國立交通大學
材料科學與工程學系
碩士論文
多孔性二氧化矽薄膜表面電漿改質在奈米積體電路
超低介電材料之應用研究
Study on Plasma Modification of ultra-Low Dielectric Constant
Nanoporous Silica Thin Films for
Nano-Scaled-Integrated-Circuit Technology
研究生:李宜芳
指導教授:潘扶民 博士
多孔性二氧化矽薄膜表面電漿改質在奈米積體電路超低介電材
料之應用研究
Study on Plasma Modification of ultra-Low Dielectric Constant
Nanoporous Silica Thin Films for
Nano-Scaled-Integrated-Circuit Technology
研 究 生:李宜芳 Student: Yi-Fang Li
指導教授:潘扶民 Adviser: Fu-Ming Pan
國立交通大學
材料科學與工程學系
碩士論文
A Thesis
Submitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Materials Science and Engineering July 2007
Hsinchu, Taiwan, Republic of China
多孔性二氧化矽薄膜表面電漿改質在奈米積體電路超低介電材料之應用研究 研究生:李宜芳 指導教授:潘扶民 國立交通大學材料科學與工程學系 摘要 當積體電路的密度增加,元件尺寸縮小,導線的電阻值與金屬間介電層的電容值 乘積所形成的功率損,將延遲訊號的傳遞時間。當製程進入深次微米領域時,元件閘 極層次的速度增益,將因增加電阻電容時間常術所引起之內連線傳導延遲而抵銷,進 而限制晶片效能的提升。為了解決內連線延遲問題,早期以線路設計的方式增加層數 及關鍵部分的線徑與空間來改善,然而如此將增大積體電路的尺寸,造成產率與成本 的負擔,為了改善這些問題,使用低介電常數材料做為導線間的介電層為必要選擇。 本研究中所使用的低介電常數材料為奈米孔洞二氧化矽薄膜。然而,由於高孔隙率 的關係,衍生出許多製程上的問題,增加實際導入生產製程上的困難,如吸水性、蝕刻 氣體滲入孔洞、以及銅鑲嵌結構製程中,氣體前驅物分子可能經由孔洞滲透擴散至介電 層中,由於前驅物中含有金屬組成,金屬原子一旦進入介電層,將會嚴重劣化材料的介 電性質。 因此,我們嘗試將開孔性薄膜表面的孔洞再次封合,來減少後續製程上所衍發的問 題。本研究將利用不同的電漿對奈米孔洞二氧化矽薄膜做前置處理,以期其離子轟擊效 應能使薄膜表面的孔洞結構崩壞,形成緻密結構,而內部的高孔隙率性質依舊存在。研 究的重點包括:經電漿處理後的奈米孔洞二氧化矽薄膜抵抗金屬原子擴散的能力同時探 討因電漿處理而造成薄膜基本性質的改變。 在本研究中發現經過電漿處理之奈米孔洞二氧化矽薄膜可於表面產生一層緻密結 構的二氧化矽薄膜,而在薄膜內部依舊維持著高孔隙率,且可抵抗後續金屬化製程中 的金屬擴散行為,而介電常數值僅些微上升,顯示電漿處理為一可用的孔洞封合技術。
Study on Plasma Modification of ultra-Low Dielectric Constant Nanoporous Silica Thin Films for Nano-Scaled-Integrated-Circuit Technology
Student: Yi-Fang Li Adviser: Fu-Ming Pan
Department of Materials Science and Engineering National Chiao Tung University
Abstract
While the semiconductor industry continues to scale down device sizes for better performance, lower power consumption and higher packing density, the interconnect delay and cross-talk between adjacent metal lines must be reduced. The performance of an IC chip can be degraded by interconnect RC delay and power consumption. In order to alleviate the problems, low dielectric constant (k) materials are used to replace the conventional intermetal dielectric (IMD), SiO2.
In this research, low-k nanoporous silica dielectrics was selected as the ultra-low k IMD material for nanoscaled integrated circuit technology. However, integration of the porous ultralow-k dielectric into Cu interconnect processes is subjected to impurity diffusion through pore channels and moisture uptake on the pore surface due to high porosity. Because of the very low mass density and enormously large and active surface area, low-k nanoporous silica dielectrics are extremely susceptible to plasma damage during etch and CVD processes. Without appropriate pore-sealing treatment, these materials are not suitable for application for Cu interconnect technology.
In the study, plasma treatments were implemented to seal open pores of porous ILDs by forming a thin dense layer on the dielectric surface. Ion bombardment on the dielectric surface during the plasma treatment will result in the collapse of the pore structure near the surface region, thereby forming a dense surface layer while making the porous bulk intact. In the surface layer, chemical and microstructure properties were dramatic different from that of the bulk. The changes in the film chemistry, mechanical properties, and resistance to metal diffusion were studied by various spectroscopies and microscopies, such as x-ray diffractometry, electron nicroscopies, and x-ray photoelectron spectroscopy.
致謝 此論文從零到有並逐漸趨近於完整,首先要感謝指導教授潘扶民博士兩年來對我 的諄諄善誘,使得我對半導體材料及奈米科技有了更進一步的認知,也因為你的指導 讓我學會了對於每個物件現象背後的學理基礎做探討,謹此致以最誠摯的謝意。 也感謝實驗室的夥伴,首先是博學多聞、文學造詣極高的大憲學長,感謝學長在 我撰寫論文期間,充當我的同義詞辭典,再我打了一百個”因此”之後,還有”引發”可 以用;在我頭昏腦脹不想google 的時候,當一個會告訴我最佳解的 google,真的很謝 謝你,祝你在美國求學一切順利!同時也要謝謝學識淵博的陳致宇博士,在辛苦的軍 旅生活的期間,仍時時關心我的進度,為我提供建言,解答我近乎無知的疑惑,心中 有萬分的感動,無法以言語表達,希望你軍旅生活事事順心,就快破百了請多加油! 還有靜雯在實驗上給予我的幫忙,為我分擔了許多工作;全雯源源不絕的愛心糖 果,讓我在實驗不順之時,亦能保持好心情;還有前後任的歐傑助教,建融、重守與 志豪學長們,遇到這麼凹的學妹真是辛苦了,謝謝你們的幫忙!還有智傑學長辛苦的 TEM 試片,如果沒有這些,我將無法完成這本論文。 也感謝交大材料94 的好朋友們,我將會珍惜在這最後階段一起奮鬥的過程。亦感 謝我的好朋友珮慈、大餅、文瑛、憶萍與慧萍,在我奮鬥的過程中,不斷地給我加油 打氣與督促,能認識你們,真的是我人生中最開心的事。要感謝的人真的很多,但為 了讓致謝保持在一頁之內,以免顯得囉嗦,只好「謝天」了。 但最後決不能忘記的,是我最親愛的家人,有了你們支持,讓我遇到任何挫折、 困難都無所畏懼,因為我知道你們永遠都在那裏,為我提供一個最佳的避風港,真的 很謝謝你們,我愛你們。
