclusions ...132
References ...133
Chapter 5 Conclusions ...145
List of Publications ...147
XI
List of Schemes
Pages
Scheme 2-1: Synthesis of phthalimidoethyl 2-bromo-2-methylpropionamide hetero-functional initiator ...61 Scheme 2-2: Synthesis of poly(N-isopropylacrylamide)-b-polypeptides...61 Scheme 3-1: Synthesis of the Fluorescent Amide-Linked Dansyl-Labeled ATRP Initiator 3
Using a Click Reaction; Preparation of Acetylene- and Azido-Functionalized PNIPAm Random Copolymers Using ATRP. ...106 Scheme 3-2: Schematic Representation of the Preparation of Covalently Stabilized
Thermoresponsive Microcapsules Through Layer-by-Layer Assembly Using Click Chemistry...107 Scheme 4-1: Synthesis of the N-(2-(2-(2-azidoethoxy)ethoxy)ethyl)acrylamide monomer ..137 Scheme 4-2: Synthetic Pathway for Preparation of Alkyne- and Azido-Functionalized
Poly(N-isopropylacrylamide) Random Copolymers via Atom Transfer Reversible Polymerization (ATRP). ...138 Scheme 4-3: Synthetic Pathway for the Aqueous Reversible Addition Fragmentation Chain
Transfer (RAFT) Polymerization of Alkyne- and Azido-functionalized Poly(N-acryloylalanine) Random Copolymer with and
4,4-Azobis(4-cyanopentanoic acid) V-501 as the Free Radical Initiator...138 Scheme 4-4: Schematic Representation of the Preparation of Covalently Stabilized Dual
XII
List of Tables
Pages
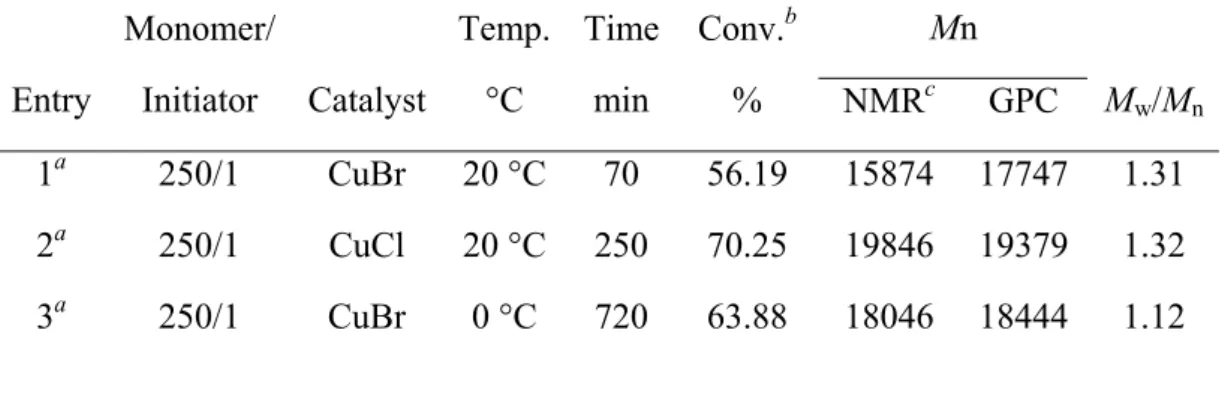
Table 2-1: Experimental Conditions and Properties of PNIPAm Prepared by ATRP: Effect of Catalyst and Temperature. ...62 Table 2-2: Results of Synthesis Diblock Copolymers PNIPA-b-PZLys and PNIPAm-b-PBLG
...63 Table 3-1: Characterization Data for the Acetylene- and Azido-Functionalized Copolymers.
...108 Table 4-1: Summary of Alkyne- and Azido-Functionalized Random Copolymers
XIII
List of Figure
Pages
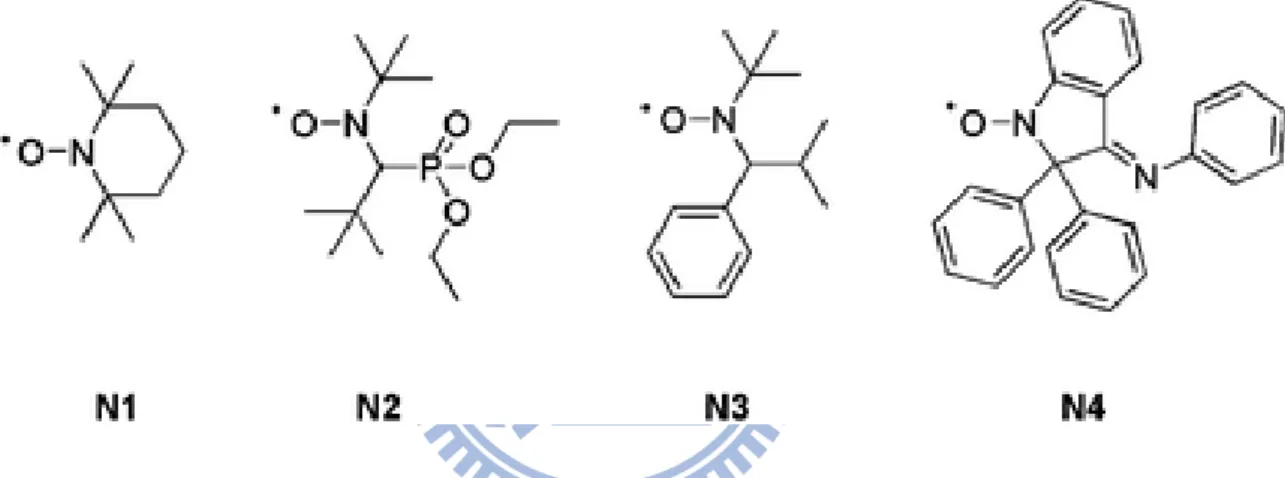
Figure 1-1: The activation–deactivation equilibrium in atom transfer radical polymerization..4 Figure 1-2: Mechanism of reversible addition fragmentation chain transfer. ...5 Figure 1-3: The activation–deactivation equilibrium in nitroxide-mediated polymerization. ...6 Figure 1-4: The structures of nitroxides used as mediators in NMP: TEMPO (N1), SG1 or
DEPN (N2), TIPNO (N3) and DPAIO (N4)...7 Figure 1-5: Examples of click reactions commonly employed in polymer synthesis and
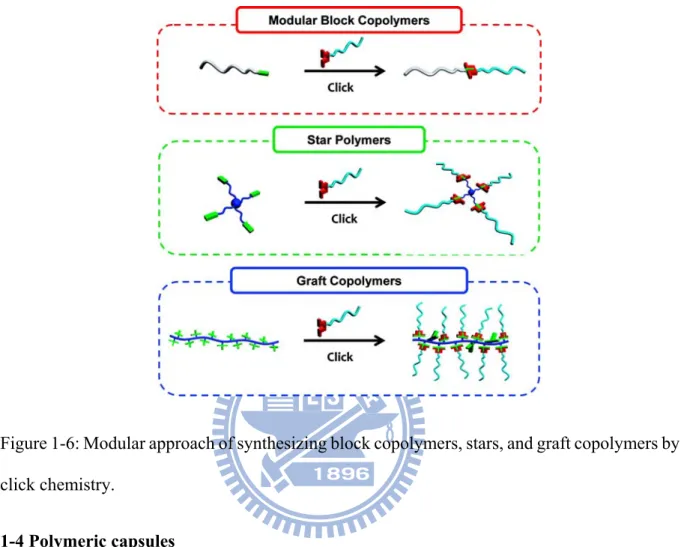
functionalization. ...9 Figure 1-6: Modular approach of synthesizing block copolymers, stars, and graft copolymers
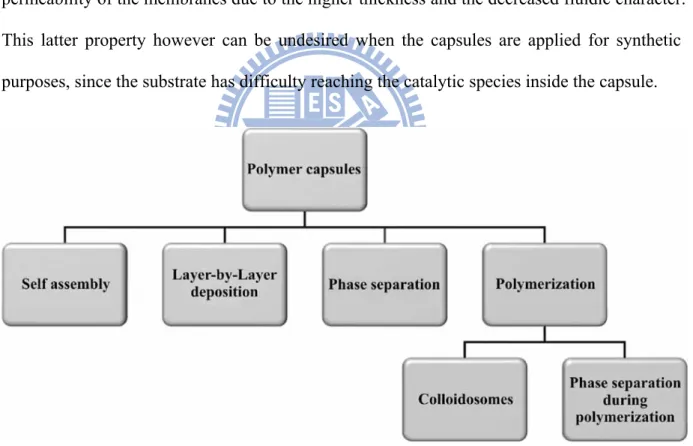
by click chemistry... 11 Figure 1-7: Overview of polymer capsules prepared via different methodologies. ...12 Figure 1-8: Different morphologies predicted by the packing parameter. For p < micelles
are predicted, vesicles are formed when < p < 1, and inverted structures are expected for p > 1. ...13 Figure 1-9: (a) Schematics of block copolymer hydrophilic fractions “f” with respective
cryogenic transmission electron microscopy images showing vesicles or worm micelles and spherical micelles. (b) Schematic scaling of polymersome membrane thickness with copolymer molecular weight (MW). ...14
Figure 1-10: Schematic representation of polyarginine-block-poly(L-leucine) with 10 mol-% randomly placed lysine residues in the arginine block that allow facile attachment of fluorescein dyes...16 Figure 1-11: Synthesis of a pair of oppositely charged block copolymers. These double
hydrophilic block copolymers can form polymersomes based on the electrostatic attraction between the oppositely charged blocks, forming polyion complexsomes, or PICsomes...17 Figure 1-12: (A) Schematic representation of the deposition of oppositely charged
polyelectrolytes. Steps 1 and 3 represent the adsorption of a polyanion and a polycation respectively. Steps 2 and 4 represent the washing steps. (B)
