複合式樹脂之唾液中釋出物固相微萃取高效液相層析儀分析方法研究
128
0
0
全文
(2) 誌. 謝. 在撰寫致謝的時候正是論文口試完那天,有口試完成的喜悅,但感 到更深的難過,在老師的教誨下,真的成長了許多,不只單單在研究方 面的發現問題、思考問題、解決問題,在思考方面也增加了邏輯能力, 難過的是對於研究的態度無法達到老師的期望及要求。 在研究所階段,體驗到收穫與付出不ㄧ定是成正比,但這是一種必 經的磨練,沒有解決不了的問題,只要你付出的夠多。 感謝王文忻老師這三年來無論是學識上的督促及指導或生活上關 心,讓生活在異鄉的我,感受到實驗室有家的感覺,感謝論文口委-陳三 餘老師在研究上總是不厭其煩的教導,讓對於牙科領域一開始一無所知 的我,順利完成研究,同時感謝蔡素珍教授對論文的指導。 另外感謝賴俊雄院長,職安系趙克平老師、蔡詩偉老師、廖宏章老 師及環醫所全體老師在課業上的指導。 感謝實驗室學弟妹在研究路上的相陪,感謝仕杭、思瑜、俊鳴學長 實驗上的教導,還有早我ㄧ步先畢業的同窗好友聖鈞、延瑜、國源、建 宗、雅萍、雅婷課業上協助,感謝對我無盡付出的家人。 老師常說不要跟別實驗室比,但還是會偷偷的比較,比完覺得真不 公平,不公平的是我學到的東西比別人多很多,感謝你王文忻老師。. I.
(3) 複合式樹脂之唾液中釋出物固相微萃取高效液相 層析儀分析方法研究 目的:複合式樹脂為牙科中常使用填補材料,其主要成分 Bis-GMA (bisphenylglycidyl dimethacrylate )、TEGDMA (triethylene glycol dimethacrylate)、 UDMA (urethane dimethacrylate ) 因聚合不完全 與唾液的侵蝕分解導致單體溶出進入人體,造成健康危害。本研究 擬開發一個以直接固相微萃取( Solid-Phase Microextraction ; SPME)吸附的方式,結合高效液相層析儀(High Performance Liquid Chromatography;HPLC)來分析唾液中樹脂溶出物,進而探討聚合 時間、溫度與 pH 影響溶出量的關係。 方法:本研究以 60 μm Polydimethylsiloxane-divinylbenzene(PDMS/DVB) 纖維作為吸附介質,以直接法萃取樹脂溶出物。完成萃取後的纖 維浸入 HPLC 的 Chamber 進行靜態脫附,脫附完後進行分析。 實驗室技術建立時,製備標準溶液進行條件探討。研究探討的參 數包括: 纖維種類、脫附溶劑、吸附時間、脫附時間、吸附溫度、 攪拌速度、及 pH 值。 於不同聚合時間: 20、40、60、80、120 秒;不同培養溫度: 0°C、 27°C、40°C;不同 pH 值: 3.0、5.0、7.6、9.0、11.0 下,以商品 Tetric Ceram 培養 7 日後分析,探討聚合時間、溫度、pH 值對溶出量的. II.
(4) 影響,並探討樹脂於不同天數下釋出量的變化。 結果:1.SPME方法為:將標準品添加於未經pH值調整唾液中,裝滿4 mL vial,瓶中加入攪拌磁石,密封後於40°C、轉速200 rpm(0.18g) 下,以60 μm PDMS/DVB纖維直接吸附20分鐘,再以移動相( 65 % 乙腈、35 %D.I.水)靜態脫附15分鐘。 2.在此萃取分析條件下,所求得的檢量線範圍為 TEGDMA: 0.3~30µg/mL;UDMA:0.5~50 µg/mL;Bis-GMA:0.5~50µg/mL, 其相關係數(r)皆在 0.995 以上;偵測極限分別為 TEGDMA:0.04 µg/mL;UDMA:0.28 µg/mL;Bis-GMA:0.06 µg/mL。 3.複合式樹脂的溶出量隨著天數增加而增加,在第7日時達到一個 較高值。 4.溫度與 pH 值皆會影響溶出,隨著溫度增加 UDMA、Bis-GMA 溶出量增加;隨著 pH 減少,溶出量隨著增加,而影響最大的是 聚合時間,增加聚合時間可降低其溶出量。 結論:以Direct-SPME-HPLC的方式來分析樹脂溶出物,可省去繁雜的前 處理步驟,同時其具有免溶劑、快速、方法簡便、以及不受樣本 基質干擾等優點。 樹脂於唾液中的溶出物快速的被酵素分解,隨即進入人體循環系 統,評估樹脂溶出物對人體健康的危害,應針對釋出物分解後產. III.
(5) 物較適合。 在填補完牙齒的第一周,以漱口來降低口腔溫度,沖淡口腔酸性 環境,盡量少吃過熱及偏酸的食物,以減少溶出量。. 關鍵字:複合式樹脂、單體溶出、固相微萃取、高效能液相層析儀. IV.
(6) SPME-HPLC Analytical Method Development for Eluted Composite Resins in Whole Saliva Objective:Dental composite resin materials are widely used for fixing teeth. The main components are BisGMA (bisphenylglycidyl dimethacrylate)、TEGDMA (triethylene glycol dimethacrylate)、UDMA (urethane dimethacrylate ). These resin monomers may be released due to incomplete polymerization and have cytotoxicity and genotoxicity to human. The purpose of this study is to develop an analytical method for determining the resin monomers in saliva using solid phase microextraction followed by high performance liquid chromatography (HPLC) analysis. Method:The 60 μm Polydimethylsiloxane-divinylbenzene(PDMS/DVB)fiber through direct extraction was tested to be the most suitable material for sample extraction. The SPME fiber was inserted into the injection chamber of HPLC and analyzed. The optimum extraction parameters investigated are pH、extraction temperature, stirring speed, extraction time、desorption time、desorption solvent. The effect of polymeric time (20、40、60、80、120s), temperature (0°C、27°C、40°C) and pH(3.0、5.0、7.6、9.0、11.0) to releasing amount are investigated. The test sample were polymerized for 120 s and stored in ethanol and saliva solution in room temperature with V.
(7) noadjustment pH for 12 h, 24 h, 3, 5, 7 or 10 days to investigate influence of immersing solution and time. Results:1. The SPME method was placing magnetic stirring-bar in an 4 mL vial, stirring 200 rpm at 40 °C, PDMS/DVB fiber was used to perform direct solid-phase microextraction for 20 min and mobile phase(65 %acetonitrile:35 %D.I. water) desorption for 15 min. 2. For the quality control calibration, the concentration ranges of TEGDMA, UDMA, and Bis-GMA were 0.3~30 µg/mL, 0.5~50 µg/mL, 0.5~50 µg/mL. The correlation coefficients(r) were all above 0.995. The method detection limit(MDL)for TEGDMA, UDMA, and Bis-GMA were 0.04 µg/mL , 0.28 µg/mL and 0.064µg/mL respectively. 3. Results showed a maximum concentration of monomers after 7 days almost. 4. Under longer polymeric time, can reducing the amount of releasing. With higher temperature and lower pH, releasing will increase. Conclusion:The analytical method of eluted composite resins in whole saliva by Direct-SPME-HPLC can be reduced complicated pretreated procedure, as well as having the advantage of solvent-free, rapidity, simplicity, and avoid interfering with a complex matrix. The enzyme of saliva will react with releasing fast and into blood circulation.. VI.
(8) Key word:composite resins, monomer, solid phase microextraction, high performance liquid chromatography. VII.
(9) 目. 錄. 第壹章 序論 ...........................................................................................1 第一節 研究背景...................................................................................1 第二節 研究目的...................................................................................4 第三節 研究內容...................................................................................4 第四節 研究假設....................................................................................4 第貳章 文獻探討...................................................................................5 第一節 填補牙材的演化.......................................................................5 第二節 複合式樹脂及聚合...................................................................6 第三節 單體溶出、唾液分解與健康危害...........................................9 第四節 物化性及毒性...........................................................................11 (一) TEGDMA..........................................................................11 (二) UDMA...............................................................................11 (三) Bis-GMA ...........................................................................11 (四)分解產物與毒性 ................................................................11 第五節 傳統萃取唾液中溶出物方法...................................................13 第六節 固相微萃取(Solid-Phase Microextraction) ..............................14 (一) 直接固相微萃取 ..............................................................14 (二) 頂空固相微萃取 ..............................................................15. VIII.
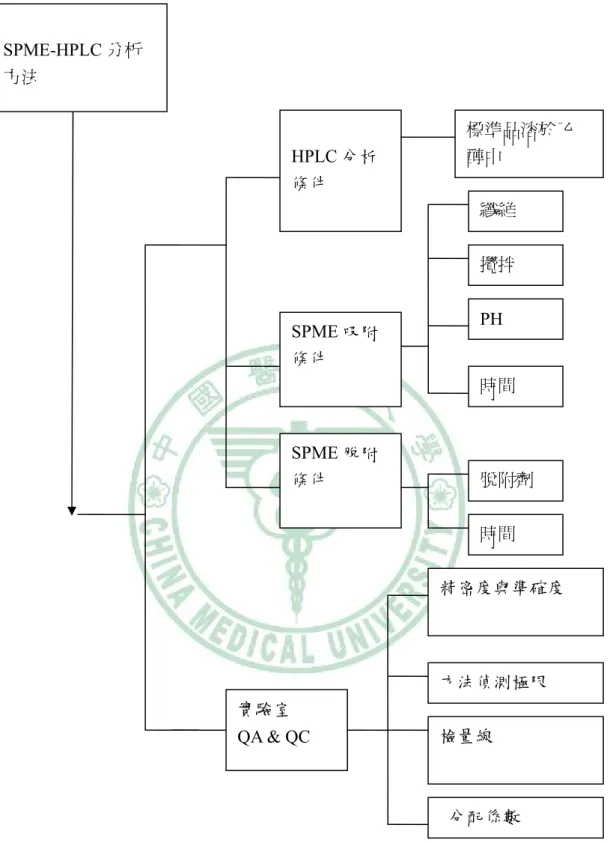
(10) (三) 固相微萃取裝置 ..............................................................16 (四) 固相微萃取操作程序......................................................16 (五) 影響固相微萃取效率的因素..........................................16 (六) 分配係數 ..........................................................................21 (七) 固相微萃取法於生物檢體的應用..................................22 第叁章. 材料設備與方法.....................................................................30. 第一節 儀器設備與試藥.......................................................................30 (一) 試藥 ..................................................................................30 (二) 設備 ..................................................................................31 第二節 儀器參數條件...........................................................................34 (一) HPLC 條件........................................................................34 (二) LC-MS 條件......................................................................35 第三節 研究架構...................................................................................36 (一) HPLC 分析方法的建立 …………………………………36 (二)直接固相微萃取方法建立................................................36 (三)分配係數 ............................................................................39 (四)分析品質管制 ....................................................................39 (五)真實樣本培養 ....................................................................40 第肆章. 結果與討論.............................................................................48. IX.