目錄 摘要...i Abstract...ii 致謝...iii 目錄...iv 表目錄...vii 圖目錄...viii 第一章 緒論...1 1.1 超大型積體電路技術現況及未來發展趨勢 ...1 1.1.1 多層內連線結構 ...1 1.1.2 內連線延遲效應 ...2 1.1.3 銅製程鑲嵌結構 ...4 1.2 研究動機與目的 ...6 第二章 文獻回顧...7 2.1 低介電常數材料 ...7 2.1.1 低介電常數材料特性 ...7 2.1.2 各種低介電常數材料特性比較 ...8 2.2 奈米孔洞二氧化矽薄膜 ...10 2.2.1 溶膠-凝膠反應...10 2.2.2 模板分子自組裝原理 ...12 2.3 孔洞封合技術 ...14 2.3.1 薄膜沉積 ...14 2.3.2 表面交聯反應與重構 ...15 2.3.3 官能基置換 ...16
2.3.4 電漿處理 ...17 2.4 電漿原理簡介 ...19 2.4.1 電漿原理 ...19 2.4.2 離子轟擊效應 ...20 2.4.3 電漿表面改質處理 ...21 2.5 原子層化學氣相沉積 ...23 第三章 實驗方法...27 3.1 試片製備 ...27 3.1.1 矽晶圓清洗 ...27 3.1.2 奈米孔洞二氧化矽薄膜前驅物的配製 ...27 3.1.3 奈米孔洞二氧化矽薄膜的沉積與乾燥 ...28 3.1.4 奈米孔洞二氧化矽薄膜的烘烤與鍛燒製程 ...29 3.1.5 電漿處理 ...30 3.1.6 疏水化改質處理 ...31 3.2 試片分析 ...32 3.2.1 表面形貌觀察 ...32 3.2.2 結構與化性分析 ...33 3.2.3 機械性質 ...35 3.2.4 介電特性 ...36 3.2.5 孔洞封合測試 ...37 第四章 結果與討論...40 4.1 奈米孔洞二氧化矽薄膜 ...40 4.1.1 表面形貌與組成化學鍵結 ...40 4.1.2 薄膜結構與機械性質 ...43 4.1.3 吸水效應與電特性 ...47
4.2 電漿處理對奈米孔洞二氧化矽薄膜特性之影響 ...50 4.2.1 電漿處理對奈米孔洞二氧化矽薄膜表面形貌影響 ...50 4.2.2 電漿處理對奈米孔洞二氧化矽薄膜對組成化學結構的影響 ...52 4.2.3 電漿處理對奈米孔洞二氧化矽薄膜結構與機械性質的影響 ...59 4.2.4 電漿處理對奈米孔洞二氧化矽薄膜介電特性的影響 ...62 4.2.5 奈米孔洞二氧化矽薄膜與氮化鉭的附著力 ...64 4.2.6 結論 ...65 4.3 孔洞封合測試 ...66 4.3.1 折射率與膜厚的改變 ...66 4.3.2 X-ray 反射率分析...68 4.3.3 熱脫附游離質譜儀吸水測試 ...71 4.3.4 歐傑電子能譜儀縱深分析 ...72 4.3.5 穿透式電子顯微鏡圖像 ...75 第五章 結論...79 參考文獻...81 附錄...84 A. X 光反射率量測法 ...84
表目錄 表1- 1 ITRS 為未來超大型積體電路的系統需求研擬的趨勢 ...4 表2- 1 各種低介電材料之性質比較...9 表2- 2 壓力及外加功率的變化對電漿的離子密度及離子能量的影響...21 表3- 1 奈米孔洞二氧化矽薄膜前驅物溶液的成分以及莫耳比...28 表3- 2 電漿處理參數...31 表4- 1 電漿處理前後奈米孔洞二氧化矽薄膜之粗糙度(Rms)...52 表4- 2 表面元素成分的原子濃度...55 表4- 3 電漿處理後的平均接觸角...57 表4- 4 電漿處理後的奈米孔洞二氧化矽薄膜的彈性係數及硬度...62 表4- 5 電漿處理後的奈米孔洞二氧化矽薄膜介電係數...63 表4- 6 電漿處理後的奈米孔洞二氧化矽薄膜漏電流值(在 2 MV/cm 電場強度下)...64 表4- 7 電漿處理後的奈米孔洞二氧化矽薄膜與氮化鉭之附著力...65 表4- 8 電漿處理的奈米孔洞二氧化矽薄膜的 Δn 與Δ(n×d)之值 ...68 表4- 9 X-ray 反射圖形經過程式的資料擬合之密度與厚度值 ...69
圖目錄 圖1- 1 超大型積體電路採用多層內連線的結構示意圖...2 圖1- 2 訊號延遲時間與技術節點的關係圖...3 圖1- 3 雙鑲嵌結構(引洞優先)的製作流程示意圖 ...5 圖2- 1 介電常數值與孔隙率的關係...9 圖2- 2 溶膠-凝膠形成的過程 ... 11 圖2- 3 有機分子模板作用示意圖...13 圖2- 4 BCN 薄膜沉積後孔洞封合的局部放大圖...15 圖2- 5 表面交聯作用示意圖...16 圖2- 6 大分子吸附在多孔性材料的孔洞開口邊緣示意圖...17 圖2- 7 表面孔洞崩壞使孔洞封合示意圖...18 圖2- 8 利用 ALCVD 成長高介電質三氧化二鋁的示意圖 ...24 圖2- 9 利用 PE-ALCVD 成長擴散阻障層氮化鉭的示意圖 ...26 圖3- 1 奈米孔洞二氧化矽薄膜製備流程圖...30 圖3- 2 AFM 量測原理 ...32 圖3- 3 光電子發生原理示意圖...35 圖3- 4 負 荷 ─ 壓 痕 位 移 關 係 圖 ...36 圖3- 5 歐傑電子產生機制示意圖...38
圖4- 1 煅燒後的奈米孔洞二氧化矽薄膜(a) SEM 與(b) AFM 平面圖 ...41
圖4- 2 烘烤後與煅燒後奈米孔洞二氧化矽薄膜的 FT-IR 光譜圖 ...42 圖 4- 3 奈米孔洞二氧化矽薄膜沉積、烘烤與煅燒後,(a)薄膜折射率與孔隙率 (b)厚 度變化趨勢...44 圖4- 4 奈米孔洞二氧化矽薄膜煅燒後之低掠角 X 光繞射圖譜...45 圖 4- 5 奈米壓痕儀(nanoindenter)量測煅燒後奈米孔洞二氧化矽薄膜的(a)彈性係數 (b)硬度 之結果 ...46
圖4- 6 奈米孔洞二氧化矽薄膜經 HMDS 處理前後的熱脫附質譜圖 ...48 圖4- 7 奈米孔洞二氧化矽薄膜經 HMDS 處理前後的 FT-IR 光譜圖...48 圖4- 8 奈米孔洞二氧化矽薄膜的(a)C-V 圖 (b)I-V 圖 ...49 圖4- 9 (a)未經電漿處理 (b) Ar (c) N2 (d) O2 (e) N2O (f) CH4 (g) CF4 電漿處理後 之奈米孔洞二氧化矽薄膜之SEM 圖...51 圖4- 10 (a)未經電漿處理 (b) Ar (c) N2 (d) O2 (e) N2O (f) CH4 (g) CF4電漿處理後 之奈米孔洞二氧化矽薄膜ESCA 能譜圖...54 圖4- 11 電漿處理後的奈米孔洞二氧化矽薄膜 FT-IR 光譜圖 ...56 圖4- 12 (a)未經電漿處理 (b) Ar (c) N2 (d) O2 (e) N2O (f) CH4 (g) CF4 電漿處理後 之奈米孔洞二氧化矽薄膜與水之接觸角...58 圖4- 13 (a)未經電漿處理 (b) Ar (c) N2 (d) O2 (e) N2O (f) CH4 (g) CF4 電漿處理後 之奈米孔洞二氧化矽薄膜X 光繞射圖 ...61 圖4- 14 電漿處理後的奈米孔洞二氧化矽薄膜的折射率與孔隙率...67 圖4- 15 (a) Ar (b) N2 (c) O2 (d) N2O 電漿處理的奈米孔洞二氧化矽薄膜其 X-ray 反射圖 形資料擬合報告...70 圖4- 16 電漿處理後的熱脫附質譜圖...72 圖4- 17 奈米孔洞二氧化矽薄膜沉積氮化鉭與銅的 SEM 斷面圖...73 圖4- 18 Cu/TaN/奈米孔洞二氧化矽薄膜經退火後之歐傑縱深成分分佈圖 ...74 圖4- 19 Cu/TaN/氧電漿處理後之奈米孔洞二氧化矽薄膜經退火後之歐傑縱深成分分 佈圖...74 圖4- 20 Cu/TaN/氮電漿處理後之奈米孔洞二氧化矽薄膜經退火後之歐傑縱深成分分 佈圖...75 圖4- 21 電漿輔助原子層沉積法沉積氮化鉭製程步驟...76 圖4- 22 奈米孔洞二氧化矽薄膜沉積 ALD-TaNX 之穿透式電子顯微鏡影像...77 圖4- 23 氧電漿處理之奈米孔洞二氧化矽薄膜沉積 ALD-TaNX之穿透式電子顯微鏡影 ...77
圖4- 24 氬電漿處理之奈米孔洞二氧化矽薄膜沉積 ALD-TaNX 之穿透式電子顯微鏡 影像(其 Ar 電漿參數為:偏壓 300W、時間 20sec ) ...78 圖A- 1 理想表面與粗糙表的 X 光反射率理論圖 ...87
第一章 緒論
1.1 超大型積體電路技術現況及未來發展趨勢
