I 摘要 吾人利用步進式時域解析霍式紅外光光譜技術,研究 O(1 D)與甲 醇及其同位素(CD3OH 與 CH3OD)之反應動態學。O(1D)由 O3於 248 nm 雷射光解產生,藉由此技術觀測產物 OH 與 OD 之振轉動放光光譜, 以分析光解產物 OH 及 OD 之內能分佈與比例。O(1 D) + CH3OH 反應 得到產物 OH 之途徑為: O(1D) + CH3OH → CH3O + OH (A) O(1D) + CH3OH → CH2OH + OH (B) 在 O(1 D) + CH3OH 實驗中,可觀測到產物 OH 分佈 P1 分支到 v≦4、 J≦2.5,P2 分支到 v≦4、J≦1.5 之放光譜線,且呈現雙轉動分佈,在 低轉動激發組成,帄均轉動能量為 5 ± 1 kJ mol-1,帄均振動能量為 44 ± 8 kJ mol-1;高轉動激發組成帄均轉動能量為 36 ± 11 kJ mol-1,帄均 振動能量為 28 ± 8 kJ mol-1。在 O(1 D) + CD3OH 實驗中,可觀測到產 物 OH 分佈 P1 分支到 v≦3、J≦8.5,P2 分支到 v≦3、J≦7.5 之放光 譜線,且呈現雙轉動分佈,在低轉動激發組成,帄均轉動能量為 6 ± 1 kJ mol-1,帄均振動能量為 42 ± 7 kJ mol-1;高轉動激發組成帄均轉動 能量為 39 ± 12 kJ mol-1,帄均振動能量為 29 ± 8 kJ mol-1;產物 OD 分 佈 P1 分支到 v≦4、J≦7.5,P2 分支到 v≦4、J≦5.5 之放光譜線,轉 動分佈呈現 Boltzmann 分佈,帄均轉動能量為 6 ± 1 kJ mol-1,帄均振
II 動能量為 43 ± 5 kJ mol-1;所觀測到產物[OH]/[OD] = 50.0/50.0。在 O(1D) + CH3OD 實驗中,可觀測到產物 OH 分佈 P1 分支到 v≦4、J≦5.5, P2 分支到 v≦4、J≦4.5 之放光譜線,轉動分佈呈現 Boltzmann 分佈, 帄均轉動能量為 12 ± 3 kJ mol-1,帄均振動能量為 59 ± 8 kJ mol-1;產 物 OD 分佈 P1 分支到 v≦4、J≦4.5,P2 分支到 v≦4、J≦7.5 之放光 譜線,且呈現雙轉動分佈,在低轉動激發組成,帄均轉動能量為 3 ± 1 kJ mol-1,帄均振動能量為 30 ± 5 kJ mol-1;高轉動激發組成帄均轉動 能量為 8 ± 1 kJ mol-1,帄均振動能量為 23 ± 4 kJ mol-1;所觀測到產物 [OH]/[OD] = 46.2/53.8。 理論計算方面,利用 Gaussian03 程式以 B3LYP/aug-cc-PVTZ 密 度函數理論計算 O(1 D) + CH3OH 在插入機制之各個過渡態(transition state)及最終產物,並優化結構,將優化結構再以 CCSD(T)/aug-cc- pvTZ 理論計算其相對能量;以及以 MRCI(8,8)//CAS(10,10)/6-311+G( 3df,2P) 密度函數理論計算 O(1D) + CH3OH 在擷取機制之各個過渡態 及最終產物。在 O(1 D) + CD3OH 實驗裡,吾人所量測到產物 OH(v=1-2) 振動態之轉動分佈皆呈現 bi-exponential 分佈,所以吾人推斷產物 OH 是氧原子經由插入機制後再斷氫氧基端之 OH,以及氧原子直接抓取 氫氧基端上之氫原子為主要反應途徑;而產物 OD 產物(v=1-3)振動態 之轉動分佈皆呈現 Boltzmann 分佈,所以吾人推斷產物 OD 為氧原子
III 直接抓取甲醇甲基端上之氘原子為主要反應途徑。在 O(1 D) + CH3OD 實驗裡,吾人所量測到產物 OD(v=1-2)振動態之轉動分佈皆呈現 bi-exponential 分佈,所以吾人推斷產物 OD 是氧原子經由插入機制後 再斷氫氧基端之 OD,以及氧原子直接抓取氫氧基端上之氘原子為主 要反應途徑;而產物 OH 產物 (v=1-3)振動態之轉動分佈皆呈現 Boltzmann 分佈,所以吾人推斷產物 OH 為氧原子直接抓取甲醇甲基 端上之氫原子為主要反應途徑。
IV 目錄 第一章 緒論………..………1 附表………8 參考資料………...10 第二章 實驗原理………13 2.1 霍式轉換紅外光譜儀………..………...…………15 2.1.1 Michelson 干涉儀基本原理………16 2.1.2 干涉譜與傳統光譜換式轉換關係………..………17 2.2 霍式轉換紅外光譜儀的優點……….…………..……….….23 2.3 時間解析-霍式轉換紅外光譜法………….………..………...….26 附圖………...33 附表………..43 參考資料………..44 第三章 實驗技術與數據處理………46 3.1 實驗裝置………..………...……46 3.1.1 雷射系統………..………46 3.1.2 反應系統………..………46 3.1.3 偵測系統………..……….………..….48 3.1.4 其他周邊儀器………..………....49 3.2 實驗前準備工作………….………..………….……….50
3.2.1 CaF2透鏡組架設與 Welsh mirror 對正……….…….50
3.2.2 氣體流速校正………....………..52 3.2.3 儀器光學響應曲線量測………...……….55 3.2.4 觸發光解雷射的反應時間………..………58 3.2.5 偵測器及相關電子儀器之響應時間………...…………..……….59 3.2.6 移動鏡穩定時間之量測………..61 3.2.7 反應物之配製與純化………..………62 3.3 實驗程序………..………...……63 3.3.1 光解雷射的準備………...………..……….63 3.3.2 光譜儀之準備與對光………...…………..……….64 3.3.3 周邊儀器設定………..………..………..65 3.3.4 進行光解實驗量測產物之放光訊號………...……..….66 3.4 數據處理………..………..68 附圖………..75 附表………..85 參考資料………...…….125 第四章 結果與討論……….……….126
V 4.1 O3 + CH3OH 在 248 nm 雷射光解之放光光譜….…………..………126 4.1.1 2950-3650 cm-1光區 OH 分子放射光譜之分析……….…..126 4.1.2 產物 OH 之轉動分佈……….……….…..127 4.1.3 轉動弛緩之影響……….……….…..129 4.1.4 產物 OH 之振動分佈及振轉帄均能量……….……….…..131 4.2 O3 + CD3OH 在 248 nm 光解之放光光譜………..132 4.2.1 3800-2170 cm-1光區 OH 與 OD 放射光譜………..…………134 4.2.2 產物 OH 之轉動分佈……….135 4.2.3 轉動弛緩之影響………...……….137 4.2.4 產物 OH 之振動分佈及振轉帄均能量……….……….…..139 4.2.5 產物 OD 之轉動分佈……….……….…..141 4.2.6 轉動弛緩之影響……….……….…..143 4.2.7 產物 OD 之振動分佈及振轉帄均能量……….……….…..144 4.3 O3 + CH3OD 在 248 nm 雷射光解之放光光譜………..…….147 4.3.1 3800-2170 cm-1光區 OH 與 OD 放射光譜………..…………147 4.3.2 產物 OH 之轉動分佈……….147 4.3.3 轉動弛緩之影響………...……….148 4.3.4 產物 OH 之振動分佈及振轉帄均能量……….……….…..149 4.3.5 產物 OD 之轉動分佈……….……….…..151 4.3.6 轉動弛緩之影響……….……….…..152 4.3.7 產物 OD 之振動分佈及振轉帄均能量……….……….…..154 4.4 理論計算 O(1D) + CH3OH 光解之過渡態與反應途徑………..157 4.5 O(1D) + CH3OH 及其同位素反應之討論………159 4.5.1 反應途徑………159 4.5.2 各光解途徑之能量計量………160 4.5.3 OH 與 OD 分子之分支比率…..……….…………..…….…161 4.5.4 由反應速率常數來比較產物 OH 與 OD 之分支比率……….167 4.5.5 其他連續放光譜帶之討論………...………….168 4.6 O(3 P) + CH3OH 在 193 nm 光解之放光光譜……….……….169 4.6.1 3000-3700 cm-1光區 OH 分子放射光譜之分析………..170 4.7 結論…………..……….172 附圖………176 附表………240 參考資料………...……….258
1 第一章 緒論 臭氧分子(O3)的基態和振動激發態,在帄流層中和中層大氣都扮 演著關鍵性的角色。臭氧層(Ozone Layer)中,臭氧的濃度並不高 (不到 1 ppm),而且非均勻地分布於地球各地區。因為臭氧能吸收 太陽光中大部分的紫外線,將這些波長很短的輻射線,轉換成熱能, 僅允許少量紫外線能到達地表,並帄衡生態系統。在 1930 年,查浦 曼(Sydney Chapman)首先提出大氣中臭氧變化的四個步驟,定為查 浦曼循環(Chapman cycle)[1]: (1a) (1b) (1c) (1d) 其中第二步驟生成臭氧,第三與第四步驟卻進行破壞臭氧的反應。如 無催化劑,此循環的第四步驟並不迅速,但如果有氫氧自由基(HO),
一氧化氮(NO),二氧化氮(NO2),氯(Cl)以及氯氧自由基(ClO)
等催化劑存在,則可加速此步驟而使臭氧的存量減少。當 O3吸收一
個波長λ小於 310 nm 的光子時,會解離產生一個氧原子與一個氧分
子,
2 兩產物都是處於電子激發態,其中,O(1 D2)是第一電子激發態的氧原 子,具有的激發能量相較電子基態 O(3 P2)高出約 190 kJ mol-1,O(1D2) 是 啟 動 大 氣 中 許 多 反 應 的 重 要 起 始 物 [2] 。 化 學 氧 化 碘 雷 射
(oxygen-iodine lasers, OIL)中的活性介質[3-5]。了解 O3光解產生 O(1D)
的產率如何[6],尤其對於大氣化學有其重要性。
甲醇(Methanol, CH3OH)在地球的大氣[7]、彗星[8-10]以及在星際
介質[11]中是很重要的物種。甲醇是最小的醇類分子,他擁有甲基