Polyelectrolytes adsorbed on the surface following the steps 1-4. (C) Chemical structure of poly(styrene sulfonate) (PSS) and poly(allylamine hydrochloride) (PAH), which are often used for LbL films. ...18
XIV
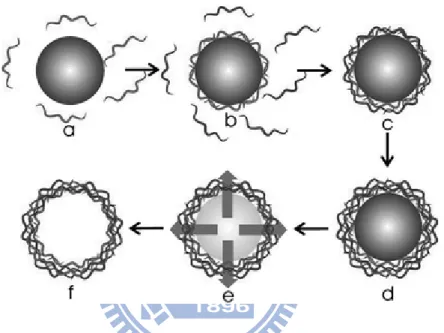
Figure 1-13: Schematic illustration showing the preparation of hollow polyelectrolyte capsules. The initial steps (a through d) involve stepwise film formation by repeated exposure of the colloids to polyelectrolytes of alternating charge.
Between each step the excess polyelectrolytes are removed before the next layer is deposited. When the desired number of polyelectrolyte layers is obtained the core is decomposed (e) resulting in a suspension of hollow polyelectrolyte capsules (f). ...19 Figure 1-14: Schematic representation of several steps during LbL encapsulation of a liquid
core. Explanation of abbreviations: DODAB: didodecyldimethylammonium bromide, PSS: poly(sodium 4-styrenesulfonate), PDADMAC:
poly(diallyldimethylammonium chloride)...20 Figure 1-15: Examples of molecular structures responsive to temperature (A) and pH (B)....23 Figure 1-16: Examples of molecular structures of photo-responsive monomers: cis–trans
isomer of azobenzene (A); ionization monomers (B) of leucos (B′) and spiropyran (B′′); and dimerization monomer of cinnamate (C)...24 Figure 1-17: Examples of molecular structures of photo-responsive monomers: Orientation
changes (A) and molecular structures (B) of liquid crystalline molecules...25 Figure 2-1: Kinetic plots for the polymerization of NIPAm at different temperature and
catalyzed by different copper catalyst. ...64 Figure 2-2: Molecular weights and polydispersities of PNIPAm as degree of conversion.
Experimental conditions are given in Table 1. ...64 Figure 2-3: Evolution of the molecular weight GPC traces in different polymerization
conditions (a) CuBr/20 °C, (b) CuCl/ 20 °C, and (c) CuBr/0 °C. ...65 Figure 2-4: 1H NMR spectrum of phthalimide end-capped poly(N-isopropylacrylamide) in
CDCl3 (*, CDCl3; #, water). ...66
Figure 2-5: GPC profiles comparison of two macroinitiators: (a) PNIPAm-NH2 dialyzed at
ambient temperature 20 °C and (b) PNIPAm-NH3+Cl- dialyzed in cold water at 5
°C...66 Figure 2-6: 1H NMR spectra of (a) amine hydrochloride-functionalized PNIPAm
macroinitiator and (b) amine-functionalized PNIPAm in D2O at 25 °C; inset shows
the amide methylene protons (f) and amine or amine hydrochloride methylene protons (g). ...67 Figure 2-7: FT-IR spectra of (a) HOBrPA, (b) PIBrPA, (c) Phthalimide end-capped PNIPAm,
XV
Figure 2-8: (a) GPC traces of monomer Z-Lys-NCA and PNIPAm90-b-PZLys90 (expt.2), (b)
GPC signals of PNIPAm90-b-PZLys38, PNIPAm90-b-PZLys71, and
PNIPAm-NH3+Cl- macroinitiator...68
Figure 2-9: GPC signals of PNIPAm-b-polypeptides and amine-functionalized PNIPAm macroinitiator. Ratio of reactants: [NIPAm]0/[NCA]0 = 1/100. ...69
Figure 2-10: 1H NMR spectra of (a) PNIPAm90-b-PZLys71 in dimethyl-d6 sulfoxide at 25 °C
and (b) PNIPAm90-b-PLys72 in D2O at 25 °C(*, dimethyl sulfoxide; #, water)..70
Figure 2-11: CPC traces of monomer BLG-NCA and PNIPAm90-b-PBLG90 (expt.4). ...71
Figure 2-12: GPC signals of PNIPAm90-b-PBLG37, PNIPAm90-b-PBLG55 and
PNIPAm-NH3+Cl-macroinitiator...71
Figure 2-13: TEM images of PNIPAm197-b-PZLys44 (a, b) and PNIPAm90-b-PZLys71 (c, d). (a)
a spherical micelles morphology, (b) a mixed spherical and wormlike micelles morphology, (c) a giant vesicles morphology, and (d) a compact vesicles
morphology...72 Figure 2-14: (a) SAXS and (b) WAXS patterns of the PNIPAm90-b-PZLys71 recorded at room
temperature. ...73 Figure 2-15: (a) TEM image of the PNIPAm90-b-PZLys71 lamellar phase separation
morphology. Note that polypeptide regions appear black due to staining with RuO4.
