經濟部智慧財產局 103年度委託研究報告
美國專利連結與橘皮書登錄制度研究
執行期間:自103年11月4日至103年12月31日止
主辦單位:經濟部智慧財產局 受委託單位:理律法律事務所
中 華 民 國 103 年 12 月 29 日
經濟部智慧財產局委託研究報告摘要
委託題目:美國專利連結與橘皮書登錄制度研究 計畫執行廠商:理律法律事務所
計畫主持人:蔣大中 研究報告摘要:
本研究係針對美國專利連結制度進行全面性的詳細了解,以分析該制度引進 我國之影響。
美國的專利連結制度源起於 1984 年 Hatch-Waxman 法案,該法案之主要內容 包括橘皮書之登錄、學名藥之上市申請程序、30 個月停止發證期與學名藥廠之 180 天銷售獨占權等。自該法案施行以來,發生相當多爭議,美國國會迺於 2003 年就該法案進行大幅度的修法,該次修法解決 Hatch-Waxman 法案施行近 20 年以 來的部分爭議,但仍存有部分未決的問題。
本研究特別針對橘皮書之登錄項目、應/不應登錄之專利,以及登錄制度下之 爭議問題(例如消除登錄)進行詳細的探討,接著針對 2003 年修法前後的爭議 為闡述、說明,並就現行專利連結制度運作下仍存在之問題為分析,最後回歸到 我國的現行制度面,分析專利連結制度引進我國可能產生或必須克服的問題,並 給予具體建議。
I
目 錄
第壹章、緒論... 1
第一節、研究範圍與方法 ... 1
第二節、研究架構 ... 2
第貳章、美國專利連結橘皮書登錄制度之相關法規範、登錄相關實務以及相關爭 議 ... 4
第一節、美國專利連結制度之介紹 ... 4
第一項、藥品之登錄 ... 5
第二項、學名藥之上市申請與學名藥廠之 180 天銷售獨占權 ... 8
第三項、30 個月停止發證期 ... 22
第四項、Section 505(b)(2)之上市申請(Paper NDA) ... 23
第二節、美國專利連結橘皮書登錄制度相關法規範、實施現況及執行 情形 ... 27
第一項、可登錄的事項及活性成分的認定 ... 27
第二項、登錄不實、漏列、誤列之處罰、救濟或相關訴訟 ... 34
第三項、消除登錄的相關法律程序說明及相關判決 ... 45
第四項、小結 ... 57
第參章、2003 年修法前後 Hatch-Waxman Act 之爭議問題探討 ... 59
第一節、修法前之爭議問題暨修法前後差異比較 ... 60
第一項、橘皮書可登錄事項之爭議 ... 60
第二項、FDA 就橘皮書登錄之審查與 ANDA/Section 505 (b)(2) 申請人提起反訴之權限 ... 63
第三項、30 個月停止發證期之次數 ... 65
第四項、ANDA 或 Section 505(b)(2)提出申請後通知原開發藥廠 及專利權人之法定期限 ... 67
第五項、學名藥廠之 180 天銷售獨占權相關爭議問題 ... 68
第六項、通知後 45 日內如受通知人未提起專利侵權訴訟,學名 藥廠或 Section 505(b)(2)申請人得提起確認之訴 ... 72
第七項、逆向和解協議 ... 74
第二節、修法後仍存在之爭議與問題 ... 89
第一項、180 天銷售獨占權得否移轉 ... 89
第二項、不同劑型與劑量之學名藥分別享有 180 天銷售獨占權 89 第三項、原廠授權學名藥(Authorized Generics)... 90
第四項、主張未構成專利侵權之 Paragraph IV 之法律效果再思考 ... 94
II
第五項、第一位以 Paragraph IV 提出 ANDA 申請者須待原開發
藥廠起訴始能得知 ... 94
第六項、專利連結制度對公共利益可能造成之損害 ... 95
第七項、修法後有阻礙創新之虞 ... 96
第三節、小結 ... 96
第肆章、國內學名藥廠與擁有專利之原開發藥廠之美國專利訴訟案例分析... 98
第一節、國內學名藥廠在美國被訴概況 ... 98
第二節、具體案例 ... 99
第一項、Par Pharmaceuticals, Inc. and Alkermespharma Ireland Ltd.v. TWi Pharmaceuticals, Inc. ... 99
第二項、Takeda Pharmaceutical Co., et al. v. TWi Pharmaceuticals, Inc. ... 101
第伍章、生物製劑... 103
第一節、美國的生物製劑產品價格競爭與創新法案 ... 104
第二節、BPCIA 與 Hatch-Waxman Act 之差異說明 ... 105
第三節、小結 ... 107
第陸章、綜合分析與建議... 109
第一節、看待橘皮書及專利連結政策問題應有之視角 ... 109
第二節、具體結論與建議 ... 114
第一項、我國專利法應新增擬制侵權規定 ... 115
第二項、橘皮書之登錄 ... 115
第三項、藥政主管機關是否實質審查提出登錄申請之專利 ... 117
第四項、不實登錄之救濟 ... 118
第五項、停止發證期間 ... 119
第六項、學名藥廠之銷售獨占期間 ... 121
第七項、暫時許可制度 ... 123
第八項、Section viii 聲明之制度設計 ... 124
第九項、美國法上 Section 505(b)(2)之制度 ... 125
第十項、原廠授權學名藥於我國法之規範 ... 126
第三節、總結 ... 127
附件 1:專利用途編號表(節錄)
附件 2:橘皮書檢索頁面
III
附件 3:FDA 表 3542a、表 3542
附件 4:21 U.S.C. §355 「標題 21:食品與藥品—第 9 章:聯邦食品、藥品與 化妝品法規—第 5 節:藥品與醫療器材—A 部:藥品與醫療器材,第 355 條新 藥」(Title 21-Food and Drugs, Chapter 9-Federal Food, Drug, and Cosmetic Act, Subchapter V-Drugs and Devices, Part A-Drugs and Devices §355 New Drugs)
附件 5:42 U.S.C. §262 「標題 42:公眾健康與福利—第 6A 章:公眾健康服 務—第 2 節:一般權力與責任—F 部:生物產品與臨床實驗室之授權—F-1 部:
生物產品,第 262 條生物產品之規範」(Title 42-The Public Health and Welfare, Chapter 6A-Public Health Service, Subchapter II-General Powers And Duties, Part F-Licensing of Biological Products and Clinical Laboratories, subpart 1-biological products § 262. Regulation of biological products)
附件 6:「美國專利連結制度與訴訟研討會」簡報中文翻譯版
1
第壹章、緒論
第一節、研究範圍與方法
本研究分為六章,主要研究內容分布於第貳章至第陸章。第貳章係就美國 專利連結橘皮書(Orange Book)登錄制度之相關法規範、登錄相關實務以及相 關爭議,透過研究美國相關法規、判決與分析國內外相關文獻進行詳細的研究 與建構。本章首先就美國專利連結制度下包括藥品登錄、學名藥之上市申請、
學名藥廠之 180 天銷售獨占權、30 個月停止發證期與 Section 505(b)(2)之上市申 請為討論,接著特別針對橘皮書登錄制度之相關法規範、實施現況及執行情形 進行研究,尤其是新藥上市申請人就橘皮書登錄所應提交之資料、應登錄與不 應登錄之專利類型進行相關法規之研究與分析。
就登錄不實的部分則以登錄不實態樣之具體分析、相關規定、救濟方式與 實際案例作為研究面向。至於消除登錄(Delisting),同樣先從相關法律程序、
規定著手進行研究,接著特別以三個與消除登錄法規發展相關之重要判決為具 體說明。
第參章則以 Hatch-Waxman Act 於 2003 年修法前後之爭議問題為研究主軸,
特別著重於橘皮書可登錄事項、不實登錄之救濟、30 個月停止發證期次數、學 名藥廠之 180 天銷售獨占權等問題進行研究,並以相關報告、文獻與比較修法 前後之法規作為主要之研究基礎,其中並特別針對近年來在美國引起廣泛討論 之「逆向和解」議題為論述。接著再探究修法後仍未解決之問題,並以此作為 第陸章提出具體建議之基礎。
第肆章就國內學名藥廠與擁有專利之原開發藥廠之美國專利訴訟部分,首
2