(methyl group)且具有內轉動運動(internal rotation),或稱扭轉運動
(torsional motion)。由於內轉動運動的低振動頻率以及其和分子內其 他振動模有很強的耦合作用,因此很早即被科學家發現此內部轉動運
動 可 促 進 分 子 內 之 能 量 重 新 分 配 (internal vibrational energy
redistribution,IVR)的效率。在燃燒化學中,甲醇可用於引擎內部經 過直接燃燒或用於燃料電池經過催化電解反應,是一很重要的替代燃 料。甲醇經由燃燒後會產生有毒的甲醛,且甲醛樹脂被用於各種建築 材料,包括膠合板、毛毯、隔熱材料、木製產品、地板、煙草、裝修 和裝飾材料,且因為甲醛樹脂會緩慢持續放出甲醛,因此甲醛成為常 見的室內空氣污染之一。從甲醇氧化的機制我們可以知道,O(1 D) + CH3OH 是此過程中的起始反應。其反應之後可以產生不同的產物 CH2OH 或 CH3O,此兩者繼續被氧化後才會產生甲醛。如果我們想要
3 了解甲醛在燃燒過程中扮演的角色,就得先從甲醇和氧原子的反應著 手。 Heicklen 研究組[12]以 213.9 nm 光子光解前驅物 N2O 得到 O(1D) 再與 CH3OH 反應,研究 N2O 以 213.9 nm 光解後得到 O(1D)的產率, 以及 O(1 D)與 CH3OH 反應後得到 OH 的產率以及 O(1D)+CH3OH 之反 應速率。由實驗結果得知,O(1 D)+CH3OH 反應所觀測到產物 OH 的 分支比率為 46% ± 10%,而觀測到 O(3 P)則小於 5%。對於 O(1D) + CH3OH 與 O(1D) + N2O 相 對 速 率 常 數 為 k[O(1D)+CH3OH]/k[O(1D)+N2O] = 5.5 ± 2.0;因為 O(1D) + N2O 反應速 率常數 k[O(1 D)+N2O] = 1.35 × 10-10,即表示 O(1D) + CH3OH 的速率常 數為 k = 7 × 10-10 cm3molecule-1s-1。
Goldstein 和 Wiesenfeld [13]利用雷射誘發螢光法(laser-induced
fluorescence,LIF),將分子激發至其電子激發態,再量測其放射之螢 光。從螢光光譜之譜強度及躍遷能態間之 Frank-Condon 因子即 Honl-London 因子,可計算得之分子基態之相對分佈資訊。以四倍頻 的摻釹釔鋁柘榴石(Nd/YAG)雷射光,波長為 266 nm,作為激發光源 光解 O3產生 O(1D),所產生之 O(1D)即刻與甲醇及其同位素(CH3OD、 CD3OH)反應,主要研究 OH 與 OD 之分支比。O(1D)+CH3OH 反應所 得到產物 OH 可能之反應途徑,如下列所示:
4
(1) O(1D)原子直接擷取(abstraction)甲基端或氫氧基端上之氫原子。 O(1D) + CH3OH’ → OH + CH2OH’ (3a)
→ CH3O + OH’ (3b)
(2) O(1D)原子插入(insertion)甲基端的 C-H 鍵後再進行各種分解之反 應(elimination)。
O(1D) + CH3OH’ → HOCH2OH’ → OH + CH2OH’ (4a)
→ CH2OH+ OH’ (4b)
→ CH2O + H2O (4c)
(3) O(1D)原子插入 O-H 鍵或 C-O 鍵後再產生 O-O 鍵裂解(scission)。 O(1D) + CH3OH’ → CH3OOH’ → CH3O + OH’ (5)
由實驗得到 O(1 D) + CD3OH 和 O(1D) + CH3OD 反應裡所觀測到的產 物 OH(OD)分支裡有 70 %來自於氫氧基位置,另外 30 %來自於甲基 位置,且反應機制傾向 O(1 D)原子插入 CH3OH 的氫氧基位置。在 O(1D) 與甲醇同位素實驗裡並沒有觀測到產物是由反應途徑(3c)而來的,除 非是 O(1 D)與較大的醇類反應才會有極小產率是經由反應途徑(3c)。 另外,在 O(1 D) + CD3OH 實驗裡觀測到產物 OH 振動能階 v = 1 與 v = 0 比率為[OH(v=1)]/[OH(v=0)] = 0.68 且產物 OD/OH 之比率為 0.4;在 O(1D) + CH3OD 實驗裡觀測到產物 OH 振動能階 v = 1 與 v = 0 比率為 [OH(v=1)]/[OH(v=0)] = 0.76 且產物 OD/OH 之比率為 2.3,列於表 1-1。 Kawasaki 研究組[14]以 193 nm 光解前驅物 N2O 或以 248 nm 光 解前驅物 O3 得到 O(1D),所產生之 O(1D)即刻與甲醇及其同位素
5
(CH3OD、CD3OH)反應,利用 Lyman-α 波長(121.1 nm)激發產物 H 和
D 原子至其電子激發態,再量測其放射之螢光,經由計算得到 H 和 D
原子之移動能及產物 H/D 分支比。他們認為 O(1
D) + CH3OH 可能的
反應途徑及其標準反應焓如下列所示[15]:
ΔH0f (kJ mol-1)
O(1D) + CH3OH→(HOCH2OH)*→HOCHOH + H
-242.7
(6a)
→ HOCH2O + H -196.6 (6b) → HOCH2 + OH -224.3 (6c) → (CH3OOH)* → CH3OO + H -4.2
(7a) → CH2OOH + H
8.4 (7b) → CH3O + OH -183.7 (7c) 此研究組實驗結果觀測到之產物 OH 主要經由反應途徑(6c)。另一方 面由標準反應焓來看,觀測到產物 H 原子主要經由反應途徑(5a),因 為其大量的放熱,且反應 O(1 D)經由插入甲醇甲基端之 C-H 鍵,所得 到產物 H 為經由中間產物 HOCH2OH 斷 O( 1 D)所產生之新之 O-H 鍵 是斷原本甲醇本身 O-H 鍵之五倍。此研究組另外使用 193 nm 激發混 合反應物 N2O/CD3OH 及 CH3OD/Ar,觀測得到產物 H 的動能為 39.7 kJ mol-1,所以產物 H 原子的反應途徑必為經由放熱反應的式(5a)。此研 究組利用擬一階(pseudo-first-order)實驗條件測量得到 O(1 D) + CH3OH 反應速率常數為 (5.1 ± 0.1) × 10-10 cm3 molecule-1 s-1,基於此反應速率 常 數 及 氘 (D) 的 反 應 速 率 常 數 kD=(0.94 ± 0.5) × 10 -10
6 cm3molecule-1s-1[16],以及實驗 O(1D)與 CH3OH/D2 (1:1)反應光解後 得到的產物[H]/[D]的分支比為 0.97 ± 0.10,經過計算得到 H 原子的量 子產率ΦH= 0.18 ± 0.02,因為在其實驗條件下 O(1D)經 CH3OH 淬熄 (quenching)至 O(3P)的效率相當低[17],如果忽略 O(3P)對產物 OH 的 貢獻,則 OH 的量子產率為 ΦOH= 0.82 ± 0.02。比較這實驗和同研究 組另一 O(1 D)+CH4實驗[16]以及 DeMore 研究組的 O(1D)+H2O 實驗 [15(a)],所得到的不同反應速率,列於表 1-2,由表 1-2 可得到 O(1D) 插入氫氧基(O-H 鍵)的反應速率常數是 O(1 D)插入甲基(C-H 鍵)的 3 倍。 Lee 和 Lin 的研究組[18]以活塞型衝擊波管-原子共振吸收光譜系 統測量 O(3 P) + CH3OH 之反應,並利用模型適解法(modeling fit method)求得此反應在 989-1450 K 的速率常數,以及理論計算B3LYP/ 6-311+G(3df ,2p)密度函數理論計算O(3 P) + CH3OH可能之光解途徑上 各個過渡態(transition state)及最終產物,以及利用CCSD(T)/6-311+G(3 df ,2p)優化結構及位能圖。此研究組以SO2為氧原子的光解前驅物。 將待測的CH3OH/SO2/Ar混合氣體送進衝擊波管偵測區,在反射衝擊 波通過偵測區,系統達成穩定的高溫狀態約100 μs後(衝擊波到達最後 一個偵測器約200 μs後),再導入193 nm ArF準分子雷射(248 nm KrF 準分子雷射),SO2分子會被光解成O及SO,此時產生的氧原子即刻和
7 甲醇分子反應。藉由量測光電倍增管之信號隨時間的變化情形,可推 算出氧原子消失的速率,進而得知O(3 P) + CH3OH反應的速率常數。 O(3P) + CH3OH反應可能產生OH之兩種解離途徑分別為: O(3P) + CH3OH → CH2OH + OH (8a) O(3P) + CH3OH → CH3O + OH (8b) 在反應途徑(7a)為O(3 P)抓取甲醇之甲基端上之氫原子形成一過渡態
TS1,最後斷C-H鍵產生CH2OH + OH,其反應能障為6.3 kcal mol-1,
其最終產物之能量相對於反應物為-5.5 kcal mol-1;反應途徑(7b)為
O(3P)抓取甲醇之氫氧基端上之氫原子形成一過渡態TS2,最後斷O-H
鍵產生CH3O + OH,其反應能障為10.6 kcal mol-1,其最終產物之能量
相對於反應物為2.4 kcal mol-1。而實驗所得到之反應速率常數在 986-1450 K的溫度範圍內,表示如下: K7a(T)=(8.831.26)×10-11exp[-(3476175)/T] cm3 molecule-1 s-1 (1-1) 且於1450 K得到之反應速率常數為8.09×10-12 cm3 molecule-1 s-1。 本實驗使用 248 nm 作為激發光源光解 O3 + CH3OH 及其同位素 O3 + CD3OH、O3 + CH3OD,研究 OH 和 OD 之內能分佈及分支比, 進而了解 O(1 D) + CH3OH 反應之動態學。另使用 193 nm 作為激發光 源光解 O3 + CH3OH,研究其內能分佈。
8 表 1-1. O(1
D)與不同氘取代之 CH3OH 分子反應之產物 OH 與 OD 的
9 表 1-2. O(1 D)和 CH3OH、CH4以及 H2O 的反應速率[14] Reactant Products k×10 -10 cm3molecule-1s-1 CH3OH HOCH2O+H CH3O+OH 0.9 ± 0.01 4.2 ± 0.1 CH4 CH3O+H CH3+OH CH2O+H2 0.2 1.2 0.14 H2O 2OH 2.2
10 參考資料:
