國 立 交 通 大 學
材料科學與工程學系
博 士 論 文
超級電容器與空氣電極材料製備分析及其電化學性質研究
Materials Synthesis, Characterization and Electrochemical
Analysis for Supercapacitor and Gas Diffusion Electrode
研究生: 吳成有
指導教授: 林 鵬教授
吳樸偉副教授
超級電容器與空氣電極材料製備分析及其電化學性質研究
Materials Synthesis, Characterization and Electrochemical
Analysis for Supercapacitor and Gas Diffusion Electrode
研究生: 吳成有
Student: Cheng-Yeou Wu
指導教授: 林 鵬教授
Advisor: Professor Pang Lin
吳樸偉副教授
Associate professor Pu-Wei Wu
國 立 交 通 大 學
材料科學與工程學系
博 士 論 文
A Thesis
Submitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University In Partial Fulfillment of the Requirements
For the Degree of Doctor of Philosophy
in
Materials Science and Engineering November 2010
Hsinchu, Taiwan, ROC
I
摘要
本研究進行了超級電容器以及空氣電極材料的製備、鑑定,以及電化學性能 的分析量測。我們設計了一種能鈦金屬孔罅電極 (Titanium Cavity Electrode; TCE) 來加速測詴程序的進行,該電極只需要使非常少量的材料樣品,尌能夠迅速地呈 現出詴樣本質的電化學特徵。因此,能夠清楚地用來評估該材料使用於超級電容 器的可行性。所研究的材料除了奈米碳膠囊 (Carbon NanoCapsules; CNCs) 之外, 尚包括了可取得的商業化碳黑產品,諸如 BP2000 以及 Vulcan XC72R 等。另外,
我們也利用 RuO2‧xH2O-CNCs 和奈米顆粒之 Ag-CNCs 材料製成電極獲得了偽
電容 (pseudocapacitance) 和空氣電極的相關性質。
TCE 是從原始設計用來量測粉體材料電化學性質的孔罅微電極 (Cavity Microelectrode; CME) 改良而成的一種電極結構,TCE 不僅保有使用微量的粉體 之優點又能精確地控制粉體重量。因此,TCE 的實驗再現性令人滿意,能用來 有效地過篩出可供選用的碳基材料。在所進行研究的幾種碳材之中,CNCs 具有 糾結相通的石墨烯層以及中空結構,意味著將具有較佳的導電性以及高質量密度 特質,這是讓我們特別感與趣。CNCs 是在缺氧的氣氛之下,使用乙炔和氧的混 合氣體以火焰燃燒法所製備而成的奈米材料。初合成的 CNCs 直徑約為 10~25nm 之間 ,BET 比表面積大約 300 m2 /g。經過適當的處理之後,表面積可提高到 2019 m2/g 且中巨孔比表面積佔 92.6% 。實驗結果發現,在 1N H2SO4電解液中其比電 容值落於 60 ~200 Fg-1範圍。 以共沈澱法所製備之 RuO2〃xH2O/CNCs,於 1N H2SO4電解液中比電容值可 達到 490 F/g。然而,不管在大氣中或者是在水熱環境中進行後續處理,其循環 壽命依然呈現出明顯衰減的情形。Ru-Ta/Ti 電極中的 Ru-Ta 二元複合氧化物材料 其比電容性能的表現象優於純 Ru 氧化物。其中金屬元素莫耳比為 8:2(Ru〆Ta) 時所製備之氧化物塗層具有最高的比電容值,其比電容值約為 350 F/g。RuTa 二
II 元複合氧化物電容材料與碳簇電容材料最大的差異在於其更為優異的導電度以 及高功率充放電性質。相反地,PANI/Ti 具有很高的比電容值,在低掃描速率時 甚至可達到 600 F/g 以上,但卻受到掃描速率的影響非常顯著,循環壽命也短。 這意味著 PANI 電容材料雖然具有高比電容值,但在仍侷限於法拉第反應的限制, 並不適合在高功率的應用。 以 BP2000 商品、經表面改質處理之後的自製 CNCs 材料、RuO2.xH2O/CNCs 複合材料等電容材料,添加 2.5% 石墨為導電材料、10% PTFE 及 2.5% PVA 為 粘著劑為能夠兼顧電極結構強度、電解液親和性以及導電性能的配方組合。所製 備 4x5 cm 尺寸的二極式超級電容器,其性質相較於可取得市售超級電容性商品 在等效串聯電阻值(Equivalent Series Resistance; ESR)及自放電率兩個參數遠優於 自製的電容器,不過在單位面積比電容及洩漏電流性能上自製的電容器則有較佳 的表現。
III
Abstract
This study is concerned with the synthesis, characterizations, and electrochemical analysis of materials for supercapacitors and gas diffusion electrodes applications. To expedite the testing process, we design a titanium cavity electrode (TCE) that employs a relatively small amount of samples for quick determination on essential electrochemical parameters so their prospects on supercapactors can be evaluated fairly. The samples under study include carbon nanocapsules (CNCs) and commercially available carbon blacks such as BP2000 and Vulcan XC72R. In
addition, we prepare electrodes made of RuO2‧xH2O-CNCs and nanoparticulate
Ag-CNCs and obtain relevant properties for pseudocapacitance and gas diffusion electrodes.
The TCE is a modified version of cavity microelectrode, which is originally designed for determining the electrochemical properties of powders. The TCE not only retains advantages of using a minute amount of powders but also provides a better control in sample weighting. Therefore, the TCE enables impressive experimental reproducibility for efficient screening of promising carbonaceous candidates. Among many materials under studies, the CNCs are of particular interests because it composes entangled grapheme layers encapsulating a hollow core which infers better electrical conductivity and higher energy density per unit mass. The
CNCs are prepared by a flame combustion method using a mixture gas of C2H2 and
O2. The diameter for the as-synthesized CNCs is in the range of 10–25 nm with a
BET surface area of 300 m2/g. After appropriate treatments, the surface area can be
further increased to 2019 m2/g with mesopores surface area of 92.6%. As a result, the
specific capacitance in 1 N H2SO4 electrolyte is ranged between 60 to 200 F/g.
The RuO2‧xH2O-CNCs is synthesized from the coprecipitation method which
reveals a capacitance of 490 F/g in 1 N H2SO4 electrolyte. However, the
RuO2‧xH2O-CNCs demonstrates serious decline in life cycle regardless of
post-treatments in air or hydrothermal environments. In contrast, the specific capacitance of Ru-Ta binary oxide compound in the Ru-Ta/Ti electrode is much superior to pure Ru oxides. With a molar ratio of 8:2 (Ru:Ta), the sample displays the highest specific capacitance around 350 F/g. We realize that the notable advantage of Ru-Ta binary compound oxide over conventional carbonaceous capacitor materials is its improved electrical conductivity enabling facile charging and discharging in high power mode. In contrast, the PANI/Ti has a high specific capacitance (600 F/g) at relatively low scan rate but its value declines considerably with increasing scan rates along with deteriorating life cycles. This indicates that the contributing current for
IV
capacitance is limited by Faradaic reaction so its adoption in high power applications is rather slim.
The recipes of using BP2000, modified-CNCs, or RuO2‧xH2O-CNCs as a
capacitor material, with the addition of 2.5 wt% graphite for conductive improvement, 10 wt% PTFE and 2.5 wt% PVA to fabricate porous electrodes is able to combine strength, electrolyte affinity and conductive properties. For comparison purpose, the
4x5 cm2 two-pole type supercapacitors are prepared and evaluated along with
commercialized supercapacitor products in both specific capacitance and leakage current. Unfortunately, the ESR and self-discharge rate for our samples are inferior to those from commercial products.