先說明目前我國學名藥廠於美國因提出學名藥上市申請案而被原開發藥廠控以 專利侵權之概況,接著再就目前一審法院已做出判決之案件為簡要介紹與說 明。由於第貳章至第肆章所論述之美國專利連結制度僅針對小分子藥品,屬於 大分子藥品之生物製劑專利連結與登錄則另闢專章說明,列於第伍章。
第陸章則以第貳至第伍章所研究之內容為基礎,針對美國專利連結制度提 出檢討,並點出專利連結制度於我國未來立法時可能面臨之問題與爭議,並進 一步提出具體建議。
第二節、研究架構
本論文共分為六章,概述如下:
第壹章、緒論:說明研究範圍、研究方法與研究架構。
第貳章、美國專利連結橘皮書登錄制度之相關法規範、登錄相關實務以及相關 爭議:首先就美國專利連結制度為說明,接著特別針對美國專利連結橘皮書登 錄制度上可登錄的事項及活性成分的認定、登錄不實、漏列、誤列之處罰、救 濟或與該核准上市藥品的相關訴訟,以及消除登錄為詳盡的論述與說明。
第參章、2003 年修法前後 Hatch-Waxman Act 之爭議問題探討:首先直接討論數 個 Hatch-Waxman Act 修法前之爭議問題,並比較其修法前後之差異,其中並包 含逆向和解議題之探討;接著再以探究修法後仍未解決之爭議問題作結。
第肆章、國內學名藥廠與擁有專利之原開發藥廠之美國專利訴訟案例分析:本 章涵蓋我國學名藥廠目前於美國提出學名藥上市申請而被原開發藥廠控以專利 侵權之概況,以及兩個一審法院已做出判決之具體案件說明。
3
第伍章、生物製劑:本章主要內容為生物製劑法規之介紹,以及生物製劑之生 物相似性產品∕可替代產品與小分子藥品之學名藥規範上的差異。
第陸章、綜合分析與建議:本章就未來我國專利連結制度之立法面向提出將面 臨之問題與具體建議。
4
第貳章、美國專利連結橘皮書登錄制度之相關法規 範、登錄相關實務以及相關爭議
第一節、美國專利連結制度之介紹
所謂的「專利連結」係指將學名藥的上市審查程序與其參考之原廠專利藥 的專利有效性資訊加以連結,以藉此確認該學名藥的上市申請是否有侵害原開 發藥廠專利權的疑慮1,確保學名藥不會因在藥品的專利保護期間內上市而被控 侵權敗訴,進而面臨藥品被回收、銷毀,造成患者用藥供給斷絕所生的公共衛 生問題2。
專利連結制度最早乃見於美國 1984 年制定之「藥價競爭及專利期間回復法」
( Drug Price Competition and Patent Term Restoration Act ) , 即 著 名 的 Hatch-Waxman Act(以下提及該法規均一律以 Hatch-Waxman Act 稱之),然於該 法施行約 20 年後,美國聯邦貿易委員會(Federal Trade Commission,以下簡稱
「FTC」)於 2002 年提出一份關於在該專利連結制度下藥廠間實務運作問題的報 告「Generic Drug Entry Prior to Patent Expiration」,並就 Hatch-Waxman Act 運作 下 之 漏 洞 提 出 修 法 建 議 , 因 此 美 國 食 品 藥 品 管 理 局 ( Food and Drug Administration,以下簡稱「FDA」)於 2003 年提出「Final Rule on Generic Drugs」
3,該規則並於同年 8 月 18 日開始施行;美國國會同樣於 2003 年通過「Medicare
1陳蔚奇,論美國專利連結制度於我國實行之妥適性,國立交通大學科技法律研究所碩士論文,
頁 6,2010 年。
2黃慧嫻,專利連結(Patent Linkage)—藥品研發與競爭之阻力或助力?談藥品查驗登記程序與
專利權利狀態連結之發展(上),科技法律透析,第 21 卷第 02 期,頁 26,2009 年 2 月。
3即 21 C.F.R. § 314.
5
Prescription Drug, Improvement, and Modernization Act of 2003」(以下簡稱 2003 MMA),其中即涵蓋專利連結制度之修正,以解決過去 Hatch-Waxman Act 施行 下的問題4。經過修法後,整個專利連結制度的內容主要包含橘皮書上藥品之登 錄、學名藥之上市申請與學名藥廠之 180 天銷售獨占權(180-day Exclusivity)、
30 個月停止發證期間(30-month stay)與 Section 505(b)(2)之上市申請,以下將 分別說明。
第一項、藥品之登錄
由於藥品安全性的要求,在 Hatch-Waxman Act 立法前,不論是專利新藥或 是學名藥都必須經過臨床試驗不斷確認藥品的療效與安全性,如此一來市面上 所能選擇的藥品自然相當少,即使是學名藥,其研發成本與專利新藥相較下亦 無法產生明顯差距而將成本上的差異反映在藥價上。在橘皮書建立後,學名藥 廠得藉橘皮書中所登錄之專利新藥的藥品療效、安全性與專利資訊,免去如新 藥 上 市 申 請 所 應 進 行 的 大 規 模 臨 床 試 驗 , 僅 須 提 供 包 括 生 體 相 等 性
(bioequivalent, BE)等資訊之實驗報告5,而以較簡易的程序加速其上市,此即 Hatch-Waxman Act 之 下 的 「 簡 易 新 藥 申 請 程 序 」 ( Abbreviate New Drug Application,簡稱 ANDA),大幅減省學名藥的試驗成本並加速學名藥的上市審 查。
依據 Hatch-Waxman Act 的規定,原開發藥廠向 FDA 提出專利新藥上市申請
(New Drug Application,簡稱 NDA)時,除了藥品療效、安全性等與取得藥品
4廖柏豪,論專利連結與資料保護對我國醫藥產業之影響,國立臺北大學法律學研究所碩士論
文,頁 6,2013 年。
5陳昭華、鐘鏡湖、張乃文、鄭耀誠,新藥監視期、資料專屬及專利連結制度對學名藥上市之
影響,基因體醫學研發創新與智慧財產權,頁 237,元照出版有限公司,2010 年。
6
上市許可相關之資料以外,尚須提出與該藥品相關的專利資訊6,包括專利號碼、
專利保護期限等,待 FDA 核准該藥品之上市許可,且與該藥品相關之專利亦經 過 FDA 之形式審查認屬可登錄之專利資訊,FDA 將核發該藥品之上市許可,並 將與該藥品相關之專利資訊登錄於橘皮書上。若 NDA 仍在審核中而有相關藥品 或專利資訊需補充,得隨時提出「修正」(amendment);若核准後原開發藥廠擬 變更該已取得藥證之藥品本身或相關專利資訊,亦應提交變更後的專利資訊予 FDA,此稱為「補充」(supplement)7
此外,由於新藥的上市申請與專利之申請本為不同的程序,若與該藥品相 關之專利於提出新藥上市申請後始經美國專利與商標局(United States Patent and Trademark Office,以下簡稱 USPTO)核准公告,藥廠得在該相關專利核准後 30 日內向 FDA 補提相關專利資訊8,以供 FDA 登錄於橘皮書上,若逾期(即相關 專利核准後 30 日內)未提,將來若發生 Paragraph IV 之專利侵權訴訟,原開發 藥廠或專利權人將無法享有 30 個月停止發證期的利益9。
綜上所述,若以 NDA 申請日作為基準點,在申請時已獲 USPTO 核准之相 關專利應於申請時提出給 FDA,若在 FDA 審核中尚有相關專利資訊需補充,一 樣要再以「修正」提出給 FDA。在 FDA 審核完畢給予 NDA 申請人新藥上市許 可後,若有與該新藥相關之專利獲得 USPTO 之核准,NDA 持有人(NDA holder)
應於專利核准後 30 日內向 FDA 請求登錄該專利,否則若日後發生 Paragraph IV 之專利侵權爭議,FDA 即不因此受 30 個月停止發證期之拘束。另外,若該相關 專利係在學名藥廠提出 Paragraph IV 之 ANDA 後才獲得核准,原開發藥廠仍得
621 U.S.C. § 355(b);Id. § 355(c); 21 C.F.R. § 314.50(h).
7「補充」(supplement)的內容規定於 21 C.F.R. § 314.3(b) 「Efficacy supplement」,由其可補 充之項目觀之,並不包含相關專利之新增,僅涵蓋「既有專利資訊」之補充。
821 C.F.R. § 314.53(c)(2)(ii).
9FTC, Biovail Corporation (Administrative), avaliable at
http://www.ftc.gov/enforcement/cases-proceedings/011-0094/biovail-corporation (last visited Dec. 15, 2014).