(b) Proposed schematic representation of the hexagonal-in-lamellar solid-state morphology of PNIPAm-b-PZLys block copolymer. ...73 Figure 2-16: (a) 1H NMR spectra recorded in D2O: (i) PNIPAm197-b-PLys44 at pH=13, (ii)
PNIPAm197-b-PLys44 at pH=7, and (iii) PNIPAm197-b-PLys44 at 45 °C. (b)
Proposed schematic representation of the PNIPAm197-b-PLys44 stimuli-responsive
behavior of the pH-induced coil-to-helix and thermo-induced coil-to-globule transitionin aqueous solution. ...74 Figure 3-1: 1H NMR spectra of (a) PNIPAm-Ace9.8 and (b) PNIPAm-Az10 in D2O at 20 °C.109
Figure 3-2: IR spectra of (a) PNIPAm-(TMS)Ace, (b) PNIPAm-Ace and (c) PNIPAm-Az. . 110 Figure 3-3: GPC traces of click-functionalized PNIPAm copolymer samples... 111 Figure 3-4: DSC thermorgrams of click-functionalized PNIPAm copolymers and a PNIPAm
homopolymer in aqueous solution (1 wt %; heating rate: 2 °C/min). ... 112 Figure 3-5: (a) Fluorescence intensity of 3-μm SiO2 particles plotted as a function of the
number of deposited PNIPAm-Ace9.8/PNIPAm-Az10 bilayers. (b) CLSM image of
XVI
bilayersin aqueous solution; the scale bar: 20 μm. (c) Tapping-mode AFM image of a collapsed PNIPAm microcapsule (dried state) featuring five
PNIPAm-Ace9.8/PNIPAm-Az10 bilayers; the AFM-derived thickness of the
multilayer was measured in terms of the difference in heights at the points marked by the arrows. (d, e) TEM images of (PNIPAm-Ace9.8/PNIPAm-Az10)5
microcapsules prepared in the (d) dried state and (e) thermally dried state (50 °C); the scale bar: 2 μm... 113 Figure 3-6: PNIPAm microcapsules multilayer thicknesses—measured using tapping-mode
AFM—plotted with respect to the number of bilayers, the cross-linking degree, and the reaction temperature... 114 Figure 3-7: AFM images of PNIPAm microcapsules fabricated at various temperatures and
with different compositions: (a) (PNIPAm-Ace22/PNIPAm-Az10)2 at 25 °C. (b)
(PNIPAm-Ace22/PNIPAm-Az22)2 at 20 °C. (c) (PNIPAm-Ace22/PNIPAm-Az10)1 at
30 °C... 114 Figure 3-8: (a, c) AFM and (b, d) TEM images of the (PNIPAm-Ace22/PNIPAm-Az10)2
microcapsules assembled closely to LCST at 30 °C. Samples prepared at 25 °C (a, b) and at 50 °C (c, d); the scale bar: 2 μm. ... 115 Figure 3-9: TEM images of the two-bilayer (PNIPAm-Ace22/PNIPAm-Az10)2 microcapsules
assembled at 30 °C after five heating/cooling cycles process. (a) swelling capsules prepared at 25 °C and (b) de-swelling capsules prepared at 50 °C; the scale bar: 5 μm... 115 Figure 3-10: CLSM images of (a, b) three-layer
(PNIPAm-Ace22/PNIPAm-Az22)PNIPAm-Ace22 and (c, d) four-layer
(PNIPAm-Ace22/PNIPAm-Az22)2-coated silica particles functionalized with
azido-modified lissamine rhodamine dye. The insets of panels (b and d) show photographs of vials containing lissamine rhodamine surface modified
PNIPAm-coated silica particles suspension. Excitation wavelength (a, c) 403 and (b, d) 561 nm; the scale bar: 20 μm... 116 Figure 3-11: CLSM and DIC images of the two-bilayer (PNIPAm-Ace22/PNIPAm-Az10)2
PNIPAm microcapsules assembled at 30 °C after mixing with probe molecule solutions of (a, d, g) rhodamine 6G and TRITC-dextran (b, e, h) Mw ~ 4.4 kDa,
(c, f, i) Mw ~ 6.5 – 7.6 kDa. Excitation wavelength (a, b, c) 403 nm and (d, e, f)
561 nm; the scale bar: 5 μm... 117 Figure 4-1: Chemical structures of the thermo and pH sensitive of (a, b) alkyne-functionalized
XVII
and (c, d) azido-functionalized random copolymers prepared by ATRP and RAFT polymerization...141 Figure 4-2: 1H NMR (500 MHz, D2O) spectrum of PNIPAm-r-PEOAm...141
Figure 4-3: 1H NMR (500 MHz, D2O) spectra of (a) PAAL-r-PAAm and (b) PAAL-r-PEOAm.
...142 Figure 4-4: Confocal laser scanning microscopy (CLSM) images of PNIPAm/PLAA/PNIPAm click capsules obtained from 5 μm diameter silica particles at (a) pH 3 and (b) pH 11. ...143 Figure 4-5: (a–c) AFM and (d–f) TEM images of the three-bilayer PNIPAm/PAAL/PNIPAm
click capsules. Samples prepared at 25 °C (a, b, d, e) and at 50 °C (pH 3) (c, f). The AFM images for scanned areas of 20 × 20 μm2. The scale bar in TEM images: 2μm...143 Figure 4-6: (a – d) AFM images and section analysis of the three-bilayer
PNIPAm/PAAL/PNIPAm click capsules in (a) pH 3, (b) pH 11, (c) at 50 °C (pH 3), and (d) at 50 °C (pH 11). ...144
1
Chapter 1
Introduction
1-1 Recent advances in the design of functional polymers from controlled/living radical polymerization
To sustain life and maintain biological function, nature requires selectively tailored molecular assemblies and interfaces that provide a specific chemical function and structure, and which change in their environment. Synthetic materials that change properties in response to local environmental stimuli with very similar attributes are often prepared for a broad range of biomedical applications. For example, physical or chemical hydrogels loaded with drug molecules may release their payload, only when and where required, in response to changes in the local environmental conditions, such as pH, temperature, presence of small molecules or enzymes, and oxidizing/reducing environment, among others.
Although Mother Nature provides many inspirations for designing and developing new materials, creating synthetic systems capable of responding to stimuli in a controllable and predictable fashion represents significant challenges. Particular challenges lie in mimicking biological systems where structural and compositional gradients at various length scales are necessary for orchestrated and orderly responsive behaviors.
Applications for these advanced stimuli-responsive soft materials that possess precisely engineered properties and functional groups have been expanding significantly with the development of nanotechnology and the growing need to address resource, health, and energy issues. Demand in a range of industrial and research settings are increasing the necessity to construct soft material systems with precise control over architecture, domain size, functionality, polarity, solubility, and reactivity. In addition to these materials and structural
2
requirements, researchers have also sought to prepare these systems via high yielding, simple covalent chemistry. Much of the inspiration for constructing such complex, multifunctional materials can be found in a combination of fundamental organic chemistry, biology, and bioconjugation chemistry; these motivations in tandem with the recent advances in controlled/living radical polymerization techniques have created a growing research area with concentration on robust, efficient, and orthogonal approaches for new soft material preparation.
1-2 Controlled/living radical polymerization (CLRP)
CLRP techniques have emerged as simple routes for preparing well-defined polymers of predetermined molecular weight (MW) and narrow molecular weight distribution (MWD), as well as various block copolymers and a wide range of complex macromolecular architectures.1 Prior to the development of CLRP, all these features were hardly achievable. Indeed, only with living ionic polymerization2 could such a degree of structural uniformity be reached but under very drastic polymerization conditions and with a limited choice of monomers.