1. S. C. Chapman, Memoirs of the Royal Meteorological Society 3, 103-25 (1930)
2. R. P. Wayne, J. Photochem. 25, 345 (1984).
3. W. E. McDermott, N. R. Pchelkin, D. J. Benard and R. R. Bousek, Appl. Phys. Lett. 32, 469 (1978).
4. S. J. Davis, W. E. McDermott and M. C. Heaven, Opt. Sci. Eng. 121, 413 (2007).
5. M. C. Heaven, Ad. Ser. Phys. Chem. 11, 138 (2001).
6. H. A. Michelsen, R. J. Salawitch, P. O. Wennberg and J. G. Anderson, Geophys. Res. Lett. 21, 2227 (1994)
7. E. Murad, W. Swider, R. A. Moss, and S. Toby, Geophys. Res. Lett. 11, 147 (1984).
8. J. M. Greenberg, Astron. Astrophys. 330, 375 (1998).
9. J. Davies and T. Kerr, Int. Astron. Union Circ. No. 7201, 1 (1999). 10. Y. L. Yung and W. B. DeMore, Photochemistry of Planetary
Atmospheres (Oxford University Press, New York, 1999), pp. 99–101.
11. T. Jacq, C. M. Walmsley, R. Mauersberger, T. Anderson, E. Herbst, and F. C. de Lucia, Astron. Astrophys. 271, 276 (1993).
11 (1975).
13. N. Goldstein and J. R. Wiesenfeld, J. Chem. Phys. 78, 6725 (1983). 14. Y. Matsumi, Y. Inagaki and M. Kawasaki, J. Phys. Chem. 98,
3777-3781 (1994).
15. Heats of reactions are calculated based on the heat of formation ΔH0f
(kcal/mol) and the bond dissociation energies D0 (kcal/mol) reported in (a) W. B. DeMore, S. P. Sander, D. M. Golden, M. J. Molina, R. F. Hampson, M. J. Kurylo, C. J. Howard and A. R. Ravishankara Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling; JPL Publication 90-1; NASA: Pasadena, 1990. (b) Benson,S. W. "Thermochemical Kinetics; John Wiley & Sons: New York, 1968. (c) Danvent, B. deB. Eond Dissociation Energies in Simple Molecules; NSRDS-NBS (USA) 3 1; 1970. (d) D. D. Wagman, W. H. Evans, V. B. Parker, I. Halow, S. M. Bailey and R. H. Schumm, NES Tech. Nore 27 & 3, US. Government Printing Office; Washington D.C., 1968. ΔH0f (kcal/mol) values used here
are 104.9 (O(lD)), -48.2 (CH3OH), 52.1 (H), 3.8 ± 2 (CH3OO), -6.2
(CH2OH), 3.5 (CH3O). -54* (HOCHOH),-42* (HOCH2O), and 7*
(CH2COOH). The asterisked values are estimated from ΔH0f
(CH2(OH)2) = - 93.5, ΔH 0
f (CH3OOH) = -31.3, D*(RO-H) = 103.6,
D0(CH(OH)-H) = 92, and D0(CH2(COOH)-H) = 90 kcal/mol. 16. Y. Matsumi, K. Tonokura, Y. Inagaki and M. Kawasaki, J. Phys.
Chem. 97, 6816 (1993)
12
18. C. W. Lu, S. L. Chou, Y. P. Lee, S. Xu, Z. F. Xu and M. C. Lin, J. Chem. Phys. 122, 244314 (2005)
13 第二章 實驗原理 當分子經由光激發至其電子激發後,以物理層次而言,其能量之 轉移可藉由放光機制,如躍遷至較低之相同電子自旋態所產生之螢光 輻 射 (fluorescence) 或 躍 遷 至 較 低 之 不 同 電 子 自 旋 態 所 產 生 燐 光 (phosphorescence)輻射過程,或藉由非輻射方式之能量轉移方式,如: 與鄰近之分子碰撞而 relaxation、分子內振動能轉移(intramolecular
vibrational energy redistribution,IVR)、內轉移(internal conversion,
IC)及系統間轉移(intersystem crossing,ISC)過程而使能量衰減。在化 學層次上,分子受光後可能經躍遷至其排斥能態而直接斷鍵,或躍遷 至預解離之位能面而間接斷鍵,或異構化(isomerization)形成其他異構 物。因此,光分解之動態學研究可使吾人能於分子之層級深入探討其 光化學過程,如:斷裂碎片之移動能分佈、角分佈、內能及反應過程 中結構變化,亦可深入地探討初始母分子之振動模式與產物能態分配 之關聯性。 在 光 分 解 的 實 驗 中 , 光 解 碎 片 移 動 能 光 譜 法 (photofragment
translational spectroscopy : PTS)常搭配之偵測技術如飛行時間譜(time
of flight)、氫原子雷德堡態飛行時間譜(H-atom Rydberg tagging time of
flight)、都卜勒位移光譜(Doppler spectroscopy)及離子成像(ion imaging) 等方式,以上各種技術可提供吾人詳盡之化學反應其產物動能分佈及
14
角分佈資訊,對於簡單的系統,較有可能利用能量守恆間接地估計產
物之內能,但對於較複雜的系統,以此技術來研究產物內能的分佈較
為困難。
研究產物之內能分佈則多由下幾種常用之量測技術,如雷射誘發 螢光(laser-induced fluorescence, LIF)、共振加強多光子游離光譜
(resonance-enhanced multiphoton ionization spectroscopy,REMPI)、紅 外放光或吸收光譜等。除了同核雙原子分子外,大部分的分子均會吸 收紅外光。由於不同分子的紅外光譜如人的指紋般各不相同,因此紅 外光譜法常用於定性或定量分析。 若欲偵測之物種具有較高之振動能,則觀測 的振動能階之 紅外放光是最直接的方法,但無法得到 能階之資訊。利用傳統 分光光譜法可鎖定特定波長作檢測,但掃描波長極度費時,靈敏度低, 太強或太弱的訊號都無法測出,光譜解析度差,並不適合進行高解析 度 光 譜 的 研 究 。 霍 式 紅 外 光 光 譜 儀 (Fourier-transform infrared spectrometer,FTIR)具有高偵測靈敏度、高波長準確度、高解析度及 多重波長(multiplex)偵測性等特性,因此,幾乎已經取代傳統分光式 光譜儀。 近 年 來 所 發 展 之 步 進 式 掃 描 霍 式 轉 換 光 譜 儀 (step-scan Fourier-transform spectroscopy,ss-FTS),結合脈衝式雷射,使舊領域
15 不再受限於僅能鑑定穩定分子之結構,對於生命期短暫或不穩定之物 種可利用此技術成功地監測。此技術最大特點為一次實驗中即能獲得 有關紅外光放光或紅外吸收之時域解析及波長解析之訊息。因此,已 被應用於偵測瞬態物種之紅外放光[1-3]、凝態[4,5]及氣態[6-10]之 時域解析吸收光譜,可進行化學動力學、動態學及光譜學等研究。 2.1 霍式轉換紅外光譜儀 西元 1891 年,Michelson 發明干涉儀(interferometer)[11],基本 原 理 為 藉 由 光 程 差 使 入 射 光 產 生 干 涉 現 象 以 紀 錄 其 干 涉 圖 譜 (interferogram),再利用理論推算將干涉圖譜轉換成一般傳統光譜。礙 於當時技術有限,且轉換程序相當粗略,故僅能解析鈉原子線光譜的