1.1.1 多層內連線結構 隨著半導體製程技術的進步,元件尺寸不斷地縮小,目前已進入深次微米的領域。 當積體電路的積集度增加,使得晶片表面無法提供足夠的面積來製作所需的內連線 (Interconnects)時,為了配合元件縮小後增加的內連線,多層金屬導線的設計,如圖 1-1 所示,便成為現今超大型積體電路(Ultra-Large Scale Integration, ULSI)所必須採 用的方式。在高電晶體容量與運算速度的需求快速增加下,隨著元件尺寸縮小,內連線的尺寸也必須相對縮小,現在最先進的製程技術將探討65 nm 以下的尺寸範疇。然 而,當導線的線寬縮小後,晶片的運作速度不再受限於元件的操作速度,而是取決於
電子訊號在其導線間之傳遞速度【1】。舉例而言,當閘極長度為 250 nm 或更小時, 高達50%的時間延遲是肇因於較長的內連線【2】。所以 ULSI 中,內連線的連結網路 將成為影響如元件速度、信號串音(cross talk)、及 ULSI 電路中的功率耗損等晶方性 能的限制因素。因此業界的傾向以低電阻的導線與超低介電常數的金屬間介電層(inter layer dielectrics, ILD)結合,以達到降低訊號傳遞延遲與損耗的目的【3】。
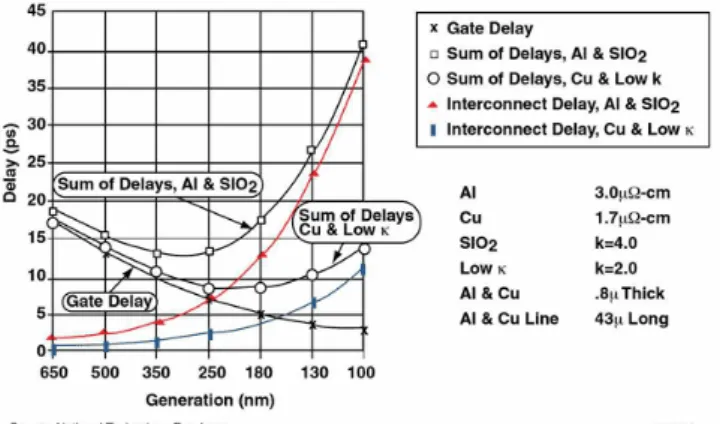
圖1- 1 超大型積體電路採用多層內連線的結構示意圖。 1.1.2 內連線延遲效應 當積體電路的密度增加,元件尺寸縮小,導線的電阻值與金屬間介電層的電容值 乘積所形成的功率損,將延遲訊號的傳遞時間。如圖 1-2 為閘極延遲(intrinsic gate delay)與電阻電容延遲(RC delay)的關係,顯示當製程進入深次微米領域時,元件 閘極層次的速度增益,將因增加電阻電容時間常數所引起之內連線傳導延遲而抵銷, 進而限制晶片效能的提升。 電阻電容之時間延遲效應的關係,基本上可用簡易的一階方程式來做初步的估算: m m Wt L R=ρ
(
)
⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ + = + = m m m m V L t W L W t L C C C 2 2εε0 ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ + = = 2 0 22 22 m m m t L W L RC ρεε τ C fV fV P 2 0 2 2 tan 2 π δ εε π ∝ ∝ 其中,R 為導線電阻,C 為金屬間介電層的電容,ρ 為電阻率,L 為內連線長度,m tm Cu-M8 Cu-M6 Cu-M4 Cu-M2 Vi a 7 Vi a 5 Vi a3 Vi a1 Cu-M1 W Low k (k≦2.8) Low k (k≦2.8) STI Cu-M8 Cu-M6 Cu-M4 Cu-M2 Vi a 7 Vi a 5 Vi a3 Vi a1 Cu-M1 W Low k (k≦2.8) Low k (k≦2.8) STI為金屬層厚度,W 為線寬,ε及ε0分別為介電層與真空的介電常數,P則為功率損。 由以上方程式可知,影響電阻電容時間延遲因素不外有二:其一為內金屬導線的幾何 結構,如內層連接線長度、線寬及金屬層厚度;另一則為材料本身的特性,即電阻或 電容率。為了解決內連線延遲問題,早期以線路設計的方式增加層數及關鍵部分的線 徑與空間來改善,然而如此將增大積體電路的尺寸,造成產率與成本的負擔,故有效 的改善方法仍需從材料本身的電阻與電容部分著手。在電阻方面,由於銅的電阻係數 只有1.67 μΩ-㎝,遠小於鋁的 2.66 μΩ-㎝ 與鎢的 5.65 μΩ-㎝,而且其抗電遷移能力佳, 故以銅金屬做為主要的導線材料,已是產業界共同發展的趨勢【4】。在低電容材料部 分,諸多人力已試著研發比傳統製程技術所使用之二氧化矽(介電常數值約為3.9)介 電常數值低的材料。如表 1-1 所示,根據 2005 年國際半導體技術藍圖(International Technology Roadmap for Semiconductors, ITRS) 為未來超大型積體電路的內連線系統 需求所研擬的趨勢,預計西元2010 年後邏輯電路進入 45nm 技術時,金屬間介電層的 等效介電常數值必須在2.5 左右,因此低介電常數材料的體介電常數必須小於 2.2【5】。 近年來低介電常數材料以電漿輔助化學氣相沉積的含氟二氧化矽(Siliconoxyfluoride, FSG),旋轉塗佈的含氫矽酸鹽(Hydrogen silsesquioxane, HSQ)、含甲基矽酸鹽(Methyl silsesquioxane, MSQ),以及多孔性二氧化矽(porous silica)等材料最具應用價值【6 ~10】。
表1- 1 ITRS 為未來超大型積體電路的系統需求研擬的趨勢。
Year of Production 2005 2006 2007 2008 2009 2010 2011
DRAM 12 Pitch(nm) 80 70 65 57 50 45 40 MPU/ASIC1 2Pitch (nm) 90 78 68 59 52 45 40 No. of metal levels 11 11 11 12 12 12 12 Barrier/Cladding thickness (nm) 6.5 5.6 4.8 4.3 3.7 3.3 2.9 Intermetal insulator-keff 3.1-3.4 3.1-3.4 2.7-3.0 2.7-3.0 2.5-2.8 2.5-2.8 2.5-2.8 Intermetal insulator-kbulk ≦2.7 ≦2.7 ≦2.4 ≦2.4 ≦2.2 ≦2.2 ≦2.2
1.1.3 銅製程鑲嵌結構
傳統的積體電路之多層金屬連線(multilevel interconnection)是以金屬層的乾蝕刻 方式來製作金屬導線,然後進行介電層的填充(dielectric gap fill)。而鑲嵌技術則是先在 介電層上蝕刻金屬導線用的圖膜,然後再填充金屬。鑲嵌技術最主要的特點是不需要 進行金屬層的蝕刻。當金屬導線的材料由鋁轉換成電阻率更低的銅的時候,由於銅的 乾蝕刻較為困難,因此鑲嵌技術對銅製程來說極為重要。 鑲嵌結構一般常見兩種:單鑲嵌結構(single damascene)以及雙鑲嵌結構(dual damascene)。單鑲嵌結構如前所述,僅是把單層金屬導線的製作方式由傳統的(金屬層 蝕刻+介電層填充方式)改為鑲嵌方式(介電層蝕刻+金屬填充),較為單純。而雙鑲嵌 結構則是將孔洞(hole)及金屬導線結合一起都用鑲嵌的方式來做。如此只需一道金屬填 充的步驟,可簡化製程,不過製程也較為複雜與困難。一般完整的雙鑲嵌製程如圖1-3 所示,先沉積介電層並以乾蝕刻完成雙鑲嵌結構之圖形後,接著需沉積一層擴散阻障 層(diffusion barrier)。然後進行金屬沉積,最後,再進行化學機械研磨即告完成。 在銅製程中,由於銅原子的擴散係數較高,極易擴散進入二氧化矽的結構中導致 深層能階等問題,因此銅導線填入前,必須在引洞與溝渠表面沉積一層可阻絕銅原子
擴散的疊層,傳統採用氮化矽等材料做為擴散阻障層,近年來則以低介電常數的碳化 矽【11】或低電阻的氮化鉭【12】最具應用潛力。 圖1- 3 雙鑲嵌結構(引洞優先)的製作流程示意圖。 via PR IMD IMD etch stopper
metal line metal line metal line
metal line metal line metal line
metal PR Cu Cu barrier (a)沉積金屬間介電層與蝕 刻終止層+引洞微影曝光 (b)蝕刻引洞+溝渠微影 曝光 (c)蝕刻溝渠+去除光阻 (d)沉積擴散阻障層 (e)填入銅導線 (d)平坦化 via PR IMD IMD etch stopper
metal line metal line metal line
metal line metal line metal line
metal PR Cu Cu barrier (a)沉積金屬間介電層與蝕 刻終止層+引洞微影曝光 (b)蝕刻引洞+溝渠微影 曝光 (c)蝕刻溝渠+去除光阻 (d)沉積擴散阻障層 (e)填入銅導線 (d)平坦化
1.2 研究動機與目的
本研究中所使用的低介電常數材料為奈米孔洞二氧化矽薄膜。然而,由於高孔隙率 的關係,衍生出許多製程上的問題,增加實際導入生產製程上的困難,如吸水性、蝕刻 氣體滲入孔洞以及銅鑲嵌結構製程中,氣體前驅物分子可能經由孔洞滲透擴散至介電層 中,由於前驅物中含有金屬組成,金屬原子一旦進入介電層,將會嚴重劣化材料的介電 性質。 因此,我們嘗試將開孔性薄膜表面的孔洞再次封合,來減少後續製程上所衍發的問 題。本研究將利用不同的電漿對奈米孔洞二氧化矽薄膜做前置處理,以其離子轟擊效應 能使薄膜表面的孔洞結構崩壞,形成緻密結構,而內部的高孔隙率性質依舊存在。研究 的重點包括:經電漿處理後的薄膜抵抗金屬原子擴散的能力、薄膜基本性質的改變,其 中包括:表面形貌、物化性、結構、機械性質與介電特性等。 上述的研究結果將有助於了解多孔性二氧化矽薄膜於現有的金屬化製程中的整合 性,有利於後續製程整合研究上之結合與進行。第二章 文獻回顧