7
於專利核准後 30 日內登錄該專利,只是 FDA 就該「早於」專利登錄提出之 ANDA 核發上市許可,並不會受到 30 個月之拘束;設若該專利登錄後仍有其他學名藥 廠提出 Paragraph IV 之 ANDA,在此情況下,FDA 就該「晚於」專利登錄提出 之 ANDA 核發上市許可,即受 30 個月之拘束。
上述藥廠提出之專利資訊由 FDA 統整後列於「具相同藥效之核准藥品目錄」
(Approved Drug Products with Therapeutic Equivalence),在昔日僅有紙本可以 查詢的年代,該目錄的封面為橘色的,因此又被泛稱為橘皮書,如今已可透過 FDA 之網站直接查詢登錄於橘皮書上之藥品專利資訊。
除了原開發藥廠必須在提出新藥上市申請時提交該藥品之專利資訊外,由 於橘皮書建立的目的之一在於提供醫師、藥劑師或消費者評估是否選擇具同等 療效學名藥的依據,因此橘皮書中也包括學名藥的療效、安全性等藥品資訊,
亦即所有經過 FDA 許可上市之藥品,均須登錄於橘皮書中10。
此外,橘皮書登錄制度的建立使學名藥廠得透過檢索橘皮書及早進行迴避 他人專利權的預防措施,對於原開發藥廠而言,由於 FDA 於審查學名藥之上市 申請時將一併形式審查如專利到期日等資訊,若學名藥廠不擬透過 Paragraph IV 挑戰原開發藥廠或專利權人,亦可發揮防止侵害原開發藥廠專利權之功能;此 外,由於所有經過 FDA 核准上市之藥品均須登錄與藥品相關之專利於橘皮書 上,因此橘皮書上的公開資訊即得作為原開發藥廠與學名藥廠間發生專利侵權 訴訟時的有力參考資料11。關於橘皮書登錄制度之相關法規範、實施現況及執行 情形詳參本章第二節。
10John R. Thomas, Pharmaceutical Patent Law 15 (2nd ed. 2010).
11黃慧嫻,淺析美國藥品上市審查程序之專利連結機制及期實施對我國可能的影響(上),科技法
律透析,第 18 卷第 6 期,頁 4,2006 年 6 月;John R. Thomas, supra note 10, at 15-16.
8
第二項、學名藥之上市申請與學名藥廠之 180 天銷售獨占權
根據 Hatch-Waxman Act 之規定,學名藥廠於提出學名藥上市申請時,由於 其援引原開發藥廠所提交之藥品安全性等資料,因此其必須提出四種證明其一 或是排除已受專利保護使用方法之聲明,始符合提出學名藥上市申請之要件。
若學名藥廠以 Paragraph IV 提出上市申請,第一位成功取得上市許可者將可享 有 180 天銷售獨占權,以下將分別說明之。
一、學名藥上市申請(ANDA)之證明(Certification)/
聲明(Statement)
所謂學名藥(generic drugs),依照美國 FDA 的定義,係指不論就劑型、安全 性、藥品強度、給藥途徑、品質與服用後產生效果的特性等各方面,均與原開 發新藥相同之藥品,學名藥就如同是原開發新藥的複製版本12(“They are copies of brand-name drugs and are the same as those brand name drugs in dosage form, safety, strength, route of administration, quality, performance characteristics and intended use.”),學名藥之上市申請所須提出之證明/聲明係規定於 21 U.S.C.
355 (j)以下,即「簡易新藥上市申請」(Abbreviated New Drug Applications,簡稱 ANDA),以下將就證明/聲明分別說明之。
(一)Paragraph I 至 IV 四種證明(certification):
依 21 U.S.C.§355(j)(2)(A)(vii)之規定,學名藥廠於提出 ANDA 申請時,須提 出下列四種證明其中一種:
12FDA,Understanding Generic Drugs, available at
http://www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/Understanding GenericDrugs/default.htm (last visited Dec. 9, 2014).
9
1、 Paragraph I:被援引的原廠藥無相關專利登錄於橘皮書。
2、 Paragraph II:被援引的原廠藥雖有相關專利,但專利保護期間已屆滿。
3、 Paragraph III:被援引的原廠藥尚在專利保護期間內,惟 FDA 將在專利保護 期間屆滿後始核准學名藥之上市。
4、 Paragraph IV:被援引的原廠藥雖仍有相關專利保護,惟學名藥之上市申請 藥廠主張該專利權無效,或其上市申請未侵害相關專利權。
在學名藥廠提出證明或聲明後,學名藥即與專利藥品產生連結13,其得否獲 得上市許可,除藥品本身的療效與安全性外,亦取決於其是否具有專利侵權之 疑慮,在 FDA 之審查權限與範圍下,由於 Paragraph I 的情況係無相關專利登錄 於橘皮書上,FDA 無從形式審查學名藥之上市申請是否有侵權疑慮;至於 Paragraph II,則因相關專利已無專利權之保護,自無侵害藥品相關專利權之疑 慮,因此只要該學名藥之安全性與療效獲得 FDA 認可並符合相關申請程序,FDA 將直接發出藥證予學名藥廠;至於 Paragraph III 與 Paragraph IV 均係在原廠藥仍 在專利保護期間內提出 ANDA,此兩種證明的差異就在於,以 Paragraph III 提 出上市申請之學名藥,雖然 FDA 將發給學名藥廠暫時許可(tentative approval),
但該學名藥仍須等到原廠專利藥品之專利保護期經過,FDA 始核發「終局許可」
(final approval),學名藥始得上市販售,因此原開發藥廠與學名藥廠間也不會 產生如同 Paragraph IV 的侵權爭議。
至於 Paragraph IV,由於學名藥廠係主張專利無效或未侵害所援引藥品之相 關專利權,其上市申請之提出即被擬制為對原開發藥廠專利權之挑戰,為了在 保障原開發藥廠權利與鼓勵學名藥提前上市的政策間取得平衡,Paragraph IV 在
13陳蔚奇,同前註 1,頁 6。
10
制度設計上透過 180 天銷售獨占權給予學名藥廠挑戰原開發藥廠的誘因,同時 也賦予學名藥廠於其 Paragraph IV 之 ANDA 申請獲得 FDA 接受後 20 日內通知 原開發藥廠與專利權人的義務14,在原開發藥廠或專利權人得知學名藥廠提出 Paragraph IV 證明並於接獲通知後 45 日內提出專利侵權訴訟,該學名藥的上市 許可程序將自專利權人或原開發藥廠接獲學名藥廠之通知日起算而自動停止 30 個月15以待法院判決之作成,以確認該學名藥是否侵權。
在 Paragraph III 與 Paragraph IV 的程序中,均有所謂的「暫時許可」,按 21 U.S.C. §355(j)(5)(B)(iv)關於暫時許可之定義,其係 FDA 用以通知 ANDA 申請人 其申請案符合 21 U.S.C. §355(j)(2)(A)之要件,但其尚無法取得「終局許可」,
理由在於該申請案尚未符合 21 U.S.C. §355(j)(5)(B)之要件,即該學名藥所援引之 藥品尚存有專屬權期間,因此提出 ANDA 之學名藥廠仍須待 FDA 核發終局許可 始得於市場上販售學名藥。
根據 FDA 進一步的解釋16,FDA 之所以核發暫時許可給申請人,係為等到 所有與相關專利侵權爭議(例如一審法院作成學名藥廠不侵權的判決)、資料專 屬權或市場專屬權之爭議17均獲得解決後,才會核發終局許可予學名藥廠。
另外,根據 WTO 2003 年執行杜哈公衛宣言第六段之決議,美國已表明不 會利用暫時許可之制度進口藥品;至於以美國 FDA 之「暫時許可」出口學名藥 品至其他國家,由於美國專利法並未參照 2005 年之 TRIPS 協定修正草案進行修 法,以暫時許可出口藥品欠缺法律基礎,因此美國境內之學名藥廠無法因取得
1421 U.S.C. § 355(j)(2)(B)(ii), Id. § 355(j)(2)(B)(iii).
1521 U.S.C. § 355(c)(3)(C).
16FDA, Drugs@FDA Glossary of Terms, available at
http://www.fda.gov/Drugs/InformationOnDrugs/ucm079436.htm#T (last visited Dec. 13, 2014).
17在 FDA 的 Drugs@FDA Glossary of Term 網頁上,關於暫時許可之說明僅述及「專屬權」
(exlusivity),專屬權包含資料專屬權與市場專屬權,因此本文於此增補說明之。關於此部分 之原文謹列如下以供參考:"…FDA delays final approval of the generic drug product until all patent or exclusivity issues have been resolved. A tentative approval does not allow the applicant to market the generic drug product."