細微結構(fine structure)。西元 1949 年 Fellget[12]、Jacquinot[13]等人
提出利用 FTS(Fourier-transform spectroscopy)方法,可在短時間內得
到比傳統上利用光柵或稜鏡等方法所得具有更高解析度的光譜。至西
元 1965 年,Cooley 和 Tukey 等人[14]提出一種快速霍氏轉換(fast
Fourier transform,FFT),使計算時間減少,提升干涉圖譜的轉換效率。 其後,由於微電腦與小分子氣體雷射兩大技術的發展,藉由使用氦氖
雷射(He-Ne laser)[15]精確地量測干涉儀中移動鏡造成的光程差,並結
合新進之微電腦處理系統,使計算的時間減少,尤其當序列長度較長
16
逐漸取代利用分光技術的傳統紅外光譜儀。
2.1.1 Michelson 干涉儀基本原理
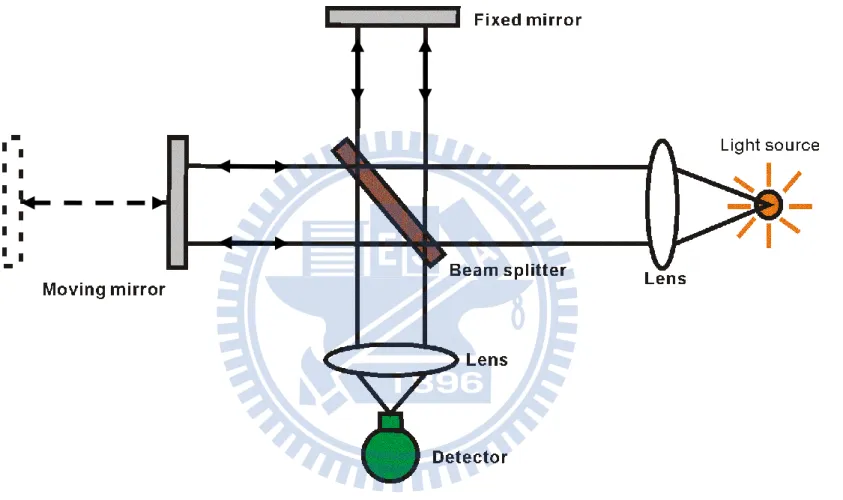
霍式轉換紅外光譜儀的核心部分包含 Michelson 干涉儀及訊號擷 取處理系統。Michelson 干涉儀主要由分光片(beamsplitter)、移動鏡
(moving mirror)及固定鏡(fixed mirror)所組成[14],其光學結構如 圖 2-1 所示。光源經過聚焦鏡後,形成帄行光進入干涉儀並導向分光 片。理想情況下,分光片將入射帄行光帄均分成強度相同的兩道光束, 其中一道光束穿透分光鏡,經由移動鏡反射後再經由分光片反射導向 偵測器,另外一道光束則由分光片反射至固定鏡,之後再經由固定鏡 反射後穿過分光片並導向偵測器。當移動鏡沿著光軸移動時,匯集於 偵測器之兩道光束所經過的光程不同,造成相位差(phase difference) 的改變,因而產生干涉現象。 當移動鏡與分光片之距離和固定鏡與分光片的距離相等時,此時 兩道光束經過相同的光程,其光程差 δ 為 0,偵測器量測到很強的訊 號。對於單色光而言,當光程差為半波長的偶數倍( ; )時,此兩道光束抵達偵測器時為同相位(in-phase),形成 建設性(constructive)干涉,此時匯集之光強度最強;若光程差為半波 長之奇數倍( ; ),則兩道光束抵達偵測器時 為反相位(out-of-phase),形成破壞性(destructive)干涉,光束強度最弱。
17 因此當移動鏡來回移動時,由於光程差的改變,使得匯集之光束重複 地經過建設性及破壞性干涉,故偵測器所量測之訊號會隨之變強變弱; 當移動鏡以定速(v)移動時,光程差之變化率隨時間成正比,為時間 之函數(δ = 2vt),因此,可由偵測器測得一隨時間變化之干涉圖譜 (interferogram)。 2.1.2 干涉譜與傳統光譜換式轉換關係 如圖(2-1)所示,當一波數為 之單色光經過分光片後,分成兩束 光,此兩光束在分別經移動鏡和固定鏡反射後,其電場變化可分別表 示為: (2-1) (2-2) 其 中 R 為 分 光 片 之 反 射 率 (reflectance) 、 T 為 分 光 片 之 穿 透 率
(transmittance)、C 為與偏振相依(polarization dependent)之常數、 為 電場強度振幅、 為電磁波之角頻率(angular frequency, ),t 為時間點(time)、~為波數(wavenumber)、 為光程差、 為固定鏡 與分光片的距離。 因為光束強度 I 與 成正比,故光束經過分光片分開最後匯集 於偵測器之光束強度為:
18 (2-3) 其中 為與波數相依(wavenumber dependent)之常數,即傳統之光譜 函數, 為單色光之干涉譜。對單色光而言,其干涉譜為餘弦曲線 圖形,因為每當光程差為波長的整數倍時,光強度最強。圖 2-2 為 不同光源之傳統光譜及其對應的干涉圖譜,其中(a)顯示為單色光源 的干涉圖譜,為一簡單的餘弦波,(b)顯示兩道頻率相近之單色光源 的干涉圖譜相互干擾,(c)顯示一連續光源之干涉圖譜,除了δ 具 有高強度外,其餘光程差強度均極弱。 利用霍氏轉換方式即可將干涉圖譜轉換成傳統光譜,其數學式如 下:
~ Im ~ Re ~ 2 sin ~ 2 cos ~ 2 ~ i d I i d I d e I B i
(2-4) 式(2-4)之實數部分可描述干涉圖譜經霍氏餘弦轉換所得到之傳統光 譜,如下表示:
I
d B
cos 2 ~ ~ (2-5) 理想之干涉圖譜應為一左右對稱圖形,因此可將(2-5)式改寫成兩 倍的 到 積分表示:
I
d B
0 ~ 2 cos 2 ~ (2-6) 亦即理想之干涉圖譜必頇記錄光程差由零至無限大的強度,但實際上,19
移動鏡移動的距離有限,光程差無法達到無限大。假設實驗上只能得
到 到 之間的干涉圖譜,以 表示,則該干涉圖譜如同理想
之干涉圖譜 受到一匣式截斷函數(boxcar truncation function)
的作用[16],而 的定義如下:
L D L D 當 當 0 1 (2-7) 因此,偵測器所測得的光束強度隨光程差的變化函數可改寫成下列的 式子:
I D I (2-8) 即傳統光譜 為
I
D
d B
0 ~ 2 cos 2 ~ (2-9) 根據霍氏分析卷積定理(the convolution theorem of Fourier analysis),兩個函數之乘積的霍氏轉換為此兩個函數個別霍氏轉換後之卷積
(convolution)。匣式截斷函數 作霍氏餘弦轉換後為一 sinc 函數
,而此函數稱為儀器譜線形狀函數(instrumental line shape function,
ILS),其數學表示式如下: L L c L f 2 sin 2 ~ ~ ~ 2 sin ~ (2-10) 其圖形如圖 2-3(a)所示。而 作霍氏餘弦轉換後為 ,因此,理 想傳統光譜和儀器譜線形狀函數卷積的結果為:
20
d D I f B G
0 ~ 2 cos 2 ~ ~ ~ (2-2) 其中 為實驗所得到的真實光譜,*表示卷積。對於單色光 而言, 上式可簡化為:
c
L
LB f B G ~ ~ 2 sin ~ 2 ~ ~ ~ 1 1 (2-3) 其圖形如圖 2-3 所示,原本應為單一波數~1且頻寬無限窄的圖譜,由 於移動鏡無法移動至無限遠處而經匣式截斷函數修正,使得譜線變寬,主峰之半高寬(full width at half maximum,FWHM)為0.605
L ,此半高 寬常被用來表示霍氏紅外光譜的理論解析度(theoretical resolution)。被 截斷的干涉譜經轉換後的傳統光譜,在主峰兩側會有小側波(side lobe),常會和鄰近的微弱吸收混淆。 為了除去匣式截斷函數造成的側波干擾,可用其他函數取代匣式 截斷函數,其作用彷彿削去主峰旁的足部一樣,故稱此類函數為削足 函數(apodization function)。表 2-1 列出幾種簡單的削足函數[17],從 中可發現削足函數雖然可以降低測波之干擾,但卻也導致主峰的頻寬 增加。因此,如果頻寬不是重要的考量,則可選擇 值較小的削足 函數;反之,若頻寬是主要的考量因素,則可選用 FWHM 較小的削 足函數。除了這些考慮因素外,還要考量解析度與譜線密度,再選擇 適合該實驗條件的削足函數。
21 本實驗使用的削足函數為 Hamming(又稱 Happ-Genzel)函數,其 定義如下:
L A L L A 當 當 0 cos 46 . 0 54 . 0 (2-13) 經由霍氏轉換之後,其儀器譜線形狀函數的數學表示式如下:
2 2
3~
3
8 ~ 2 ~ 2 sin ~ 64 . 0 08 . 1 ~ L L L L L f (2-4) 由於電子濾波器、光學元件及不當取樣等因素會造成相位差 (phase error),影響干涉圖譜之對稱性。如光學元件對不同頻率的光有 不同之響應,對不同頻率的光也會產生不同的相位延遲( ,phase lag)效應,因此,必頇利用相位修正(phase correction)來修正此誤差。 亦即(2-5)式必頇加上 以進行相位修正,才能描述真實之干涉圖譜:
~ ~ sin ~ 2 sin ~ cos ~ 2 cos ~ ~ ~ ~ 2 cos ~ d B d B I
(2-5) 上式中之 效應相當於原餘弦函數中引入一正弦函數成分,使原本 以 對稱之干涉譜變得稍不對稱。如果只是以餘弦霍氏轉換將會 導致光譜上的誤差,因此,(2-4)式中,將干涉圖譜進行霍氏餘弦及正 弦轉換後即可得相位角:
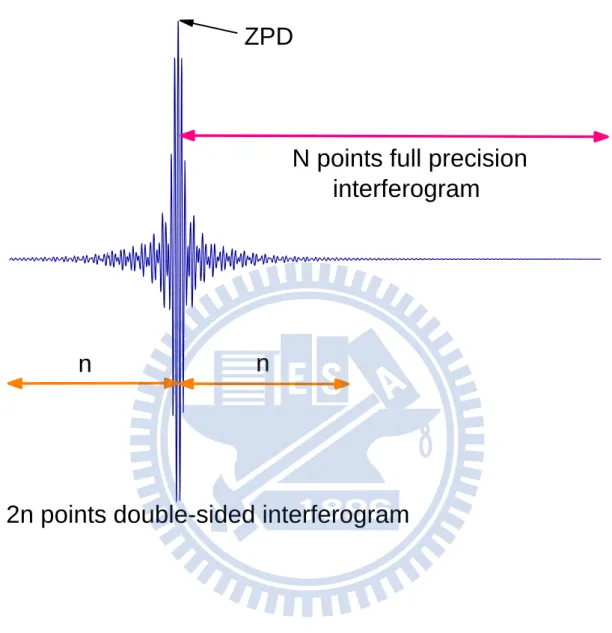
~ Re ~ Im arctan ~ (2-6) 最後,可進一步得到修正後之傳統光譜。22 此外,不當取樣也會影響所量測之干涉圖譜。當理想的干涉圖譜 對稱於 ,但如果第一個取樣點並非於 ,而是在 時, 實際之干涉圖譜應修正表示為[14]:
0 ~ ~ 2 cos ~ B d I (2-7) 此因素所造成的誤差也和有相位差類似。因此,無論是不當取樣或濾 波等因素所產生干涉譜的相位誤差均可以相位修正之數學步驟加以 修正,以避免光譜轉換後所得之傳統光譜發生嚴重的誤差。本實驗所 使用的相位修正方法為 Mertz method[18][19]。 通常實驗僅擷取單邊之干涉圖譜(single-sided interferogram),以 節省掃瞄時間及縮短霍氏轉換運算量及時間,故相位校正程序相當重 要。實驗時於干涉圖譜 左側多取 n 個數據點,得到一個含 2n 個 數據點的雙邊干涉圖譜,如圖 2-4 所示,再將此區域進行 FFT 轉換, 以取得相位誤差資訊,作為相位校正。而 n 值的大小取決於實驗時所 選擇的相位解析度(phase resolution),一般而言,n 值必頇大於 250。 一般的霍氏轉換紅外光譜儀有三組不同光源之干涉儀,三組干涉 儀共用分光鏡和移動鏡。三組光源包括連續波長的紅外光源、氦氖雷 射以及連續白光光源,分別做為偵測樣品光譜、測量取樣之相對光程 差、定義零光程差點之用途,圖 2-5 為其干涉圖譜及傳統光譜。氦氖 雷射可提供頻率極為穩定之單色光源(波長= 632.8 nm),故其干涉圖23 譜為一餘弦函數。如圖 2-6 所示,餘弦波每段波長有兩個零交叉點, 其固定間隔為 316.4 nm。霍氏轉換紅外光譜儀以氦氖雷射干涉圖譜的 零交叉點(zero-crossing)做為定位點,建立一個固定光程差的量度法, 並以之作為取樣的間隔。由於此餘弦波之頻率與移動鏡速率成正比, 若移動鏡速率稍有變動時,則餘弦波之頻率亦隨之改變。因此,電腦 係依光程差之改變(即:He-Ne 之 zero-crossing)而非時間的改變來取 樣,才能確保每一個取樣點之光程差的準確性。由於氦氖雷射只能定 位移動鏡位移每段距離的相對位置,故利用白光光源干涉圖譜的最高 點作為零光程差位置的訂定。如圖 2-5 (c)所示,連續波長的白光, 其干涉圖譜在 時,為完全建設性干涉,強度最大,而 時強 度迅速減弱,故可產生一個強而窄的訊號,以此定位取樣的起始點。 然而,本實驗所使用的 Bruker FTIR 是利用步進式馬達來驅動移動鏡, 可精準的定位移動鏡的位置,因此僅使用兩組干涉儀,不需使用連續 白光干涉儀的部分。在實驗正式擷取數據前,先利用光譜儀的紅外光 源(globar)對正(align)干涉儀,並儲存干涉圖譜的波峰相對之光程差位 置,作為零光程差的參考基準點,以確保每個干涉圖譜擷取訊號的起 始點一致,才可多次掃瞄光譜而不失真。 2.2 霍式轉換紅外光譜儀的優點 霍式轉換光譜儀相較於傳統分光光譜儀有下列之優點:
24 1. 高光通量之優點(throughput advantage): 由於傳統分光儀的解析度受到狹縫開口的限制,而干涉儀無頇使 用光狹縫及光柵等裝置,只是利用光圈來限制光的散射角,因此光通 量遠高於傳統分光儀。相較於傳統分光儀,偵測器所測得訊號較強, 靈敏度較高,也有更好的訊雜比,此優點由 Jacquinot 提出,故又俗 稱 Jacquinot 優點[13]。 2. 多重波數之優點(multiplex advantage): 傳統掃描式之光譜儀只能掃描一小段波長,並需要分光掃描,故 容易受到光源或吸收物濃度變化之影響,而且單一光柵並不適用於所 有光區,所以在更換時會有光譜不連續的情形。而 FTIR 是利用干涉 現象來分光,故能同時偵測整個光區,且在更短的時間內擷取整個光 區的光譜。因此,對於相同光譜範圍及相同解析度之光譜,FTIR 所 需的偵測時間遠小於傳統的分光儀。此外,在相同時間下,干涉儀可 藉由多次掃瞄帄均來提高訊雜比。如果雜訊是以隨機的形式出現,則 該訊雜比(signal-to-noise ratio,SNR)與掃瞄次數 N 的關係如下: N SNR (2-18) 此優點由 Fellgett 提出,亦稱 Fellgett 優點[12]。
3. 高波數精確性之優點(spectral accuracy advantage):
25 擇的波數,必頇利用標準樣品產生的已知譜線來校正其絕對波數位置。 而干涉儀則是利用氦氖雷射精確地標定光程差,經由霍氏轉換干涉圖 譜得到波數準確度可達 0.001 cm-1。因此,干涉儀在波數的準確度上 遠高於傳統分光儀,且不需要另外進行波數的校正工作。此優點由 Connes 提出,又稱 Connes 優點[20]。 4. 高解析度之優點(high-resolution advantage): 傳統分光儀的解析度主要是受到光柵刻痕密度的限制,一般傳統 分光儀之解析度不易優於 0.1 cm-1。而干涉儀的解析度是與最大光程 差max成反比,關係式如下: max 2 1 1 ~ ~ (2-19) 其中~1、~2為兩道恰可完全解析的波數, 即為完全解析度[14]。而 最大光程差max即為移動鏡移動距離的兩倍;因此,解析度亦可與兩 倍的移動鏡移動距離成反比。目前市售 FTIR 之最大解析度可達到 0.001 cm-1的解析度,遠較於傳統分光儀高。 5. 抑制散逸光之優點(stray-light control): 波長與單光儀設定不同之光子穿越入口狹縫或光圈後,理論上不 應由出口狹縫射出,但因為光學零件之不完美,極少部分仍可由出口 狹縫散逸而被偵測器偵測到,此稱為散逸光(stray-light)。對於傳統分 光儀而言,欲降低此散逸光並不容易。然而就干涉儀而言,對於每一
26 波數~的單光光源,若移動鏡的移動速率為 v,則偵測器可測得餘弦 干涉訊號頻率為 。選用適當的電子濾波器將其他頻率範圍的 訊號過濾除去,則偵測器僅能測到特定波段的訊號,便可有效抑制其 他波段的散逸光。 6. 靈活且應用廣泛(versatile)之優點: 只要選擇適當的光源、分光鏡、鏡片及偵測器等光學元件,便 可將霍氏轉換光譜儀應用在遠紅外光、中紅外光、近紅外光、可見光 或紫外光區的測量。此外,若再對光譜儀搭配其他元件,例如:氣相 層析儀(GC)、液相層析儀(HPLC)、質譜儀(MS)、多重反射吸收槽 (White cell)、衰減式全反射(ATR)等,即可應用於其他定量、定性的 分析。 2.3 時間解析-霍式轉換紅外光譜法 一般的霍氏轉換紅外光譜儀取樣模式並不適用於觀測瞬態訊號, 因此對一些生命期較短的物種,例如:自由基、反應中間物、微弱鍵 結分子及高激發態的分子等,都無法進行鑑定。為了改進此不足,目 前已發展出許多技術,使得霍氏轉換紅外光譜儀具有時間解析的功能。 常見的各種方法介紹如下: 一. 連續式掃瞄模式(continuous-scan mode): 未將霍氏轉換紅外光譜儀取樣模式做任何重大的改變,只是結合
27
適當之反應系統。連續式掃瞄模式又可分成幾種的掃瞄技術:
1. 氣流管法(flow tube method):
此實驗方式乃將 FTIR 與裂解氣流管(discharge flow)結合。於管
中任一位置化學反應的程度皆維持在 steady state 的狀態下,藉由改變 氣流管內伸縮管導入反應物開始反應與偵測器(FTIR)的距離,以觀察 不同反應時間的紅外放光之變化。儘管此實驗裝置已可觀測產物隨時 間變化的放光光譜,但受限於氣流管內伸縮管可改變的距離及氣流管 的抽氣速率,此實驗方式的時間解析度只達數十毫秒範圍,無法觀測 更短時間之化學變化[21-24]。 2. 快速掃瞄 (rapid scan): 直接利用移動鏡快速掃瞄一次或數次的時間為時間的解析度,故 時間的解析度主要受限於移動鏡的移動速率。而移動鏡在快速移動中, 其穩定度也間接限制了移動速率,因此利用快速掃瞄可達到的時間解 析度也只能有數十毫秒(ms)範圍[25],且訊雜比通常受到限制。 3. 同步式掃瞄(synchronous scan): 移動鏡需保持固定速率持續的移動,通過零交叉點時送出脈衝以 觸發反應,產生瞬態放光,並同時在固定延遲時間擷取訊號[1-3,5, 26]。以一般市售之 FTIR 為例,如果其移動鏡的最小移動速率為 0.05 cm s-1,則每秒會通過 3161 個零交叉點。對目前進行光解的高能量脈
28 衝雷射來說,是很難產生如此高重複頻率但強度足夠又穩定的之雷射 光;而且在高重複頻率的操作下,反應系統內氣體的更新速率也相對 要提高,但不易有幫浦可滿足如此高抽氣速率的條件。此外,在同步 式掃瞄模式下,移動鏡移動速度是否能長時間維持其穩定性,亦是造 成誤差的大問題之一。 4. 非同步式掃瞄(asynchronous scan): 非同步式掃瞄是利用移動鏡反覆穩定地掃瞄,光解雷射以另一頻 率重覆觸發。每次觸發雷射後,在固定延遲時間擷取訊號,經過多次 掃瞄之後,將訊號點集合在一起所成的干涉圖譜轉換成傳統光譜。所 以雷射之觸發與干涉圖譜掃瞄之間並非同步進行,即氦氖雷射干涉圖 譜和反應起始時間並沒有關連。此方法的優點是反應觸發無頇與移動 鏡到達零交叉點的時間同步,可避免同步式掃瞄對光解雷射的高重複 頻率之要求。然而,非同步式掃瞄的缺點是每一次實驗只能得到某單 一時間下的光譜,無法一次得到所有觸發後不同時間下的光譜[5]。 二. 步進式掃瞄模式(step-scan mode): 步進式掃瞄模式中移動鏡並非如傳統 FTIR 般連續式地移動,而 是利用電子儀器控制移動鏡精準地定位在氦氖雷射零交叉點上,後才 觸發反應,開始擷取時間解析的訊號。當每次移動鏡移動到下一個定 位點時,需要時間待其穩定靜止,此時間稱為定位時間(settling time),