(11) 第一節 HPLC 分析................................................................................48 第二節 固相微萃取方法建立...............................................................49 (一) 固相微萃取纖維的選擇..................................................49 (二) 萃取時間 ..........................................................................49 (三) 脫附溶劑 ..........................................................................49 (四) 脫附時間探討 ..................................................................50 (五) 萃取溫度探討 ..................................................................50 (六) 攪拌速度對萃取的影響..................................................51 (七) 樣本 pH 值探討 ...............................................................51 第三節 分配係數...................................................................................52 第四節 分析品質管制...........................................................................53 (一) 檢量線建立 ......................................................................53 (二) 方法偵測極限(MDL).......................................................53 (三) 分析之精密度與準確度 ..................................................53 第五節 真實樣本培養...........................................................................54 (一) 市售複合式樹脂 ..............................................................54 (二) LC-MS-MS 質譜...............................................................54 (三) 聚合時間探討 ..................................................................54 (四) 溫度探討 ..........................................................................55. X.
(12) (五) pH 值探討 .........................................................................55 第六節 討論 ...........................................................................................56 (一) 固相微萃取 ......................................................................56 (二) 複合式樹脂 ......................................................................59 第伍章. 結論與建議.............................................................................104 (一) 結論與建議 ......................................................................104 (二) 未來發展 ..........................................................................105. 參考文獻 .................................................................................................106. XI.
(13) 圖. 目. 錄. 圖 2-1 TEGDMA, UDMA, Bis-GMA 結構圖 .......................................24 圖 2-2 固相微萃取裝置分解圖 ............................................................25 圖 2-3 SPME-HPLC Chamber 圖解.......................................................26 圖 2-4 固相微萃取操作圖解 ................................................................27 圖 3-1 唾液樣本 SPME-HPLC 分析方法建立 .....................................44 圖 3-2 真實樣本培養架構 .....................................................................45 圖 3-2 SPME 實驗操作流程 ..................................................................46 圖 4-1 HPLC 分析圖 ..............................................................................62 圖 4-2-1a 纖維的探討 ...........................................................................63 圖 4-2-1b 不同纖維的 RSD 圖 .............................................................63 圖 4-2-2a 吸附時間對吸附量的影響 ...................................................64 圖 4-2-2b 吸附時間曲線圖 ...................................................................64 圖 4-2-3a 脫附劑探討 ...........................................................................65 圖 4-2-3b 不同脫附劑下之 RSD 值 .....................................................65 圖 4-2-4a 脫附時間探討 .......................................................................66 圖 4-2-4b 脫附時間曲線圖 ...................................................................66 圖 4-2-4c 不同脫附時間下之 RSD 值 ..................................................67 圖 4-2-5a 萃取溫度探討 ........................................................................68. XII.
(14) 圖 4-2-5b 不同萃取溫度下之 RSD 值 ..................................................68 圖 4-2-6a 攪拌速度探討 .......................................................................69 圖 4-2-6b 不同攪拌速度下之 RSD 值 .................................................69 圖 4-2-7a pH 值探討...............................................................................70 圖 4-2-7b 不同 pH 值下之 RSD 值 .......................................................70 圖 4-5-1a 商品 Tetric Ceram..................................................................71 圖 4-5-1b 商品 Spectrum........................................................................71 圖 4-5-1c 商品 Palfique Estelite .............................................................72 圖 4-5-1d 商品 Compoglass flow ...........................................................72 圖 4-5-1e 商品 Dyract.............................................................................73 圖 4-5-1f 商品 7 日釋出量比較.............................................................73 圖 4-5-2a 標準溶液 TEGDMA LC-MS-MS 質譜圖 .............................74 圖 4-5-2b 樹脂培養液 TEGDMA LC-MS-MS 質譜圖.........................74 圖 4-5-2c 標準溶液 UDMA LC-MS-MS 質譜圖.................................75 圖 4-5-2d 樹脂培養液 UDMA LC-MS-MS 質譜圖.............................75 圖 4-5-2e 標準溶液 Bis-GMA LC-MS-MS 質譜圖 .............................76 圖 4-5-2f 樹脂培養液 Bis-GMA LC-MS-MS 質譜圖 .........................76 圖 4-5-3 聚合時間影響釋出量探討 ......................................................77 圖 4-5-4 溫度影響釋出量探討 ..............................................................78. XIII.
(15) 圖 4-5-5 pH 影響釋出量探討 ................................................................79. XIV.
(16) 表. 目. 錄. 表 2-1 市售固相微萃取纖維 ................................................................28 表 2-2 各種纖維的耐受溫度 .................................................................29 表 3-1 市售複合式樹脂商品 .................................................................47 表 4-2-2 吸附時間探討 .........................................................................80 表 4-2-3 脫附劑探討 .............................................................................81 表 4-2-4 SPME 脫附時間探討..............................................................82 表 4-2-5 SPME 萃取溫度探討...............................................................83 表 4-2-6 攪拌與否探討 .........................................................................84 表 4-2-7 pH 值探討 ................................................................................85 表 4-3-a TEGDMA 低濃度分配係數 ....................................................86 表 4-3-b TEGDMA 高濃度分配係數 ....................................................86 表 4-3-c UDMA 低濃度分配係數 .........................................................87 表 4-3-d UDMA 高濃度分配係數.........................................................87 表 4-3-e Bis-GMA 低濃度分配係數 .....................................................88 表 4-3-f Bis-GMA 高濃度分配係數......................................................88 表 4-4-1 檢量線製作 ..............................................................................89 表 4-4-2 方法偵測極限 ..........................................................................90 表 4-4-3 準確度與精密度測試 ..............................................................92. XV.
(17) 表 4-5-3 聚合時間對溶出量影響 ..........................................................95 表 4-5-4 溫度對溶出量影響 ..................................................................98 表 4-5-5 pH 值對溶出量影響 ................................................................101. XVI.
(18) 第壹章 第一節. 序論. 研究背景. 銀汞合金為早期齲齒填補主要材料,銀汞合金充填材料在牙科填補 的使用已超過一百五十年,由於銀汞合金在室溫下是一種可塑性的合 金,可放到牙齒窩洞內進行充填,經過一段時間銀汞合金即可固化,但由 於銀汞金屬會因口腔中之物理、化學作用而逐漸滲出,有汞危害之虞,危 害人體健康,於是發展出聚合式複合樹脂。 聚合物(polymer)乃指分子量在 104~106 間,由許多的小分子彼此接合 所產生的巨大分子,組成聚合物的單位分子稱為單體(monomer)而由小分 子相互結合的過程稱為聚合作用(polymerization)。 聚合作用的發生需藉由單體彼此間相互結合而形成大分子,分子間的接 合須靠單體中的共價雙鍵(covalent double bond)進行聚合,當能量加入(列 如:光、熱或化學能)而促使聚合反應發生,就可形成分子量較大的聚合 物 1,牙科樹脂的形成就是單體與單體間發生聚合反應後的產物 2。 牙科樹脂廣泛應用於牙科材料,如牙科臨時牙橋、人工牙齒及義齒 基底材料等,填補牙齒所使用的複合式樹脂主要由不同成份材質所組 成,聚合本體由丙烯酸酯類為主要原料並與其他單體聚合而成 3,例如 Bis-GMA (bisphenylglycidyl dimethacrylate), TEGDMA(triethylene glycol dimethacry-late), UDMA(urethane dime-thacrylate )等。. 1.
(19) 複合樹脂充填在早期是以化學自聚式硬化方式,由於硬度較差,只 能用於前牙充填。在最近十年內發展非常快,主要的優點是和牙齒的顏 色一樣,可以比色選擇,便宜、製作方便、修補容易而且安定性高 4。 目前牙材樹脂應用於補牙技術是採用光聚合機使複合樹脂在瞬間硬 化,硬度也和牙齒一樣,因此可用於牙齒的充填,因銀汞合金的金屬滲 出導致健康危害,發展出取代的複合式樹脂材料,大量的使用了複合式 樹脂一段時間之後,學者對於複合樹脂的研究陸續投入,諷刺的是,這 些研究指出,複合式樹脂在聚合過程中,會因聚合條件不同:聚合方式、 聚合溫度、樹脂材料等因素,有不同程度的單體溶出 5。 在樹脂聚合的技術上,聚合不完全的問題一直存在 6,研究指出聚合 的程度範圍約 52-77 %7. 、8、9、10. ,因聚合不完全或唾液的侵蝕分解 11 導致單. 體溶出進入人體,造成健康危害,且具細胞及基因毒性 12,此外,牙科技 術人員接觸這些物質也會造成接觸性皮膚炎或過敏現象 13。 目前研究文獻中,針對牙材聚合物的釋出物分析 14,以及溶出物與唾 液分解後產物分析 15,主要以高效液相層析儀法(High Performance Liqud Chromatography;HPLC)分析 TEGDMA、UDMA、Bis-GMA,主要原因 是分子量大及沸點高,另有文獻係使用氣相層析火燄離子化偵檢器分析 TEGDMA、Bis-GMA16。 大部分的研究,使用乙腈、乙醇、水、甲醇等溶劑當作複合樹脂溶. 2.
(20) 出培養液,唾液中含有特殊酵素會影響溶出的量. 17. ,評估健康效應以唾. 液當浸泡液是最適當的 18。 傳統以唾液為浸泡液分析需複雜瑣碎的離心、溶液萃取等前處理步 驟 19 相較於傳統的液-液相萃取,萃取程序複雜、費時且需要大量有機溶 劑萃取造成日後廢液處理及環境污染問題,針對釋出單體 TEGDMA, UDMA,. Bis-GMA. 的 分 析 , 應 用 固 相 微 萃 取 (Solid-Phase. Microextraction ;SPME)之技術,進行水基質的樣本萃取,結合液相層 析儀分析,可降低樣品處理的繁複手續,得到更有效率的分析結果, 以固相微萃取的方式,可整合取樣、萃取、濃縮及樣品注入等四個 步驟於一個分析方法中,本研究擬開發直接浸入式固相微萃取(SPME)之 高效液相層析儀(HPLC)分析方法,針對溶出單體 TEGDMA, UDMA, Bis-GMA 進行唾液中的樹脂溶出物檢測。. 3.
(21) 第二節. 研究目的. (一) 開發一個以直接浸入式固相微萃取結合液相層析儀的方法來分 析唾液中牙材樹脂溶出物檢測的方法。 (二) 探討影響樹脂溶出量的參數: 浸泡液溫度、浸泡液 pH 值、聚合時間。 (三) 將探討完的影響釋出量參數套用於不同天數下乙醇浸泡 (HPLC 分析)與唾液浸泡(SPME-HPLC 分析)探討。. 第三節. 研究內容. (一) 探討影響固相微萃取裝置吸附效率、脫附效率以及儀器設定等 參數,進而探討分配係數,並執行檢量線製作、精密度、準確度、 方法偵測極限。 (二) 探討影響樹脂溶出因素: 浸泡液溫度、浸泡液 pH 值、聚合時間。 (三)以開發之 SPME 方法比較唾液與非唾液浸泡液相同天數下釋出 之差別。. 第四節. 研究假設. (一) Fick’s First Law (二) Fick’s second law. 4.