11
暫時許可而將學名藥出口至其他國家。
惟有文章指出,即使學名藥所援引之藥品仍在專利保護期間內,暫時許可 得允許學名藥廠於美國境外之經濟條件落後,且面臨重大公共衛生問題的國家 進行學名藥品銷售(例如治療後天免疫缺乏症候群的藥品),然美國境內之銷售 則是被禁止的18,就此,根據 FDA 於 2012 年 7 月 23 日所發佈之新聞19,雖然取 得暫時許可之藥品不得於美國境內販售,但是透過「美國總統愛滋病緊急救援 計畫」(President's Emergency Plan for AIDS Relief,簡稱 PEPFAR),治療後天免 疫缺乏症候群的「抗反轉錄病毒藥品」(antiretroviral drugs)得透過 PEPFAR 於 美國境外販售。
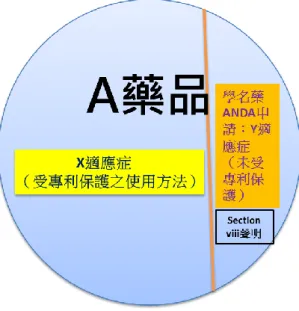
(二)「Section viii」聲明(statement):
依 21 U.S.C.§355(j)(2)(A)(viii)20規定,若學名藥廠係針對使用方法專利
(method of use patent)提出聲明,即聲明該申請上市許可學名藥之用途(適應 症)與原開發藥廠登錄於橘皮書上之使用方法專利不同而無侵權問題(如圖 1 所示)。由於提出「section viii」聲明之學名藥未涉及專利侵權之爭議,因此只 要符合要件及相關申請程序之規定,FDA 將於審查完畢後立即許可該學名藥上 市,惟由於此聲明與該學名藥所援引之原廠藥使用方法專利有效性無涉,亦不 涉及侵權爭議,因此不須如同 Paragraph IV 般,藉由給予 180 天銷售獨占權鼓勵 學名藥挑戰原開發藥廠,故學名藥廠獲得上市許可後,將無法獲得 180 天銷售
18Generics and Biosimilars Initiative, What is ‘tentative approval’ and how does it affect generics?available at
http://gabionline.net/Generics/General/What-is-tentative-approval-and-how-does-it-affect-generics (last visited Dec. 13, 2014).
19FDA, FDA: More than 150 antiretroviral drugs available through PEPFAR for worldwide HIV/AIDS relief, available at
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm313042.htm (last visited Dec.
27, 2014).
20"if with respect to the drug for which investigations described in paragraph (1)(A) were conducted information was filed under paragraph (1) or subsection (c) of this section for a method of use patent which does not claim a use for which the applicant is seeking approval under this subsection, a statement that the method of use patent does not claim such a use."
12
獨占權21。
Section viii 之具體主張方式則規定於 21 C.F.R. §314.92 (a)與(a)(12)(iii),由 於橘皮書之登錄資訊中,只要是登錄使用方法專利,即應一併登錄「專利用途 編號」(patent use code),專利用途編號係用以分類藥品之適應症,例如 U-1 為 具避孕功能者(Prevention of Pregnancy)、U-161 為用以抑制膽固醇於病人體內 進行生物合成之方法(Method of Inhibiting Cholesterol Biosynthesis in a Patient)
22。因此提出 section viii 聲明者,須向 FDA 提出「提議標示」(proposed label)
23,該標示必須刪除已有專利保護之使用方法標示資訊或將已有專利保護之使用 方法標示資訊排除,反應於橘皮書之登錄上,即允許學名藥廠得將原開發藥廠 針對該藥品之適應症自其專利用途編號中排除(carve out)。
至於「排除已有專利保護之使用方法之標示資訊或自其中排除」是否將違 反 ANDA 申請之「相同標示」要求?就 ANDA 申請人所提出之未受專利保護之 使用方法(適應症)而言,只要該學名藥之藥品安全性、藥效與其援引之新藥 相較之下非較劣,即不違反「相同標示」之要求24。
21John R. Thomas, supra note 10, at 385.
22詳參附件 1。
23John R. Thomas, supra note 10, at 384.
2421 C.F.R. § 314.127(a)(7).
13
圖 1:Section viii 聲明示意圖
針對使用方法專利,學名藥廠是否須要提出 Paragraph I-IV 四種證明其中一 種,以及「section viii 」聲明,抑或是僅提出其中一種即符合規定?FDA 目前 認為兩者互斥,亦即僅能提出其中一種,學名藥廠不得就同一方法專利提出 Paragraph I 至 IV 其中一種證明及「section viii 」聲明25;惟有學者認為,閱讀 Paragraph I 至 IV 四種證明與「section viii」聲明的條文(即§355(j)(2)(A)(vii)與
§355(j)(2)(A)(viii)間),由於兩個條文間的連接詞為「and」而非「or」,因此二者 須一併提出,可能的組合則是 Paragraph I 與 section viii,或是 Paragraph IV 與 section viii,前者係因無相關使用方法專利登錄於橘皮書上,後者則是該學名藥 之使用方法(適應症)並未侵害所援引原廠藥之相關使用方法專利,因此就二 者是否須一併提出,抑或是須擇一提出,學者認為立法者應為明確的立法26;至 於實務運作方面,若 FDA 認為學名藥廠之 ANDA 內容不符規定,例如學名藥廠
25John R. Thomas, supra note 10, at 384.
26John R. Thomas, supra note 10, at 388.
14
提出 section viii 聲明,但 FDA 認為應改成 Paragraph IV 證明,或是學名藥廠同 時提出 section viii 聲明與 Paragraph IV 證明,FDA 認為不妥,FDA 將要求 ANDA 申請人進行修正,惟實務上似尚未發生 ANDA 申請人堅持同時提出 Paragraph I-IV 之證明與 section viii 聲明而與 FDA 發生爭執的案例。
惟即使 FDA 允許學名藥廠以 section viii 聲明將受專利保護之使用方法排除 而進行學名藥之上市,學名藥廠仍不因此免於受到原開發藥廠或專利權人主張 侵權之風險,若發生專利侵權爭訟,原開發藥廠或專利權人仍可透過暫時限制 令(Temporary Restraining Order,簡稱 TRO)或暫時禁制令(Preliminary Injunction Order,簡稱 PIO)之聲請而阻止學名藥品之銷售。
二、學名藥廠之 180 天銷售獨占權
學名藥廠之 180 天銷售獨占權係指第一個因提出 Paragraph IV 之 ANDA 申 請而獲准上市的學名藥,在其學名藥首次上市後 180 天內 FDA 將不會再核准相 同的學名藥上市,該學名藥廠得保有 180 天獨占市場的利益,此銷售獨占權的 立法目的在於促進學名藥廠間的良性競爭,使之為了 180 天銷售獨占權而競先 投入試驗研發,此外,也透過獨占權賦予經濟上的誘因27,鼓勵學名藥廠挑戰原 開發藥廠之藥品專利,經由此挑戰機制排除「不應受到專利保護」之藥品專利。
就此,以下將就學名藥廠之通知義務、180 天銷售獨占權之認定、取得、起 算時點及其失權事由分別說明之。
(一)學名藥廠之通知義務
Hatch-Waxman Act 規定,申請人若以 Paragraph IV 提出 ANDA,其負有於
27陳昭華、鐘鏡湖、張乃文、鄭耀誠,同前註 5,頁 237。
15
提出學名藥上市申請後通知專利權人與原開發藥廠之義務,該通知之法定期 限,針對 ANDA 申請人規定於 21 U.S.C. §355(j)(2)(B)(ii)(I),該規定要求 ANDA 申請人必須在提出申請資料予 FDA,且 FDA 接受該申請案後之 20 日內,由 ANDA 申請人通知原開發藥廠與專利權人28。
(二)180 天銷售獨占權之認定
針對 180 天銷售獨占權係以提出 ANDA 申請之藥品個數為基礎或係以藥品 相關專利個數為基礎核發,以及若有多個 Paragraph IV 申請人時,應如何認定第 一位申請者(first applicant),以及若同時有多位第一位申請者時,180 天銷售獨 占權應如何分配的問題,將於以下簡要說明。
1. 以產品認定(product-by-product)
由於 Hatch-Waxman Act 並未規定 180 天銷售獨占權之認定方式,因此 2003 年修法前 FDA 曾採用「以學名藥援引之相關專利個數」(patent-by-patent)為基 礎核發 180 天銷售獨占權的方式,以鼓勵更多學名藥廠挑戰原開發藥廠的專利 以提早進入市場(此修法前爭議之論述詳參第參章第一節第五項之說明)。然現 行法係以提出 ANDA 之產品個數認定(product-by-product)180 天銷售獨占權,
亦即就同一學名藥品只有第一個提出 Paragraph IV 之 ANDA 申請者始得享有 180 天銷售獨占權29,然若係不同劑型、劑量之學名藥,由於仍屬不同產品,因此仍 得以不同的劑型或劑量分別提出數個主張 Paragraph IV 的 ANDA,並得分別取 得 180 天銷售獨占權30。
至於第一位申請者則係指第一個以 Paragraph IV 提出 ANDA 申請,且其提
2821 U.S.C. § 355(j)(2)(B)(iii).
2921 U.S.C. § 355(j)(5)(B)(iv)(I).
30John R. Thomas, supra note 10, at 448.