29 定位時間與移動鏡所走距離有關,通常在 20-100 ms 之範圍內。待移 動鏡穩定後,反應隨即被觸發並開始擷取訊號。在此一特定的移動鏡 位置(定位點)上,可累積多次的訊號加以帄均後,再移動到下一個定 位點擷取訊號,待完成所有的擷取訊號程序後,重新組合並轉換成光 譜,即可得到不同時間下的光譜[7][9-10][27-28]。 步進式掃瞄模式示意圖如圖 2-7 所示。當移動鏡穩定停在 x1位 置,待反應觸發之後,每隔固定的時間間隔擷取訊號,並觸發多次反
應,累加多次訊號作帄均,即可得到訊號序列 I(x1,t1)、I(x1,t2)、I(x1,
t3)、……、I(x1,tm)。接著,當移動鏡移動到下一個位置 x2,並取得
訊號序列 I(x2,t1)、I(x2,t2)、I(x2,t3)、……、I(x2,tm);重複此方式
至擷取完所有的訊號序列,將這些訊號序列組合成訊號陣列。最後, 重組訊號序列,例如:在同一時間 tk下的訊號序列 I(x1,tk)、I(x2,tk)、 I(x3,tk)、……、I(xn,tk)即相當於在時間 tk下所測到的干涉圖譜,再 轉換成傳統光譜,即可得到相當於時間 tk之傳統光譜;對所有之時間 點 tj作相同的轉換,即可得到時間解析的光譜。 與同步式掃瞄模式相比較,步進式掃瞄可以經由掃瞄一次得到各 個不同反應時間下的光譜;取樣過程中,在每個訊號擷取點可進行多 次訊號的累計以提高訊雜比,對於能量不穩定的雷射脈衝亦有帄均其 脈衝強度的效果,其累計帄均之時間原則上亦不受限制,故不需要極
30 高重複頻率之雷射光或高效率抽氣幫浦。 由於氦氖雷射波長為 632.8 nm,即每一個零交叉點相距 316.4 nm, 若移動鏡在每一個零交叉點停留取樣,則能偵測波段範圍為 15802 cm-1的訊號,即可量測光譜範圍為 0-15802 cm-1、15802-31604 cm-1… 等。但是實驗時,常常不需要如此寬廣之光區,為了節省取樣時間, 此時就可使用跳點取樣(undersampling)的方法來進行掃瞄,即移動鏡 不在每一個零交叉點都停留取樣,而是固定間隔數個零交叉點才取樣。 舉例來說,若每兩個零交叉點才停留取樣,其偵測波段範圍為 7901 cm-1,則可量測之最大光譜範圍為 0-7901 cm-1或 7901-15802 cm-1;如 果每隔三個零交叉點才停留擷取訊號,能偵測波段範圍為 5267 cm-1, 可量測之光譜區間為 0-5267 cm-1、5267-10534 cm-1 或 10534-15082 cm-1等光譜訊號,以此類推。因此,在相同的解析度下,欲測量的光 譜範圍越窄,則其可跳過的零交叉點越多,使得取樣點減少,故所需 的時間也就越少。但使用跳點取樣必頇注意在偵測訊號時,不能有非 偵測光區的光線進入偵測器,所以必頇加入濾光片(optical filter)將欲 偵測光區以外的光源濾掉,以免造成偵測光區以外的光源訊號疊合 (folding)或失真(aliasing),造成不必要的譜線干擾。 除了訊號的再現性、偵測器本身的雜訊干擾會影響光譜資訊之精 確度之外,進行步進式掃瞄時,受限於移動鏡位置之準確度與穩定性,
31 其取樣定位點的誤差會影響光譜的訊雜比。即移動鏡位置的不準確度 將造成訊雜比降低,其關係式如下:
(2-20) 其中 (cm)為移動鏡位置的誤差, (cm-1)為光譜最大的波數[13]; 當 越大時,所得到的訊雜比越差。目前技術使得移動鏡停留的位 置的準確度可達到± 0.2 nm [29],若 ,則 SNR = 18000。所以吾人目前實驗之訊雜比並不受限於移動鏡停留位置的準 確度,而是受限於偵測器與類比/數位轉換器反應時間(response time) 以及偵測器本身的雜訊干擾。 另外,由連續式掃描模式(continuous-scan mode)轉成步進式掃瞄 模式(step-scan mode)中,移動鏡並非來回移動使偵測器偵測到光源 經調制(modulate)後的頻率 ,而是停留在固定位置上,訊號由
交流耦合模式(ac-coupled mode)轉成直流耦合模式(dc-coupled mode)。
因此,在步進式掃描模式上觀測連續式的放光是要測直流耦合訊號。 但是在偵測瞬間產生的放光時,如果其信號之變化率極快時,則可以 利用交流耦合模式以減少儀器的長時間的信號偏移。此時,ac 訊號要 在下一發雷射觸發前能夠回到零點,故需依雷射的重覆率(一般雷射 低於 100 Hz,即 10 ms)來決定偵測器之 RC 電路(RC circuit)充放電之 時間週期。原則上,偵測反應時間之上限只受限於偵測器、放大器及
32 干涉儀數位化電子元之響應時間(response time)。但是如果因為訊號 強度不夠,則頇累積較長時域以增進訊雜比,因而使得時間解析度變 差。 此外,在實驗前應利用相同的偵測系統及相同的實驗條件下,量 測偵測器及其他相關電子儀器之響應時間,實驗取得時間解析之光譜 後,才能精確地推得有效的紅外光訊號之起始時間,關於本系統電子 儀器之響應時間的量測,請參見本論文第三章第二節。
33
圖 2-1. Michelson 干涉儀之示意圖。其主要由分光片(beam splitter)、移動鏡(moving mirror)及固定鏡(fixed mirror) 所
34
Retardation ( ) Wavenumber ( )
圖 2-2. 不同光源之傳統光譜(右側)及其對應之干涉圖譜(左側)。
35 (a) 0.605/L Hm Hs 2L 1/L (a) f( )~ (cm-1) ~ (b) 1/L 1 ~ ~1 2L 1 (cm- 1) ~ ~1 2L 1 B( )~ (b) 圖 2-3. 在有限之位移(L)下量測單色波數之頻寬變化:(a) 以匣式截 斷函數進行霍氏轉換後之圖譜,其波形為 sinc x 函數;(b) 單色光波 數為1之正弦干涉圖譜以匣式截斷函數進行霍氏轉換後之圖譜,此光 譜的最大位移距離為 L cm。
L c
L
f ~ 2 sin 2~
LB
c
L
B~ 2 ~1 sin 2 ~1~36 圖 2-4. 干涉圖譜之取樣示意圖。實驗擷取單邊之 N 點干涉圖譜,並 以零光程差點為中心,相位校正時,左右各取 n 個點數以進行相位校 正。
n
n
N points full precision
interferogram
2n points double-sided interferogram
ZPD
37 圖 2-5. 干涉圖譜及其對應之傳統光譜。(A)連續波長的紅外光源;(B) 氦氖雷射;(C) 連續白光光源。 (A) IR 光譜 (B) He-Ne laser (C) 白光光譜 632.8 nm visible
38 圖 2-6. 氦氖雷射之干涉圖譜。圖中實心方格為零光程差點,實心圓 點為零交叉點。 -1000 0 1000 2000 3000 4000 0 path difference (nm) Int ens it y
zero path difference
39 圖 2-7. 步進式時間解析掃瞄模式取樣示意圖。X 為光程差,t 為反應 時間,I為訊號強度。 I:intensity of the interferogram intensity of the interferogram optical path difference of the interferometer (Xn)
T
X
t
time evolution of the process (tm)t
1t
2t
3t
4 Data acquisition43
表 2-1:數種簡單削足函數對儀器譜線形狀函數的影響[16]。1 2為主峰之
半高寬(full width at half maximum),實驗可測得之光程差 δ 的範
圍為 -a≦δ≦a。而 為側波最大振幅值H (side lobe amplitude S
maximum)與主峰高度H 之百分比絕對值。 m
Apodization Function Instrument Function
Bartlett Blackman Cosine Gaussian Hamming Boxcar(Uniform) Welch
44 參考資料:
1. E. L. Woodbridge, T. R. Fletcher, and S. R. Leone, J. Phys. Chem. 92, 5387 (1988).
2. P. W. Seakins and S. R. Leone, J Phys. Chem. 96, 4478 (1992).
3. P. W. Seakins, E. L. Woodbridge, and S. R. Leone, J Phys. Chem. 97, 5633 (1993).
4. J. M. Preses, G. E. Hall, J. T. Muckerman, T. J. Sears, R. E. Weston, Jr., C. Guyot, J. C. Hanson, G. W. Flynn, and H. J. Berstein, Rev. Sci. Instru. 64, 95 (1993).