In contrast to conventional free-radical polymerization where propagating radicals exhibit a very short lifetime ( 1 s) that hampers the conception of well-defined architectures, the concept of CLRP is to increase this lifetime up to the timescale of the polymerization reaction, via the establishment of a reversible equilibrium between active species, which can propagate, and dormant/capped species, which cannot propagate. CLRP then proceeds in such a manner that all polymer chains grow at the same rate and the detrimental impact of irreversible termination events over the MW and the MWD are made almost negligible.3 An ideal living polymerization system should exhibit the following features: (i) a linear evolution of the logarithmic conversion (ln [1/(1 − conversion)]) with time, accounting for a constant
3
propagating radicals concentration; (ii) a linear increase in the number-average molar mass, Mn, with monomer conversion, where the degree of polymerization, DPn, is predetermined by
the consumed monomer to initially introduced initiator molar ratio; (iii) low polydispersity indexes, Mw/Mn, close to a Poisson distribution (Mw/Mn ~1 + 1/DPn); (iv) a quantitative -
and -functionalization and (v) the possibility for polymer chains, after monomer consumption, to further grow when additional monomer is introduced which allows block copolymer synthesis to be performed by sequential monomer addition.3
A plethora of well-established CLRP methods offers the polymer chemist an impressive toolbox for making advanced macromolecular architectures of increasing complexity. Among them, nitroxide-mediated polymerization (NMP),1a atom transfer radical polymerization (ATRP)1b,1c,1f,1h and reversible addition-fragmentation chain transfer (RAFT),1d,1e,1j,1k the latter including macromolecular design via the interchange of xanthates (MADIX), represent the three most well-known CLRP techniques. In addition to these famous approaches, one can find the iniferter4 system and cyanoxyl-mediated polymerization5 as well as recently emerged
CLRP techniques, such as iodine transfer polymerization (ITP),6 single electron transfer-living radical polymerization (SET-LRP),7 organotellurium-mediated polymerization (TERP),1k organostibine-mediated polymerization (SBRP)1k and cobalt-mediated polymerization (CMRP).8
1-2.1 Atom transfer radical polymerization (ATRP)
ATRP was discovered independently by Sawamoto9 and Matyjaszewski.10 The ATRP process is based on a rapid exchange of a halide atom (especially Cl or Br) between a growing radical and a dormant species, via a redox process involving a transition metal complex as shown in Figure 1-1.
4
Figure 1-1: The activation–deactivation equilibrium in atom transfer radical polymerization. Various transition metals can be employed in ATRP (Cu, Ru, Fe, Ni, etc.). In the first ATRP process, called direct ATRP, the transition metal complex in a lower oxidation state (Mtn/Lm, where Mt is the metal and L is the ligand) is directly added to the reaction as an
activator and reacts reversibly with the dormant species (P-X, with X a halogen atom) to form a deactivator (Mtn+1X/Lm) and the active species P . In contrast, when the polymerization is
initiated by a conventional initiator and a metal complex at the higher oxidation state, the process is called reverse ATRP. The simultaneous reverse and normal initiation (SR&NI) process takes advantage of both normal and reverse ATRP as Cu(II) (which is tolerant to oxygen), an alkyl halide and a radical initiator are initially present in the reaction medium.11 It provides a way to reduce the amount of copper complex and to prepare more complex macromolecular architectures.
Recently, new ATRP processes have been developed, namely activators generated by electron transfer (AGET)12 and activators regenerated by electron transfer (ARGET).13 The AGET process employs a reducing agent (e.g. ascorbic acid or tin(II) 2-ethylhexanoate) which reacts with Mtn+1X/Lm to generate the active catalyst (Mtn/Lm). The process then
follows a direct ATRP process. AGET ATRP allows the preparation of pure block copolymers with no homopolymer of the second monomer. The ARGET process uses an excess of reducing agent which allows a significant reduction of the amount of metal in the media.
5
This powerful and versatile technique can be used under mild experimental conditions and in various polymerization media with a wide range of monomers including styrenics, alkyl (meth)acrylates, acrylonitrile, (meth)acrylamides as well as water-soluble monomers to give tailor-made polymers. An additional flexibility is provided by the possibility of using commercially available functionalized initiators and to functionalize chain ends. However, if ATRP can be used with a large range of monomers, the polymerization of functional monomers bearing acid or amine function remains hardly achievable.
1-2.2 Reversible addition fragmentation chain transfer (RAFT)
RAFT polymerization is governed by a reversible transfer reaction between a growing (macro)radical (active species) and a (macro)RAFT agent (dormant species).14 RAFT agents,
denoted Z–C( S)SR, act as transfer agents by a two-step addition-fragmentation mechanism as shown in Figure 1-2.
Figure 1-2: Mechanism of reversible addition fragmentation chain transfer.
The RAFT group is typically a thiocarbonylthio group such as dithioester (Z = alkyl), trithiocarbonate (Z = S-alkyl), xanthate (O-alkyl) or dithiocarbamate (Z = N(alkyl)2). The
RAFT process using thiocarbonylthio compounds, including dithioesters and trithiocarbonates, was reported by the CSIRO laboratory in early 199815 whereas a similar process using
xanthates as RAFT agents (the so-called MADIX) was reported in late 1998.16 RAFT is potentially universal and can be applied to a wide range of functional monomers (styrenics,
6
alkyl (meth)acrylates, acrylic acid, vinyl acetate etc.), which allows polymers with precisely controlled structural parameters to be prepared such as random, block, gradient, grafted and star copolymers.1d,1j,1k
Even though Moad, Rizzardo and co-workers recently succeeded in elaborating a switchable RAFT agent,17 one of the major drawback of this technique was the lack of a
universal RAFT agent. In particular, dithioesters or trithiocarbonates were suitable for controlling polymerization of more activated monomers such as styrene (S) and derivatives, methacrylic esters (e.g. methyl methacrylate MMA), methacrylic acid (MA), methacrylamide (MAM), acrylic acid (AA), acrylamide (AM) or acrylonitrile (AN). However they inhibit or retard the polymerization of less activated monomers such as vinyl acetate (VAc),
N-vinylpyrrolidone (NVP) or N-vinylcarbazole (NVC), for which xanthates or
dithiocarbamates are more suitable.1d,1j,1k The choice of R and Z groups is thus crucial to achieve a good control of the polymerization.
1-2.3 Nitroxide-mediated polymerization (NMP)
NMP is based on a reversible termination reaction between a growing radical, P., and a free nitroxide, N., to form a (macro)alkoxyamine, P–N (Figure 1-3).
7
This equilibrium between active and dormant species presents the advantage of being a purely thermal process: the (macro)alkoxyamine regenerates the propagating radical and the nitroxide by homolytic cleavage at high temperature (usually > 70 °C).
A typical NMP can be initiated following two different pathways: (i) by using a bicomponent initiating system (i.e. a conventional radical initiator and a free nitroxide) or (ii)
via a monocomponent initiating system (i.e. a preformed alkoxyamine). Georges and
co-workers first reported the controlled radical polymerization of styrene with (2,2,6,6-tetramethylpiperidinyl-1-oxy) (TEMPO, N1, Figure 1-4) as the mediator.18
Figure 1-4: The structures of nitroxides used as mediators in NMP: TEMPO (N1), SG1 or DEPN (N2), TIPNO (N3) and DPAIO (N4).