2.1 低介電常數材料
隨著半導體製作技術的進步,元件尺寸已進入深次微米的領域,積體電路所需要的 金屬疊層層數越來越多。當線寬和間距縮小時,必須使用低介電材料來因應電阻電容延 遲效應所造成的問題。 要得到低介電常數,材料的結構必須有以下效應之一種: (1) 設法降低材料本身的極性(polarization),這包括了降低材料中的電子極化 (electronic polarization)、離子極化(ionic polarization)以及分子極化(dipolar polarization)。 介電常數與極化率的關係可經由 Clausius-Mossotti equation 來表示【13】, ) ( 3 2 1 0 o i e r r N α α α ε ε ε = + + + − , 其中,εr 及ε0 分別為介電層與真空的介電常數,N 為每立方公尺的分子數,αe, αi, αo 分別為分子的電子極化率,離子極化率及方向極化率。由此方程式可知,材料中的極性 分子越少,則此材料的介電常數值就越低。 (2) 結構開放,或是使材料多孔。材料結構鬆散可增加材料內的自由空間,而空 氣的介電常數定義為1,因此能藉由此種方法得到低介電材料的介電常數。 2.1.1 低介電常數材料特性 在將低介電常數材料應用於積體電路的整合製程時,對於低介電常數材質特性的要 求【14】,除了要具備有低的介電常數之外,還需具有以下的材料特性:(1) 高熱穩定性:玻璃轉移溫度(glass transition temperature, Tg)大於400℃。短期在425 ℃以上安定,低膨脹度。
(2) 電性高可靠度:漏電電流和崩潰電場均和二氧化矽相似,低殘餘電流。
巨大剪應力。
(4) 良好的化學性質:低水氣吸附率,高蝕刻選擇性,化學安定性,儲存期長。
2.1.2 各種低介電常數材料特性比較
目前產學界已研發出的多種低介電常數材料,與傳統介電層材料SiO2相較,都具
有較低的介電常數。其中以電漿輔助化學氣相沉積的含氟二氧化矽(Siliconoxyfluoride, FSG),旋轉塗佈的含氫矽酸鹽(Hydrogen silsesquioxane, HSQ)、含甲基矽酸鹽(Methyl silsesquioxane, MSQ),以及多孔性二氧化矽(porous silica)等材料最具應用價值,表 2-1 為現有的低介電常數材料的性質比較。 從表中我們發現高分子薄膜其介電常數較低,但熱穩定性差﹔無機薄膜其熱穩定性 較佳,但介電常數較高﹔而多孔性二氧化矽除了具有不錯的熱穩定外,其材料性質與傳 統介電材料(SiO2)相近,對於製程設備之相容性高。且文獻中多有闡述介電值可調式材 料的諸多特性,可將介電常數值降至 2.5 以下抑或更低,如圖 2-1。因此多孔性二氧化 矽為目前最具潛力的低介電材料,而本研究即選用此材料作為研究對象。
表2- 1 各種低介電材料之性質比較。
圖2- 1 介電常數值與孔隙率的關係。
介電材料 介電常數值 沉積方式 熱穩定性(oC)
二氧化矽(SiO2) 3.9~4.9 CVD 或 PECVD >500
氟化二氧化矽(SiOF) 2.8~3.75 PECVD >500 Porous Silica 1.1~2.4 Sol-gel 900 無機矽烷氧類高分子 HSQ MSQ 2.7~3.8 ~2.7 Spin on Spin on >400 >400 Benezocyclobutene(BCB) 2.7 Spin on >350 聚亞芳香醚高分子 PAE FLARETM1.0 FLARETM2.0 2.4~2.8 2.4 2.75 Spin on Spin on Spin on 450 280 450 聚亞醯胺(PI) 3.2~3.6 Spin on 450 氟化聚亞醯胺(FPI) 2.6~2.8 Spin on 450 Parylene 2.2~2.3 CVD 450 氟化非結晶性碳膜(a-C:F) 2.3~2.5 PECVD >400 Rel a tiv e die lectric constant Porosity (%) 0 0 1 2 3 4 20 40 60 80 100 Oxide k~4.0 Oxide k~4.0 Low k~2.5 Low k~2.5 Rel a tiv e die lectric constant Porosity (%) 0 0 1 2 3 4 20 40 60 80 100 Oxide k~4.0 Oxide k~4.0 Low k~2.5 Low k~2.5
2.2 奈米孔洞二氧化矽薄膜
多孔性二氧化矽薄膜,可利用矽源、水及醇類混合,並以酸或鹼的催化合成二氧 化矽溶膠凝膠,經時效後旋塗於基材表面,再以熱處理除去溶劑後獲得。以傳統氣凝 膠法(Aerogel)與乾凝膠法(Xerogel)所製備的二氧化矽薄膜雖可得到極高的孔隙率 (>85%)與超低的介電常數(k=1.1-2.5),但在熱處理的過程中有收縮與殘留應力的 問題,因此其孔洞大小不均勻、機械性質差、不符合實際製程需求【15~18】。 而本研究中所使用的奈米孔洞二氧化矽薄膜是在適當溫度與 pH 條件下,經由溶 膠–凝膠水解縮合反應(sol-gel reaction),將四氧烷基矽 TEOS (tetraethyl orthosilicate, Si(OC2H5)4)、H2O、HCl 及乙醇合成二氧化矽溶膠前驅物,再添加兩相高分子(一種界 面活性劑)於酸催化二氧化矽溶膠凝膠內作為孔洞的模板(template)分子。模板分子在乾 燥時會因溶劑的揮發而連帶產生自組裝(self-assembly)排列【19】,並於煅燒的過程中 分解揮發,最後在 SiO2 薄膜內留下具規則排列的孔洞結構【20,21】,以下將分別就 溶膠–凝膠反應與模板分子的自組裝原理加以討論。 2.2.1 溶膠-凝膠反應 溶膠-凝膠法【22】首次被提出可以應用於無機光學材料,如氧化矽玻璃,是由法 國化學家M. Ebelmen 在1845 年的研究論文提到。在實驗中M. Ebelmen 發現矽酸酯可 以被水氣緩慢水解而形成含氫氧基(-OH)的化合物,此氫氧化合物可以進一步互相反應 而形成透明氧化矽聚合體,當時就被認為可以用來製作氧化物材料。而所謂溶膠凝膠法係利用膠質懸浮物(colloidal suspension, submicron particles) 來製 備無機高分子化合物材料。簡單地說,在製備的過程中前趨物反應先產生有微小固體散
佈於其中的膠狀溶液,稱之為溶膠(sol)。因固體顆粒通常介於1-100 nm,所以可以忽略 其重力效應,並無沉澱的發生。這些微小粒子繼續反應互相連結在一起,凝固化後就成
時也可加入其他的物質(dopant),在凝膠形成的過程中被包入所形成網狀氧化物的孔洞 中,如圖2-2所示【23】。 (a) (b) (c) (d) 圖2- 2 溶膠-凝膠形成的過程。 圖 2-2(a)為含無機氧化物顆粒的溶膠,溶膠顆粒以「○」表示,「■」或藍色正 方形為掺雜物 (dopant)。而圖 2-2(b)、2-2(c)中,溶膠進行聚合產生凝膠,乾燥後形成 多孔性的材料,掺雜物被包藏在孔洞中。圖 2-2(d)中「▲」或紅色三角形為其他的分 子,可擴散到孔洞中與掺雜物作用 (如紅色圈圈所示)。 以溶膠-凝膠的方法製備高分子化合物時是先將前趨物、掺雜物溶於溶劑中,加入 適量的水及催化劑後啟動水解及縮合反應以形成最後的產物,所形成高分子化合物的結 構及形態受水解及縮合反應的影響。以本研究中使用的奈米孔洞二氧化矽薄膜為例,前 趨物為Si-OR,在酸或鹼的催化下形成含SiOxHy 的網狀結構,整個製程所牽涉的反應如 下【24】: ROH OH Si O H OR Si− + ⎯hydrolysis⎯⎯⎯→≡ − + ≡ 2 .(2-1) ROH Si O Si Si HO OR Si− + − ≡⎯alcohol⎯⎯⎯condensati⎯⎯⎯on→≡ − − ≡+ ≡ _ . (2-2) O H Si O S Si HO OH Si water condensation 2 _ ⎯⎯⎯ ≡ − − ≡+ ⎯ ⎯ → ⎯ ≡ − + − ≡ . (2-3) 其中R為烷系官能基。如式(2-1)所示,水解反應利用水的氫氧基取代TEOS的烷氧 基,生成矽醇基(silanol group)。而所生成之矽醇基(-Si-OH),其氫氧基會繼續與矽烷氧
化之氧基或氫氧基進行水縮合和醇縮合反應,生成矽氧烷鍵(Si-O-Si),並釋出醇類或水, 如方程式(2-2)和(2-3)所示。通常縮合反應在水解反應尚未完成時即開始進行。此外,由 於TEOS與水不互溶,因此添加乙醇作為均質劑(homogenizing agent)來調和分離的兩 相,同時在水解以及醇縮合反應中將不斷釋放醇類,可確保均質化的效果。所形成膠的 性質如孔洞的大小及分佈狀況、表面積的大小可以藉由控制反應的各項參數來決定,如 反應時所加入的水與前趨物的比例、催化劑的濃度及性質、所使用前趨物的種類等。一 般而言當酸鹼度低、水較少時所得的凝膠質地較緻密、孔洞較小,相反地,當酸鹼度及 水較高時所產生的材質孔洞多,質地較疏鬆。 整體來說,溶膠凝膠法具有以下的優點【25】:(1) 均勻性好、(2) 純度高、(3) 組 成成份易控制、(4) 可降低製程中的溫度、(5) 具有流變特性,可用於不同用途產品的 製備。 2.2.2 模板分子自組裝原理 所謂自組裝顧名思義就是無任何外力介入自發地組裝,通常藉由分子間的鍵結相互 作用,在一定的條件下自發性的形成某種特定的有序結構。 高分子塊體共聚合物,其由於塊體間化學成分的不相同,造成彼此的不互容效應, 自組裝而形成所謂的微觀相分離(microphase separation),最終將形成穩定有序微結構, 同時其微結構尺寸約在數十奈米之間。根據體積分率的組成變化,具有多樣化之結構形