5. K. Matsutani, A. Yokota, and M. Tasumi, Appl. Spectrosc. 46, 560 (1992).
6. R. E. Murphy and H. Sakai, Aspen. Int. Cof. Fourier Spectrosc. [Proc.], Aspen, Colorado, 1970, p. 301.
7. R. E. Murphy, F. Cook, and H. Sakai, J. Opt. Soc. Am. 65, 600 (1975). 8. D. E. Heard, D. G. Weston, and G. Hancock, Appl. Spectrosc. 47,
1438 (1993).
9. G. V. Hartland, D. Qin, and H. –L. Dai, A. Simon and M. J. Anderson, Rev. Sci. Instrum. 63, 3261 (1992).
10. G. V. Hartland, W. Xie, and H. –L. Dai, J. Chem. Phys. 100, 7832 (1994).
11. A. A. Michelson, Phil. Mag., Ser. 5, 31, 256 (1891). 12. P. Fellgett, J. Phys. Radium 19, 187 (1958).
13. P. Jacquinot, Rep. Progr. Phys. 23. 267 (1960).
45
Spectroscopy (John Wiley & Sons, Inc., New York, 1986).
15. A. Javan, W. R. Bennet, Jr., and D. R. Herriott, Phys. Rev. Lett. 6, 106 (1961).
16. J. Kauppinen and J. Partanen, in “Fourier Transform Infrared Spectroscopy”, 1st edition (Berlin, Germany, 2001)
17. http://mathworld.wolfram.com/ApodizationFunction.html 18. L. Mertz, Transformations in Optics, Wiley, New York (1965). 19. L. Mertz, Infrared Phys. 7, 17 (1967).
20. J. Connes, and P. Connes, J. Opt. Soc. Am. 56, 896 (1966). 21. D. J. Donaldson and J. J. Sloan, J. Chem. Phys. 82, 1873 (1985). 22. E. Arunan, G. Manke II, and D. W. Setser, Chem. Phys. Lett. 207, 81
(1993).
23. N. I. Butkovskaya, and D. W. Setser, J. Chem. Phys. 106, 5028 (1996).
24. N. I. Butkovskaya, and D. W. Setser, J. Phys. Chem. 102, 9715 (1998).
25. L. Mertz, Astron. J. 70, 548 (1965).
26. C. A. Carere, W. S. Neil, and J. J. Sloan, Appl. Opt. 35, 2857 (1996). 27. R. A. Palmer, C. J. Manning, J. A. Rzepiela, J. M. Widder, and J. L.
Chao, Appl. Spectrosc. 43, 193 (1989).
28. D. E. Heard, R. A. Brownsword, D. G. Weston, and G. Hancock, Appl. Spectrosc. 47, 1438 (1993).
46 第三章 實驗技術與數據處理 3.1 實驗裝置 本實驗之系統裝置主要分成三個部分:雷射系統、反應系統、偵 測系統,如圖 3-1 所示。 3.1.1 雷射系統
本實驗利用氟化氪準分子雷射( KrF excimer laser;Lambda Physik,
model LPX 240i ),產生波長為 248 nm 的無偏極性雷射光,以反射 鏡及 Ca 透鏡將雷射光束經 S1UV 材質的兩吋光窗導入反應槽中央, 利用透鏡調整光解區域之光解截面積,控制光解通量( fluence )。本實 驗中,吾人控制雷射以 19 Hz 之重複頻率操作,將氟化氪準分子雷 射導入反應槽中並聚焦至 1.5 × 0.8 = 1.2 cm2 ,雷射能量約為 60-65 mJ, 估算光通量約為 50-54 mJ cm-2 (約 6.5 × 1016 photons pulse-1 cm-2 )。 利 用 氟 化 氰 雷 射 (ArF excimer laser ; Lambda Physik , model
CompexPro 50 F-Version),產生波長為 193 nm 的無偏極性雷射光,吾 人控制雷射以 19 Hz 之重複頻率操作,並利用可調焦距式的聚光透 鏡組將氟化氪準分子雷射導入反應槽中並聚焦至 1.0 × 1.0 = 1.0 cm2 , 雷射能量約為 24-25 mJ,估算光通量約為 24-25 mJ cm-2 (約 2.4 × 1016 photons pulse-1cm-2 )。 3.1.2 反應系統
47 反應槽為一個六通之不鏽鋼腔體,如圖 3-2 所示。在雷射光束通 過之 X 軸方向的兩面腔體封蓋上各裝上材質為 S1UV 材質的兩吋光 窗(λ>180 nm,T>90 %),使雷射可以入射腔體;垂直於雷射光束行 進 Y 軸方向之腔體內部裝置 2 組已切割之鍍金球面鏡(Welsh mirror, 直徑 2 吋,焦距 4 吋,2 組距離相距 4 吋),利用多重反射來收集產物 之放光。鍍金球面鏡在紅外光區反射率約 96 %,於本系統中,Welsh mirror 至少可以收集八次反射之放光,理論上可以使收集的訊號強度 增加約 6-7 倍[1,2]。所收集之放光在經由材質為 Ca 的兩吋光窗(在 800-5000 cm-1光區中,穿透度 )射出反應槽,導入光譜儀中。 Z 軸方向為氣體進氣方向,反應氣體從反應槽下方經一狹長隙縫流入, 利用針閥(needle valve)及流量計(flow meter)來調節監控反應氣體之
流量。
在反應槽上方的管路連接至乾式真空幫浦(dry pump,TAIKO,
model BEH-1800,抽氣速率為 30000 L min-1),從反應槽上方 (Z 軸 上方) 抽出光解反應後之氣體。在幫浦與反應槽間裝置有閘式真空閥
(gate valve)及碟形真空閥(butterfly valve)以調節抽氣速率用以控制
反應槽內之總壓。反應槽之壓力以電容式壓力計(MKS Baratron,
model 622A,10 Torr)測量;各反應物之分壓乃由反應槽內總壓乘上 反應物之相對流量比而得。
48 3.1.3 偵測系統 偵 測 系 統 主 要 為 霍 式 轉 換 紅 外 光 譜 儀 (Fourier transform spectrometer,Bruker,model IFS-66v/s),其移動鏡為步進掃描式。為 了保持移動鏡之穩定,光譜儀放置在光學防震桌上,且光譜儀之抽氣 幫浦的前段抽氣管路亦需以重物將之固定於地上,以避免幫浦的震動 經由管路傳至系統。 吾人於反應槽與光譜儀間架設一組 CaF2透鏡組(直徑 2 吋,焦距 分別為 4 吋及 6 吋),可將 Welsh mirror 收集之產物放光經過光圈 (aperture)有效率地引導至 FTIR 偵測。光束在光譜儀中先後經由干 涉儀、光圈及濾光片(optical filter),最後到達偵測器。 本實驗選用之 FTIR 內部相關光學元件如下: 一. 分光片:材質為 CaF2之分光片,可穿透之光區為 1200–10000 cm-1。
二. 偵測器:InSb(Kolmar Technologies,model KISDP-1-LJ2,rise
time = 220 ns,Bandwidth = 1.6 MHz,Responsivity (AC) = 7.9 ×
105 V/A,Responsivity (DC) = 3.95 × 105 V/A)紅外光偵測器,偵
測範圍為 1666-10000 cm-1。偵測器本身內建前置放大器可將電流
訊號放大轉換為電壓訊號,偵測器可輸出 ac 與 dc 耦合兩種訊號,
49 三. 光圈:光譜儀在一特定的解析度下,可使用的最大光圈直徑為: (3-1) 其中 D 為光圈直徑(mm),F 為光譜儀光源區內部拋物面鏡之焦 距(150 mm),R 為解析度(cm-1 ),S 為偵測光區之最大波數(cm-1)。 當欲擷取光區為 2950–3650 cm-1及解析度為 0.5 cm-1時,可使用 最大光圈直徑則為 3.51 mm。 四. 濾光片:因實驗中僅需測量小段光區之光譜,為節省實驗時間, 使用跳點取樣方式。為避免訊號摺疊(folding)的現象,需加入適當 的濾光片,只讓欲偵測光區的光線通過,本實驗所使用的濾光片 光區分別為 2650-3650 cm-1 (OCLI,WO3071-4),1170-3900 cm-1 (Spectrogon,LP2500 nm),2170-4400 cm-1 (Spectrogon,SP4730 nm)。 3.1.4 其他周邊儀器
利用脈衝產生器(Standford Research System,DG535)產生一道脈
衝以觸發光解雷射,並控制光譜儀進行數據擷取。由於雷射經觸發後
需要經過延遲時間(delay time)才會放出雷射光抵達反應槽,因此必頇
再產生另外一道脈衝至光譜儀經延遲時間後觸發放光訊號之擷取。放