(22) 第貳章. 文獻探討. 第一節 填補牙材的演化 從西元1930年,各式各樣的材料開始被應用在移動性假牙的製作 上,1937提出熱聚合樹脂Biolon商品來製作修補牙床(Prosthodontics gums)。到了1947年,一些用在製作臨時假牙的自聚式聚甲基丙烯甲酯樹 脂( Polymethyl methacrylate ) 紛紛問市,在臨床上使用過後,發現聚甲基 丙烯甲酯樹脂雖然有低的體積收縮度,但是有硬度不佳、容易斷裂與變 色等問題,到了1960年代,學者以增加分子量形成共聚合物的觀點提出 了乙烯聚乙基丙烯酸甲酯(Vinyl polyethyl methacrylate) 的材料20。 1969年,針對聚合過程中發生的高放熱與收縮現象可能造成的牙髓 刺激與尺寸改變,提出了不同化學結構的乙基亞胺衍生物( Ethyl imine derivatives ) 之材料Scutan,此材料聚合後放熱較低、有較低的體積收縮 度與反應後較少的單體含量等優點,但是它不像共聚合物可任意添加材 料,且表面硬度不佳,所以在市場上只風行一陣子21。 之後1980年推出了複合樹脂材料,宣稱以多種材料複合而成的結 構,可以提供較高的斷裂強度,近幾年為了操作方便,出現了同時具有 光聚合與自聚合的複合樹脂材料22。. 5.
(23) 第二節 複合式樹脂及聚合 複合式樹脂顧名思義就是由兩種以上不同化學物質單體聚合而成 的樹脂,而現今市售複合式樹脂材料都是經過調配好的原料及比例而成 的商品,主要的原料包括 TEGDMA, UDMA, Bis-GMA等,不同的原料比 例,會有不同的強度效果,列如增加Bis-GMA 的比例會增加混合樹脂的 強度,但是改變此比例也降低樹脂的滲透能力23。 現今聚合複合式樹脂的方式大部份為光聚式,所謂的光聚合複合樹 脂通常使用 camphoroquinone 或 benzyl作為聚合起始劑,而三級胺 (amine)則作為加速劑。當聚合作用不完全時,除了影響樹脂本身的機械 性質、體積穩定度、水份吸收及溶解度外,也造成邊緣染色及變色、二 次蛀牙、釋出的單體與牙本質接觸,甚至對牙髓造成刺激24。 一般牙科樹脂依聚合反應的發生方式,主要可分為三類,分別為自聚 式 (autopolymerization)、熱聚式 (heat-polymerization) 及光聚式 (light-polymerization)。 自聚式的材料內之聚合物粒子、單體及増塑劑 (plasticizer) 的比例 含量經適當調配,施與壓力作用即可自行聚合。熱聚式的樹脂是在粉劑 與液劑混合均勻後、須加熱或置於熱水中使聚合反應完成。光聚式的材 料通常作成凝膠式 (gel type) 的包裝,暴露於可見光下即可起始聚合反 應,而本研究所使用的光聚型複合樹脂的聚合程度受到三方面的影響,. 6.
(24) 如材料因素:包括樹脂的化學成分、填料的大小;光學因素:樹脂的色度、 透光性、折射係數等,以及可見光照射的強度和時間 24。 三種不同的聚合方式,在顏色穩定性、邊緣密合度、表面硬度、斷 裂強度、抗磨損度、生物相容性等特性上,各有優缺點:1996 年 Wang 等研究熱聚式與自聚式樹脂在衰退處理後之顏色變化發現,熱聚式比自 聚式產生的顏色變化較小 25;1998 年 Stavros A.等比較三種聚合方式的 樹脂浸泡在水、咖啡與茶後之顏色變化發現,熱聚式較自聚式顏色變化 少,光聚式樹脂是顏色變化最多的 26。 1998 年 Michele 等比較光聚式與自聚式複合樹脂的斷裂強度,發現 光聚式複合樹脂表現較佳 21。 1989 年 Russell 等對自聚式與光聚式複合樹脂作牙刷磨耗實驗,發 現光聚式複合樹脂的抗磨損度較好 27;1990 年 Mohd. Zainal 等針對牙科 常用材料的抗磨損度,作一資料整理評估,複合式樹脂的部份,光聚式 較自聚式佳 28。 在生物相容性部分,樹脂的釋出單體會對牙周組織與口腔組織產生 刺激而引起過敏反應,2001 年 Tai 等以人類口腔上皮細胞株 (Human oral epithelial KB cell) 與初代口腔纖維母細胞 (Primary human oral fibroblast) 觀察三種聚合方式的樹脂釋出細胞毒性,其中自聚式的細胞毒 性是最高的 29。. 7.
(25) 由以上文獻回顧可知三種聚合方式各有優缺點自聚式樹脂在顏色穩 定度、橫向斷裂強度表現較佳,而光聚式樹脂在表面硬度、抗磨損力、 生物相容性方面表現較佳,此外如自聚式樹脂較其他聚合方式經濟,光 聚式的聚合讓操作者有足夠的時間塑形,且光聚式操作方便,目前修補 大部分是使用光聚式,所以本研究所使用的複合樹脂皆為光聚式樹脂。. 8.
(26) 第三節 單體溶出、唾液分解與健康危害 樹脂的聚合是不完全的,以自聚式材料而言, 1977 年 Ruyter 等以 紅外線光譜儀研究,六種自聚式樹脂聚合程度,得到了聚合程度只有 52-75%8; 1982 年 Asmussen 等以紅外線光譜儀分析自聚式複合樹脂的 聚合程度,發現聚合程度僅達 57-77%9; 1984 年 Ferracanca 等亦對自聚 式無填料複合樹脂聚合程度作研究,經傅立葉紅外線光譜儀分析( Fourier Transform Infrared Spectrometer :. FTIR)得到 55-72%的聚合程 10。. 2000 年 Huang 等針對自聚式、熱聚式、光聚式的樹脂泡水七天的釋 出單體作一比較,顯示自聚式樹脂溶出量最高,熱聚式其次,而光聚式 最低 30。 對於單體釋出量的檢測方法有傅立葉紅外線光譜法10、氣相色層分析 法(Gas chromatography)16、高效能液相層析法(High performance liquid chromatography). 15. 與碳十四核磁共振法(Carbon- 14 Nuclear Magnetic. Resonance : NMR)31,傅立葉紅外線光譜法偏向於定性分析,碳十四核磁 共振法的設備昂貴,高效能液相層析技術較氣體色層分析廣為人使用。 除了溶出單體,研究更指出聚合好的樹脂於口腔環境中,有水解及 酵素分解的反應14,如TEGDMA會與pseudocholinesterase (PCE)分解產生 methacrylic acid (MA) 以及triethylene glycol (TEG) ;Bis-GMA 會與 cholesterol esterase (CE) 分解產生methacrylic acid (MA)、bis-hydroxy –. 9.
(27) propoxyphenyl propane (bis-HPPP)以及ethoxylated bisphenol A (E-bisPA) 15. 。 口腔中的環境複雜,牙科複合樹脂在口腔中除了處於潮濕的狀態. 外,還有多種酵素及微生物,因此釋出單體發生氧化或水解的機會更大, 也就更容易對細胞有不同程度影響。 溶出單體與分解後產物,與人體口腔有直接接觸之虞,且直接進入人 體循環體系,對於健康造成的更大的引響,1999對於丙烯酸的動物實驗 中,讓懷孕母鼠每天6小時吸入0~300ppm丙烯酸6~20天,發現會造成出生 老鼠體重不足,也會造成慢性胃炎,引起過敏反應及改變代謝路徑32,此 外唾液與樹脂的反應機制、溶出樹脂單體經酵素反應後之產物物種及產 量,對於人體健康影響甚巨15。. 10.
(28) 第四節 物化性及毒性 (一)TEGDMA33 Triethylene glycol dimethacrylate (TEGDMA)俗稱3G,在複合樹脂中 可稀釋Bis-GMA、UEDMA的黏度,常溫下為輕微的紅褐色,分子量為 286.33g/mol、沸點為162゚C(760torr)、閃火點為167゚C(閉杯)、密度為1.072 g/cm3、分子式為C14H22O6、結構式如附圖2-1;在動物實驗部份,LD50 : 10837 mg/Kg(大鼠,吞食),對肺部及呼吸系統有損害,此外大鼠在500 UMOL/L的暴露下,對於肺部細胞有致突變性。 (二)UDMA33 Urethane dimethacrylate (UDMA),為複合樹脂主要材料之ㄧ,以不同 比例的UDMA與TEGDMA混合,產生不同強度的複合樹脂,分子量為470 g/mol , 常 溫 下 為 黏 稠 液 體 , 蒸 氣 壓 < 1mmHg 、 沸 點 200 ゚ C、 密 度 1.11 g/mL at 25 °C分子式為C25H38O6N2、結構式如附圖2-1。 (三)Bis-GMA33 2,2-bis[4(2-hydroxy-3-methacryloyloxy-propyloxy) -phenyl] propane (Bis-GMA)與UDMA的功能相似,在常溫下幾乎成為固狀,分子 量為512.59 g/mol、密度1.161 g/mL at 25 °C,分子式為C29H36O8、結構式 如附圖2-1。 (四)分解產物與毒性 在 牙 科 發 現 釋 出 的 MA 會 造 成 口 腔 黏 膜 的 刺 激 (irritation) 、 發 炎 11.
(29) (inflammation)及過敏反應(allergic response)32,此外MA經細胞內溶小體 (microsome)中的脫酸酯脢(carboxylesterase)水解作用,會生成甲醇,甲醇 經氧化作用的結果可生成福馬林,可造成患者的過敏症(hypersensitivity)。 TEGDMA, UDMA, Bis-GMA這些由丙烯酸所衍生成的單體會發生水 解反應,產生丙烯酸34,造成慢性胃炎及改變代謝途徑,且以不同的丙烯 酸甲酯聚合物與口腔纖維母細胞培養,發現對細胞具有毒性35。 2000年Cimpan等以牙科樹脂材料進行研究,結果發現隨著材料劑量 的增加,人類淋巴球細胞株U-937發生細胞凋亡與細胞壞死的比例隨之增 加36,在1995年Ratanasathien等針對釋出單體作毒性比較,其中Bis-GMA 的毒性最高,UDMA次之,TEGDMA最低37,雖然TEGDMA的毒性最低, 但在1988年Fujisawa等研究中指出,TEGDMA會誘發微粒體的脂質層進 行過氧化反應38,毒性更高的UDMA, Bis-GMA對人體的毒性危害可想而 知。. 12.
(30) 第五節 傳統萃取唾液中溶出物方法 傳統方法是利用有機溶劑將浸泡好之樹脂微量溶出物萃取出來,經 離心、濃縮後,再以注射針取出唾液上清液。如 Larsen 等 1991年的研 究就是將唾液樣本先離心後再取100 μL的唾液樣本上清液加上50 μL的 乙醇進行液-液相萃取,經離心後以注射針取出上清液,之後再以液相層 析儀來進行分析39; Atkinson 等 2002年的研究是將經離心之後1mL的唾 液樣本上清液加入1 mL的正己烷及1mL乙酸乙酯進行液-液相萃取,離心 後以注射針取出上清液,再以氣相層析儀來進行分析40。但是液-液萃取 法是一種複雜且多步驟的前處理方式,容易造成樣品流失或受到污染, 此外,方法中使用有機溶劑,則會造成分析者的暴露,以及溶劑污染、 廢棄處理等問題。. 13.