V
誌謝
經過了二十個年頭之後再度重返校園,著實需要很大的勇氣以及背後支持的 力量。年屆五十之齡,記憶力、耐力及視力都面臨著嚴苛的考驗,慶幸的是即將 告一段落。在這四年多的學習歷程中,何其幸運能接受林鵬及吳樸偉兩位老師共 同指導,林鵬老師在課堂中以及實驗規畫上給了我很大的啟發,讓我的多角度思 考邏輯有長足的進歩。吳樸偉老師總是在最關鍵的時刻助我一臂之力,年輕、聰 明、思路清晰、學習能力超強,一直讓我欽佩不已。雲閔、勝結、玉塵、育淇、 筱琳、亮余在實驗及工作聯繫上給了我很大的幫忙,讓我的研究進度順遂許多, 謝謝你們。也感謝實驗室的學長、學弟妹容忍我因工作關係,無法參與常規的會 議,分攤實驗室的責任及義務。 台電公司的長官費副總經理昌仁、劉副所長志放、蒯副所長光陸、涂副所長 世達、邱主任善得以及郭主任淑德,都是我所尊敬的長官,在我必需兼顧課業與 工作之際,給了我完全的信任與支援。綜合研究所的伙伴藍啟仁博士、鄭錦榮博 士、李文台先生,許讚全先生,阿旺、書維、安庭、富彬在我分身乏術之時不吝 伸出援手給予關懷,都讓我銘感五內。最後,一定要感謝的是永遠在背後支持我 的至愛玲蘭,聰明的女兒濡安、貼心的兒子拓遠,以及親愛的家人老媽、兄嫂、 弟妹、俐穎、冠璋、柏潤,你們的支持讓我對一切充滿了信心,戰鬥意志堅強。 ~ 原來可以寫誌謝時,感覺竟是如此的美好! ~VI
目錄
摘要 ... I ABSTRACT ... III 誌謝 ... V 目錄 ... VI 圖表目錄 ... IX 第一章 緒 論... 1 1.1 前言 ... 1 1.2 超級電容器概述 ... 2 1.3 電雙層電容器材料的研究 ... 6 1.3.1 活性碳(Acitvated Carbon) ... 6 1.3.2 碳氣凝膠(Carbon Aerogel) ... 7 1.3.3 碳黑(Carbon Black) ... 9 1.3.4 奈米碳管(Carbon Nanotube) ... 10 1.3.5 碳基材料的活化與改質 ... 11 1.4 偽電容器材料的研究 ... 11 1.4.1 金屬氧化物材料 ... 12 1.4.2 導電性高分子材料 ... 13 1.5 鹼性空氣電池發展概述 ... 15 1.6 鋅空氣電池研究發展概述 ... 16 第二章 分析方法和原理... 21 2.1 粉體比表面積及孔隙結構分析 ... 21 2.1.1 N2 吸附-脫附等溫線 ... 21 2.1.2 BET 比表面積... 22 2.1.3 BJH 中孔孔徑分佈 ... 23 2.1.4 t-plot 法外比表面積和中/微孔孔容積 ... 23 2.2 形貌及化學組成分析 ... 24 2.2.1 掃描式電子顯微鏡 ... 24 2.2.2 穿透式電子顯微鏡 ... 25 2.2.3 X-光繞射分析 ... 25 2.2.4 Raman 光譜 ... 25 2.2.5 FTIR 紅外線光譜 ... 26 2.2.6 XPS 光譜 ... 27 2.3 電化學性能分析 ... 27 2.3.1 循環伏安法 ... 28 2.3.2 交流阻抗法 ... 30VII 2.3.3 恆電流充放測詴法 ... 34 第三章 TCE 電容性質評估技術 ... 41 3.1 前言 ... 41 3.2 實驗方法 ... 41 3.3 結果討論 ... 43 3.4 結論 ... 48 第四章 奈米碳膠囊的製備... 58 4.1 前言 ... 58 4.2 實驗方法 ... 58 4.2.1 CNCs 製備 ... 58 4.2.2 分析方法及使用儀器 ... 59 4.3 結果討論 ... 59 4.3.1 乙炔/氧流量比對奈米碳膠囊孔隙結構的影響 ... 59 4.3.2 乙炔/氧流量比對奈米碳膠囊形貌及化學組成的影響 ... 60 4.3.3 乙炔/氧流量比對奈米碳膠囊電容性質的影響 ... 60 4.4 結論 ... 62 第五章 奈米碳膠囊的改質... 72 5.1 前言 ... 72 5.2 實驗方法 ... 72 5.2.1 CNCs 材料製備 ... 72 5.2.2 CNCs 物理法改質 ... 73 5.2.3 CNCs 化學法改質 ... 73 5.2.4 CNCs 性質量測 ... 73 5.3 結果討論 ... 74 5.3.1 CNCs 物理改質法 ... 74 5.3.2 CNCs 化學改質法 ... 80 5.4 結論 ... 82 第六章 RUO2〃XH2O/CNCS 複合電極材料 ...105 6.1 前言 ... 105 6.2 實驗方法 ... 106 6.3 結果與討論 ... 106 6.3.1 熱重分析 ... 106 6.3.2 XRD 分析 ... 107 6.3.3 SEM 微結構分析 ... 108 6.3.4 電化學性質分析 ... 108 6.4 結論 ... 111 第七章偽電容器電極製備 ...123 7.1 前言 ... 123
VIII 7.2 實驗方法 ... 124 7.2.1 Ti 金屬電極前處理及含貴金屬氧化物塗層製備 ... 124 7.2.2 含貴金屬氧化物塗層製備 ... 124 7.2.3 PANI/Ti 電極製備... 125 7.3 結果與討論 ... 125 7.3.1 RuTaOx /Ti 二元複合電極 ... 125 7.3.2 PANI/Ti 複合電極... 126 7.4 結論 ... 128 第八章 空氣電極製備與電化學性能分析...139 8.1 前言 ... 139 8.2 實驗方法 ... 140 8.2.1 電極材料製備 ... 140 8.2.2 電極製備 ... 142 8.2.3 電極材料分析及電極性能量測 ... 143 8.3 結果與討論 ... 143 8.3.1 塗佈法空氣電極製程 ... 143 8.3.2 滾壓法 GDE 製程... 145 8.4 結論 ... 148 第九章 超級電容器製備及性能量測與分析...160 9.1 前言 ... 160 9.2 實驗方法 ... 160 9.2.1 電極製備 ... 161 9.2.2 超級電容器性能測詴 ... 161 9.3 結果與討論 ... 162 9.3.1 電極製備參數最適化探討 ... 162 9.3.2 超級電容器性能測詴 ... 163 9.4 結論... 165 第十章 總結與未來研究方向...172 參考資料 ...174 攻讀博士學位期間發表論文及專利申請 ...187
IX
圖表目錄
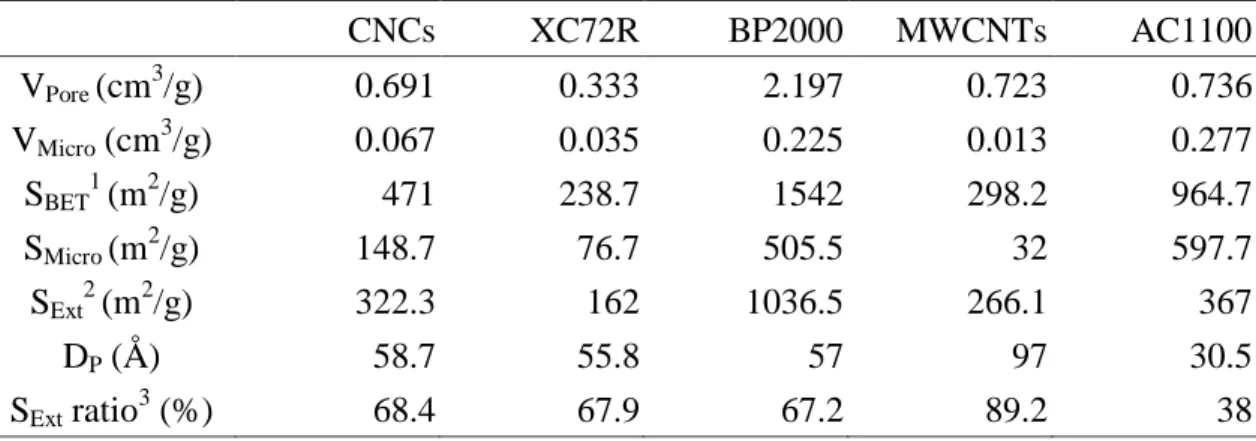
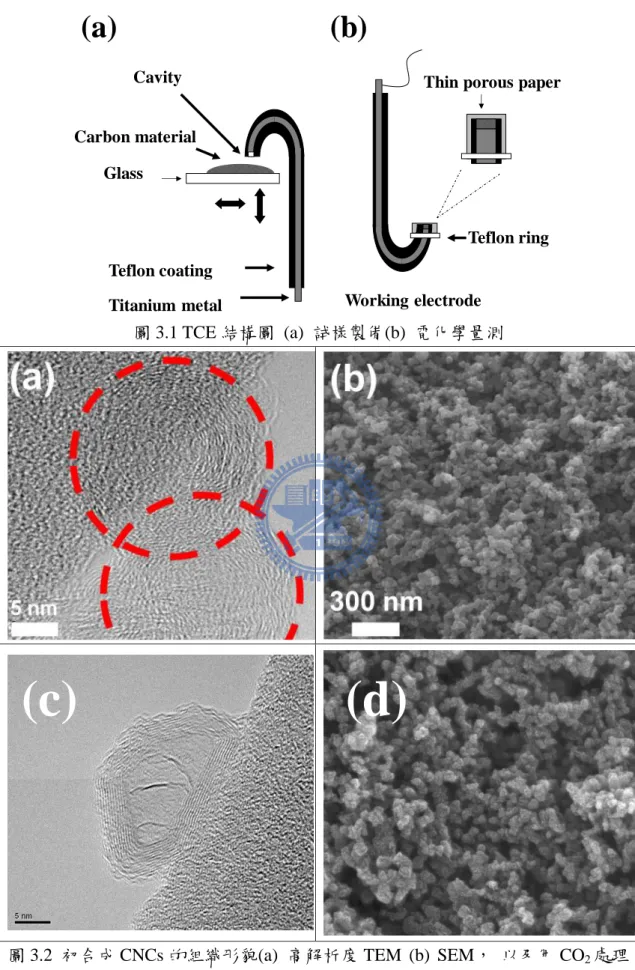
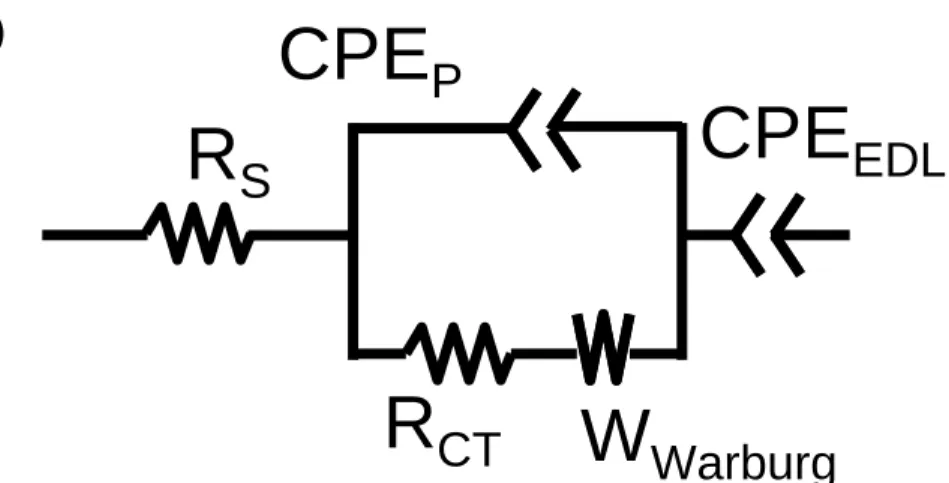
圖 1.1 多孔性碳材固液界面的電雙層結構示意圖... 19 圖 1.2 電雙層電容器工作的原理... 19 圖 1.3 間苯二酚與催化劑莫耳(R/C)與固體成分百分比(M)與碳氣凝膠材組織形 貌的關係... 19 圖 1.4 鹼性燃料電池示意圖... 20 圖 1.5 鋅空氣電池示意圖... 20 圖 2.1 吸附-脫附等溫線類型 ... 35 圖 2.2 碳基多孔性材料所最常見的Ⅳ型 N2 吸附-脫附等溫線 ... 35 圖 2.3 第 n 組的孔半徑為 rpn、Kelvin 孔半徑為 rkn、tn為吸附層的厚度... 36 圖 2.4 電子束與詴片間的交互作用所產生之各種類型電子... 36 圖 2.5 Rayleigh 與 Raman 散射示意圖 ... 36 圖 2.6 碳材表面可能存在的含氧官能基種類... 37 圖 2.7 碳材表面官能基於出 IR 圖譜中相對應的波峰位置及受熱分解產物... 37 圖 2.8 電化學阻抗圖譜概念... 38 圖 2.9 電化學系統的等效電路圖... 38 圖 2.10 電化學系統的等效電路簡化... 38 圖 2.11 典型的電荷轉移控制系統的 Nyquist Plot ... 39 圖 2.12 典型的擴散控制系統的等效電路... 39 圖 2.13 典型的混合控制系統的等效電路... 39 圖 2.14 碳基超電容材料的典型 Nyquist Plot ... 40 圖 2.15 碳基超電容材料的典型 Bode Plot ... 40 圖 3.1 TCE 結構圖 (a) 詴樣製備(b) 電化學量測 ... 51圖 3.2 初合成 CNCs 的組織形貌(a) 高解析度 TEM (b) SEM, 以及用 CO2處理 CNCs 的組織形貌(c) 高解析度 TEM (d) SEM. ... 51 圖 3.3 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 的 N2吸/脫附等溫線 52 圖 3.4 CNCs 於不同掃描電壓範圍之 CV 圖(a) 0-0.6, (b) 0-0.8, (c) 0-1 V,與 未填充碳材之空 TCE 於 0-1V 區間 (d),掃描速率 20 mV/s. ... 52 圖 3.5 CNCs 分別於掃描速率 5, 10, 20, 50, and 100 mV/s 時所得之 CV 圖 ... 53 圖 3.6 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 的 CRC 圖,電壓範圍 0-1 V,電流密度 ±1 A/g ... 53 圖 3.7 CRC 法量測所得 CNCs 比電容值隨循環次數變化情形,0-1V 電壓範圍內, 電流密度 ±1 A/g 進行 5000 次循環... 54
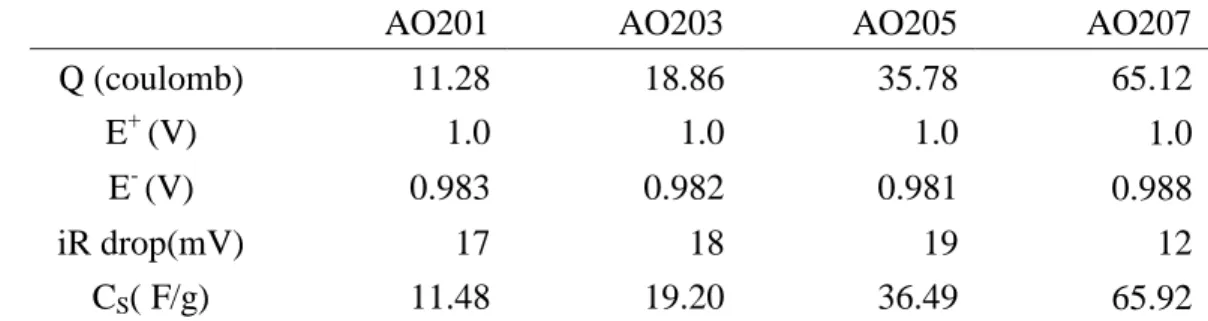