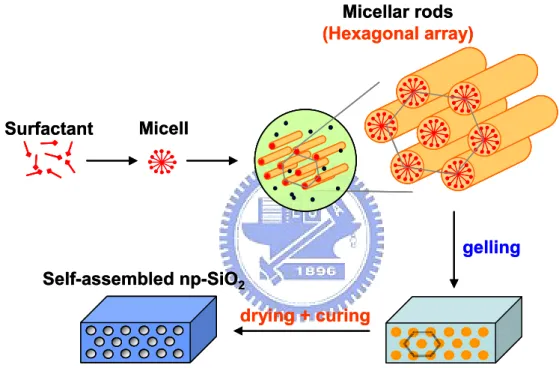
態如圓球体心立方堆積(body center cubic, BCC)結構、六角圓柱堆積(hexagonal cylinder, HC)結構等。由於其結構排列方式的多樣化,兼具一維、二維、三維之排列整齊性,且 藉由其聚合度(degree of polymerization)調控可控制其排列結構之尺寸大小。 本研究中,奈米孔洞二氧化矽薄膜沉積時形成孔洞結構的驅動力,主要來自於前 驅物中二氧化矽溶膠與有機模板分子間的微弱作用力(如凡得瓦耳力、偶極-偶極力以 及微弱的氫鍵),加諸有機模板分子本身的自組構能力。研究中我們使用三塊狀共聚高 分子EO20PO70EO20 (P-123)做為模板分子。利用 EO 區塊為親水性,PO 區塊為疏水性
的特性,在前驅物中匯聚成微胞(micell),進一步形成微胞桿(micellar rod)。當前驅物 旋塗於矽晶片表面時,藉由溶劑的揮發,誘導有機模板分子在數秒內完成自組裝,生
成具六角對稱(hexagonal symmetry)形式規則排列的孔洞陣列特定相,稱為揮發誘導自 組構(evaporation-induced self-assembly)【19】,如圖2-3 所示。但在旋轉塗佈的過程(圓 周運動),由於載台提供試片的向心力,因此將無可避免衍生機械應力導致薄膜微結構
的變化,使自組構的薄膜失去長程有序(long range ordering)的特性。
圖2- 3 有機分子模板作用示意圖。 gelling Self-assembled np-SiO2 drying + curing Surfactant Micellar rods (Hexagonal array) Micell gelling Self-assembled np-SiO2 drying + curing Surfactant Micellar rods (Hexagonal array) Micell
2.3 孔洞封合技術
為了因應後續世代半導體製程技術的需求,多孔性介電材料成為未來超低介電常 數材料的最終選擇已逐漸成為一般研發者的共識。多孔性介電材料是一種材質內部充 滿孔洞的物質,由於孔洞內部充滿了空氣,而空氣的介電係數值定義為1.0,因此其整 體介電常數值將可被有效地降低。但由於多孔特性衍生出許多不利於現今半導體製程 技術整合與材料間相容性的問題,至今多孔性低介電材料尚未能應用在實際的產品上。 首先,多孔性介電材料因具有很高的孔洞密度且因此引致極大的表面面積,所以 導致多孔性低介電常數材料有很強的吸水性,並且使氣體及溶液分子極易在材料內部 滲透擴散,這多孔的特性對低介電常數材料整合至半導體銅金屬鑲嵌連線製程中有非 常不良的影響。此外,為了在深窄的結構裡得到較佳的覆蓋率,化學氣相沉積(CVD) 是不可避免的鍍膜方法,在 CVD 的沉積過程中,反應氣體可能循著孔徑擴散進入薄 膜內部,進而改變材質特性。而在蝕刻過程中,蝕刻所使用的反應性氣體亦會經由孔 洞進入薄膜內部。而在微影、蝕刻與機械研磨製程後,晶圓必須加以清洗,如果採用 濕式清洗法,洗淨液不僅會吸附於孔壁表面,更有可能改變薄膜化性。 為了解決上述多孔性低介電常數材料與半導體製程的整合問題,目前文獻上較為 可行的方之法是將多孔性薄膜表面的孔洞再次封合,來減少後續製程上所衍發的問 題。以下將分別介紹現行文獻中較為常見的表層孔洞封合技術。 2.3.1 薄膜沉積 許多文獻利用 CVD 在多孔性低介電材料上,沉積一層厚度為數奈米至數十奈米 不等的薄膜做為孔洞封合的覆蓋層。在薄膜的選擇上,一般均以本身即為介電材料、 介電常數較低、熱穩定性良好且結構緻密為基本條件,常見的覆蓋層薄膜有非晶相氫 化碳化矽薄膜(α-SiC:H)與硼氮碳(BCN)。 非晶相氫化碳化矽薄膜的介電常數約在4-6之間,隨著其沉積條件的不同而有所改變【26】。其擁有良好的機械性質,對於一般的化學物質以及水氣具備絕佳的阻隔能力, 同時在銅雙鑲嵌結構中,非晶相氫化碳化矽亦可滿足製程所需的蝕刻選擇比,因此非常 適合應用於多層內連線的製作。在Jousseaume【27】等人的研究中,其在300 nm的MSQ 上沉積7.5 nm的非晶相氫化碳化矽薄膜,利用二次離子質譜儀比較有沉積與未沉積的Ta 的擴散情形並發現同時可以改善薄膜的疏水性,而有效k值僅有小幅度的上升。 而硼氮碳薄膜的介數常數值約在2-6之間,亦可利用改變沉積條件來做調整【28】。 相關文獻指出此薄膜具有良好的物化性質,例如:高硬度,高熱傳導,寬能隙,低介電 常數等優質特性,而在此薄膜中提高碳原子含量可有效減低薄膜之介電常數【29】,亦 可藉此調整薄膜之介電常數。在Ahearn【30】等人的研究中,發現沉積3.9 nm的BCN薄 膜於MSQ介電材料上,就可抵抗後續金屬化製程的擴散與汙染,圖2-4為BCN薄膜沉積 後其表面孔洞封合的情形。利用化學氣相沉積法於深寬比大的孔洞其階梯覆蓋性差之特 性,使薄膜沉積於洞口達到孔洞封合的效果。 圖2- 4 BCN 薄膜沉積之孔洞封合部分的局部放大圖。 2.3.2 表面交聯反應與重構 為了不影響到有效k 值,必須使孔洞封合的覆蓋層在最佳均勻度之最小厚度,在 沉積技術的限制之下, Iacopi【31】等人研究試著以材料表面的化學反應來減少表面
封合層的厚度。當低介電常數材料表面具有含碳之鍵結時,利用物理氣相沉積技術 (PVD)沉積 TaN,可藉由表面的交聯反應與重構,產生孔洞封合的效果。隨著碳含量 的增加,封合的效果越好,當沉積10 nm 的 TaN 於碳含量 24 % 的 MSQ 薄膜上,會 形成一完全封合的覆蓋層;而當一條件完全相同的 TaN 薄膜沉積於碳含量 18% 的 MSQ 薄膜時,薄膜的表面依舊存在著孔洞。此外,當 10 nm 的非晶氫化碳化矽薄膜先 行沉積於HSQ 薄膜上再沉積 10 nm 的 TaN 薄膜可有效地封合其表面的孔洞,若純粹 沉積TaN 或非晶氫化碳化矽薄膜則需 30 nm 的厚度才能徹底地將表面孔洞封合。 這是由於TaN中的Ta原子與表面的碳反應形成了Ta-C的鍵結,增強了TaN與介電材 料表面的附著力,但由於鄰近金屬原子誘發的電荷影響,使得原本的低介電材料或是非 晶氫化碳化矽中的C-H鍵結變弱,因此當PVD沉積TaN時,較高的沉積溫度亦或是離子 轟擊的能量釋放,會造成C-H鍵結的斷裂,而失去氫原子的碳,彼此間產生交聯作用, 形成C-C聚合鏈,如圖2-5所示。 圖2- 5 表面交聯作用示意圖。 2.3.3 官能基置換 在文獻中,也有嘗試利用大分子吸附在多孔性材料的孔洞開口邊緣,以達到孔洞 封合的效果,如圖 2-6 所示。圖中吸附的大分子間相互糾結產生一個類似過濾性薄膜
的功能,可以阻擋外來的雜質汙染物進入孔洞通道中。相對於孔洞尺寸,一般無機分 子的體積略嫌太小,因此文獻中多利用大的有機分子來進行此官能基置換的反應。 一般利用分子或是微結晶工程概念的有機-無機混成奈米結構製備方法,牽涉到利 用如氯化有機物或者烷氧基物等反應物,嫁接於位矽酸鹽有序平面中的矽醇基,如式2-2 【32】。文獻中指出一般的奈米孔洞二氧化矽薄膜,煅燒後會在薄膜表面及孔壁充斥大 量 未 交 聯 的 矽 醇 基 。 在Beck 【 33 】 等 人 的 研 究 中 , 利 用 氯 化 三 甲 基 矽 烷 (Trimethylchlorosilane, TMCS)中的三甲基取代位於MCM41的二氧化矽薄膜孔洞表面的 矽醇基,但因薄膜中孔洞尺寸大,分子將無可避免的擴散進入孔道中。因此,Nawal【34】 等人研究出在MCM41薄膜煅燒之前,先藉由香豆素的衍生物附著於孔道口上,再移除 模板分子以防止分子擴散進入孔道中,但是由於模板分子一般都是經由高溫煅燒來移 除,因此在模板分子移除的過程中,官能分子可能受高溫而分解,失去原有孔洞封合的 性質,在此研究中改用特定波長的紫外線煅燒的方式,可移除模板分子又可保留官能基 分子。 圖2- 6 大分子吸附在多孔性材料的孔洞開口邊緣示意圖。 2.3.4 電漿處理 利用電漿對多孔性材料做表面處理,不管是物理或化學的反應均會有孔洞封合的 效果。在物理反應方面,電漿作用帶來的離子轟擊效應,造成表面孔洞結構的崩壞並 填滿孔洞,如圖2-7【35】。許多研究指出,控制電漿的偏壓與作用時間等參數,可使 得離子轟擊的效應只影響薄膜表面的數奈米至數十奈米間。因此,可預期有效k 值並
結產生交聯作用,我們可以藉由此一特性,在某種條件下,於孔洞表面產生聚合,以
達成孔洞封合的效果。
