15
第二章 桂皮單寧酚之分析
第一節 桂皮單寧成分之高效能液相層析
2-1-1 前言
2-1-1-1 桂類藥材
桂皮以菌桂及牡桂之名,收載於神農本草經,唐蘇敬謂:古方用筒桂,
即二或三重卷縮者為良品,神農本草經所謂菌桂可能為筒桂之誤。肉桂當 作藥物使用的歷史甚久,古代埃及文獻已有記載,如西方最古老的本草書 De Materia Medica,書中已收錄桂皮類藥物-Kinnamono 及Kassia[49]。
市面上桂皮類藥材因使用部份、出產地點不同,種類頗多,如清化桂、板 桂、桂通、桂心、肉桂、桂枝等。
桂枝、桂皮為最常用的中藥材,分別是樟科植物(Lauraceae)中肉 桂或其同屬植物的嫩枝和皮,來源包括[49]:
(Ⅰ)Cinnamomum cassia BLUME(肉桂) (Ⅱ)C. obtrssifolium NEES(越桂) (Ⅲ)C. zeylanicum NEES(錫蘭桂)
因取用部位不同而分成樹皮(桂皮)與嫩枝(桂枝)兩大類,根據歷 代文獻的記載,桂皮藥性味辛、性大熱,歸肝、腎、脾經,為芳香健胃劑,
是下行溫補之品。而桂枝味甘、辛、性溫,歸心、肺、膀胱經,為解肌發 表興奮藥,是上行發表之品,二者藥效不盡相同[49]。
桂皮、桂枝屬最常用藥材,近來藥理研究[49,50,51]顯示,肉桂的精油 有健胃作用,肉桂醛能擴張皮膚血管,促進血液循環至體表,有發汗解熱 作用;肉桂醛能抑制小白鼠的中樞神經系統,有顯著的鎮靜作用;促進循 環作用、抗菌作用、抗過敏作用、對中樞神經系統影響、對心血管系統影 響。
圖 2-1-1 桂皮植物之外觀圖[64] 圖 2-1-2 桂皮藥材之外觀圖[64]
桂類藥材含有多種精油成分,最主要的是肉桂醛(Cinnamaldehyde), 含量高達精油的 50﹪∼77﹪[52],以外尚有肉桂醇(Cinnamyl alcohol)
[53],肉桂酸(Cinnamic acid)[54],乙酸肉桂酯(Cinnamyl acetate)
與 香 豆 素 ( C oumarin ) 等 成 分 [55] 。 而 桂 類 中 亦 含 有 少 量 的 單 寧 類
(tannins)[54-56]及倍 類(diterpenes)化合物[59-62]。
1980 年 Yagi 等人自桂皮中分離出一系列之倍 類,經試驗發現其可 能具有 anti-complement 活性,進而分離並鑑定出下列六種化合物:
cinnzeylanine , cinnzeylanol , anhydrocinnzeylanine , anhydrocinnzeylanol , cinncassiol A , cinncassiol A 19-O- β -D-glucopyranoside[59]。
同年, Nohara 等人也自桂皮中分離出一系列具有抗過敏活性之倍 類,其中 cinncassiol C 已經由 x-ray 結構解析,確定結構[60]。
而另外的七個化合物,則只鑑定二個化合物,分別為 cinncassiol B , cinncassiol B 19-O-β-D-glucopyranoside[61]。到了 1985 年他們又分 離出 cinncassiol E [62]。
近年來, Nishioka 等人自肉桂中分離出一些單寧類化合物,計有七
17
種 flavan-3-ol 的甲基衍生物,一種 1’3-diaryl propan-2-ol 的甲基衍生 物[56];1986 年又自廣南桂皮中分離出六種 flavan-3-ol 的衍生物,分別 為:cinnamtannins A2、A3、A4,(-)-epicatechin 3-O、8-C、6-C β-D-gluco -pyranosides cinnamtannins[57]。;自東興桂皮中分離出二種單寧類化 合物:procyanidin B-2、8-C、6-C β-D-glucopyranosides;自越南桂 皮 中 分 離 出 (+)-catechin 5-O- β -D-(2”-O-feruloyl-6”-O-p -coumaroyl)-glucopyanosides[58]。
1987 年, Oshima 等人以 HPLC 分析桂皮的成分,定量香豆素、肉桂 醇、肉桂酸、肉桂醛、乙酸肉桂酯的含量,該文較 GC 方法多定出肉桂酸 的含量[54]。1987 年高橋邦夫等利用 GC 可同時偵測十種化合物,在該篇論 文中並測定 23 種含桂皮方劑的肉桂酸、肉桂醛的含量[63]。1988 年,
Archer 以 HPLC 分析桂皮的肉桂酸、香豆素、肉桂醇、肉桂醛、丁香酚、
乙酸肉桂酯、2-甲氧基肉桂醛等成分;同時比較不同品種藥材中香豆 素、肉桂醇、肉桂醛三成分的含量[55]。
1992 年 Nishioka 等人利用 HPLC 分析大戟屬植物的成分,以 5C18-AR 管柱及磷酸鹽流動相定量水解型 tannins 的含量[69]。1998 年 E.Hagerman 等人利用 C18 管柱及 1%冰醋酸沖提液分析柳樹的葉子、枯枝的單寧成 分,並發現這類化合物萃取液經 2∼4 週會迅速減損[70]。
2-1-1-2 單寧
單寧(tannins)原指植物體內具收斂作用的物質;近年來,凡植物體 內之多酚類成分(polyphenols),其易與纖維素(cellulose)等一些天然 高分子化合物、蛋白質、重金屬離子及鹽基性等物質結合[65-67],形成難 溶性複合物均稱之。單寧廣布於植物體中,與日常生活相關,例如飲料、
食物及一些藥用植物均富含單寧成分[68,71,72]。單寧常應用在墨水及接著 劑之製造[73];亦可作整腸、止血、止瀉等藥劑[74]。由於單寧對酸、鹼及 熱不安定,在植物界中也會和構造相似之化合物呈混合狀態,造成分離上 的困難,使得有關單寧方面之研究報告較少。有關單寧化學成分研究的歷 史,溯自 1785 年由沒食子(Quercus infectoria Oliver)中分離出沒食子 酸(gallic acid)開始;1796 年 Seguin 將 oak gall 收斂的成分以單寧
(tannin)稱之;1945 年以後,Schmidt 等人繼續進行單寧方面的研究,
並以當時的研究方法決定了 chebulinic acid, chebulagic acid 等單寧之 化學構造[75]。
有關單寧成分的研究,自 1980 年以來,由於儀器的發展及分離方法 的 改 良 [76-78]而有很大進展,例如:液滴逆流分配層析法(droplet counter-current chromatography,DCCC)、離心分配層析法(centrifugal partition chromatography,CPC )、 膠 體 管 柱 層 析 法 ( gel column chromatography,GCC)、高效能液相層析法(high-performance liquid chromatography,HPLC ) 以 及 各 種 光 譜 技 術 ( two-dimensional NMR spectrum,fast atom bombardment mass spectrum (FAB-MS),circular dichroism (CD) spectrum 等)[79-81]的開發,因而突飛猛進。尤其自牛 兒苗(Geranium thumbergii Sieb. et Zucc.)[Geraniaceae]中分離出 結晶性之 geraniin [73] (屬於 ellagitannin) 後,單寧成分之化學構造 逐漸解明。
單寧屬於多酚類化合物,是指分子量 500 以上的類黃酮酚類化合物
(flavonoid phenolic compounds),可以網狀結構或氫鍵與蛋白質結合 的物質,主要可分為縮合單寧(condensed tannins)與水解性單寧 (hydrolysable tannins) 二 類 。 縮 合 單 寧 是 由 flavan-3-ols 或 flavan-3,4-diols 縮合而成的聚合物[82]。水解性單寧則分成沒食子單寧