(31) 第六節 固相微萃取(Solid-Phase Microextraction)41~45 相較於傳統液-液相萃取的方法,固相微萃取法不僅免溶劑使用並且 結合了取樣、萃取、濃縮及樣本導入等四個步驟於一個方法中,可大幅 減少分析時間、降低分析物基質干擾、降低人為誤差、裝置簡單、能快 速有效的處理樣本等優點。其最大的特色在於免溶劑使用,不但省去許 多繁雜的前處理步驟,更能減少有機溶劑的使用及避免污染產生。 固相微萃取依不同的取樣方式又可分為直接固相微萃取法(Direct immersed SPME)和頂空固相微萃取法(Headspace SPME)兩種,直接固相 微萃取法是將萃取纖維直接浸入樣本基質中,待纖維塗布物質、樣本間 達成固液相平衡後即完成萃取,有較佳的吸附效果;而頂空固相微萃取 法是以萃取纖維吸附揮發至頂空之分析物,待纖維塗布物質、樣本、樣 本頂空空間達成固、液、氣三相平衡後即完成萃取,其可避免樣本基質 干擾以及保護纖維。. (一) 直接固相微萃取42. ﹑45. 固相微萃取主要是利用分析物質在樣本基質與纖維上塗布之固定靜 相進行分配反應,當分配達到平衡時,吸附在固定靜相上的量可以下列 方程式(1)來表示: n=. KfsVfC 0Vs KfsVf + Vs. 14. (1).
(32) n:吸附在固定靜相上分析物之質量 Vf:固定靜相之體積 Vs:樣品體積 Kfs:分析物在固定靜相與樣品間之分配係數 Co:樣品中分析物之原始濃度. 由於固定靜相的體積相對於樣本體積來説是相當小的(Vs»KfsVf),因此 方程式(1)可以簡化為方程式(2): n = KfsVfC 0. (2). 由方程式(2)可得知,SPME纖維固定靜相上所吸附的量與樣本體積沒 有關係,只與固定靜相的體積、分析物在固定靜相與樣品間之分配係數、 以及樣品中分析物之原始濃度相關。 然而此萃取技術達平衡的時間受限於披覆固定靜相與分析物擴散速 率來決定,當萃取液態樣本時,披覆固定靜相與樣本間存在一薄的水層 膜造成分析物與披覆固定靜相之擴散速率減緩,取得平衡的時間相對的 拉長。 (二) 頂空固相微萃取42. 、45. 面對分析物或基質較為複雜污穢的樣本而言,為避免污染SPME纖維 影響其使用壽命,遂由直接固相微萃取法延伸出另一種萃取法,即頂空 固相微萃取法。 頂空固相微萃取法即先將欲分析之物質先揮發到樣本上方的頂部空 間,再將吸附纖維伸入到樣本頂空進行吸附,此方法針對分配係數較高 或基質複雜之生物檢體由於氣體擴散係數大,所以能縮短達平衡的時 15.
(33) 間,同時不會有汙染纖維的問題44,但對於揮發性低的分析物,即分配係 數較低,較適合直接萃取。 (三) 固相微萃取裝置 SPME 裝置是由兩個部份所組成,其一為纖維固定器(SPME holder), 另一為塗佈有吸附介質的固相微萃取纖維(SPME fiber),纖維必須經由纖 維固定器推出與樣本接觸。此外,由於 SPME 纖維上塗布之吸附介質相 當脆弱易脫落,所以纖維外部以不鏽鋼針管保護之,於使用前才將纖維 推出。詳細分解圖見附圖 2-247。 (四) 固相微萃取操作程序 進行樣品萃取時,首先將設定好長度之不鏽鋼針頭穿過樣品瓶墊片, 再將推桿推至底後固定,使纖維由不鏽鋼管中伸出並固定於樣品瓶中。 當分析物與纖維分配反應達平衡時,便可將纖維縮回,並抽離樣品瓶, 即完成萃取樣品的步驟;分析時,只需將不鏽鋼管插入HPLC –SPME Chamber中圖2-3,再將纖維以相同的方式推出進行溶劑脫附即可49。詳細 步驟如附圖2-449。 (五) 影響SPME-HPLC直接萃取效率的因素42. ﹑44﹑45﹑48. 固相微萃取技術已發展多年,已有許多文獻說明了影響固相微萃取 吸附效率的因素,統整文獻上曾提及的因素,一般而言影響SPME-HPLC 直接萃取效率的因素有(1) SPME纖維的種類 (2) 萃取時間 (3) 脫附溶劑. 16.
(34) (4) 脫附時間 (5) 萃取溫度 (6) 攪拌之影響 (7) pH值 (8)動態脫附及靜 態脫附,綜合以上的因子詳細敘述如下: (1) SPME纖維對萃取效率的影響46 在進行直接萃取較佳分析方法條件討論之前,必須先針對分析物的 特性,選擇一適合的吸附介質。目前已商業化的吸附纖維共有七種如附 表 2-146,不同的纖維塗布著不同的固定靜相,固定靜相的極性、厚度、 表面積、耐溫溫度皆會影響萃取效率。 其中 Polyacrylate (PA), Carbowax-divinylbenzene(CW/DVB), Carbowax-templated resin (CW/TPR ) 纖維屬於極性纖維,主要用於萃取 極性分析物;Polydimethylsiloxane (PDMS)纖維屬於非極性纖維,主要是 用於萃取非極性分析物;其他的三種纖維都是由兩種以上不同性質的聚 合物所塗布而成;Carboxen-polydimethylsiloxane(CAR-PDMS), Polydimethylsiloxane-divinylbenzene(PDMS/DVB), Divinylbenzene-Carboxen-PDMS(DVB/CAR/PDMS)這三種纖維皆可用來 同時吸附不同性質的分析物。 從 SPME 眾多的研究之中,針對分析物的特性選取 FIBER 種類的重 點,有以下六點: 1. 極性分析物選擇極性纖維,非極性分析物選擇非極性纖維。 2. 多種分析物,且特性不同時,選擇雙極性纖維。 3. 極性對於小分子量(<150)分析物影響不大,對於大分子量分析物影響 17.
(35) 較大。 4. 萃取小分子量分析物,CAR/PDMS 是萃取效率最好的纖維。 5. 有塗布 Carboxen 的纖維,萃取大分子量和有極性的分析物的效率較 差。 6. CW/DVB 和 Polyacrylate 纖維最適合萃取大分子量和有極性的分析物。 所以在選擇纖維時應針對分析物的極性、分子量、揮發性等特性,選擇 一較佳萃取纖維,而本研究中所分析的四種化學物質中,TEGDMA 較具 揮發性且分子量低,故挑選 50 μmCW/TPR 為測試纖維,而 UDMA, Bis-GMA 較不具揮發性且分子量大,針對三種分析物極性、分子量差異 大的特性,挑選 60 μmPDMS/DVB, 50/30 μmDVB/CAR/PDMS 為測試纖 維,此外挑選與分析物分子結構式相似的 85 μmPolyacrylic 為測試纖維, 附表 2-2 為四種測試纖維的使用溫度以及操作時間 46。 (2)萃取時間42. ﹑45. 萃取時間主要是和所選擇吸附纖維的種類以及分析物性質有關,吸 附纖維的分配係數越大,吸附量越多,其吸附平衡時間越長;而膜越厚, 吸附量越多,同樣的吸附平衡時間也越長。 而在進行吸附時,吸附量並不是隨著時間的增長而無限量的增加; 其吸附量增加的最大量,是依據分析物在固相微萃取纖維的靜相與樣本 之間質量的傳遞,傳遞達平衡,即表示達吸附平衡,即使吸附時間再增. 18.
(36) 長,吸附量並不會再增加。 (3) 脫附劑48 SPME-HPLC的纖維脫附是以脫附劑來進行的,所以脫附劑的選擇對 於脫附效率影響甚巨,一般脫附劑選擇LC移動相,或其他可溶解樣品的 溶劑,以不影響分析及損害纖維為原則。 (4) 脫附時間42. 、45. 當固相微萃取纖維完成吸附萃取後,繼之的就是以溶劑脫附的方式 將分析物從靜相上脫附下來。當脫附時之脫附時間太短會使得吸附在靜 相上之分析物無法完全脫出殘留在纖維上,會對定量上產生誤差以及造 成纖維下一次使用時連續污染現象;若脫附時間太長則會對纖維造成損 害而影響其使用壽命。而不同的纖維材質對脫附時間也有不一樣的要 求,故慎選脫附時間不僅可確保分析之準確性,更能延長萃取纖維的使 用壽命。 (5) 萃取溫度41. 、44、45. 固相微萃取纖維吸附分析物質會受到萃取時溫度的影響,可從兩方 面來進行討論。在動力學上當增加吸附溫度時會使樣本擴散速率增加, 進而對分析物的吸附量也會增加,同時達平衡所需的時間亦會縮短,若 以相同的吸附時間來比較,增加萃取溫度,纖維所吸附的分析物也會呈 現增加趨勢。以熱力學的角度來看,由於吸附是屬於放熱反應,分配係. 19.
(37) 數K會隨著溫度上升而降低,故樣本吸附量會下降。 因此在整個吸附的過程中這兩個因素會相互影響,一般而言在吸附 未達平衡前,當溫度較高時會增加纖維的吸附量,而達平衡後熱力學繼 而影響整個吸附,增加吸附溫度使分配係數K下降,反而使纖維上已吸附 之分析物再度被脫附,故會使吸附量下降,因此分析時選擇一適當的萃 取溫度是非常重要的44. 、45. 。. (6) 攪拌之影響44. 、45. 一般而言,萃取時當樣本處於靜止狀態時,與達吸附平衡時間有相 關者為分配係數K以及固定靜相的體積;若樣品處於攪動的狀態時,由於 質傳性質不同,對於平衡時間、偵測極限及萃取回收率的相對標準偏差 造成影響,故理論上對唾液樣品進行攪拌,可縮短達平衡的時間。而造 成攪動的方式有magnetic stirring、vortex mixing、fiber moving、sonication 等,而以magnetic stirring較常應用在固相微萃取上44。 (7) pH值45 分析樣本之 pH 值對萃取效率的影響取決於分析物質本身的種類。 從文獻中發現, pH 值的改變對一般的分析物並不會有所影響,如鹵化 醚類、硝基苯類或芳香族碳氫化合物等,pH值的改變對萃取效率並無明 顯影響,唯有對酚類化合物時,若將 pH 值調整至較低時會使酚類解離 而提升萃取量42. 、49. 。. 20.
(38) (8) 動態脫附及靜態脫附48 一般 SPME-HPLC 脫附分為動態及靜態脫附,動態脫附即所謂的移動 相直接脫附,將吸附好的纖維置入脫附chamber中後,直接以移動相進行 脫附且直接進入分析管柱進行分析,也就是脫附與分析同時進行,優點 在於可省去脫附時間;而靜態脫附是先將脫附chamber注入選定好的脫附 劑,再將吸附完成後之纖維置入其中進行脫附,待脫附完成後,取出纖 維後才進行分析,與動態脫附比較起來有較佳的脫附效果,考慮脫附效 率及纖維的使用壽命,研究選擇靜態脫附。 (六) 分配係數 選定較好的SPME參數即可得一較高分配係數,也就是較佳的吸 附效率,吸附在固定靜相上的量可以下列方程式(1)來表示: n=. KfsVfC 0Vs KfsVf + Vs. (1). n:吸附在固定靜相上分析物之質量 Vf:固定靜相之體積 Vs:樣品體積 Kfs:分析物在固定靜相與樣品間之分配係數 Co:樣品中分析物之原始濃度. 由於固定靜相的體積相對於樣本體積來説是相當小的(Vs»KfsVf),因此 方程式(1)可以簡化為方程式(2): n = KfsVfC 0 n K fs = C0V f. (2) (3). 由方程式(3)可知分配係數可經由固定靜相吸附量、固定靜相體積、及樣 21.