16
供之資料已符合主管機關所有要求而足以進行上市核准審查之人,亦可稱該人 為第一個實質完成(substantially complete)申請之人31。
2. 180天銷售獨占權於存在多個第一位申請者之適用
若有多個申請者同時均為第一位申請者(first applicant),為達成鼓勵學名 藥廠挑戰原廠藥品相關專利與促成學名藥提早上市之目的,在此情況下,此多 個第一位申請者得共同享有 180 天銷售獨占權,並以第一位上市者的上市日期 為起算日32,此即「共享銷售獨占權」(shared exclusivity)33的概念。
(三)180 天銷售獨占權之取得
在「原開發藥廠或專利權人未於接到通知後 45 日內對學名藥廠起訴」、「30 個月停止發證期屆滿」,或是「在 30 個月期間內,一審法院作出對學名藥廠有 利的判決」的情況下,第一個因提出 Paragraph IV 之 ANDA 申請而獲准上市的 學名藥,FDA 在 180 天內將不會再核准相同的學名藥上市,該學名藥廠得保有 180 天獨占市場的利益。
在「原開發藥廠或專利權人未於接到通知後 45 日內對學名藥廠起訴」或「30 個月停止發證期屆滿」的情況下,第一位提出 Paragraph IV 之 ANDA 申請人於 FDA 審查完成後即可獲得含有 180 天銷售獨占權之上市許可,此在現行法規運 作下並無疑義,然過去 FDA 曾在 1994 年 10 月施行的登錄規則34中規定,180 天銷售獨占權的授與必須符合「成功防禦原則」(successful defense),即 Paragraph IV 的主張者必須在專利侵權訴訟中成功說服法院系爭專利無效或是其學名藥未
3121 U.S.C. § 355(j)(5)(B)(iv)(II)(bb).
32John R. Thomas, supra note 10, at 451.
33John R. Thomas, supra note 10, at 449, 459.
341994 年 10 月施行的 21 C.F.R. § 314.107(c)(1),參自 John R. Thomas, supra note 10, at 441。
17
侵權,始能獲得 180 天銷售獨占期間。
FDA 認為須採成功防禦原則之理由在於,若學名藥廠未被起訴(即「原開 發藥廠或專利權人未於接到通知後 45 日內對學名藥廠起訴」的情況),「以法 院判決起算」(court-decision trigger)35之起算方式無法適用,如此一來,因為僅 適用「學名藥之上市銷售首日」作為 180 天銷售獨占權之起算時點,第一位申 請者即可自行決定何時將學名藥上市,若第一位申請者與原開發藥廠聯手,例 如透過逆向和解或其他契約之約定延後或不開啟 180 天銷售獨占期間之起算,
由於 FDA 必須等到 180 天銷售獨占期間經過始得許可其他學名藥上市,第一位 申請者得將學名藥之上市銷售首日「無限延期」,此舉將導致其他學名藥無法進 入市場之時間延長,或甚至無法進入市場36;至於學名藥廠尚未取得有利判決的 情況,若僅因 30 個月期滿即給予包含 180 天銷售獨占權的上市許可,FDA 亦認 為學名藥廠會容易受到原開發藥廠的利誘而共同聯手阻止或延後其他學名藥廠 進入市場的時間37。
然此規則卻被法院38認為嚴重違背 Hatch-Waxman Act 鼓勵學名藥儘早進入 市場的立法意旨,因此最終 FDA 取消「成功防禦原則」之規定39 40,故自此之 後,只要原開發藥廠或專利權人未於接到通知後 45 日內對學名藥廠起訴,或 30 個月停止發證期屆至,即使並未存有對第一家提出 Paragraph IV 申請之學名藥廠 有利之判決,FDA 仍可逕行核准第一位申請者之 ANDA 並給予 180 天銷售獨占 權。學名藥廠於取得藥證後即可將其學名藥品上市,惟其亦可等待專利侵權訴
35FDA 採成功防禦原則的時點是 2003 年修法之前,因此當時仍適用「以法院判決起算」
(court-decision trigger)的 180 天銷售獨占期間起算方式。
36John R. Thomas, supra note 10, at 440-441.
37FTC, Generic Drug Entry Prior to Patent Expiration: An FTC Study 58-59 (2002).
38Mova Pharmaceutical Corp. v. Shalala, 140 F.3d 1060, 46 USPQ 2d 1385 (D.C. Cir. 1998).
39FTC, Generic Drug Entry Prior to Patent Expiration: An FTC Study 58-59 (2002); John R. Thomas, supra note 10, at 440.
40關於 FDA 原認為第一個 Paragraph IV 之 ANDA 申請人若未於專利侵權訴訟中盡一切努力取得 有利判決,將可能容易與原開發藥廠聯手延後或阻止其他學名藥廠進入市場的憂慮,已在 2003 年修法後透過 180 天銷售獨占權失權事由之增定加以處理。
18
訟之法院判決作成或是提起「確認之訴」41,以確認原廠藥相關專利之有效性或 其提出 ANDA 之行為是否構成專利侵權,否則其若貿然於專利侵權爭訟結果未 定或不知是否構成侵權的情況下將學名藥上市,一旦專利侵權訴訟敗訴或學名 藥上市後才被控侵權,因學名藥廠已將侵權產品上市販售,將可能必須面對鉅 額的損害賠償,但其等待期間仍須注意不要造成失權事由之成就(就失權事由 之論述,詳參「(五)180 天銷售獨占權之失權事由」)。
(四)180 天之起算時點
180 天銷售獨占權之起算時點,依現行 21 U.S.C. 355 (j)(5)(B)(iv)規定,係
「FDA 核准上市後的銷售首日」42,即 ANDA 申請人於獲得含有 180 天銷售獨 占權之上市許可後,將該學名藥上市之首日。180 天的銷售獨占權之起算時點於 2003 年修法前曾發生爭議,但修法後僅餘「FDA 核准上市後的銷售首日」一種 起算時點,過去的爭議已告終結。關於此修法前之爭議詳參第參章第一節第五 項之說明。
(五)180 天銷售獨占權之失權事由
為避免原開發藥廠為了維持自己在市場上的獨占利益而與取得銷售獨占權 的學名藥廠達成協議不將學名藥上市販售,因而導致其他學名藥均無法取得上
41在「原開發藥廠或專利權人未於接到通知後 45 日內對學名藥廠起訴」的情況下,FDA 是否發
給第一位 Paragraph IV 之 ANDA 申請者含有 180 天銷售獨占權之上市許可並不受學名藥廠是否 提起確認之訴影響,但確認之訴之結果卻可能與失權事由起算時點相關,關於失權事由之說明,
詳參「(五)180 天銷售獨占權之失權事由」。
4221 U.S.C. § 355(j)(5)(B)(iv):"if the application contains a certification described in paragraph (2)(A)(vii)(IV) and is for a drug for which a first applicant has submitted an application containing such a certification,the application shall be made effective on the date that is 180 days after the date of the first commercial marketing of the drug (including the commercial marketing of the listed drug) by any first applicant."; John R. Thomas, supra note 10, at 459; Narinde Banait, Authorized Generics:
Antitrust Issues and the Hatch-Waxman Act 3 (Nov.4, 2005), available at
http://www.fenwick.com/fenwickdocuments/authorized_generics.pdf (last visited Dec.10, 2014).
19
市許可的情況,國會於 2003 年特別針對 Hatch-Waxman Act 中的 Paragraph IV 增 加 180 天銷售獨占權之失權事由(forfeiture of 180-day exclusivity period),若符 合失權的要件,學名藥廠之獨占權即失其效力,FDA 將可核發藥證予第二家學 名藥廠及後續其他提出上市申請之學名藥廠43。該失權事由整理如下44:
1. 與學名藥上市時點相關之失權事由45:
若下列兩類時點發生前學名藥仍未上市販售,即產生失權效果,並以發生在後 者作為認定失權的時點:
(1) 下列兩個時點視何者發生於前:
A.取得含有 180 天銷售獨占權之上市許可後,75 天內未將學名藥上市販售;
B.提出上市許可申請後,30 個月內未將學名藥上市販售。
(2) 下列三個時點發生後 75 日內:
A.若法院就專利侵權訴訟或由 ANDA 申請人所提之確認之訴作成對 ANDA 申請人有利之判決(即系爭專利無效或 ANDA 申請人未構成侵權),該判 決因當事人均未上訴而登錄為終局確定判決;
B.專利侵權訴訟或確認之訴之當事人達成經法院簽署之相關專利無效或學名 藥廠不侵權之和解命令或雙方合意之判決(a court signs a settlement order
43李素華,從公共衛生之觀點論醫藥專利權之保護與限制,國立臺灣大學法律學研究所博士論
文,頁 49,2006 年;Narinde Banait, supra note 42 , at 3;Matthew Avery, Continuing Abuse of the Hatch-Waxman Act by Pharmaceutical Patent Holders and the Failure of the 2003 Amendments, 60 HSTLJ 171, 186 (2008).