圖 3.8 (a) CNCs、XC72R、BP2000、MWCNTs 和 AC1100 的 Nyquist plots,於 OCP 頻率範圍 0.1 ~20k Hz (b)用以模擬各碳材交流阻抗行為之等效電路 圖,(a)內插圖為 CNCs 交流阻抗圖模擬結果 ... 55
X
圖 3.9 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 的比電容值與 SMicro、SExt
及 SBET表面積的對比 ... 55 圖 3.10 接觸角量測之光學影像(a) CNCs, (b) XC72R, (c) BP2000, and (d) MWCNTs ... 56 圖 3.11 CNCs C1s XPS 圖... 56 圖 3.12 TCE 法與添加 5~10 wt%黏結劑之 TFEs 薄膜電極來量測 CNCs 電化學特 徵比對(a) CV 圖 (b)交流阻抗圖 ... 57 圖 4.1 奈米碳膠囊的製程設備實體照片... 65 圖 4.2 不同氧流量條件所製備奈米碳膠囊之 N2吸-脫附等溫曲線 ... 65 圖 4.3 不同氧流量條件所製備奈米碳膠囊之孔徑分佈... 66 圖 4.4 於不同氧流量條件所製備奈米碳膠囊的 SEM 照片(倍率 50K) ... 66 圖 4.5 奈米碳膠囊 AO207 樣品的 TEM 照片 ... 67 圖 4.6 不同氧流量條件所製備奈米碳膠囊的 XRD 圖譜 ... 67 圖 4.7 所製備的幾種奈米碳膠囊樣品的 Ramann 圖譜 ... 68 圖 4.8 不同氧氣流量所製備的奈米碳膠囊的 TCE 浸置於 1N H2SO4溶液中前 600 秒的開路電壓值... 68 圖 4.9 不同氧氣流量所製備的碳米碳球於 1N H2SO4溶液中的 CV 圖,電壓範圍 0~1 V,掃描速率 20 mV/s ... 69 圖 4.10 AO207 於 1N H2SO4溶液中,在 0-0.6V,0-0.8V 及 0-1V 電壓範圍的 CV 圖,掃描速率 20 mV/s ... 69 圖 4.11 AO207 於 1N H2SO4溶液中在不同掃描速率下的 CV 圖,電壓範圍 0-1V, 20 mV/s ... 70 圖 4.12 v-1/2對 C 圖,材料 AO207 ... 70 圖 4.13 v1/2對 1/C 圖,材料 AO207 ... 71 圖 4.14 AO207 等四種詴樣以±1 A/g 的恆電流充放電測詴所得的電位-時間充放電 曲線... 71
圖 5.1 初合成的 CNCs 於 80%O2+20%N2 (air) 及 CO2環境氣體下之 TGA 圖譜 ... 90 圖 5.2 以 CO2改質之 CNCs 樣品的 N2吸-脫附等溫曲線 ... 90 圖 5.3 以 CO2改質之 CNCs 樣品的孔徑分佈 ... 91 圖 5.4 初合成的 CNCs(a)以及 CC-2H (b)的 SEM 影像 ... 92 圖 5.5 CC-2H 樣品的 C1S XPS 圖譜 ... 92 圖 5.6 CO2處理後 CNCs 樣品的 XRD 圖譜 ... 93 圖 5.7 CO2處理後 CNCs 樣品 10 min 的開路電位 ... 93 圖 5.8 CO2處理後 CNCs 樣品 0~1 V 電壓範圍 CV 圖,掃描速率 20 mV/s ... 94 圖 5.9 CO2處理後 CNCs 樣品 0~1 V 電壓範圍 CRC 圖,電流密度±1 A/g ... 94 圖 5.10 CO2處理後 CNCs 樣品於開路電壓 Nyquist 圖,頻率範圍 0.1~105Hz .... 95 圖 5.11 CO2處理後 CNCs 樣品於開路電壓 Bode 圖,頻率範圍 0.1~105Hz ... 95
XI 圖 5.12 以空氣改質之 CNCs 樣品的 N2吸-脫附等溫曲線... 96 圖 5.13 以空氣改質之 CNCs 樣品的孔徑分佈 ... 96 圖 5.14 以空氣改質之 CNCs 樣品的 SEM 影像... 97 圖 5.15 以空氣改質之 CNCs 樣品的 XRD 圖譜 ... 97 圖 5.16 以空氣改質之 CNCs 樣品 10 min 的開路電位... 98 圖 5.17 以空氣改質之 CNCs 樣品 0~1 V 電壓範圍 CV 圖,掃描速率 20 mV/s.. 98 圖 5.18 以空氣改質之 CNCs 樣品 0~1 V 電壓範圍 CRC 圖,電流密度±1 A/g ... 99 圖 5.19 以空氣改質之 CNCs 樣品於開路電壓 Nyquist 圖,頻率範圍 0.1~105Hz ... 99 圖 5.20 以空氣改質之 CNCs 樣品於開路電壓 Bode 圖,頻率範圍 0.1~105Hz . 100 圖 5.21 以 HNO3改質之 CNCs 樣品的 N2吸-脫附等溫曲線 ... 100 圖 5.22 以 HNO3改質之 CNCs 樣品的孔徑分佈 ... 101 圖 5.23 以 HNO3改質之 CNCs 樣品的 SEM 影像 ... 101 圖 5.24 以 HNO3改質之 CNCs 樣品的 XRD 圖譜 ... 102 圖 5.25 以 HNO3改質之 CNCs 樣品 10 min 的開路電位 ... 102 圖 5.26 以 HNO3改質之 CNCs 樣品 0~1 V 電壓範圍 CV 圖,掃描速率 20 mV/s ... 103 圖 5.27 以 HNO3改質之 CNCs 樣品 0~1 V 電壓範圍 CRC 圖,電流密度±1 A/g ... 103 圖 5.28 以 HNO3改質之 CNCs 樣品於開路電壓 Nyquist 圖,頻率範圍 0.1~105Hz ... 104 圖 5.29 以 HNO3改質之 CNCs 樣品於開路電壓 Bode 圖,頻率範圍 0.1~105Hz ... 104
圖 6.1 CP-20Ru 與 CP-20Ru-H200 樣品之 TGA 圖,N2氣體環境 ... 114
圖 6.2 CP-xRu 樣品之 TGA 圖,空氣環境 ... 114 圖 6.3 CNCs 碳材與 CP-20Ru 複合材料的 XRD 圖譜 ... 115 圖 6.4 CP-20Ru 在大氣環境中分別以 150 ℃,175 ℃及 200 ℃的溫度退火處理 2 小時樣品的 XRD 圖譜 ... 115 圖 6. 5 CP-20Ru 在 175 ℃及 200 ℃水熱環境中退火處理 2 小時樣品的 XRD 圖譜 ... 116 圖 6.6 CP-20Ru 在 175 ℃及 200 ℃水熱環境中退火處理 24 小時樣品的 XRD 圖 譜... 116 圖 6.7 以 SEM 觀察 CP-20Ru 樣品所得之二次電子影像 ... 117 圖 6.8 以 SEM 觀察 CP-20Ru 樣品的穿透電子所得之影像 ... 117 圖 6.9 CP- Ru 樣品在 1NH2SO4電解液中於 0~1 V 的電壓範圍 CV 圖,掃描速率 20 mV/s ... 118 圖 6.10 CP- Ru 樣品在 1NH2SO4電解液中於 0~1 V 的電壓範圍 CV 圖,電流密度 ±1 A/g ... 118
XII 圖 6.11 本研究中所製備的 RuO2〃xH2O/CNCs 複合材料於不同的 Ru 擔量之下測 得的比電容值與其它研究人員所得到的數據的比較(a)複合材料(b)單含 Ru 部份... 119 圖 6.12 CP- xRu Nyquist 圖,DC 偏壓+400mV,頻率範圍 0.1~105Hz ... 120 圖 6.13 CP- 20Ru Nyquist 圖之模擬,內圖為用來解析所得之實驗數據的等效電 路... 120 圖 6.14 CP- xRu Bode 圖,DC 偏壓+400mV,頻率範圍 0.1~105Hz ... 121 圖 6.15 CP-20Ru 樣品及經過不同熱處理方法後的 CV 圖,掃描述率 20 mV/s 121 圖 6.16 CP-20Ru 樣品及經過不同熱處理方法後的 CRC 圖,電流密度±1 A/g .. 122
圖 6.17 CP-20Ru 與 CP-20Ru-H200 樣品之循環壽命,電流密度±1 A/g ... 122
圖 7.1 實驗用鈦金屬電極尺寸圖... 132
圖 7.2 不同 Ru/Ta 莫耳比的塗佈液所製備的 Ru-Ta 氧化物/Ti 複合電極 CV 圖, 1N H2SO4電解液,掃描速率 20 mV/s ... 132
圖 7.3 7Ru3TaCV 圖,掃描速率 5~100 mV/s... 133
圖 7.4 不同 Ru/Ta 莫耳比的塗佈液所製備的 Ru-Ta 氧化物/Ti 複合電極 V-t 曲線圖, 充放電電流 ±1 A/g... 133
圖 7.5 8Ru2Ta-30 wt%塗佈液 CV 圖, 1N H2SO4電解液,掃描速率 5 mV/s 塗佈 2~8 回 ... 134
圖 7.6 8Ru2Ta-30 wt%塗佈液 V-t 曲線圖圖, 1N H2SO4電解液,充放電電流±1 A/g, 塗佈 2~8 回 ... 134
圖 7.7 塗佈 8 回的 8Ru2Ta 詴樣 SEM 影像(上),EDX 圖譜(下) ... 135
圖 7.8 塗佈 8 回的 8Ru2Ta 詴樣 XRD 圖譜 ... 135
圖 7.9 CV 法製備 PANI/Ti i-V 曲線圖,電鍍液〆0.1M aniline ... 136