However, as TEMPO was almost exclusively limited to styrenic monomers (only a sterically hindered TEMPO derivative allowed the control of the n-butyl acrylate polymerization),19 new acyclic nitroxides have been designed to improved the range of polymerizable monomers under controlled/living conditions. More precisely,
N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide (SG1 or DEPN, N2,
Figure 1-4)20 and N-tert-butyl-N-[1-phenyl-2-(methylpropyl)]nitroxide (TIPNO, N3, Figure 1-4)21 are now able to control the polymerization of styrenics, alkyl acrylates, acrylic acid, acrylamides and dienes.20a,21a,22 The polymerization of methacrylic esters can be controlled
8
either: (i) by using a particular nitroxide such as 2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl nitroxide (DPAIO, N4, Figure 1-4),23 specific to methacrylates or (ii) by a copolymerization approach under SG1 control with a small amount of comonomer such as styrene24 or acrylonitrile.25
1-3 Click chemistry reactions employed in polymer science
Recent advances in living/controlled polymerization techniques have facilitated access to (co)polymers with controlled molecular weights, complex architectures, and precisely positioned functional groups. However, even the most robust polymerization methods are not sufficient for the synthesis of many interesting macromolecules. Postpolymerization modification is still an essential method of incorporating functionality not compatible with polymerization, characterization, or processing conditions. Polymer−polymer conjugation is often the only viable means to prepare complex chain topologies or copolymers that contain monomer units not polymerizable by the same methods. However, transformations on polymers are often inefficient and may lead to side reactions with other groups within the polymer. Therefore, efficient and specific reactions are needed to ensure successful postpolymerization modification.
As usual, Nature provides inspiration by example, since many natural molecules of the most sophisticated function, such as proteins and nucleic acid, are made by the seemingly simple repetition of reliable bond-forming operations. In 2001, a group of chemists led by K. Barry Sharpless at The Scripps Research Institute gave the name of click chemistry to the very best chemical reactions.26 They are easy to perform, give rise to their intended products in very high yields with little or no byproducts, work well under many conditions, and are unaffected by the nature of the groups being connected to each other. Click reactions share the following attributes:
9
(1) Many click components are derived from alkenes and alkynes, and thus ultimately from the cracking of petroleum. Carbon–carbon multiple bonds provide both energy and mechanistic pathways to be elaborated into reactive structures for click connections.
(2) Most click reactions involve the formation of carbon–heteroatom (mostly N, O, and S) bonds. This stands in contrast to the march of modern synthetic organic chemistry, which has emphasized the formation of carbon–carbon bonds.
(3) Click reactions are strongly exothermic, either by virtue of highly energetic reactants or strongly stabilized products.
(4) Click reactions are usually fusion processes (leaving no byproducts) or condensation processes (producing water as a byproduct).
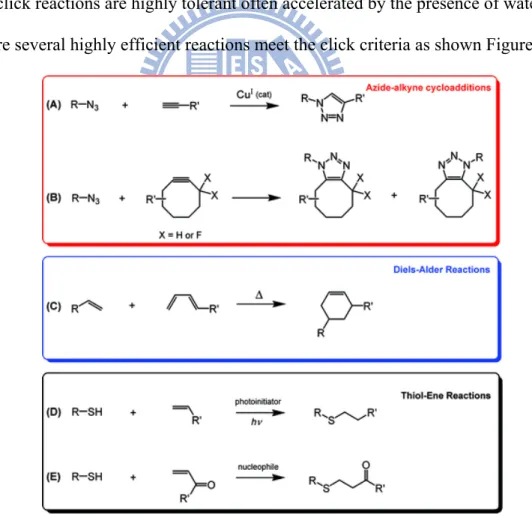
(5) Many click reactions are highly tolerant often accelerated by the presence of water. There are several highly efficient reactions meet the click criteria as shown Figure 1-5.
Figure 1-5: Examples of click reactions commonly employed in polymer synthesis and functionalization.
10
1-3.1 Copper-Catalyzed Azide−Alkyne Cycloadditions (CuAAC)
In 2002, the groups of Meldal27 and Sharpless28 independently reported the use of a copper(I) catalyst to allow azide−alkyne cycloadditions to be conducted at low temperatures with high rates, efficiency, and (regio)specificity (Figure 1-5A). This coupling process reaches near-quantitative conversion in both aqueous and organic media with little or no side reactions being observed. The efficiency afforded by CuAAC has influenced the field of macromolecular engineering in many ways. For example, block copolymer synthesis commonly relies on living/controlled polymerization methods to grow one block from another; however, many click reactions have proven capable of coupling preexisting homopolymers to prepare block copolymers in a modular and highly efficient manner (Figure 1-6). Several benefits arise from employing a modular method of this type. Each block can be individually characterized prior to coupling, and block copolymers can be prepared from monomers that do not polymerize by a common mechanism. Opsteen and van Hest first demonstrated it was possible to prepare a library of AB29 or ABC30 block copolymers by coupling various combinations of azido- or alkyne-terminated polymers by CuAAC. Barner-Kowollik and Stenzel et al.31 successfully coupled RAFT-generated polymers by a similar approach. Macromolecular architectures of greater intricacy have been prepared in an analogous manner, with CuAAC facilitating efficient preparation of various types of graft32 and star33 copolymers via modular grafting-to approaches.
While the large majority of reports to date have relied on CuAAC to enhance macromolecular engineering capabilities, several other highly efficient reactions also meet the click criteria.34 Additional cycloaddition reactions such as strain-promoted azide−alkyne coupling35 (SPAAC) (Figure 1-5B) and Diels−Alder reactions36 (Figure 1-5C) have allowed many new polymers to be efficiently prepared or functionalized. Thiol−ene reactions37 (radical- or nucleophile-mediated) have proven particularly useful for polymer synthesis
11
under extremely mild conditions, often with no solvent and little-to-no product cleanup (Figure 1-5D,E).
Figure 1-6: Modular approach of synthesizing block copolymers, stars, and graft copolymers by click chemistry.
1-4 Polymeric capsules
Nature serves as the model for stimuli-responsive polymers, but it remains a challenge to create materials that interact with, or respond to, biological environments.38 A typical cell in Nature contains several types of organelles. These compartments, which are mostly surrounded by a single bilayer phospholipid membrane, each fulfill a unique role within the cellular environment. Different organelles comprise different collections of specific enzymes that catalyze requisite chemical reactions. The process of compartmentalization to ensure the integrity of catalytic pathways has inspired chemists to mimic Nature and create artificial environments for reactions to take place in.
12
In recent years a different type of vesicular architecture, composed of macromolecules instead of small organic compounds, has become topic of investigation. A wide variety of polymeric capsules has been developed, such as polymersomes,39 layer-by-layer capsules,40 and hollow microspheres,41 or hollow microcapsules.42 (Scheme 1-7) The use of polymers instead of organic molecules to construct the membrane makes these systems more stable and robust compared to liposomes. Additionally a vast amount of monomers as well as many different techniques to build up the polymeric structures are available; it is better possible to tune the properties of polymeric capsules for the desired purpose. Another effect of the use of polymers instead of small phospholipids for the build-up of the capsule shell is the reduced permeability of the membranes due to the higher thickness and the decreased fluidic character. This latter property however can be undesired when the capsules are applied for synthetic purposes, since the substrate has difficulty reaching the catalytic species inside the capsule.
Figure 1-7: Overview of polymer capsules prepared via different methodologies.