(gallotannins)及鞣花單寧(ellagitannins),為沒食子酸(galic acid)
或鞣花酸(ellagc acid)與糖類及其他類似的醇類形成之聚酯類化合物,
易為酸所水解[83]。
單寧易氧化成 類(quinones), 類化性活潑,可進行下列反應:
作為氧化劑,促進其他物質的氧化;快速聚合,形成深色高分子聚合物;
藉氫鍵、共價鍵、離子鍵等鍵結或疏水性聚合與蛋白質結合[84-86]。
影響蛋白質與單寧作用的因素包括單寧分子大小、溫度及離子強度、
pH、溶劑種類;Kumar and Horigome 指出,縮合單寧在 pH7~8 以下與蛋 白質形成氫鍵,主要以未離子化的氫氧基與蛋白質胜 鍵上的羰基作用,
19
而水解性單寧在 pH3~4 時與蛋白質鍵結最強[87]。
單寧類化合物普遍存於植物界,其對生化上的影響,主要即因為其易 與蛋白質作用引起。目前研究對象以葡萄、柿、高梁及飼料等較多。當農 作物本身含高量單寧時,便有防鳥類吃食、抗黴菌生長、防蟲及減少收穫 前發芽等功用,但此種化合物不但味道較差,且不易消化、營養價值低,
在飼料中甚至有因食用過量而中毒的例子[87]。1987 年范明仁認為單寧酚 基氧化會形成苯 (quinone)而抑制黴菌生長,已知單寧與苯 化合物對 微生物具有毒性,使其分泌之酵素失去活性[88]。
單寧類化合物,在工業上除用於皮革業外,亦可利用化學反應作成修 飾單寧分子以作為沉澱水中污泥、黏著劑、木材表面塗敷(coatng)、助濾 劑等[82],在食品上則可作為去蛋白質劑(de-proteining agent),最常用 於沉澱酒中蛋白質。
單寧具相當重要的生物活性,有關具含量測定的報告不多[69,70],本 文擬用七個桂皮單寧化合物[106]為指標,開發 HPLC 及 CE 分析方法。
2-1-1-3 定量分析之指標成分結構
本研究擬開發-高效能液相層析法以分析桂皮中的七個桂皮單寧成分
【 (+) – catechin (1),(-) – epicatechin (2) , Procyanidin B-1 (3) , Procyanidin B-2 (4) , Arecatannin A1 (5) , Cinnantannin B2 (6) , Cinnantannin C2 (7) 】[106],並評估此分析方法之適宜性。
七個指標成分結構如下:
1. (+) – catechin 2. (-) – epicatechin
3. Procyanidin B-1 4. Procyanidin B-2
O
OH
OH
OH
OH HO
OH
OH
OH OH
HO O
OH
OH OH
O
OH
OH O OH
HO HO
HO
OH HO HO
HO
O
OH
OH
OH
OH O
OH
OH
OH
21
5. Arecatannin A1 6. Cinnantannin B2
7.Cinnantannin C2 IS Gallic acid
圖 2-1-3 桂皮指標成分結構圖
OH
OH OH O
OH OH
OH OH O
HO HO
HO
HO HO
HO
O
OH O
OH OH OH
HO OH OH
HO
OH
OH
OH
OH
O
HO HO HO
O
OH
OH
OH OH
HO
HO
HO O
OH OH OH
O HO O
HO
OH
OH
OH
OH
O
HO HO HO
O
OH
OH
OH OH O
O HO
O
HO
OH OH
HO
HO
COOH
OH OH HO
2-1-2 實驗部分
2-1-2-1 藥品與儀器
2-1-2-1-1 實驗藥品
(1) 桂皮的指標成分(+) – catechin (1) , (-) – epicatechin (2) Procyanidin B-1 (3) , Procyanidin B-2 (4) ,Arecatannin A1 (5) ,Cinnantannin B2 (6) , Cinnantannin C2 (7)為大仁技術學院林大禎博士提供 [106]。
(2) 內標準品gallic acid購自Merck (Darmatadt, Germany)。
(3) 磷酸鹽及磷酸購自Acros (New Jersey, USA),甲醇(Methanol)及氰甲 烷(Acetonitrile)購自Merck(Darmatadt,Germany)。
(4) 配 置 樣 品 及 流 動 相 的 去 離 子 水 (deionized water) 取 自 於 Milli-Q System (Millipore, Bedford,MA,USA)。
2-1-2-1-2 實驗儀器
(1)高效能液相層析(HPLC) 泵:Waters 510 pump 兩個
注射器:Microliter type 802 (25µL loop) 偵測器:Waters 990 photodiode array detector
資料處理器:Shimadzu SPD-M10AVP and 宏碁-586電腦 (2)酸鹼測定儀( pH meter ):Suntex SP-2200
(3)超音波震盪器:Elma Transsonic Sigital
(4)離心機:Hettic Universal 轉盤為10 mL(15mL)¯12 (5)pipette:Nichiryo (Japan) 0-1 mL,0-5 mL兩支
23
2-1-2-2 分析條件
2-1-2-2-1 最佳分析條件
前置管柱:Nova-Pak C18(Millipore, Milford, MA, USA)
分離管柱:Cosmosil 5C18-MS, 5µm, 25cm×4.6mm (Nacalai tesque, Kyoto, Japan)
流動相: (A) 20 mM KH2PO4(以 10%H3PO4 調整至 pH = 4.0) (B) CH3CN/H2O=80/20
梯度沖提程式:
表2-1-1 桂皮分析方法之梯度沖提程式 時間(min)
流速
(mL/min) A% B% Curve initial 1.0 100 0
15 1.0 85 15 linear 25 1.0 70 30 linear 35 1.0 50 50 linear 45 1.0 0 100 linear 50 1.0 0 100 linear 60 1.0 100 0 linear
分析時間:60分鐘 平衡時間:15分鐘 偵測波長:210 nm
2-1-2-2-2 分析條件的選擇
(1)分離管柱的選擇
選擇四種不同填充材料之管柱(表2-1-2),以表2-1-1之梯度沖提程式及 流動相分析條件進行分析,各組進行兩次重覆注射,以探討不同填充材料 之管柱對各成分的分離效果。
表2-1-2 分離管柱的種類及其材質表 管柱品名a 孔徑
(Å)
表面積 (m2/g)
含碳比 (%)
鍵結型式b 110 330 20 mono 120 300 16 mono 120 300 16 polymeric Cosmosil 5C18
Cosmosil 5C18-MS Cosmosil 5C18-AR
Cosmosil 5C18-AR II 120 300 17 polymeric
a.四種管柱皆是出自Nacalai tesque, Kyoto, Japan。
填充粒子大小 5 µm,長度 25cm,內徑 4.6mm。
b.
(CH2)17CH3
(2)流動相(A)中不同鹽類濃度的探討
選定最適當的分離管柱後,將流動相(A)分別配成20、30、40、50 mM 磷酸二氫鉀溶液,利用磷酸將pH值調至4.0,以前述之梯度沖提程式進行 分析,各組重覆二次注射。
(3)流動相(A)中不同酸鹼值的探討
取最適當的磷酸鹽濃度,在流動相(A)中加入不同量10%的磷酸,配製 不同酸鹼度的緩衝溶液,其pH值範圍由2.50至5.00,以前述之梯度沖提程 式進行分析,各組重覆二次注射。
(4)流動相(B)中不同氰甲烷/水之比例的探討