在Hua-Gen Peng【35】等人的研究中,利用氨電漿對MSQ做表面處理,並使用穿透 式電子顯微鏡與正子消散時間光譜儀(Positron annihilation lifetime spectroscopy)證實了 離子轟擊確實在薄膜表面形成數奈米的緻密層;而二次離子質譜儀也證明氨電漿亦與 MSQ反應產生薄且緻密的氮化矽薄膜,有效的達到孔洞封合的效果。除此之外,氨電漿 處理亦增強了介電材料的機械性質,並改善了材料的吸水性質。 圖2- 7 表面孔洞崩壞使孔洞封合示意圖。 在半導體製程中多有電漿製程,包括:乾式蝕刻、薄膜沉積、光阻去阻等,使用電 漿處理與現今製程相容性高。且電漿處理可藉由參數的改變控制影響層的厚度,可以最 小的厚度達到最好的封合效果,並可同時改善薄膜的電性、機械性質與化學性質等。因 此在本研究中,我們選用電漿處理做為孔洞封合的製程,並改變電漿種類做為比較。
2.4 電漿原理簡介
物質三態中,當給予固體適當能量則可使其相轉換為液體,再提高能量,則液體亦 可以轉成氣體,但是當氣體又持續被供給能量之後,將會導致部分氣體之離子化,而形 成電漿態,因此也稱電漿為物質的第四態。而所謂的電漿,指的是一個遭受部分離子化 的氣體。氣體裡面的組成,包括各種帶電荷的電子、離子、以及不帶電的分子與原子團 等,而電漿本身就是這些粒子的集體行為。由於這些粒子不論帶電與否,其活性皆很強, 因此電漿被廣泛的運用在各個半導體製程技術上,如:薄膜沉積、乾式蝕刻、離子植入 技術等。 2.4.1 電漿原理 對氣體施加一個強度足夠大的電場時,自由電子經電場的加速而獲得極高的動能, 與氣體分子產生多次的碰撞,便可將氣體游離及解離而形成電漿。式(2-4~6)【36】是這 一連串的碰撞反應中,最為相關的三個反應式。 離子化碰撞:e- + A → A+ + 2e- (2-4) 激發-鬆弛碰撞:e- + A → A* + e- (2-5) 分解碰撞:e- + AB → A + B + e- (2-6) 式(2-4) 為 氣 體 分 子 遭 電 子 撞 擊 後 , 產 生 帶 正 電 荷 的 氣 態 離 子 的 離 子 化 反 應 (Ionization Reaction)。由於此反應需使電子完全脫離原子核的束縛,因此發生所需要的 能量較高,一般電子需具備約12ev以上的能量,才足以讓以上的反應發生。而當氣體分 子與能量較低的電子相碰撞時,撞擊電子無法提供足夠的能量供軌道電子逃脫原子核的 束縛,只使得軌道電子躍遷到能量較高的能態,此過程稱為激發(excitation,如式(2-5)), 激態既不穩定且短暫,在激發態軌道的電子無法停留太久,便會掉回最低的能階或基態,並且以光子的型態把他從電子撞擊中所獲得的多餘能量釋放出來,便產生了電漿獨 特的輝光(Glow)現象。此外,當電子和分子碰撞時,如果因撞擊而傳到分子的能量比分 子的鍵結能量要高時,便會破壞分子間的化學鍵並產生自由基(如式(2-6)),這個過程稱 之為裂解分應(Dissociation Reaction)。以上這些反應式反應出藉著與氣體分子撞擊引發 的離子同時伴隨著自由電子的產生,而釋放出來的自由電子將再從電場獲得動能,重複 上述步驟,使反應持續不斷地發生。因此,當外加的電壓大過於崩潰位能時,一個自我 維持式的電漿就會形成在整個反應腔體中。 2.4.2 離子轟擊效應 在電漿產生的過程中,電場變化的非常快。其中,電子可以快速地加速且開始碰 撞,離子太重無法立即對交流的電場作出反應;由於離子的碰撞截面較大所以有較多 的碰撞,也減緩了離子的運動速度。因此,在電漿中電子的移動速度比離子快得多, 導致當電漿製程一開始產生後,任何接近電漿的東西,包括反應腔體與電極,都會帶 上負電。帶負電的電極會排斥帶負電的電子而吸引帶正電的離子,而正電荷與負電荷 的差值導致在電極附近形成一個電場,稱為鞘層電位(Sheath Potential)。鞘層電位會加 速離子朝向電極移動,並造成離子轟擊(Ion Bombardment)。若將晶圓放置於電極上方, 就可利用鞘層電位所造成的離子加速而使晶圓表面受到轟擊。 影響離子轟擊程度多寡的主要變數有二:其一為離子在電場內的密度,或稱為離
子密度(Ion Density),另一為離子在電漿裡所獲得的能量,亦稱離子能量(Ion Energy)。 不管是離子密度亦或是離子能量均與外部的功率供給、反應室壓力、電極間的間距以
及製程所使用的氣體有關【37】。表2-2 為壓力及外加功率的變化對電漿的離子密度及 離子能量的影響趨勢。
碰撞頻率下降,所以離子的密度也隨之降低。但因平均自由徑增加,所以離子在電場 內所獲得的能量也就比較高。綜合這些結果可推論,電極將遭受少量但劇烈的離子轟 擊。至於電力的影響就比較直接,因為電能是電漿內各粒子獲得能量的來源,所以電 力增強,通常代表離子轟擊的現象將比較劇烈。 表2- 2 壓力及外加功率的變化對電漿的離子密度及離子能量的影響。 2.4.3 電漿表面改質處理 有別於傳統化學反應僅以自由基或特殊官能基作用的結果,由於電漿中具有高能量 的電子、高轟擊能力的離子及高反應性的自由基,因此在低溫下可以達到許多一般化學 反應難以達到的效果。且電漿可控制其作用僅止於表面,因此電漿經常被使用為表面改 質的工具,其獨特的優點是,可針對表面特性進行有選擇性的強化,而使材料本體特性 保持不變,且可利用適當的電漿,強化材料表面的化性、電性、機械性質等特性。在電 漿改質方法裡,可簡單區分為下列三種反應: (1)電漿聚合反應:使用聚合性氣體(如:NH3、CF4)產生電漿,電漿中高能電子撞 擊氣體分子而分裂為各種活性化學物質,接著發生許多複雜的化學反應。反應的生成 物會沉積在基材上,聚合形成薄膜,此一過程稱為電漿沉積聚合反應。由於電漿聚合 離子密度 離子能量 備註 壓力上升 增加 下降 粒子平均自由徑變短 壓力降低 降低 上升 粒子平均自由徑變短 外加功率增加 增加 下降 粒子碰撞頻率增加 外加功率減低 降低 上升 粒子碰撞頻率降低
反應中,反應物的化學結構會被電子或高能量的粒子完全破壞,所以這層薄膜其結構 通常是以高度分支結構而且是交聯的形態存在。在Hua-Gen Peng【35】的研究中,將 MSQ 薄膜做氨電漿處理,由於氨電漿的聚合反應,處理過後薄膜表面多了一層氮化 物,此氮化物薄膜改善了 MSQ 薄膜的吸水性,並改善了薄膜的吸水性質,並可以抵 擋後續金屬製程的擴散,大大提升薄膜的性質。 (2)非聚合性電漿反應:以非聚合性氣體經輝光放電產生的電漿氣體處理高分子材料 表面時,材料會與電漿中各種活性化學性質進行蝕刻或化學斷鍵反應,離子分解與受激 化學反應物種間的平衡,決定電漿處理材料表面鍵結能量。在Ting-Chang Chang【38】 研究中,使用氫電漿處理MSQ薄膜,發現薄膜不管是在電性、熱穩定性等方面均有顯著 的改善,這是因為在氫電漿處理中,氫自由基能填補或鈍化(passivation) 薄膜中因前處 理所導致的許多懸鍵(dangling bonds) 與殘餘井(residual traps) 而形成穩定的鍵結,進而 改善薄膜品質。 (3)電漿誘導接枝共聚合反應:以惰性氣體(Ar、He)或可反應性氣體(reactive gas, 如:NH3、O2)經電漿輝光放電產生的離子化氣體處理材料表面,其可在基材表面產生 自由基、化學斷鍵和交錯連結等的活性表面,再與特定物接觸後產生鍵結置換,進行 接枝共聚反應,此方法稱為電漿誘導接枝共聚合反應。此優點是使接枝點只限於材料 表面活化區域,表面產生一層薄膜,但本體結構不會受到破壞,得以維持原有材料的 特性。在A. T. Cho【39】等人的研究中,利用氨電漿對多孔性二氧化矽薄膜做表面處 理,在電漿處理中其所產生的氫原子(或氫自由基) 可將薄膜中的 S i - O H 還原成 S i - H 以增加其薄膜之疏水性,進而改善薄膜品質並且降低薄膜的介電常數值和漏電流。 而經過氨電漿處理後,再將薄膜暴露在充滿HMDS (hexamethyldisilazane) 蒸氣的腔體 中,讓電漿處理後所產生的懸鍵與HMDS 反應,產生三甲基矽基,可再更進一步降低 介電常數與漏電流。
2.5 原子層化學氣相沉積
原子層化學氣相沉積法(atomic layer chemical vapor deposition, ALCVD)又名原 子層沉積(ALD),1970 年代中期起源於芬蘭【40】。早期應用在Ⅲ-Ⅴ族與Ⅱ-Ⅵ族等 化合物半導體(Compound Semiconductor )的應用。但隨著半導體製程技術往下推進到 100 nm 以下之後,部分製程對 CVD 薄膜的要求,將超越現有設備與技術所能提供的 能力。因此,可以達到超薄、厚度均勻且階梯覆蓋性極佳的 ALD 技術,將成為新一 代的CVD 製程。 顧名思義,ALCVD是一種逐層沉積原子層級厚度的薄膜沉積技術,ALCVD利用氣 體前驅物在基材表面進行選擇性化學吸附反應時,在達到單一飽和吸附層狀態後,即不 再產生表面吸附反應,因此在薄膜成長過程中,利用此一自我限制的反應特性,即可以 實現原子級厚度的薄膜成長【41】。在ALD製程中,交互通入前驅物與低反應性氣體藉 由表面化學吸附與表面化學反應的鍍膜技術,依序通入前驅物A、清潔氣體、前驅物B、 清潔氣體完成單一反應循環。在適當的條件下,每一循環皆使表面形成飽和狀態,如此 可製作大面積精準控制膜厚且披覆極佳之平坦薄膜,其厚度僅與反應循環次數有關,單 一循環成長之厚度為單一原子層厚度。以利用Al(CH3)3與H2O作為反應物以成長高介電 質三氧化二鋁的 ALCVD為例,如圖2-9所示,首先在適當的溫度使基材表面產生氫氧 基,以Al-OH*來表示,接著將Al(CH 3)3 前驅物通入反應腔,將進行如下反應:
Al-OH* + Al(CH3)3 → Al-O-Al-CH3* + CH4 (2-7)
表面的-OH 中的 H 將與 Al(CH3)3中的甲基(CH3)鍵結產生甲烷氣體並由於高 溫 脫 附 離 開 表 面 , 而 剩 下 來 的 氧 原 子 將 與 前 趨 物 中 的 Al 產 生 鍵 結 , 形 成 Al-O-Al-CH3*。當表面層的OH 基完全反應完畢, Al(CH3)3 便不再吸附在基材表面 , 再利用Ar 氣體流過表面,以帶走殘留表面的雜質,此時表面吸附狀態從原本的 Al-OH* 轉變成Al-CH3*。接著再通入氧化劑H2O,進行如下反應: Al-CH3* + H2O → Al-OH* + CH4 (2-8)
H2O 與表面上的甲基(CH3)反應生成 OH 表面官能基與 CH4氣體分子,兩個相鄰 的OH 基交互作用而發生脫水反應,鋁原子於是與氧原子形成化學鍵結,當表面所有 的CH3 基全被 OH 基取代,再通入 Ar 清除殘餘的 H2O 和 CH4,如此表面又回到原 先 Al-OH*的狀態,如此便完成一個循環(cycle) ,理論上,現在表面上已經成長出了 一層非常均勻的原子級厚度的薄膜生成物,要增加薄膜厚度,只須重覆前述的循環步 驟。 圖2- 8 利用 ALCVD 成長高介電質三氧化二鋁的示意圖。
由於ALCVD上述的飽和吸附成長機制,薄膜厚度只與製程時所選定之循環數有 關,且原子層成長方式對於膜厚能有相當準確的控制,可精準到奈米尺度的範圍。飽和 吸附的影響,再複雜的基板表面型態,都能有良好的階梯覆蓋性,這對於未來深寬比日 益增加半導體製程將會是個非常有利的技術。且由於反應前趨物分別通入的影響,可避 免反應物在還未到達基板表面時就先行裂解進行反應,在反應前趨物的選擇上也較自 由。ALCVD 最大的的缺點,即來自於一層一層成長薄膜的步驟,造成鍍膜成長速度非 常緩慢。在平均鍍膜速率約 0.8Å/cycle 的情形下,此種鍍膜速率與傳統的CVD 製程或 濺鍍製程比起來,很難使其與量產畫上等號。所幸目前在半導體製程技術不斷微縮的情 況下,所需介電薄膜厚度已降至幾十奈米的尺度,因此利用ALCVD 成長介電薄膜所需 的時間也隨之下降,如此便能與前、後段的製程相整合,在量產上也慢慢發揮其優勢。 由於優異的均勻覆蓋率與順應性,ALCVD 製程技術被認為是未來 IC 超薄與極小 結構之薄膜沉積製成中極重要之 CVD 技術,可被應用的製程模組包括高介電係數閘 極介電層與銅金屬連線技術裡的銅晶種層與阻障層。目前半導體業界採行的 Cu 阻障 層為 Ta/Ta(N)的結構,主要是附著性的考量,由於金屬薄膜厚度尺寸縮小的同時,所 引發額外的困擾就是薄膜電阻的上升。而ALCVD 沉積法一般可分為加熱式爐管原子 層 化 學 氣 相 沉 積 法(thermal ALCVD) 與 電 漿 輔 助 原 子 層 化 學 氣 相 沉 積 法 (PE-ALCVD)。因此,有研究利用 PE-ALCVD 成長諸如 TaN 等擴散阻障層,其沉積步 驟如圖 2-10。首先利用 Ar 等惰性氣體連帶出金屬前驅物的揮發氣體,使其吸附於基 材表面上;持續一段短暫時間後,進行腔體抽真空與Ar 淨化(purge);第三階段則是通 入H2及N2,並導入RF 電漿進行反應;最後關閉 RF 電漿,停止通入 H2及N2,再進 行腔體抽真空與Ar 淨化。所有的過程與一般加熱式 ALCVD 一樣,但多了電漿輔助的 部份。以此法所沉積的TaN 薄膜,其含 N 量較少,因此薄膜的電阻率(resistivity)較小, 以此來減小電阻上升的問題。
圖2- 9 利用 PE-ALCVD 成長擴散阻障層氮化鉭的示意圖
而在Maeng【42】等人的研究中,也分別利用 thermal ALCVD 與 PE-ALCVD 來沉 積 Ta2O5。研究發現由於電漿中包括許多自由基與高能分子,能在溫度較低的狀態下反 應,因此在同樣條件下PE-ALCVD 可於較低溫時沉積薄膜;而在相同溫度下,PE-ALCVD 的沉積速度可比thermal ALCVD 快約 50%,這對一向因沉積速度過慢而被排除於量產 之外的ALCVD,無疑是一大進步。此外,研究並發現,PE-ALCVD 所沉積之薄膜,不 論在薄膜缺陷或汙染都比thermal ALCVD 輕微,薄膜的平整度好,因此所量得的電性較 佳。
第三章 實驗方法
3.1 試片製備
試片的製備過程包含基材的前處理、奈米孔洞二氧化矽前驅物的配製、薄膜的沉 積與乾燥、烘烤與鍛燒處理、以及疏水化改質處理處理等程序。 3.1.1 矽晶圓清洗 基材所使用的六英吋P-type (100)矽晶片,必須經過清洗蝕刻工作站標準清洗流程 (RCA clean)的清潔以去除表面的汙染物。在晶圓的洗淨過程中,需要用到很多高純度 的化學品來清洗、高純度的去離子純水(DI Water)來洗濯(rinse),最後再用高純度的氣 體(如:氮氣)在高速下脫水旋乾。其程序概要如下:(1) 浸入 120℃的硫酸水溶液 (H2SO4:H2O=3:1)10 分鐘,氧化並去除矽晶片表面殘留的有機物;(2) 浸入常溫 的氫氟酸水溶液(HF:H2O=1:50)直至晶背不沾水,去除原生的二氧化矽(nativeoxide);(3) 浸入 75℃的氨水與過氧化氫水溶液(NH4OH:H2O2:H2O=0.25:1:5)
10 分鐘,去除矽晶片表面殘留的細微顆粒、有機物以及 1A、2A 族金屬離子;(4)浸入 75℃的鹽酸與過氧化氫水溶液(HCl:H2O2:H2O=1:1:6)10 分鐘,去除矽晶片表 面殘留的1A 族金屬離子;(5) 再次浸入常溫的氫氟酸水溶液(HF:H2O=1:50)直 至晶背不沾水,去除因前述步驟所導致的二氧化矽。上述五項步驟,在每一項結束後 皆需以去離子水浸泡清洗(rinse)七個循環,最後置入旋乾機乾燥,完成標準清洗流 程。 3.1.2 奈米孔洞二氧化矽薄膜前驅物的配製 奈米孔洞二氧化矽薄膜前驅物的製備,係以溶膠-凝膠法為基礎。主要為酸催化二
氧化矽溶膠凝膠並添加兩相高分子(Binary Polymer)做為模板分子。於是,前驅物的配 製一般可分為兩個步驟: (1) 二氧化矽溶膠凝膠的製備:利用 TEOS 溶液與水分別作為矽源以及反應物, 乙醇作為主要的溶劑,用以製造酸性催化環境的鹽酸水溶液以滴定方式加入。將上述 溶液以特定比例混合後,置於 70℃的持溫系統進行 90 分鐘的回流,合成酸催化二氧 化矽溶膠凝膠。 (2) 兩 相 高 分 子 的 添 加 : 先 利 用 特 定 比 例 的 乙 醇 將 三 區 塊 共 聚 物 ( triblock copolymer)Pluronic P123 加以溶解後,做為有機模板分子(template molecular),導入 酸催化二氧化矽溶膠凝膠充份混合。最後,在室溫下靜置四小時進行時效處理(aging) 配製而成。奈米孔洞二氧化矽薄膜前驅物的成分與莫耳比如表3-1 所示。 表3- 1 奈米孔洞二氧化矽薄膜前驅物溶液的成分以及莫耳比。 3.1.3 奈米孔洞二氧化矽薄膜的沉積與乾燥 本研究採用旋轉塗佈(spin-on)的方式將所製備的前驅物溶液沉積於矽晶片基材 上。在旋轉塗佈的過程中,矽晶片表面上溶液中的溶劑(EtOH)不斷揮發,最後奈米孔 洞二氧化矽薄膜在矽晶片上形成。 首先,將六英吋P-type(100)矽晶片固定於旋塗機(spin coater)上,同時沉積系統必 成分 莫耳比
TEOS (tetraethyl orthosilicate)
1
P123 (triblock copolymer)
0.008~0.03
H
2O 3.5~5.0
HCl 0.003~0.03
EtOH 10~34
需維持在富含乙醇氣氛中,確保旋塗過程中前驅物的性質。滴入適量的前驅物並使其 佈滿矽晶片表面。設定旋塗機轉速為1700 rpm,時間 26 秒,將前驅物溶液旋轉塗佈 於矽晶片上。 剛成形的奈米孔洞二氧化矽薄膜其結構並不穩定,所以塗佈後的試片需靜置於烘 箱中,進行40℃兩小時的乾燥步驟,讓殘留的溶劑可以緩慢揮發而不破壞薄膜內部結 構,使薄膜有充分的時間進行反應,同時奈米孔洞二氧化矽的骨架結構得以獲得初步 的穩定。 3.1.4 奈米孔洞二氧化矽薄膜的烘烤與鍛燒製程 為了使奈米孔洞二氧化矽薄膜的結構更趨穩固,必須將試片再經過 110℃一小時 的烘烤過程。其目的是為了加速剩餘溶劑的揮發,使溶質密度增加,且使薄膜內Si-OH 基進行固化,彼此間進行交聯(crosslink)反應,形成以 Si-O-Si 鍵結為主的骨架結構。 此時,薄膜結構已十分穩固,但有機模板分子尚存於薄膜內部,所以必須再經由以下 的鍛燒(calcination)製程,奈米孔洞二氧化矽薄膜才算完成。 鍛燒製程是為了移除薄膜內部的有機模板分子,使原本被微胞所佔據的空間為空 氣所取代,形成奈米級的孔道(pore channel)。在此製程中,試片被送入 400℃充滿氮 氣氛圍的高溫常壓爐管,持續作用30 分鐘。利用高溫使有機模板分子分解並揮發,奈 米尺寸的孔道於是形成。 經過前述步驟,可製得奈米孔洞二氧化矽薄膜(as-calcined),其製備流程如圖 3-1 所示。