1-4.1 Polymersomes
Polymer vesicles, commonly called polymersomes, are spherical shell structures in which an aqueous compartment is enclosed by a bilayer membrane made of amphiphilic block
13
copolymers. Because of the high molecular weight of these block copolymers, polymer vesicles have superior stability and greater toughness43 than liposomes, their low molecular weight analogues. More interestingly, their membrane properties can be extensively tailored using polymer chemistry (e.g. variation of chemical structure44 and length of each polymer component,45 crosslinking,46 conjugation with biomolecules such as membrane proteins,47 etc).
For the aggregation of small amphiphiles such as phospholipids, Israelachvili developed a model which is based on the geometry of the molecules.48 A packing parameter is defined in this model, which is determined by the surface area of the polar headgroup and the length and volume of the hydrophobic tails as depicted in Figure 1-7. This packing parameter can predict whether the formation of micelles, vesicles or inverted structures is expected.
Figure 1-8: Different morphologies predicted by the packing parameter. For p < micelles are predicted, vesicles are formed when < p < 1, and inverted structures are expected for p > 1.
14
This theory was adapted by Discher et al. for block copolymers with hydrophilic and hydrophobic character. They introduced a theory in which the effective interaction energy between hydrophobic blocks is the key to aggregate stability, whereas the relative mass or volume fraction of each block is the key to morphology (Figure 1-9).49 The molecular shape of an amphiphile, whether cylinder, wedge or cone shape, dictates whether membrane, cylindrical, or spherical morphologies will respectively form. This shape is simply a reflection of the hydrophilic fraction f. If large hydrophilic blocks are used, f 50%, this leads to conically shaped amphiphiles, which give micellar structures. When a hydrophilic fraction of 40% > f > 50% is used, wedge shaped amphiphiles are self-assembled into rod-like aggregates. For vesicles it is ideal to have a hydrophilic fraction of 25% > f > 40%, in which the amphiphile has a cylindrical shape and assembles into a vesicular morphology.
Figure 1-9: (a) Schematics of block copolymer hydrophilic fractions “f” with respective cryogenic transmission electron microscopy images showing vesicles or worm micelles and spherical micelles. (b) Schematic scaling of polymersome membrane thickness with copolymer molecular weight (MW).
Polymer vesicles have mostly been produced from three families of amphiphilic block copolymers-regular (coil-coil) block copolymers, block copolymers with rigid blocks (rod-coil and rod-rod) and block copolymers with intermolecular interactions (charge
15
interactions, ligand binding, H-bonds, dipolar interactions, etc).50 Of these families, coil-coil block copolymers have been the most studied. In dilute solution, the shape and size of the supramolecular assemblies of coil-coil block copolymers, such as poly(acrylic acid)-b-polystyrene (PAA-b-PS)51 and poly(ethylene oxide)-b-polybutadiene (PEO-b-PBD),52 are typically determined by the respective lengths of the polymer blocks, their affinity for each other and for the solvents, the temperature, and also the ionic concentration in the case of charged systems.53
1-4.2 Polypeptide-based vesicles
Besides the well known vesicles that are composed of synthetic block copolymers, vesicles also have been prepared, which consist solely of polypeptides as the hydrophilic and hydrophobic block, or which contain one block that is comprised of a polypeptide structure. This class of peptide containing polymersomes has recently drawn much attention, since new methods have emerged in which the molecular weight of block copolypeptides can be precisely controlled and increasingly complex polypeptides can be synthesized yielding polymers having superior properties than ill-defined homopolypeptides.54
Deming et al. showed vesicle formation using either poly(L-lysine)-block-poly(L-leucine) or poly(L-glutamic acid)-block-poly(L-leucine) diblock copolypeptides.55 These vesicles could be prepared with diameters ranging from 50 to 1000 nm and which retained their polar contents without leakage. Recently they reported on a polypeptide block copolymer56 with a small variation to the previously described polypeptides. Polyarginine-block-poly(L-leucine), depicted in Figure 1-10, has an α-helical hydrophobic block which favors the formation of flat membranes and a highly charged hydrophilic segment, which increases membrane fluidity.
Vesicles could also be prepared from hybrid polymer-polypeptide systems. The formation of spherical micelles and large vesicles ( peptosomes ) in an aqueous solution of a block
16
copolymer of polybutadiene-block-polyglutamate was described by Kukula et al.57 Also Checot et al. reported vesicular aggregates formed by polybutadiene-block-polyglutamate and showed the occurrence of a transition of the hydrophilic block from an α-helical conformation to a random coil conformation.58
Figure 1-10: Schematic representation of polyarginine-block-poly(L-leucine) with 10 mol-% randomly placed lysine residues in the arginine block that allow facile attachment of fluorescein dyes.
Kataoka et al. developed a new entity of hybrid peptide based polymer vesicles with a polyion complex (PIC) membrane, so called PICsomes.59 These PICsomes (Figure 1-11) were prepared by mixing the positively charged poly(ethylene glycol)-block-poly[(5-aminopentyl)-α, β-aspartamide] [PEG-P(Asp-AP)] with the negatively charged poly(ethylene glycol)-block-poly(α, β-aspartic acid) [PEG-P(Asp)] in a Tris-HCl buffer and subjecting them to sonication. By matching the chain length of block copolymer pairs with oppositely charged segments and compensating for the counter charge in a stoichiometric manner, semipermeable vesicles were formed.
17
Figure 1-11: Synthesis of a pair of oppositely charged block copolymers. These double hydrophilic block copolymers can form polymersomes based on the electrostatic attraction between the oppositely charged blocks, forming polyion complexsomes, or PICsomes.
1-4.3 Layer-by-layer vesicles
In the beginning of the nineteen nineties the layer-by-layer (LbL) technique was introduced by Decher and was originally used for sequential adsorption of oppositely charged polymers, polyelectrolytes, on a charged surface.60 When a polyelectrolyte is adsorbed on a glass surface, the charge of the polymer overcompensates, leading to a reversal of the surface charge, thereby promoting the adsorption of the next oppositely charged polyelectrolyte. This procedure is schematically illustrated in Figure 1-12.
Currently a large number of components, other than charged polymers, have been included in between the multilayers, like DNA, proteins, nanoparticles, lipids and viruses, yielding thin films with tailor made properties. Besides electrostatic interactions, other interactions such as H-bonds, covalent bonds, biospecific interactions and stereocomplex formation have also been used to build up these multilamellar systems.40a
18
Figure 1-12: (A) Schematic representation of the deposition of oppositely charged polyelectrolytes. Steps 1 and 3 represent the adsorption of a polyanion and a polycation respectively. Steps 2 and 4 represent the washing steps. (B) Polyelectrolytes adsorbed on the surface following the steps 1-4. (C) Chemical structure of poly(styrene sulfonate) (PSS) and poly(allylamine hydrochloride) (PAH), which are often used for LbL films.
In the late 1990s Möhwald et al.61 extended the LbL technique to polymeric capsules by depositing polyelectrolytes onto charged colloidal particles as templates. Various multilayers can be formed with a variety of constituents. The process for assembly of these multilayers is depicted in Figure 1-13. For the fabrication of hollow polyelectrolyte capsules, a template is used which can be removed later. First PS or cross-linked melamine formaldehyde (MF) microcapsules were used as core material. However, the MF capsules proved difficult to remove after the formation of the multilayer and in case of PS an osmotic pressure could be built up in the capsule due to the fast dissolution of the polymer core, which could destroy the
19
polymeric shell. To overcome these problems inorganic substrates such as CaCO3, MnCO3
and CdCO3 are now used more frequently.62 These inorganic carbonates have the advantage
that upon dissolution with an ethylenediamine tetraacetate (EDTA) solution the metal ions are complexed and can pass the membrane of the polyelectrolyte shell. These capsules are known for their permeability of molecules with a molecular weight below 5 kDa and should therefore have no osmotic stress.63
Figure 1-13: Schematic illustration showing the preparation of hollow polyelectrolyte capsules. The initial steps (a through d) involve stepwise film formation by repeated exposure of the colloids to polyelectrolytes of alternating charge. Between each step the excess polyelectrolytes are removed before the next layer is deposited. When the desired number of polyelectrolyte layers is obtained the core is decomposed (e) resulting in a suspension of hollow polyelectrolyte capsules (f).