4421 U.S.C. § 355(j)(5)(D).
4521 U.S.C. § 355(j)(5)(D)(i)(I).
20
or consent decree),該和解命令或判決並被登錄為終局判決(enters a final judgment)46;
C.原開發藥廠撤回相關專利之登錄。
茲舉一例說明之,若 A 學名藥廠於 2014 年 1 月 1 日提出 Paragraph IV 之 ANDA 申請,提出申請後之 30 個月為 2016 年 6 月 30 日。設若 FDA 於 2016 年 5 月 1 日發給 A 學名藥廠含有 180 天銷售獨占權之上市許可,那麼即自 2016 年 5 月 1 日起算 75 日,即 2016 年 7 月 15 日,惟因提出申請後之 30 個月時間為 2016 年 6 月 30 日,早於 2016 年 7 月 15 日,因此即以 2016 年 6 月 30 日作為第 一類時間之基準點,接著再以之與第二類時間比較何者為後。若原開發藥廠於 2016 年 5 月 10 日撤回相關專利之登錄,自此日起算 75 日為同年 7 月 24 日,與 第一類時間 2016 年 6 月 30 日相較之下,2016 年 7 月 24 日晚於 2016 年 6 月 30 日,因此 2016 年 7 月 24 日即為學名藥廠 180 天銷售獨占權之失權效發生時點,
即 A 學名藥廠必須在 2016 年 7 月 24 日前將學名藥上市販售,否則將失去 180 天銷售獨占權。
2. 其他失權事由規定:
(1) 撤回申請(withdrawal of application)47:第一位提出 Paragraph IV 之 ANDA 申 請 者 主 動 撤 回 其 申請 , 或 FDA 之 審 查 者 認 為 該 申 請 者 不 符合 核 發 Paragraph IV 藥證之條件或要求,而作成「要求該申請者撤回其申請」之決 定。
4621 U.S.C. § 355(j)(5)(D)(i)(I)(bb)(BB)“In an infringement action or a declaratory judgment action described in subitem (AA), a court signs a settlement order or consent decree that enters a final judgment that includes a finding that the patent is invalid or not infringed.”
4721 U.S.C. § 355(j)(5)(D)(i)(II).
21
(2) 修改證明(amendment of certification)48:該第一位提出 Paragraph IV 之 ANDA 申請者雖符合取得 180 天銷售獨占權之要件,但其主動修改(例如原提出 Paragraph IV 的申請,後改成 Paragraph I 的申請)或撤回其申請。
(3) 無 法 於提 出申 請後 30 個 月內 取得 暫時 許可 ( failure to obtain tentative approval)49:若第一位提出 Paragraph IV 之 ANDA 申請者未能於申請後 30 個月內取得 FDA 之暫時許可,原則上其將失去 180 天銷售獨占權,即使未 來其仍可能取得上市之許可,但該許可不包含 180 天銷售獨占權;除非未能 取得暫時性許可之原因係因提出上市申請後發生與許可核准與否相關之事 件,始得阻卻此失權條件之成就。
(4) 該提出 Paragraph IV 之 ANDA 申請者與其他申請者、原開發藥廠或專利權人 達成違反競爭法規之協議(agreement with another applicant, the listed drug application holder, or a patent owner)50:若提出 Paragraph IV 之 ANDA 申請 者與其他申請者、原開發藥廠或專利權人簽訂之協議被 FTC 或檢察總長
(Attorney General)起訴主張違反競爭法規,且當事人並未對 FTC 之違反競 爭法規之終局決定主張不服,或該訴已由法院做出違反競爭法規之終局判決 且當事人未上訴。
(5) 第一位提出 Paragraph IV 之 ANDA 申請者於證明中所提之所有登錄於橘皮書 上之相關專利期間均屆至(expiration of all patents)51。
若第一位成功取得 Paragraph IV 上市許可者之 180 天銷售獨占期間屆至,或 失權條件成就,FDA 即可核發上市許可給後續提出申請之學名藥廠,惟後續取 得許可之學名藥廠也無法擁有 180 天銷售獨占權,因此將可能發生多家學名藥
4821 U.S.C. § 355(j)(5)(D)(i)(III).
4921 U.S.C. § 355(j)(5)(D)(i)(IV).
5021 U.S.C. § 355(j)(5)(D)(i)(V).
5121 U.S.C. § 355(j)(5)(D)(i)(VI).
22
廠同時取得上市許可的情況。
第三項、30 個月停止發證期
學名藥廠之所以在原廠藥之專利保護期間內對該原廠藥提出專利權無效或 未侵害專利權的挑戰,係為了取得提前進入市場的優勢與伴隨該優勢而來的利 益,此舉對於原開發藥廠的影響甚鉅,因此在學名藥廠向 FDA 提出 Paragraph IV 的上市申請並獲得 FDA 許可進行審查後,申請人即應在 20 日內通知原開發藥 廠與專利權人52,受通知者必須在受通知後的 45 天內決定是否對該學名藥廠提 出侵害專利權的訴訟,一旦原開發藥廠或專利權人在 45 天期間內起訴,FDA 對 於學名藥的上市核准將依 Hatch-Waxman Act 自動延後至法院作出判決為止,該 停止期間最長達 30 個月(30-month stay)53,30 個月停止發證期之起算時點為 原開發藥廠或專利權人接獲通知之日54。如此一來,在涉及 30 個月停止發證期 的情況下55,學名藥廠除非在「30 個月停止發證期屆滿」或是「在 30 個月期間 內,法院作出對學名藥廠有利的判決」始可能獲得 FDA 之含有 180 天銷售獨占 權上市許可56;至於原廠藥之專利保護期間若於 30 個月停止發證期內屆至,學 名藥廠仍可取得上市許可,惟其無法獲得 180 天之銷售獨占權,若此時有多家 學名藥廠提出上市申請,FDA 得准許多家學名藥廠之學名藥上市。
有認為 30 個月停止發證期其實如同法院的禁制令57,因為在這段期間內,
學名藥廠不可能取得藥證,原開發藥廠得繼續獨占市場,此依法自動停止核發
5221 U.S.C. § 355(b)(3)(Section 505 (b)(2) application); Id. 21 U.S.C. § 355(j)(2)(B)(ANDA).
53蘇建智,藥品查驗登記制度與智慧財產權保護,東吳大學法律學系法律專業碩士班碩士論
文,頁 15,2004 年。
5421 U.S.C. § 355(c)(3)(C)(Section 505(b)(2)application); Id. § 355(j)(5)(B)(iii)(ANDA).
55另外,在學名藥廠提出 Paragraph IV 之 ANDA 申請並通知原開發藥廠或專利權人,經過 45 天 原開發藥廠或專利權人未起訴學名藥廠的情況下,學名藥廠亦可在 FDA 就 ANDA 審核完成後 取得 180 天銷售獨占權。
56李素華,同前註 43,頁 11。
57John R. Thomas, supra note 10, at 17.
23
藥證之規定,對於原開發藥廠而言甚至比獲得法院之禁制令更為有利,原開發 藥廠完全無庸對法院提出任何聲請,30 個月停止發證期於其接獲學名藥廠提出 Paragraph IV 證明,並對學名藥廠提出專利侵權訴訟時即自動開啟,專利連結制 度因伴隨著 30 個月停止發證期,原開發藥廠可能因此獲有利益。針對 30 個月 停止發證期,其實在 2003MMA 施行前,曾存有原開發藥廠透過獲得多次 30 個 月停止發證期的方式擴張其專利權權利保護範圍之問題58,因而引發爭議與批 評,此將於第參章第一節第三項詳細探究之。
第四項、Section 505(b)(2)之上市申請(Paper NDA)
59新藥研發流程(參下列圖 2)之初始係從約 5000 至 10000 種化合物
(compounds)中篩選出新的有效標的化合物或新成分,接著進行臨床前試驗
(preclinical testing),於此階段透過電腦模擬與動物試驗,找出對抗目標疾病 的化合物,此階段所篩選出的化合物將降至約 250 種,經過完整的臨床前試驗 階段以後,藥廠即可向 FDA 提出進行人體臨床試驗的申請,藥廠必須提出臨床 試驗階段進行的計畫、以及前階段生物作用、藥品療效、藥理試驗與毒性試驗 報告的文件,由人體試驗委員會(Institutional Review Board,簡稱 IRB)審核通 過後始得開始進行人體臨床試驗(clinical trials),進入人體臨床試驗階段的化 合物或成分約僅剩 5 種,經過人體試驗階段之藥品安全性、療效等檢驗後,藥 廠最終將對 FDA 提出 1 種化合物或成分之 NDA 上市許可申請60。
整個藥品研發的流程平均耗時可長達 15 年,甚至更久,期間的研發費用則 高達 10 幾億美元。然除了最終提出 NDA 的 1 種化合物或成分外,整個藥品研
58John R. Thomas, supra note 10, at 391.