由於流動相A中含有適當濃度的鹽類,流動相(B)若含有適量的水,則 可防止鹽類析出又能獲得較平整的基線,所以配製不同氰甲烷/水比例的流 動相(B),比例分別為氰甲烷/水 (V/V) = 95/5,90/10,85/15,80/20,75/25。
依上述適當的磷酸鹽緩衝溶液及梯度沖提程式進行分析,每組重覆操作兩
25
2-1-2-2-3 配製標準品溶液及製作檢量線
稱取11.2 mg 的gallic acid 以70%甲醇配成50 mL溶液,做為內標準品 溶液(IS)。
分別稱取 (+) – catechin (1) 3.1 mg, (-) – epicatechin (2) 3.2 mg,
Procyanidin B-1 (3) 3.5 mg, Procyanidin B-2 (4) 3.2 mg,Arecatannin A1 (5) 3.1 mg,Cinnantannin B2 (6) 1.1 mg, Cinnantannin C2 (7) 3.2 mg,溶 於 70%甲醇,分別配成 10 mL 標準品母液 1-7 【I】。
分別以吸量管取標準母液【I】中之(+) – catechin (1)6.0mL、Procyanidin B-1 (3) 6.0 mL為標準母液【II】(各成分濃度分別為:1.0.155mg/ml,
3.0.175mg/ml),分別取標準母液【II】5.0、3.0、2.0、1.0、0.5 mL之標準 品於量瓶中,各加入1.0 mL之內標準品溶液,再以70%甲醇配成10 mL溶液。
分 別 以 吸 量 管 取 標 準 母 液 【I】 中 之 (-) – epicatechin (2)4.0mL 、 Procyanidin B-2 (4) 4.0 mL、Arecatannin A1(5) 8.0 mL為標準母液【III】(各 成分濃度分別為:2.0.080mg/ml,4.0.080mg/ml,5.0.155mg/ml),分別取標 準母液【III】4.5、3.5、3.0、2.5、1.5、0.5 mL之標準品於量瓶中,各加入 1.0 mL之內標準品溶液,再以70%甲醇配成10 mL溶液。
分 別 以 吸 量 管 取 標 準 母 液 【I】 中 之 Cinnantannin B2 (6) 8.0 mL Cinnantannin C2 (7) 4.0mL為標準母液【IV】(各成分濃度分別為:
6.0.073mg/ml,7.0.107mg/ml),分別取標準母液【IV】5.0、3.0、2.0、1.0、
0.5 mL之標準品於量瓶中,各加入1.0 mL之內標準品溶液,再以70%甲醇 配成10 mL溶液。
各濃度的標準品1-7,分別在210 nm波長下,以2-1-2-2-1之最佳分析條 件重覆兩次分析,每次注入10 µL ,取其平均值作檢量線。
2-1-2-2-4 分析條件之適宜性評估
(1) 再現性(Reproducibility)
取標準母液【II】1.0 mL、標準母液【III】1.0 mL及標準母液【IV】
1.0 mL於量瓶中,加入1.0 mL之內標準品溶液,以70%甲醇水溶液稀釋至 10mL , 作 為 檢 液 。 每 次 注 射 10 μ L 進 行 分 析 ; 同 一 天 內 重 覆 六 次 ( intraday ),不同天總計重覆六次 ( interday ) 。
(2)回收率(Recovery)
精稱桂皮粉末 1.0 g,以70%甲醇10 mL作為萃取溶劑,超音波震盪 15 分鐘後離心,重覆三次,合併萃取液,濃縮至10 mL,取5mL於10mL量瓶,
加入0.5 mL標準母液【II】、0.5 mL標準母液【III】及0.5 mL標準母液【IV】, 再加入1.0 mL之內標準品溶液,以70%甲醇水溶液稀釋至10 mL,經 0.45 μm濾膜過濾,作為檢液。以2-1-2-2-1之最佳分析條件重覆兩次分析,每 次注射10μL進行分析,定量值為兩次注射的平均分析結果。
(3)偵測極限
逐步稀釋標準品溶液,注入偵測,直到 signal /noise = 3 / 1以下,計算 其注入量及其濃度。
2-1-2-2-5 桂皮單寧之定量分析
精稱桂皮粉末 1.0 g,以 70%甲醇 10 mL 作為萃取溶劑,超音波震盪 15 分鐘後離心,重覆三次,合併萃取液,濃縮至 10 mL,取 5mL 於 10mL 量瓶中,加入 1 mL 的內標準品溶液,再以 70%甲醇稀釋至 10 mL,經 0.45 μm 濾膜過濾,作為檢液。以 2-2-1 之最佳分析條件重覆三次分析,每次 注射 10μL 進行分析,定量值為三次注射的平均分析結果。
2-1-3 結果與討論
2-1-3-1 分析條件的選擇
(1)偵測波長的選擇
由附錄一發現1-7化合物的最大吸收波長在210nm 附近【(+) – catechin (1) 203nm、(-) – epicatechin (2) 203nm、Procyanidin B-1 (3) 201nm、
27
Procyanidin B-2 (4) 203nm、Arecatannin A1 (5) 203nm、Cinnantannin B2 (6) 201nm、Cinnantannin C2 (7) 201nm】,故以210nm進行樣品分析。
(2)分離管柱的選擇
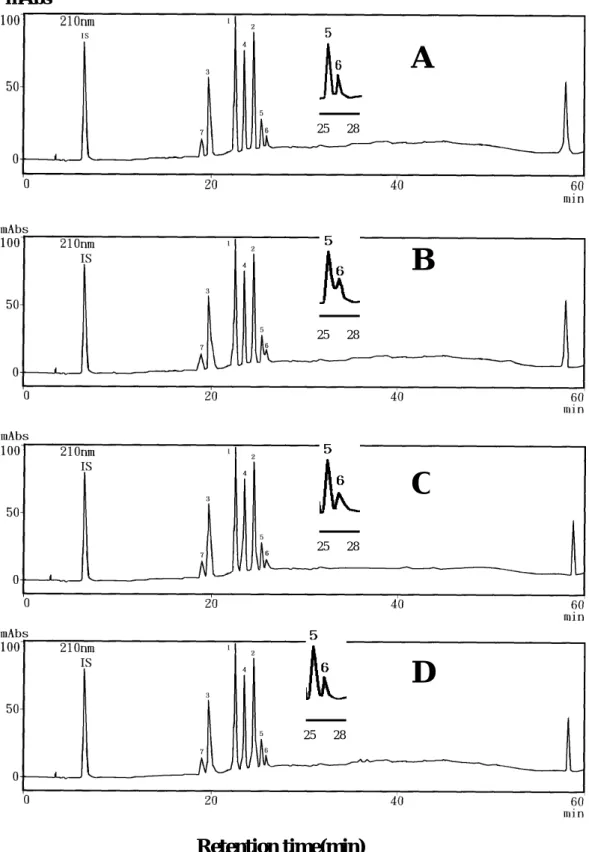
以本實驗室備有的四種不同材質管柱,進行篩選,分析圖譜如圖 2-1-4。在桂皮單寧成分分析上,本文選取該四種管柱,加上近年來新開發 的5C18-AR II管柱等四種來比較。
實驗結果如表2-1-3、圖2-1-5及圖2-1-6所示,發現5C18-MS管柱分析成 分1 –7結果不錯,其理論板數最好。
表 2-1-3 不同分析管柱與各成分 N 值(N×104)的關係表 化合物 5C18-MS 5C18 5C18-AR 5C18-AR II
1 13.83 4.37 2.29 5.15 2 11.66 5.50 2.18 2.47 3 11.78 4.80 1.37 3.94 4 10.51 4.86 2.67 5.93 5 4.14 * 4.10 4.08
6 3.95 * 3.52 2.80
7 3.50 0.67 0.85 1.37 (*代表和相鄰成分重疊)
若由表2-1-4解析度來做比較, 5C18管柱無法分離5和6; 5C18-AR及 5C18-AR II在5/6的解析度甚佳,但還是以5C18-MS在5/6的解析度最好;對 全部指標成分而言,5C18-MS、5C18-AR及5C18-AR II均可有效分離,但 綜合理論板數及解析度兩因素,則以5C18-MS最佳。
表 2-1-4 不同管柱與 Rs 值的關係表
Rs 5C18-MS 5C18 5C18-AR 5C18-AR II 5/6 1.999 0 1.081 1.660
(3)流動相(A)中不同鹽類濃度的探討
桂皮、桂枝是樟科植物中肉桂或其同屬植物的嫩枝和皮,本實驗室作
過桂枝成分分析,皆採用磷酸鹽系統作為分析流動相(A),故本研究擬以磷 酸鹽作為流動相(A)之系統來探討分析效果。
為了探討磷酸鹽對各化合物的影響,在流動相(A)中加入不同濃度 之磷酸鹽及適當量之磷酸,使pH=4.0,作一系列比較。結果如圖2-1-7。由 圖中可看到,對大部分成分而言,磷酸鹽濃度由20~50 mM皆有不錯的分離 效果。當磷酸鹽濃度增加時,對於3、7、5和6等成分解析度不佳以外,其 它化合物皆能明顯分離,且其解析度隨磷酸鹽濃度增加而增加。由圖2-1-7 的K’值可以看出在水相中加入磷酸鹽後,化合物的滯留時間縮短,這主要 是因為磷酸鹽可覆蓋管柱表面殘餘矽羥基基團,因而阻止吸附樣品及參與 離子樣品的滯留,因此分析時間縮短。
然而,濃度過高可能造成鹽類析出,降低管柱壽命,對於泵的損耗較 大;由圖中可看出磷酸鹽濃度對大部分成分的分離並無太大影響,唯獨對 於3、7、5和6等成分影響較大,因為這四種成分受磷酸鹽濃度影響不同,