(39) 本濃度求得。 (七) 固相微萃取法於生物檢體的應用 固相微萃取技術除了環境分析上的應用外,應用於基質複雜的生物 檢體時由固相微萃取所衍生出的頂空固相微萃取法由於固定靜相僅於樣 本頂部空間來進行萃取,避免了樣本基質污染纖維同時亦能保護並延長 纖維使用壽命。 目前固相微萃取法於生物檢體的技術最常應用於藥物分析以及刑事 科學上的應用,如從尿液檢體中檢驗 MDMA、安非他命藥物濫用的情形 50. ;從血液樣本中檢測酒精中毒以及麻醉劑的使用51;從血漿及唾液樣本. 中檢測類固醇及安眠藥52;以及牛乳中戴奧辛的汙染等,由以上的應用例 子可知 SPME 對於生物檢體的檢測方法是多元且可行的,牙科材料的危 害評估相關研究略顯不足,以 SPME 的特性應用於唾液檢體檢測,是可 行的,2004年Ortengren等針對HQ(Hydroquinone monomethyl ether)、 bisphenol-A(4,4-Isopropylidenediphenol)、TEGDMA、EGDMA(Ethylene glycoldimethacrylate)、MMA(Methyl acrylate)、MA(Methacrylic)六種低分 子物質開發SPME-GC-MS方法,而主要高分子量材質UDMA、Bis-GMA 無法同時分析,以HPLC結合SPME進行分析是較適合的53。. 22.
(40) 第貳章 圖表附錄. 23.
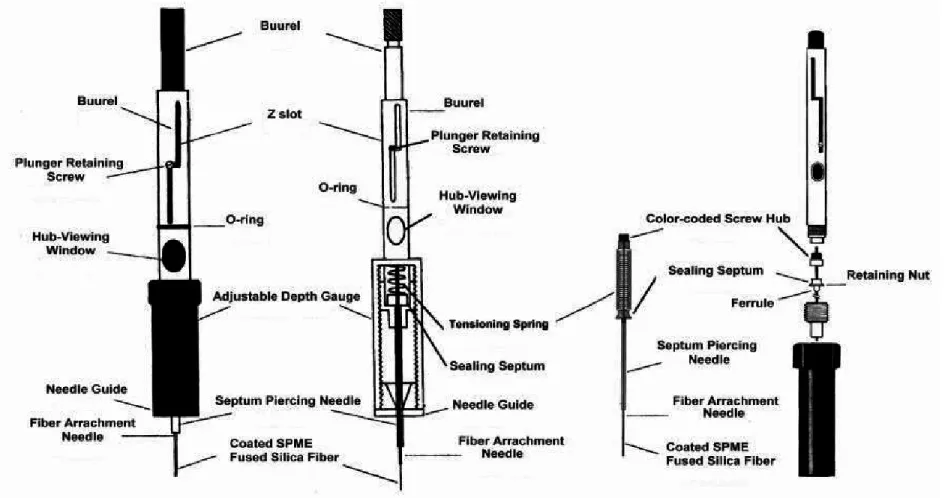
(41) 圖2-1 TEGDMA, UDMA, Bis-GMA結構圖. 24.
(42) 圖 2-2 固相微萃取裝置分解圖(SUPELCO 2000)47. 25.
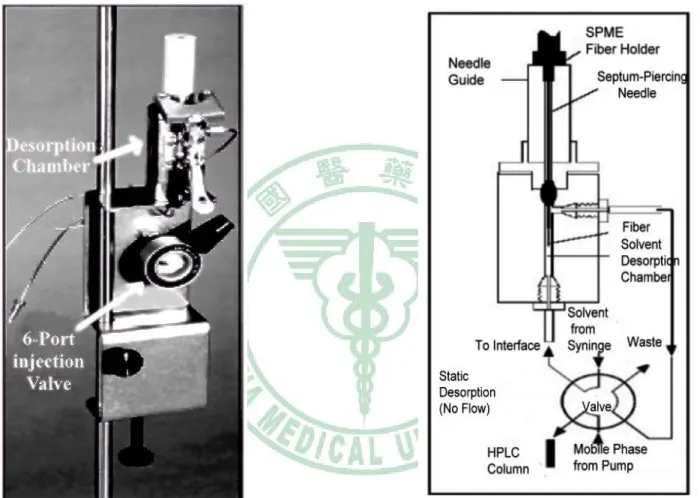
(43) 圖 2-3 SPME-HPLC Chamber 圖解 49. 26.
(44) 針頭刺穿墊片. 伸出纖維進行. 收回纖維並. 萃取. 抽離注射針頭. 針頭插入. 伸出纖維進入. 收回纖維並. HPLC Interface Chamber 進行脫附 抽離針頭. 圖 A 為吸附過程. 圖 B 為脫附過程 圖 2-4 固相微萃取操作圖解 49. 27.
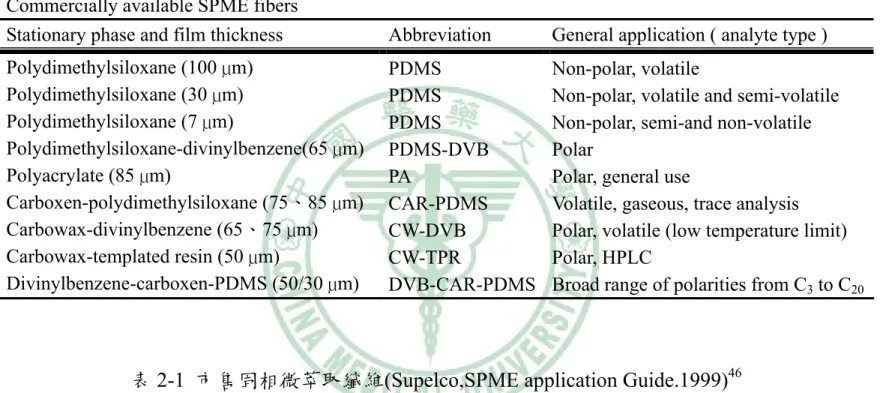
(45) Commercially available SPME fibers Stationary phase and film thickness. Abbreviation. General application ( analyte type ). Polydimethylsiloxane (100 µm) Polydimethylsiloxane (30 µm) Polydimethylsiloxane (7 µm) Polydimethylsiloxane-divinylbenzene(65 µm) Polyacrylate (85 µm) Carboxen-polydimethylsiloxane (75、85 µm) Carbowax-divinylbenzene (65、75 µm) Carbowax-templated resin (50 µm) Divinylbenzene-carboxen-PDMS (50/30 µm). PDMS PDMS PDMS PDMS-DVB PA CAR-PDMS CW-DVB CW-TPR DVB-CAR-PDMS. Non-polar, volatile Non-polar, volatile and semi-volatile Non-polar, semi-and non-volatile Polar Polar, general use Volatile, gaseous, trace analysis Polar, volatile (low temperature limit) Polar, HPLC Broad range of polarities from C3 to C20. 表 2-1 市售固相微萃取纖維(Supelco,SPME application Guide.1999)46. 28.
(46) Stationary Phase PDMS PDMS/DVB PA CW/TPR DVB/CAR/PDMS. Film Thickness. pH. 100µm 2-10 65 µm 85 µm 50 µm 50/30 µm. 2-11 2-11 2-11 2-11. Maximum Temp. Recommended Operating Temp. Conditioning Temp. Time (Hrs.). 280 ℃. 200-280 ℃. 250 ℃. 0.5. 270 ℃ 320 ℃ 260 ℃ 270 ℃. 200-270 ℃ 220-310 ℃ 200-250 ℃ 230-270 ℃. 250 ℃ 300 ℃ 220 ℃ 270 ℃. 0.5 2 0.5 1. Source:Supelco,SPME application Guide. 1999. 表 2-2 各種測試纖維的耐受溫度 46. 29.
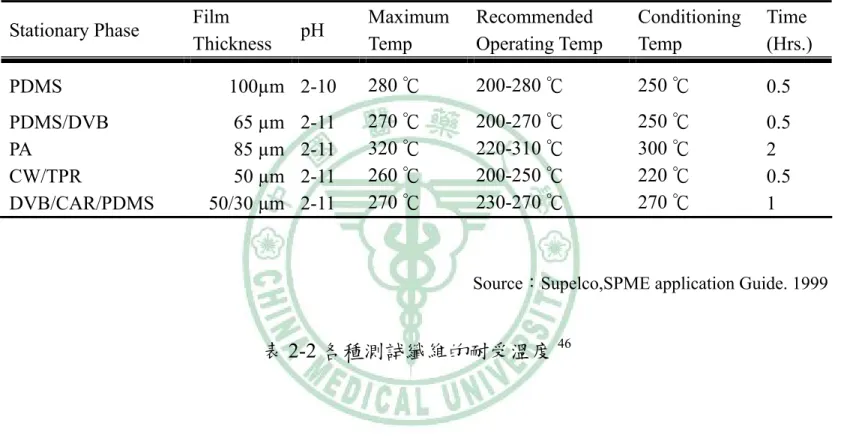
(47) 第叁章 材料設備與方法. 第一節 儀器設備與試藥 (一) 試藥 1.乙醇(Ethanol): Anhydrous 99.5%, NASA, Lot : 014937( Fair Lawn, New Jersey, USA.) 2.乙腈(Acetonitrile): HPLC solvent, J.T. Baker, Lot: A36808 ( Phillipsburg, New Jersey, USA.) 3. TEGDMA (Triethylenglykol-dimethacrylat): 95%, ALDRICH, Lot: 09004BC-184(Aldrich Chemie Gmbh, Riedstr. 2, D-89555. Steinheim, Germany) 4. UDMA (Urethane dimethacrylate): Shinnakamura Kagaku. (Wakayama, Japan) 5. Bis-GMA (2, 2-bis [4(2-hydroxy-3-methacryloyloxy-propyloxy) -phenyl] propane): Shinnakamura Kagaku. (Wakayama, Japan) 6.硫酸(Sulfuric acid):ACS Grade, 96.4 %, Tedia, Lot:703029 (Tedia Co., Fairfield, OH 45014 USA) 7.氫氧化鈉(Sodium hydroxide):ACS Grade, 50.4 %, Fisher, Lot:920682-24(Fisher Scientific, Fair Lawn, New Jersey 07410 USA) 8. 複合樹脂 Tetric Ceram 顏色 B3 廠商: Ivoclar Vivadent AG ( FL-9494 Schaan, Liechtenstein) 30.
(48) 9.複合樹脂 Palfique Estelite 顏色 A2 廠商: Tokuyama dental (38-9, Taitou 1-chome, Taitou-ku, Tokyo, Japan) 10.複合樹脂 Compoglass flow 顏色 A3 廠商: Ivoclar Vivadent AG (FL-9494 Schaan, Liechtenstein) 11.複合樹脂 Dyract 顏色 A3 廠商: Dentsply Dmbh (78467 Konstanz, Germany) 12.複合樹脂 Spectrum 顏色 A3 廠商:Dentsply Dmbh (78467 Konstanz, Germany) (二) 設備 1. 高效液相層析儀(HPLC)機型:Perkin Elmer Series 200 (Norwalk, Connecticut, USA) 2.偵測器機型 : Perkin Elmer 785A UV/VIS Detector (Norwalk, Connecticut, USA) 3. 層析管柱:25 cm x 4.6 mm , 5 μm SUPELCO C-18 (Supelco Co., Bellefonte, PA 16823 USA) 4. SPME-HPLC Interface: 250 μL chamber interface (Supelco Co., Bellefonte, PA 16823 USA) 5. pH Meter:pH/mv/Temp meter, SP-701;(Suntex Instruments Co.,Taiwan R.O.C) 6. 微量注射器 syringe:10 µL、50 µL、500 µL、1000 µL, glass; (Hamilton Co., Reno, Nevada 89502 USA) 7. 固相微萃取採樣器 SPME holder:Supelco 57330-U;(Supelco. 31.