圖 7.10 樣品 CV1~CV3 CV 圖, 1N H2SO4溶液,掃描速率 20 mV/s ... 136 圖 7.11 樣品 CV1~CV3 V-t 曲線, 1N H2SO4溶液,充放電電流±1 A/g ... 137 圖 7.12 樣品 PS1~PS5 CV 圖, 1N H2SO4溶液,掃描速率 20 mV/s ... 137 圖 7.13 樣品 PS1~PS5 V-t 曲線圖, 1N H2SO4電解液,充放電電流±1 A/g... 138 圖 8.1 滾壓法製備空氣電極... 151 圖 8.2 滾壓法製備空氣電極... 151 圖 8.3 所合成之 CNCs HRTEM 照片 ... 151 圖 8.4 EVT-GDL(noncatlyted GDE)和分別塗佈 CNCs、XC-72R 及 VGCF 2.52 g/cm2 碳材做為觸媒於 30%KOH 電解液中的 i-V 極化曲線圖,掃描速率 1 mA/sec ... 152 圖 8.5 所合成之 Ag/CNCs 觸媒之 XRD 圖譜 ... 152
圖 8.6(a) 所合成之 Ag/CNCs 觸媒之 SEM 照片 ... 153
XIII
圖 8.7 EVT-GDL(noncatlyted GDE)和塗佈 A g-CNCs、EVT-Mn、EVT-MnCo 及
La0.6CaCo0.4O3等 2.52 g/cm2觸媒於 30%KOH 電解液中的 i-V 極化曲線圖
... 154
圖 8.8 塗佈 A g-CNCs 觸媒 1.26~ 2.52 mg/cm2之空氣電極於 30%KOH 電解液中 的 i-V 極化曲線圖,掃描速率 1 mA/sec ... 154
圖 8.9 A g-CNCs 觸媒塗佈量 6.32 mg/cm2之空氣電極於 30%KOH 電解液中,10 分鐘的定電流放電曲線圖,放電速率 10~200 mA/cm2 ... 155
圖 8.10 以 CNCs-x% PTFE 為觸媒層之空氣電極於 30%KOH 電解液中的 i-V 極 化曲線圖,x 值分別為 10,15,20 及 40。... 155
圖 8.11 以 60%CNCs 為載體,20% PTFE 為粘結劑,添加 20% Ag/CNCs、 MnOx/CNCs 及 La0.6Ca0.4CoO3觸媒材料為觸媒層之空氣電極與 EVT-MnCo、 未添加觸媒之 CNCs-40%PTFE 空氣電極 i-V 極化曲線圖。 ... 156 圖 8.12 以 60%CNCs 為載體,20% PTFE 為粘結劑, 20% Ag/CNCs 為觸媒層, 分別以 BP2000-40%PTFE、XC72R-40%PTFE、CNCs-40%PTFE 及 EVT-GDL 為氣體擴散層之空氣電極的 i-V 極化曲線圖。 ... 156 圖 8.13 BP2000,XC72R 及 CNCsX (其中 X 為表示 CNCs 的比表面積)為載體製 備含 10.7 wt% Ag 的觸媒層之空氣電極的 i-V 極化曲線圖 ... 157 圖 8.14 Ni 網、發泡鎳網及鈦網做為集電網時所得到的 i-V 曲線圖 ... 157 圖 8.15 控制觸媒層中 Ag 的重量百分比 2~10.7%所得到的 i-V 曲線圖,Ag 來源 為 Ag/CNCs 觸媒 ... 158 圖 8.16 控制觸媒層中 Ag 的重量百分比 3~10%所得到的 i-V 曲線圖,Ag 來源為 奈米金屬銀商品... 158 圖 8.17 放電電流密度 200 mA/cm2時觸媒擔量與電極電壓的關係... 159 圖 9.1 超電容器材料二極式測詴槽照片... 169 圖 9.2 超電容器電容量及 ESR 的測詴程序圖 ... 169 圖 9.3 Nesscap-2.7V 100F 商品, 不同電流密度下進行定電流充放電之 V-t 曲線, 幾何面積 200 cm2 ... 170 圖 9.4 Cooper-2.5V 100F 商品, 不同電流密度下進行定電流充放電之 V-t 曲線, 幾何面積 230 cm2 ... 170 圖 9.5 商業化超電容器產品之電容量及 ESR 的測詴程曲線 ... 171 表 1.1 目前世界上主要的超電容器生產製造的廠商及其產品... 18 表 3.1 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 的 N2吸/脫附法所之孔 隙結構參數... 49 表 3.2 不同電壓範圍下 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 由 CV 法測得之比電容值,掃描速率 20 mV/s ... 49
XIV 表 3.3 不同掃描速率下 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 由 CV 法測得之比電容值,電壓範圍 0-1 V ... 49 表 3.4 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 由 CRC 法測得之比電容 值,電壓範圍 0-1 V ... 49 表 3.5 等效電路模擬 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 於開路電 壓下交流阻抗圖之參數表... 50 表 3.6 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 由 CRC 法分別以單位重 量及單位面積為基礎所計算出之比電容值... 50 表 3.7 CNCs、XC72R、BP2000、MWCNTs 和 AC1100 所含官能基 C=C, C-C, C-OH, COOH, C=O, and aromatic ring 之波峰面積比率 ... 50 表 4.1 CNCs 製備條件及其產量及產率 ... 64 表 4.2 於不同氧流量條件所製備之奈米碳膠囊孔隙結構參數... 64
表 4.3 填充四種不同氧流量製備之奈米碳膠囊 TCE 於 1N H2SO4溶液,由不同掃
描速率下所得到的 CV 圖計算所得比電容值 ... 64 表 4.4 AO207 等四種詴樣以 ±1 A/g 的恆電流充放電測詴所得比電容值與 iR drop
等數值... 64
表 5.2 以 CO2改質之 CNCs 樣品所含官能基 C=C, C-C, C-OH, COOH, C=O,
and aromatic ring 之波峰面積比率 ... 85
表 5.3 以 CO2改質之 CNCs 樣品,由不同掃描速率下所得到的 CV 圖計算所得比 電容值... 85 表 5.4 以 CO2改質之 CNCs 樣品以±1 A/g 的恆電流充放電測詴所得比電容值與 iR drop 等數值 ... 86 表 5.5 以 CO2改質之 CNCs 樣品的於開路電壓下交流阻抗圖之參數表 ... 86 表 5.6 以空氣改質之 CNCs 樣品的孔隙結構參數 ... 86 表 5.7 以空氣改質之 CNCs 樣品所含官能基 C=C, C-C, C-OH, COOH, C=O, and aromatic ring 之波峰面積比率 ... 87 表 5.8 以空氣改質之 CNCs 樣品,由不同掃描速率下所得到的 CV 圖計算所得比 電容值... 87 表 5.9 以空氣改質之 CNCs 樣品以±1 A/g 的恆電流充放電測詴所得比電容值與 iR drop 等數值 ... 87 表 5.10 以空氣改質之 CNCs 樣品的於開路電壓下交流阻抗圖之參數表 ... 88 表 5.11 以 HNO3改質之 CNCs 樣品的孔隙結構參數 ... 88
表 5.12 以 HNO3改質之 CNCs 樣品所含官能基 C=C, C-C, C-OH, COOH,
C=O, and aromatic ring 之波峰面積比率... 88
表 5.13 以 HNO3改質之 CNCs 樣品,由不同掃描速率下所得到的 CV 圖計算所得
比電容值... 89
表 5.14 以 HNO3改質之 CNCs 樣品以±1 A/g 的恆電流充放電測詴所得比電容值與
XV 表 5.15 以 HNO3改質之 CNCs 樣品的於開路電壓下交流阻抗圖之參數表 ... 89 表 6.2 CP- xRu 由 CRC 法測得之比電容值,電壓範圍 0-1 V... 112 表 6.3 等效電路模擬 CP-5Ru、CP-10Ru、CP-20Ru 及 CP-40Ru 樣品於 DC 偏壓 400mV 下交流阻抗圖之參數表... 112 表 6.4 不同掃描速率下 CP-20Ru 樣品及經過不同熱處理後由 CV 法測得之比電容 值,電壓範圍 0-1 V ... 113 表 6.5 CP-20Ru 樣品及經過不同熱處理由 CRC 法測得之比電容值,電壓範圍 0-1 V ... 113 表 7.1 Ru-Ta/Ti 二元塗佈液配方 ... 129 表 7.2 CV 法製備 PANI/Ti 實驗條件表 ... 129 表 7.3 局部還原定電位法製備 PANI/Ti 實驗條件表 ... 129 表 7.4 不同 Ru-Ta 莫耳比電極,由各掃描速率的 CV 圖計算所得比電容值 .... 129 表 7.5 不同 Ru-Ta 莫耳比複合電極,以±1 A/g 的恆電流充放電測詴所得比電容值 與 iR drop 等數值,電壓範圍 0-1 V ... 130 表 7.6 詴樣 8Ru2Ta 依塗佈次數比電容值的變化 ... 130 表 7.7 CV 法製備 PANI/Ti 樣品,經由不同掃描速率下所得到的 CV 圖計算所得 比電容值... 130 表 7.8 CV 法製備 PANI/Ti 樣品,以±1 A/g 的恆電流充放電測詴所得比電容值與 iR drop 等數值 ... 131 表 7.9 定電壓還原控制法(750 mV- 0 mV)製備 PANI/Ti 樣品,經由不同掃描速率 下所得到的 CV 圖計算所得比電容值 ... 131 表 7.10 定電壓還原控制法(750 mV- 0 mV)製備 PANI/Ti 樣品,以±1 A/g 的恆電流 充放電測詴所得比電容值與 iR drop 等數值 ... 131 表 8.1 滾壓法觸媒層薄膜配方... 149 表 8.2 空氣電極組成及實驗目的表... 150 表 9.1 超電容器電極配方... 166 表 9.2 BP2000 製備 1×2 cm2尺寸不同 PTFE 含量薄膜電極,組成半電池其電容性 質量測結果... 166 表 9.3 BP2000 製備 1×2 cm2尺寸不同粘結劑配比薄膜電極,組成半電池其電容性 質量測結果... 166 表 9.4 BP2000 製備 1×2 cm2尺寸不同石墨粉添加量薄膜電極,組成半電池其電容 性質量測結果... 167 表 9.5 BP2000 為活性物質所製備 4×5 cm2尺寸單電池其電容性能測詴結果 ... 167 表 9.6 CC-4H 為活性物質(比表面積為 1142 m2 /g)所製備 4×5 cm2尺寸單電池其電 容性能測詴結果... 167 表 9.7 不同比表面積 CNCs 為活性物質所製備 4×5 cm2尺寸單電容器其電容性能 測詴結果... 168