且滯留時間非常接近,故在磷酸鹽濃度的選擇上,係以這四種成分的解析 度來考量,其解析度如表2-1-6所示,其中可看到濃度20mM、50mM對於3、7、
5和6等成分之解析度較差,但濃度20mM對於3、7、5和6等成分化合物之解 析度皆佳且理論板數較優(1.99×104至1.38×105),所有化合物之理論板數 如表2-1-5及圖2-1-8所示,因而顧及管柱壽命和成分分離解析度二方面,故 選擇適當20mM磷酸鹽作為流動相(A)的濃度。
表2-1-5 不同濃度磷酸鹽與各成分N值(N×104)的關係表 化合物 0 mM 10 mM 20 mM 30 mM 40 mM 50 mM
pH 4.0 4.0 4.0 4.0 4.0 4.0 1 6.45 8.02 13.83 8.24 7.99 6.84 2 6.05 9.12 11.66 10.26 7.10 8.54 3 * 8.04 11.78 6.71 5.66 4.86 4 5.23 6.78 10.51 9.09 6.26 7.55 5 * 2.56 4.14 2.60 0.63 0.91 6 * 2.43 3.95 1.08 0.96 0.69 7 * 1.80 3.50 1.80 0.52 0.76
(*代表和相鄰成分重疊)
29
表2-1-6 不同濃度磷酸鹽與Rs值的關係表 Rs 0 mM 10 mM 20 mM 30 mM 40 mM 50 mM 3/7 0 0.901 2.181 1.531 0.926 0.898 5/6 0 0.986 1.999 0.936 0.786 0.778
(4)流動相(A)中不同酸鹼值的探討
在 20 mM 磷酸二氫鉀溶液中,加入不等量的 10%磷酸,製備不同 pH 值 的流動相(A),分別為 2.5、3.0、3.5、4.0、4.5、5.0 來探討 pH 值對分離 的影響,所得的酸鹼值及 k’值關係,結果如圖 2-1-9。事實上,當 pH 低於 3.5 時,不僅部分吸收峰(3、7、5、6)會重疊,參見表 2-1-8,且理論 板數也大幅下降,參見表 2-1-7 及圖 2-1-10。因此,考量解析度與滯留時 間等因素,本研究方法選擇以 20mM 磷酸二氫鉀水溶液,用 10%磷酸調整 pH 值至 4.00 作為流動相(A)的組成。
表2-1-7不同酸鹼值與各成分N值(N×104)的關係表 化合物 2.50 3.00 3.50 4.00 4.50 5.00
1 8.03 8.96 7.71 13.83 6.44 7.75 2 8.60 1.22 8.88 11.66 9.60 10.56 3 * * 5.98 11.78 4.09 2.76 4 10.47 8.79 9.06 10.51 7.58 8.26 5 * * 3.34 4.14 1.32 1.60 6 * * 2.90 3.95 0.98 1.50 7 * * 2.30 3.50 0.72 1.65
(*代表和相鄰成分重疊)
表2-1-8不同酸鹼值與Rs值的關係表
Rs 2.50 3.00 3.50 4.00 4.50 5.00 3/7 0 0 0.862 2.181 0.988 1.321 5/6 0 0. 0.701 1.999 1.401 0.801
(5)流動相(B)中不同氰甲烷/水之比例的探討
由於流動相(A)中含有鹽類,當與完全為有機溶劑之流動相(B)混 合時很容易析出鹽類,且考量當二流動相混合時會有溫度的變化,導致基 線的不穩,故主體為有機溶劑之流動相(B)應含適量的水,以防止鹽類 析出並有平整基線。在此改變氰甲烷/水的體積比例為95/5、90/10、85/15、
80/20、75/25來探討流動相(B)比例對k’值的影響,結果如圖2-1-11所示。
由圖可看出氰甲烷比例愈高,各成分之滯留時間愈短。如同pH值影響的探 討所述,由於滯留時間相當接近,在此仍以考量3與7及5、6等兩組成份,
各成分之間的解析度如表2-1-10。
由表2-1-10可知在80/20比例時有不錯的解析度,再由理論板數來看(表 2-1-9及圖2-1-12),在流動相B中增加氰甲烷比例,理論板數會降低,且以 80/20的比例為最佳。但是當氰甲烷比例太高時,容易造成鹽類析出而對管 柱有不利的影響。因此,本研究方法選擇氰甲烷/水之體積比例=80/20作 為流動相(B)之組成。
表 2-1-9 不同氰甲烷/水比例與各成分 N 值(N×104)的關係表 化合物 95/5 90/10 85/15 80/20 75/25
1 2.98 2.82 7.57 13.83 9.15 2 5.30 3.82 8.46 11.66 10.2 3 1.77 1.81 6.15 11.78 5.80 4 3.89 4.00 6.63 10.51 6.35 5 1.38 1.52 1.41 4.14 1.48 6 0.37 0.55 0.96 3.95 1.00 7 0.45 0.70 1.57 3.50 1.00
表 2-1-10 不同氰甲烷/水比例與 Rs 值的關係表 Rs 95/5 90/10 85/15 80/20 75/25 3/7 0.765 0.742 0.991 2.181 1.913 5/6 0699 0.776 0.874 1.999 1.201
31
mAbs
Retention time(min)
圖 2-1-4 使用不同分析管柱的桂皮分析圖譜 (A)5C18-MS (B)5C18 (C)5C18-AR (D)5C18-AR II
(成分結構圖,如圖 2-1-3 pp.20-21)
A
B
C
D
25 28
25 28
25 28
25 28
4 4.5 5 5.5 6 6.5 7 7.5
5C18-MS 5C18 5C18-AR 5C18-ARII
Columns
k' (capacity factor)
1 2 3 4 5 6 7
圖 2-1-5 不同管柱與各成分 k’值的關係圖
33
0 2 4 6 8 10 12 14 16
5C18-MS 5C18 5C18-AR 5C18-ARII Columns
N(theoretical plate number)
1 2 3 4 5 6 7
圖 2-1-6 不同管柱與各成分 N 值的關係圖
3 3.5 4 4.5 5 5.5 6 6.5 7 7.5 8
0 10 20 30 40 50
KH2PO4 cono. (mM)
K' (capacity factor)
1 2 3 4 5 6 7
圖 2-1-7 流動相 A 中磷酸鹽濃度與各成分 k’值的關係圖
35
0 2 4 6 8 10 12 14 16
0 10 20 30 40 50
KH2PO4 cono. (mM)
N (theoretical plate number) x 104
1 2 3 4 5 6 7
圖 2-1-8 流動相 A 中磷酸鹽濃度與各成分 N 值的關係圖
3.5 4 4.5 5 5.5 6 6.5 7 7.5 8
2.5 3 3.5 4 4.5 5
pH value
k' (capacity factor)
1 2 3 4 5 6 7
圖 2-1-9 流動相 A 中不同酸鹼值與各成分 k’值的關係圖
37
0 2 4 6 8 10 12 14 16
2.5 3 3.5 4 4.5 5
pH value
N (theoretical plate number) x 104
1 2 3 4 5 6 7
圖 2-1-10 流動相 A 中不同酸鹼值與各成分 N 值的關係圖
4 4.5 5 5.5 6 6.5 7
75% 80% 85% 90% 95%
CH3CN(%)
k' (capacity factor)
1 2 3 4 5 6 7
圖 2-1-11 流動相 B 中氰甲烷/水比例與各成分 k’值的關係圖
39
0 2 4 6 8 10 12 14 16
75% 80% 85% 90% 95%
CH3CN (%)
N (theoretical plate number) x 104
1 2 3 4 5 6 7
圖 2-1-12 流動相 B 中氰甲烷/水比例與各成分 N 值的關係圖