圖3- 1 奈米孔洞二氧化矽薄膜製備流程圖。
3.1.5 電漿處理
經過煅燒後的奈米孔洞二氧化矽薄膜,利用高密度電漿系統做電漿處理。高密度
電 漿 化 學 氣 相 沉 積 系 統(Duratek Multiplex Cluster System, High Density Plasma Chemical Vapor Deposition),係利用高功率的感應偶合式電漿(Inductively Coupled Plasma) , 同 時 進 行 沉 積 (deposition) 、 濺 蝕 (sputtering) , 以 及 濺 蝕 物 質 的 再 沉 積 (redeposition)來達到沉積薄膜的目的,同時主要因為具有離子轟擊(Ion Bombardment) 的效應,可使基材近表面的區域強迫性地得到廣泛的離子植入,得到良好的電漿處理 酸催化二氧化矽凝膠,置於 70℃ 回流系統 90 分鐘 添加 P123 有機模板分子 時效處理 ,成為前驅物 1700 rpm 26 秒,旋塗前驅物於 矽晶片表面 40℃ 120 分鐘的乾燥 110 ℃ 60分鐘的烘烤 鍛燒處理 奈米孔洞二氧化矽薄膜 酸催化二氧化矽凝膠,置於 70℃ 回流系統 90 分鐘 添加 P123 有機模板分子 時效處理 ,成為前驅物 矽晶片表面 40℃ 120 分鐘的乾燥 110 ℃ 60分鐘的烘烤 奈米孔洞二氧化矽薄膜
效果。其電漿處理參數如表3-2: 表3- 2 電漿處理參數。 Ar、N2、O2、N2O、CH4、CF4 ICP Power(W) 750 Bias(V) 150 Time(s) 10 Pressure(mtorr) 20 Temperature(oC) 25 Flow rate(sccm) 70 3.1.6 疏水化改質處理 鍛燒後的奈米孔洞二氧化矽薄膜其孔洞表面帶有大量的矽醇基,易與水氣結合, 其中水的介電常數值為78,所以其吸水性質將導致介電常數大幅上升,為了確保低介 電常數值的穩定,必須將薄膜表面改質成疏水性質,以符合實際應用之需求。 其處理方法為將液態 HMDS 置入加溫至 165℃之密閉容器中,由於 HMDS 的沸 點為 124~127℃,因此 HMDS 將會揮發形成一充分的 HMDS 氛圍,對奈米孔洞二氧 化矽薄膜進行一小時的改質處理。藉由HMDS 的三甲基矽化作用,將疏水性的官能基 嫁接(grafting )到孔洞表面,將原本親水性的薄膜表面轉換成疏水。
3.2 試片分析
試片的分析包含表面形貌的觀察、結構與化性分析、機械性質及介電特性,以及
孔洞封合測試等。
3.2.1 表面形貌觀察
我們分別利用原子力顯微鏡(Atomic Force Microscope, AFM)與場發射掃描式電 子顯微鏡(Field Emission Scanning Electron Microscopy, SEM)來鑑定薄膜表面的形貌。
(1) 原子力顯微鏡:其量測原理如圖 3-2 所示,係利用懸臂(cantilever)端點的探 針,逼近試片表面,探針與試片之間的作用力大小反應於懸臂的形變上,並將偏折量 轉換為電流輸入回饋系統,將訊號送至回饋控制電路處理並輸出至Z 掃描器,可以得 到等作用力的高度輪廓,加以 X-Y 掃描器做探針-試片間相對性位移,即描繪出試片 微區的表面形貌。經由表面形貌數據的統計,可以得到表面粗糙度、粒徑大小等參數。 圖3- 2 AFM 量測原理。
(2) 場發射掃描式電子顯微鏡:利用場發射掃描式電子顯微鏡對經過電漿處理之試 片表面做形貌觀察,同時對試片的劈裂面形貌作觀測,鑑定各薄膜疊層的厚度。場發射 係利用高電場扭曲靶材功函數,使電子穿隧通過能障自陰極尖端發射,因此可獲得高電 流密度的電子束,經過電磁透鏡的聚焦以及電場的加速,照射在試片表面,試片受到入 射電子撞擊,使外層軌域鍵結較弱的電子得到動能而脫離原子,稱為二次電子。二次電 子屬於試片表面所發散的低能電子,並且發散數目受到表面形貌影響,而掃描式電子顯 微鏡即利用二次電子訊號,得到具有亮度與對比的影像,得知試片表面形貌。SEM主要 用來觀察物體的表面狀態,其影像解析度極高,放大倍率可達一萬倍以上,並具有景深 長的特點,可以清晰的觀察起伏程度較大的物體,如破斷面等。 3.2.2 結構與化性分析 我們利用X光繞射儀(X-ray Diffractometer, XRD)鑑定薄膜內部孔洞排列的規則度; X光反射率量測(X-ray reflectivity, XRR)則用來分析薄膜的密度;利用n&k系統,測量奈 米孔洞二氧化矽薄膜與各疊層所擁有的折射指數以及膜厚;而傅立葉轉換紅外線光譜儀
(Fourier Transform Infrared Spectroscopy, FT-IR)與化學分析電子儀(Electron Spectroscopy for Chenical Analysis, ESCA)則用來偵測薄膜的化學組成。
(1) X光繞射儀:XRD是相當有利於進行材料結構的分析技術之一,由於X光的波長 在0.01-100 Å之間,相當於晶格中原子間的距離,因此X光會對晶體產生繞射,此繞射現 象可以視為入射光被晶面反射。此種反射如同鏡面反射,入射角(θ)等於反射角。在某些 散射角下,相鄰晶面散射波彼此相位相同,光程差為波長(λ)的整數倍(n),因而產生建設 性干涉。滿足此條件便可產生繞射,並可用來量測結晶性材料的原子平面間距(d)。此一 繞射現象可以布拉格定律(Bragg’s law)來描述: 2dsinθ=nλ (3-1)
對於薄膜的結構分析,通常使用低掠角X光繞射法(Grazing Incident X-ray Diffraction, GIXD)。由於X光入射光束與試片表面的夾角很小,所以在進入試片內部時,X光的行徑
路線主要在表面的薄層內,因此會得到較顯著的薄膜繞射訊號。而於本研究中,我們是 將入射角設為0.5度,以此法來鑑定奈米孔洞二氧化矽薄膜內孔洞排列的規則性。 (2) X光反射率量測:而XRR是目前用來測量薄膜厚度、電子密度及其表層和介面粗 糙度等參數的有利工具。當X光在一非常小的入射角時,會被固體物質表面完全反射, 而在臨界角以上時,X光反射率下降得很迅速。而當表層或介面具有一些粗糙度時,X 光反射率則下降得更快。當表層沒有其他堆疊層時,X光反射率從臨界角θC開始急遽下 降;當試片為多層薄膜時,X光沿著每一介面反射與入射之X光產生干涉,而使反射率 曲線強度呈現多重週期。因此所測試片之詳細的電子密度分布、各層薄膜厚度以及介面 與表層粗糙度皆可從X光反射率曲線上獲得【43】。XRR對於所測樣品之型態並未有特 別限制,不論是單晶、多晶或非晶質材料。我們利用此方法來分析經過電漿處理後之薄 膜的密度變化。 (3) n&k 系統:n&k 分析儀係利用已知波長的光入射至試片表面,藉由反射光譜
( )
λ R 可得知試片的本質(intrinsic)訊息,如:膜厚d、薄膜的折射指數nf( )
λ 、消光 指數kf( )
λ 、能隙E ,以及基材的折射指數g ns( )
λ 、消光指數ks( )
λ 等。 (4) 傅立葉轉換紅外線光譜儀:FTIR 藉由吸收光譜中的特性吸收頻率(波數的倒 數)鑑定試片含有的官能基(functional groups)種類,被吸收的輻射頻率係分子振動 所吸收的能量,並根據分子結構而呈現不同的振動模式。傅立葉轉換紅外線光譜儀擁有高輸出能量,因此在分析時可獲得較強的訊號雜訊比(signal to noise ratio),並具備 高解析度、頻率再現性等優點。 自紅外線光譜可得知試片成份、分子結構以及鍵結型態等相關訊息,文獻中經常 利用光譜鑑定鍵結形式以及相關定量、定性分析。首先將矽晶片批次建立背景訊號值, 依序量測各薄膜疊層,針對各階段處理造成的化學性質進行定性分析,觀察特性訊號 峰,藉此印證薄膜的鍵結組成。 (5) 化學分析電子儀:ESCA之基本原理是為光電效應,利用X-Ray照射固態表面可
以游離發射光電子,量測光電子的動能,並從而推算該光電子的束縛能。如圖3-3所示, 當原子受到X-ray照射,若照射能量大於內層Z軌域電子與原子核之束縛能(Binding Energy, EZ),則Z軌域電子可被撞擊而游離成為自由電子,即為光電子,其動能為KE, 基於能量守恆原理,則可表式為: KE=hν-EZ (3-2) 其中h跟ν分別為浦朗克常數與X光頻率。若光電子發射自固態表面,則必須將電子脫離 固體表面位能束縛的功函數(work function, ω)考慮進去,於是 KE=hν-EZ-ω (3-3) 由於各元素有不同的特定電子束縛能,KE亦將因元素種類的變化而不同,所以檢測光 電子的動能可以鑑定試品的元素種類。 圖3- 3 光電子發生原理示意圖。 3.2.3 機械性質 在材料的機械性質方面,我們利用奈米壓痕儀(型號為美國MTS公司Nano Indenter XP System)來量測薄膜的硬度與彈性係數。 材料硬度檢測的方法是利用一壓痕器打入材料,藉由壓痕的幾何形狀,得知材料抵 抗塑性變形的能力。一般量測薄膜硬度都使用微硬度計,但因為微硬度計無法提供更小