光訊號經由偵測器偵測後,利用偵測器的內建之前置放大器將電流訊
50
訊號連接至電壓放大器(Standford Research System,SR560)以適當倍
率做二次放大,再將放大後的訊號由 SR 560 之輸出端(50 Ω)輸出至
FTIR 的類比/數位轉換器(analogue to digital converter,ADC),進行數 據擷取程序。實驗所使用類比/數位轉換器有兩種:光譜儀內部配置
類比/數位轉換器,其 A/D 解析度為 16 bit,取樣間隔最快為 5 μs,
輸入訊號上限為 10V 。另外插置於電腦主機板的類比/數位轉換器
PAD 1232,其 A/D 解析度為 12 bit,取樣間隔最快為 25 ns,輸入訊 上限為 1V。本實驗亦使用示波器(oscilloscope,Tektronix,model TDS
2014B,Bandwidth = 100 MHz,Sample rate = 1GS/s)觀測放光訊號強 度隨時間之變化,以及雷射輸出能量之強度。 3.2 實驗前準備工作 實驗前的準備工作可分為下列幾個部分:CaF2 透鏡組架設與 Welsh cell 對正、氣體流速校正、光譜儀與周邊儀器之連接及時序設 定、儀器之響應時間的量測、移動鏡穩定時間之量測及反應物之配製 與純化。
3.2.1 CaF2透鏡組架設與 Welsh mirror 對正
對正主要原理為利用光的可逆性,將光束由偵測器位置導向反應
51 的位置。
(1) 架設鹵素燈
開啟 NIR 光源,利用 OPUS 軟體中將光源轉換至 NIR,偵測器
轉換至 DTGS。調整 DTGS 前方的拋物面鏡,使入射的 NIR 光束為 一對稱圓形,且均勻的聚焦於偵測器之光窗,並根據 DTGS 偵測器所 偵測到的訊號大小微調拋物面鏡,調整到最佳訊號後移走 DTGS 偵測 器。架設鹵素燈於 DTGS 偵測器之位置,使 NIR 光束確實聚焦於鹵 素燈之燈絲中心處。完成後,關閉 NIR 光源。 (2) 架設 CaF2透鏡組 開啟 OPUS 軟體將光源轉換至 emission,使光源選擇鏡轉換至外 在光源位置。開啟鹵素燈光源,調整 FTIR 內部樣品槽中的光圈大小, 以調整鹵素燈出光強度大小,光束通過干涉儀組件,逆向地導出 FTIR ,並聚焦在光譜儀放光入口處外 1 吋之位置。在此焦點位置架設一光 圈,使鹵素燈光束的聚焦點通過光圈正中央。於反應槽與光譜儀間架 設一組透鏡組,如圖 3-1 所示。其中一片 L1(直徑 2 吋,焦距 4 吋) 架於距光譜儀光入口處 5 吋的位置,另一片 L2(直徑 2 吋,焦距 6 吋) 則架於距反應槽出口之 Welsh mirror 處 6 吋的位置。使鹵素燈光束通 過光圈發散後經過 L1 形成帄行光。擺置一帄面鏡架設於 L1 前並與 鹵素燈之光束成 45 度角導出,將光束導向至少 1 公尺處,微調 L1
52 之位置使導出之光束為一亮度均勻的帄行光。移去帄面鏡,微調 L2 之焦距使鹵素燈之光束聚焦於架於反應槽出口位置之 Welsh mirror 中 間,如此即可完成 CaF2透鏡組架設程序。 (3) 裝置 Welsh mirror Welsh mirror (直徑 2 吋,焦距 4 吋) 為一組鍍金球面反射鏡。將 兩個鍍金球面反射鏡對半切開,其中一組係由中間切開約 5 mm 之間 隙,背面研磨一缺口,黏貼在放光出口位置之鏡座,如圖 3-3 之 M1、 M2 上;另一組鍍金球面反射鏡則在中間僅切掉 2.5 mm 之間隙,黏 貼於距離 M1、M2 4 吋遠位置之鏡座 M3、M4。鏡座上均有調整桿, 可以調整球面鏡之角度。 (4) 微調 Welsh cell 由 CaF2透鏡組所聚焦於 M1、M2 中間之光束會再發散並覆蓋於 M4 反射鏡的中央,旋轉 M4 鏡座的調整桿以調整 M4 的反射角度, 使反射光聚焦於 M2 距切口約 1 mm 處;接著旋轉 M2 鏡座調整桿, 使光線得以覆蓋於 M3 反射鏡的中央,接著再調整 M3 鏡座,使光聚 焦於 M1 中間距切口約 1 mm 處,如此反覆微調 M1-M4 使 M1、M2 鏡面上均各出現至少一排 4 點以上之聚焦光點,且各光點大小、高度 一致,如圖 3-3 所示。如此即可完成 Welsh mirror 對正程序。 3.2.2 氣體流速校正
53 實驗前必頇量測各反應氣體的流速,並轉換成標準狀態(T = 273.15 K,P = 1 atm)下之流量(以 STP 下 cm3 s-1表之)。實驗上所使用 的氣體流速校正法有兩種:定容下壓力對時間變化率(dP/dt)及等壓下 體積對時間變化率(dV/dt)兩種校正法。 (1)定容下 dP/dt 校正法 將校正球 (體積約 1088 cm3) 接在氣體管路上,紀錄所能抽至壓 力最低值,打開氣體閥門,旋轉針閥到某特定刻度讓氣體以一定流速 流進校正球及管路內。待流量穩定後,關閉抽氣閥門,同時以馬錶計 時於定容下,上升壓力對時間之變化量(dP/dt,Torr s-1),每一特定流 速至少重複三次測量,並利用下列公式將流速轉換成標準狀態下之流 量 (FSTP): FSTP = (dP/dt)×(273.15/Troom)×(V/760) (3-2) 其中 V 為氣體校正球與所接氣體管路體積(從針閥到抽氣閥門)之總 和(cm3),T room=室溫(K)。將旋轉針閥刻度與相對之 FSTP作一對應圖, 即 可 完 成 校 正 程 序 。 此 法 適 用 於 流 量 ≦10 sccm (standard cubic
centimeter per minute,標準狀態下每分鐘的氣體流量,例如:1 sccm 代表在標準狀態(溫度 273 K, 壓力 760 torr)下, 每分鐘有 1 cm-3的氣
體流量) 之氣體校正,流量>10 sccm 時,因壓力計反應時間跟不上
54 (2)定壓下 dV/dt 校正法
常用測量方法有氣泡法(bubble method)及濕式流量計(drum-type
wet gas meter)校正。 a. 氣泡法校正 將欲測量之無毒性且不溶於水的氣體先流經含蒸餾水之錐形瓶, 使其富含飽和水蒸氣壓,再導入裝有肥皂水之玻璃瓶,玻璃瓶上端出 口裝置一支有體積刻度之玻璃管,調整肥皂水液面與玻璃管底部相隔 小於 5 mm 的距離。打開氣體閥門,旋轉針閥到某特定刻度讓氣體以 特定流速流經錐形瓶及玻璃瓶。此時擠壓玻璃滴管的吸球使玻璃管底 端產生一層肥皂膜,以馬錶計時於定壓下肥皂膜上升特定體積所需時 間以求出體積對時間之變化量(dV/dt)。每一特定流速至少重複三次測 量,並利用下列公式將流速轉換成標準狀態下之流量(FSTP):
FSTP = (dV/dt)×(273.15/Troom)×[(Proom-Pwater)/760] (3-3)
其中 Proom為當時氣壓,Pwater=室溫下的飽和水蒸氣壓(in Torr)。將旋
轉針閥刻度對 FSTP作一對應圖,即可完成校正程序。此法適用於流量
介於 10–100 sccm 之氣體校正,對於流量小於 10 sccm 的氣體,會因
水的蒸氣壓及不同肥皂膜的表面張力而使得流速測量的不準度增
加。
55
流速介於 33-2000 sccm 間之無毒性且不溶於水之氣體(如 Ar、
He、N2 ...等)可採用濕式測量儀(Sinagawa Corporation;W-NK-0.5)校
正,其誤差僅於 0.2 %之內。此測量儀乃利用排水測定的原理,量測 排出氣體體積隨時間的變化。吾人依所欲量測之氣體流速以調整適當 之量測體積(即 gas meter 指針轉的圈數)及時間,減小其量測誤差。 此方法之精確度較 bubble calibration 方法高。在定壓下,吾人通入不 同流速之氣體,量測其體積與時間的變化率(dV/dt),並依 3-3 式將其 轉換成標準狀態(STP)下之流量(VSTP)。最後,將流量計讀數對測量所 得流量值作一對應圖即完成校正。 3.2.3 儀器光學響應曲線量測 待測光束必需藉由許多光學元件才能導入偵測器,但每個光學元 件對於不同波長的光之穿透率及反射率不盡相同,且所使用的偵測器 對於不同波長的靈敏度也不同,因此在測量光譜之前,必頇使用一個 標準的黑體輻射光源進行校正,以得到所有元件對於不同波長的響應 曲線,才能反映真正的放光強度。雖然黑體輻射校正程序並無法測量 Welsh 球面鏡之反射響應值,但根據測詴報告可得知,在測量光區內 球面鏡的反射率幾乎為定值,故其對實驗的影響可忽略不計。黑體輻 射校正程序如下:
56 1. 架設鹵素燈光源於 DTGS 偵測器位置,如本文 3.2.1 之步驟 1 所 述。 2. 移去反應槽,將黑體輻射校正儀(Graseby,model SR20)擺設至原 反應槽的位置。微調黑體輻射校正儀的位置,使鹵素燈光源經 CaF2透鏡組聚焦後,其聚焦點位置正好在黑體輻射源的輸出小孔, 並固定黑體輻射校正儀的位置。 3. 關閉鹵素燈,開啟 OPUS 軟體將偵測器位置更改為 InSb。 4. 設定黑體輻射源溫度,使其逐漸增溫至 1273 K 並穩定之(約費時 1.5 小時)。
5. 利用 FTIR 對正程序,觀測黑體輻射源的強度(ADC counts),再調 整黑體輻射源的出射光束之方向及輸出光圈大小,使訊號最佳化, 但應避免訊號過飽和(saturate)而造成誤差。 6. 掃瞄全光區(以 InSb 偵測器為例,其偵測光區範圍為 1666-10000 cm-1)光譜,不放任何濾光片,先做低解析度(8 cm-1)放光光譜量測。 在熱帄衡之條件下,黑體輻射源其單位波數(cm-1 )區間內之輻射密 度ρ 為:
![表 2-1:數種簡單削足函數對儀器譜線形狀函數的影響[16]。 1 2 為主峰之 半高寬(full width at half maximum),實驗可測得之光程差 δ 的範 圍為 -a≦δ≦a。而 為側波最大振幅值 H (side lobe amplitude S maximum)與主峰高度 H 之百分比絕對值。 m](https://thumb-ap.123doks.com/thumbv2/9libinfo/8245901.171511/45.892.74.820.98.927/半高寬實驗可測得之光程δ的範圍為≦δ大振幅值S與主峰高.webp)