2-1-3-2 檢量線的製作
選擇分析波長上,以選擇最強吸收峰時的波長為原則,由附錄一中的 各成分UV圖可以看到,1-7化合物的最大吸收波長在210nm,故以210 nm 做檢量線,結果如表2-1-11。
依實驗2-2-2-2所述,以吸收峰面積與內標準品面積的比值(y)與各成分 的濃度(x, mg/mL)關係作圖,所得檢量線為:
表2-1-11 桂皮單寧成分在210nm之檢量線 y=ax+b
線性範圍 Slope Intercept R2 Compound
(µg/mL) a b 1 (+) – catechin 3.0 – 35.0 53.784 -0.009 0.9996 2 (-) – epicatechin 4.0 – 72.0 71.047 -0.067 0.9996 3 Procyanidin B-1 4.0 – 40.0 22.482 +0.036 0.9996 4 Procyanidin B-2 4.0 – 72.0 42.864 -0.086 0.9996 5 Arecatannin A1 7.8 – 139.5 16.894 -0.010 0.9999 6 Cinnantannin B2 3.7 – 36.7 3.008 -0.012 0.9996 7 Cinnantannin C2 5.3 – 53.4 2.811 +0.002 0.9996
*y = peak-area ratio,x = conc.(mg/mL)
2-1-3-3 分析條件之適宜性評估
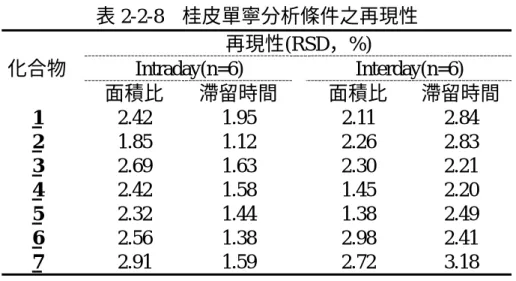
(1)再現性
以 最 佳 分 析 條 件 對 各 成 分 進 行 定 量 , 在 同 一 天 內 連 續 注 射 六 次 ( intraday )及不同天總計注射六次 ( interday )。分別以吸收峰面積比及遷移
41
時間,來計算相對標準偏差 ( RSD, % ),見表2-1-12。
一般而言,在有機相比例較高時,管柱內表面張力較小,再現性良好,
滯留時間之RSD值均在3%以內,同一天滯留時間的相對標準偏差可在 0.11-0.55%,不同天滯留時間的相對標準偏差可在1.67-2.19%之間。
同一天面積比的相對標準偏差在0.78-2.78%之間,不同天面積比的相 對標準偏差在0.70-2.76%之間,兩者差異不大,可以看出再現性良好。
表 2-1-12 桂皮單寧分析條件之再現性 再現性(RSD,%)
化合物 Intraday(n=6) Interday(n=6) 面積比 滯留時間 面積比 滯留時間 1 1.03 0.28 0.87 2.05 2 0.78 0.44 0.70 1.91 3 1.48 0.11 2.09 2.19 4 0.80 0.44 0.99 2.05 5 2.31 0.44 2.26 1.81 6 2.71 0.55 2.38 1.67 7 2.78 0.15 2.76 2.17
(2)回收率(Recovery)
以2-2-1之最佳分析條件重覆兩次分析,每次注射10μL進行分析,定 量值為兩次注射的平均分析結果。結果顯示在已知含量成分的桂皮藥材中 添加各成分純品,如表2-1-13所示。各成分的回收率95-101﹪之間。
(3)偵測極限
逐步稀釋標準品母液,每次注入10μL予以分析,直到signal/noise = 3/1 以下,並記錄此時的濃度值及注入量。結果如表2-1-13:
表2-1-13 桂皮單寧分析條件之回收率與偵測極限 偵測極限
化合物 回收率
(%) µg/mL ng 1 98 0.14 2.80 2 98 0.16 3.20 3 99.7 0.16 3.20 4 95 0.79 5.80 5 99.7 1.25 5.00 6 101 1.67 7.40 7 101 3.30 10.6
2-1-3-4 桂皮藥材定量結果
取市售桂皮藥材三批(n=3)萃取液進行定量,每一樣品重覆注射3次,
求各成分含量之平均值,結果如表2-1-14。層析圖譜見圖2-1-15,圖中X、
Y吸收峰分別為X = (肉桂醇、肉桂酸);Y = (肉桂醛、乙酸肉桂酯),由於 這四個成分極性較低所以在35 min ~ 45 min才被沖提出。
表 2-1-14 桂皮藥材單寧成分之定量結果(n=3)
化合物 代碼 mean±SD
(mg/g)
(+) – catechin 1 5.180 ± 0.002 (-) – epicatechin 2 7.995 ± 0.002 Procyanidin B-1 3 0.014 ± 0.009 Procyanidin B-2 4 0.852 ± 0.003 Arecatannin A1 5 18.91 ± 0.008 Cinnantannin B2 6 12.69 ± 0.011 Cinnantannin C2 7 31.76 ± 0.006 Total 77.40 ± 0012
43
中藥成分繁多複雜,具有許多結構相似、物理性質相同的物質,因此 在定量分析上,常會受到許多未知成分的干擾而成效不佳。桂皮單寧類成 分即有此困擾,利用 HPLC 分析桂皮之單寧成分,在標準品的分析上雖有 不錯的分離效果,但應用到藥材則由於組成成分太多而使吸收峰無法回到 基線,另外單寧 Procyanidin B-1 (3)、Procyanidin B-2 (4)兩成分在桂皮藥 材中含量很少,加上單寧的吸收峰在 210nm,使得基線非常不平整,對定 量結果的精確性有很大影響。因而嘗試進行萃取方法的探討,希望藉由提 高分析成分的濃度或去除未知物的干擾,來改善桂皮藥材的分析條件,萃 取方法用不同種類的萃取液進行,以丙酮、乙酸乙酯、乙醚、正己烷、70%
甲醇之五種萃取液分別萃取。
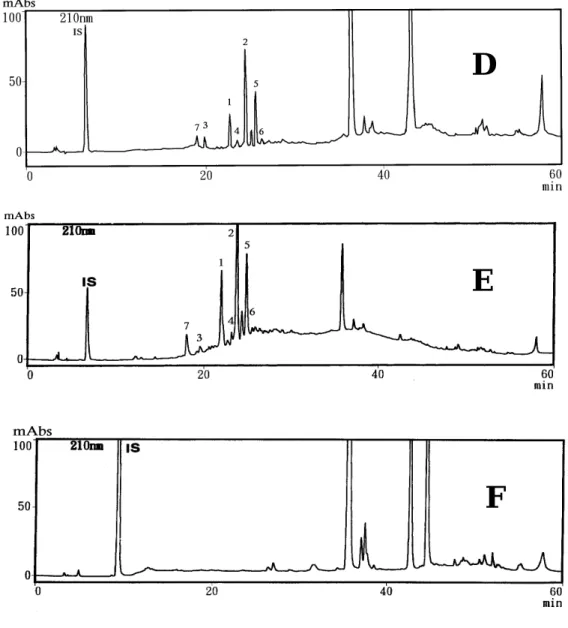
由於桂皮之單寧為高極性化合物,所以在高極性的萃取液中萃取桂皮 藥材之單寧含量較多,但因為萃出物多以致於基線不平,其中1、2、4、5、
6化合物吸收峰無法回到基線;乙醚無法萃取桂皮藥材之單寧成分,而丙 酮和正己烷之萃取液則是無法萃取4之單寧化合物;乙酸乙酯和70%甲醇萃 取液可萃出1-7成分,其中又以70%甲醇萃取液含量較高。雖然乙酸乙酯萃 取液之1-7成分的含量較70%甲醇萃取液少,但乙酸乙酯萃取液之1-7成分 吸收峰較可回到基線,如圖2-1-13、圖2-1-14。綜合以上比較,若要基線較 平整,則選擇乙酸乙酯為萃取液;若要定量則選擇70%甲醇為萃取液。
mAbs
Retention time (min)
圖 2-1-13 不同萃取液之 HPLC 層析圖【1】
(A) 七個標準品 (B)70%甲醇 (C)丙酮
(成分結構圖,如圖 2-1-3 pp.20-21)
B
C A
mAbs
45
Retention time (min)
圖 2-1-14 不同萃取液之 HPLC 層析圖【2】
(D)乙酸乙酯 (E)正己烷 (F)乙醚
(成分結構圖,如圖 2-1-3 pp.20-21)
F D
E
Retention time (min)
圖 2-1-15 桂皮藥材之 HPLC 層析圖 (A) 七個標準品(B)市售藥材
(成分結構圖,如圖 2-1-3 pp.20-21)
A
B
mAbs
mAbs
47
第二節 桂皮單寧成分之毛細管電泳分析
2-2-1 前言
近年來,毛細管電泳因其具有高效、快速且可用於微量樣品分析的優 點,在生物與化學分析領域上大放異彩,發展十分迅速,將毛細管電泳應 用於中藥的分析,也已有很多論文發表,1998 年本實驗室黃明星學長成功 開發了桂枝藥材小分子成分之 HPLC 及 MEKC 分析方法[89],本文嘗試以 毛細管電泳來分析桂皮中單寧成份,並藉此比較 HPLC 和 CE 在桂皮單寧 分析上的優缺點及適用性。
桂皮單寧的藥效和成分說明如本章之前 2-1-1 所述。第一節的高效能 液相層析法已成功分離桂皮中的七種指標成分,但所費時間冗長(60 分 鐘),故希望能開發更快速有效的方法。由該節之敘述中可得知桂皮單寧 成分之結構以苯酚居多,在高 pH 值環境下很容易解離成陰離子,但因分 子量大相對帶電量偏低,故考慮以微膠粒電動毛細管電泳(MEKC)分析 這些成分。