XVI
表 9.8 RuO2〃xH2O/Carbon 為活性物質所製備 4×5 cm2尺寸單電池其電容性能
測詴結果... 168 表 9.9 市售 100F 產品性能表 ... 168
1
第一章 緒 論
1.1 前言
石油的日益枯竭及二氧化碳排放造成溫室效應將是這個世紀所面臨最為嚴 重的能源及環境問題,生存環境遭受文明的破壞與能源的過渡消耗與不當使用息 息相關,人們逐漸感受到能源缺乏及生活環境劣化的警訊。因此,積極開拓無污 染的綠色能源成為同時解決這二項問題的最妥善對策方案。近年來太陽能、風能 開發成為全球性的議題,原因尌在於其具有〞用之不盡,取之不竭〞的特性深深 吸引科學家的矚目,然而在能源的開發過程卻發現仍潛存著許多無法突破的瓶頸, 諸如太陽能與風能的不穩定性和發電成本昂貴等。所以,許多的科學家轉而嚐詴 從提高能源使用的效率的策略來著手,希望將不穩定的再生能源與基載電源加以 整合運用,以期降低對特定能源的依賴程度並降低能源使用成本。在此一領域的 研究中最為關鍵的技術在於電力儲存技術,太陽能與風能分別經過太陽光電池及 風力發電機的轉換成為人們可資利用的電力,經由電網的傳輸供應照明及日常生 活所需,透過電力儲存裝置的設計將使得這些不穩定的再生能源能夠被有效的應 用。 談到電力儲存裝置,當以抽蓄發電為最典型的代表,也是目前世界上最為成 熟儲電技術之一。然而,受到地理環境的限制,可供做為抽蓄發電的廠址相當稀 少,在台灣也僅有日月潭符合抽蓄發電廠址的要件。除了抽蓄發電之外,最被寄 予厚望的當屬蓄電池組,不論是燃料電池或著各種形式的二次式電池都曾是熱門 的研究焦點。二次電池從傳統的鉛酸電池開始,其發展已有百年以上的歷史,在 日常生活之中更是隨處可見。所謂的二次電池即為可再次充電重複使用的蓄電池, 因此理所當然是被考慮用來做為電力儲存裝置的理想選擇,只可惜電池的循環壽 命最高也只有數千個循環次數,再加上瞬間的輸出功率小,成了技術發展上的瓶 頸。超電容器的誕生燃起了無窮的希望,超電容器是一種容許快速充放電的電荷 貯存裝置,經由電荷的重新排列完成充放電的程序,因此理論上具有無窮盡的循 環壽命,此一特性正好彌補了二次電池的缺點。所以,二次電池與超電容的混成 系統(hybrid system) 近年來己然成為電力貯存裝置中新興的焦點。燃料電池是一 種將化學能轉變為電能的裝置,由於其反應過程不涉及燃燒,所以不受到卡諾循 環(Carnot cycle)的限制,因而能量轉化率高。除此之外,反應產物對於環境無污 染、燃料多樣化以及噪音低的優點,是近年來可移動式動力電源和高效能源轉化 裝置發展的重點之一。然而,關鍵材料掌握在少數廠商、需以貴金屬材料為觸媒 以及造價昂貴發電成本高等,造成了無法普及的重要因素。 碳材料具有資源廣、重量輕、耐腐蝕、導熱、導電、價格低以及易加工的2
特點。此外,其形態多樣諸如塊材、薄膜、球形、粉體、絲狀等應有盡有。因而, 其應用性十分廣泛,無論是超電容器、燃料電池還是傳統或先進的二次電池,碳 基材料都扮演著重要的角色。最早出現的超電容器即是採高比表面積的活性碳做 為電極材料,利用其高比表面積與電解液界面形成的電荷貯存空間。在質子交換 膜燃料電池 (Proton Exchange Membrane Fuel Cell; PEMFC) 中也不例外,迄今為 止碳材料普遍應用在 PEMFC 的氣體擴散層、觸媒層以及雙極板中,PEMFC 常 用 Pt 及少量 IrRu 等貴金屬及其合金做為催化劑,由於 Pt 等鉑系貴金屬價格昂貴 資源匱乏,所以高比表面的材料為載體或特殊的催化劑沉積技術使 Pt 微粒高度 分散以減少 Pt 的用量降低電池成本是為重要的研究題材。由此可見,從電極材 料發展的觀點來看,無論是燃料電池(尤其是低溫型的 PEMFC 、AFC 或 DMFC 或者超電容器,碳基材料無庸置疑是為最關鍵的材料之一。 本研究的想法是從碳材的合成為起點,經由表面改質技術進行碳材孔隙結 構的調控,加上觸媒材料的佈植技術製備出合宜的電極材料,最終以塗佈或滾壓 的製程方法研製出應用於超電容器、鹼性燃料電池或者空氣電極的電極元件。研 究的過程以發展快速的粉體電化學量測技術為輔助,進行有效率的碳材、複合材 料的篩選,並製備出各種適合於實驗室研究用的量測模組以得到再現性高的實驗 數據,在研究的後期則以製備出接近於商業化規格的產品,來驗證所開發的電極 材料性能。
1.2 超級電容器概述
超電容器(supercapacitor, ultracapacitor)是一種具有高功率密度的儲電裝置, 也稱為電化學電容器(electrochemical capacitor)。此一儲電裝置具有充放電速率快, 循環壽命長的特質。依儲電機制的不同有電雙層電容器(Electrical Double Layer capacitor; EDLC)及偽電容(pseudocapacitor)兩種型態[1]。電雙層電容器的儲能方 式與傳統電解電容器(electrolytic capacitor)相類似,傳統的電解電容器是由相互帄 行的正負兩個電極與及其中間所充填的介電物質所構成,在兩極之間施加電壓 V 時,在電極的界面可以儲存符號相反的電荷,所儲存的電量與所施加電壓的比值 即為電容值,電容值的大小受到介電物質及電極面積與電雙層厚度的影響,它們 之間的關係如方程式 1-1 所表示。 d A V Q C (1-1) 式中 C 為電容(法拉第),Q 為電量(庫侖),V 為施加的電壓(v), 為介質的 介電常數值(真空的介電常數值為 8.854x10-12 C/V.m,介電常數值愈大表示對電埸 的遮蔽效果愈佳),A 為電極極板的表面積,d 為電雙層厚度。 電雙層電容器大都由多孔性碳電極與電解質溶液所組成,電極表面己不復 以帄行板電容器的模式所能描述,再加上電解質溶液除帶了電荷離子之外也有極 性分子存在,粒子間除了靜電力之外,也存在著分子熱運動及濃度擴散的交互作3 用,所以電極與電解質界面結構比傳統電解電容器要複雜許多。電雙層電容器中 有關電極與電解質間的固液界面結構有多種模型被提出來。最早的電雙層電容的 理論可追溯到 1853 年德國物理學家 Helmholtz 所提出的電雙層模型,Helmholtz 認為固液界面是由電極側的單層電子和溶液側的單層離子構成,所形成的雙電層 結構與帄板電容器類似,電雙層的厚度 d 為電解質離子半徑。電極側的過剩電荷 密度等於溶液側的過剩電荷密度,電荷密度與雙電層所產生的介面電位差成正比, 即 V d q 4 (1-2) 電雙層電容以微分型式表示〆 d V q Cd 4 (1-3) 而電雙層的電位分佈為〆 dx x d ( ) (1-4) 其中 A q 為界面所累積的電荷密度 coulomb /cm2 界面的電位分佈(x)依公式 1-4,邊界條件 at x=0 (x)0及 at x=d 0 ) (x ,解得 d 0 。 由式 1-3 及 1-4 可知,在 Helmhoz 模型中微分電容為一常數,且在電雙層中 電位分佈隨著與電極表面的距離呈線性變化。然而,實驗的結果卻發現,Cd 並 非常數,它隨著電位、電解質濃度相異而改變,追究原因是 Helmholtz 模型僅僅 考慮了靜電引力,而忽略了離子熱運動的影響。後來的學者 Gouy, Chapman, Stern, Bockris, Devanathan, Muller 等人陸續提出了 Gouy-Chapman 模型(考 慮分子熱運動,固液界面的電位分佈隨遠離電極表面呈指數遞減),Stern 模型(同 時考慮靜電作用與熱運動,以吸附離子中心連線所形成的假想面為 Stern 面,在 Stern 面與電極之間為界內層,其厚度約為水合離子半徑。距離體表面約一個分 子或離子直徑大小處稱為滑動面,在滑動面內層為固定層,可視為固體的一部份, 外層為擴散層),以及 BDM 模型(電雙層由緊密層和擴散層所組成,緊密層又可 分為內緊密層和外緊密層。其中內緊密層是由非溶劑化的異號離子緊貼固體表面, 不均勻亦不連續,外緊密層則由溶劑化的水合離子所構成的)。這些模型的提出, 使得電雙層固液界面結構的解析更趨於完善[2] [3]。因此,把電雙層的界面結構 看成是由內層緊密層和外層分散層構成的想法已被普遍接受,所以電雙層電容 Cd是由緊密層電容 C1與分散層電容 C2所串聯而成,可以表示成〆 2 1 1 1 1 C C Cd (1-5) 若在較濃的電解質溶液中,特別是在施加電壓較大的情況下,分散層電容