(49) Co., Bellefonte, PA 16823 USA) 8. 固相微萃取纖維 SPME fiber: 85 µm PA, Supelco 5-7306; (Supelco Co., Bellefonte, PA 16823 USA) 9. 固相微萃取纖維 SPME fiber:50 µm CW/TPR, Supelco 57315;(Supelco Co., Bellefonte, PA 16823 USA) 10. 固相微萃取纖維 SPME fiber:50/30 µm DVB/CAR/PDMS, Supelco 57348-U;(Supelco Co., Bellefonte, PA 16823 USA) 11. 固相微萃取纖維 SPME fiber:60 µm PDMS/DVB, Supelco 57317;(Supelco Co., Bellefonte, PA 16823 USA) 12. 加熱攪拌器:Barnstead/Thermolyne, SP46925;(Dubuque, Iowa, 52001 USA) 13. 攪拌子:magnetic stirring-bar, 8 mm × 3 mm;Spinbar (made in USA) 14. 取樣瓶 Vial:Amber Vial, Screw Top 4mL with Hole Cap PTFE/Silicone Septa; (Supelco Co., Bellefonte, PA 16823 USA) 15. 玻璃滴管:Pasteur pipette, glass; (Kimble Glass Inc., USA) 16. Pipette:Transferpipette 100~1000 µl;(Brand GMBH Co., Germany) 17. Micro-Pipette:Fixed Volume Micropipette 1000 µl;(Socorex ISBA S.A., Switzerland) 32.
(50) 18. 定量瓶 volumetric flack:10、25、50、100 mL, glass;(Brand GMBH Co., Germany) 19. 定量天平 Electronic Balance:AY220 220 g~0.1 mg(Shimadzu Co., Japan) 20. 0.45μm HPLC- NYLON 濾紙:Lida Manufacturing Corp. (9115 Avenue Kenosha, WI) 21. 2.5mL 塑膠注射針筒;台灣特浦股份有限公司 高雄縣大寮 鄉華中路 40 號 22. 樹脂可見光聚合機: Visible Light Curing Unit;Visilux 2(3M), model 5520AA (St. Paul, MN 55144-1000 West Germany) 23. LC-MS 機型: LCQ DECA XPplus,廠商: Thermo Finnigan (Milan, Italy). 33.
(51) 第二節 儀器參數條件 (一) HPLC 條件: HPLC 機型. : Perkin Elmer Series 200. 移動相. : Acetonitrile 65% : D.I Water 35%. 流速. : 1 mL/min. 層析管柱. : 25 cm x 4.6 mm , 5 μm SUPELCO C-18. SPME-LC Interface : 250 μL chamber interface 偵測器機型. : Perkin Elmer 785A UV/VIS Detector. Solvent delay time :5 分鐘 吸收波長. : 215 nm14. 、15. Noise threshold. : 1μV. Area threshold. : 100 μV. Bunching factor. :3. 34.
(52) (二) LC-MS 條件: 移動相. :40%(D.I.水+0.1% 甲酸):60%乙腈. 流速. : 0.2 mL/min. 層析管柱. : 150 mm x 0.2 mm , 4μm Meta Chem Technologies.Inc C-18. LC-MS mode. : postitive mode. Capillary temp. : 350° C. Source voltage. : 4.5 KV. Capillary voltage. : 21 V. Sheath gas flow. : 35 arb (0.525 L/min). 35.
(53) 第三節 研究架構 本研究係在實驗室中開發以直接固相微萃取結合高效液相層析儀 的方法針對牙科填補之複合式樹脂,進行其唾液培養之溶出物的定量分 析,研究架構如附圖 3-1、3-2 所示。 (一) HPLC 分析方法的建立 建立 HPLC 分析方法,參考 HPLC-UV 為分析方法的文獻 13﹑14﹑15. 找出適當的分析條件,於乙醇中配置標準溶液,以直接注. 入法建立檢量線;於唾液中配置標準溶液,取 0.5mL 唾液溶液與 0.5mL 乙醇 1:1 混合,24°C、5000rpm 下離心 10 分鐘,取上清液 10μl 注射分析,建立唾液溶劑萃取檢量線。 (二) 直接固相微萃取方法建立 SPME-HPLC 直接固相微萃取法受到下列因素的影響,(1) SPME 纖維的種類 (2) 萃取時間 (3) 脫附溶劑 (4) 脫附時間 (5) 萃取溫度 (6) 攪拌之影響 (7) pH 值 48. 、49. ,研究係針對上述參數逐. 一進行探討,實驗操作步驟見附圖 3-3。 (1) 固相微萃取纖維的選擇 於唾液中配製濃度分別為 10 µg/mL 的 TEGDMA,10 µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 之標準溶液,混合均勻後裝至 4 mL vial 中,依據分析物的分子量及極性等特性來選擇適當的分析纖. 36.
(54) 維。研究所選定的纖維有 85 µm PA 纖維、50µm CW/TPR 纖維、60 µm PDMS/DVB 纖維以及 50µmDVB/CAR/PDMS 等四種性質不同的 纖維來進行試驗,樣本於室溫下,以直接法吸附 20 分鐘,以移動 相(65%乙腈、35%D.I.水)脫附 20 分鐘,進行萃取有效性探討,選擇 最佳者作為研究之微萃取纖維。 (2) 萃取時間探討 於唾液中配製濃度分別為 10µg/mL 的 TEGDMA,10µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 之標準溶液,混合均勻後裝至 4 mL vial 中,樣本於室溫下,以 PDMS/DVB 纖維分別直接吸附 5、10、 15、20、25 分鐘,脫附 20 分鐘來進行萃取時間探討,選擇最佳者 作為研究之最適萃取時間。 (3) 脫附溶劑探討 於唾液中配製濃度分別為 10µg/mL 的 TEGDMA,10 µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 之標準溶液,混合均勻後裝至 4 mL vial 中。於室溫下,以 PDMS/DVB 纖維直接吸附 20 分鍾,分別以 移動相(65%Acetonitrile 35%D.I.Water)、100%Acetonitrile、100%乙 醇脫附 20 分鐘作脫附溶劑比較,選擇最佳者作為研究之最適脫附 溶劑。 (4) 脫附時間探討. 37.
(55) 將萃取完成之纖維以移動相靜態脫附 5、10、15、20 分鐘,進行脫 附時間探討;於未經 pH 值調整之唾液中配製 10µg/mL 的 TEGDMA,10 µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 之標準溶液, 混合均勻後裝至 4mL vial 中,於室溫下,以 PDMS/DVB 纖維直接 吸附 20 分鐘來進行探討,選擇最佳者作為研究之最適脫附時間。 (5) 萃取溫度探討 加熱分別調為室溫(約 27 ℃)、40 ℃、60 ℃、80 ℃來測試不同 樣本溫度下對萃取效率的影響;於未經 pH 值調整之唾液中配製 10µg/mL 的 TEGDMA,10µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 之標準溶液,混合均勻後裝至 4mL vial 中,調整不同的加熱溫度, 以 PDMS/DVB 纖維直接吸附 20 分鐘,以移動相靜態脫附 15 分鐘 來進行吸附溫度影響探討,選擇最佳者作為研究之最適樣本加熱溫 度。 (6) 攪拌速度對萃取的影響 在瓶中加入一攪拌磁石,並將攪拌器轉速調整為不攪拌、200 rpm(0.18g)、500 rpm(1.11g)、800 rpm(2.86g),藉由改變不同的攪拌 速度來探討相同吸附時間下攪拌與否對纖維上吸附量的影響;於未 經 pH 值調整之唾液中配製 10µg/mL 的 TEGDMA,10 µg/mL 的 UDMA,10µg/mL 的 Bis-GMA 標準溶液,混合均勻後裝至 4mL vial. 38.
(56) 中,樣本於 40 ℃下,調整不同轉速,以 PDMS/DVB 纖維直接吸附 20 分鐘,以移動相靜態脫附 15 分鐘來進行攪拌速度對萃取的影響 探討,選擇最佳者作為研究之最適攪拌速度。 (7) 樣本 pH 值探討 用 citrate-phosphate buffer 將未經 pH 值調整之唾液調整至 pH=3、5、9 以及 pH=11,未經調整之唾液 pH 值約為 7.65;於不同 pH 值之唾液中配製 10µg/mL 的 TEGDMA,10 µg/mL 的 UDMA, 10µg/mL 的 Bis-GMA 標準溶液,混合均勻後裝至 4mL vial 中,加 入攪拌磁石。樣本於 40 ℃下,轉速調整為 200rpm,以 PDMS/DVB 纖維直接吸附 20 分鐘,以移動相靜態脫附 15 分鐘來進行 pH 值對 吸附效率影響探討,選擇最佳者作為研究之最適樣本 pH 值。 (三)分配係數 於唾液中配製濃度高低的標準溶液,分別為 1.5、7.5μg/ml 的 TEGDMA;2.5、12.5µg/mL 的 UDMA;2.5、12.5µg/mL 的 Bis-GMA 標準溶液,混合均勻後裝至 4mL vial 中,加入攪拌磁石。樣本於 40 ℃下,轉速調整為 200rpm,以 PDMS/DVB 纖維直接吸附 20 分 鐘,以移動相靜態脫附 15 分鐘,得到纖維吸附量後,來探討高低 濃度的分配係數。 (四)分析品質管制. 39.
(57) (1) 檢量線建立 標準溶液的配製是在未經 pH 值調整之唾液中配製 TEGDMA、 UDMA、Bis-GMA 標準溶液,混合均勻後裝至 4 mL vial 中,加入 攪拌磁石,樣本於 40 ℃下,轉速調整為 200 rpm,以 PDMS/DVB 纖維直接吸附 20 分鐘,以移動相靜態脫附 15 分鐘。製作異日三條 檢量線。 檢量線的配製應至少配製五種不同的濃度,其所配製之標準溶 液,線性相關係數(r)>0.995,各濃度之相對預測偏差(RPD)應小於 10 %。檢量線配製濃度範圍如下 TEGDMA:0.3~30 µg/mL;UDMA: 0.5~50 µg/mL;Bis-GMA:0.5~50µg/mL。 (2) 方法偵測極限 以檢量線最低點濃度,分別為 TEGDMA:0.3 µg/mL;UDMA: 0.5 µg/mL;Bis-GMA:0.5 µg/mL,重複分析 7 次,所得 7 次儀器 訊號代入迴歸方程式即得相當之濃度,取其 3 倍標準偏差(standard deviation;SD),即為各物質之分析方法偵測極限。 (3) 分析之精密度與準確度 取檢量線範圍內低濃度及中間偏高濃度各一點,分別為 TEGDMA:1.5、7.5µg/mL;UDMA:2.5、12.5µg/mL;Bis-GMA: 2.5、12.5µg/mL,於樣本分析的同時,進行標準品分析,以確認分. 40.