2-2-2 實驗部分
2-2-2-1 實驗藥品
1. 桂皮的指標成 1-7 為大仁技術學院林大禎博士提供。
2. 內 標 準 品 bezoic acid 購 自 Acros (New Jersey, USA) ; sodium borate(Na2B4O7) 購 自 Acros (New Jersey, USA) ; 界 面 活 性 劑 sodium cholate (SC)、sodium dodecylsulfate (SDS)購自Sigma (St. Louis, MO, USA);磷酸購自Acros (New Jersey, USA)。
3. 異丙醇(isopropyl alcohol)、甲醇(Methanol)、氰甲烷(Acetonitrile)購自 Merck(Darmatadt,Germany)。
4. 配 置 樣 品 及 流 動 相 的 去 離 子 水 (deionized water) 取 自 於 Milli-Q System(Millipore, Bedford,MA,USA)。
2-2-2-2 實驗儀器
1. 毛細管電泳系統:Spectra PHORESIS 1000
管柱:70 cm× 75 μm I.D.uncoated (J&W Scientific, USA),管柱長度70 cm,有效長度62 cm。
偵測器:on-line UV detector 210 nm 數據積分處理系統:OS/2 system
PHORESIS 1000 v1.05b PC1000
2. 酸鹼度計(pH meter):SP 2000(Suntex Instruments Co.,LTD) 3. 高速離心計:Hettich Universal D-7200 10 mL× 12
4. 超音波震盪器:Elma Transsonic Digital
2-2-2-3 最佳分析條件
毛細管沖洗模式:
1.新毛細管使用時,要先用1 M NaOH清洗5分鐘(60℃),再以0.1 M NaOH 5分鐘(60℃)及去離子水10分鐘(60℃)清洗毛細管,才能開始使 用。
2.每天開始實驗時,沖洗時間分別為:以去離子水5分鐘, 0.1 M NaOH 5分鐘及去離子水10分鐘沖洗毛細管。
3.在每次樣品分析前,去離子水3分鐘(30℃),0.1 M NaOH 3分鐘(30℃) 及緩衝溶液4分鐘(25℃)。
4.在每次樣品分析後,去離子水5分鐘(30℃),MeOH 7分鐘(30℃)及去 離子水7分鐘(30℃)。
緩衝溶液:
含20mM SC、25 mM硼酸鈉,以10% H3PO4調pH值至7.00的溶 液,和異丙醇(7%,v/v)混勻。
分析時間:40分鐘 電壓:25 kV 溫度:25.0℃
樣品注射模式:注射時間 3 sec。
49
2-2-2-4 緩衝溶液的選擇
(1)不同硼酸鈉濃度的探討
配製含 20mM SC 及一系列不同濃度的硼酸鈉溶液(15、20、25、30 和 35 mM),並以 10% H3PO4調 pH 值至 7.00,再加入小量異丙醇(7%),
配製成系列緩衝溶液,以探討硼酸鈉濃度對桂皮單寧成分分離的影響,各 緩衝溶液重覆操作三次。
(2)不同pH值的探討
在 20mM SC 及 25mM 硼酸鈉溶液中,加入不等量的 10% H3PO4使 pH 值分別為 6.50、7.00、7.50、8.00 和 8.50,再加入異丙醇(7%),配製成 系列緩衝溶液,以探討不同 pH 值對桂皮單寧成分分離的影響,各緩衝溶 液重覆操作三次。
(3)不同種類有機修飾劑的探討
在20mM SC及25mM硼酸鈉溶液中,以10% H3PO4調pH值至7.00,分 別加入異丙醇、甲醇、氰甲烷,依7%、15%不同比例,配製成系列緩衝溶 液,以探討不同種類有機修飾劑對桂皮單寧成分分離的影響,各緩衝溶液 重覆操作三次。
(4)不同比例有機修飾劑的探討
在 20mM SC 及 25mM 硼酸鈉溶液中,以 10% H3PO4調 pH 值至 7.00,
再加異丙醇,依 10%、7%、5%、3.5%、2.5%和 0%不同比例(V/V),配 製成系列緩衝溶液,以探討不同比例有機修飾劑對桂皮單寧成分分離的影 響,各緩衝溶液重覆操作三次。
(5)不同 SC 濃度的探討
配製含 25mM 硼酸鈉濃度及加入 SC 配製分別為 20、25、30 和 35 mM 等不同濃度的 SC 溶液,並以 10% H3PO4調 pH 值至 7.00,再加異丙醇
(7%),配製成系列緩衝溶液,以探討不同 SC 濃度對桂皮單寧成分分離 的影響,各緩衝溶液重覆操作三次。
2-2-2-5 配製標準品溶液及製作檢量線
稱取7.8 mg 的benzoic acid 以70%甲醇配成25 mL溶液,做為內標準品 溶液(IS)。
分別稱取 (+) – catechin (1) 3.1 mg, (-) – epicatechin (2) 3.2 mg,
Procyanidin B-1 (3) 3.5 mg, Procyanidin B-2 (4) 3.2 mg,Arecatannin A1 (5) 3.1 mg,Cinnantannin B2 (6) 1.1 mg, Cinnantannin C2 (7) 3.2 mg,溶 於 70%甲醇,分別配成 10 mL 標準品母液 1-7 【I】。
分別以吸量管取標準母液【I】中(+) – catechin (1)6.0mL、Procyanidin B-1 (3) 6.0 mL為標準母液【II】,分別取標準母液【II】5.0、3.0、2.0、1.0、
0.5 mL之標準品於量瓶中,各加入1.0 mL之內標準品溶液,再以70%甲醇 配成10 mL溶液。
分別以吸量管取標準母液【I】中(-) – epicatechin (2)4.0mL、Procyanidin B-2 (4) 4.0 mL、Arecatannin A1(5) 8.0 mL為標準母液【III】,分別取標準 母液【III】4.5、3.5、3.0、2.5、1.5、0.5 mL之標準品於量瓶中,各加入1.0 mL之內標準品溶液,再以70%甲醇配成10 mL溶液。
分 別 以 吸 量 管 取 標 準 母 液 【I】 中 Cinnantannin B2 (6) 8.0 mL Cinnantannin C2 (7) 4.0mL為標準母液【IV】,分別取標準母液【IV】5.0、
3.0、2.0、1.0、0.5 mL之標準品於量瓶中,各加入1.0 mL之內標準品溶液,
再以70%甲醇配成10 mL溶液。
各濃度的標準品1-7,分別在210 nm波長下,以2-2-2-3之最佳分析條 件,各濃度重覆三次注射,取其平均值作檢量線。
2-2-2-6 分析條件之適宜性的評估
(1)再現性(Reproducibility)
取標準母液【II】1.0 mL、標準母液【III】1.0 mL及標準母液【IV】
1.0 mL於量瓶中,加入1.0 mL之內標準品溶液,以70%甲醇水溶液稀釋至
51
10mL,作為檢液。同一天內重覆六次 ( intraday ),不同天總計重覆六次 ( interday ) 。
(2)回收率(Recovery)
精稱桂皮粉末 1.2 g,以70%甲醇10 mL作為萃取溶劑,超音波震盪 15 分鐘後離心,重覆三次,合併萃取液,濃縮至10 mL,取5mL於10mL量瓶,
加入0.5 mL標準母液【II】、0.5 mL標準母液【III】及0.5 mL標準母液【IV】, 再加入1.0 mL之內標準品溶液,以70%甲醇水溶液稀釋至10 mL經 0.45μm 濾膜過濾,作為檢液。以2-2-2-3之最佳分析條件重覆三次分析,取其平均 值,計算回收率。
(3)偵測極限
將各檢量線之最低濃度檢測液,逐步稀釋,每次稀釋一倍,注入分析,
直至S/N < 3/1為止,計算其偵測極限。
2-2-2-7 藥材之定量分析