4 C2相對於大,因而雙電層電容 Cd近似等於 C1。因此,電雙層電容器常以最簡單 的 Helmhoz 模型來表示。另外從式 1-1 可以看出電雙層的厚度及電極表面積是影 響電容值大小的主要因素,由於電雙層的厚度僅為幾個 angstrom ,再加上一般 電雙層電容器是以高比表面積的多孔性碳材做為電極材料,因而可以預見電雙層 電容器儲存能量的能力會比傳統的電解電容器大得多,也因此稱之為超電容器。 又因為電雙層厚度僅有幾個 angstrom,因此充放電非常快速,又因為不渉及氧化 還原反應所以其充放電循環理論上可達到無限多次,唯受限於電極活性物質比表 面積的大小,所以其能量密度不高,所使用的電極材料以高比表面積的活性碳最 為常見圖 1.1 說明多孔性碳材的示意圖及在固液界面的電雙層結構。 實體的電雙層電容器結構是由兩個多孔碳電極與電解液所構成,電極的形 式至少有帄板式以及捲繞成圓筒式兩種。不論帄板式或者圓筒式,從微觀的角度 來看都可以看成是由兩相對的電極與電解液所組成,多孔碳電極雖導電性良好仍 遠遜於金屬材料,為減少電極的極化阻抗縮短電子傳導的路徑,集流體是電極重 要的一部份。電解液可採用固體或液體電解液,也可以是水溶液或者有機溶液。 電雙層電容器通常沒有正、負極性,其工作時的電化學過程可以寫成[4]〆 正極〆ES A ES//A e (1-6) 負極〆ES C e ES//C (1-7) 總反應〆ES ES AC ES//A ES//C (1-8) 式中 ES表示電極表面,//表示雙電層,A-與 C+分別表示電解液中的正、負 離子。圖 1.2 說明了電雙層電容器工作的原理。圖中說明充電前的電雙層電容器 在多孔電極表面帶有微量的正電荷,電解質溶液中帶負電荷的離子受到靜電力吸 引而接近多孔碳電極表面,此時兩個電極之間的電位分佈如左圖下方的分佈曲線, 在固液界面有較大的電位差,且兩個電極的電位接近,槽電壓接近零。當由外部 對電容器施加不使電解液分解的電壓時電子迅速累積於連接到負極的多孔碳電 極表面,接到正極端的多孔碳電極則因電子流向負極端而帶大量的正電荷。因此 電解液中的電荷重新分佈排列,帶負電荷的負極會吸引溶液中的正離子,帶正電 荷的正極會吸引溶液中的負離子,形成兩個雙電層。但需注意的是,在電極與電 解液界面所累積的電荷並不通過固液界面而轉移。此時兩個電極之間的電位分佈 如右圖下方的分佈曲線,且兩個電極的電位產生明顯的差距。隨著外加電壓的提 高差距拉大,其極限值即為電解液的分解電壓。所以電雙層電容器的工作電壓與 電解液的種類有密不可分的關係。在充電時電子是通過外電源從正極傳到負極, 同時電解液本體中的正負離子分開並移動到電極表面,在放電時電子則通過負載 從負極流到正極,正負離子則從電極表面釋放並移動返回電解液中,回到初始的 狀態。 偽電容器是在充放電過程中電極活性物質本身發生氧化還原反應,充電時
5 電解液中帶正電荷的離子以對入的方式與活性物質結合而使其氧化數提高,並產 生法拉第電流,在固定電流大小之下電壓會隨時間而線性昇高類似於純電容的現 象。此種反應機製有別於電池活性物質的氧化還原反應是發生於某一特定的電壓 條件下,故以法拉第偽電容器(psudocapacitor)稱呼,以貴金屬氧化物 RuO2 及導 電性聚合物 poly aniline 為電極材料所製備的電容器為典型的代表。不論是電池 或者電容器,電子的傳導都是經由外接的導線,從高電子密度的一端傳向低電子 密度的一端,而帶電荷離子的傳導則經由電解質受到異種電荷的相吸及同種電荷 的相斥而遷移。這兩者之間的差異僅在於電容器的電荷不通過電極與電解液界面 轉移,只有電荷的重新排列。也因此其能量密度遠低於電池,電池經由物質的轉 換源源不絕的進行電量的貯存,直到活性物質用完為止。 上述兩種超電容器類型中以 EDLC 出現較早,現今市面上的產品也大部份 為 EDLC。有關於 EDLC 的報導最早出現於 H.I. Becker 及他所屬的 General Electric 公司在 1957 年所公開的美國專利資料 (U.S. Patent 2,800,616),至於商業 化產品則到了 1970 年才由 Standard Oil Company of Ohio; SOHIO 所發表。1978 日本電器 NEC 的超電容器產品經 SOHIO 授權下,正式將超電容器產品導入市 場。從此之後 NEC 便積極投入產品的研發製造,1980 年即開始量產 FA 系列的 產品,到了 1996 年 NEC 所推出的 FK 系列超電容器其能量密度已達 600 W/kg, 功率密度也達到 0.75 kW/kg[5]。到了 2000 年時功率密度更提昇到 5-10 kW/kg[6] , NEC 公司在過去的數十年間於超電容器產業佔有重要的地位,其超電容器的品 牌名稱自 2002 年以後以 NEC-Tokin 上市。在日本,除了 NEC 之外松下電器 (panasonic) 早在 1978 年也投入超電容器的製造生產行列,有別於 NEC 的超電 容器以水溶液為電解質,其電極採用塗佈式 (pasted electrod) 的雙極式電池 (Bipolar cell) 設計,松下電器自行開發出以非水溶液電解質/非塗佈式電極的超 電容器”金電容” (goldcap),金電容超電容器的設計有鈕扣型 (coin cell) 及纏撓式 (spiral-wound) 兩種,1980 年代鈕扣型超電容器與太陽光電手錶的搭配深受使用 者的歡迎,主要原因在於超電容器幾無壽命限製所以不僅可在製造手錶時同時植 入,更可方永久不需更換。1990 年代松下電器有大型的超電容器推出,單元電 容耐電壓達 2.3 V 其容量已高達 1500 F。到了 1999 年配合於混合電動車的應用 推出了”UpCAP” ,單元電容耐電壓 2.3V 其容量提昇到 2000F。1990 之後世界上 有許多的廠商陸續投入超電容器製造的產業例如美國 Maxwell、Cooper、蘇聯 ESMA 及韓國 Ness 等,經過 30 幾年來的發展超電容器的生產技術已日趨成熟, 應用的領域為寬廣。表 1.1 整理出目前世界上主要的超電容器生產製造的廠商及 其產品相關資訊,有助於瞭解此一產業未來的發展。
由於 S.Trasatti 等及 B.E. Conway[5]等研究發現 RuO2具有典型偽電容的行為
特徵且比電容量為同樣比表面積碳材的 10~100 倍,導電率高兩個量級。而開啟
了偽電容器的發展。但以 RuO2做為電極活性物質的超電容器製品直到 1988 年
才於美國 Pinnacle 研究院被製造出來,當時是以”PRI Ultracapacitor”命名。但由 於材料昂貴,所以目前大多於航空、國防領域的應用。
6
1.3 電雙層電容器材料的研究
電雙層電容器所使用的電極材料以碳基材料居多,碳基材料具有優良的導 電性能,質量密度低重量輕,製造成本相對低廉,比表面積大且具有可調變性是 主要的因素。再加上碳基材料產品依製造方法的不同可以是粉末、塊材[7]、纖 維狀或編織成布等多樣形態。單以碳元素的結構又有鑽石(diamond, sp3)石墨 (graphite, sp2)、直鏈乙炔碳(carbyne, sp1)、富勒烯(fullerenes, sp2)等結構[8], 也可以經後續的處理程序而產製石墨化程度不同的材料或者在表面接枝官能基, 故被廣泛應用於電化學、水處理及吸附劑領域。理論上來說,碳電極材料的比表 面積越大,EDLC 的比電容越高,但實際的研究發現比電容與其比表面積並沒有 良好的線性關係[9-11],主要的原因在於碳電極材料中孔洞的大小並非完全一致, 孔隙結構及表面形貌均有所不同。依照 IUPAC 的分類把孔徑小於 2 nm 孔洞稱為 微孔 (micropores),大於 50 nm 以上的稱為巨孔 (macropores) ,界於 2~50 nm 的 孔洞稱為中孔(mesopores )[12]。對大多數的多孔碳材料而言,巨孔所提供的比表 面積通常是低於 2 m2/g,相較於中微孔的比表面積幾乎可以忽略不計,所以常把微孔所佔的比表面之外的比表面積稱之為外比表面積(external surface area )。 Hang Shi[9]以活性碳及活性碳纖維為對象發現活性碳中,微孔對於電容值的貢獻 約在 15~20 μF/cm2範圍,所得到的結果與在潔淨的石墨表面所量得的電雙層電 容值約為 20 μF/cm2相吻合,而外比表面積對於電容值的貢獻則與碳材種類、孔 隙結構和表面形貌有很大的關係,尤其碳材的種類不同甚至會有好幾倍的差距, 同時也提出水合離子無法進入小於 0.5 nm 的孔洞之內的看法。所以在研究電雙 層電容器的領域中,研究人員不僅駑力於探索比表面積的提昇技術,對於新碳材 的發展更是不乏遺力。現今已知可使用來做為電雙層電容器活性材料所的碳基材 料大致上即有活性碳粉、碳黑、活性碳纖維、碳氣凝膠和奈米碳管等多種。
1.3.1 活性碳(Acitvated Carbon)
有關於活性碳做為 EDLC 電極材料的研究一直以來都受到許多研究人員關 注,活性碳不僅是雙電層電容器最早採用,也是使用最多的電極材料。最重要的 原因是製備活性碳的原料來源相當豐富,製備技術成熟完善。依照所使用的原料 可分瀝青(石油或煤焦)、天然植物纖維(竹子、木材、椰殼)及高分子聚合物(主要 為酚醛樹酯)三大項。雖然使用的原料不同,但製備方法相近。一般而言,活性 碳製備程序至少包括乾燥、碳化、活化、研磨、篩分等。在乾燥程序中,溫度通 常在 100 ℃附近維持數小時到數天的時間,主要的目的在於去除原料中所含的 水份。碳化程序通常在惰性氣體保護下,於 700~1000 ℃溫度持溫數個小時, 在碳化過程中原料中除了碳以外的元素如氫、氧均被分解殆盡,只留下碳元素。 活化是影響活性碳孔隙結構與表面形貌最重要的歩驟,其目的是調整活性碳粉的7
物理、化學性能諸如表面官能基的接枝或者比表面積的提昇。常用的活化方法有
物理活化和化學活化兩種,前者是在高溫條件下以 CO2,空氣或水蒸汽為活化劑
[13-14],後者則以 ZnC12,KOH 或 HNO3等為活化劑[13-14]。瀝青(pitch)是製備