(58) 析之精密度與準確度。 (五)樹脂樣本 (1) 聚合時間影響溶出量探討 針對 5 種商品中(如表 3-1),具代表性的商品 Tetric Ceram 進 行聚合時間探討。在光聚合機照射 20、40、60 、80、120 秒下聚 合 6.5±0.5 mm × 1 mm 大小樹脂份,聚合後於室溫下放置 24 小時自 然風乾,再分別浸入裝滿乙醇的 4 mL vial 中,於室溫下培養 7 天 後進行分析,探討聚合時間與溶出量的關係。 (2) 溫度影響溶出量探討 針對 5 種商品中,具代表性的商品 Tetric Ceram 進行溫度探討。 在光聚合機照射 80 秒下分別聚合 6.5±0.5 mm × 1 mm 大小樹脂, 聚合後於室溫下放置 24 小時自然風乾,再分別浸入裝滿乙醇的 4 mL vial 中,分別於 0 ℃、室溫(27 ゚ C)、40 ℃下培養 7 天後進行分析, 探討溫度對溶出量影響。 (3) pH 值影響釋出量探討 針對 5 種商品中,具代表性的商品 Tetric Ceram 進行 pH 值探 討。在光聚合機照射 80 秒下聚合 6.5±0.5 mm × 1 mm 大小樹脂, 聚合後於室溫下放置 24 小時自然風乾,再浸入裝滿經調整 pH 值為 3、5、7.65(不調整)、9、11 乙醇的 4 mL vial 中培養 7 天後進行分. 41.
(59) 析,探討 pH 值對溶出量影響。 (4) 市售商品天數探討 將市售 5 種光聚式複合式樹脂(如表 3-1),在光聚合機照射 120 秒下分別聚合 6.5±0.5 mm × 1 mm 大小樹脂,聚合後於室溫下放置 24 時自然風乾,再分別浸入裝滿乙醇及唾液的 4 mL vial 中,於室 溫下浸泡 0.5、1、3、5、7、10 天後進行分析,探討浸泡天數的關 係,且比較唾液與乙醇之間的差異及傳統分法與 SPME 的關係。 (5)浸泡液質譜定性 將配置好的標準溶液與浸泡好的溶液以 LC-MS-MS 作質 譜確認,以定性釋出物。. 42.
(60) 第叁章 圖表附錄. 43.
(61) SPME-HPLC 分析 方法. HPLC 分析 條件. 標準品溶於乙 醇中 纖維 攪拌. SPME 吸附 條件. PH. 時間. SPME 脫附 條件. 脫附劑 時間 精密度與準確度. 方法偵測極限 實驗室 QA & QC. 檢量線. 分配係數. 圖 3-1 唾液樣本 SPME-HPLC 分析方法建立. 44.
(62) 複合樹脂樣本 浸泡. 聚合時間: 20 40 60 80 120 s. 溫度 : 0 ゚ C 室溫 40 ゚ C﹑. PH : 3、5 、7.65、 9、11 溶出 因素. 浸泡天數. 圖 3-2 真實樣本培養架構. 45. 分別浸泡於乙醇及唾 液浸泡液中 0.5 、1、 3、5、7、10 日.
(63) Fiber condition. 將配製好濃度之標準品放至 4 mL vial 中,瓶內置入磁石. 將樣本置於加熱攪拌器上,攪拌 2 小時. 將 SPME 穿刺過墊片,推出纖維,進行吸附. 待吸附達平衡後,收回纖維. 將 SPME 置於 Interface 上,推出纖維於 Chamber 中進行脫附. 數據分析. 圖 3-3SPME 實驗操作流程. 46.
(64) 商品名. TEGDMA. UDMA. Bis-GMA. Tetric Ceram. O. O. O. Palfique Estelite. O. ×. O. Compoglass flow. O. O. ×. Dyract. ×. O. ×. Spectrum. O. ×. O. 表 3-1 市售複合式樹脂商品. 47.
(65) 第肆章. 結果與討論. 第一節 HPLC 分析 於乙醇中配製 TEGDMA、UDMA、Bis-GMA 標準溶液,乙腈 65 % D.I.水 35 % 固定比例下,流速 1 ml/min 下,分析物分別於 5.29 分、7.17 分、8.92 分時出現波峰(圖 4-1a),完成整個分析時間約 15 分鐘。 直接注入法 TEGDMA 檢量線濃度範圍為 2.5~200µg/mL;UDMA 檢量線濃度範圍為 6.5~260 µg/mL;Bis-GMA 檢量線濃度範圍為 3.2~ 320µg/mL 異日線性相關係數 r 皆大於 0.995,RPD 值皆在 13 %以下。 溶劑萃取唾液 TEGDMA 檢量線濃度範圍為 3.125~250µg/mL; UDMA 檢量線濃度範圍為 3.325~270 µg/mL;Bis-GMA 檢量線濃度範圍 為 7.5~300µg/mL 異日線性相關係數 r 皆大於 0.995,RPD 值皆在 12 % 以下。. 48.
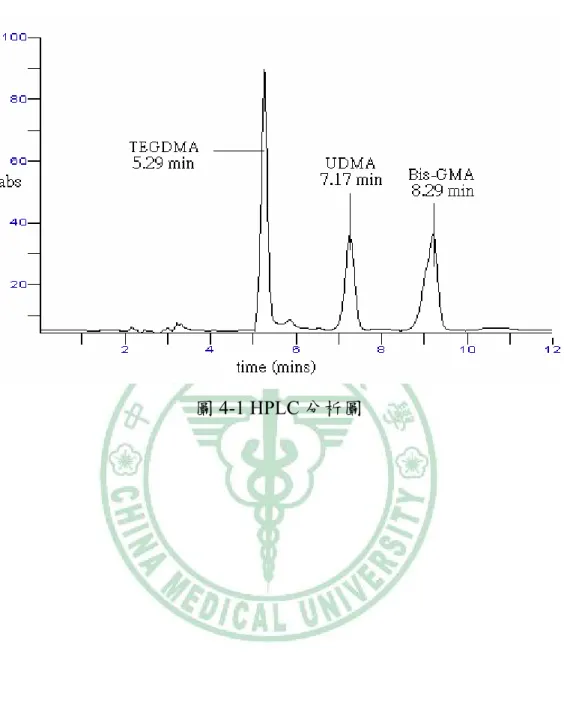
(66) 第二節 固相微萃取方法建立 (一) 固相微萃取纖維的選擇 在 相 同 的 濃 度 、 相 同 的 分 析 條 件 下 分 別 以 85 μmPA 、 50 μmCW/TPR 、60 μmPDMS/DVB、50/30 μmDVB/CAR/PDMS 等四 種 纖 維 來 進 行 吸 附 測 試 , 由 研 究 結 果 顯 示 ( 圖 4-2-1a) , 60 μmPDMS/DVB 能同時吸附 4 種分析物,雖面積值小於 50/30 μmDVB/CAR/PDMS 纖維,考慮分析時之穩定性,以相對標準偏差 (Relative Standard Deviation;RSD)來判定,60 μmPDMS/DVB 較為 穩定(圖 4-2-1b),故選定以 60 μmPDMS/DVB 為此次研究中 SPME 吸附介質。 (二) 萃取時間 以 60 μmPDMS/DVB 纖維直接浸入吸附 5、10、15、20、25 分 鐘,其餘條件參數相同下來進行萃取時間探討。 由結果顯示(圖 4-2-2a、4-2-2b)吸附 20 分鐘時,纖維吸附值大 約已呈現飽和狀態,吸附 25 分鐘面積值不再增加且有下降趨勢,, 選定以吸附 20 分鐘為最適吸附時間。(數據詳見表 4-2-2) (三) 脫附劑 以 60 μmPDMS/DVB 纖維直接浸入吸附 20 分鐘,分別以移動 相(65% acetonitrile 35% D.I.Water)、100% acetonitrile、100% 乙醇脫. 49.
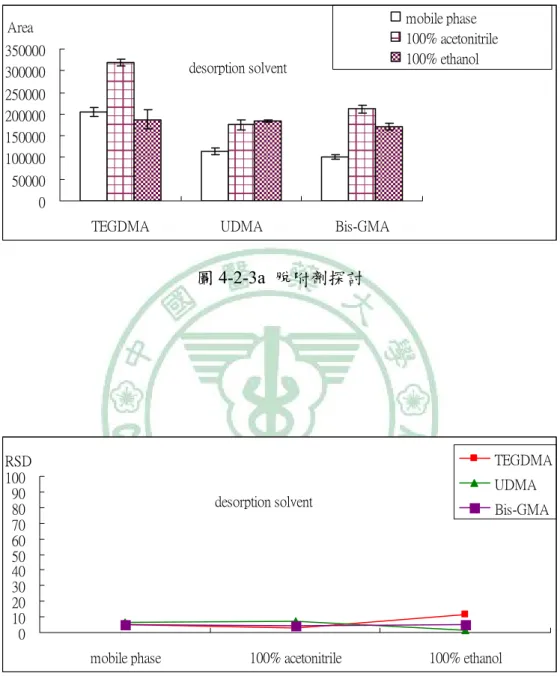
(67) 附,其餘條件參數相同下來進行脫附溶劑探討。 由結果顯示(圖 4-2-3a)面積值以 100% acetonitrile 最大,100% 乙醇次之,而 65% acetonitrile 35% D.I. Water 最小。在考慮分析時 之穩定性,以相對標準偏差(Relative Standard Deviation;RSD)來判 定(圖 4-2-3b),100%乙醇 RSD 值大於 10%以上,而 65% acetonitrile 35% D.I. Water 移動相的 RSD 值均小於 10%(圖 4-2-3b),雙重考量 下最後選定 65% acetonitrile 35% D.I. Water 移動相為較佳脫附劑。 (數據詳見表 4-2-3) (四) 脫附時間探討 以 60 μmPDMS/DVB 纖維直接浸入吸附 20 分鐘,以移動相(65% acetonitrile 35% D.I.Water) 分別脫附 5、10、15、20 分鐘,其餘條 件參數相同下來進行脫附時間探討。 由結果顯示(圖 4-2-4a、4-2-4b)在脫附達 15 分鐘時,面積值已 有趨緩現象,20 分鐘時有明顯下降趨勢,考慮分析時之穩定性(圖 4-2-4c)脫附 15 分鐘的 RSD 值均小於 10%,雙重考量下最後選定以 脫附時間 15 分鐘為較佳脫附時間。(數據詳見表 4-2-4) (五) 萃取溫度探討 萃取溫度分別為室溫(約 27 ℃)、40 ℃、60 ℃、80 ℃其餘條件 參數相同下來測試不同萃取溫度下對萃取效率的影響。. 50.
(68) 由研究結果顯示(圖 4-2-5a),有加熱分析物之面積值明顯大於 不加熱,但隨著溫度上升而明顯減少(40 ℃>60 ℃>80 ℃),且 40 ℃ 的 RSD 值均小於 10 %(圖 4-2-5b)。在此兩種因素考量下,遂決定以 40 ℃為研究最適萃取溫度。(數據詳見表 4-2-5) (六) 攪拌速度對萃取的影響 將攪拌器轉速調整為不攪拌、200 rpm(0.18 g)、500(1.11 g) rpm、800(2.86 g) rpm 來進行攪拌速率對吸附量的探討。 由結果顯示(圖 4-2-6a),有攪拌者之面積值明顯優於不攪拌。 在考慮分析時之穩定性下,200rpm 的 RSD 值均小於 10 %(圖 4-2-6b),在此兩種因素考量下遂決定以 200 rpm 為最適萃取攪拌速 度。(數據詳見表 4-2-6) (七) 樣本 pH 值探討 未經調整之唾液 pH 值約為 7.65,以 citrate-phosphate buffer 將 未經 pH 值調整之唾液調整至 pH=3、pH=5、pH=9 以及 pH=11,來 測試 pH 值改變是否會對萃取造成影響。 由研究結果顯示,調整 pH 值對整個萃取並沒有正面的提升, 並且在未經調整之樣本,無論是面積值(圖 4-2-7a)或 RSD 值(圖 4-2-7b)皆優於其他者,故選定不調整 pH 值為最適萃取 pH 值。(數 據詳見表 4-2-7). 51.