精稱桂皮粉末 1.3 g,以 70%甲醇 10 mL 作為萃取溶劑,超音波震盪 15 分鐘後離心,重覆三次,合併萃取液,濃縮至 10 mL,取 5mL 於 10mL 量瓶中,加入 1 mL 的內標準品溶液,再以 70%甲醇稀釋至 10 mL,經 0.45 μm 濾膜過濾,作為檢液。以 2-2-2-3 之最佳分析條件重覆三次分析,取 其平均值。
2-2-3 結果與討論
2-2-3-1 最佳分析條件之選擇
以本實驗室在 2001 年用於分析桂枝的 MEKC 條件為基礎,進行桂枝 分析方法開發[89],該論文用 30mM SDS(sodium dodecyl sulfate)、18 mM 硼酸鈉和 12.5%的異丙醇作為緩衝溶液。結果發現該分析條件並無法將 1-4 分離,但添加 SDS 的多寡對各成分之遷移時間確有很大的影響,SDS 濃度 越高,遷移時間越往後移;用 SC(sodium cholate)取代 SDS,能成功的將桂
皮之單寧指標成分 1-7 分離。分析圖形如圖 2-2-6。
2-2-3-1.1緩衝溶液的條件選擇
(1)不同硼酸鈉濃度的探討
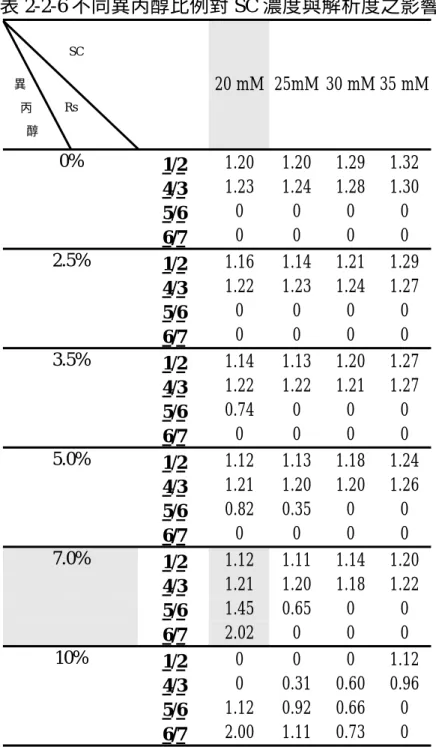
為了探討硼酸鹽濃度對桂皮單寧成分分離的影響,在 20 mM SC 中,
加入硼酸鈉配製 15、20、25、30 和 35 mM 等不同濃度的硼酸鈉溶液,並 以 10% H3PO4調 pH 值至 7.00,再加入異丙醇(7%),配置成一系列緩衝 溶液,分析結果如圖 2-2-1。
硼酸鈉濃度增加,會使離子強度增加,電流變大,增加 EOF 速度,減 少各成分的遷移時間;此外,當硼酸鈉濃度由 15mM 增加到 35mM 時,電 流強度會由 58μA 增加到 86μA 。
各成分的離子強度不同,往後遷移的速度亦不同,所以會造成分析物 與未知物間有所重疊;不同硼酸鈉濃度下,分析物間的解析度,整理如表 2-2-1;1/2、3/4的解析度,隨硼酸鈉濃度增加而減少,但5/6和6/7的解析 度,隨硼酸鈉濃度增加也增加,綜合比較之下,以硼酸鈉濃度為 25mM 時 的解析度最佳。
表 2-2-1 不同硼酸鈉濃度對解析度的影響 濃度 15 mM 20 mM 25 mM 30 mM 35 mM 電流(μA) 58 62 68 75 86
1/2 1.11 1.10 1.12 0.89 0.57 4/3 1.27 1.28 1.27 0.93 0.67 5/6 0 0 1.45 1.46 1.67 6/7 0 0 2.02 2.72 2.60
(2)不同pH值的探討
為了探討不同 pH 值對分析成分的影響,在 20Mm SC 及 25mM 硼酸 鈉中,分別以 10 % H3PO4調 pH 值至 6.50、7.00、7.50、8.00 和 8.50,再
53
加入異丙醇(7%),配置一系列的緩衝溶液。圖 2-2-2 為不同酸鹼值下,
各成分的遷移時間圖。
採不同的無機酸配製 pH 值會影響單寧成分的分離,用 HCl 調配 pH 值時化合物 1~7解析度較差,改用 10%的 H3PO4配製 pH 值就可使桂皮之 單寧成分有效分離。
緩衝溶液的酸鹼值會影響醇基的解離,造成不同的淌度,並產生不同 的分離效果。由圖 2-2-2 來看,隨著 pH 值增加,EOF 延後,會導致所有 化合物的遷移時間往後延。
遷移時間延後,理論上各成分應該可以分離,但因各成分淌度不同,
往後遷移速度亦有不同,會造成解析度的差異。下表 2-2-2 可以發現,當 pH 值高於 7.50,1/2、4/3、5/6無法分離,pH=6.50 的 5/6、6/7解析度不理 想;綜合比較之下,以 pH=7.00 為最佳分析條件的酸鹼值。
表 2-2-2 不同 pH 值對解析度的影響 pH 值 6.50 7.00 7.50 8.00 8.50
1/2 1.12 1.12 0 0 0 4/3 1.14 1.27 0 0 0 5/6 0.53 1.45 1.59 1.11 0 6/7 0.71 2.02 1.82 1.60 0.73
(3) 不同種類有機修飾劑的探討
為了探討不同有機修飾劑對成分分離的影響,在 20mM SC 及 25mM 硼酸鈉中,以 10%的 H3PO4調 pH 值至 7.00,再與異丙醇、氰甲烷和甲醇,
依 7%和 15%等不同比例,配成一系列的緩衝溶液,以探討有機修飾劑對 桂皮單寧成分分離的影響;各分析成分在不同比例的有機修飾劑下。有機 修飾劑比例越高,各成分遷移時間越往後延,反而不易分離,由下表 2-2-3
可知當氰甲烷和甲醇比例越高時,雖然 1/2、3/4 解析度非常好但 5/6、6/7 卻無法分離。所以綜合比較之下,還是以異丙醇比例為 7%時最佳。
表 2-2-3 不同有機修飾劑對解析度的影響
Rs 異丙醇 異丙醇 氰甲烷 氰甲烷 甲醇 甲醇
7.0% 10% 7.0% 15% 7.0% 15%
1/2 1.12 0 1.21 2.04 1.12 1.45 4/3 1.21 0 1.53 2.01 1.33 1.32
5/6 1.45 1.12 0 0 0 0
6/7 2.02 2.00 0 0 0 0
(4)不同比例有機修飾劑的探討
為了探討有機修飾劑對分析成分的影響,在 20mM SC 及 25mM 硼酸 鈉中,以 10%的 H3PO4調 pH 值至 7.00,再加異丙醇,依 0%、2.5%、3.5%、
5%、7%和 10%等不同比例,配成一系列的緩衝溶液,以探討有機修飾劑 濃度對桂皮單寧成分分離的影響;各分析成分在不同比例的有機修飾劑 下,遷移時間的變化,整理如圖 2-2-3。
添加有機修飾劑會增加緩衝溶液的黏滯性[90]及降低偶電層的 zeta 勢,因而降低了 EOF 速度,使遷移時間往後面延遲出現;由下表 2-2-4 當 異丙醇的比例由 0%增加到 5%時,成分 6/7 仍無法分離,增加到 7%各成 分的解析度均大於 1,但比例增加到 10%時,成分1/2、3/4反而無法分開;
綜合以上探討,以異丙醇 7%之比例為最佳分析條件。
表 2-2-4 不同異丙醇比例對解析度的影響
Rs 0 % 2.5 % 3.5 % 5.0 % 7.0 % 10 % 1/2 1.20 1.16 1.14 1.12 1.12 0 4/3 1.23 1.22 1.22 1.21 1.21 0 5/6 0 0 0.74 0.82 1.45 1.12
6/7 0 0 0 0 2.02 2.00
55
(5)不同SC濃度的探討
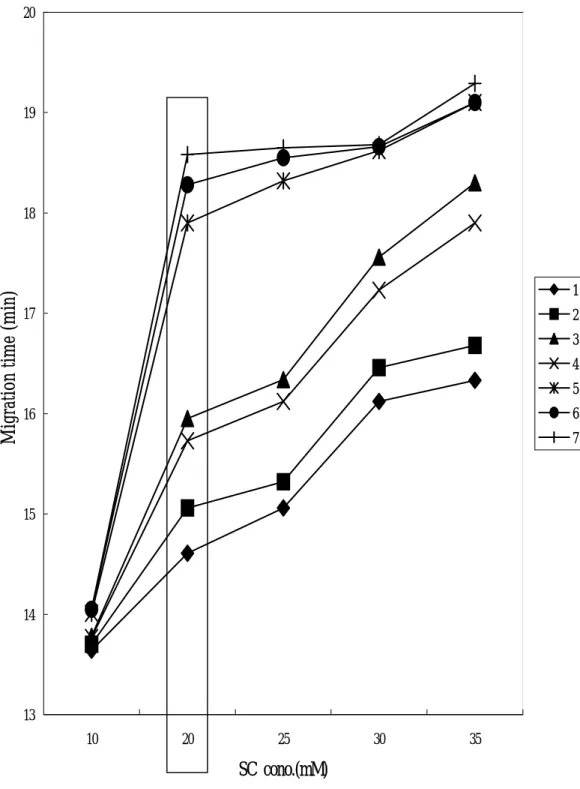
為了探討 SC 濃度對桂皮單寧成分分離的影響,在 25 mM 硼酸鈉中,
加入 SC 配製 20、25、30 和 35 mM 等不同濃度的 SC 溶液,並以 10% H3PO4
調 pH 值至 7.00,再加入異丙醇(7%),配置成一系列緩衝溶液,分析結 果如圖 2-2-4。
本實驗曾嘗試以 SDS 進行分析,發現無法分離 1、2、3 和 4 四種成分,
因此仍以 SC 進行 MEKC 分析。
SC 為一種膽酸鹽,其 CMC(臨界微膠粒濃度)為 14.0 mM [91],從 圖亦可看出 SC 濃度為 10 mM 時,根本無法分離 5、6 和 7 三種成分,也 無法分離 1、2、3 和 4,這是因為要形成微膠粒,界面活性劑濃度必須在 CMC 以上方可進行 MEKC 的分離。由圖可看出幾乎所有成分的遷移時間 皆隨 SC 濃度增加而增加。各成分在不同 SC 濃度下的解析度列於表 2-2-5,
SC 濃度增加至 20 mM 時,即有不錯的分離效果,當濃度增加至 35 mM 時,
除了分析時間加長,對於分離效果幫助不大,因此採取 20 mM 之 SC 進行 分析。
不同異丙醇比例對 SC 濃度對解析度之影響,由表 2-2-6 可看出 SC 濃 度越大對1/2、3/4之解析度越好,但對5/6、6/7之解析度則越差;而異丙 醇的比例越小對5/6、6/7解析度越好,但對 1/2、3/4之解析度越差,綜合 比較之後取決於 7%異丙醇和 20 mM SC 為最佳分析條件。