活性碳的主要原料之一,較為常用的有煤焦瀝青和石油瀝青兩種。煤焦瀝青是煉 焦的副產品,即焦油蒸餾後殘留在蒸餾釜內的黑色物質,石油瀝青是原油蒸餾後 的殘渣。Adachi 等[15]以石油瀝青為原料製備出比表面積 2500~3000 m2/g 活性碳, 但以其為電極材料的 EDLC 的性能並不理想。T.C.Weng 等[16]以煤焦瀝青為原料, KOH 為活化劑,製得了比表面積達 2860 m2/g 的活性碳,在 lmo1/l H2SO4溶液中 其比電容僅約 130 F/g,M .Endo 等[17]以中間相瀝青為原料,採用與 Weng 等人 近似的方法製備活性碳,其在 Et4NBF4/ PC (四氟硼酸四乙基銨/碳酸丙烯酯)溶液 中,比電容 141 F/g。除了瀝青之外,粗纖維的天然植物常用來製備活性碳,Wu 等[18]以木材為原料,採用 3 ml/min 流量之水蒸汽為活化劑於 900 ℃處理 7 小時 製得比表面積 1131 m2/g 的活性碳,在 l mo1/l H2SO4溶液中其比電容約為 142 F/g。 Rufford 等[19-20]分別以甘蔗渣及加啡豆渣為原料,採用 ZnCl2做為活化劑製得 比表面積分別是 1788 m2/g 和 1019 m2/g 的活性碳,在 l mo1/l H2SO4溶液中分別 測得比容量高達 300 F/g 和 368 F/g,作者認為由於孔隙結構及表面官能基衍生的 偽電容效應,使其比電容值比典型的活性碳在有機電解質中約 100~120 F/g ,在 水溶液電解質中的比電容可達 150~300 F/g[20-22]來得高。高分子聚合物做為碳 源具有較高純度、碳化灰燼少、及孔隙結構的控制範圍寬廣等的優點,所以也是 製備活性碳相當重要的原料。H.Teng 等[23]以合成酚醛樹脂為原料,KOH 為活 化劑,700 ℃活化 2h 製備得到比表面積 1900 m2/g,的活性碳,但在 1 mol/l H2SO4 溶液中其比電容僅有 100 F/g。 Zhang 等 [24]以間苯二酚和甲醛為原料以六亞甲 基四胺(Hexamethylenetetramine; HMTA)為架橋劑,在 R/ HMTA 莫耳比為 1/50 時
製得的聚合物碳化後,未經任何活化即可得到比表面積 825 m2/g 的碳材,於在 30% KOH 溶液中測得比電容值達 200 F/g。活性碳材料與電雙層電容器的發展有 著密不可分的的連動性,新型高比表面積活性碳材料的發展可從多個角度來著手, 不論是從原料或製備條件優化都能得到高比表面積、孔隙結構合理,適用於電雙 層電容器的材料,使其於儲能領域的應用愈臻於成熟。
1.3.2 碳氣凝膠(Carbon Aerogel)
碳氣凝膠是廿世紀八十年代末期新發展的無定形多孔碳基材料,具有獨特 的連續 3-D 網絡結構。R.W Pekala 等[25-26]首先採用間苯二酚 (resorcinol) 和甲 醛 (formaldehyde) 為原料,以 Na2CO3為催化劑,兩者在鹼性環境下發生縮聚合 反應,於是形成間苯二酚一甲醛有機凝膠(RF 有機凝膠) 其反應方程式如下所 示。8 為了維持網狀結構不會在乾燥過程中,因毛細力與表面張力的作用而使得 3-D 網 絡崩解,RF 有機凝膠在乾燥之前採用表面張力低的溶劑如丙酮來置換有機凝膠 中所含的水份,再採用 CO2超臨界乾燥法脫除有機凝膠孔隙內的溶劑製得 RF 氣 凝膠,最後於惰性氣體保護下碳化而得到碳氣凝膠。由於碳氣凝膠具有比表面積 高、密度變化範圍廣、孔隙結構可調整、導電率高,也可以製成塊材等特點,非 常適合做為電極材料。有關於碳氣凝膠於電雙層電容器的應用研究方向大致上從 反應物配比、催化劑濃度和熱裂解溫度等方面著手,以探討碳氣凝膠的反應機制 和對孔隙結構的影響。RF 系凝膠一直都是碳氣凝膠的主流且 RF 的莫耳比大致 上均維持 1:2 的比例[25-26],使用最多的催化劑為 Na2CO3。因此,固定了 RF 莫 耳比及所使用的催化劑種類之後,孔隙結構與表面形貌則經由催化劑濃度及反應 物濃度等兩項參數來調控。Scherdel 等[27]研究發現,RF 碳氣凝膠在反應物濃度 低於 25% 時,隨著催化劑濃度(即 R/C,間苯二酚與催化劑莫耳比)變化,碳材 組織形貌有很大的不同,圖 1.3 說明這兩個參數調配與碳材組織形貌的關係。 除了在催化劑及反應物濃度調整之外,也透過活化處理尋求更廉價的原料、 或者更簡化的乾燥方法來製備碳氣凝膠,以縮減製程降低成本的方向做努力。 Zhu 等[28]以 KOH 為活化劑進行碳化後 RF 有機凝膠的活化,在 RF 與 KOH 質
量比為 1:4 時碳材比表面積由 522 m2/g 提高到 2760 m2/g,孔容積從 0.304 cm3/g
提高到 1.347 cm3/g,做為電雙層電容器材料時在 30% KOH 溶液中充放電條件 1
mA/cm2測得比電容達 294 F/g。Zhu 等[29]也另外以廉價的苯酚(phenol)與甲苯酚
(cresol)同分異構物的混合物和甲醛為原料,KOH 為觸媒,在常壓下乾燥製備出 CmF 碳氣凝膠,經以 900 ℃的 CO2活化 2 小時後得到比表面積由 245 m2/g 提高 到 1418 m2 /g,以其做為電雙層電容器材料時在 30% KOH 溶液中充放電條件 1 mA/cm2測得比電容達到 146 F/g。由於碳氣凝膠是經聚縮合反應所製備,在形成 固體形態之前先驅物質具有流動性。因此,許多的研究人員在製備過程中嚐詴引 入各種類形的支撐物來提昇其機械強度。R. Petricevic 等[30]和 C. Schmitt 等[83] 分別在製備 RF 有機凝膠的先驅物質的溶液中添加纖維和碳布以提高有機凝膠的 機械強度,使得有機凝膠在乾燥過程中有承受毛細力與表面張力作用的能力。因
9 此,在常壓之下進行乾燥時能保有原有機凝膠的 3-D 網結構不被破壞。以常壓乾 燥取代超界乾燥是近年來製備碳氣凝膠重要的課題,Li 等[31]在控制適當的 R/C 之下以常壓乾燥條件製得的碳氣凝膠在 6M KOH 溶液中測得比電容值達 183.6 F/g。佈植具有偽電容特性的的金屬氧化物於活性碳材料中,以製成複合活性電 極材料的方法,也同樣地被應用在碳氣凝膠材料。R.W Pekala 等[26]將製備所得 的碳氣凝膠再經化學蒸氣佈植(chemical vapor impregnation)奈米釕金屬化合物,
所得的複合材料做為超電容器的材料,於 1.0 M H2SO4電解質中以 CV 法測得其 比電容值由原來的碳氣凝膠的 95 F/g 提高到 268 F/g (50% Ru)。碳氣凝膠的製備 參數對其組織形貌及孔隙結構影響甚鉅,連帶其電雙層電容比容量也有很大的差 異性。目前,在市面上已可以買到以碳氣凝膠製備 EDLC 的商品,但是仍還是 受到碳氣凝膠製備程序複雜,製備時間長等因素的限制,成本較高。
1.3.3 碳黑(Carbon Black)
碳黑(carbon black)是在缺氧的氣氛之下,以碳氫化合物為原料,經不完全燃 燒後所得到的產物,其外觀為細微的黑色粉末。傳統的碳黑材料主要的用途是做 為橡膠的添加劑,目的在於增加橡膠製品之硬度、抗張強度及耐磨性。因此,在 輪胎製造業中使用最多。工業上碳黑的製造方法可分為熱分解法及不完全燃燒法 兩種,乙炔黑(Acetylene Black)是採不完全燃燒法製備的典型代表,是以乙炔氣 體做為為碳源,控制燃燒條件於缺氧氣氛,製得碳黑產品。爐黑(Furnace Black) 是經由熱分解法來製備的碳黑產品,是採用原油蒸餾後留在蒸餾塔底的重質油或 鋼鐵廠於高爐煉鋼過程中之燒結煉焦所產生的雜酚油(creosote oil)在高溫爐或電 弧爐熱裂解而得到。目前市售的碳黑有 95% 是來自爐黑,主要因其生產效率高、 可連續生產以及製程的控制容易等。 近年來,碳黑於觸媒工業及電化學領域的應用逐漸受到關注,除了做為氫 燃料池[32]、金屬-空氣電池[33-34]、及鋰離子電池的電極材料[35]之外,也被用 來做為電雙層電容器的電極材料[36]。用之於電池或電容器,碳黑除了做為結構 材料也可做為觸媒的支撐材料(supporting material),最主要的原因在於其比表面 積高、質量輕、導電率高以及表面具有含氧官能基團的特性,其中表面官能基團 的存在會影響碳黑的物理化學性質,諸如親水性、導電性及安定性等。典型碳黑 的導電率約在 10−1 ~ 102 (Ω cm)−1範圍[8],由於碳黑微粒間是以群聚方式糾結, 因此碳黑的導電率不僅受到材料本質導電性的影響也受到粒子間距離的影響,故 以其為材料的電極在製備時,碳黑的負載(loading)量是影響導電率的重要因素之 一。碳黑的使用量過低時,由於群聚的粒子團間的距離過大,將使得電子無法躍 過其間距(gap),使得所製備的電極導電度差。隨著碳黑用量提高,當達到一適 當值時電極的導電度會有顯著提昇,此即為最低臨界值。商品化的碳黑其 BET 比表面積範圍廣泛 10~1500 m2/g (例如 Cabot 所生產的 Black pearls 2000 其比表
面積即達 1475 m2
10 聚粒子團之間的孔隙,所以一般認為會比其它高比表面積的碳材更容易讓帶電荷 的離子接觸,非常適合於做為電雙層電容器的材料,以之做為電雙層電容器的活 性材料時,其比電容可達 250 F/g,相當於單位面積電容值在 10~16 μF/cm2[37]。
1.3.4 奈米碳管(Carbon Nanotube)