59在 Hatch-Waxman Act 立法前稱為 Paper NDA,立法後改列於 Hatch-Waxman Act 之 Section 505 (b) (2)而得名。
60Pharmaceutical Research and Manufacturers of America, The Research and Development Process, available at http://www.phrma.org/research-development-process (last visited Dec. 10, 2014).
24
發過程亦對候選之化合物或成分投入高額的研發成本,並取得試驗之數據或資 料仍具有應用價值,若後續有人擬進行候選化合物或成分之研發,似無庸使之 重新進行 NDA 申請之試驗流程,現有的試驗成果應得被更充分的應用,在此種 想法下產生了 Section 505(b)(2)的上市申請程序。
Section 505(b)(2)申請係針對與 NDA 產品有些微差異者,因此,其不歸類為 NDA 產品也不歸類為 ANDA。一般而言,Section 505(b)(2)申請案用於下列產品 的上市許可61:
新劑型(new dosage formulation):例如已上市藥品為膠囊,Section 505(b)(2) 申請為錠劑。
新適應症(new indication):例如已上市藥品治療病況 A,Section 505(b)(2) 申請為治療病況 B。
改變活性成分劑量(change strength of the drug substance):例如已上市藥品 為 20 毫克錠劑投藥 2 個錠劑,Section 505(b)(2)申請為單一 40 毫克錠劑。
新投藥途徑(changed route of administration):例如已上市藥品為吸入投藥,
而 Section 505(b)(2)申請為非吸入投藥。
改變投藥療程(changed dosing regimen):例如已上市藥品為每日 3 次,而 Section 505(b)(2)申請為每日 1 次。
不同活性成分(different active ingredient):例如改變鹽、酯或異構物的種類。
取代組合藥品中的一成分為另一成分(swap of one active ingredient in a
61Shashank Upadhye, Generic Pharmaceutical Patent and FDA Law 566-567 (2014 ed.).
25
combination drug for another):例如已上市藥品為 X+Y 的組合,而 Section 505(b)(2)申請以 Z 取代 Y。
要求從處 方藥變 更為 非處方藥 (request to switch from prescription to over-the-counter (OTC) status)。
Section 505(b)(2)申請可基於部分上市產品的已公開資料,這也是 Section 505(b)(2)申請曾被稱為 Paper NDA 之原因;然而,其也必須提出申請上市產品 不同於 NDA 申請的之安全性與療效資料。上開已公開資料的來源包括(1)已公開 的資訊,例如討論過去已進行的臨床試驗、動物試驗等的文獻;(2)FDA 過去已 發現的安全性及療效資料,但申請人嘗試由這些過去已發現的安全性及療效資 料作改變62。
關 於 Section 505(b)(2) 申 請 所 須 提 出 的 證 明 或 聲 明 規 定 於 21 U.S.C.
§355(b)(A)與§355(b)(B),前者為四種證明,後者為切割受專利保護使用方法之 聲明,其內容與學名藥 ANDA 之 Paragraph I-IV 證明與 section viii 聲明之內容完 全相同。簡言之,雖然學名藥之 ANDA 與 Section 505(b)(2)申請提出產品之「性 質」並不同,但簡化之程序實際上係相同的,後續關於提出 Paragraph IV 證明之 通知義務、通知之法定期限為 20 日、遭原開發藥廠或專利權人以專利侵權為由 起訴之 30 個月停止發證期、以及提起反訴(登錄不實的救濟)與確認之訴(原 開發藥廠或專利權人未於接獲通知後 45 日內起訴)之規定均相同63。
62Id. at 568.
63John R. Thomas, supra note 10, at 382-383;
關於 Section 505(b)(2)申請是否有四種證明/聲明之適用、Paragraph IV 證明之通知義務、通知之 法定期限為 20 日、30 個月停止發證期等規定之適用,詳參以下法條:
1. 四種證明/聲明:21 U.S.C. § 355(b)(2)(A); Id. § 355(b)(2)(B) 2. Paragraph IV 證明之通知義務:21 U.S.C. § 355(b)(3)(A) 3. 通知之法定期限為 20 日:21 U.S.C. § 355(b)(3)(B) 4. 30 個月停止發證期:21 U.S.C. § 355(c)(3)(C)
5. 提起反訴:21 U.S.C. § 355(c)(3)(D)(ii),關於反訴之說明,詳參本章第二節第三項「二、消 除登錄(Delisting)之程序」。
6. 提起確認之訴:21 U.S.C. § 355(c)(3)(D)(i),關於確認之訴之說明,詳參第參章第一節第六
26
惟應注意,自 21 U.S.C. 355 之體系觀之,由於 180 天銷售獨占權僅於 21 U.S.C. §355(j) Abbreviated new drug applications 規範64,因此 Section 505(b)(2)之 申請應無 180 天銷售獨占權相關規定之適用,故即使以 Paragraph IV 證明提出 Section 505(b)(2)申請,該申請獲得 FDA 核准後仍無 180 天銷售獨占權,180 天 銷售獨占權係 ANDA 申請者所獨有。Section505(b)(2)之申請性質上實為「新 藥」,因此其雖無 ANDA 之 180 天銷售獨占權規定之適用,若其申請人成功取得 上市許可,該申請人仍可享有與新藥申請人相同之待遇,例如同新藥申請人享 有專屬權65,即新成分新藥享有 5 年專屬權、孤兒藥享有 7 年專屬權、含有先前 已核准的活性成分但有新的臨床研究的藥品享有 3 年專屬權及小兒科用藥享有 原專屬權再加 6 個月的專屬權。
圖 2:新藥研發流程示意圖66
在對於專利連結制度有一個簡要、全面的認識後,接下來將特別針對橘皮 書之登錄進行詳細的說明。
項。
64至於 180 天銷售獨占權,僅於 21 U.S.C. § 355(j)(5)(iv)規範之,在 Section 505(b)(2)申請之相關 條文無法找到相同或類似之規定。
65Shashank Upadhye, supra note 61, at 563.
66Pharmaceutical Research and Manufacturers of America, The Research and Development Process, available at http://www.phrma.org/research-development-process (last visited Dec. 10, 2014).
27
第二節、美國專利連結橘皮書登錄制度相關法規範、實施 現況及執行情形
新藥申請人提出 NDA 申請並經核准後,FDA 會將相關專利資訊登錄於橘皮 書,除了相關專利資訊外,尚有藥品安全性與療效等資訊。2005 年後,FDA 亦 每年出版橘皮書(Annual Edition)及每月補充版(Cumulative Supplement)的文 本及電子檔,每年的橘皮書即係 FDA 詳列所有藥品之安全性及療效資訊,而每 月補充版則提供 FDA 新核准藥品、藥品之相關修正、補充治療相等性(therapeutic equivalence)、專利資訊更新及專屬權等資料,另外也建構得以電子方式搜尋的 網站67,可用活性成分(Active Ingredient)、商品名稱(Proprietary Name)、申請 人姓名(Applicant Holder)、申請號(Application Number)及專利(Patent)等 條件搜尋,該網站每日更新學名藥及相關專利資料,每月更新新藥之資料68。
第一項、可登錄的事項及活性成分的認定
一、Hatch-Waxman Act
根據 21 U.S.C §355 (b)(1)(G)69規定,NDA 申請人應向 FDA 提出申請上市藥 品之相關專利,以供 FDA 登錄於橘皮書,且根據法條文義,申請人是「應」
("shall")向 FDA 提出相關專利之專利號碼與專利到期日,在 AaiPharma Inc. v.
Tompson 判決(關於此判決詳參本節第二項)中,法院亦認為,根據法條文義,
67FDA,Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations, http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm(網站頁面詳參附件 2)
68FDA, Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book), available at http://www.fda.gov/Drugs/InformationOnDrugs/ucm114166.htm (last visited Dec.14, 2014);廖柏 豪,同前註 4,頁 7-8。
6921 U.S.C. § 355(b)(1)(G):"…the applicant shall file with
the application the patent number and the expiration date of any patent which claims the drug for which the applicant submitted the application or which claims a method of using such drug…"
28
NDA 持有人有「義務(obligation)」登錄相關專利70,而 FDA 的責任則在於公 開 NDA 持有人所提供之專利資訊71。雖然依法條文義 NDA 持有人有登錄相關 專利之義務,但是依現行法並無關於未盡登錄義務之處罰72,可能產生的法律效 果也僅在與 Paragraph IV 相關之專利侵權訴訟發生時,原開發藥廠或專利權人無 法以未登錄專利所涵蓋之藥品取得 30 個月停止發證期的利益而已。
關於何者屬應登錄之專利,21 U.S.C §355 (b)(1)(G)73規定之要件為:(1)請求 該提出上市申請之藥品或藥品使用方法之專利,且該專利為(2)若有人在未取得 專利授權的情況下就該藥品為製造、使用或銷售,該專利之相關權利人因此得 主張專利侵權。因此,符合上述兩要件者即屬應登錄之專利。
二、FDA 制定登錄規則
(一)制訂登錄規則之背景
雖然 Hatch-Waxman Act 就橘皮書之登錄有所規範,但實務運作上尚無法據 以進行登錄,因此 Hatch-Waxman Act 授權 FDA 制訂登錄規則。1994 年 10 月 3 日,FDA 制定新藥申請相關規則,其中即包括所有新藥申請人或 NDA 持有人應 提供之藥品相關專利資訊。在該規則中,FDA 即闡明專利連結之目的在於-禁 止主管機關於該藥品之所有相關專利失效或保護期間屆至前為學名藥之藥證核 准,除非學名藥廠能夠證明其學名藥未侵害專利權或該專利權無效74。
在 2003 年 6 月 18 日,FDA 發布新登錄規則,就登錄事項方面,主要在於
70AaiPharma Inc. v. Tompson, 296 F.3d 227, 235 (Fed. Cir. 2002).