These layer-by-layer techniques describe above using solid particles in an aqueous medium. A novel development is the layer-by-layer deposition on a liquid core. Grigoriev et al. have recently described a general method for the encapsulation of a broad range of emulsions comprised of various hydrophobic substances and oil-soluble compounds.64 To an
20
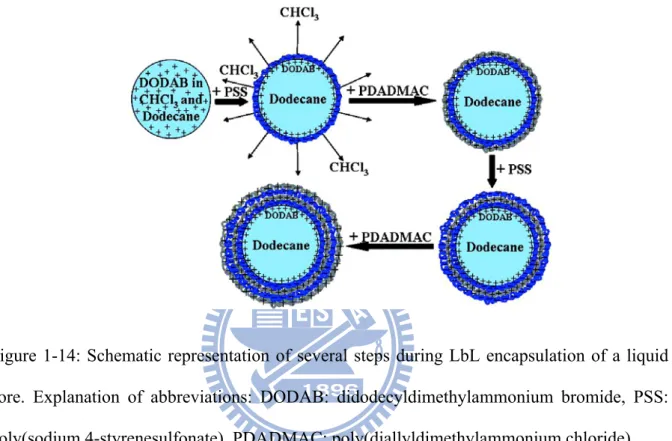
emulsion of didodecyldimethylammonium bromide (DODAB), chloroform and dodecane in water was gently added a polyelectrolyte solution. After deposition of a layer, a washing step was carried out, before the next layer was deposited, resulting in microcapsules as shown in Figure 1-14.
Figure 1-14: Schematic representation of several steps during LbL encapsulation of a liquid core. Explanation of abbreviations: DODAB: didodecyldimethylammonium bromide, PSS: poly(sodium 4-styrenesulfonate), PDADMAC: poly(diallyldimethylammonium chloride).
1-5 Stimuli-responsive polymer materials
The human body is fascinating for its ability to respond to its environment from the molecular to the macroscopic level. Stimuli response is crucial for maintaining normal function as well as fighting disease. At the molecular level, for example, the body releases insulin to initiate glycogen formation in response to higher glucose levels in the blood. Molecular interactions also take place at cellular surfaces to stimulate a number of events including cell signalling and endocytosis. At the macroscopic level, the body responds to external stimuli with a cascade of events, such as when nerve cells transmit signals to the brain in response to a pain-causing stimulus and subsequently cause muscle contraction. These examples have
21
inspired scientists to fabricate 'smart' materials that respond to light, pH, temperature, mechanical stress or molecular stimuli. Synthetic polymeric materials capable of responses to external or internal stimuli represent one of the most exciting and emerging areas of scientific interest and unexplored commercial applications. While there are many exciting challenges facing this field, there are a number of opportunities in design, synthesis, and engineering of stimuli-responsive polymeric systems and Mother Nature serves as a supplier of endless inspirations.
1-5.1 Stimuli-responsive solutions
Stimuli-responsive behavior is easily obtained in polymeric solutions as the Brownian motion of solvent molecules requires relatively low energies for macromolecular segments to displace solvent molecules. For an ideal system, the kinetic energy required for the Brownian motion at room temperature is 1.5kBT (where the kB is Boltzman constant (1.38 × 10−23 J/K), and T is
the temperature).One example of these transitions in polymeric solutions is lower critical solution temperature (LCST), which is the lowest temperature of phase separation on the concentration–temperature phase diagrams. Below LCST, polymer chains and solvent molecules are in one homogenous mixing phase, and exhibit favorable free energy (ΔG < 0), which is believed to be facilitated by hydrogen-bonding interactions between the two phases. Above LCST, a phase separation occurs as enthalpic (ΔH) energy overcomes the entropic (ΔS) contributions resulting in unfavorable free energy (ΔG > 0) of the entire system. For the majority of polymeric solutions65 temperature induced LCST transitions result in the particle or aggregate size decreases above LCST, and the reported size changes are substantial, typically in the range from 250 to 3000 nm below LCST, to 100 to 1000 nm above.
22
1-5.2 Temperature and pH responses
Energetic rearrangements are driven by molecular entities capable of responsiveness to stimuli in a given environment. For polymeric solutions to exhibit stimuli-responsiveness, it is necessary to provide suitable molecular building blocks capable of responses. Figure 1-15, A and B, illustrates several examples of temperature and pH-responsive monomeric blocks which, upon polymerization maintain stimuli-responsiveness. A well known polymer with the LCST behavior is poly(N-isopropylacrylamide) (PNIPAm)65a,b,c which exhibits coil-to-globule phase transition at 32 °C. Poly(N-vinylcaprolactone) (PVCL),65d,e,f poly(N-(DL)-(1-hydroxymethyl) propylmethacrylamide) (p(DL)-HMPMA),66 and
poly(N,N′-diethylacrylamide) (PDEAAm)67 are also temperature-responsive, and their LCSTs
are about 32, 37, 33 °C, respectively. Thus, molecular designs of a polymer backbone allow one to control temperature at which a given system is responsive. It is well established that the LCST phase transition is a nanometer scale event, where the particle or aggregate dimensions change.68 However, for individual polymer chains, the coil-to-globule transitions can be thermodynamically controlled by adjusting polymer compositions,69 as determined by
atomic force microscopy (AFM). When copolymerized with hydrophilic or hydrophobic comonomers, LCST transitions may shift to higher or lower temperatures, respectively. Block copolymers of poly(ethylene oxide)–poly(propylene oxide) (PEO–PPO) also exhibit thermal responses in solutions,70 but it is believed that the driving forces for these transitions originate from amphiphilic balance.
pH-responsive polymer solutions represent another group, in which chemical structures of pH-responsive compounds have ionizable functional groups capable of donating or accepting protons upon environmental pH changes. In this case, electrostatic repulsions between generated charges cause alternations of the hydrophobic volume along a polymer chain, which is capable of extending or collapsing. Polyacids, such as poly(acrylic acid)
23
(PAAc),71 and poly(methacrylic acid) (PMAAc)72 with pKa values in the range of 5 will release protons and swell under basic pH values. In contrast, pH-responsive polybases accept protons and extend under acidic pH conditions, where amino and amine functional groups in poly(N,N′-dimethyl aminoethyl methacrylate) (PDMAEMA)73 and poly(vinyl pyridine) (PVP),74 respectively, are responsible for these transitions. One of the common trends in designing stimuli-responsive polymers is to copolymerize monomers with different stimuli-responsiveness in order to achieve multiple-responsive behavior.
Figure 1-15: Examples of molecular structures responsive to temperature (A) and pH (B).
1-5.3 Electromagnetic-responsiveness
Incorporation of photo-chromic molecules provides opportunity to develop polymers capable of responding to electromagnetic radiation.