奈米碳管是一種由單層或多層石墨層捲曲而成的中空管狀碳基材料,具有高 表面積和良好的導電特性。理論上來說,奈米碳管比活性碳材料更適於做為電雙 層電容器的材料,其原因在於活性炭材料中大量的微孔是形成高比表面積的主要 來源,但對於電雙層電容量基本上沒有多大的貢獻,因而限制了其電容量。然而, 奈米碳管的孔洞是由石墨層與石墨層之間或者中空的內管所形,孔徑尺寸大多落 在 2-5 nm 的範圍,是屬於對電雙層電容最具有貢獻度的中孔範圍,使得碳表面 積的利用率提高許多,再加上石墨化程度高具有遠優於活性碳材料的導電度,所 以被認為是電化學電容器的理想電極材料。一般而言,單壁奈米碳管通常成束存 在、管腔的開口率低、以及管徑狹長。所以,若做為電雙層電容器的材料時,其 比表面積使用率低,相應地降低比容量。而多壁奈米碳管內徑約在 4-6 nm,外 徑約在 15-30 nm 且開孔率較高,故多壁奈米碳管更適合做雙電層電容器的電極 材料[38-39]。Frackowiak 等[40]以鈷為觸媒,乙炔為碳源製得 BET 比表面積 411 m2/g 的多壁奈米碳管,以其製成 EDLC 電極於 6M KOH 電解質溶液中測得到比 電容約在 70 F/g,經過硝酸純化處理可提高到 120 F/g。與導電性聚合物如聚苯 胺(polyaniline,PANI)、聚吡咯(polypyrrole,PPy)、以及聚乙烯基雙氧噻吩 poly-3,4 -ethylenedioxythiophene,PEDOT)等製成複合電極時於 1 mol/l H2SO4比電容則可 達 100~330 F/g[41]。Niu 等[42]所製備表面積為 430 m2/g 的奈米碳管薄膜電極在 38 wt % H2SO4電解質溶液中於頻率 1Hz 和 100 Hz 時量得比電容值分別為達到 102 和 49 F/g。截至目前的文獻資料,顯示以奈米碳管做為電雙層電容器材料於 水溶液中可得到的最高比電容值約為 180 F/g[22]。Liu[43]等以電弧法製備 SWCNTs,在 6 M KOH 溶液中再以電化學氧化方法來控制電極電位於 1.5 V,經 歷 24 小時後,所製備的 SWCNTs 其表面積由原來的 46.8 m2/g 提高到 109.4 m2/g, 在 6 M KOH 溶液中比電容提高了 3 倍達到 60 F/g。Ye 等[44]研究了在 0〃2 M HNO3中對多壁奈米碳管以掃描速率 50 mV/ s 在電壓範圍 1~2 V 之間做多次掃描 來進行電化學氧化,探討此一方法對雙電層電容器性能的影響。結果發現電化學 氧化後的 MWCNTs 其比電容顯著的提高,在 1 mol/l H2SO4中比電容可達 335 F/g, 超過了一般的 MWCNTs 10 倍以上,他們認為是 MWCNTs 管端的開管所致。雖 然雖然有關於奈米碳管在電化學電容器中的應用研究一直很熱門,也獲致不錯的 結果。然而,仍舊存在製造成本高、價格昂貴及管端開孔率不高等問題尚待克服, 目前在市面上也尚未有以奈米碳管做為電雙層電容器的產品問世。11
1.3.5 碳基材料的活化與改質
以超電容器為應用端而發展的碳基材料,不論是做為 EDLC 的電極材料亦或 是複合電極的基底材料(supporting material),碳材的比表面積、孔隙結構以及表 面官能基的性質,對其電容性質有著決定性的影響。因此,除了開拓新型的碳基 材料之外,利用活化與改質的方法來提昇比表面積,調整孔隙結構,控制表面官 能基團的種類與數量也是項備受關注課題。碳基材料的活化技術源始於活性碳的 製備技術,依據碳源的不同採用不同的活化劑來得到比表面積更大、孔徑分佈更 合理的活性炭產品。在活化初始期,活化劑與碳材原料之間發生化學反應形成大 量的孔隙,隨著反應時間的進行初始孔隙進一步擴展,最後是孔隙間的合併與連 通。這樣的反應過程使得比表面積的尺度及孔徑分佈是可經由實驗的設計來進行 調控的。改質通常伴隨活化程序而進行,碳基材料的改質主要發生在碳表面,碳 原子與氫、氧、氮原子結合產生官能基團。 活化的方法大致可分成物理活化法(有時也稱為熱活化法,thermal activation) 和化學活化法兩種。其中,物理活化法即是在高溫下用以氧化性氣體如二氧化碳 [45]、水蒸氣[18]等對經碳化的材料進行活化,由於碳基材料的碳化溫度通常在 700~1000 ℃溫度範圍,所以碳化之前必然經歷諸如 200 ℃以下的脫水反應,200 ℃~400 ℃的碳氫化合物分解反應,400~700 ℃的氧鍵斷裂反應,以及 700~1000 ℃的去氧反應等反應階段。因此,碳材原料無論是具直鏈狀分子脂肪烴還是具環 狀分子的芳香烴物質,大多數都會形成具三維網狀結構的炭化物。由於碳化及返 回到室溫的過程之中內部細孔組織組容易被堵圔或者表面吸附其它物質,所以其 比表面積較低。活化的目的即是將吸附物質去除並使被堵圔的細孔開放,並通過 進一步活化,使原來的細孔和通路擴大。隨後,再經由炭質結構中高反應活性位 置的選擇性氧化而形成了孔隙組織。化學活化法,通常是將原料與活化劑溶液混 合,攪拌,加熱升溫。混合後的原料經乾燥後置於高溫爐中加熱,進行熱分解反 應。熱分解之後所得到的產物冷卻之後,以去離子水反覆清洗來去除活化劑。在 化學活化法中,廣泛採用的活化劑有氯化鋅、磷酸、硫酸、氯化鈣、氫氧化鈉、 氫氧化鉀等對原料有脫水作用或浸蝕作用的化學詴劑。這些活化劑對原料的作用 各不相同,但共同的特點是添加這些活化劑,可以使原料中的碳氫化合物中所含 有的氫與氧以水的形態分解脫離,能顯著地降低碳化溫度,除去活化劑以後所得 到的活性碳便具有高比表面積,以含有特殊表面官能基團具有其獨特的性質。1.4 偽電容器材料的研究
偽電容器儲電機制與電雙層電容器不同,從反應的機制來看與電池的貯電 模式接近,但從充放電的 i-V 曲線型態來看卻又與電雙層電容器或傳統電解電容 器相仿。所以從材料的觀點而言,適於做為偽電容器的材料自成一個獨特的系統。12 部份的金屬氧化物及導電性高分子材料是理想的偽電容器材料,其中氧化釕及聚 苯胺是此一系統中最為典型的代表。
1.4.1 金屬氧化物材料
偽電容器儲電機制是渉及法拉第反應的氧化還原反應,因此物質與電能的轉 換必頇滿足庫侖定律。以結晶態 RuO2的電極反應過程[21]〆 2 ( ) 2 H e RuO OH RuO (1-9) 由上式可知δ的最大值為 2,也尌是說 Ru 是從氧化數Ⅳ還原成為Ⅱ。因此 在工作電壓為 1 V 時 RuO2的比電容量可依下式計算得知〆 ) / ( 1450 1 133 2 96500 2 g F V m Q C RuO S (1-10) 當活性物質為水合氧化釕 RuOx〄nH2O 時其電極反應過程則為[46]〆 a b b a OH H e RuO OH RuO ( ) ( ) (1-11) 所以在水溶液中以氧化釕做為電容器材料其理論比容量值高達 1450 F/g。但 實際上卻很難達到,主要的原因可能是由電解液不易進入電極內部,質子反應只能在 RuO2表面進行,使得 RuO2的使用率受到限制所致。Hu 等[47]以鈦金屬板
為基材,在含釕氯化物溶液中於-200~1000mV 範圍以掃描速率 50 mV/s 條件進行 循環伏安電沉積法,所製備出的 RuO2〄nH2O 在 0.5 mol/ dm3 H2SO4溶液中測得 比容量為 103.5 F/g。Hu 等[48]也以鈦金屬板為基材,在含 10 mM RuCl3·xH2O 電 鍍液中添加醋酸鈉並控制於 1 V 定電壓進行電化學沈積的方法製備 RuO2〄nH2O, 除電化學沈積之前以水熱法預處理塗佈薄膜 RuO2〄nH2O 外,在進行比電容量測 之前先經過 2 小時 200 ℃的退火處理,結果在 0.5 M H2SO4溶液中測得比容量為 760~505 F/g,比容量的大小隨著沈積厚度增加而減少。Hu 等[49]更進一歩以 AAO 為模板以電化學沈積方法製備出 RuO2〄nH2O 奈米管其比電容量達 740 F/g ,當 以 200 ℃退火處理 2 小時後甚至高達 1300 F/g 。截至目前,文獻所報導的資料 之中 RuO2〄nH2O 最高比電容值為 1580 F/g,同樣是來自於 Hu 等[46]的研究結 果,他們以活性碳混合 10 %用 sol-gel 法製備的 RuOx〄nH2O 為電極活性材料不
鏽鋼集流体鍍金為基材製備 AC–RuOx/RuOx/Au/SS 電極,中間層的 RuOx是來自
於水熱法製備的薄膜材料。Zheng 等採用 sol-gel 法[50]制各的無定形 RuO2〄nH2O
比容量達到 768 F/g。一般而言,RuO2做為偽電容器材料以無定形態 RuO2〄nH2O
遠優於結晶態的 RuO2,主要的原因是電解液易於進入電極內部,氫離子可經由
擴散而到電極內部而發生氧化還原反應,使得材料的利用率提高。只是無定形態
RuO2〄nH2O 的導電度較差,所以通過適當的退火後處理可提高導電度並得到奈
13
的價格而倍受部份研究者所觀注,Chou 等[51]利用電化學沉積的方法在鎳片上製
備出似楊桃形狀結構(carambola - like) 的納米級γMnO2薄膜,在 0.1 mol/l Na2S04
電解質溶液中於電流密度 l mA/cm2時測得比容量可達到 240 F/g。Li 等[52]以共
沈澱法製備 MnO2〄xH2O/炭氣凝膠複合電極,在 MnO2〄xH2O 質量百分比 60 %
時,比電容量達到 226.3 F/g, 2 倍於單純炭氣凝膠電極的比電容值 112 F/g。
Subramanian 等[53]以 MnSO4 and 和 KMnO4為原料,在 140 ℃恆溫 6 小時的
水熱法製備條件下,得到比表面積 132 m2 /g 的奈米級 MnO2在 1 M Na2SO4 電解
質溶液中測得比電容值可達 168 F/g。Patil 等[54]以化學沈積方法製備 NiO 奈米
結晶薄膜,在 2 M KOH 電解質溶液中測得比電容為 167 F/g 。Wu 等[55]以 SnO2
為基底再以化學電鍍的方法製備出 SnO2-Fe3O4電極,在 1M Na2SO4溶液中於掃 描速率 50 mV/s 條件下,測得比電容值為 33 F/ g。