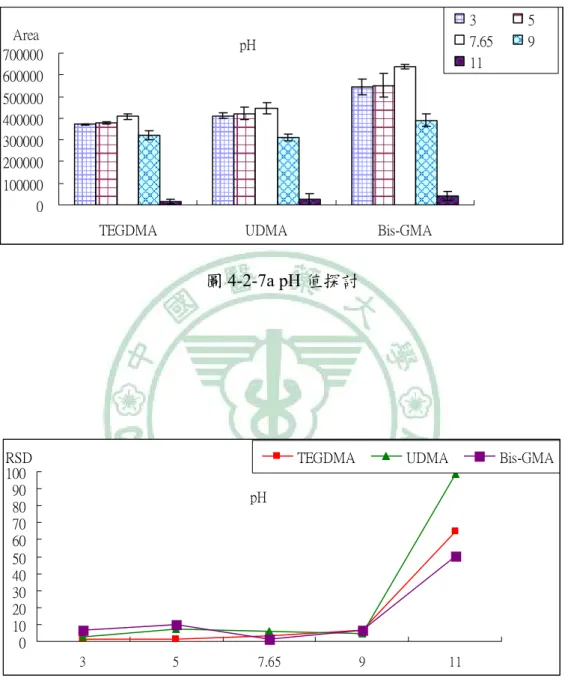
(69) 第三節分配係數 於唾液中分別配製濃度 1.5、7.5 μg/ml 的 TEGDMA;2.5、 12.5 µg/mL 的 UDMA;2.5、12.5 µg/mL 的 Bis-GMA 標準溶液,以 探討的 SPME 參數, 分析得到纖維吸附量後,即可得到分配係數 由研究結果顯示,重複 7 次高低濃度的分配係數皆相近且穩定,分 別為;TEGDMA: 70.17、72.12;UDMA: 85.27、84.56;Bis-GMA: 151.01、152.05。(數據詳見表 4-3). 52.
(70) 第四節 分析品質管制 (一) 檢量線建立 TEGDMA 檢量線濃度範圍為 0.3~30µg/mL;UDMA 檢量線濃 度範圍為 0.5~50 µg/mL;Bis-GMA 檢量線濃度範圍為 0.5~50µg/mL 異日線性相關係數 r 皆大於 0.995,RPD 值皆在 12 %以下。(表 4-4-1) (二)方法偵測極限(MDL) 以檢量線最低點濃度,分別為 TEGDMA:0.3 µg/mL;UDMA: 0.5 µg/mL;Bis-GMA:0.5 µg/mL,重複分析 7 次,所得 7 次儀器 訊號代入迴歸方程式即得相當之濃度,取其 3 倍標準偏差(standard deviation;SD),即為各物質之分析方法偵測極限。(表 4-4-2) (三) 分析之精密度與準確度 分別取檢量限範圍內中低、高濃度值 2 點,分別為 TEGDMA: 1.5、7.5 µg/mL;UDMA:2.5、12.5 µg/mL;Bis-GMA : 2.5、12.5 µg/mL,於樣本分析的同時,進行標準品分析,結果顯示無論在低 濃度或高濃度的樣本,其變異係數(CV %)皆小於 10 %,且準確度 皆介於 100±10 %之間,表示此分析方法之再現性良好。(表 4-4-3). 53.
(71) 第五節 真實樣本培養 (一) 市售複合式樹脂 市售商品中, 5 種商品的溶出物皆隨天數增加而溶出量增加 (圖 4-5-1a~e),乙醇培養的樣品,大部分在第 7 天時,明顯大量溶 出,且到第 10 天並無明顯增加,其中商品 Tetric Ceram 是含分析 物種類最多的商品,適合做為聚合時間、溫度及 pH 探討的樹脂。 針對 5 種商品,以乙醇培養 7 日溶出量做一比較,TEGDMA 溶出最多為商品 Palfique Estelite;UDMA 釋出最多為商品 Compoglass flow;Bis-GMA 溶出最多為商品 Palfique Estelite (圖 4-5-1f)。 唾液浸泡的樣本中,所有天數皆無測到分析物。 (二) LC-MS-MS 質譜 將配製好的標準溶液與培養好的乙醇溶液以 LC-MS-MS 作質 譜確認,以定性溶出物。 標準溶液 LC-MS-MS 的質譜圖(圖 4-5-2 a、c、e)與乙醇培養液 的質譜圖(圖 4-5-2 b、d、f)特殊質荷皆相同。 (三) 聚合時間探討 以樹脂 Tetric Ceram 在光聚合機照射 20、40、60 、80、120 秒, 於室溫下浸泡 7 天後進行分析,探討聚合時間與溶出量的關係。 由結果顯示(圖 4-5-3),隨著聚合時間的增加,溶出量隨著減. 54.
(72) 少,尤其在 60 秒前更明顯。(數據詳見表 4-5-3) (四) 溫度探討 以樹脂 Tetric Ceram 在 0 ℃、室溫(27 ゚ C)、40 ℃下浸泡 7 天 後分析,進行溫度對溶出量影響的探討。 由結果顯示(圖 4-5-4), UDMA、Bis-GMA 隨著溫度的上升溶 出量大量增加,而 TEGDMA 較不受溫度的的影響,只有些微的增 加。(數據詳見表 4-5-4) (五) pH 值探討 以樹脂 Tetric Ceram 於經調整 pH 值為 3、5、7.65、9、11 乙醇 溶液中浸泡 7 天後分析,進行 pH 值對溶出量影響的探討。 由結果顯示(圖 4-5-5),隨著 pH 的上升,TEGDMA、UDMA、 Bis-GMA 溶出量減少,。(數據詳見表 4-5-5). 55.
(73) 第六節 討論 (一) 固相微萃取 纖維的材質種類和厚度對萃取效率影響最大,需根據待萃取成 分的性質,綜合考慮其極性、沸點、分子量來選擇萃取纖維,不同 的纖維塗布著不同的固定靜相,固定靜相的極性、厚度、表面積、 耐受溫度皆會影響萃取效率,選擇的基本原則是同性相容原理:極 性纖維萃取極性化合物,非極性纖維萃取非極性化合物。 分析物之萃取絕對量會隨纖維體積增加而增加,較長較厚的纖 維可萃取更多的分析物,但過長纖維容易折斷,故一般纖維長度介 於 1~2 公分,越厚的纖維雖可以萃取較多分析物,但相對的會使擴 散速率減慢,延長平衡時間,因此高沸點的分析物通常因擴散速率 較慢,因此選擇厚度較薄之纖維縮短平衡時間,兩種以上不同性質 裹附纖維對不同待測物有較佳的同時吸附能力,DVB 材質適合極 性物質,PDMS 材質適合非極性物質 54,本實驗因分析物分子量差 異大,且極性不同而選擇了 PDMS/DVB 纖維,因分析物沸點較高, 選擇較薄的 60 μmPDMS/DVB 纖維。 吸附時間是指從開始萃取到達到萃取平衡所需的時間。在萃取 初始階段,分析物很容易被吸附到纖維固定相中,隨著時間的增加, 吸附速度越來越慢,接近平衡狀態時,即使延長吸附時間,吸附量. 56.
(74) 不會有太大的增加,本實驗在吸附初始階段 10 分鐘已吸附大量分 析物,隨著吸附時間到了 20 分鐘,吸附量呈現趨緩。 萃取溫度的差異使分析物吸附於纖維的量有所不同,以動力 學的角度而言,溫度上升加速質量傳輸、提供分析物脫離樣品所需 的能量;但就熱力學而言,溫度上升則亂度增加且分配細數下降, 而本實驗在萃取溫度探討上,隨溫度上升而纖維上所吸附之分析物 含量有明顯的增加,但大量的提高溫度( > 40°C),吸附量不增反 減。41. 、44、45. 樣本攪拌之目的主要在於加速分析物的擴散速率,縮短萃取平 衡的時間,不同的攪拌方式各有其優缺點:樣品旋轉(纖維需承受 壓力)、纖維移動(僅限小體積樣品)、超音波(會加熱樣品),由於 磁力轉動攪拌設備簡單,對纖維較不會有損害,因此磁力轉動攪拌 是常用的攪拌方法在攪拌測試上,本實驗在相同吸附時間下,有攪 拌者(200 rpm)之面積比值明顯大於未攪拌者,因攪拌可縮短達平衡 的時間,但已達平衡之後的攪拌,增加其攪拌速度,對吸附量無增 加的效果。 以直接萃取模式採樣時,水樣的 pH 值改變,對分析物的分配 常數會造成改變,而影響萃取效果,通常分析物為酸鹼化合物時, 調整 pH 可影響分析物是以離子態或分子態存在於水相中而影響到. 57.
(75) 吸附,對於在水中不解離的芳香族碳氫化合物、鹵化醚類化合物, pH 值的改變對萃取並無影響,而本實驗 pH 值的改變對吸附效率並 沒有正面的提升 45。 選定較好的 SPME 參數即可得一較高且穩定的分配係數,也就 是較佳的吸附效率,以探討好的 SPME 參數進行分配係數探討,不 管是高低濃度,其分配係數皆穩定。 早期關於複合式樹脂釋出的研究,將樹脂培養於有機溶劑或水 中,釋出濃度較高,重點放於釋出物的定量,近期研究發現唾液中 的酶會與釋出物反應,樹脂於唾液環境中原釋出單體的量將大大減 少,開發之 SPME 方法可分析唾液中釋出單體,分析方法其方法偵 測極限分別為 TEGDMA:0.035µg/mL;UDMA:0.276 µg/mL; Bis-GMA:0.064 µg/mL,可應用於研究原釋出物於唾液中反應機制 探討。 目前將 SPME 方法應用於牙科材料分析上的研究尚少,2004 年 Ortengren 等針對 HQ(Hydroquinone monomethyl ether)、 bisphenol-A(4,4-Isopropylidenediphenol)、TEGDMA、EGDMA (Ethylene glycoldimethacrylate)、MMA(Methylacrylate)、MA (Methacrylic)六種低分子物質開發 SPME-GC-MS 方法,而其 SPME 的參數無一完整探討,以開發 SPME 方法而言是不足的,相對本研. 58.
數據


+7
Outline
相關文件
Hedonic Price method is used features variable of housing to assay the housing price , in this study, we designated a range for 6 km radius effect sphere of High Speed Rail
By using Balanced Scorecard (BSC), the purpose of this study is to construct indicators of school management with Analytic Hierarchy Process (AHP) for L junior high school in
本研究採用的方法是將階層式與非階層式集群法結合。第一步先運用
In order to serve the fore-mentioned purpose, this research is based on a related questionnaire that extracts 525 high school students as the object for the study, and carries out
The purpose of this research is to develop an approach that uses the triangular distribution with the Fractile Method to estimate the optimistic and pessimistic duration of
This purpose of study was to realize, as well as the factors of influence of information technology integrated in teaching by junior high school special education teachers in
The isothermal and anisothermal mechanical behavior were analyzed by using finite element method (FEM) in this study to simulate the stress/strain behavior of the solder balls
The main purpose of this study is to explore the status quo of the food quality and service quality for the quantity foodservice of the high-tech industry in Taiwan;