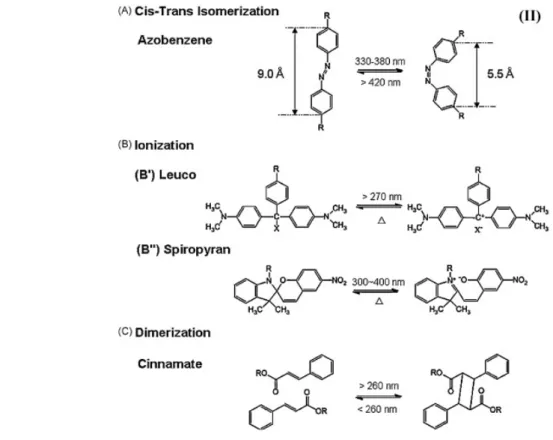
Figure 1-16 illustrates most common photo-sensitive molecules, which are classified into the following categories: cis–trans isomers (A), ionization monomers (B), and dimerization monomers (C). As shown in Figure 1-16A, photo-responsive azobenzene is a molecule that exhibits trans-to-cis photoisomerization with sufficiently low energy (2–3 kcal/mol) to induce
24
photo-chromic transitions.75 The rearrangement mechanism for lecuo76 and spiropyran77 derivatives shown in Figure 1-16B is based on ionization upon exposure to electro-magnetic irradiation. When exposed to UV light, dissociated ion pairs are generated, which can be further neutralized when heated in the dark. As illustrated in Figure 1-16B′ and B″, photo-induced polymer chains of lecuo and spiropyran derivatives shrink and expand, which is attributed to the reversible exchange of the electrostatic repulsion between ionic states. This process typically requires less than 5 kcal/mol.78 Figure 1-16C illustrates another photo-reactive molecule cinnamate79 which is able to dimerize upon UV irradiation with the energy barrier of about 7 kcal/mol.80 These molecular entities can be utilized as photo-reversible covalently crosslinkers in polymers, thus offering potential applications as switching segments in shape memory systems and other devices.
Figure 1-16: Examples of molecular structures of photo-responsive monomers: cis–trans isomer of azobenzene (A); ionization monomers (B) of leucos (B′) and spiropyran (B′′); and dimerization monomer of cinnamate (C).
25
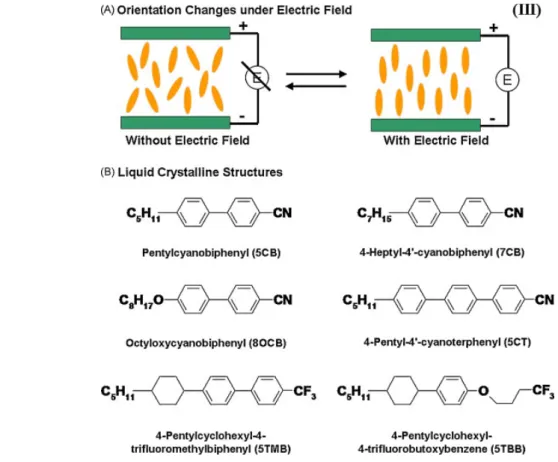
Liquid crystalline materials are stimuli-responsive polymers that have been known for a few decades. These are molecules with permanent dipole moments embedded in polymer matrices, which due to optical and geometrical anisotropies are able to respond to electromagnetic field by aligning their mean optic axis parallel to the external field,81 which results in orientation changes.
As shown in Figure 1-17A, liquid crystalline molecules are freely dispersed between the two electrodes with no electric field. When an electric field is applied, the molecules align along the electric field axis and the driving force for the alignment results from the electrostatic interactions. Figre 1-17B summarizes selected examples of chemical entities capable of responsiveness, and their common feature is the permanent dipole moment, in this case generated by electron-withdrawing groups nitrile (CN) and trifluoromethyl (CF3) groups.
Figure 1-17: Examples of molecular structures of photo-responsive monomers: Orientation changes (A) and molecular structures (B) of liquid crystalline molecules.
26
1-5.4 Multiple-responsive systems
As pointed out earlier, a growing trend in designing stimuli-responsive polymeric solutions is toward creating systems with multiple-responsive components, with covalently or physically bonded segments responsive to different stimuli. One example of the multi-responsive switchable copolymer is based on temperature-responsive NIPAAm and pH- and photo-responsive spirobenzopyran which exhibits temperature, pH, and photo-responsiveness in aqueous solutions. Along the same lines, acrylic acid, NIPAAm, and cinnamoyloxyethyl acrylate have been copolymerized and exhibit four responses.82 Acrylic acid component responds to pH and ionic strength, while the NIPAAm and cinnamoyloxyethyl acrylate species exhibit temperature and UV responses, respectively. There are many opportunities for designing multiple-responsive polymeric solutions, in particular using colloidal synthesis where surfaces of colloidal particles may serve as protein or cell adhesion sites,83 or stabilized with bioactive materials.84 Due to spatial restrictions significantly greater challenges exist in the development of multiple-responsive systems at surfaces and interfaces, in polymeric gels, and in thermoplastic and thermosetting polymeric networks.
27
References
1. (a) Hawker, C. J.; Bosman, A. W.; Harth E. Chem. Rev. 2001, 101, 3661–3688. (b) Kamigaito M.; Ando, T.; Sawamoto, M. Chem. Rev. 2001, 101, 3689–3745. (c) Matyjaszewski K.; Xia, J. Chem. Rev. 2001, 101, 2921–2990. (d) Perrier S.; Takolpuckdee P.; J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 5347–5393. (e) Favier, A.; Charreyre, M. T. Macromol. Rapid Commun. 2006, 27, 653–692. (f) Ouchi, M.; Terashima, T.; Sawamoto, M. Chem. Rev. 2009, 109, 4963–5050. (g) Yamago, S. Chem.
Rev. 2009, 109, 5051–5068. (h) Tsarevsky, N. V.; Matyjaszewski, K. Chem. Rev. 2007, 107, 2270–2299. (i) Braunecker, W. A.; Matyjaszewski, K. Prog. Polym. Sci. 2007, 32,
93–146. (j) Moad, G.; Rizzardo, E.; Thang, S. H. Aust. J. Chem. 2005, 58, 379–410. (k) Moad, G.; Rizzardo, E.; Thang, S. H. Aust. J. Chem. 2006, 59, 669–692.
2. Szwarc M. Nature, 1956, 178, 1168–1169.
3. Matyjaszewski, K.; Davis, T.P. ed., Handbook of Radical Polymerization, John Wiley & Sons, Inc., Hoboken, 2002.
4. (a) Otsu, T.; Yoshida, M. Makromol. Chem. Rapid Commun. 1982, 3, 127–132. (b) Otsu, T.; Yoshida, M.; Tazaki, T. Makromol. Chem. Rapid Commun. 1982, 3, 133–140. (c) Otsu, T. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2121–2136. (d) Otsu T.; Matsumoto, A. Adv. Polym. Sci. 1998, 136, 75–137.
5. Druliner, J. D. Macromolecules 1991, 24, 6079–6082.
6. David, G.; Boyer, C.; Tonnar, J.; Ameduri, B.; Lacroix-Desmazes, P.; Boutevin, B. Chem.
Rev. 2006, 106, 3936–3962.
7. Percec, V.; Guliashvili, T.; Ladislaw, J. S.; Wistrand, A.; Stjerndahl, A.; Sienkowska, M. J.; Monteiro, M. J.; Sahoo, S. J. Am. Chem. Soc. 2006, 128, 14156–14165.
8. (a) Debuigne, A.; Caille, J.-R.; Detrembleur C.; Jérôme, R. Angew. Chem., Int. Ed. 2005,