71Id. at 234.
72Natalie M. Derzko, The Impact of Recent Reform of the Hatch-Waxman Scheme on Orange Book Strategic Behavior and Pharmaceutical Innovation, 45 IDEA 165, 221 (2005).
7321 U.S.C. § 355(b)(1)(G):"…any patent which claims the drug for which the applicant submitted the application or which claims a method of using such drug and with respect to which a claim of patent infringement could reasonably be asserted if a person not licensed by the owner engaged in the manufacture, use, or sale of the drug."
74Abbreviated New Drug Application Regulations; Patent and Exclusivity Provisions, 59 Fed. Reg.
50363 (October 3,1994), available at
http://www.gpo.gov/fdsys/pkg/FR-1994-10-03/html/94-24052.htm (last visited Dec. 15, 2014).
29
透過明訂應登錄專利及不應登錄專利限縮登錄之範圍,並提出更詳細之專利登 錄宣誓聲明,該聲明係用以宣誓藥品相關專利之登錄均合乎法定要求,倘有不 實願接受刑事處罰,以減少不當登錄之情事,促進公平競爭75。其後並有多次修 正,以下就最新版76之登錄規則為析論。
(二)登錄規則相關內容
1. 表 3542、3542a 須登載之項目
FDA 發布 21 C.F.R. §314 規則,詳列各新藥提出 NDA 上市申請所應注意之 事項(Application For FDA Approval To Market A New Drug)。其中第§314.53(a) 規定,向 FDA 提出新藥上市申請之申請人必須提出專利資訊,俾便 FDA 得於 橘皮書登錄專利資訊。第§314.53(b)進一步規範必須提出及不能提出之專利資 訊;而第§314.53(c)規定,提出新藥上市申請(NDA)或 Section 505(b)(2)77申請之 人,必須以表 3542、3542a 提出所應提出之資訊。
表 3542 中,須提出新藥申請號、新藥試驗委託者名稱、新藥品名、新藥活 性成分、新藥劑量(strength)、新藥劑型(dosage form)、新藥核准日、美國 專利號、公告日(issue date)、到期日(expiration date)、專利權人基本資料、
代表人基本資料。若提出上市申請之新藥為原料藥(drug substance;即活性成 分)、藥品(drug product;即調配物(formulation)或組合物(composition))
或使用方法(method of use),申請人須提交表 3542a;復按§314.53(ii)之規定於 該新藥上市申請或 Section 505(b)(2)申請核准後 30 日內,申請人須再提出表 3542,FDA 將根據表 3542 進行橘皮書登錄,若該專利資訊之聲明不完整,或經 FDA 形式審查後認非屬得登錄之適格專利,FDA 將不登錄此類專利資訊。
若有相關專利權係於提出 NDA 申請後、獲得 FDA 核准前取得,該 NDA 申 請人須於取得該專利權後 30 日內向 FDA 就該申請案以表 3542a 提出相關專利
75Natalie M. Derzko, supra note 72, at 214-215.
76更新日期為 2014 年 4 月 1 日。
77有關 Section 505(b)(2)申請之說明,詳參第貳章第一節第二項。
30
資訊更正之修正78;在 FDA 核准 NDA 申請後,若該 NDA 持有人擬變更調配物
(formulation)、增加新的適應症(包括改變用藥的方式)、改變劑量,或就關 於該藥(drug)、藥品(drug product)或任何使用方法(method of use)有任何 變更,均須向 FDA 提出相關專利資訊之補充(Supplement)79,該補充仍須以 表 3542a、表 3542 提出。
2. 應登錄之專利
申請人須以宣誓書方式提出專利資訊。至於應登錄之專利則包括:
(1) 原料藥專利(drug substance patent),即活性成分(active ingredient):
申請人應僅提出請求與新藥上市申請案(申請中或已核准)中活性成分相同 的原料藥專利。關於請求與新藥上市申請案中活性成分相同之多形體(polymorph) 專利,申請人應該在宣誓書中保證申請人具有試驗數據,說明含該多形體的藥 品產品的表現與新藥上市申請案中的藥品產品相同。所謂多形體係相同活性成 分 但 具 不 同 物 理 型 態 的 物 質 , 其 包 括 具 不 同 結 晶 結 構 、 不 同 水 合 物 (water-of-hydration)、溶劑化物及無晶型式的化合物80。
至於一化合物之異構物(鏡像異構物或立體異構物)、鹽類或酯類專利81 82, 在實務上亦可登錄於橘皮書,其條件為該專利之請求項請求(claim)藥品且專 利持有人聲稱專利為真83。
7821 C.F.R. § 314.53(d)(1).
7921 C.F.R. § 314.53(d)(2).
80Applications for FDA Approval to Market a New Drug: Patent Submission and Listing Requirements and Application of 30-Month Stays on Approval of Abbreviated New Drug Applications Certifying That a Patent Claiming a Drug Is Invalid or Will Not Be Infringed, 68 Fed. Reg. 36678 (June 18, 2003).
81關於鹽類與酯類專利,在很多情況下,藥的活性成分(active pharmaceutical ingredient,簡稱 API)是無法單就其本身為使用,其必須與其他分子結合始可使用,而該與活性成分結合之分子 有助於穩定活性成分(stabilize the API)或調配活性成分(formulate the API),因此專利權人 可能會以活性成分與分子之結合提出專利申請,例如化合物之鹽類、酯類專利即屬之。參自 Shashank Upadhye, supra note 61, at 16.
82就鹽類、酯類專利可否登錄於橘皮書,此乃實務操作問題,Shashank Upadhye 所著之法規釋義
書籍(Generic Pharmaceutical Patent and FDA Law)也未說明鹽類、酯類及化合物之異構物(鏡 像異構物或立體異構物)專利可否登錄。
83此為詢問美國實務律師之結論。
31
(2) 藥 品 產 品 專 利 (drug product patent) : 調 配 物 與 組 合 物 (formulation and composition)
申請人應僅提出請求新藥上市申請案(申請中或已核准)中之藥品產品的專 利。實務上,該藥品產品專利可涵蓋各種組合物(composition)、組合(combination) 或製劑(preparation)專利;其為特定的藥品製劑,包括原料藥/藥品活性成分及 其他化合物,例如其他活性成分、安定劑、溶劑或 pH 緩衝劑等。
(3) 使用方法專利(method-of-use patent):
申請人應僅提出描述於新藥上市申請案(申請中或已核准)中的適應症或其 他使用情況的專利。申請人應分別指明每ㄧ申請案中的使用方法及其相關的專 利。針對已核准的申請案,提出使用方法專利的申請人應在核准標籤(label)
中特別指出對應到專利的使用方法。
(4) 方法界定物(product-by-process)專利:
方法界定物專利亦可登錄,但有其限制,方法界定物專利登錄的條件為「須 提出相關資料證明該物具有新穎性」,蓋就方法界定物而言,其仍為物的專利,
具新穎性者理應為該「物」而非該方法;而該規則亦規定得登錄者僅限於方法 界定物,排除製成該藥品方法專利之登錄。
(5) 醫療器材專利:
至於醫療器材專利,亦可登錄於橘皮書,惟其條件限於醫療器材中的活性 成分或藥品產品為新藥上市申請案(申請中或已核准)中之活性成分或藥品產 品。美國 FDA 認同藥品遞送裝置及其相關的包裝(為 NDA 核准事項的一部份) 係核准的藥品產品不可或缺的部份,因此,在實務上認為符合藥品產品之定義
84。如判斷醫療器材專利是否可登錄於橘皮書中,須先判斷是否該專利請求藥物
84FDA, Draft Guidance For Industry: Bioavailability And Bioequivalence Studies For Nasal Aerosols And Nasal Sprays For Local Action 7 (2003); FDA, MDI Draft Guidance 157-59 (1998) (‘‘MDIs and