表 2-2-5 不同 SC 濃度對解析度之影響 Rs 10 mM 20 mM 25mM 30 mM 35 mM 1/2 0.64 1.12 1.11 1.14 1.20 4/3 0.50 1.21 1.20 1.18 1.22 5/6 0 1.45 0.65 0 0
6/7 0 2.02 0 0 0
表 2-2-6 不同異丙醇比例對 SC 濃度與解析度之影響
20 mM 25mM 30 mM 35 mM
0% 1/2 1.20 1.20 1.29 1.32 4/3 1.23 1.24 1.28 1.30
5/6 0 0 0 0
6/7 0 0 0 0
2.5% 1/2 1.16 1.14 1.21 1.29 4/3 1.22 1.23 1.24 1.27
5/6 0 0 0 0
6/7 0 0 0 0
3.5% 1/2 1.14 1.13 1.20 1.27 4/3 1.22 1.22 1.21 1.27
5/6 0.74 0 0 0
6/7 0 0 0 0
5.0% 1/2 1.12 1.13 1.18 1.24 4/3 1.21 1.20 1.20 1.26 5/6 0.82 0.35 0 0
6/7 0 0 0 0
7.0% 1/2 1.12 1.11 1.14 1.20 4/3 1.21 1.20 1.18 1.22 5/6 1.45 0.65 0 0 6/7 2.02 0 0 0
10% 1/2 0 0 0 1.12
4/3 0 0.31 0.60 0.96 5/6 1.12 0.92 0.66 0 6/7 2.00 1.11 0.73 0
SC
Rs 異
丙 醇
57
8 10 12 14 16 18 20
15 20 25 30 35
Sodium Borate conc.(mM)
Migration time (min)
1 2 3 4 5 6 7
圖 2-2-1 硼酸鹽濃度與遷移時間的關係
13 14 15 16 17 18 19 20
6.5 7 7.5 8 8.5
pH value
Migration time (min)
1 2 3 4 5 6 7
圖 2-2-2 pH 值與遷移時間的關係
59
12 13 14 15 16 17 18 19
0 2.5 3.5 5 7 10
isopropanol cono.(%)
Migration time (min)
1 2 3 4 5 6 7
圖 2-2-3 異丙醇比例(v/v)與遷移時間的關係
圖 2-2-4 SC 濃度與遷移時間的關係
13 14 15 16 17 18 19 20
10 20 25 30 35
SC cono.(mM)
Migration time (min)
1 2 3 4 5 6 7
61
2-2-3-2 檢量線之製作
依實驗部分 2-2-2-5 所述,以吸收峰面積與內標準品面積之比值(y)
與各成分濃度(x,mg/mL)之關係,作圖得檢量線如下:
表 2-2-7 桂皮各單寧成分在最佳條件下的檢量線 y=ax+b
線性範圍 Slope Intercept R2 Compound
(µg/mL) a b 1 (+) – catechin 7.1 – 71.0 10.064 +0.497 0.9994 2 (-) – epicatechin 24.0 – 72.0 83.041 -1.419 0.9996 3 Procyanidin B-1 8.0 – 82.0 4.929 +0.199 0.9991 4 Procyanidin B-2 24.0 – 72.0 53.289 -0.736 0.9992 5 Arecatannin A1 46.5 – 148 18.282 -0.678 0.9991 6 Cinnantannin B2 22.0 – 60.8 1.029 +0.009 0.9991 7 Cinnantannin C2 10.7 – 74.4 1.092 +0.114 0.9994
*y=peak-area ratio,x=conc.(mg/mL)
2-2-3-3 分析條件之適宜性之評估
(1)再現性(reproducibility)
以最佳分析條件對各成分1-7進行定量,同一天內連續重覆注射六次 (intraday)及不同天總計重覆六次(interday),記錄其面積比(面積/內標準品 面積)及遷移時間,計算其相對標準偏差。同一天的滯留時間相對標準偏 差可在1.12 – 1.95%,不同天的滯留時間相對標準偏差可在2.20 – 3.18%之 間。
同一天的面積比相對標準偏差在1.85 – 2.91%之間,不同天的面積比 相對標準偏差在1.38 – 2.98%之間。
表 2-2-8 桂皮單寧分析條件之再現性 再現性(RSD,%)
化合物 Intraday(n=6) Interday(n=6) 面積比 滯留時間 面積比 滯留時間 1 2.42 1.95 2.11 2.84 2 1.85 1.12 2.26 2.83 3 2.69 1.63 2.30 2.21 4 2.42 1.58 1.45 2.20 5 2.32 1.44 1.38 2.49 6 2.56 1.38 2.98 2.41 7 2.91 1.59 2.72 3.18
(2)回收率(Recovery)
在已知含量成分的桂皮藥材中添加各成分純品,以最佳分析條件重覆 三次分析,求平均值,結果如表2-2-9所示。各成分的回收率在96-102 %之 間。
(3)偵測極限
逐 步 稀 釋 標 準 品 母 液 , 每 次 注 射 3 秒 ( 約 7.5 µl)予以分析,直到 signal/noise = 3/1以下,記錄此時的濃度及注入量。結果如表2-2-9。表中亦 附上最佳分析條件時的理論板數。
表2-2-9 桂皮單寧分析條件之回收率、理論板數與偵測極限
理論板數 偵測極限
化合物 回收率
(%) (N) µg/mL ng 1 99 203411 0.28 0.0021 2 99 131123 0.32 0.0024 3 101 137811 0.32 0.0024 4 96 116712 1.58 0.0119 5 102 131411 1.55 0.0116 6 101 85986 7.34 0.0551 7 98 63567 10.6 0.0795 injection volume=2.5 µL/sec,injection time=3 sec
column diameter=75µm
63
2-2-3-4 桂皮藥材定量結果
取市售桂皮藥材三批(n=3)萃取液進行定量,每一樣品重覆注射3次,
求各成分含量之平均值,結果如表2-2-10。層析圖譜如圖2-2-5。
表 2-2-10 桂皮藥材單寧成分之定量結果(n=3)
化合物 代碼 mean±SD
(mg/g)
(+) – catechin 1 - (-) – epicatechin 2 - Procyanidin B-1 3 0.011 ± 0.008
Procyanidin B-2 4 0.552 ± 0.006 Arecatannin A1 5 15.91 ± 0.008 Cinnantannin B2 6 13.69 ± 0.015 Cinnantannin C2 7 29.76 ± 0.008 Total 59.92 ± 0022
(-為低於偵測極限)
圖 2-2-5 桂皮單寧之電泳層析圖 (A)標準品 (B)藥材
(成分結構圖,如圖 2-1-3 pp.20-21)
B
A
B
65
圖 2-2-6 桂皮單寧之電泳層析圖 (A)添加 SC (B)添加 SDS
(成分結構圖,如圖2-1-3 pp.20-21)
A
B
![圖 2-1-1 桂皮植物之外觀圖[64] 圖 2-1-2 桂皮藥材之外觀圖[64]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7258891.67385/2.892.175.748.199.454/圖211桂皮植物之外觀圖64圖212桂皮藥材之外觀圖64.webp)